

Abstract
Rubinstein-Taybi syndrome (RSTS) is an autosomal dominant genetic syndrome characterized by distinct facial features, broad thumbs, growth restriction, microcephaly, intellectual disability, and developmental delay. Pathogenic variants in both CREBBP and EP300 have been associated with RSTS. Here we present a case of a female with hyperinsulinism and features consistent with RSTS, found to have a pathogenic variant in EP300. While there have been a few rare case reports of hyperinsulinism in RSTS, we suggest that hyperinsulinism might be a more prominent feature in EP300 variant RSTS than previously recognized.
Keywords: EP300, hyperinsulinism, hypoglycemia, Rubinstein Taybi
1 |. INTRODUCTION
Rubinstein-Taybi Syndrome (RSTS) is an autosomal dominant genetic syndrome characterized by distinct facial features, broad thumbs, growth restriction, microcephaly, intellectual disability (ID), and developmental delay (Rubinstein & Taybi, 1963). Other features commonly seen in RSTS include congenital cardiac disease, renal anomalies, hearing loss, and eye anomalies (Hamilton et al., 2016; Solomon et al., 2015). While a pathogenic variant is only identified in about 63% of individuals with suspected RSTS, pathogenic variants in both CREBBP and EP300 have been associated with RSTS. Pathogenic variants in CREBBP account for around 55% of cases, while pathogenic variants in EP300 account for about 8% of cases of suspected RSTS (Hamilton et al., 2016). Although CREBBP and EP300-mediated RSTS have significant phenotypic overlap, facial features are less striking and neurodevelopmental outcomes are more typical in EP300-mediated disease, with most children displaying mild to moderate intellectual disability and some exhibiting borderline or low-normal intellectual ability (Hamilton et al., 2016).
Hyperinsulinism has only been previously reported in a few rare case reports of individuals with molecularly confirmed RSTS, the majority of whom have EP300 variant RSTS (Costain, Kannu, & Bowdin, 2018; Welters, El-khairi, Dastamani, et al., 2019). Hyperinsulinism is the most common cause of persistent hypoglycemia in infants and children (Rosenfeld, Ganguly, & De Leon, 2019). There are currently at least nine known monogenic forms of hyperinsulinism (ABCC8, KCNJ11, GLUD1, GCK, HADH, UCP2, HNF4A, HNF1A, MCT1), in addition to several syndromic forms (Rosenfeld et al., 2019). The most common syndromic forms include Beckwith-Wiedemann syndrome, Kabuki syndrome, Turner syndrome, and various congenital disorders of glycosylation (Rosenfeld et al., 2019). Molecular diagnosis is important to predict therapeutic options including responsiveness to medical treatment, yet up to 50% of individuals do not currently have a genetic etiology of their hyperinsulinism identified (Kapoor et al., 2013). The genetic basis for syndromic forms of hyperinsulinism may be well understood, but the mechanisms responsible for hyperinsulinism remain unknown (Rosenfeld et al., 2019).
Here we present a case of a female with hyperinsulinism and features consistent with RSTS, found to have a pathogenic variant in EP300, consistent with a diagnosis of RSTS. These cases illustrate that EP300 variant RSTS should be considered in individuals with hyperinsulinism for whom another genetic etiology is not identified as the facial features can be less striking and neurodevelopmental outcomes can be less severe in EP300 variant RSTS compared to other forms of RSTS. Although hyperinsulinism has been reported in rare case reports of RSTS, hyperinsulinism is not yet considered to be a known association. With these cases, we suggest that hyperinsulinism might be a more prominent feature in EP300 variant RSTS than previously recognized.
2 |. CLINICAL REPORT
A 19-month-old female born at a gestational age of 27 weeks and 1 day was referred to Genetics for persistent hyperinsulinism and dysmorphic facial features. Her medical history was notable for persistent hypoglycemia and an atrial septal defect, as well as several sequelae of extreme prematurity, including chronic lung disease, bilateral grade I intraventricular hemorrhages, retinopathy of prematurity, and swallowing difficulty requiring gastrostomy tube feeds.
