

Abstract
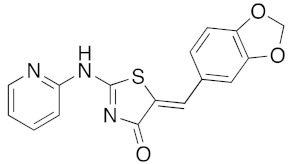
Here, we report on the synthesis of libraries of new 5-arylidene-2-thioxo-1,3-thiazolidin-4-ones 3 (twenty-two compounds) and new 2-amino-5-arylidene-1,3-thiazol-4(5H)-ones 5 (twenty-four compounds) with stereo controlled Z-geometry under microwave irradiation. The 46 designed final compounds were tested in order to determine their activity against four representative protein kinases (DYR1A, CK1, CDK5/p25, and GSK3α/β). Among these 1,3-thiazolidin-4-ones, the molecules (5Z) 5-(4-hydroxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one 3e (IC50 0.028 μM) and (5Z)-5-benzo[1,3]dioxol-5-ylmethylene-2-(pyridin-2-yl)amino-1,3-thiazol-4(5H)-one 5s (IC50 0.033 μM) were identified as lead compounds and as new nanomolar DYRK1A inhibitors. Some of these compounds in the two libraries have been also evaluated for their in vitro inhibition of cell proliferation (Huh7 D12, Caco2, MDA-MB 231, HCT 116, PC3, and NCI-H2 tumor cell lines). These results will enable us to use the 1,3-thiazolidin-4-one core as pharmacophores to develop potent treatment for neurological or oncological disorders in which DYRK1A is fully involved.
Keywords: microwave; Knoevenagel condensation; sulphur/nitrogen displacement; 1,3-thiazolidin-4-one; inhibition; protein kinase; DYRK1A; cell lines
1. Introduction
Protein kinases represent an important class of enzymes that play an important role in the regulation of various cellular processes. These enzymes catalyze protein-phosphorylation on serine, threonine, and tyrosine residues, which are frequently deregulated in human diseases. Only a small fraction of the 538 human kinases have been investigated as potential therapeutic targets [1]. Consequently, the search of protein kinase inhibitors represents an interesting target for the pharmaceutical industry for new therapeutic agents. Over the past decade, drug discovery has consisted of the empirical approach of random screening of large synthetic molecule libraries (or derivative of natural products) using high-throughput cell-based cytotoxicity assays to screening against clinical validated molecular targets. This approach has been successful in the area of oncology with small-molecule inhibitors of kinases, particularly in cancer progression and metastasis [2]. Among the 538 human kinases, DYRKs (dual-specificity tyrosine phosphorylation regulated kinases, 5 members) are a family of eukaryotic kinases that belong to a larger CMGC family of proline/arginine directed serine/threonine kinases. In this DYRK family, there are five mammalian subtypes (1A, 1B, 2, 3, and 4). The Dyrk1A gene is located within the human chromosome 21 Down Syndrome Critical Region (DSCR) [3]. According to recent literature, DYRK1A appears to be involved in various diseases, including Alzheimer’s disease (AD), Down syndrome (DS) [4], and cancer [5,6,7], and many efforts have evolved over the last five years toward the identification of potent and selective DYRK1A inhibitors [8,9]. Few potent and selective Dyrks inhibitors have been described (Figure 1). As example, TG003 (I) has been demonstrated as a potent inhibitor of DYRK1A (12 nM), as well as DYRK1B (130 nM) [10,11]. In 2011, KH-CB19 (II) was identified as a specific and interesting inhibitor of DYRK1A (33 nM) [12]. Harmine (III), a plant-derived β-carboline alkaloid, is an ATP-competitive DYRK1A (55 nM) and DYRK1B (166 nM) [13]. A series of thiazolone-substituted aminopyrimidines (IV) has been patented as DYRK1A inhibitors with potencies < 100 nM [14]. A structure activity relationship (SAR) study around the quinazoline core was carried out at the NIH, and their efforts culminated in the identification of compound (V) as a potent inhibitor of DYRK1A (62 nM) [15]. Revisiting the quinazoline chemistry, the same authors have also identified a new derivative (VI) (compound 46: 14 nM) [16]. Recently, a series of tricyclic pyrimidines (VII) was evaluated for their ability to inhibit DYRK1A (0.16 nM) and DYRK1B [17]. Meridianines are marine alkaloids isolated from the South Atlantic tunicate Aplidium meridanium, and the synthesis of new analogues [18] substituted at the C-4 to C-7 position either by iodide atom or by nitro or amino groups induced the identification of meridianine 74 (VIII) as an interesting inhibitor of DYRK1A (compound 74: 66 nM). The chemical structure resemblance between the marine alkaloids meridianines and variolin B encouraged the synthesis of a hybrid structure named meriolins [19], and compound (IX) provided good inhibition against DYRK1A (meriolin 2: 29 nM). The V-shaped 2-(pyridin-3-yl)-1H-indol-6-ol (X) was discovered to inhibit DYRK1A (60 nM) [20].
Figure 1.

Reported small molecules as DYRK1A kinase inhibitors with their IC50 values in nM.
Efforts for the synthetic preparation of new chromeno[3,4-b]indoles as Lamellarin D isosteres [21] have been developed, and compound (XI: 67 nM) emerged among the Lamellarin D derivatives. In 2013, the first successful synthesis and separation of the atropoisomeric (aR)- and (aS)-16-methyl lamellarins N and their different kinase inhibitory action against DYRK1A were reported [22]. The isomer (aR) (XII) showed potent but non-selective inhibition of DYRK1A (42 nM). The 2-amino imidazolone core is present in several marine sponges [23], such as polyandrocarpamines [24], dispacamides [25,26], aplysinopsine [27,28], clathridine [29], hymenialdisine [30,31,32,33,34,35,36], and Leucettamine B. This last alkaloid was isolated in 1993 from the marine calcareous sponge Leucetta microraphis Hæckel of the Argulpelu Reef in Palau [37], and no significant biological activity has been reported. From a biological screening, we discovered that the Leucettamine B analogues [38], named leucettines, exerted a selective inhibition toward protein kinases. A detailed structure-activity relationship (SAR) led to the identification of leucettine L41 (XIII) as good inhibitor of DYRKs and cdc2-like kinases (CLKs), which are involved in Alzheimer’s disease/Down syndrome [39]. We also demonstrated that leucettine L41 inhibits DYRK1A and CLKs in a cellular context and displayed neuropropective properties [40].
The 2-amino 5-arylidene-5H-thiazol-4-ones and their 5-arylidene rhodanine precursors are a class of five-membered heterocyclic rings (FMHRs) considered as “privileged scaffolds” in the medicinal chemistry community [41], and considerable work has been published over decades about their chemistry and biology. Some of them exhibited anti-inflammatory [42], anti-tumor effects [43], or have been identified as selective inhibitors of the dual specificity phosphatase (DSP) family [44].
Continuing in the effort to identify new DYRK1A inhibitors, we decided to explore the synthesis of new 5-arylidene-5H-thiazol-4-ones and new 5-arylidene-2-thioxo-1,3-thiazolidin-4-ones. We also studied their effects on some protein kinases (DYRK1A, CK1, CDK5-p25, and GSK-3α/β) and on six representative tumor cell lines. Herein, we present the building of this thiazolone/thiazolidinone library, mostly carried out under microwave irradiation, and the biological activities of these compounds.
2. Results and Discussion
2.1. Chemistry
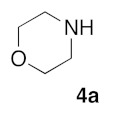
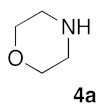
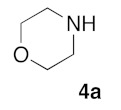
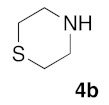
Retrosynthetic strategy toward new (5Z) 2-amino-5-arylidene-1,3-thiazol-4(5H)-ones 5a–q is based on the use of secondary cyclic amines building blocks 4a–h in sulfur/nitrogen displacement reaction and the use of various 2-thioxo-1,3-thiazolidin-4-ones 1a–i as starting platforms in Knoevenagel condensation (Figure 2 and Figure 3).
Figure 2.

Retrosynthetic strategy toward new (5Z) 2-amino-5-arylidene-1,3-thiazol-4(5H)-ones 5a–q based on the use of secondary cyclic amines building blocks 4a–h in sulfur/nitrogen displacement reaction and the use of various 2-thioxo-1,3-thiazolidin-4-ones 1a–i as starting platforms in Knoevenagel condensation.
Figure 3.

Retrosynthetic strategy toward new (5Z) 2-N-heteroarylamino-5-arylidene-1,3-thiazol-4(5H)-ones 5r–x based on the use of aromatic aldehydes 2a–c and various 2-N-heteroarylamino-1,3-thiazolidin-4-one 8a–e as other starting platforms in Knoevenagel condensations.
2.1.1. Synthesis of 5-Arylidene-2-thioxo-1,3-thiazolidin-4-one Library
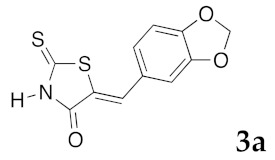
In order to synthesize a library of 5-arylidene-2-thioxo-1,3-thiazolidin-4-ones 3 (Scheme 1) for a systematic structure-activity relationship (SAR) study, we have examined the reactivity of 2-thioxo-1,3-thiazolidin-4-ones 1a–i with a series of various aromatic aldehydes 2a–p. We selected commercial aldehydes substituted by phenol function at various positions (2f, 2g, 2i, and 2n) and/or methoxy groups (2d,e, 2h) and cyclic ether function (2a–c, 2l, 2o, 2p) to evaluate how the protic character and steric nature of the substituents on the phenyl ring will influence the biological activity of compounds 3. We also examined the bio-isosteric replacement of the benzene ring of 3 with pyridine (2k) or benzofuran (2m) moieties. Compounds 3 are designed to evaluate whether the NH function in position N-1′ or a functional group with similar H-bond interactions will retain the activity.
Scheme 1.

