

Abstract
Background and purpose:
In this study, some new cytotoxic hybrid structures were synthesized by combining pyrazolinone and imidazolinone rings with quinazoline pharmacophores.
Experimental approach:
The benzoxazinone, pyrazolo-quinazoline fused ring, and imidazolinone anchored quinazoline derivatives were synthesized by simple ring-opening, ring expansion, and ring closure strategies from oxazolones. The molecular docking studies of the final derivatives were accomplished on the epidermal growth factor receptor enzyme. The cytotoxic effect of the final compounds on the MCF-7 cell line was evaluated by MTT assay.
Findings/Results:
The docking results confirmed the optimized electrostatic, H-bonding, and hydrophobic interactions of structures with the key residues of the active site (ΔGbin< -9Kcal/mol). The derivatives have been obtained in good yield and purity, and their structures were confirmed by different methods (FT-IR, 1H-NMR, 13C-NMR, and CHNS analysis). The IC50s of all final derivatives against the MCF-7 cell line were lower than 10 μM, and between all, the IXa from pyrazolo-quinazolinone class (IC50: 6.43 μM) with chlorine substitute was the most potent. Furthermore, all derivatives showed negligible cytotoxicity on HUVEC normal cell line which would be a great achievement for a novel cytotoxic agent.
Conclusion and implications:
Based on the obtained results, pyrazolo[1,5-c] quinazolin-2-one series were more cytotoxic than imidazolinone methyl quinazoline-4(3H)-ones against MCF-7 cells. Chlorine substitute in the para position of the aromatic ring improved the cytotoxicity effect in both classes. It could be related to the polarizability of a chlorine atom and making better intermolecular interactions. Further pre-clinical evaluations are required for the promising synthesized cytotoxic compounds.
Keywords: Anticancer activity, Molecular docking, Oxazolone, Pyrazoline, Quinizolinone
INTRODUCTION
To date, the development of antitumor agents to treat various types of cancers has been one of the most important medical achievements. Increasing the incidence of cancer due to the aging of the world’s population, led to the efforts made by medicinal chemists to develop new therapeutic anti-cancer compounds. In this regard, natural and synthetic sources could be used as lead compounds for the design of novel and potent anticancer agents.
Five and six-membered nitrogen heterocycles such as pyrazoles and quinazolines have been recognized as potential scaffolds for therapeutic investigations (1). Several synthetic compounds containing pyrazoles have shown different biological properties such as antibacterial, antifungal, antiproliferative, anti-inflammatory, anti-tubercular, antiviral, anti-Alzheimer, anti-diabetic, and anti-leishmanial activities (2).
Also, quinazolines nucleus have been extensively used in different compounds to get a broad spectrum of pharmacological activities, comprising antibacterial, antifungal, anticancer, anticonvulsant, antimalarial, anti-inflammatory as well as anti-ulcer activities (3).
Tricyclic rigid moieties have been used commonly in pharmaceutical chemistry as an attractive template for drug design. Among the tricyclic systems, pyrazolo-quinazoline core showed different pharmaceutical activities such as gamma-aminobutyric acid A (GABAA) receptor modulator (4), anti-inflammatory (5), and xanthine oxidase inhibitory (6).
A series of pyrazolo[4,3-h] quinazoline derivatives have been synthesized by Zaho et al. (7), and their antiproliferative activities were evaluated through inhibition of the CDK4/6 pathway. Also, Mohareb et al. reported the synthesis and antiproliferative evaluation of a new series of pyrazolo[5,1-b]quinazoline (8). Based on cytotoxic results, their derivatives were found to inhibit significantly Pim-1 kinase and indicated a good antiproliferative activity against the human prostatic cancer PC-3 cell line (8). In another attempt, pyrazolo[1,5-a] quinazolines were synthesized, and their antitumor activities were determined against HepG-2 and MCF-7 cell lines (9). The results revealed that quinazoline core showed profound antitumor activity probably via the epidermal growth factor receptor (EGFR) inhibition mechanism (10,11).
The importance of pyrazole and quinazoline scaffolds as pharmacological sub-segments encouraged us to synthesize a series of pyrazolo and imidazolo quinazoline-2-one derivatives. The general structures of all synthesized compounds have been illustrated in Figure 1. Also, the MTT assay was used to determine the initial in vitro activity of designed cytotoxic agents against the MCF-7 cell line.
Fig. 1.
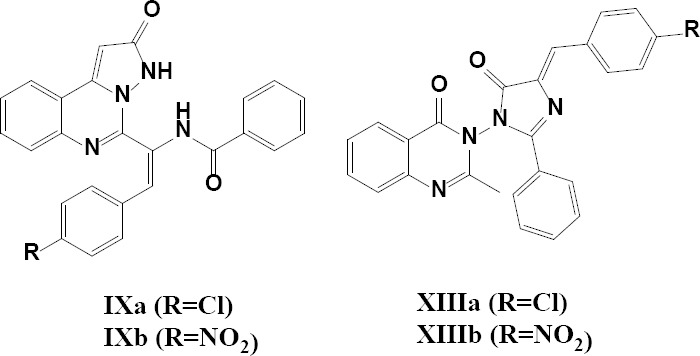
General structure of the synthesized compounds.
MATERIALS AND METHODS
Materials and instruments
All chemicals were purchased from Merck and Sigma-Aldrich chemical companies and were used without further purification. Thin-layer chromatography (TLC) was accomplished on Merck silica gel (60F254) sheets. Proton and carbon-13 nuclear magnetic resonance (1HNMR and 13CNMR) spectra were recorded in either dimethyl sulfoxide (DMSO) or CDCl3 solvents on Bruker 400 MHz spectrometers (Germany) using tetramethylsilane (TMS) as an internal reference. Chemical shifts are expressed in δ (ppm). Infrared (IR) spectra were recorded as KBr pellets on a WQF-510 Fourier-transform (FT)-IR spectrophotometer (China). Elemental analysis was carried out using Elemental Analyzer for CHN (Perkin Elmer, Germany). Melting points were determined on a Mettler capillary melting point apparatus (Electrothermal 9200, England).
Synthesis methods
Hippuric acid (III)
Glycine (10 g, 130 mmol) was added to water (100 mL) in a round-bottomed flask and then it was cooled in an ice bath to dissolve glycine. Then, a solution of benzoyl chloride (15.2 mL, 130 mmol) in a sodium hydroxide (40%, 22.6 mL) was added dropwise via a dropping funnel for 20 min. The reaction mixture was heated and stirred for half an hour. The mixture was then cooled slightly, and the solution was acidified with concentrated HCl to obtain a white solid. The resulting solid was washed with chloroform (10 mL) and dried in a vacuum oven at 40 °C for 12 h to yield hippuric acid (III) as a pure white powder (93%). The obtained product was identified by comparing its melting point and appearance with the value reported in the literature (12).
