

Abstract
Hepatitis B e antigen (HBeAg) is a soluble viral protein in plasma of patients with hepatitis B virus infection. HBeAg loss is an important first stage of viral antigen clearance. We determined the rate and predictors of HBeAg loss in a North American cohort with chronic hepatitis B viral infection (CHB). Among children and adults with CHB and without HIV, HCV or HDV co-infection enrolled in the Hepatitis B Research Network prospective cohort studies, 819 were HBeAg positive at their first assessment (treatment naïve or >24 weeks since treatment). Of these, 577 (200 children, 377 adults) were followed every 24–48 weeks. HBeAg loss was defined as first HBeAg negative value; sustained HBeAg loss was defined as ≥2 consecutive HBeAg negative values ≥24 weeks apart. During a median follow-up of 1.8 years, 164 participants experienced HBeAg loss, a rate of 11.4 (95% CI, 9.8–13.3) per 100 person-years. After adjustment for confounders, HBeAg loss rate was significantly higher in males than females, in older than younger individuals, in Whites or Blacks than Asians, in those with genotype A2 or B versus C, and in those with basal core promoter/precore mutations versus wildtype. Additionally, during follow-up, an ALT flare and a lower quantitative HBsAg, quantitative HBeAg or HBV DNA level predicted higher rates of HBeAg loss. The majority (88%) with HBeAg loss had sustained HBeAg loss. In conclusion, a number of specific demographic, clinical and viral characteristics impacted rate of HBeAg loss and may prove useful in design and interpretation of future therapeutic studies.
Keywords: hepatitis B, prospective cohort, e antigen
Introduction
Hepatitis B e antigen (HBeAg) is a soluble viral protein produced from the preCore/Core gene of hepatitis B virus (HBV) that has been associated with higher levels of viremia, as well as increased infectivity and hepatocellular carcinoma (HCC) risk (1–6). The clearance or loss of detectable HBeAg from the circulation is considered a crucial first step towards eventual HBV immune control (1). As a group, HBeAg negative patients have much lower HBV DNA and aminotransferase levels compared to HBeAg positive patients and are perceived to have better clinical outcomes over time (3). Therefore, HBeAg loss and hepatitis B e antibody (anti-HBe) development is a desired therapeutic goal and can lead to HBV (nucleoside analogue) medication discontinuation in certain circumstances (7,8). However, detailed prospective studies of host and viral factors associated with HBeAg loss among North American chronic hepatitis B (CHB) patients are lacking (9,10).
Cross-sectional analyses from the Hepatitis B Research Network (HBRN), identified features associated with HBeAg positivity (11), and quantitative HBeAg level among those with HBeAg positivity (12), respectively, among children and adults with CHB, but were unable to establish causality or evaluate associations between time-varying viral markers and HBeAg loss. The present observational study represents a longitudinal analysis of this large, well-characterized, multiracial HBeAg positive group. Our primary aim was to determine the rate and clinical predictors of HBeAg loss. Secondary aims were to determine the durability of HBeAg loss in the presence or absence of HBV medication and to describe the serological outcomes after HBeAg loss.
Participants and Methods
The HBRN is a National Institutes of Health-funded clinical research network of 21 adult and 7 pediatric clinical sites throughout the US and Canada, that enrolled hepatitis B surface antigen (HBsAg) positive pediatric (6 months to <18 years) and adult (≥18 years old) patients who were not currently on antiviral medication into prospective cohort studies between 2012 and 2017 (13). Participants underwent initial evaluation and then returned for follow-up assessments every 24 weeks for adults and 48 weeks for children, with a ± 12-week data collection window. The study protocols were approved by the institutional review boards of participating institutions and participants provided written, informed consent. For minors, written informed consent was provided by their parent/guardian and assent from patient was obtained whenever possible.
Both symptomatic and asymptomatic HBeAg positive participants of the HBRN Adult or Pediatric Cohort studies were eligible for inclusion in this study; those with acute HBV, co-infection with human immunodeficiency virus, the hepatitis C virus or hepatitis delta virus were excluded. In general, the first available HBeAg measurement after study enrollment was used to determine baseline status. However, among participants who took HBV nucleos(t)ide analogue medication within 24 weeks prior to enrollment, their first HBeAg measurement at least 24 weeks after HBV medication stopped was used.
The primary outcome was HBeAg loss, defined by a single HBeAg negative result (i.e., below lowest detectable value: 0.30 IU/mL) via serological testing performed at the central or local laboratory. If both central and local laboratory results were obtained on the same date, the central result was used. The secondary outcome, sustained HBeAg loss, was defined as at least two consecutive HBeAg negative results at least 24 weeks apart. Development of anti-HBe was not required for categorization of either outcome. However, anti-HBe was measured when possible.
Date of birth was used to calculate age on the day of baseline HBeAg status and age at baseline was categorized by decade; age groups above 50 years were collapsed due to the similarity in HBeAg prevalence and small numbers of participants (11). Race was self-reported. Presumed (i.e., most likely) mode of transmission was recorded by physician investigators after participant interview/assessment. The start and end dates of pregnancies, assessed throughout follow-up, were used to determine pregnancy status at time of HBeAg measurement. The start and stop dates of HBV medication were used to determine if participants had been on HBV medication at least 24 weeks in the prior 36 weeks (considered ‘yes’ to medication) at each assessment.
Measurement of serum aspartate aminotransferase (AST), alanine aminotransferase (ALT) and platelet counts were done at local laboratories. The AST to platelet ratio index (APRI) was calculated [(AST (U/L)/upper limit normal AST (U/L)) / PLT x 109] x 100 (14). The upper limit of the normal range (ULN) for ALT was “standardized” based on sex and age (i.e., ≤33 U/L for males and females ages < 1 year; ≤25 U/L for males and females ages 1 year-<13 years; ≤25 U/L for males and ≤22 U/L for females ages 13 years-<18 years (4); ≤30 U/L for adult males and ≤20 U/L for adult females) (8). An ALT flare was defined as ≥10xULN (15).
Genotyping of HBV was performed by the Centers for Disease Control and Prevention using mass spectrometry (MALDI-TOF) of a 441bp fragment of the S gene containing the a-determinant (16). Pre-core (PC) and basal core promoter (BCP) variants were similarly determined by sequencing of the relevant regions of the viral genome, to detect the following variants: A1762T and G1764A (BCP), and G1896A (PC) (17). CHB phenotype was determined from HBeAg status, ALT and HBV DNA as described in the Appendix (18).
HBV DNA and quantitative HBsAg and HBeAg testing was done centrally at a HBRN-funded virology laboratory (University of Washington, Seattle, WA) as previously described (11). Briefly, quantitative HBeAg and HBsAg levels were measured by Elecsys HBeAg II Quant and Elecsys HBsAg II Quant assay, respectively (Roche Molecular Systems, Inc) (19,20). The lowest detectable value for HBV DNA was 10 IU/mL, for HBsAg was 0.05 IU/mL and for HBeAg was 0.30 IU/mL; the lowest quantifiable value for HBV DNA was 20 IU/mL Qualitative assays for HBsAg, hepatitis B surface antibody (anti-HBs), HBeAg and anti-HBe were performed locally using commercially available ELISA assays. To supplement missing local anti-HBe results, anti-HBe testing was performed centrally at UT Southwestern Medical Center at Dallas, specifically for assessments near the time when participants first met the definition of HBeAg loss, as well as the following assessment and the last assessment, when stored sera were available.
The database was maintained by the data coordinating center at the University of Pittsburgh. The authors had all the study data available to them while developing this manuscript.