General synthetic approach for the (5Z) 5-arylidene-2-thioxo-1,3-thiazolidin-4-one derivatives 3(a–v) and for the (5Z) 2-amino-5-arylidene-1,3-thiazol-4(5H)-one 5a–q. Reagents and reaction conditions: (i) n-PrNH2 2 eq, 80 °C, 30 min for compounds 3a–l in open vessel using Synthewave® 402 reactor for MWI; Et3N 0.1 eq or piperidine 0.1 eq or AcONa 1 eq, AcOH 0.1–6.6 eq, 85–150 °C, 20–210 min for compounds 3m–v in closed vessel using Explorer 24® CEM or Monowave® 300 Anton-Paar reactors for MWI. (ii) 4a–h 1.2 eq, MWI in Synthewave® 402 microwave reactor, 80 °C, 20–60 min.
For the 2-thioxo-1,3-thiazolidin-4-one platforms 1, rhodanine 1a, 2-(4-oxo-2-thioxo-thiazolidin-3-yl)acetic acid 1b and 3-amino rhodanine 1d are commercially available. Access to the 3-(4-oxo-2-thioxo-thiazolidin-3-yl)propanoic acid 1c could be accomplished by a recent patented protocol [45]. For introduction of the arylsulfonamide functionality on the rhodanine moiety in compound 1e, we used the Power’s method [46]. Starting from commercial 3-amino-rhodanine 1d, compounds 1f–i were synthesized easily, according to a recent published procedure developed in our laboratory [47].
With the aim to develop a more efficient synthetic process for the synthesis of highly functionalized 5-arylidene rhodanines 3a–v from the building blocks 1a–i and 2a–p, microwave irradiation was employed to shorten the reaction times in comparison to conventional heating. For the reported methods, the Knoevenagel condensations have been realized in the presence of catalyst with various solvents: sodium acetate in glacial acetic acid [48] or in ethanol [46], piperidinium benzoate in toluene [49], tetrabutylammonium bromide in water [50], and piperidine in ethanol [51], using conventional heating in an oil bath. These older protocols suffer from one or more limitations, such as requiring harsh reaction conditions, low moderate yields, cumbersome experimental process, and long reaction time. Considering the latter aspect, we reasoned that microwave dielectric heating assisted chemistry might be an interesting approach for the synthesis of 5-arylidene rhodanine derivatives as potential biological molecules.
For this study, we investigated the use of three microwave reactors: the Synthewave® 402 and the Explorer® 24 apparatus with continuous irradiation power from 0 to 300 W and the Monowave® 300 with continuous power from 0 to 800 W. These three microwave irradiation reactors comprise a mono-mode microwave cavity that operates at a frequency of 2.45 GHz; the reaction temperature in the microwave cavity was controlled with a calibrated infrared sensor, and the microwave irradiation parameters (power and temperature) were monitored by a software package. For optimization of the experimental reaction conditions (selection of the appropriate reactor, reaction temperature and irradiation power), we have not realized a comparative study between these three reactors because we focused only our work on the microwave apparatus, which lead us to better results.
As can be seen in Table 1, the experiments revealed that the optimal reaction conditions for the 5-arylidene-2-thioxo-1,3-thiazolidin-4-ones 3a–l were obtained in an open vessel after 60 min at 80 °C (with a power of 90 W) in the Synthewave® 402 apparatus with a stoichiometry of 1/1–4 of aryl aldehyde 2 and the volatile propylamine in order to be able to develop one-pot tandem reactions (aldimine synthesis/Knoevenagel condensation) [52]. According to this microwave method, the compounds 3a–l were synthesized in yields ranging from 62 to 89%. For the other compounds, 3m, 3n, and 3p–v, we screened a range of another reaction condition parameters in a closed vessel (a tube sealed with a snap cap) with the appropriate microwave reactors. With the conditions shown in Table 1, AcONa (0.1 or 1–1.2 eq) and 0.1 eq of piperidine in glacial acetic acid (0.1–6.6 eq) were identified to execute the Knoevenagel reactions in good yields (62–98% excepted for 3p). Unfortunately, we are not able to use this protocol for the preparation of compound 2o. It was obtained in moderate yield (2o: 26%) using classical heating in an oil bath. The structures of the desired 5-arylidene-2-thioxo-1,3-thiazolidin-4-ones 3a–v were substantiated by 1H, 13C NMR (Supplementary Materials), and HRMS analyses, and only the thermodynamically more stable Z-isomers were obtained [50].
Table 1.
Results for the preparation under microwave irradiation of (5Z) 5-arylidene-2-thioxo-1,3-thiazolidin-4-one derivatives 3a–v from aromatic aldehydes 2a–p and 2-thioxo-1,3-thiazolidin-4-ones 1a–i.
| Compound 3 | Reagents | Reactions Conditions Used under Microwave a | Yield b of 3 (%) d | ||||
|---|---|---|---|---|---|---|---|
| Starting Aldehyde 2 | Reaction Temp. (°C) | Reaction Time (min.) | Power (W) | ||||
| 3a |
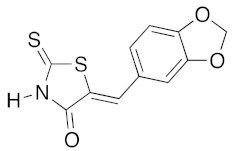
|
n-PrNH2 2 eq | 2a | 80 | 60 | 90 c | 79 |
| 3b |
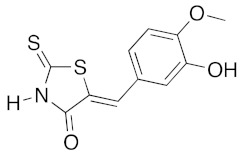
|
n-PrNH2 2 eq | 2d | 80 | 60 | 90 c | 81 |
| 3c |
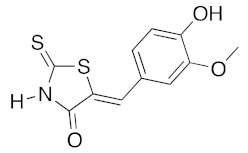
|
n-PrNH2 4 eq | 2e | 80 | 60 | 90 c | 80 |
| 3d |
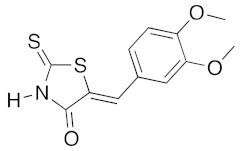
|
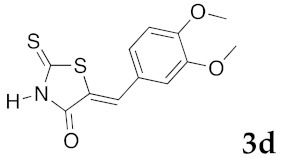
n-PrNH2 2 eq | 2h | 80 | 60 | 90 c | 83 |
| 3e |
|
n-PrNH2 2 eq | 2i | 80 | 40 | 90 c | 79 |
| 3f |
|
n-PrNH2 2 eq | 2c | 80 | 60 | 90 c | 89 |
| 3g |
|
n-PrNH2 2 eq | 2b | 80 | 60 | 90 c | 79 |
| 3h |
|
n-PrNH2 2 eq | 2k | 80 | 60 | 90 c | 68 |
| 3i |
|
n-PrNH2 1 eq | 2l | 80 | 60 | 90 c | 70 |
| 3j |
|
n-PrNH2 2 eq | 2f | 80 | 60 | 90 c | 79 |
| 3k |
|
n-PrNH2 2 eq | 2n | 80 | 60 | 90 c | 79 |
| 3l |
|
n-PrNH2 2 eq | 2m | 80 | 60 | 90 c | 62 |
| 3m |
|
AcONa 1.2 eq AcOH 1.2 eq |
2o | 140 | 20 | - e | 93 |
| 3n |
|
AcONa 1.2 eq AcOH 1.2 eq |
2p | 140 | 30 | - e | 94 |
| 3o |
|
Et3N 0.1 eq AcOH 0.1 eq AcOEt |
2a | 85 | 210 | -f | 26 |
| 3p |
|
piperidine 0.1 eq AcOH 0.1 eq |
2a | 150 | 20 | 200 d | 27 |
| 3q |
|
AcONa 1 eq AcOH 6.6 eq |
2a | 120 | 20 | 100 d | 98 |
| 3r |
|
AcONa 1 eq AcOH 6.6 eq |
2a | 120 | 20 | - e | 68 |
| 3s |
|
AcONa 1 eq AcOH 6.6 eq |
2a | 120 | 20 | 100 d | 83 |
| 3t |
|
piperidine 0.1 eq AcOH 0.1 eq |
2a | 150 | 20 | 200 d | 64 |
| 3u |
|
AcONa 0.1 eq AcOH 0.1 eq |
2a | 150 | 20 | - e | 72 |
| 3v |
|
piperidine 0.1 eq AcOH 0.1 eq |
2a | 150 | 20 | - e | 62 |
a Microwave irradiation (MWI) of the reaction mixture was realized in an open glass microwave reactor or in tube sealed with a snap cap (closed vessel). b Isolated yield after work up, crystallization, and washing using appropriate solvent. c Synthewave® 402 (Merck Eurolab Prolabo France) used as microwave reactor (Power max.: 300 W). Reaction under microwave was realized in an open glass reactor. d Explorer® 24 (CEM μWave France) used as a microwave reactor (Power max.: 300 W). Reaction under microwave was realized in a tube sealed with a snap cap. e Monowave® 300 (Anton Paar France) used as a microwave reactor (Power max.: 800 W). Reaction under microwave was realized in a tube sealed with a snap cap. f Compound 3o was synthesized in an oil bath, and initial attempts to obtain 3o under microwave irradiation were unsuccessful.