General procedure for oxazolone synthesis (Va and Vb)
A mixture of arylaldehydes (1 mmol), sodium acetate (0.082 g, 1 mmol), and hippuric acid (0.21 g, 1.2 mmol) in acetic anhydride (5 mmol) was refluxed for 10 h, and the progress of the reaction was monitored by TLC (n-hexane/EtOAc: 7/3). After the completion of the reaction, the mixture was cooled slightly to yield a solid precipitated. The resulting solid was washed with ethanol and then neutralized with an aqueous solution (20%) of NaHCO3 (10 mL). The obtained product was recrystallized from ethyl acetate to yield Va (85%) as pale-yellow solid or Vb (91%) as deep-yellow solid. The obtained product was identified by 1HNMR, FT-IR spectra, and melting point (13).
Benzoxasinone derivatives (VIIa and VIIb)
An equimolar mixture of anthranilic acid and related oxazolones was refluxed in glacial acetic acid for 3 h. The obtained solid was hot filtered and dried under vacuum at 40 °C for 12 h. In the following reaction, the solid material was transferred into acetic anhydride and refluxed for 3 h. Finally, the mixture was cooled to room temperature, and the precipitates were filtered and washed with cold ethanol. The products were further purified by recrystallization in ethanol-dioxane to give VIIa (83%) as white solids or VIIb (79%) as pale-orange solid. The obtained product was identified by 1HNMR, 13CNMR, and FT-IR spectra (14).
Pyrazolo[1,5-c] quinazolin-2-one derivatives (class A; IXa and IXb)
The related Va or VIb (1 mmol) and hydrazine hydrate (0.03 mL, 1 mmol) was refluxed in glacial acetic acid (10 mL) for 6 h. After the completion of the reaction confirmed by TLC (n-hexane/EtOAc: 7/3), the solvent was evaporated to give a solid. The product was recrystallized from ethanol to give IXa (59%) as pale-yellow solid or IXb (53%) as deep-orange solid in vacuo at 40 °C for 12 h. The obtained product was identified by 1HNMR, 13CNMR, and FT-IR spectra (15).
2-Methyl-benzo[d] [1,3] oxazin-4-one (XI)
Anthranilic acid (1.37 g, 10 mmol) was added to acetic anhydride (10.2 mL, 100 mmol), and refluxed for 1 h. After the completion of the reaction obtained by TLC (n-hexane/EtOAc: 7/3), the excess of acetic anhydride was evaporated under reduced pressure, and the obtained solid was dissolved in petroleum ether and kept at room temperature for 1 h. The resulting solid was filtered off and dried in a vacuum oven at 40 °C for 12 h to produce a pure solid product (90%). The pure product was used in the next step without further purification. The obtained product was identified by melting point and 1HNMR spectra (16).
3-Amino-2-methyl-3H-quinazolin-4-one (XII)
Compound XI (1.61 g, 10 mmol) was added to a solution of hydrazine hydrate (1.5 g, 30 mmol) in ethanol and was refluxed for 2 h. After the completion of the reaction (recognized by TLC, n-hexane/EtOAc: 7/3), the mixture was cooled through iced water to give a solid product. The separated solid was recrystallized from ethanol and dried in a vacuum oven to produce a pure solid product (86%). The obtained product was identified by melting point and FT-IR spectra (16).
2-methylquinazolin-4(3H)-one derivatives with anchored imidazolinone moiety (class B: XIIIa, XIIIb)
A mixture of XII and Va or Vb in the presence of n-butanol was heated under reflux conditions for 6 h. After the completion of the reaction (recognized by TLC, n-hexane/EtOAc: 7/3), the solvent was evaporated in vacuo, the resulting product was filtered and washed with cold ethanol. The solid product was dried in a vacuum oven to give a pure product (XIIIa, 49%; XIIIb, 45%). The obtained product was identified by 1HNMR, 13CNMR, and FT-IR spectra.
Docking studies
The computer-simulated docking studies were performed using AutoDock 4.2 (17). The three-dimensional (3D) crystal structure of the EGFR enzyme, determined by X-ray crystallography, was retrieved from RCSB Protein Data Bank (PDB code 1M17). Then, the protein was corrected by removing extra crystallized water molecules using Accelrys discovery studio visualizer 4.0 (18) and adding polar hydrogens and Kollman charges by Autodock software (17). The structures of ligands were optimized using the MM+ force field and PM3 semi-empirical techniques in HyperChem8 software. Then, the Gasteiger-Marsili partial charges were added, and rotatable bonds were assigned using the AutoDockTools version1.5.6rc3 (19). To calculate atomic affinity grid maps for each atom type in the ligand by AutoGrid, a grid map with 60 grid points in X, Y, and Z directions was built and constructed centered on the ligand’s binding site. A grid spacing of 0.375 Å and a distance-dependent function of the dielectric constant for the energetic map calculations were employed. The genetic algorithm of the AutoDock 4.2 program was used as the search algorithm through a protocol with an initial population of 150 randomly placed individuals, a maximum number of 250 million energy evaluations, a mutation rate of 0.02, a crossover rate of 0.8 (17). Fifty independent docking runs were carried out for each compound, and the structures were ranked by energy, as were the clusters, and binding free energy of each run was provided in the docking log file (dlg file). Ligand-receptor interactions were all visualized based on docking results using Discovery Studio Visualizer 3.5.
In vitro cytotoxicity assessment
The cytotoxic effects of prepared compounds were studied against HUVEC and MCF-7 cell lines as described previously (20). Roswell Park Memorial Institute (RPMI)-1640 culture medium was purchased from Biosera (Germany) and completed with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin antibiotic solution (10000 Unit/mL penicillin and 10 mg/mL streptomycin). Briefly, 180 μL of cell suspensions (5-7 × 104 cells per well) were cultivated in 96-well microplates, and incubated in standard culture conditions (37 °C, 5% CO2 air humidified). For initial screening, 20 μL of each compound with different concentrations of 10, 40, 60, 80, 100, 400, 600, 800, and 1000 μM were added to each microplate wells and followed by 48 h of incubation. The final concentrations of the 1, 4, 6, 8, 10, 40, 60, 80, and 100μM, respectively then were selected as treating concentration in the next evaluations for all derivatives, after incubation time in each study, MTT solution (20 μL in phosphate-buffered saline (PBS), 5 mg/mL) was added into each well and incubated for 3 h. Formazan crystals were dissolved in DMSO (150 μL per well), and its absorbance was measured by an ELISA plate reader at 570 nm. Doxorubicin (20 μM) was used as the positive control. A mixture of cell suspension (180 μL) with RPMI (20 μL) was considered as the negative control with 100% viability, while RPMI medium (200 μL) was used as the blank. The percentage of cell viability was calculated using the following equation:
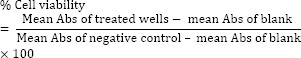
Each test was performed in triplicate and repeated on three different days independently.