2.1 |. Birth history
She was born at a gestational age of 27 weeks and 1 day weighing 794 g (30th percentile for 27 weeks gestation) following an otherwise uncomplicated pregnancy. She was conceived via in vitro fertilization with a sperm donor. The mother presented in spontaneous preterm labor. The neonatal course was complicated by chronic lung disease of prematurity requiring a prolonged neonatal intensive care unit hospitalization. She was discharged home on oxygen, which she was weaned off of several months later. Of note, there were multiple documented low plasma glucose concentrations (plasma glucoses concentrations ranging in the 40 mg/dl range shortly after birth and as late as at 2 months after birth) during the neonatal intensive care unit stay, which were managed with continuous feedings. An evaluation for a persistent hypoglycemia disorder was not pursued and neonatal hypoglycemia was not listed among her discharge diagnoses.
2.2 |. Hypoglycemia evaluation
Following discharge from the neonatal intensive care unit, hypoglycemia was first recognized at 10 months of age. At that time, she presented with acute gastroenteritis and severe hypoglycemia with a plasma glucose of 29 mg/dl. The hypoglycemia was attributed to the acute illness and not investigated. The infant was discharged home and parents were instructed on how to check her glucose levels if she presented any symptoms of hypoglycemia. Of note, although this child had a gastrostomy tube, she did not have a fundoplication as a potential cause for hypoglycemia. Two months later, at age 13 months, she was admitted with hypoglycemia again in the setting of persistent vomiting, diarrhea, and fever. On admission, her plasma glucose was 13 mg/dl. There were limited laboratories obtained at the time of presentation, but of note, a bicarbonate level was 23 mEq/L and urine ketones were large. The local endocrinology team was consulted and a diagnostic fast was performed. A critical sample obtained when the plasma glucose was 39 mg/dl was remarkable for inappropriately suppressed betahydroxybutyrate (0.61 mmol/L) and free fatty acids (0.83 mmol/L), an appropriately elevated cortisol (16.8 mcg/dl), a normal ammonia (13 μmol/L), a normal acylcarnitine profile, and a low growth hormone (4.3 ng/ml). Insulin and C-peptide were not measured. Growth factors were measured and were appropriate for age (IGF-I 56 ng/ml and IGFBP-3 1990 ng/ml). The pituitary gland was reported to be normal in morphology and signal intensity on an MRI. Based on the low growth hormone level during hypoglycemia, a diagnosis of growth hormone deficiency was made. She was discharged home on a regimen of uncooked cornstarch. Growth hormone was prescribed at a follow-up outpatient appointment. She continued to have episodes of hypoglycemia and was admitted for additional evaluation. A glucagon stimulation test was done when her glucose was 54 mg/dl and 10 min later her glucose increased to 142 mg/dl. The diagnosis of hyperinsulinism was made and the patient was started on treatment with diazoxide 3 mg/kg/day. Because of persistent hypoglycemia, the patient was referred to our institution for a second opinion. Diazoxide was discontinued and a diagnostic fast was performed 4 days later. Plasma glucose was maintained >70 mg/dl for 15 hr, the fast was terminated at 18 hr when plasma glucose was 48 mg/dl and betahydroxybutyrate was 1.2 mmol/L. Critical laboratory testing obtained during the diagnostic fast is shown in Table 1. Insulin was undetectable, C-peptide was 0.4 ng/mL, IGFBP-1 was 106 ng/ml, ammonia was 13 μmol/L, cortisol was 7.1 mcg/dl, free fatty acids were 0.8 mmol/L, and lactate 1.8 mmol/L. In response to 1 mg IV of glucagon, plasma glucose increased from 47 to 109 mg/dl within 40 min. An oral protein tolerance test was performed, which demonstrated protein-induced hypoglycemia with a nadir glucose of 45 mg/dl and a peak insulin of 42.4 uU/ml. Because she was able to fast for 12 hr without hypoglycemia, diazoxide was not restarted. Feedings were modified to be administered over 2 hr.