2.1.2. Synthesis of (5Z) 2-Amino-5-arylidene-1,3-thiazolidin-4-one Library
With the compounds 3a–v in hand, we investigated the preparation of novel 2-amino-5-arylidene-1,3-thiazolidin-4-ones 5, according to Scheme 1. Transformation of 5-arylidene-rhodanine derivatives 3 into their 2-amino-5-arylidene-1,3-thiazolidin-4-ones 5 involved a sulfur/nitrogen displacement in the presence of a primary amine [53] or a secondary amine [54]. Commonly, the synthesis of 2-amino-1,3-thiazol-4-one involved activation of the C=S bond of the starting 5-arylidene rhodanine, and reaction with an halogenoalcane (i.e., EtI or MeI) potentially gave the desired S-alkyl-5-arylidene-1,3-thiazol-4(5H)-one, together with the N-alkyl-5-arylidene rhodanine derivative, as a side product [55]. In this context, we preferred to directly investigate the sulfur/nitrogen displacement in a faster and more efficient way by using microwave dielectric heating for the synthesis of targeted compounds 5 from the intermediate 3 and a variety of polar cyclic secondary amines 4. In order to obtain interesting information for the structure-activity relationship (SAR) study on these compounds 5 as inhibitors of the protein kinase DYRK-1A, compounds 5 were designed to evaluate how the electrostatic and steric nature of the substituents on the phenyl ring and/or the presence of various amino moieties in position C-2 will influence the biological activity. For this study, we have employed eight polar cyclic secondary amines 4a–h as elements of the second point of molecular diversity for the construction of compounds 5. After a preliminary screening of the microwave irradiation conditions (open or closed vessel mode, temperature, power of irradiation, time), the experiments revealed the optimal ratio of 3/4 under microwave irradiation was obtained with 1.5 eq of amino reagent 5 using the “open vessel mode”. For the other parameters (temperature: 80–100 °C, power: 80–150 W, time: 20–60 min), the results for each product 5 are presented in Table 2. To verify the versatility and efficiency of this open vessel microwave protocol, different starting 5-arylidene rhodanine derivatives 3 and commercially available cyclic secondary amines 4a–h were used as building blocks to generate a collection of seventeen new 2-amino-5-arylidene-1,3-thiazolidin-4-ones 5a–q: in all cases, the products were easily isolated in good to high yields (72–96%) after a simple precipitation from EtOH. The structure identification of all these new compounds 5 were based on the 1H and 13C assignments and was performed extensive 1D and 2D NMR spectroscopy. For the exocyclic double bond (CH=C) of all the 2-amino-5-arylidene-1,3-thiazolidin-4-ones 5, 1H NMR spectra show only one signal for the methylene proton (CH=) in the range of 7.56–7.61 ppm at lower field values than those expected or previously reported in the literature for the E-isomers which, strongly indicates that the compounds 5a–q have the Z-configuration because of the high degree of thermodynamic stability of this isomer [56].
Table 2.
Results for the solvent-free synthesis under microwave irradiation of (5Z) 2-amino-5-arylidene-1,3-thiazol-4(5H)-one derivatives 5a–q by sulfur/nitrogen displacement reaction from (5Z) 5-arylidene-2-thioxo-1,3-thiazolidin-4-one derivatives 3 and cyclic secondary amines 4a–h.
| Compound 5 | Starting Compound 3 | Starting Compound 4 | Reactions Conditions Used under Microwave a | Yield b of 5 (%) | |||
|---|---|---|---|---|---|---|---|
| Temp. (°C) | Time (min.) | Power c (W) | |||||
| 5a |
|
|
|
80 | 20 | 80 | 96 |
| 5b |
|
|
|
80 | 25 | 80 | 64 |
| 5c |
|
|
|
80 | 20 | 80 | 88 |
| 5d |
|
|
|
80 | 20 | 80 | 86 |
| 5e |
|
|
|
80 | 60 | 80 | 78 |
| 5f |
|
|
|
80 | 60 | 80 | 84 |
| 5g |
|
|
|
80 | 30 | 80 | 80 |
| 5h |
|
|
|
80 | 35 | 80 | 78 |
| 5i |
|
|
|
80 | 20 | 80 | 80 |
| 5j |
|
|
|
100 | 30 | 80 | 75 |
| 5k |
|
|
|
120 | 20 | 150 | 72 |
| 5l |
|
|
|
100 | 25 | 150 | 80 |
| 5m |
|
|
|
80 | 20 | 90 | 80 |
| 5n |
|
|
|
80 | 20 | 90 | 84 |
| 5o |
|
|
|
80 | 20 | 90 | 86 |
| 5p |
|
|
|
80 | 20 | 90 | 88 |
| 5q |
|
|
|
80 | 20 | 90 | 80 |
a Microwave irradiation (MWI) of the reaction mixture was realized in an open glass microwave reactor. b Isolated yield after work up, crystallization, and washing using appropriate solvent. c Synthewave® 402 (Merck Eurolab Prolabo Fr) used as a microwave reactor (Power max.: 300 W).
To extend the molecular diversity on the C-2 position of the rhodanine platform, we have also examined introduction of aromatic primary amine (i.e., aniline, C-substituted aniline, amino pyridine, etc.) using the microwave dielectric heating protocol for sulfur/nitrogen displacement from 5-arylidene-2-thioxo-1,3-thiazolidin-4-one 3 and appropriate aromatic primary amine. After screening of the microwave irradiation conditions based on the reaction temperature (90–160 °C), the power for irradiation (90–150 W), the reaction time (30–60 min), the use of polar or non-polar solvent (EtOH, hexane), the use of open or closed vessel mode, and the ratio of the reagents (ratio 3/aromatic amine: 1–3.5), it was not possible to obtain the desired 2-N-arylamino-5-arylidene-1,3-thiazolidin-4-one 5 after analysis of the crude reaction mixture by 1H NMR. Accordingly, we decided to drop this synthetic strategy for access to 2-N-arylamino-5-arylidene-1,3-thiazolidin-4-one and to focus now on the use of 2-N-heteroarylamino-1,3-thiazolidin-4-ones 8a–e in Knoevenagel condensation (Scheme 2). The designed compounds 8a–e were easily prepared from N-aryl thiourea 6a–d and ethyl bromoacetate according to a protocol developed in our laboratory [55]. As shown in Table 3, the “closed vessel” microwave irradiation procedure involved the use of piperidine (0.1 eq) or AcONa (0.1–1 eq) in glacial acetic acid for the synthesis of 2-N-heteroarylamino-5-arylidene-1,3-thiazolidin-4-one 5r–x from the building blocks 8a–e and aromatic aldehydes 2a–c after 20–40 min at 120 or 150 °C with a power of 100–200 W. This protocol is also very simple to execute because the desired products 5r–x were isolated after crystallization from ethanol in moderate to good yields (34–90%). The expected Z-geometry of these compounds 5 was confirmed by the chemical shift of the exocyclic methylene proton (CH=) obtained in 1H NMR [56].
Scheme 2.

General synthetic approach for the (5Z) 2-N-heteroarylamino-5-arylidene-1,3-thiazol-4(5H)-one derivatives 5r–x from the starting 2-N-heteroarylamino-1,3-thiazolidin-4-one 8a–e and aromatic aldehydes 3(a–c). Reagents and reaction conditions: (i) 7 1.3 eq, EtOH, 55 °C, 3.5–22 h; (ii) piperidine 0.1 eq or AcONa 0.1 eq, AcOH 0.1–6.6 eq, 120–150 °C, MWI in closed vessel (Explorer 24® CEM μWave reactor), 20–40 min.
Table 3.
Results for the preparation under microwave irradiation of (5Z) 2-N-heteroarylamino-5-arylidene-1,3-thiazol-4(5H)-one derivatives 5r–x from 2-N-heteroarylamino-thiazolidin-4-one 8a–e and aromatic aldehydes 3a–c.
| Compound 5 | Reaction Conditions Used under Microwave a | Yield c of 5 (%) | ||||
|---|---|---|---|---|---|---|
| Reagents | Reaction Temp. (°C) | Reaction Time (min.) | Power b (W) | |||
| 5r |
|
piperidine 0.1 eq AcOH 0.1 eq |
150 | 20 | 200 | 34 |
| 5s |
|
AcONa 0.1 eq AcOH 6.6 eq |
120 | 20 | 100 | 90 |
| 5t |
|
AcONa 0.1 eq AcOH 6.6 eq |
120 | 40 | 100 | 43 |
| 5u |
|
AcONa 0.1 eq AcOH 6.6 eq |
120 | 20 | 100 | 90 |
| 5v |
|
AcONa 0.1 eq AcOH 6.6 eq |
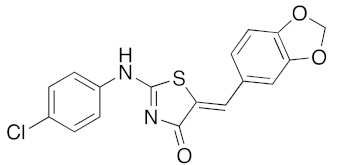
120 | 20 | 100 | 79 |
| 5w |
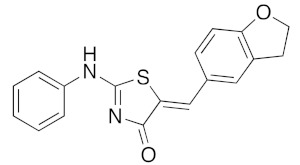
|
piperidine 0.1 eq AcOH 0.1 eq |
150 | 20 | 200 | 64 |
| 5x |
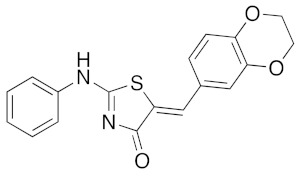
|
AcONa 0.1 eq AcOH 6.6 eq |
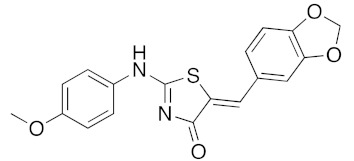
120 | 20 | 100 | 98 |
a Microwave irradiation (MWI) of the reaction mixture was realized in a glass tube sealed with a snap cap (closed vessel). b Explorer® 24 (CEM μWave France) used as a microwave reactor (Power max.: 300 W). c Isolated yield after work up, crystallization, and washing using appropriate solvent.
2.2. Biological Activities
The twenty-two (5Z) 5-arylidene-2-thioxo-1,3-thiazolidin-4-ones 3a–v and the twenty-four (5Z) 2-amino-5-arylidene-1,3-thiazolidin-4(5H)-ones 5a–x were evaluated against four different and representative in vitro kinase assays DYRK1A (dual specificity tyrosine phosphorylation regulated kinase), CK1 δ/e (caseine kinase 1), CDK5/p25 (cyclin dependant kinase) [57], and GSK3 α/β (glycogen synthase kinase 3) to evaluate their inhibition potency. These kinases were purified and assayed in the presence of 15 μM ATP and appropriate substrates (GS-1 peptide for GSK3 α/β, CKS peptide for CK1 δ/ε, histone H1 for CDK5/p25) as previously described [39]. All the compounds were initially tested at a 10 μM concentration. Those displaying less than 50% inhibition were considered inactive. Compounds showing more than 50% inhibition at 10 μM were next evaluated over a wide range of concentrations (usually 0.01, 5 and 10 μM), and the IC50 values were calculated from the dose-responses curves (Sigma–plot). (R)-Roscovitine and harmine (a natural β-carboline alkaloid known to be a potent inhibitor of DYRK1A [12]) were also used as positive control, and its IC50 values were compared with those obtained for the compounds 3 and 5. Results are summarized, respectively, in Table 4 for the (5Z) 5-arylidene-2-thioxo-1,3-thiazolidin-4-one 3a–v, and Table 5 for the (5Z) 2-amino-5-arylidene-1,3-thiazol-4(5H)-one 5a–x.