Statistical analyses
The full analysis plan is provided in the Appendix. Briefly, the HBeAg loss rate was estimated by dividing the number of participants’ first HBeAg negative result known to have occurred by the number of person-years of observation. Follow-up was censored 1) the day after a participant’s last HBeAg measurement which was followed by 60 or more weeks without another HBeAg measurement, or 2) the day after a participant’s first HBeAg negative measurement, whichever came first. Kaplan-Meier curves were used to visualize the cumulative probability of HBeAg loss over time by sex, age, race, genotype, BCP/PC mutations and baseline phenotype. Cox Proportional Hazards regression was used to report the hazard ratio (HR) of HBeAg loss by static and time-varying participant characteristics. Time-varying characteristics included pregnant (yes/no), HBV medication (yes/no), ALT flare (yes/no), quantitative HBeAg (log10 IU/mL), quantitative HBsAg (log10 IU/mL) and HBV DNA (log10 IU/mL). Because we were interested in predicting the outcome rather than measuring associations, the value of each time-varying characteristic at the prior assessment (i.e., approximately 24 weeks before HBeAg measurement) was utilized.
Results
Study cohort and follow-up
Study flow from HBRN enrollment to inclusion is reported in Figure 1. Among the 819 HBeAg positive participants without acute HBV or co-infection, 242 did not meet the follow-up criteria, leaving 577 participants in the study sample. Prior to censorship participants were followed for a median of 4.7 years (IQR: 2.2–6.5; range: 0.5–7.9). After censorship, participants were followed for a median of 1.8 years (IQR: 1.0–3.7; range: 0.1–7.7), with a median of 4 HBeAg measures (IQR: 3–8; range: 2–21).
Figure 1.
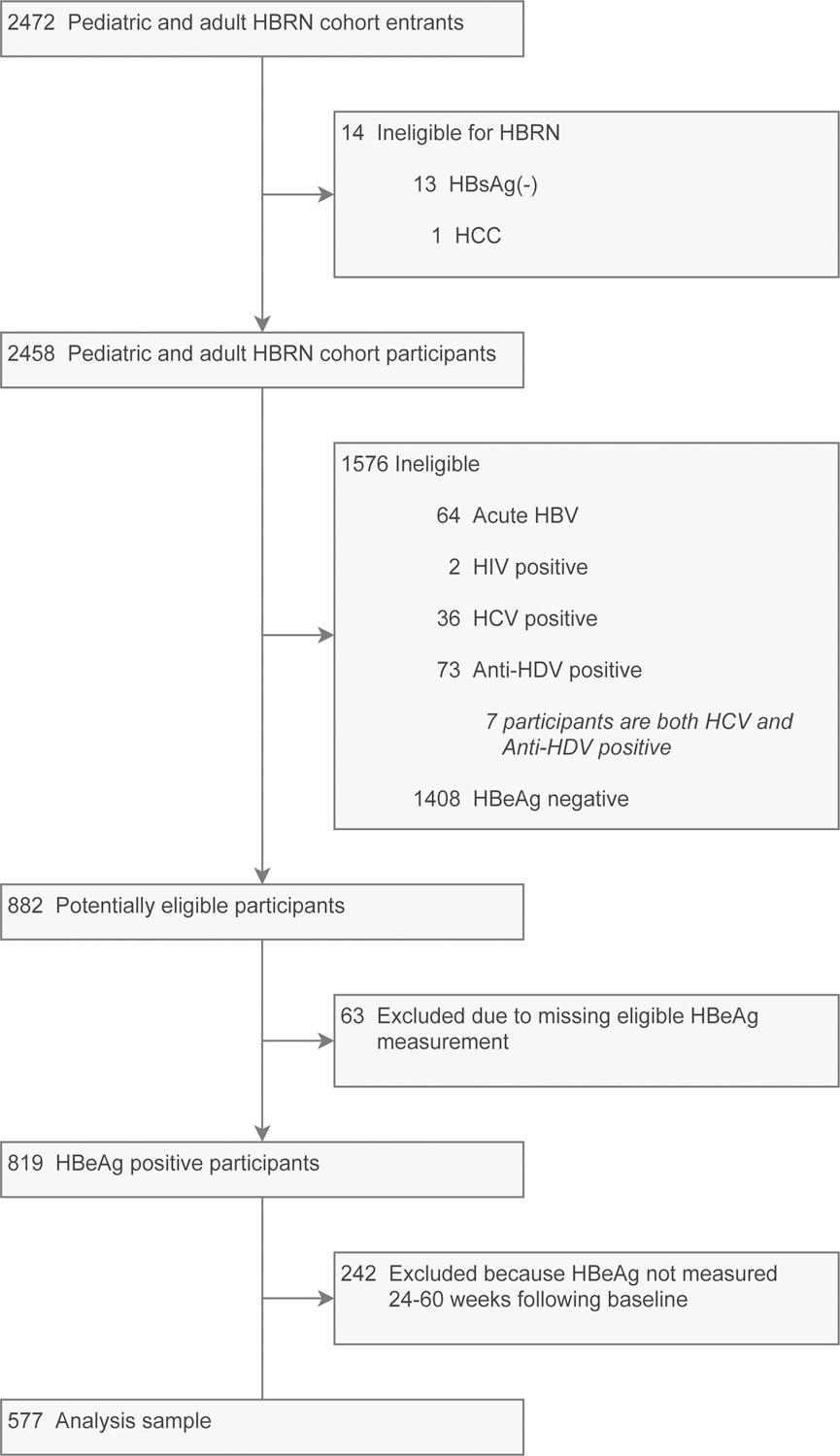
Flow of participants from study enrollment to analysis sample.
Participant characteristics
Table 1 outlines the participant baseline characteristics, which included 200 children (35%) and 377 adults (65%). Median age was 27.2 years and 58% were females. Asians were the largest racial group at 85%; 80% of participants were presumed to have vertical HBV transmission. HBeAg+ immune active phenotype was most common (78%), followed by immune tolerant (15%)(13). Genotypes B (38%) and C (44%), most common in East and Southeast Asia, predominated, likely reflecting the sample’s racial distribution. One-third (33%) of participants, despite being HBeAg positive, had either one or both PC or BCP mutations. Eighty-two percent had elevated ALT levels. As part of regular clinical care, HBV medication was initiated in one third (195; 34%) of participants during follow-up; 188 participants received nucleoside analogue medication alone, while 4 received pegylated interferon alone and 3 received both.
Table 1.
Baseline characteristics of North American children and adults with HBeAg positive CHB.