TABLE 1.
Critical sample during diagnostic fast
| Parameter | Value in critical sample | Expected value in the setting of hypoglycemia | Diagnostic value for hyperinsulinism |
|---|---|---|---|
| Glucose (mg/dl) | 48 | <50 | <50 |
| Bethydroxybutyrate (mmol/L) | 1.2 | ≥2 | <1.8a |
| Free fatty acids (mmol/L) | 0.8 | ≥1.7 | <1.7 |
| Insulin (uU/ml) | Undetectable | Undetectable | Above the level of detection |
| C-peptide (ng/ml) | 0.4 | <0.5 | ≥0.5 |
| IGFBP-1 (ng/ml) | 106 | >110 | ≤110 |
| Growth hormone (ng/ml) | N/A | ≥7 | N/A |
| Cortisol (mcg/dl) | 7.1 | ≥10 | N/A |
Note: Reference: Ferrara, Patel, Becker, et al. (2015).
100% sensitivity and specificity.
A hyperinsulinism gene panel sent to Prevention Genetics, which included the following genes: ABCC8, APPL1, BLK, GCK, GLUD1, HADH, HNF1A, HNF1B, HNF4A, INS, KCNJ11, KLF11, NEUROD1, PAX4, PDX1, PGM1, SLC16A1, and UCP2, was negative. She had not had any other genetic testing.
2.3 |. Genetics evaluation
During genetics evaluation at 19 months chronological age and 16 months adjusted age for prematurity, she was crawling and pulling to stand, but was not yet walking. She sat independently at 11 months chronological age, 8 months adjusted age for prematurity. She would babble and had a few words. She was receiving physical therapy and was in the process of getting occupational therapy and speech therapy. Family history was non-contributory. Her biological mother had no significant past medical history. She was born via the assistance of a sperm donor who had a normal karyotype and no chronic medical conditions. There was no family history of hypoglycemia. Growth