Table 4.
Effects of the (5Z) 5-arylidene-2-thioxo-1,3-thiazolidin-4-one 3a–v intermediates on the catalytic activity of four purified protein kinases a.
| Compound | DYRK1A | CK1 | CDK5/p25 | GSK3α/β |
|---|---|---|---|---|
| 3a | 0.070 | - | >10 | 8.5 |
| 3b | 1.4 | - | - | >10 |
| 3c | 0.064 | ≥10 | 8 | 8 |
| 3d | >10 | - | - | - |
| 3e | 0.028 | >10 | >10 | >10 |
| 3f | 0.056 | - | - | ≤10 |
| 3g | 0.090 | - | - | ≥10 |
| 3h | 0.35 | - | - | 1.6 |
| 3i | 0.7 | - | - | >10 |
| 3j | >10 | - | - | >10 |
| 3k | >10 | - | >10 | >10 |
| 3l | 15 | - | - | >100 |
| 3m | 6.5 | - | - | >10 |
| 3n | >10 | - | - | >10 |
| 3o | >10 | - | - | >10 |
| 3p | >10 | - | - | >10 |
| 3q | >10 | - | - | >10 |
| 3r | >10 | - | - | >10 |
| 3s | >10 | - | - | >10 |
| 3t | >10 | - | - | >10 |
| 3u | 0.16 | - | - | >10 |
| 3v | 0.70 | - | - | >10 |
| Harmine b | 0.029 | 1.5 | >10 | >10 |
| (R)-Roscovitine b | 11.0 | 2.3 | 0.2 | 130 |
a Compounds were tested at various concentrations on each kinase as described in Experimental Section. IC50 values, calculated from the dose-response curves, are reported in μM. -, inactive at the highest concentration tested (10 μM); >10, inhibitory, but IC50 > 10 μM. b IC50 values; see refs. [39,40].
Table 5.
Effects of the (5Z) 2-amino-5-arylidene-1,3-thiazol-4(5H)-one derivatives 5a–x on the catalytic activity of four purified protein kinases a.
| Compound | DYRK1A | CK1 | CDK5/p25 | GSK3α/β |
|---|---|---|---|---|
| 5a | 0.63 | - | - | 7.4 |
| 5b | >10 | - | - | - |
| 5c | - | - | - | - |
| 5d | >10 | - | - | - |
| 5e | 1.8 | - | - | ≤8.8 |
| 5f | 2.3 | - | - | - |
| 5g | 0.4 | - | - | ≥10 |
| 5h | - | - | - | - |
| 5i | 0.053 | - | - | 7.4 |
| 5j | ≥10 | - | - | - |
| 5k | 1.6 | - | - | - |
| 5l | 1.8 | - | - | >10 |
| 5m | 0.6 | >10 | - | ≥10 |
| 5n | 0.070 | >10 | - | 5.3 |
| 5o | 0.090 | ≥10 | - | 4.7 |
| 5p | 0.94 | ≥10 | - | >10 |
| 5q | 0.17 | - | - | 3.7 |
| 5r | 0.61 | - | - | - |
| 5s | 0.033 | - | - | ≤10 |
| 5t | >10 | - | - | >10 |
| 5u | 0.9 | - | - | >10 |
| 5v | 9.0 | >10 | >10 | >10 |
| 5w | 0.12 | - | - | - |
| 5x | 0.27 | - | - | - |
| Harmine | 0.029 | 1.5 | >10 | >10 |
| (R)-Roscovitine | 11 | 2.3 | 0.2 | 130 |
a Compounds were tested at various concentrations on each kinase as described in Experimental Section. IC50 values, calculated from the dose-response curves, are reported in μM. -, inactive at the highest concentration tested (10 μM); >10, inhibitory, but IC50 > 10 μM.
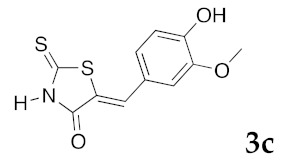
Results given in Table 4 showed that, among these products 3, the molecules 3j–l and 3n–t were completely inactive on the four kinases, DYRK1A, CK1, CDK5/p25, and GSK3, for which IC50 values > 10 μM were observed. Some interesting results were obtained for products 3h,i, 3m, and 3u,v with micromolar IC50 values for DYRK1A. Among these compounds, the presence of the aminoacetic moiety on N-3 position of 3u leads to enhancement of the inhibitory kinase activity for DYRK1A (3u: IC50 0.16 μM). The most promising results were obtained with products 3a, 3c, and 3e–g in this series, which exhibited selective, and sub-micromolar inhibitory activity toward DYRK1A (0.090 μM > IC50 > 0.028 μM). The molecule 3c bearing a hydroxyl group at the C-4 position of the exocyclic phenyl ring was strongly considered as the most interesting compound with an IC50 value of 0.028 μM. In compound 3j, the presence of a supplementary hydroxyl group in the C-3 position of the phenyl ring completely decreases inhibition of kinase activity (3j: IC50 > 10 μM), which confirms that the presence of only one hydroxyl group on the phenyl ring of 3c seems to be crucial for an optimal inhibition activity. In product 3e, a small methoxy group was partially tolerated, but the DYRK1A inhibition activity decreases slightly (3c: IC50 0.064 μM). The introduction of more bulky groups at the 5-ylidene position as 1,3-benzodioxol-5-yl in 3a (IC50 0.070 μM) or 2,3-dihydro-1,4-benzodioxin-5-yl in 3f (IC50 0.056 μM) or 2,3-dihydro-benzofuran-5-yl in 3g (IC50 0.090 μM) maintained partially the kinase inhibition activity.
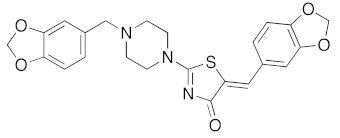
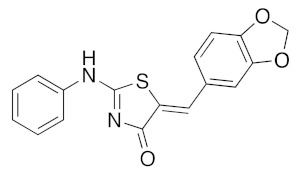
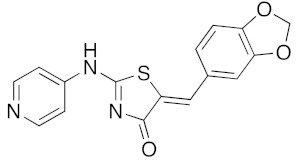
Concerning the library of the twenty-four (5Z) 2-amino-5-arylidene-1,3-thiazolidin-4(5H)-one 5a–x derivatives, the results are given in Table 5. The main part of their activity was focused on DYRK1A (and GSK3 α/β for some of them) for fourteen products in this second series (5a, 5e–g, 5k–m, 5p–r, and 5u–x), which exhibited inhibitory activity in the micromolar and sub-micromolar ranges (2.3 μM > IC50 > 0.12 μM). For the three compounds 5i, 5n, and 5o, we observed low nano-molar inhibition activity of DYRK1A (IC50 values for 5i: 0.053 μM, 5n: 0.070 μM, and 5o: 0.090 μM) with moderate selectivity because they are also able to inhibit GSK3 α/β. It is interesting to note that these three compounds 5i, 5n, and 5o are, respectively, substituted with bulky groups at the 5-ylidene position (1,3-benzodioxol-5-yl or 2,3-dihydro-1,4-benzodioxin-5-yl) and piperidin-1-yl or thiomorpholin-1-yl moieties in the C-2 position of the 1,3-thiazolidin-4(5H)-one platform. Finally, introducing a pyridin-2-yl group in N-2 position of the molecule 5s resulted in selective and sub-micromolar affinity for DYRK1A (5s: IC50 0.033 μM). Replacement of the pyridin-2-yl group in N-2 position by a pyridin-4-yl at the same position in compound 5t resulted in a complete loss of kinase inhibitory activity on DYRK1A (5t: IC50 > 10 μM). Again, the replacement of the pyridin-2-yl or pyridin-4-yl moiety in N-2 position by a phenyl group, as example in molecule 5r, maintained a moderate inhibition activity at a micromolar range (5r: IC50 0.61 μM).
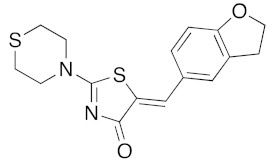
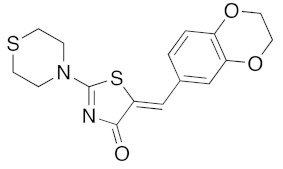
Comparison of all the results obtained in the two series showed the difficulties to define any role for the various substituents on the exocyclic phenyl ring (arylidene moiety in C-5 position), as well on the amino substituents in N-2 position, in order to obtain sub-micromolar affinity for DYRK1A with good selectivity. However, it is interesting to note that the presence of hydroxyl group in para–position of the phenyl ring of 5-arylidene-2-thioxo-1,3-thiazolidin-4-one derivative 3c is very important to access of a sub-micromolar inhibitory activity for DYRK1A. However, the presence of the bulky 1,3-benzodioxol-5-yl group on the 5-arylidene moiety is not a crippling factor when it is associated with a thiomorpholin-1-yl substituent in N-2 position, with is the case for the compound 5n (IC50 0.070 μM).
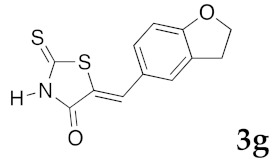
To evaluate the potency of selected compounds 3 and 5 for their in vitro inhibition of cell proliferation, we used a panel of 6 representative tumoral cell lines, namely Huh7 (differential hepatocellular carcinoma), Caco2 (differentiating colorectal adenocarcinoma), HCT (actively proliferating colorectal carcinoma), PC3 (prostate carcinoma), NCI-H2 (lung carcinoma), MDA-MB 231 (breast carcinoma), and diploid skin fibroblasts, as normal cell lines for control. Results of the in vitro antiproliferative data activity are reported in Table 6. The most active compound is clearly the (5Z)-5-(2,3-dihydro-benzofuran-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3g, which exhibits antitumor activities in the Caco2 and HCT 116 cell lines with IC50 values lower than 10 μM (Caco2: IC50 8 μM and HCT 116: IC50 6 μM). Compound 3g also presents a selective and sub-micromolar inhibitory activity on DYRK1A (IC50 0.090 μM). It is interesting to note that 3c is also moderately active in the HCT 116 cell line (IC50 12 μM) but showed a more efficient inhibitory activity for DYRK1A (IC50 0.064 μM). Compounds 3c and 3g also did not exhibit the growth of normal fibroblasts (IC50 > 25 μM). In the selected (5Z) 2amino-5-arylidene-1,3-thiazol-4(5H)-one derivatives 5, none of these compounds presented a significant activity against the six representative tumoral cell lines.