| Variable | Total (n=577) |
|---|---|
| Female, n/total (%) | 333/577 (57.7%) |
| Age group, years, n (%) | n=577 |
| 0.5–10 | 100 (17.3%) |
| >10–20 | 109 (18.9%) |
| >20–30 | 117 (20.3%) |
| >30–40 | 122 (21.1%) |
| >40–50 | 77 (13.3%) |
| >50 | 52 (9.0%) |
| Race, n (%) | n=577 |
| White | 35 (6.1%) |
| Black | 36 (6.2%) |
| Asian | 490 (84.9%) |
| Other/Mixed | 16 (2.8%) |
| Presumed mode of HBV transmission, n (%) | n=453 |
| Vertical | 363 (80.1%) |
| Horizontal | 89 (19.6%) |
| Other | 1 (0.2%) |
| Phenotype, n (%) | n=538 |
| Immune tolerant | 82 (15.2%) |
| HBeAg+ active CHB | 419 (77.9%) |
| Indeterminant A | 37 (6.9%) |
| HBV Genotype, n (%) | n=537 |
| A1 | 18 (3.4%) |
| A2 | 30 (5.6%) |
| B | 202 (37.6%) |
| C | 237 (44.1%) |
| D | 40 (7.4%) |
| E | 9 (1.7%) |
| Other or multiple | 1 (0.2%) |
| BCP/PC Mutation (A1762T, G1764A, G1896A), n (%) | n=440 a |
| Wild type | 294 (66.8%) |
| BCP only | 111 (25.2%) |
| PC only | 26 (5.9%) |
| BCP & PC | 9 (2.0%) |
| ALT, xULN | n=572 |
| Median (25th%-ile:75th%-ile) | 1.8 (1.2: 3.1) |
| Min: Max | 0.4: 56.6 |
| ALT flare (≥10xULN), n/total (%) | 30/572 (5.2%) |
| Prior antiviral medication, n/total (% | 80/577 (13.9%) |
| HBV DNA, log10 IU/mL2 | n=546 |
| Median (25th%-ile:75th%-ile) | 8.1 (7.2: ALD) |
| Min: Max | BLD: ALD |
| Serum HBsAg, log10 IU/mL | n=505 |
| Median (25th%-ile:75th%-ile) | 4.5 (3.9: 4.8) |
| Min: Max | −0.6: 5.9 |
| Serum HBeAg, log10 IU/mL2 | n=512 |
| Median (25th%-ile:75th%-ile) | 3.2 (2.2: 3.3) |
| Min: Max | BLD: 4.1 |
Abbreviations: ALD, above the level of detection; ALT, Alanine aminotransferase; APRI, aspartate aminotransferase to platelet ratio index; BCP, basal core promoter; BLD, below the limit of detection; CHB, chronic Hepatitis B virus; DNA, deoxyribonucleic acid; HBeAg, hepatitis B e-antigen; HBsAg, hepatitis B surface antigen; PC, Pre-core; ULN, upper limit of normal.
BCP/PC mutation could not be amplified in 115 participants.
HBV DNA BLD ≤1.0 log10 IU/mL; HBV DNA ALD >8.23 log10 IU/mL; serum HBeAg BLD ≤−0.5 log10 IU/mL.
HBeAg loss rates
Among the 577 participants, 164 experienced HBeAg loss during 1433 person-years (PY) of follow-up, a rate of 11.4 (95% CI, 9.8–13.3) per 100 PY (Table 2). In a sensitivity analysis, in which data was censored the day after HBV medication use was initiated (no matter the duration), 91 participants experienced HBeAg loss during 977 PY of follow-up, a rate of 9.3 (95% CI, 7.6–11.4) per 100 PY.
Table 2.
Rates of HBeAg loss among North American children and adults with CHB by baseline characteristics.
| # participants contributing | # HBeAg negative | Person-years of follow-up | HBeAg loss per 100 person-yrs (95% CI) |
|
|---|---|---|---|---|
| Overall | 577 | 164 | 1433.4 | 11.4 (9.8–13.3) |
| Sex | ||||
| Male | 244 | 82 | 576.3 | 14.2 (11.5–17.7) |
| Female | 333 | 82 | 857.1 | 9.6 (7.7–11.9) |
| Age groups, years | ||||
| 0.5 – 10 | 100 | 9 | 209.9 | 4.3 (2.2–8.2) |
| >10 – 20 | 109 | 19 | 234.8 | 8.1 (5.2–12.7) |
| >20 – 30 | 117 | 35 | 333.3 | 10.5 (7.5–14.6) |
| >30 – 40 | 122 | 39 | 314.5 | 12.4 (9.1–17.0) |
| >40 – 50 | 77 | 35 | 193.5 | 18.1 (13.0–25.2) |
| >50 | 52 | 27 | 147.5 | 18.3 (12.6–26.7) |
| Race | ||||
| Asian | 490 | 132 | 1260.3 | 10.5 (8.8–12.4) |
| Black | 36 | 12 | 59.2 | 20.3 (11.5–35.7) |
| White | 35 | 16 | 71.2 | 22.5 (13.8–36.7) |
| Genotype | ||||
| A1 | 18 | 5 | 30.8 | 16.2 (6.8–39.0) |
| A2 | 30 | 18 | 47.4 | 37.9 (23.9–60.2) |
| B | 202 | 63 | 492.7 | 12.8 (10.0–16.4) |
| C | 237 | 62 | 651.7 | 9.5 (7.4–12.2) |
| D | 40 | 8 | 123.3 | 6.5 (3.2–13.0) |
| E | 9 | 2 | 21.1 | 9.5 (2.4–38.0) |
| BCP/PC mutation | ||||
| Wild type | 294 | 48 | 778.5 | 6.2 (4.6–8.2) |
| BCP only | 111 | 39 | 289.5 | 13.5 (9.8–18.4) |
| PC with or without BCP | 35 | 21 | 68.7 | 30.6 (19.9–46.9) |
| Baseline Phenotype a | ||||
| HBeAg+ immune active | 419 | 117 | 1104.8 | 10.6 (8.8–12.7) |
| Immune tolerant | 82 | 12 | 203 | 5.9 (3.4–10.4) |
| HBeAg+ indeterminate | 37 | 27 | 56.6 | 47.7 (32.7–69.6) |
Abbreviations: BCP, basal core promoter; CHB, chronic Hepatitis B virus; PC, Pre-core.
Immune tolerant: HBV DNA ≥105 IU/mL and ALT normal; HBeAg+ immune active: HBV DNA ≥105 IU/mL and ALT elevated; HBeAg+ indeterminate: HBV DNA <105 IU/mL, regardless of ALT level.
HBeAg loss rate per 100 PY was higher in males (14.2, 95% CI, 11.5–17.7) versus females (9.6, 95% CI, 7.7–11.9), older individuals (e.g., 18.3, 95% CI, 12.6–26.7, for those over 50 years, versus 4.3, 95% CI, 2.2–8.2, for those 10 years and younger), and Whites (22.5, 95% CI, 13.8–36.7) and Blacks (20.3, 95% CI, 11.5–35.7) versus Asians (10.5, 95% CI, 8.8–12.4). HBeAg loss rate among those with vertical transmission (9.1, 95% CI, 7.3–11.3) was similar to Asians, and the rate among those with horizontal transmission (16.3, 95% CI, 11.9–22.4) was similar to Whites and Blacks. Given the strong association between race and genotype (supplemental material, sTable 1), we were unable to estimate the effect of genotype and race, independent of the other. Those with genotype A2 (57% White and 32% Black) demonstrated higher HBeAg loss rate than those with genotypes B and C (both 100% Asian), as well as D (59% Asian) and E (100% Black). Comparisons with genotypes A1 (61% Black) and E were limited by low case frequencies. Those having either BCP or PC mutations, but particularly PC, had higher HBeAg loss rates (13.5, 95% CI, 9.8–18.4, for BCP only and 30.6, 95% CI, 19.9–46.9, for PC, regardless of whether BCP was also present) versus wild type HBV (6.2, 95% CI, 4.6–8.2). Finally, those with phenotype indeterminate A (HBeAg positive, HBV DNA < 105 IU/L; 13) had a higher HBeAg loss rate (47.7, 95%CI, 32.7–69.6) than either active CHB (10.6, 95%CI, 8.8–12.7) or immune tolerant participants (5.9, 95%CI, 3.4–10.4).
Figure 2 shows the cumulative probability of HBeAg loss during follow-up according to sex (A), age group (B), race (C), HBV genotype (D), presence of PC/BCP mutations (E) and phenotype (F). Estimates were truncated once groups had fewer than 10 participants at risk.