Statistical analysis
One-way ANOVA followed by LSD was implemented using SPSS software to evaluate the differences between the groups. Furthermore, for checking the differences between two groups paired-wise comparison t-test, and for the differences among more than two groups, the Post Hoc test was used. All data were presented as means ± SD. The P-value lower than 0.05 was considered significant.
RESULTS
Chemistry and synthesis
The chemical procedure for the synthesis of all different derivatives Va, Vb, VIIa, VIIb, IXa, IXb, XIIIa, and XIIIb have been summarized in Schemes 1-3.
Scheme 1.
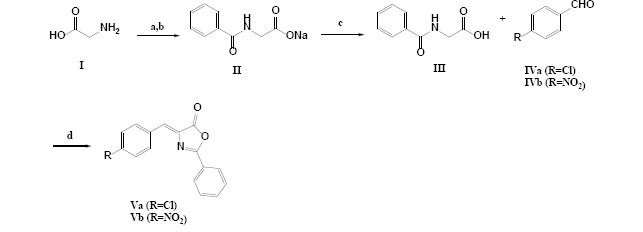
General procedure applied for the synthesis of oxazolone derivatives. Reagents and conditions: (a) H2O/ ice bath; (b) benzoyl chloride, NaOH, 30 min; (c) concentrated HCl; (d) (CH3CO)2O, 10-h reflux.
Scheme 3.
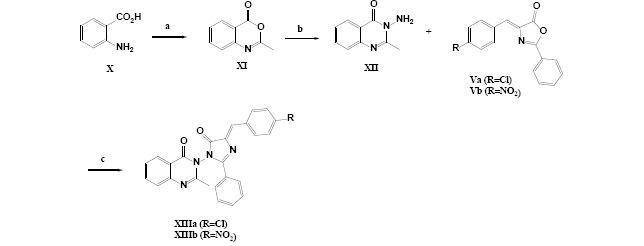
General procedure applied for the synthesis of 2-methylquinazolin-4(3H)-one derivatives with anchored imidazolinone moiety. Reagents and conditions: (a) (CH3CO)2O, 10-h reflux; (b) NH2NH2, EtOH, 2-h reflux; (c) n-butanol, 6-h reflux.
Scheme 2.
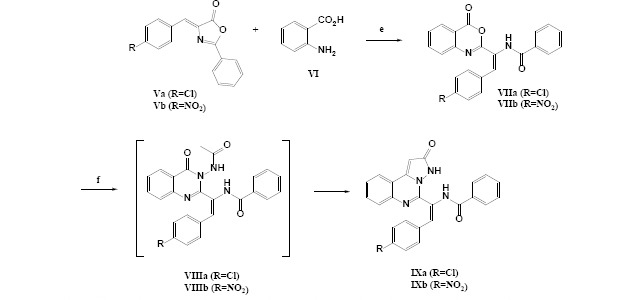
General procedure applied for the synthesis of pyrazolo[1,5-c] quinazolin-2-one derivatives. Reagents and conditions: (e) 1: CH3CO2H, 10-h reflux, 2: (CH3CO)2O, 3-h reflux; (f) glacial CH3CO2H, NH2NH2, 6-h reflux.
The chemical structure of all derivatives was confirmed using spectroscopic data as follows:
(Z)-4-(4-chlorobenzylidene)-2-phenyloxazol-5(4H)-one (Va): yellow solid, mp. 187-189.5 °C; FTIR (KBr): υ (cm-1) 3049, 1793, 1654, 1552, 1486, 1449, 1325, 1160, 824; 1HNMR (400 MHz, CDCl3) δ (ppm): 7.19 (S,1H, H7), 7.45 (d, J = 8.0 Hz, 2H, H10, 12), 7.53-7.58 (m, 5H, H9,13,16,17,18), 7.94-7.97 (m, 2H, H15,19).
(Z)-4-(4-nitrobenzylidene)-2-phenyloxazol-5(4H)-one (Vb): yellow solid, mp. 238-240 °C; FTIR (KBr): υ (cm-1) 3063, 1794, 1661, 1559, 1537, 1457, 1352, 1284; 1HNMR (400 MHz, CDCl3) δ (ppm): 7.3 (S,1H, H7), 7.59-7.65 (m, 3H, H16,17,18), 7.80 (d, J = 8.0 Hz, 2H, H4, 6), 8.11-8.13 (m, 2H, H15, 19), 8.27 (d, J = 8.0 Hz, 2H, H1, 3).
(E)-N-(2-(4-chlorophenyl)-1-(4-oxo-4H-benzo[d][1,3]oxazin-2-yl)vinyl)benzamide (VIIa): yellow solid, mp. 101-102 °C; FTIR (KBr): υ (cm-1) 3289, 3087, 1763, 1664, 1599, 1482, 1271; 1HNMR (400 MHz, DMSO) δ (ppm): 7.30 (S,1H, H14), 7.47-7.62 (m, 8H, H1, 16, 17, 19, 20, 26, 27, 28), 7.75 (dd, J = 8.3 and 1.1 Hz, 1H, H3), 7.76-7.90 (m, 1H, H2), 7.99 (d, J = 8.1 Hz, 2H, H25, 29), 8.13-8.15 (dd, J = 7.8, 1.3 Hz, 1H, H6), 9.87 (s, 1H, H13); 13CNMR (100 MHz, DMSO) δ (ppm): 125.6, 126.4, 128.04 (2C), 128.3, 128.6 (2C), 129.0, 129.5 (2C), 130.0 (2C), 132.0, 135.3.
(E)-N-(2-(4-nitrorophenyl)-1-(4-oxo-4H-benzo[d][1,3] oxazin-2-yl)vinyl)benzamide (VIIb): yellow solid, mp. 101-102 °C; FTIR (KBr): υ (cm-1) 3279, 3081, 1764, 1665, 1599, 1521, 1477, 1344, 1264, 1111; 1HNMR (400 MHz, DMSO) δ (ppm): 7.25 (S, 1H, H14), 7.45-7.63 (m, 4H, H1, 16, 17, 19), 7.76 (dd, J = 8.3 and 1.1 Hz, 1H, H3), 7.84-8.03 (m, 5H, H2, 20, 26, 27, 28), 8.13 (dd, J = 7.8, 1.3 Hz, 1H, H6), 8.26 (d, J = 8.4 Hz, 2H, H25, 29), 10.00 (s, 1H, H13).