Table 6.
Effects of the (5Z) 2-amino-5-arylidene-1,3-thiazol-4(5H)-one derivatives 5r–x a and (5Z) 5-arylidene-2-thioxo-1,3-thiazolidin-4-one 3a–c, 3f,g derivatives b on six representative tumoral cell lines.
| Compound | Cell Lines IC50 (μM) | ||||||
|---|---|---|---|---|---|---|---|
| Huh7 D12 | Caco2 | MDA-MB231 | HCT116 | PC3 | NCI-H727 | Fibroblasts | |
| 5(r–x) | >25 | >25 | >25 | >25 | >25 | >25 | >25 |
| 3a | >25 | >25 | >25 | >25 | >25 | >25 | >25 |
| 3b | >25 | >25 | >25 | >25 | >25 | >25 | >25 |
| 3c | >25 | >25 | >25 | 12 | >25 | >25 | >25 |
| 3f | >25 | >25 | >25 | >25 | >25 | >25 | >25 |
| 3g | >25 | >25 | >25 | >25 | >25 | >25 | >25 |
| Roscovitine | 10 | 8 | 15 | 6 | 20 | >25 | >25 |
| DMSO | >25 | >25 | >25 | >25 | >25 | >25 | >25 |
a The other derivatives 5a–q have not been tested, or some of them are not soluble in the conditions for the preparation of the biological test. b The other derivatives 3d,e and 3h–v have not been tested, or some of them are not soluble in the conditions for the preparation of the biological test.
3. Material and Methods
3.1. Chemistry–General Remarks
Melting points were determined on a Kofler melting point apparatus and were uncorrected. 1H NMR spectra were recorded on BRUKER AC 300 P (300 MHz) spectrometer, 13C NMR spectra on BRUKER AC 300 P (75 MHz) spectrometer. Chemical shifts are expressed in parts per million downfield from tetramethylsilane as an internal standard. Data are given in the following order: δ value, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad), and number of protons, and coupling constants J is given in Hertz. The mass spectra (HRMS) were taken, respectively, on a MS/MS ZABSpec Tof Micromass (EBE TOF geometry) at an ionizing potential of 8 eV and on a VARIAN MAT 311 at an ionizing potential of 70 eV in the “Centre Régional de Mesures Physiques de l’Ouest” (CRMPO, Rennes, France). Reactions under microwave irradiations were realized in the Synthewave® 402 apparatus (Merck Eurolab, Div. Prolabo, France) or in the Explorer® 24 CEM microwave reactor (CEM France, Saclay, France), as well as in the Anton Paar Monowave 300® microwave reactor (Anton Paar France, Les Ulis, France). Microwave irradiation reactions were realized in open cylindrical quartz reactor (Ø = 1.8 cm) with the Synthewave® 402 apparatus or in borosilicate glass vials of 10 mL equipped with snap caps (at the end of the irradiation, cooling reaction was realized by compressed air) with the Explorer® 24 or Monowave® 300 reactors. The microwave instrument consists of a continuous focused microwave power output from 0 to 300W for the Synthewave® 402 or the Explorer® 24 CEM apparatus and from 0 to 800W for the Anton Paar Monowave 300® apparatus. All the experiments in each microwave reactor were performed using the stirring option. The target temperature was reached with a ramp of 2–5 min, and the chosen microwave power stays constant to hold the mixture at this temperature. The reaction temperature is monitored using calibrated infrared sensor and the reaction time included the ramp period. The microwave irradiation parameters (power and temperature) were monitored by the ChemDriver software package for the Explorer® 24 CEM apparatus and by the Monowave software package for the Anton Paar Monowave 300® reactor. Solvents were evaporated with a BUCHI rotary evaporator. All reagents and solvents were purchased from Acros (Geel, Belgium), Sigma-Aldrich Chimie (Saint-Quentin-Fallavier, France), and Fluka France and were used without further purification.
3.1.1. Standard Procedure for the Preparation of 5-Arylidene-2-thioxo-1,3-thiazolidin-4-one (3a–l) in the Synthewave® 402 Microwave Reactor
In a cylindrical quartz reactor (Ø = 1.8 cm), successively commercial rhodanine 1a (1 eq), aromatic aldehyde 2 (1 eq), and n-propylamine (2 eq) were placed. The reactor was then introduced into the Synthewave® 402 Prolabo microwave cavity (power: 300 W). The stirred mixture was irradiated at 80 °C (after a ramp of 2 min. from 20 to 80 °C) for 60 min (power level: 30%, 90 W). After microwave dielectric heating, the crude reaction mixture was allowed to cooling down at room temperature, and ethanol (20 mL) was added in the cylindrical quartz reactor. The resulting insoluble product 3 was filtered, recrystallized in EtOH, and dried under high vacuum (10−2 Torr) for 1 h.
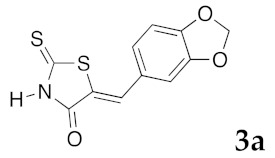
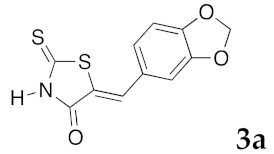
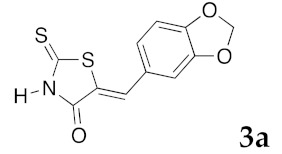
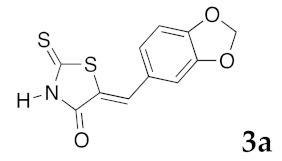
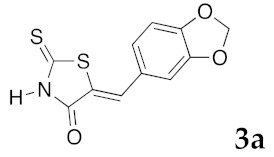
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one (3a). According to the standard procedure, 3a was synthesized from commercial rhodanine 1a (958 mg, 7.2 mmol), piperonaldehyde 2a (1.081 g, 7.2 mmol), and n-propylamine (1.17 mL, 839 mg, 14.2 mmol) in 79% yield as light-yellow powder; mp = 246–250 °C. 1H NMR (DMSO-d6) δ: 6.13 (s, 2H, OCH2O); 7.11 (d, 3H, J = 8.7 Hz, H-2, H-5, H-6, Ar); 7.54 (s, 1H, =CH); 13.74 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 102.1 (OCH2O); 109.2 (C-2′); 109.4 (C-5′); 122.8 (C=, C-5); 126.6 (C-6′); 127.1 (CH=); 131.8 (C-7′); 148.2 (C-4′); 149.6 (C-3′); 169.3 (C=O, C-4); 195.3 (C=S, C-2). HRMS, m/z: 264.9864 found (calculated for C11H7NO3S2, M+. requires 264.9867).
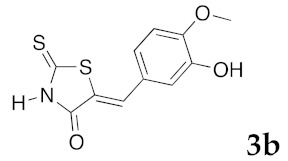
(5Z)-5-(3-Hydroxy-4-methoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (3b). According to the standard procedure, 3b was synthesized from commercial rhodanine 1a (958 mg, 7.2 mmol), 3-hydroxy-4-methoxybenzaldehyde 2d (1.095 g, 7.2 mmol), and n-propylamine (1.17 mL, 839 mg, 14.2 mmol) in 81% yield as light-yellow powder; mp = 222–224 °C. 1H NMR (DMSO-d6) δ: 3.82 (s, 3H, OCH3); 6.94 (d, 1H, J = 6 Hz, H-6, Ar); 7.04 (d, 1H, J = 7.9 Hz, H-5, Ar); 7.11 (s, 1H, H-2, Ar); 7.53 (s, 1H, =CH); 10.06 (br s, 1H, OH); 13.65 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 56.0 (OCH3); 114.7 (C-2′); 116.7 (C-5′); 121.5 (C=, C-5); 124.8 (C-6′); 125.5 (C-1′); 133.1 (=CH); 148.5 (C-4′); 150.4 (C-3′); 169.8 (C=O, C-4); 195.8 (C=S, C-2). HRMS, m/z: 267.0023 found (calculated for C11H9NO3S2, M+. requires 267.0024).
(5Z)-5-(4-Hydroxy-3-methoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (3c). According to the standard procedure, 3c was synthesized from commercial rhodanine 1a (665 mg, 5 mmol), 4-hydroxy-3-methoxybenzaldehyde 2e (1.52 g, 10 mmol), and n-propylamine (1.64 mL, 1.18 g, 20 mmol) in 80% yield as light-yellow powder; mp = 220–222 °C. 1H NMR (DMSO-d6) δ: 3.82 (s, 3H, OCH3); 7.00 (d, 1H, J = 8.9 Hz, H-6, Ar); 7.07 (d, 1H, J = 8.6 Hz, H-5, Ar); 7.13 (s, H-2, Ar); 7.48 (s, 1H, =CH); 9.57 (br s, 1H, OH); 13.62 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 56.1 (OCH3); 112.9 (C-2′); 116.5 (C-5′); 122.4 (C=, C-5); 124.8 (C-6′); 126.1 (C-1′); 132.7 (=CH); 147.5 (C-4′); 150.9 (C-3′); 169.9 (C=O, C-4); 196.0 (C=S, C-2). HRMS, m/z: 267.0022 found (calculated for C11H9NO3S2, M+. requires 267.0024).