Figure 2:
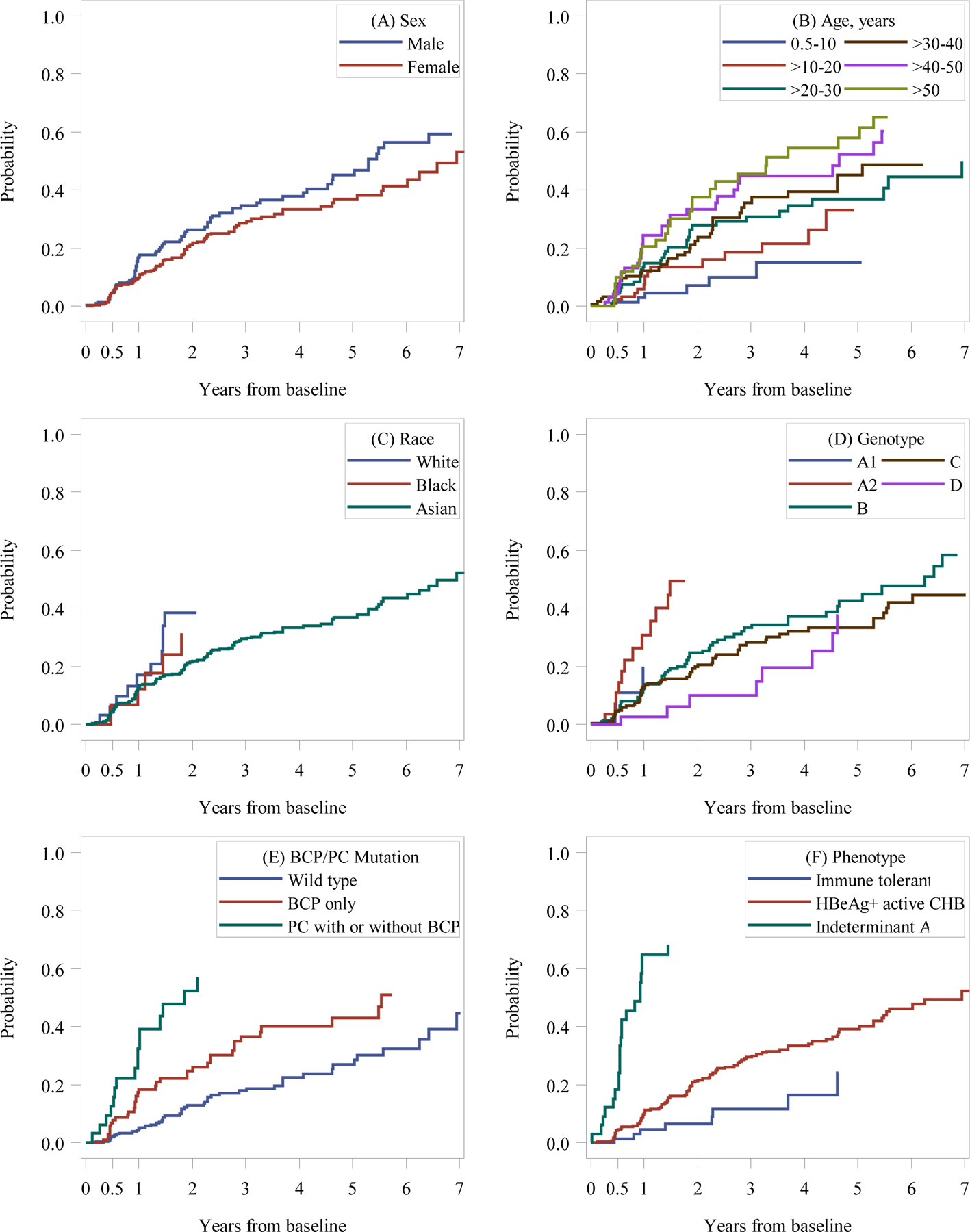
Cumulative probability of HBeAg loss among North American children and adults with CHB by baseline characteristics.
Footnote: Abbreviations: BCP, basal core promoter; CHB, chronic Hepatitis B virus; PC, Pre-core.
Kaplan-Meier curves are truncated when fewer than 10 participants are at risk.
Predictors of HBeAg loss
Hazard Ratios (HRs) for HBeAg loss (i.e., ratios of HBeAg loss rate between groups, such that a HR >1 indicates less time to HBeAg loss) are shown in Table 3. Adjustment for potential confounders (e.g., HBV medication use) had minimal effects on estimated associations. For example, with or without adjustment, the HBeAg loss HR was 1.4–1.5 times higher in males versus females, approximately 4 times higher in those who were >40–50 or >50 versus ≤ 10 years old, and approximately 2 times higher for White or Black versus Asian race. With adjustments, the HBeAg loss HR was approximately 1.5 times higher in genotype B versus C, and 3 times higher in genotype A2 versus C, approximately 2 and 6 times higher in the presence of BCP-only and PC (±BCP) mutations respectively, versus wildtype, and approximately 6 times higher in those with phenotype HBeAg+ indeterminate versus HBeAg+ immune active. Additionally, having an ALT flare, and lower versus higher quantitative HBsAg, quantitative HBeAg or HBV DNA level throughout follow-up, predicted a higher HBeAg loss rate in the 24 weeks that followed. For example, the HBeAg loss HR was approximately 4 times higher in the 24 weeks following an ALT flare and reduced by one third in the 24 weeks following HBsAg assessment for every log10 IU/mL higher HBsAg.
Table 3.
Hazard ratios (HRs)a of HBeAg loss among North American children and adults with CHB by participant characteristics.
| Unadj. HR (95% CI) | Adj. HR (95% CI)b N=532 |
Adj. HR (95% CI)c | |
|---|---|---|---|
| Sex (ref=Female) | n=577, P=0.01 | P=0.04 | |
| Male | 1.49 (1.10–2.02) | 1.44 (1.02–2.02) | |
| Age group, years (ref=0.5–10) | n=577, P<0.01 | P<0.01 | |
| >10 – 20 | 1.89 (0.86–4.19) | 1.75 (0.71–4.27) | |
| >20 – 30 | 2.56 (1.23–5.34) | 2.64 (1.14–6.10) | |
| >30 – 40 | 2.95 (1.43–6.10) | 3.29 (1.44–7.49) | |
| >40 – 50 | 4.36 (2.10–9.09) | 4.33 (1.87–10.03) | |
| >50 | 4.49 (2.11–9.55) | 3.94 (1.62–9.60) | |
| Pregnant (ref=no/unknown/NA) | n=577, P=0.54 | P=0.45 | |
| Yes, 24 weeks prior | 1.43 (0.46–4.50) | 1.58 (0.48–5.15) | |
| Race (ref= Asian) | n=561d, P<0.01 | n=556, P<0.01 | |
| Black | 1.93 (1.07–3.51) | 1.90 (1.01–3.57) | |
| White | 2.16 (1.28–3.63) | 2.32 (1.32–4.07) | |
| Genotype (ref=C) | n=536d, P<0.0001 | P<0.01 | |
| A1 | 1.64 (0.66–4.09) | 1.52 (0.61–3.83) | |
| A2 | 3.93 (2.32–6.68) | 2.98 (1.67–5.30) | |
| B | 1.33 (0.94–1.89) | 1.46 (1.02–2.09) | |
| D | 0.69 (0.33–1.44) | 0.95 (0.44–2.07) | |
| E | 1.03 (0.25–4.21) | 1.34 (0.33–5.56) | |
| BCP/PC mutation (ref=wildtype) | n=440, P<0.0001 | n=416, P<0.0001 | |
| BCP only | 2.19 (1.43–3.34) | 2.34 (1.46–3.75) | |
| PC with or without BCP | 4.91 (2.94–8.22) | 5.89 (3.34–10.39) | |
| Phenotype (ref= HBeAg+ immune active) | n=538, P<0.0001 | n=508, P<0.0001 | |
| Immune tolerant | 0.55 (0.30–.99) | 0.58 (0.31–1.08) | |
| HBeAg+ indeterminant | 4.59 (3.10–7.01) | 5.74 (3.63–9.08) | |
| HBV treatment (ref=No) | n=577, P=0.53 | P=0.16 | |
| Yes ≥24wks in past 36 weeks | 1.11 (0.80–1.53) | 0.78 (0.55–1.10) | |
| ALT flare (≥10xULN) (ref=no) | n=572, P<0.0001 | P<0.0001 | |
| Yes, 24 weeks prior | 4.47 (2.76–7.23) | 4.40 (2.65–7.31) | |
| Serum HBsAg (log10 IU/mL) | n=510, P<0.0001 | n=484, P<0.0001 | |
| 24 weeks prior | 0.74 (0.66–0.84) | 0.67 (0.58–0.77) | |
| Serum HBeAg (log10 IU/mL) | n=512, P<0.0001 | n=486, P<0.0001 | |
| 24 weeks prior | 0.52 (0.46–0.59) | 0.46 (0.39–0.53) | |
| HBV DNA (log10 IU/mL) | n=567, P<0.0001 | n=527, P<0.0001 | |
| 24 weeks prior | 0.90 (0.85–0.94) | 0.73 (0.67–0.80) |
Abbreviations: ALT, Alanine aminotransferase; BCP, basal core promoter; CHB, chronic Hepatitis B virus; PC, Pre-core.