parameters were notable for weight at the 19th percentile and length at the 50th percentile, however head circumference was less than the 1st percentile and was 50th percentile for 6 months of age. Physical exam was notable for a scooped bridge of the nose, normal placement of the eyes with prominent eyelashes and bilateral epicanthus, a thin upper lip, relatively broad square fingers bilaterally with persistent fetal pads on the fingers of both hands, and a relatively broad great toe on both feet. Her pictures are shown in Figure 1.
FIGURE 1.
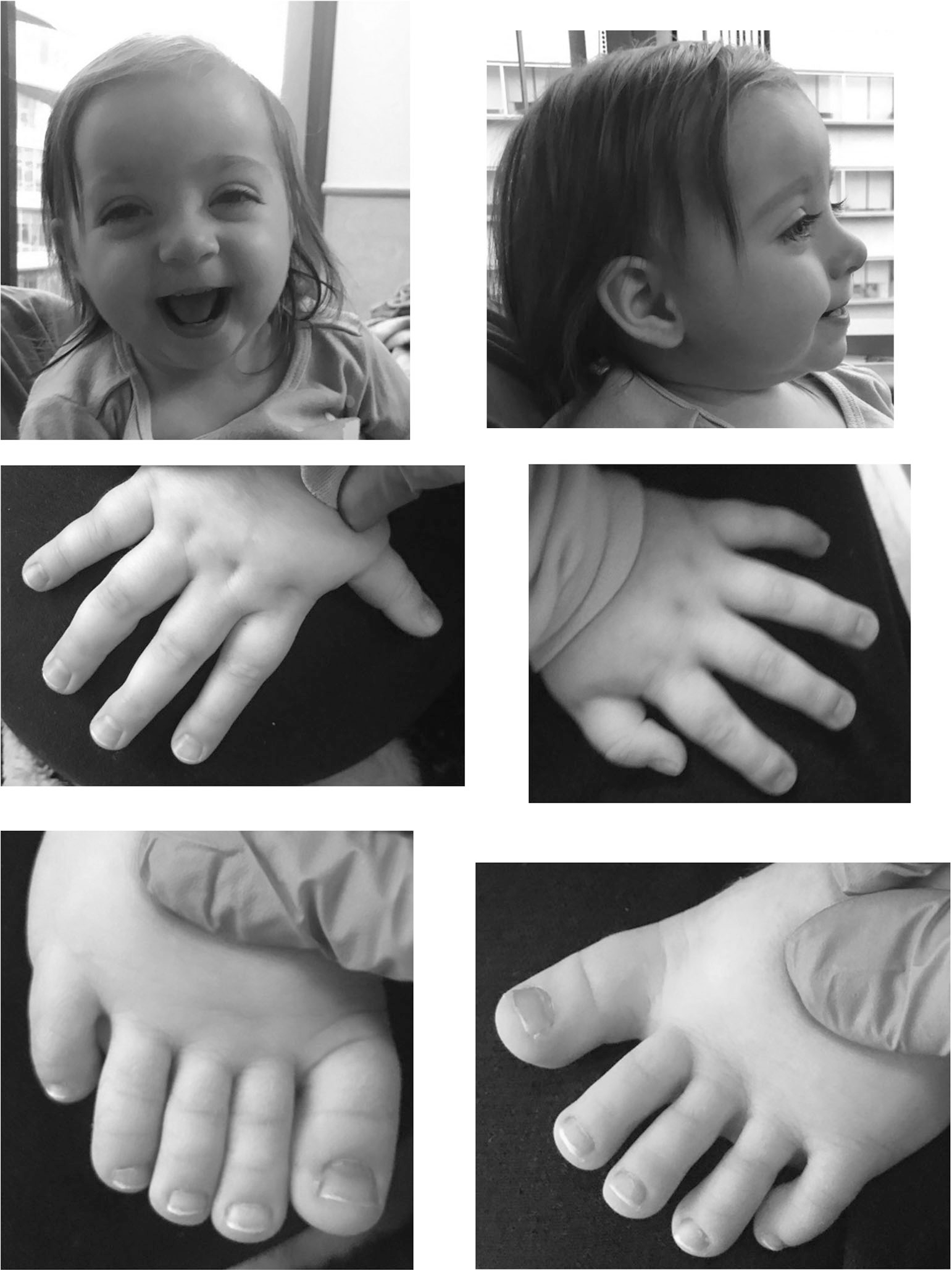
Features shown here include a scooped bridge of the nose, normal placement of the eyes with prominent eyelashes and bilateral epicanthus, a thin upper lip, relatively broad square fingers bilaterally with persistent fetal pads on the fingers of both hands, and a relatively broad great toe on both feet
While syndromes that classically have hyperinsulinism as a feature were considered including Beckwith-Wiedemann syndrome (BWS), Kabuki syndrome, and Sotos syndrome, the child did not have the classic features any of these syndromes. The only features of Kabuki syndrome seen in our child were long eyelashes, microcephaly, and persistent fetal finger pads. Our child’s exam findings of broad fingers and toes with persistent fetal pads, prominent eyelashes, and a relatively scooped nose, made us concerned for RSTS. While development was not as delayed as in other cases with RSTS, she did have developmental delay and microcephaly. However, her constellation of clinical findings was also more difficult to interpret given her extreme prematurity.