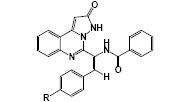
(E)-N-(2-(4-chlorophenyl)-1-(2-oxo-2,3-dihydropyrazolo[1,5-c]quinazolin-5-yl)vinyl)benzamide (IXa): yellow solid, mp. > 300 °C; FTIR (KBr): υ (cm-1) 3224, 3079, 1693, 1650, 1597, 1536, 1501, 1365, 1260, 1017; 1HNMR (400 MHz, DMSO) δ (ppm): 6.22 (S, 1H, H13), 7.01 (s, 1H, H18), 7.35 (d, J = 8.0 Hz, 2H, H22, 26), 7.46 (d, J = 8 Hz, 2H, H23, 25), 7.49-7.61 (m, 5H, H1, 2, 30, 31, 32), 7.78 (m, 1H, H3), 7.96 (m, 2H, H29, 33), 8.19 (m, 1H, H6), 9.98 (s, 1H, H16), 11.06 (s, 1H, H11); 13CNMR (100 MHz, DMSO) δ (ppm) 168.9, 162.3, 152.5, 146.3, 144.4, 135.2, 135.1, 132.7, 131.9, 131.8, 131.5, 128.9, 128.4, 126.6, 126.2, 122.2, 119.7, 119.5, 100.2; elemental analysis: theoretical (%): C, 68.11; H, 3.89; N, 12.71, found experimentally (%): C, 68.20; H, 3.96; N, 12.58.
(E)-N-(2-(4-nitrophenyl)-1-(2-oxo-2,3-dihydropyrazolo[1,5-c]quinazolin-5-yl)vinyl) benzamide (IXb): pale-yellow solid, mp. > 300 °C; FTIR (KBr): υ (cm-1) 3312, 3224, 3064, 1696, 1652, 1585, 1506, 1469, 1338, 1260, 1016; 1HNMR (400 MHz, DMSO) δ (ppm): 6.21 (S, 1H, H13), 7.00 (s, 1H, H18), 7.50-7.59 (m, 5H, H1, 2, 29, 30, 31), 7.67 (d, J = 8.0 Hz, 2H, H22, 26), 7.76-7.79 (m, 1H, H3), 7.95-7.97 (m, 2H, H29, 32), 8.18-8.20 (m, 1H, H6), 8.23 (d, J = 8.0 Hz, 2H, H23, 25), 9.84 (s, 1H, H16), 10.89 (s, 1H, H11); 13CNMR (100 MHz, DMSO) δ (ppm) 103.2, 119.9, 120.2, 120.9, 121.3, 123.8 (2C), 126.1, 126.6, 127.9 (2C), 128.5 (2C), 128.8 (2C), 131.9, 132.2, 132.7, 134.1, 147.6, 149.9, 151.5, 152.9, 165.3, 166.3.
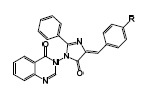
(Z)-3-(4-(4-chlorobenzylidene)-5-oxo-2-phenyl-4,5-dihydro-1H-imidazol-1-yl)-2-methylquinazolin-4(3H)-one (XIIIa): pale-yellow solid, mp. 122-125 °C; FTIR (KBr): υ (cm-1) 3039, 2929, 1718, 1648, 1599, 1510, 1467, 1315, 1255; 1HNMR (400 MHz, DMSO) δ (ppm): 2.68 (S, 3H, CH3), 7.44-7.53 (m, 6H, H28, H27,29,H1, 21, 23), 7.58 (d, J = 7.9 Hz, 2H, H20, 24), 7.65-7.67 (dd, J = 8.3 Hz, J = 1.2 Hz, 1H, H3), 7.75-7.79 (m, 1H, H2), 7.82-7.84 (m, 3H, H18, 26, 30), 8.16-8.18 (m, 1H, H6); 13CNMR (100 MHz, DMSO) δ (ppm) 22.2, 119.4, 123.9, 126.3, 127.0 (3C), 127.4, 128.8 (2C), 129.6 (2C), 130.4, 130.7 (2C), 131.9, 132.6, 133.6, 133.8, 147.1, 148.7, 150.0, 161.3, 165.7.
(Z)-2-methyl-3-(4-(4-nitrobenzylidene)-5-oxo-2-phenyl-4,5-dihydro-1H-imidazol-1-yl) quinazolin-4(3H)-one (XIIIb): pale-yellow solid, mp. 126-129 °C; FTIR (KBr): υ (cm-1) 3071, 2960, 2934, 1719, 1647, 1598, 1514, 1476, 1339, 1311, 1257; 1HNMR (400 MHz, DMSO) δ (ppm): 2.68 (S, 3H, CH3), 7.44-7.53 (m, 4H, H1, 27, 28, 29), 7.64 (d, J = 8.0 Hz, 1H, H3), 7.77 (m, 1H, H2), 7.82-7.84 (m, 3H, H18,26,30), 7.98 (d, J = 8.0 Hz, 2H, H20, 24,), 8.17 (d, J = 8.0 Hz, 1H, H6), 8.27 (d, J = 8.0 Hz, 2H, H21, 23).
In vitro cytotoxicity assessment
The results of anti-proliferation studies are given in Table 1. IXa, IXb, XIIIa, and XIIIb derivatives had lower IC50s against the MCF-7 cell line compared to doxorubicin. Among all synthesized compounds, IXa had the best effect among all and Va had the minimum activity.
Table 1.
Cytotoxicity results for tested compounds in MCF-7 and HUVEC cell lines.
| Compounds | IC50 (μM)a | |
|---|---|---|
|
| ||
| MCF-7 | HUVEC | |
| IXa | 6.43 ± 0.54 | 79.04 ± 3.71 |
| IXb | 6.73 ± 0.37 | 96.15 ± 4.66 |
| VIIa | 20.45 ± 0.91 | 72.18 ± 1.5 |
| VIIb | 25.4 ± 1.11 | 79.23 ± 2.3 |
| XIIIa | 8.49 ± 0.35 | 94.41 ± 0.89 |
| XIIIb | 8.70 ± 0.50 | 81.2 ± 6.17 |
| Va | 85.88 ± 4.41 | 96.43 ± 2.22 |
| Vb | 56.57 ± 3.07 | 93.32 ± 4.11 |
| Doxorubicin | 17.18 ± 0.07 | 21.17 ± 0.05 |
aIC50 Values from three independent experiments were given as means ± SD and the standard deviations were less than 10%.
Docking study
The free binding energies (ΔGb) and inhibition constants (Ki) as calculated by AutoDock have been summarized in Table 2, and the best position of docked ligands within the receptor active site has been presented in Fig. 2.
Table 2.