The ratio of the rate at which participants experience HBeAg loss where a faster rate suggests a shorter time of HBeAg positivity. Thus, a value >1 indicates less time to HBeAg loss.
Estimate are from one multivariable model that includes sex, age group, pregnancy, genotype, treatment and ALT flare.
Each estimate is from a unique model with adjustment for sex, age group, pregnancy, genotype, treatment and ALT flare with two exceptions; race is not adjusted for genotype; phenotype is not adjusted for ALT flare.
Does not include those in “other/mixed” race (n=16), “other” genotype categories (n=1).
Approximately one-fifth (695; 19%) of follow-up HBeAg measurements were taken among participants who had been on HBV medication at least 24 weeks in the preceding 36 weeks. HBV medication use was not associated with HBeAg loss rate (Table 3). To address the possibility that inclusion of females who were pregnant might impact the estimated effect of treatment (since many pregnant females are placed on nucleos(t)ide analogues solely for lowering viral load prior to delivery) or other factors, as a sensitivity analysis, modeling was repeated excluding females who were pregnant at baseline (n=36) and censoring follow-up data of females once they became pregnant (n=19). To address a concern that the definition of HBV medication use might impact the estimated effect of treatment, modeling was repeated with alternative definitions of HBV medication use (e.g., any HBV medication use in the past 24 weeks). The lack of an association between HBV medication use and HBeAg loss held with all definitions (data not shown).
Sustained HBeAg loss
Among the 164 participants with HBeAg loss on at least one occasion, 15 (9%) did not have a second HBeAg value at least 24 weeks following their first HBeAg negative result. Among the 149 participants who did, HBeAg was measured at least every 60 weeks for a median of 2.4 years (IQR: 1.0–4.5; range: 0–7.2). In this timeframe, 131 (88%) participants met the definition of sustained HBeAg loss: in follow-up, 106 had no HBeAg positive values following their first HBeAg negative value (group 1), 20 had at least one later HBeAg positive value before eventually sustaining HBeAg loss through follow-up (group 2), and 5 were HBeAg positive at the end of follow-up (group 3). The percentage of participants in each of the these groups, as well as among those who never achieved sustained HBeAg loss (group 4; n=18), whose HBeAg loss occurred spontaneously versus following HBV medication is reported in supplemental material sTable 2.
Associations with sustained HBeAg loss mimicked associations with HBeAg loss (supplemental material sTable 3). As with HBeAg loss, HBV medication use did not predict sustained HBeAg loss rate (adjHR=1.04, 95%CI, 0.71–1.53).
Most of the 56 participants who experienced spontaneous sustained HBeAg loss (i.e., from groups 1 and 2), had undetectable viral loads and quiescent disease by end of follow-up: 5 were on HBV medication, 3 had experienced HBsAg loss, 22 were inactive, 7 demonstrated HBeAg- immune active hepatitis and 19 were HBeAg- indeterminate in phenotype, mostly HBV DNA low. Participants who were on HBV medication prior to HBeAg loss were generally maintained on medication with low ALT/DNA levels throughout follow-up.
Anti-HBe status among those with HBeAg loss and sustained HBeAg loss
Two-thirds (67%) of participants with HBeAg loss (107 of 160 tested) were anti-HBe positive at the time of initial HBeAg loss. An additional 17% (28 of 162 tested) were anti-HBe positive at least once during follow-up, totaling 83% (135 of 162) of participants. However, at participants’ final anti-HBe assessment following HBeAg loss, only 70% (104 of 148 tested) were anti-HBe positive. Anti-HBe positivity was not significantly different at time of first HBeAg negative value by whether HBeAg loss was spontaneous or followed HBV medication use (e.g., 74% vs. 60% positive, respectively, at time of initial HBeAg loss; p=.17). Anti-HBe positivity at least once during follow up was similar among participants with sustained HBeAg loss: 89% (115 of 130 tested).
HBsAg loss
Among the 577 HBeAg positive participants, 15 (2.5%) had one or more HBsAg negative values during follow-up. Eleven of these 15 participants’ first HBsAg negative value was measured a median of 2.4 years (IQR: 1.8–4.2) following their first HBeAg negative value (supplemental material sTable 2). Of the remaining four participants, one was first detected as HBsAg and HBeAg negative on the same day, and three while HBeAg positive. However, in all four cases, the positive HBeAg values were close to the lower limit of detection, and some of the negative HBsAg values were followed by HBsAg positive values (sFigure 1).
Discussion
In this large North American cohort of children and adults with HBeAg positive chronic HBV who were not on HBV medication at study entry, the overall HBeAg loss rate was 11.4 per 100 person-years but varied greatly by participant characteristics. For example, the rate was 4 times higher in those over 40 years old versus those 10 years old or younger. HBeAg loss also occurred at a faster rate in males versus females, White or Black versus Asian race, genotype A2 and B versus C, and in those with PC and BCP mutations versus wildtype HBV. Additionally, an ALT flare was associated with four times greater rate of HBeAg loss in the ensuing 24 weeks. Lower quantitative HBsAg, serum HBV DNA and HBeAg also predicted faster rate of HBeAg loss; these associations were independent of sex, age group, genotype, pregnancy, HBV medication and ALT flare.
Since HBV medication use was an exclusion at study entry, this observational study largely represents a natural history of CHB in children and adults. Over the course of the observational study, however, and at the discretion of site clinicians, one third of participants were placed on HBV medication for a minimum of 24 weeks and approximately one-fifth of follow-up HBeAg measurements were taken while participants were on HBV medication. Medication use did not predict HBeAg loss or sustained HBeAg loss in unadjusted or adjusted analyses. Furthermore, a sensitivity analysis demonstrated that HBeAg loss rate was similar (the point estimate was slightly lower, but the 95% CIs overlapped) with censorship of assessments following HBV medication initiation. However, it is difficult to evaluate the effect of HBV medication in an observational study design in which participants start and stop various regimens of HBV medication based on differing criteria, including patient preference, and at varying time intervals (21,22). Clinical trials have shown that interferon and nucleos(t)ide analogues increase HBeAg loss rate compared to placebo or no treatment among patients with immune active CHB (23).