(5Z)-5-(3,4-Dimethoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (3d). According to the standard procedure, 3d was synthesized from commercial rhodanine 1a (958 mg, 7.2 mmol), 3,4-dimethoxybenzaldehyde 2h (1.19 g, 7.2 mmol), and n-propylamine (1.18 mL, 851 mg, 14.4 mmol) in 83% yield as light-yellow powder; mp = 232–234 °C. 1H NMR (DMSO-d6) δ: 3.74 (s, 3H, OCH3); 3.81 (s, 3H, OCH3); 7.14 (s, 1H, H-2, Ar); 7.32 (d, 1H, J = 8.6 Hz, H-5, Ar); 7.44 (d, 1H, J = 6.3 Hz, H-6, Ar); 7.59 (s, 1H, =CH); 13.72 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 57.0 (OCH3); 57.1 (OCH3); 112.4 (C-2′); 112.7 (C-5′); 123.6 (C=, C-5); 124.8 (C-6′); 127.5 (C-1′); 128.7 (=CH); 150.4 (C-4′); 150.7 (C-3′); 169.5 (C=O, C-4); 195.8 (C=S, C-2). HRMS, m/z: 281.0167 found (calculated for C12H11NO3S2, M+. requires 281.0184).
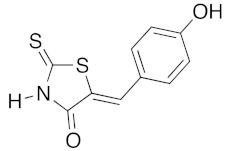
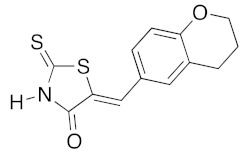
(5Z)-5-(4-Hydroxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (3e). According to the standard procedure, 3e was synthesized from commercial rhodanine 1a (1.33 g, 10 mmol), 4-hydroxybenzaldehyde 2i (1.22 g, 10 mmol), and n-propylamine (1.64 mL, 1.18 g, 20 mmol) in 80% yield as light-yellow powder; mp = 240–242 °C. 1H NMR (DMSO-d6) δ: 6.91 (d, 2H, J = 8.6 Hz, H-3, Ar); 7.47 (d, 2H, J = 8.6 Hz, H-2, Ar); 7.58 (S, 1H, =CH); 10.48 (br s, 1H, OH); 13.68 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 116.7 (C-3′); 123.9 (C-2′); 123.8 (C=, C-5); 129.1 (C-1′); 130.7 (=CH); 159.9 (C-4′); 169.7 (C=O, C-4); 195.7 (C=S, C-2). HRMS, m/z: 236.9918 found (calculated for C10H7NO2S2, M+. requires 236.99182).
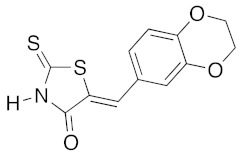
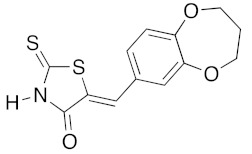
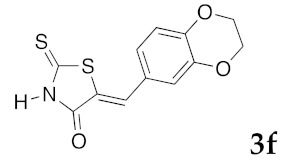
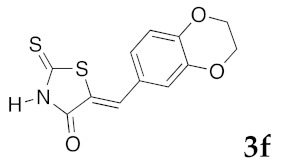
(5Z)-5-(2,3-Dihydro-1,4-benzodioxin-6-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one (3f). According to the standard procedure, 3f was synthesized from commercial rhodanine 1a (958 mg, 7.2 mmol), 2,3-dihydro-1,4-benzodioxin-6-carboxaldehyde 2c (1.18 g, 7.2 mmol), and n-propylamine (1.18 mL, 851 mg, 14.4 mmol) in 89% yield as light-yellow powder; mp = 250–252 °C. 1H NMR (DMSO-d6) δ: 4.29 (t, 2H, OCH2CH2O); 4.30 (t, 2H, OCH2CH2O); 7.02 (d, 1H, J = 8.1 Hz, H-5, Ar); 7.08 (s, 1H, H-2, Ar); 7.10 (d, 1H, J = 8.2 Hz; H-6, Ar); 7.53 (s, 1H, =CH); 13.76 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 63.8 (OCH2CH2O); 64.4 (OCH2CH2O); 118.6 (C-5′); 119.7 (C-2′); 123.5 (=CH); 124.7 (C-6′); 126.7 (C-1′); 132.1 (C=, C-5); 144.2 (C-4′); 146.5 (C-3′); 170.0 (C=O, C-4); 195.9 (C=S, C-2). HRMS, m/z: 279.0016 found (calculated for C12H9NO3S2, M+. requires 279.0024).
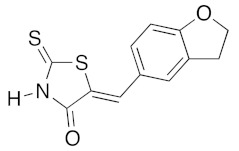
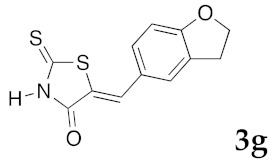
(5Z)-5-(2,3-Dihydro-benzofuran-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one (3g). According to the standard procedure, 3g was synthesized from commercial rhodanine 1a (1.357 g, 10.2 mmol), 2,3-dihydro-1-benzofuran-5-carbaldehyde 2b (1.067 mg, 7.2 mmol), and n-propylamine (1.67 mL, 1.205 g, 20.4 mmol) in 79% yield as light-yellow powder; mp = 250–252 °C. 1H NMR (DMSO-d6) δ: 4.02 (t, 2H, J = 6.8 Hz; CH2CH2O); 6.02 (t, 2H, J = 6.9 Hz; CH2CH2O); 6.84 (d, 1H, J = 8 Hz, H-3, Ar); 7.12 (dd, 1H, J = 8, 1.5 Hz, H-2, Ar); 7.37 (d, 1H, J = 1.4 Hz, H-6, Ar); 8.16 (s, 1H, =CH); 13.66 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 29.2 (CH2CH2O); 71.7 (CH2CH2O); 107.0 (C-5′); 108.4 (C-2′); 123.5 (=CH); 124.7 (C-6′); 126.7 (C-1′); 132.1 (C=, C-5); 144.2 (C-4′); 148.6 (C-3′); 170.0 (C=O, C-4); 195.9 (C=S, C-2). HRMS, m/z: 263.0063 found (calculated for C12H9NO2S2, M+. requires 263.0075).
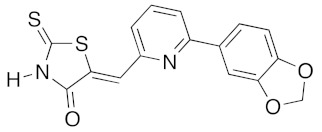
(5Z)-5-{[6-(1,3-Benzodioxol-5-yl)pyridin-2-yl]methylene}-2-thioxo-1,3-thiazolidin-4-one (3h). According to the standard procedure, 3h was synthesized from commercial rhodanine 1a (18 mg, 0.134 mmol), 6-(1,3-benzodioxol-5-yl)-2-pyridinecarbaldehyde 2k (30 mg, 0.134 mmol), and n-propylamine (22 mL, 15.8 mg, 0.268 mmol) in 79% yield as light-yellow powder; mp = 246–250 °C. 1H NMR (DMSO-d6) δ: 6.05 (s, 2H, OCH2O); 6.94 (d, 1H, J = 8 Hz, H-3′); 7.55 (dd, 1H, J = 8, 1.5 Hz, H-2′); 7.59 (d, 1H, J = 8 Hz, H-3); 7.66 (dd, 1H, J = 8, 1.5 Hz, H-5); 7.77 (t, 1H, J = 8 Hz, H-4); 7.93 (dd, 1H, J = 1.4 Hz, H-6′); 8.46 (s, 1H, =CH); 12.46 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 101.8 (OCH2O); 107.0 (C-5″); 107.5 (C-6′); 108.4 (C-2″); 114.0 (C-4′); 116.0 (C=, C-5); 118.0 (C-5″); 124.5 (C-6″); 13.,6 (C-1″); 131.9 (=CH); 132.1 (C-3″); 132.4 (C-2′); 148.6 (C-3″); 150.0 (C-4″); 169.9 (C=O, C-4); 192.4 (C=S, C-2). HRMS, m/z: 342.0131 found (calculated for C16H10N2O3S2, M+. requires 342.0133).
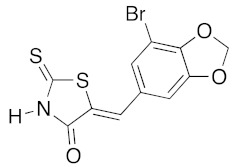
(5Z)-5-[(7-Bromo-1,3-benzodioxol-5-yl)methylene]-2-thioxo-1,3-thiazolidin-4-one (3i). According to the standard procedure, 3i was synthesized from commercial rhodanine 1a (1.31 g, 9.8 mmol), 7-bromo-1,3-benzodioxole-5-carbaldehyde 2l (2.24 g, 9.8 mmol), and n-propylamine (1.6 mL, 1.15 g, 19.6 mmol) in 70% yield as light-yellow powder; mp > 260 °C. 1H NMR (DMSO-d6) δ: 6.18 (s, 2H, OCH2O); 6.37 (s, 1H, H-6); 7.39 (s, 1H, H-2); 7.88 (s, 1H, =CH); 12.10 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 100.7 (C-Br); 102.7 (OCH2O); 109.4 (C-2′); 110.7 (=CH); 127.7 (C-6′); 128.1 (C-1′); 128.7 (C=, C-5); 146.8 (C-4′); 148.7 (C-3′); 166.4 (C=O, C-4); 192.6 (C=S, C-2). HRMS, m/z: 342.8923 found (calculated for C11H1679BrNO3S2, M+. requires 342.8972).
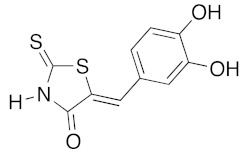
(5Z)-5-(3,4-Dihydroxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (3j). According to the standard procedure, 3j was synthesized from commercial rhodanine 1a (1.385 g, 10.4 mmol), 3,4-dihydroxybenzaldehyde 2f (1.44 g, 10.4 mmol), and n-propylamine (1.7 mL, 1.23 g, 20.8 mmol) in 79% yield as light-yellow powder; mp = 210–212 °C. 1H NMR (DMSO-d6) δ: 6.82 (s, 1H, H-2); 6.89 (d, 1H, J = 8.6 Hz, H-6); 7.05 (d, 1H, J = 8.2 Hz, H-5); 7.47 (s, 1H, =CH); 9.54 (br s, 1H, OH); 9.96 (br s, 1H, OH); 13.66 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 115.5 (C-2′); 117.5 (C-5′); 123.2 (C=, C-5); 128.51 (C-6′); 129.8 (C-1′); 132.7 (=CH); 148.7 (C-4′); 149.4 (C-3′); 169.6 (C=O, C-4); 195.8 (C=S, C-2). HRMS, m/z: 252.9862 found (calculated for C10H7NO3S2, M+. requires 252.9867).