Given our exam, a chromosomal microarray was sent, which was normal, as well as a RSTS gene panel that included CREBBP and EP300 sequencing. EP300 sequencing revealed a pathogenic variant c.4066C>T (p.Arg1356*). The specific variant of EP300 detected (p.Arg1356*) has been seen in two other individuals, both of whom had moderate intellectual disability (Fergelot, Van belzen, Van gils, et al., 2016; Hamilton et al., 2016). Participant 8, as described by Hamilton et al. had global developmental delay and required special education classes. At the age of 13 years, he was working at an 8-year-old level for reading, 6–7-year-old level for numeracy, and a 5 year-old level for writing (Hamilton et al., 2016). Hypoglycemia was not discussed in a cohort of 52 individuals (Fergelot et al., 2016) or 9 individuals (Hamilton et al., 2016) with EP300 variant RSTS.
3 |. DISCUSSION
Hyperinsulinism is the most common cause of persistent hypoglycemia in infants and children. The list of known monogenic causes of hyperinsulinism has grown significantly in the last several years (Rosenfeld et al., 2019), however, in up to 50% of affected patients, genetic testing through multiple gene panels is negative (Snider, Becker, Boyajian, et al., 2013). For these cases, a syndromic cause of hyperinsulinism should be considered. Hyperinsulinism has only been previously reported in a few case reports of individuals with molecularly confirmed RSTS. Costain et al. reported a case of EP300 variant RSTS with hyperinsulinism requiring diazoxide (Costain et al., 2018). Welters et al. described 4 individuals with severe hyperinsulinism, 3 of whom had EP300 mutations and 1 who had a CREBBP mutation. To our knowledge, this is the only case of hyperinsulinism reported in an individual with CREBBP variant RSTS. The hyperinsulinism in this individual was reportedly transient, lasting about 2.5 months (Welters et al., 2019). Hyperinsulinism was additionally noted in a case by Wyatt who described an individual with clinically suspected RSTS, but who never had molecular confirmation. The hyperinsulinism in this individual was also transient and lasted about 2 weeks (Wyatt, 1990). In addition, a recent large meta-analysis of 102 subjects in the literature with EP300 variant RSTS showed that at least four had hypoglycemia (Cohen et al., 2020). Thus, it appears that hyperinsulinism may be disproportionately represented in EP300 variant RSTS, which only accounts for 8% of all molecularly confirmed RSTS.
The mechanism for hyperinsulinism in EP300 variant RSTS is not well understood. Wong et al. found that both CREBBP and EP300 are required for normal pancreatic islet cell development, beta cell function, and survival. However, mice lacking EP300 or CREBBP in pancreatic islet cells have reduced alpha and beta cell mass and develop hypoinsulinemia (Wong et al., 2018). This hypoinsulinemia is in contrast to the phenotype of hyperinsulinism in our case and in previously reported cases. Thus, additional mechanistic studies are required to understand the pathophysiology in CREBBP- and EP300-associated hyperinsulinism.
As evidenced by the child described here, in addition to the four previously reported individuals, hyperinsulinism is associated with EP300 variant RSTS and EP300 variant RSTS should be considered in individuals who have hypoglycemia in addition to other features of RSTS. We believe the child reported here provides the most comprehensive hyperinsulinism evaluation of a patient with RSTS and substantiates that the hyperinsulinism was not secondary to perinatal stress and is a true association with RSTS. In our case and in the cases described by Welters et al., genetic testing was negative for known non-syndromic causes of hyperinsulinism (ABCC8, KCNJ11, GCK, GLUD1, HADH, HNF1A, HNF4A, SLC16A1) (Welters et al., 2019). A genetic etiology is still not identified in approximately 50% of individuals with clinical hyperinsulinism (Kapoor et al., 2013). Thus, it is especially important to consider EP300 variant RSTS in individuals with hyperinsulinism for whom another genetic etiology is not identified as the facial features can be less striking and neurodevelopmental outcomes can be more typical in EP300 variant RSTS compared to other forms of RSTS.
ACKNOWLEDGMENTS
Research reported in this publication was in part supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number TL1TR001880. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Research reported in the publication was supported by the Institute for Translational Medicine and Therapeutics of the Perelman School of Medicine at the University of Pennsylvania.