One factor affecting the overall HBeAg loss rate in this cohort is the preponderance of Asians (85%) and consequently, genotypes B (38%) and C (44%). Specifically, HBeAg loss occurred at a faster rate in Whites and Blacks versus Asians, and in genotype A2 and B versus C. Unfortunately, given the strong association between race and genotype, we were unable to estimate the effect of each factor, independent of the other. However, when analysis was limited to Asians, HBeAg loss occurred faster in genotype B versus C (data not shown).
Age was also a key variable determining rate of HBeAg loss. For example, compared with those under 10 years of age, age >10–20 years was associated with almost a 2-fold increase, and age >50 years with a 4-fold increase in rate of HBeAg loss. Among the 200 pediatric participants, we could not determine specific age or puberty status thresholds that differentiated HBeAg loss rate better than natural decades, thus we used this simplified schema in analysis.
Our study provides the first evidence that males may have a higher rate of HBeAg loss than females. However, since the lower end of the HR 95% confidence interval was 1.02, the difference by sex may not be clinically meaningful. Given this finding, and unrelated observations suggesting that females are more likely to clear acute HBV infection compared to males (24,25), additional research is needed to determine whether significant sex differences in HBeAg loss occur in either acute or chronic infection.
The HBRN previously reported PC and BCP mutations are more common in HBeAg negative patients but occur in a proportion of HBeAg positive patients in association with older age and lower HBV DNA levels (17). In the present study we quantified the increased rate of HBeAg loss associated with PC and BCP mutations, independent of age and other potential confounders, consistent with previous work (26–29). Specifically, among the subset of participants whose BCP/PC mutation status was known, BCP alone (versus wildtype) was independently associated with > 2-fold higher HBeAg loss rate, while PC mutation with or without BCP mutation was independently associated with greater than 5-fold higher HBeAg loss rate.
While nearly one-fourth of participants with HBeAg loss had at least one later HBeAg positive result, over half of these cases appeared to be due to small changes in qHBeAg over time that were close to the limit of detection. Still, there were a minority of patients with more curious patterns of HBeAg over time, including four cases with very high HBeAg levels following HBeAg negative results (i.e., a subgroup of group 4), calling into question whether these may have represented false negatives or errors in the process of sample collection. Data from other studies have indicated that ~95% of patients remain HBeAg negative after initial HBeAg loss (28,29); however, duration of follow-up and number of subsequent HBeAg tests as well as definition of sustained HBeAg loss vary across studies. In our study, sustained HBeAg loss, defined as at least 2 negative HBeAg results at least 24 weeks apart, was observed in 85% after a median follow-up of 2.4 years from initial HBeAg loss. Additionally, among 2749 HBeAg measurements that were available from both the central laboratory and a local laboratory on the same day, kappa was 0.91 (95% CI: 0.89 – 0.93), indicating excellent or almost perfect agreement (30,31). Previous studies relying only on qualitative HBeAg testing may have been less sensitive than the quantitative assay we used. Notwithstanding, our results indicate that once HBeAg negativity is achieved it is generally well-sustained in those experiencing either spontaneous or antiviral induced HBeAg loss, independently of anti-HBe seroconversion.
Limitations of our study include the potential for patient selection bias. The current cohort, though a large one, may not be representative of the general population of mono-infected patients with HBeAg positive CHB in terms of age distribution, pregnancy status and other factors, which were likely influenced by limiting enrollment to untreated patients with evidence of CHB seeking medical care at specific medical centers, and targeted enrollment of specific adult subgroups (e.g., pregnant females, those experiencing an ALT flare)(13). The preponderance of Asian participants in the cohort reflects, in large part, the CHB community in North America, but Blacks or other minorities with CHB were likely under-represented (21), reflecting the clinic population of academic medical centers. Furthermore, approximately 30% of participants who were identified as HBeAg positive without co-infection were excluded from this report due to lack of follow-up data. Thus, the overall HBeAg loss rate reported in this study should be interpreted with caution. Conversely, the adjusted associations between participant demographic and clinical characteristics and HBeAg loss rate are likely generalizable to children and adults with HBeAg positive chronic HBV residing in North America.
Additional study limitations include that our study design precluded a clear understanding of the effect of HBV medication on HBeAg loss, and that our definition of initial and sustained HBeAg loss did not include development of anti-HBe, as these data, primarily from local laboratories, were not consistently available throughout follow-up for the entire cohort. However, 83% of participants with HBeAg loss were anti-HBe positive during follow-up. The sustained development of anti-HBe has been proposed by AASLD and EASL as required before antiviral medications can be discontinued (7,8). Finally, there were relatively few clinical outcomes (e.g., HCC, death from cirrhosis), limiting interpretation of the role of HBeAg sero-conversion in these outcomes. The strengths of our study include a large number of participants, wide age range, diverse races and HBV genotypes, and the use of quantitative HBeAg as a measure of HBeAg loss.
In conclusion, this study offers important information regarding the evolution of HBeAg positive CHB. Among HBeAg positive patients, older, and non-Asian patients evolve to HBeAg loss faster, as do those with BCP and PC mutations versus wildtype HBV. Additionally, an ALT flare, and lower levels of HBsAg, HBV DNA and HBeAg predict faster HBeAg loss. These data may prove useful in the design of future studies aiming to increase the rate of HBeAg and HBsAg clearance.
Supplementary Material
Statement of Significance.
Hepatitis B e antigen (HBeAg) is a virus-produced protein associated with high viral replication. HBeAg loss from the circulation is an essential step in hepatitis B virus clearance. In a large prospective cohort study of North American children and adults with chronic hepatitis B (N=577), rate of HBeAg loss was 11.4 per 100 person-years. Male sex, older age, white or black race versus Asian, genotype A2 or B versus C, basal core promoter/precore mutations, ALT flare, and lower HBsAg, HBeAg and HBV DNA levels predicted faster clearance. The present study establishes a framework for understanding eventual hepatitis B virus clearance.