Free binding energy (ΔGb, kcal/mol) and inhibition constants (Ki, nM) of synthesized compounds with EGFR calculated by AutoDock
|
|
||||||
|
| |||||||
|---|---|---|---|---|---|---|---|
| Class A | Class B | ||||||
|
| |||||||
| R | Ki | ΔGb | Hbond | R | Ki | ΔGb | Hbond |
|
| |||||||
| Cl | 36.3 | -10.13 | met769 | Cl | 41.99 | -10.06 | Met769 |
| NO2 | 114.32 | -9.47 | met769 | NO2 | 62.32 | -9.00 | Met769, lys692 |
Fig. 2.

The binding mode of (a) IXa, (b) IXb, (c) XIIIa, and (d) XIIIb in the active site of epidermal growth factor receptor.
DISCUSSION
The oxazolone ring plays an important role in drug discovery strategies. This ring, as a synthon, participates in the synthesis of a wide range of heterocycles and chemical compounds with biological properties (13). These compounds, with their good stability, are very valuable synthetic intermediates that allow them to be separated and subjected to more sequential reactions (21).
The final designed derivatives of the current study were in two structural categories. The first class was pyrazolo quinazoline derivatives (IXa and IXb), and the second class was imidazolinone quinazoline derivatives (XIIIa and XIIIb). In both series, the synthesis started from oxazolone (Va and Vb), which were simply synthesized in a one-pot reaction of aldehyde and hippuric acid in the presence of acetic anhydride/NaOAc as ring closure dehydration agents (22). In the next step, anthranilic acid was reacted with oxazolones in the presence of freshly distilled acetic anhydride to produce benzoxazinone derivatives (VIIa and VIIb) in a ring-opening and ring closure strategies (14). In the next step, for class A compounds, benzoxazinones (VIIa and VIIb, Scheme 5) reacted with hydrazine and were treated with glacial acetic acid to yield IXa and IXb compounds (23). The possible mechanism for the synthesis of IXa and IXb compounds is shown in Schemes 4 and 5.
Scheme 4.
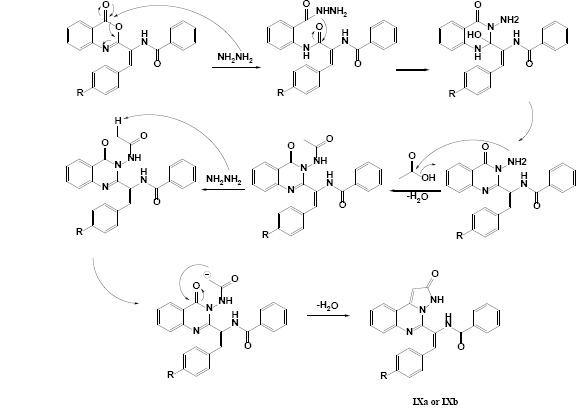
Reasonable mechanism for the synthesis of compounds IXa and IXb.
Scheme 5.
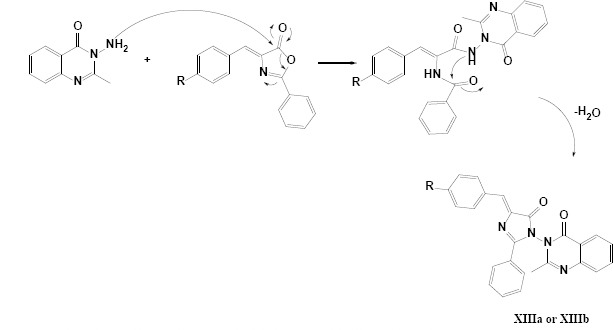
Plausible mechanism for the synthesis of compounds XIIIa and XIIIb
For the synthesis of the second class of derivatives (XIIIa and XIIIb), in the first step, anthranilic acid (X) was refluxed in acetic anhydride to get a yellow powder of benzoxazinone (XI) as a cyclic ester, after that, this fairly liable compound reacted with hydrazine hydrate to produce the hydrazine quinazoline (XII) via an early nucleophilic ring-opening and subsequent ring closure in the presence of hydrazine. Finally, upon the reaction of oxazolones (Va and Vb) with XII at the reflux condition in n-butanol, two derivations of XIIIa and XIIIb were synthesized. The general procedure applied for the synthesis of oxazolone (Va and Vb), benzoxazinone derivatives (VIIa and VIIb), pyrazolo[1,5-c] quinazolin-2-one derivatives (IXa and IXb), and 2-methylquinazolin-4(3H)-one derivatives (XIIIa and XIIIb) with anchored imidazolinone moiety are summarized in Schemes 3-5, respectively.
In the oxazolone and benzoxazinone derivatives (Va,b and VIIa,b), the specific cyclic anhydride segments were clearly seen in FTIR spectra above 1730 cm-1. In 1HNMR spectra for oxazolone and benzoxazinone the vinilic signal is a characteristic singlet signal that could be easily detected before aromatic signals. In the first class of derivatives (IXa and IXb), FTIR spectra have some characteristic features for carbonyl groups (around 1650-1700 cm-1) and aromatic rings (> 3000 cm-1). Determining evidences in these structures (IXa and IXb) was obtained from 1HNMR spectra. Special features of signals and patterns of splitting in the aromatic region of the spectrum are good pieces of evidence for structural clarification. Another spectral proof for class A of derivatives is the N-H signals around 10 and 11 ppm, which are not apparent in class B. Finally, 13CNMR complementary details successfully approved the structure of final derivatives. Two signals related to the carbonyl group are the best evidence for structure approval in both classes of derivatives.
In conclusion, the number of signals beside the characteristic signals for aromatic, aliphatic, tertiary, and vinylic carbons convinced us of the correct synthesis of structures. The same spectral details were previously reported by Hemdan et al. (15) for related derivatives in class A, and similar spectral features for compounds in class B have been reported before. In conclusion, with a precise analysis of spectrums for all synthesized derivatives, the correct structures were approved. Furthermore, as the final proof, the CHNS analysis was done for the IXa compound. The experimental results for carbon, hydrogen and nitrogen contents were in agreement with the theoretical data.
Structural hybridization in drug design and discovery is a powerful approach in making new therapeutic agents with synergistic effects arising from the stimulation of multiple targets or the aggregation of effects on a single target (24). In this study, we aimed to introduce some novel cytotoxic agents by applying this strategy to boost efficacy in cancer treatment. It was anticipated that these hybrid structures could fit themselves in EGFR active sites to act as an inhibitor with enhanced efficacy.
In vitro cytotoxic activity of prepared compounds was assayed using MTT assay in which the formation of formazan during the reduction of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide in the mitochondria of live cells in response to the succinate dehydrogenase enzyme is measured (25). The amount of produced formazan is related to the number of viable cells. The effects of the synthesized compounds on cell viability were determined against MCF7 and HUVEC cell lines. As it can be seen in Table 1, all of the newly designed derivatives (IXa, IXb, XIIIa, and XIIIb) have excellent cytotoxic effects against the MCF-7 cell line. With a simple evaluation of the results of VIIa and VIIb in comparison to their fused pyrazole hybrid (IXa and IXb), it is argued that the hybridization has improved the cytotoxicity about 3-4 times.