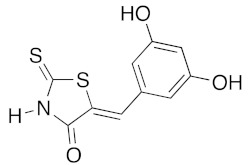
(5Z)-5-(3,5-Dihydroxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (3k). According to the standard procedure, 3k was synthesized from commercial rhodanine 1a (1.385 g, 10.4 mmol), 3,5-dihydroxybenzaldehyde 2n (1.44 g, 10.4 mmol), and n-propylamine (1.7 mL, 1.23 g, 20.8 mmol) in 79% yield as light-yellow powder; mp = 230–232 °C. 1H NMR (DMSO-d6) δ: 6.62 (s, 1H, H-3); 6.65 (d, 1H, J = 8.7 Hz, H-5); 7.46 (d, 1H, J = 5.8 Hz, H-6); 7.94 (s, 1H, =CH); 10.23 (br s, 1H, OH); 10.54 (br s, 1H, OH); 13.72 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 104.9 (C-3′); 110.1 (C-5′); 114.1 (C-6′); 122.4 (C=, C-5); 130.5 (C-1′); 130.8 (=CH); 159.8 (C-2′); 161.1 (C-4′); 169.9 (C=O, C-4); 196.1 (C=S, C-2). HRMS, m/z: 252.9859 found (calculated for C10H7NO3S2, M+. requires 252.9867).
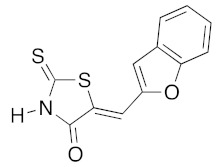
(5Z)-5-(1-Benzofuran-2-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one (3l). According to the standard procedure, 3l was synthesized from commercial rhodanine 1a (1.33 g, 10 mmol), 1-benzofuran-2-carbaldehyde 2m (1.52 g, 10.4 mmol), and n-propylamine (1.64 mL, 1.18 g, 20 mmol) in 62% yield as light-yellow powder; mp = 246–250 °C. 1H NMR (DMSO-d6) δ: 6.98 (s, 1H, C-3); 7.11–7.28 (m, 2H, H-5, H-6); 7.47 (d, 1H, J = 7.7 Hz, H-7); 7.83 (d, 1H, J = 7.5 Hz, H-4); 8.51 (s, 1H, H-2); 8.98 (s, 1H, =CH); 12.10 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 107.2 (=CH); 108.8 (C-3′); 112.6 (C-7′); 118.6 (C-4′); 121.2 (C-5′); 122.1 (C=, C-5); 123.1 (C-6′); 127.6 (C-3a′); 129.7 (C-2′); 136.4 (C-7a′); 164.3 (C=O, C-4); 194.1 (C=S, C-2). HRMS, m/z: 260.9842 found (calculated for C12H7NO2S2, M+. requires 260.9918).
7-Bromo-1,3-benzodioxole-5-carbaldehyde (2l). In a 25 mL round-bottomed flask provided with a magnetic stirrer and condenser, a mixture of commercial 3-bromo-4,5-dihydroxybenzaldehyde (217 mg, 1 mmol), potassium fluoride (529 mg, 9.1 mmol, 9.1 eq), and dibromomethane (70 mL, 173 mg, 1 mmol) in 3 mL of dimethylformamide was stirred vigorously under a stream of nitrogen for 4 h at 140 °C in an oil bath. After cooling down to room temperature, 3 mL of deionized water was added in one portion to the crude reaction mixture. The resulting solution was submitted to extraction with Et2O (3 × 5 mL), and, after decantation, the organic layer was dried over MgSO4. After filtration, the solvent of the filtrate was eliminated in a rotary evaporator under reduced pressure; then, the crude residue was dried under high vacuum (10−2 Torr) at 25 °C for 2 h. The desired 7-bromo-1,3-benzodioxole-5-carbaldehyde 2l was obtained as a light-yellow powder in 51% yield (117 mg) and was sufficiently pure to be used further without purification; mp = 123–125 °C. 1H NMR (DMSO-d6) δ: 6.17 (s, 2H, OCH2O); 7.28 (d, 1H, J = 1.4 Hz, H-2, Ar); 7.55 (d, 1H, J = 1.3 Hz, H-6); 9.78 (s, 1H, CHO). 13C NMR (DMSO-d6) δ: 100.9 (C-Br); 102.6 (OCH2O); 106.2 (C-2); 131.0 (C-6); 132.8 (C-1); 148.9 (C-4); 151.2 (C-3); 189.1 (C=O). HRMS, m/z: 227.9426 found (calculated for C8H5O379Br, M+. requires 227.9422).
3.1.2. Standard Procedure for the Preparation of 5-Arylidene-2-thioxo-1,3-thiazolidin-4-one (3m,n) in the Monowave® 300 Microwave Reactor
In a 10 mL glass tube were placed successively commercial rhodanine 1a (1 eq), aromatic aldehyde 2o,p (1.1–1.2 eq), sodium acetate (26.4 mg, 0.31 mmol, 0.1 eq), and glacial acetic acid (1.2 eq). The glass tube was sealed with a snap cap and placed in the Monowave 300® Anton-Paar microwave cavity (power = 850 W). The mixture was irradiated at 140 °C for 20–30 min under vigorous magnetic stirring. After microwave dielectric heating, the crude reaction mixture was allowed to cool down at room temperature. To this crude mixture, 4 mL of deionized water was added, and the resulting suspension was submitted to ultrasound in a Branson 1510 apparatus at 25 °C for 30 min. Then, the desired compound 3 was collected by filtration, and the crude solid was washed with 5 mL of deionized water and dried under high vacuum (10−2 Torr) at 25 °C for 2 h.
(5Z)-5-(Chroman-6-yl)methylene-2-thioxo-1,3-thiazolidin-4-one (3m). According to the standard procedure, 3m was synthesized after a reaction time of 20 min from commercial rhodanine 1a (200 mg, 1.5 mmol), 6-chromanecarbaldehyde 2o (292 mg, 1.8 mmol), sodium acetate (147 mg, 1.8 mmol), and glacial acetic acid (0.6 mL, 108 mg, 1.8 mmol) in 93% yield as light-yellow powder; mp > 260 °C. 1H NMR (DMSO-d6) δ: 1.92 (m, 2H, CH2); 2.77 (t, 2H, J = 6.3 Hz, CH2); 4.19 (m, 2H, CH2); 6.83 (d, 1H, J = 8.4 Hz, Ar); 7.27 (m, 2H, Ar); 7.47 (s, 1H, CH=); 12.06 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 21.3 (CH2); 24.1 (CH2); 66.6 (CH2); 117.5 (=CH); 121.9 (Ar); 123.6 (C=, C-5); 124.9 (Ar); 130.2 (Ar); 131.9 (Ar); 132.8 (Ar); 157.0 (Ar); 169.7 (C=O, C-4); 195.6 (C=S, C-2). HRMS, m/z: 300.0129 found (calculated for C15H11NO2NaS2, [M+Na]+. requires 300.0129).
(5Z)-5-(3,4-Dihydro-2H-1,5-benzodioxepine-7-yl)methylene-2-thioxo-1,3-thiazolidin-4-one (3n). According to the standard procedure, 3n was synthesized after a reaction time of 20 min from commercial rhodanine 1a (200 mg, 1.5 mmol), 3,4-dihydro-2H-1,5-benzodioxepine-7-carbaldehyde 2p (321 mg, 1.8 mmol), sodium acetate (147 mg, 1.8 mmol), and glacial acetic acid (0.6 mL, 108 mg, 1.8 mmol) in 94% yield as light-yellow powder; mp = 220–222 °C. 1H NMR (DMSO-d6) δ: 2.13 (m, 2H, CH2); 4.20 (m, 4H, 2xCH2); 7.05 (m, 1H, Ar); 7.15 (m, 2H, Ar); 7.50 (s, 1H, CH=); 13.68 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 30.7 (CH2); 70.4 (CH2); 70.5 (CH2); 122.4 (CH=) 123.7 (Ar); 123.8 (C=, C-5); 125.9 (Ar); 128.1 (Ar); 131.0 (Ar); 150.9 (Ar); 153.0 (Ar); 169.3 (C=O, C-4); 195.4 (C=S, C-2). HRMS, m/z: 316.0079 found (calculated for C18H13NO2NaS2, [M+Na]+. requires 316.0078).
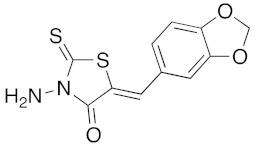
(5Z)-3-Amino-5-benzo[1,3]dioxol-5-ylmethylene-2-thioxo-thiazolidin-4-one (3o). In a 25 mL round-bottomed flask provided with a magnetic stirrer and condenser, a mixture of commercial 3-amino rhodanine 1d (197 mg, 1.33 mmol), piperonaldehyde 2a (200 g, 1.33 mmol), triethylamine (19 mL, 13 mg, 0.13 mmol, 0.1 eq), and glacial acetic acid (8 μL, 8 mg, 0.13 mmol, 0.1 eq) in 4.3 mL of ethyl acetate was stirred vigorously under a stream of argon for 3.5 h at 85 °C in an oil bath. After cooling down to room temperature, the insoluble product of the orange reaction mixture was filtered, washed with absolute ethanol (2 × 3 mL), and was dried under high vacuum (10−2 Torr) for 1 h. The desired 3-amino-5-benzo[1,3]dioxol-5-ylmethylene-2-thioxo-thiazolidin-4-one 3o was obtained as a light-yellow powder in 26% yield (97 mg); mp = 224–226 °C. 1H NMR (DMSO-d6) δ: 5.92 (s, 2H, NH2); 6.15 (s, 2H, H-1); 7.12 (d, 1H, J = 8.1Hz, H-7); 7.19–7.25 (m, 2H, H-3, H-4); 7.79 (s, 1H, H-5); 12.71 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 102.1 (C-1′); 109.3 (C-4′); 109.6 (C-3′); 117.5 (C=, C-5); 127.0 (C-6′); 127.2 (C-5′); 133.5 (CH=); 148.3 (C-2′); 149.9 (C-2′); 163.7 (C=O, C-4); 187.1 (C=S, C-2). HRMS, m/z: 302.9872 found (calculated for C11H8N2O3NaS2, [M+Na]+. requires 302.9874).