Acknowledgments:
In addition to the authors, the HBRN would like to acknowledge the contributions of the following: Harvard Consortium: Jianghe Niu, PhD, Asad Javaid, MBBS, Bilal Nasir, MBBS, Ammu Susheela, MBBS, Imad Nasser, MD (Beth Israel Deaconess Medical Center, Boston, MA), Arley Donovan, Nifasha Rusibamayila, Cara Foley (Massachusetts General Hospital, Boston, MA). Minnesota Alliance for Research in Chronic Hepatitis B: Alisha C. Stahler, Linda Stadheim, RN (Mayo Clinic Rochester, Rochester, MN), John Lake, MD, Philip Lacher (University of Minnesota, Minneapolis, MN), Shannon M. Riggs, LPN, AS (Department of Pediatrics, University of Minnesota Masonic Children’s Hospital, Minneapolis, MN). Midwest Hepatitis B Consortium: Kathryn Rushing, RN, Rosemary A. Nagy, RDN, LD, MBA, Jacki Cerkoski, RN, MSN (Saint Louis University School of Medicine, St Louis, MO), Debra DeMarco Shaw, RN, BSN, Lisa Kessels, RN, Michael K. Klebert, PhD, RN, ANP-BC (Washington University School of Medicine, St. Louis, MO). University of Toronto Consortium: Seham Noureldin, PhD, Danie La, RN, Lucie Liu, MSc, CCRP, Diana Kaznowski, RN, Jiayun Chen, Fengfei Huang, Doinita Vladutu, Orlando Cerocchi (Toronto General Hospital, Toronto, Ontario), Athena Hau, BSc (Department of Paediatrics, The Hospital for Sick Children, University of Toronto, Toronto, Ontario). HBV CRN North Texas Consortium: Debra Rowan, LVN (Division of Digestive and Liver Diseases, University of Texas Southwestern Medical Center at Dallas, Dallas, TX), Sheila Bass (University of Texas Southwestern, Dallas, TX), Barbara Lilly, BS (Baylor University Medical Center, Dallas, TX), Laurie A. Rodgers-Augustyniak, RN, Shirley Montanye, RN (Department of Pediatrics, UTSW, Dallas, TX). Los Angeles Hepatitis B Consortium: Samuel French, MD, Velma Peacock, RN (David Geffen School of Med, UCLA, Los Angeles, CA). San Francisco Hepatitis B Research Group Consortium: Marion Peters, MD, Ashley Shobe, MS, Rayshawnda Davis, Romuald Kuras, Claudia Ayala, MS, Ivy Lau, BS (University of California-San Francisco, San Francisco, CA), Veronika Podolskaya, BS, NCPT, Anna von Bakonyi, LVN, CCRC, Nata DeVole, RN (California Pacific Medical Center Research Institute, San Francisco, CA), Natasha Feier, MS, Joel Feier, BS, Camille Langlois, MS (Department of Pediatrics, UCSF, San Francisco, CA). Michigan Hawaii Consortium: Barbara McKenna, MD, Karen Choi, MD, Kelly Oberhelman, PAC, Sravanthi Kaza, Bpharm, Isabel Moran (University of Michigan, Ann Arbor, MI), Leslie Huddleston, NP, Richmond Wong (The Queen’s Medical Center, University of Hawaii, Honolulu, HI). Chapel Hill, NC Consortium: A. Sidney Barritt, M.D., Tiffany Marsh, BA, Vikki Metheny, ANP, Danielle Cardona, PA-C (University of North Carolina at Chapel Hill, Chapel Hill, NC). Virginia Commonwealth University Medical Center: Paula G. Smith, RN, BSN, Charlotte Hofmann, RN (Virginia Commonwealth University Health System, Richmond, VA). PNW/Alaska Clinical Center Consortium: Alycia Wolfstone, RN, MN (University of Washington Medical Center, Seattle, WA) Jody Mooney, Lupita Cardona-Gonzalez (Virginia Mason Medical Center, Seattle, WA), Kara L. Cooper (Center for Clinical and Translational Research, Seattle Children’s Institute, Seattle, WA). Johns Hopkins University: Hongxia Li, MBBS, MS, Robert Anders, MD, PhD, Hejab Imteyaz, Peter Lee, MD, Kiyoko Oshima, MD, Kim Kafka, RN, Naureen Islam, BS (Department of Pediatrics, Johns Hopkins Medical Institutions, Baltimore, MD). Liver Diseases Branch, NIDDK, NIH: Nancy Fryzek, RN, BSN, Elenita Rivera, BSN, Nevitt Morris, Vanessa Haynes-Williams, Amy Huang, RN, Catherine Nadal, RN, MS, Jaha Norman-Wheeler, RN, BA (National Institutes of Health, Bethesda, MD). Liver Disease Research Branch, NIDDK, NIH: Jay H. Hoofnagle, MD, Averell H. Sherker, MD, Edward Doo, MD, Rebecca J. Torrance, RN, MS, Sherry R. Hall, MS (National Institutes of Health, Bethesda, MD). Immunology Center: Mary E. Valiga, RN, Keith Torrey, BS, Danielle Levine, BS, James Keith, BS, Michael Betts, PhD (University of Pennsylvania, Philadelphia, PA), Luis J. Montaner, DVM, DPhil (Wistar Institute, Philadelphia, PA). Data Coordinating Center: Frani Averbach, MPH, Tamara Haller, Regina Hardison, MS, Stephanie Kelley, MS, Christina M. Lalama, MS, Sharon Lawlor, MBA, Hsing-Hua S. Lin, MS, PhD, Manuel Lombardero, MS, Andrew Pelesko, BS, Donna Stoliker, Melissa Weiner, MPH, Ella Zadorozny, MS, Qian Zhao, PhD (Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA).
Funding
The HBRN was funded as a Cooperative Agreement between the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and the following investigators: Lewis R. Roberts, MB, ChB, PhD (U01-DK082843), Anna Suk-Fong Lok, MD (U01-DK082863), Steven H. Belle, PhD, MScHyg (U01-DK082864), Kyong-Mi Chang, MD (U01-DK082866), Michael W. Fried, MD (U01-DK082867), Adrian M. Di Bisceglie, MD (U01-DK082871), William M. Lee, MD (U01-DK082872), Harry L. A. Janssen, MD, PhD (U01-DK082874), Daryl T-Y Lau, MD, MPH (U01-DK082919), Richard K. Sterling, MD, MSc (U01-DK082923), Steven-Huy B. Han, MD (U01-DK082927), Robert C. Carithers, MD (U01-DK082943), Mandana Khalili, MD (U01-DK082944), Kathleen B. Schwarz, MD (U01-DK082916), an interagency agreement with NIDDK: Lilia M. Ganova-Raeva, PhD (A-DK-3002–001) and support from the intramural program, NIDDK, NIH: Marc G. Ghany, MD. Additional funding to support this study was provided to Kyong-Mi Chang, MD, the Immunology Center, (NIH/NIDDK Center of Molecular Studies in Digestive and Liver Diseases P30DK50306, NIH Public Health Service Research Grant M01-RR00040), Richard K. Sterling, MD, MSc (UL1TR000058, NCATS (National Center for Advancing Translational Sciences, NIH), Norah A. Terrault, MD, MPH (CTSA Grant Number UL1TR000004), Michael W. Fried, MD (CTSA Grant Number UL1TR001111), Anna Suk-Fong Lok (CTSA Grant Number UL1RR024986, U54TR001959), and Kathleen B. Schwarz, MD (CTSA Grant Number UL1TR000423). Additional support was provided by Gilead Sciences, Inc. and Roche Molecular Systems via a CRADA through the NIDDK. The investigators recognize and appreciate the contributions of the HBRN participants without whom this research would not be possible.
Footnotes
Conflicts of interest
Dr. Lee receives research support from Merck, Conatus, Intercept, Bristol-Myers Squibb, Novo Nordisk, Synlogic, Eiger, Cumberland, Exalenz, Instrumentation Laboratory and Ocera Therapeutics, now Mallinckrodt Pharmaceuticals. He has consulted for Novartis, Sanofi and Genentech and Seattle Genetics. Dr. King receives grant support from Abbott. Dr. Feld receives research support from Abbvie, Enanta, Gilead, Janssen and consults for Abbvie, Gilead, GSK, Roche. Dr. Fontana has received research support from Gilead, Abbvie, and Bristol Myers Squibb (BMS). Dr. Janssen reports receiving consultant fees and/or grant support from BMS, AbbVie, Gilead Sciences, Novartis, Roche, Janssen, Arbutus, VIR and Merck. Dr. Sterling receives grant support from Roche, Abbott, Abbvie, and Gilead. Dr. Di Bisceglie consults for Gilead and BMS. Dr. Ghany, Dr. Mogul and Ms. Wang have no conflicts to disclose.
ClinicalTrials.gov Identifiers:
Data Availability Statement
For all HBRN manuscripts, the data set is filed with the NIDDK repository within 6 months of publication, following which it undergoes internal review and following that step it can be made available upon request to NIDDK via Dr. Edward Doo, Project Officer.