Among all the tested compounds, [1,5-c] quinazolin-2-one derivatives (IXa and IXb) showed the highest cytotoxicity against MCF-7 with IC50 values around 6 μM. The structure-activity relationship (SAR) studies revealed that the presence of pyrazolo[1,5-c] quinazolin-2-one moiety seems to play an important role in the growth inhibition as Ansari et al. reported for relative pyrazol-quinazoline fused heterocycles (26). In this regard, the presence of chlorine group (IXa, IC50= 6.43 μM) in the para position of an aromatic ring is more effective than those with electron-withdrawing groups such as the nitro group to some extent (IXb, IC50= 6.73 μM). In the second class of derivatives, (XIIIa and XIIIb) on the other hand, the IC50 values were around 8μM and they showed less cytotoxic effects in comparison with those of the pyrazolo [1,5-c]quinazolin moieties (IXa and IXb). As Abbas et al. stated in their evaluation, inserting the imidazolinone ring in the same position did not show any significant activity compared to other related structural patterns (27), but the results of our evaluation are not so much different from class A results. Also, in this series, the introduction of chlorine group (XIIIa, IC50= 8.49 μM) in the para position of the aromatic ring leads to better activity than electron-withdrawing groups such as the nitro group (XIIIb, IC50= 8.70 μM). Our results interestingly are supported by those of Ansari et al. on partially the same pyrazolo quinazoline structures as EGFR inhibitors, they evaluated the cytotoxicity of synthesized compounds against MDA-MB-231 cell line versus the erlotinib and gefitinib as positive controls (26), same as their study, chlorine and bromine groups induced supportive effects on cytotoxicity of derivatives.
Intermediate compounds (Va and Vb), with IC50 values more than 50 μM, have no remarkable cytotoxic effect on the MCF-7 cell line, which approved the synergistic effects of the two pharmacophores combination. In the case of normal cells (HUVEC), none of the synthetic compounds revealed an effective cytotoxic impact (IC50 values were more than 79 μM). It means that synthesized derivatives do not have any especial cytotoxicity against normal cells at concentration ranges of obtained IC50s for cancer cells. In general, the MTT test revealed that the lowest IC50 value for IXa between all tested derivatives, which means consequently, the highest anticancer activity against the MCF-7 cell line. Kumar et al. have reported the same study on structures partially similar to class B derivatives, but their reported IC50s against MCF-7 versus the cisplatin as the positive control were relatively higher than those obtained in this study. Indeed, substitution patterns and the arrangement of sub-segments have undeniable effects on cytotoxic effects (28).
The initial cytotoxicity results of this study strongly support the potency of both evaluated classes. High IC50 values of studied compounds on the HUVEC cell line is another prominent result of this study, which has always been the most important value to evaluate a new therapeutic agent. As it can be seen, in all tested derivatives the IC50s on the HUVEC cell line are obviously higher than the cancerous cells which could ensure minimized side effects during chemotherapy (29). Another interesting finding is about consistency in docking and cell-based results, the anticancer of the final designed derivatives IXa, IXb, XIIIa, and XIIIb were in accordance with docking results.
Since EGFR is overexpressed in many cancers, such as breast cancer (30), molecular docking studies of synthesized compounds on EGFR were performed. Quinazoline scaffold and related derivatives have always been considered as potential EGFR inhibitors (31). So, it was hoped that the designed derivatives could be introduced as novel inhibitors in this regard.
Among available X-ray structures of the EGFR-inhibitor complex, the structure (PDB ID: 1M17) with a high resolution (2.6 Å) was chosen. The docking protocol was validated by removing the co-crystallized ligand (4-Anilinoquinazoline) from the binding site of EGFR and re-docking it into the active site. The results of the re-docking indicated that the re-docked conformer of ligand accepted a similar binding approach to that seen in its parent crystal structure. The root means square deviation (RMSD) value for this coordination comparison was 1.3 Å (Table 2).
In chloro-substituted pyrazolo[1,5-c] quinazolin-2-one derivatives (IXa), the carbonyl of pyrazolone ring established the hydrogen bond with Met769 residue (Fig. 2a). Among the aromatic rings, quinazolinone provided the hydrophobic interaction with Glu738, The830, and Lys721. The benzoyl and chlorophenyl groups in IXa formed proper hydrophobic interactions with Leu694, Pro770, Leu768, Asn810, and Asp831 (Fig. 2a). Nitro substituted pyrazolo[1,5-c] quinazolin-2-one derivative (IXa) showed an H-bonds which is stabilized by the binding of the Met769 amino acids with the carbonyl of pyrazolone ring in this compound (Fig. 2b). Pyrazolo[1,5-c] quinazolin moieties established the proper hydrophobic interaction with Leu694, Leu768, and Leu820. The benzoyl and nitrophenyl groups in this compound interacted with Val702, Lys721, Thr704, Arg817, and Asp831 through hydrophobic interaction (Fig. 2b). In 2-methylquinazolin-4(3H)-one derivatives (XIIIa and XIIIb), the amino acid Met769 was found to play an important role in hydrogen bond interactions with the carbonyl of pyrazolone ring (Fig. 2c and (2d). Quinazoline ring in these compounds could foster van der Waals interactions with the gap bounded by Leu820, Thr766, Met742, and Val702. Phenyl ring also formed hydrophobic interaction with Val702, Leu768 in c3, and Gly 772 and Cyc773 in XIIIb derivatives. Chloro phenyl in XIIIa showed proper hydrophobic interactions with Gly772, Leu694, and Pro770. Nitro substitution in XIIIb was found to play an important role in hydrogen bond interactions with Lys692 (Fig. 2d).
In both pyrazolo[1,5-c] quinazolin-2-one (class A) and 2-methylquinazolin-4(3H)-one (class B) derivatives, chloro-substituted molecules (compounds IXa and XIIIa) were found to fit better in the active site of the receptor than those with nitro substituents (compounds IXb and XIIIb). This is consistent with previously reported results that halogen derivatives mostly fit better in the active site of the EGFR enzyme (32).
Generally, the synthesized compounds were accommodated well into the active site of EGFR and the van der Waals interactions, hydrogen bonds are the key factors for ligand-receptor interactions. These novel compounds could be considered for further studies as EGFR inhibitors in enzymatic evaluations.