Funding information
National Center for Advancing Translational Sciences, Grant/Award Number: TL1TR001880
Footnotes
CONFLICT OF INTEREST
The authors have no conflicts of interest to disclose.
DATA AVAILABILITY STATEMENT
Data available on request from the authors. The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Cohen JL, Schrier Vergano SA, Mazzola S, Strong A, Keena B, McDougall C, … Deardorff MA (2020). EP300-related Rubinstein-Taybi syndrome: Highlighted rare phenotypic findings and a genotypephenotype meta-analysis of 74 patients. American Journal of Medical Genetics, 182, 2926–2938. 10.1002/ajmg.a.61883 [DOI] [PubMed] [Google Scholar]
- Costain G, Kannu P, & Bowdin S (2018). Genome-wide sequencing expands the phenotypic spectrum of EP300 variants. European Journal of Medical Genetics, 61, 125–129. [DOI] [PubMed] [Google Scholar]
- Fergelot P, Van belzen M, Van gils J, Afenjar A, Armour CM, Arvelier B, … Hennekam RC (2016). Phenotype and genotype in 52 patients with Rubinstein Taybi syndrome caused by EP300 mutations. American Journal of Medical Genetics. Part A, 170, 3069–3082. [DOI] [PubMed] [Google Scholar]
- Ferrara C, Patel P, Becker S, Stanley CA, & Kelly A (2015). Biomarkers of insulin for the diagnosis of hyperinsulinemic hypogylcemia in infants and children. Journal of Pediatrics, 168, 212–219. [DOI] [PubMed] [Google Scholar]
- Hamilton MJ, Newbury-ecob R, Holder-espinasse M, Yau S, Lillis S, Hurt JA, … Suri M (2016). Rubinstein-Taybi syndrome type 2: Report of nine new cases that extend the phenotypic and genotypic spectrum. Clinical Dysmorphology, 25(4), 135–145. [DOI] [PubMed] [Google Scholar]
- Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, & Hussain K (2013). Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. European Journal of Endocrinology, 168, 557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld E, Ganguly A, & De Leon DD (2019). Congenital hyperinsulinism disorders: Genetic and clinical characteristics. American Journal of Medical Genetics Part C Seminars in Medical Genetics, 181(4), 682–692. 10.1002/ajmg.c.31737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein JH, & Taybi H (1963). Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. American Journal of Diseases of Children, 105, 588–608. [DOI] [PubMed] [Google Scholar]
- Snider KE, Becker S, Boyajian L, Shyng S-L, MacMullen C, Hughes N, … Ganguly A (2013). Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. Journal of Clinical Endocrinology and Metabolism, 98(2), E355–E363. 10.1210/jc.2012-2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon BD, Bodian DL, Khromykh A, Gomez Mora G, Lanpher BC, Ramaswamy KI, … Niederhuber JE (2015). Expanding the phenotypic spectrum in EP300-realted Rubinstein-Taybi syndrome. American Journal of Medical Genetics, 167(5), 1111–1116. [DOI] [PubMed] [Google Scholar]
- Welters A, El-khairi R, Dastamani A, Bachmann N, Bergmann C, Gilbert C, … Kummer S (2019). Persistent hyperinsulinaemic hypoglycaemia in children with Rubinstein-Taybi syndrome. European Journal of Endocrinology, 181(2), 121–128. [DOI] [PubMed] [Google Scholar]
- Wong CK, Wade-Vallance AK, Luciani DS, Brindle PK, Lynn FC, & Gibson WT (2018). The p300 and CBP transcriptional coactivators are required for beta-cell and alpha-cell proliferation. Diabetes, 67, 412–422. [DOI] [PubMed] [Google Scholar]
- Wyatt D (1990). Transient hypoglycemia with hyperinsulinemia in a newborn infant with Rubinstein-Taybi syndrome. American Journal of Medical Genetics, 37(1), 103–105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data available on request from the authors. The data that support the findings of this study are available from the corresponding author upon reasonable request.