References
- 1.Magnius LO, Lingholm A, Lundin P, Iwarson S. A new antigen-antibody system: Clinical significance in long-term carriers of hepatitis B surface antigen. JAMA 1975;231:356–359. [DOI] [PubMed] [Google Scholar]
- 2.Beasley RP, Trepo C, Stevens CE and Szmuness W. The e antigen and vertical transmission of hepatitis B surface antigen. Am J Epidemiol 1977;105:94–98. [DOI] [PubMed] [Google Scholar]
- 3.Alter HJ, Seeff LB, Kaplan PM et al. Type B hepatitis: the infectivity of blood positive for e antigen and DNA polymerase after accidental needlestick exposure. N Engl J Med 1976;295:909–913. [DOI] [PubMed] [Google Scholar]
- 4.Yang H-I, Lu S-N, Liaw Y-F et al. Hepatitis B e antigen and the risk of hepatocellular carcinoma. N Engl J Med 2002;347:168–174. [DOI] [PubMed] [Google Scholar]
- 5.Song E, Dusheiko GM, Bowyer S and Kew MC. Hepatitis B virus replication in southern Africa blacks with HBsAg-positive hepatocellular carcinoma. Hepatology 1984;4:608–610. [DOI] [PubMed] [Google Scholar]
- 6.Yang H-I, Yuen M-F, Chan HL-Y et al. Risk estimation for hepatocellular carcinoma in chronic hepatitis B (REACH-B): development and validation of a predictive score. Lancet Oncology 2011;12:568–574. [DOI] [PubMed] [Google Scholar]
- 7.Lampertico P, Agarwal K, Bert T, et al. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol 2017;67:370–398. [DOI] [PubMed] [Google Scholar]
- 8.Terrault N, Lok ASF, McMahon BJ, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 Hepatitis B Guidance. Hepatology 2018;67:1560–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song G, Rao H, Feng B, Wei L. Prediction of spontaneous HBeAg seroconversion in HBeAg positive chronic hepatitis B patients during the immune clearance phase. J Med Virol 2014;86:1838–1844. [DOI] [PubMed] [Google Scholar]
- 10.Livingston SE, Simonetti JP, Bulkow LR, et al. Clearance of hepatitis B e antigen in patient with chronic hepatitis B and genotypes A, B, C, D and F. Gastroentoerology 2007;133:1452–1457. [DOI] [PubMed] [Google Scholar]
- 11.Di Bisceglie AM, King WC, Lisker-Melman M, et al. Age, race and viral genotype are associated with the prevalence of hepatitis B e antigen in children and adults with chronic hepatitis B. J Viral Hepat 2019;26:856–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper SL, King WC, Mogul DB, for the Hepatitis B Research Network (HBRN). Clinical significance of quantitative e antigen in a cohort of hepatitis B virus infected children and adults in North America. J Viral Hepatol. Revision under review 2/9/21 [DOI] [PubMed]
- 13.Ghany MG, Perrillo R, Li R, et al. Characteristics of adults in the hepatitis B research network in North America reflect their country of origin and hepatitis B virus genotype. Clin Gastroenterol Hepatol 2015;13:183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wai CT, Greenson JK, Fontana RJ, et al. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology 2003;38:518–26. [DOI] [PubMed] [Google Scholar]
- 15.Brahmania M, Lombardero M, Hansen BE, et al. Association between severe serum alanine aminotransferase flares and hepatitis B e antigen seroconversion and HBV DNA decrease in untreated patients with chronic HBV infection. Clin Gastroenterol Hepatol 2019;17:2541–2551. 10.1016/j.cgh.2019.02.005. Epub 2019 Feb 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ganova-Raeva L, Ramachandran S, Honisch C, Forbi JC, Zhai X, Khudyakov Y. Robust hepatitis B virus genotyping by mass spectrometry. J Clin Microbiol 2010;48:4161–4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lau DY, Ganova-Raeva L, Wang J, et al. Precore and basal core promoter hepatitis B virus (HBV) variants are present from a young age and differ across HBV genotypes. Hepatology 2020. August 29. doi: 10.1002/hep.31506. Online ahead of print. [DOI] [PMC free article] [PubMed]
- 18.Di Bisceglie AM, Lombardero M, Teckman J, et al. Determination of hepatitis B phenotype using biochemical and serological markers. J Viral Hepat 2017;23:320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim H, Hur M, Bae E, Lee K-A, Lee W-I. Performance evaluation of Cobas HBV real-time PCR assay on Roche Cobas 4800 System in comparison with COBAS AmpliPrep/COBAS TaqMan HBV Test. Clin Chem Lab Med 2018;56:1133–1139. [DOI] [PubMed] [Google Scholar]
- 20.Wursthorn K, Zacher BJ, Jaroszewicz J, et al. Development of a protocol for the quantitative determination of HBeAg using the Elecsys® HBeAg immunoassay. J Viral Hep 2011;18:e179–183. [DOI] [PubMed] [Google Scholar]
- 21.Dakin H, Fidler C, Harper C. Mixed treatment comparison meta-analysis evaluating the relative efficacy of nucleos(t)ides for treatment of nucleos(t)ide-naive patients with chronic hepatitis B. Value Health 2010;13:934–945. [DOI] [PubMed] [Google Scholar]
- 22.Wiens A, Lenzi R, Venson R, et al. Comparative efficacy of oral nucleoside or nucleotide analog monotherapy used in chronic hepatitis B: a mixed-treatment comparison meta-analysis. Pharmacotherapy 2013;33:144–151. [DOI] [PubMed] [Google Scholar]
- 23.Ahn SH, Marcellin P, Ma X, et al. Hepatitis B surface antigen loss with tenofovir disoproxil fumarate plus peg-interferon alfa-2a: Week 120 analysis. Dig Dis Sci 2018;63:3487–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyams K Risks of chronicity following acute hepatitis B virus infection: A review. Clin Infect Dis 1995;20:992–1000. [DOI] [PubMed] [Google Scholar]
- 25.McKeating C, Cadden I, McDougall N, et al. Progression from acute to chronic hepatitis B is more common in older adults. Ulster Med J 2018;87:177–180. [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Y, Deng H, Peng Z, Huang Y, Long Q, Huang A. Mutations of basal core promoter and precure regions in hepatitis B genotypes B and C. Hepat Mon 2014;15:e23034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamaura T, Tanaka E, Matsumoto A, et al. A case control study of early prediction of hepatitis B e antigen seroconversion by hepatitis B virus DNA levels and mutations in the precure region and core promoter. J Med Virol 2003;70:545–552. [DOI] [PubMed] [Google Scholar]
- 28.Fung SK, Lok AS. Hepatitis B virus genotypes: Do they play a role in the outcome of hepatitis B virus infection? Hepatology 2004;40:790–792. [DOI] [PubMed] [Google Scholar]
- 29.Chan HLY. Significance of hepatitis B virus genotypes and mutations in the development of hepatocellular carcinoma in Asia. J Gastroenterol Hepatol 2011;26:8–12. [DOI] [PubMed] [Google Scholar]
- 30.Hsu Y-N, Pan CQ, Abbasi A, Xia V, Bansal R, Hu K-Q. Clinical presentation and disease phases of chronic hepatitis B using conventional versus modified ALT criteria in Asian Americans. Dig Dis Sci 2014;59:865–871. [DOI] [PubMed] [Google Scholar]
- 31.Yuen M, Yuan H, Hui C, Wong DHK, et al. A large population study of spontaneous HBeAg seroconversion and acute exacerbation of chronic hepatitis B infection: implications for antiviral therapy. Gut 2003;52:416–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fleiss JL. Statistical methods for rates and proportions (2nd ed). New York: John Wiley; 1981. 38–46 p. [Google Scholar]
- 33.Landis JR; Koch GG. The measurement of observer agreement for categorical data”. Biometrics 1977;33:159–174. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For all HBRN manuscripts, the data set is filed with the NIDDK repository within 6 months of publication, following which it undergoes internal review and following that step it can be made available upon request to NIDDK via Dr. Edward Doo, Project Officer.