CONCLUSION
In conclusion, two series of hybrid quinazoline-based compounds were synthesized in good yield and purities. Cytotoxic activity was determined by MTT assay against MCF-7 and HUVEC cell lines. The results showed that the pyrazolo[1,5-c] quinazolin-2-one series (IXa and IXb) were more cytotoxic than methyl quinazoline-4(3H)-ones (XIIIa and XIIIb) with anchored imidazolinone moiety. Based on SAR studies, chlorine substitute in the para position of the aromatic ring improved the cytotoxicity effect in class A, while a decrease in activity was observed mainly when the nitro group was employed. All the prepared compounds had no apparent cytotoxic effect on normal cells (HUVEC). Also, oxazolone derivatives (Va and Vb) had no inhibitory activity against the MCF-7 cell line, and the cytotoxicity of newly synthesized derivatives was higher than their benzoxazinone intermediates (Va and Vb). These results demonstrated the cytotoxic activity of pyrazolo[1,5-c] quinazolin derivatives, especially IXa, and make them promising cytotoxic hybrid compounds for further evaluations on cancer.
Conflicts of interest statement
The authors declared no conflicts of interest in this study.
Authors’ contribution
M. Malekzadeh contributed to the literature review, data collection and analysis, and manuscript preparation; Sh. Dadkhah collected the data and cooperated in the manuscript preparation; Gh. Khodarahmi contributed to data analyzing, supervising, and manuscript revision; P. Asadi designed and managed molecular docking studies; F. Hassanzadeh supervised the work; and M. Rostami contributed to idea developing, supervising, data analyzing, manuscript preparation, revision, and final correction. The final version of the manuscript has been approved by all authors.
Acknowledgments
This study was financially supported by the Vice-Chancellery of Research of Isfahan University of Medical Sciences, Isfahan, I.R. Iran through Grant No. 398437.
REFERENCES
- 1.Xu P, Yu B, Li FL, Cai XF, Ma CQ. Microbial degradation of sulfur, nitrogen and oxygen heterocycles. Trends Microbiol. 2006;14((9)):398–405. doi: 10.1016/j.tim.2006.07.002. DOI: 10.1016/j.tim.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 2.Khan MF, Alam MM, Verma G, Akhtar W, Akhter M, Shaquiquzzaman M. The therapeutic voyage of pyrazole and its analogs: a review. Eur J Med Chem. 2016;120:170–201. doi: 10.1016/j.ejmech.2016.04.077. DOI: 10.1016/j.ejmech.2016.04.077. [DOI] [PubMed] [Google Scholar]
- 3.Alagarsamy V, Chitra K, Saravanan G, Solomon VR, Sulthana MT, Narendhar B. An overview of quinazolines: pharmacological significance and recent developments. Eur J Med Chem. 2018;151:628–685. doi: 10.1016/j.ejmech.2018.03.076. DOI: 10.1016/j.ejmech.2018.03.076. [DOI] [PubMed] [Google Scholar]
- 4.Guerrini G, Vergelli C, Cantini N, Giavannoni MP, Daniele S, Mascia MP, et al. Synthesis of new GABAA receptor modulator with pyrazolo [1,5-a] quinazoline (PQ) scaffold. Int J Mol Sci. 2019;20((6)):1438–1458. doi: 10.3390/ijms20061438. DOI: 10.3390/ijms20061438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hussein MA. Synthesis, anti-inflammatory, and structure antioxidant activity relationship of novel 4-quinazoline. Med Chem Res. 2013;22((10)):4641–4653. DOI: 10.1007/s00044-013-0468-9. [Google Scholar]
- 6.Kumar D, Kaur G, Negi A, Kumar S, Singh S, Kumar R. Synthesis and xanthine oxidase inhibitory activity of 5,6-dihydropyrazolo/pyrazolo[1,5-c] quinazoline derivatives. Bioorg Chem. 2014;57:57–64. doi: 10.1016/j.bioorg.2014.08.007. DOI: 10.1016/j.bioorg.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 7.Zhao H, Hu X, Cao K, Zhang Y, Zhao K, Tang C, et al. Synthesis and SAR of 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline derivatives as potent and selective CDK4/6 inhibitors. Eur J Med Chem. 2018;157:935–945. doi: 10.1016/j.ejmech.2018.08.043. DOI: 10.1016/j.ejmech.2018.08.043. [DOI] [PubMed] [Google Scholar]
- 8.Mohareb RM, Abdo NYM, Wardakhan WW. Synthesis and evaluation of pyrazolo[5,1-b]quinazoline-2-carboxylate, and its thiazole derivatives as potential antiproliferative agents and Pim-1 kinase inhibitors. Med Chem Res. 2017;26:2520–2537. DOI: 10.1007/s00044-017-1951-5. [Google Scholar]
- 9.El-Naggar M, Hasan AS, Awad HM, Mady MF. Design, synthesis and antitumor evaluation of novel pyrazolopyrimidines and pyrazoloquinazolines. Molecules. 2018;23((6)):1249–1268. doi: 10.3390/molecules23061249. DOI: 10.3390/molecules23061249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haghighijoo Z, Rezaei Z, Jaberipoor M, Taheri S, Jani M, Khabnadideh S. Structure based design and anti-breast cancer evaluation of some novel 4-anilinoquinazoline derivatives as potential epidermal growth factor receptor inhibitors. Res Pharm Sci. 2018;13((4)):360–367. doi: 10.4103/1735-5362.235163. DOI: 10.4103/1735-5362.235163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.RezaeeNasab R, Mansourian M, Hassanzadeh F, Shahlaei M. Exploring the interaction between epidermal growth factor receptor tyrosine kinase and some of the synthesized inhibitors using combination of in silico and in vitro cytotoxicity methods. Res Pharm Sci. 2018;13((6)):509–522. doi: 10.4103/1735-5362.245963. DOI: 10.4103/1735-5362.245963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borsook H, Dubnoff J. The biological synthesis of hippuric acid in vitro. J Biol Chem. 1940;132((1)):307–324. DOI: 10.1016/s0021-9258(18)73418-2. [PubMed] [Google Scholar]
- 13.Rostami M, Khosropour AR, Mirkhani V, Mohammadpoor-Baltork I, Moghadam M, Tangestaninejad S. [C6(MIm)2]2W10O32.2H2O: a novel and powerful catalyst for the synthesis of 4- arylidene-2-phenyl-5(4)-oxazolones under ultrasonic condition. C R Chimie. 2011;14(10):869–877. DOI: 10.1016/j.crci.2011.02.003. [Google Scholar]
- 14.Haneen DSA, Gouhar RS, Hashem HE, Youssef ASA. Synthesis and reactions of 4H-3,1-benzoxazin-4-one derivative bearing pyrazolyl moiety as antimicrobial and antioxidant agents. Synth Commun. 2019;49((21)):2840–2855. DOI: 10.1080/00397911.2019.1646288. [Google Scholar]
- 15.Hemdan MM, Youssef AS, El-Mariah FA, Hashem HE. Synthesis and antimicrobial assessments of some quinazolines and their annulated systems. J Chem Res. 2017;41((2)):106–111. DOI: 10.3184/174751917X14858862342269. [Google Scholar]
- 16.Alagarsamy V, Murugananthan G, Venkateshperumal R. Synthesis, analgesic, anti-inflammatory and antibacterial activities of some novel 2-methyl-3-substituted quinazolin-4-(3H)-ones. Biol Pharm Bull. 2003;26((12)):1711–1714. doi: 10.1248/bpb.26.1711. DOI: 10.1248/bpb.26.1711. [DOI] [PubMed] [Google Scholar]
- 17.Morris GM, Huey R, Olson AJ. Using autodock for ligand-receptor docking. Curr Protoc Bioinformatics. 2008;8((14)):Unit 8. doi: 10.1002/0471250953.bi0814s24. 14,1-40. DOI: 10.1002/0471250953.bi0814s24. [DOI] [PubMed] [Google Scholar]
- 18.Visualizer D. Accelrys Discovery Studio Visualizer 3.0. Available from: https://accelrys-discoverystudio-visualizer.software.informer.com/3.0/
- 19.El-Hachem N, Haibe-Kains B, Khalil A, Kobeissy FH, Nemer G. AutoDock and AutoDockTools for Protein-Ligand Docking: Beta-Site Amyloid Precursor Protein Cleaving Enzyme 1 (BACE1) as a Case Study. In: Neuroproteomics. Springer. 2017:391–403. doi: 10.1007/978-1-4939-6952-4_20. [DOI] [PubMed] [Google Scholar]
- 20.Asadi P, Khodarahmi G, Jahanian-Najafabadi A, Saghaie L, Hassanzadeh F. Synthesis, characterization, molecular docking studies and biological evaluation of some novel hybrids based on quinazolinone, benzofuran and imidazolium moieties as potential cytotoxic and antimicrobial agents. Iran J Basic Med Sci. 2017;20((9)):975–989. doi: 10.22038/IJBMS.2017.9260. DOI: 10.22038/IJBMS.2017.9260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma N, Banerjee J, Shrestha N, Chaudhury D. A review on oxazolone, it’s method of synthesis and biological activity. European J Biomed Pharm Sci. 2015;2((3)):964–987. [Google Scholar]
- 22.Puterová Z, Sterk H, Krutošíková A. Reaction of substituted furan-2-carboxaldehydes and furo[b]pyrrole type aldehydes with hippuric acid. Molecules. 2004;9((1)):11–21. doi: 10.3390/90100011. DOI: 10.3390/90100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hekal MH, Abu El-Azm FS. New potential antitumor quinazolinones derived from dynamic 2-undecyl benzoxazinone: synthesis and cytotoxic evaluation. Synth Commun. 2018;48((18)):2391–2402. DOI: 10.1080/00397911.2018.1490433. [Google Scholar]
- 24.Fortin S, Bérubé G. Advances in the development of hybrid anticancer drugs. Expert Opin Drug Discov. 2013;8((8)):1029–1047. doi: 10.1517/17460441.2013.798296. DOI: 10.1517/17460441.2013.798296. [DOI] [PubMed] [Google Scholar]
- 25.Asadi P, Khodarahmi G, Jahanian-Najafabadi A, Saghaie L, Hassanzadeh F. Biologically active heterocyclic hybrids based on quinazolinone, benzofuran and imidazolium moieties: synthesis, characterization, cytotoxic and antibacterial evaluation. Chem Biodivers. 2017;14((4)):e1600411. doi: 10.1002/cbdv.201600411. DOI: 10.1002/cbdv.201600411. [DOI] [PubMed] [Google Scholar]
- 26.Ansari AJ, Joshi G, Yadav UP, Maurya AK, Agnihotri VK, Kalra S, et al. Exploration of Pd-catalysed four-component tandem reaction for one-pot assembly of pyrazolo [1,5-c] quinazolines as potential EGFR inhibitors. Bioorg Chem. 2019;93:103314. doi: 10.1016/j.bioorg.2019.103314. 1-12. DOI: 10.1016/j.bioorg.2019.103314. [DOI] [PubMed] [Google Scholar]
- 27.Abbas SE, Barsoum FF, Georgey HH, Mohammed ER. Synthesis and antitumor activity of certain 2,3,6-trisubstituted quinazolin-4(3H)-one derivatives. Bull Fac Pharm Cairo Univ. 2013;51((2)):273–282. DOI: 10.1016/j.bfopcu.2013.08.003. [Google Scholar]
- 28.Kumar D, Mariappan G, Husain A, Monga J, Kumar S. Design, synthesis and cytotoxic evaluation of novel imidazolone fused quinazolinone derivatives. Arab J Chem. 2017;10((3)):344–350. DOI: 10.1016/j.arabjc.2014.07.001. [Google Scholar]
- 29.Abandansari HS, Abuali M, Nabid MR, Niknejad H. Enhance chemotherapy efficacy and minimize anticancer drug side effects by using reversibly pH-and redox-responsive cross-linked unimolecular micelles. Polymer. 2017;116:16–26. DOI: 10.1016/j.polymer.2017.03.062. [Google Scholar]
- 30.Verbeek BS, Adriaansen-Slot SS, Vroom TM, Beckers T, Rijksen G. Overexpression of EGFR and c-erbB2 causes enhanced cell migration in human breast cancer cells and NIH3T3 fibroblasts. FEBS Lett. 1998;425((1)):145–150. doi: 10.1016/s0014-5793(98)00224-5. DOI: 10.1016/s0014-5793(98)00224-5. [DOI] [PubMed] [Google Scholar]
- 31.Thirumurugan K, Lakshmanan S, Govindaraj D, Prabu DSD, Ramalakshmi N, Antony SA. Design, and anti-inflammatory activity of pyrimidine scaffold benzamide derivatives as epidermal growth factor receptor tyrosine kinase inhibitors. J Mol Struct. 2018;1171:541–550. DOI: 10.1016/j.molstruc.2018.06.003. [Google Scholar]
- 32.Zhu ML, Wang CY, Xu CM, Bi WP, Zhou XY. Evaluation of 6-chloro-N-[3,4-disubstituted-1,3-thiazol-2(3H)-ylidene]-1,3-benzothiazol-2-amine using drug design concept for their targeted activity against colon cancer cell lines HCT-116, HCT15, and HT29. Med Sci Monit. 2017;23:1146–1155. doi: 10.12659/MSM.899646. DOI: 10.12659/msm.899646. [DOI] [PMC free article] [PubMed] [Google Scholar]