3.1.3. Standard Procedure for the Preparation of 5-Arylidene-2-thioxo-1,3-thiazolidin-4-one (3p,q) and (3s,t) in the Explorer® 24 Microwave Reactor
In a 10 mL glass tube were placed successively the 3-N-amino rhodanine derivative 1 (1 eq), piperonaldehyde 2a (1 eq), piperidine (0.1 eq) or sodium acetate (0.1 eq), and glacial acetic acid (0.1–6.6 eq). The glass tube was placed in the Explorer® 24 CEM microwave cavity (power = 300 W). The mixture was irradiated at 120 or 150 °C (with a power of 100 or 200 W) for 20 min under vigorous magnetic stirring. After microwave dielectric heating, the insoluble compound 3 was separated from the crude reaction mixture by filtration. To the collected crude compound 3, absolute ethanol or deionized water was added, and the resulting suspension was stirred vigorously under magnetic stirring for 18 h. The desired compound 3 was collected by filtration and was dried under high vacuum (10−2 Torr) at 25 °C for 1 h.
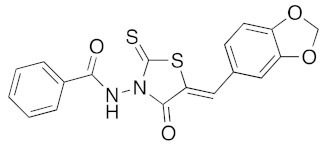
(5Z)-N-(5-Benzo[1,3]dioxol-5-ylmethylene-4-oxo-2-thioxo-thiazolidin-3-yl)-benzamide (3p). According to the standard procedure, 3p was synthesized at 150 °C with a power of 200 W from N-(4-oxo-2-thioxo-thiazolidin-3-yl)-benzamide 1f (222 mg, 1 mmol), piperonaldehyde 2a (150 mg, 1 mmol), piperidine (10 μL, 9 mg, 0.1 mmol, 0.1 eq), and glacial acetic acid (6 μL, 6 mg, 0.1 mmol, 0.1 eq) in 27% yield as light-yellow powder; mp = 182–184 °C. 1H NMR (DMSO-d6) δ: 6.64 (s, 2H, H-1); 7.56 (d, J = 8.1 Hz, 1H, Ar); 7.66 (d, J = 1.7 Hz, 1H, Ar); 7.77 (dd, J = 1.3, 8.2 Hz, 1H, Ar); 8.03 (m, 2H, Ar); 8.29 (s, 1H, CH=); 8.50 (m, 1H, Ar); 8.53 (m, 1H, Ar); 12.03 (br s, 1H, H-6′). 13C NMR (DMSO-d6) δ: 102.3 (OCH2O); 109.4 (Ar); 109.80 (Ar); 116.4 (C=, C-5); 126.9 (C=6); 127.6 (Ar); 127.8 (Ar); 128.8 (Ar); 132.9 (Ar); 135.2 (CH=); 148.4 (Cipso, Cortho, Ar); 150.3 (C-2′); 163.4 (C=O); 164.5 (C=O, C-4); 190.3 (C=S, C-2). HRMS, m/z: 385.0307 found (calculated for C18H13N2O4S2, [M+H]+. requires 385.0317).
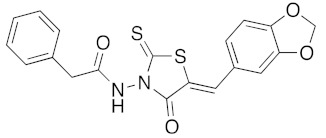
(5Z)-N-(5-Benzo[1,3]dioxol-5-ylmethylene-4-oxo-2-thioxo-thiazolidin-3-yl)-2-phenyl-acetamide (3q). According to the standard procedure, 3q was synthesized at 120 °C with a power of 100 W from N-(4-oxo-2-thioxo-thiazolidin-3-yl)-2-phenyl-acetamide 1h (50 mg, 0.19 mmol), piperonaldehyde 2a (28 mg, 0.19 mmol), sodium acetate (15 mg, 0.19 mmol, 0.1 eq), and glacial acetic acid (70 μL, 74 mg, 1.24 mmol, 6.6 eq) in 98% yield as light-yellow powder; mp = 216–218 °C. 1H NMR (DMSO-d6) δ: 3.71 (s, 2H, CH2Ph); 6.16 (s, 2H, OCH2O); 7.13 (d, 1H, J = 8.1 Hz, Ar); 7.21 (m, 1H, Ar); 7.25 (m, 1H, Ar); 7.28 (m, 1H, Ar); 7.32–7.34 (m, 4H, Ar); 7.84 (s, 1H, CH=); 11.44 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 39.5 (CH2); 102.3 (OCH2O); 109.4 (Ar); 109.7 (Ar); 116.6 (C=, C-5); 126.7 (Ar); 126.9 (Cipso, Ar); 127.4 (Ar); 128.3 (Ar); 129.1 (Ar); 134.6 (Cipso, Ar); 134.9 (Ar); 148.4 (CAr-O); 150.2 (CAr-O); 163.2 (C=O); 168.6 (C=O, C-4); 190.2 (C=S, C-2). HRMS, m/z: 421.0294 found (calculated for C19H14N2O4NaS2, [M+Na]+. requires 421.0293).
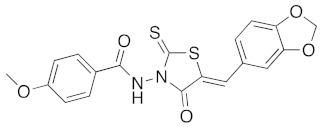
(5Z)-5-Benzo[1,3]dioxol-5-ylmethylene-3-[2-(4-methoxy-phenyl)-2-oxo-ethyl]-2-thioxo-thiazolidin-4-one (3s). According to the standard procedure, 3s was synthesized at 120 °C with a power of 100 W from 4-methoxy-N-(4-oxo-2-thioxo-thiazolidin-3-yl)-benzamide 1g (50 mg, 0.18 mmol), piperonaldehyde 2a (27 mg, 0.18 mmol), sodium acetate (15 mg, 0.18 mmol, 0.1 eq), and glacial acetic acid (70 μL, 70 mg, 1.17 mmol, 6.6 eq) in 83% yield (61 mg) as light-yellow powder; mp = 226–228 °C. 1H NMR (DMSO-d6) δ: 3.85 (s, 3H, MeO); 6.18 (s, 2H, OCH2O); 7.11 (m, 2H, Ar); 7.15 (d, 1H, J = 8.2 Hz, Ar); 7.26 (d, 1H, J = 1.7 Hz, Ar); 7.31 (dd, 1H, J = 1.5, 1.6, 8.4 Hz, Ar); 7.90 (s, 1H, CH=); 7.93 (m, 1H, Ar); 7.96 (m, 1H, Ar); 11.61 (s, 1H, Ar). 13C NMR (DMSO-d6) δ: 55.5 (CH3O); 102.3 (OCH2O); 109.4 (Ar); 109.8 (Ar); 114.0 (Ar); 116.5 (C=, C-5); 122.8 (Cipso, Ar); 126.9 (Cipso, Ar); 127.6 (Ar); 129.9 (Ar); 135.1 (CH=); 148.4 (O-CAr); 150.3 (O-CAr); 162.8 (Cipso, Ar); 163.5 (C=O); 163.9 (C=O, C-4); 190.5 (C=S, C-2). HRMS, m/z: 437.0237 found (calculated for C19H14N2O5NaS2, [M+Na]+. requires 437.0242).
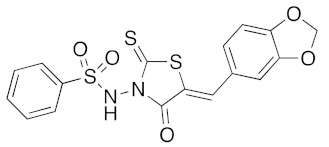
(5Z)-N-(5-Benzo[1,3]dioxol-5-ylmethylene-4-oxo-2-thioxo-thiazolidin-3-yl)-benzenesulfonamide (3t). According to the standard procedure, 3t was synthesized at 150 °C with a power of 200 W from N-(4-oxo-2-thioxo-thiazolidin-3-yl)-benzenesulfonamide 1e (100 mg, 0.35 mmol), piperonaldehyde 2a (52 mg, 0.35 mmol), piperidine (3 μL, 3 mg, 0.034 mmol, 0.1 eq), and glacial acetic acid (2 μL, 2 mg, 0.034 mmol, 0.1 eq) in 64% yield (94 mg) as light-yellow powder; mp = 214–216 °C. 1H NMR (DMSO-d6) δ: 6.16 (s, 2H, OCH2O); 7.12 (d, 1H, J = 8.1 Hz, Ar); 7.19 (d, 1H, J = 1.6 Hz, Ar); 7.24 (dd, 1H, J = 1.7, 8.2 Hz, Ar); 7.59 (m, 2H, Ar); 7.69 (m, 1H, Ar); 7.79 (s, 1H, CH=); 7.86 (m, 2H, Ar); 11.57 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 102.3 (OCH2O); 109.4 (Ar); 109.7 (Ar); 115.9 (C=, C-5); 126.8 (Cipso, Ar); 127.3 (Ar); 127.5 (Ar); 129.1 (Ar); 133.5 (Ar); 134.9 (CH=); 140.7 (Cipso, Ar); 148.3 (O-CAr); 150.2 (O-CAr); 163.4 (C=O, C-4); 189.6 (C=S, C-2). HRMS, m/z: 442.9785 found (calculated for C17H12N2O5NaS3, [M+Na]+. requires 442.9806) and 301.9780 found (calculated for C11H7N2O3NaS2, [M-SO2Ph + Na]+. requires 301.9796).
3.1.4. Standard Procedure for the Preparation of 5-Arylidene-2-thioxo-1,3-thiazolidin-4-one (3r) and (3u,v) in the Monowave® 300 Microwave Reactor
In a 10 mL glass tube, the 3-N-amino rhodanine derivative 1 (1 eq), piperonaldehyde 2a (1 eq), piperidine (0.1 eq) or sodium acetate (0.1 eq), and glacial acetic acid (0.1–6.6 eq) were successively placed. The glass tube was placed in the Monowave® 300 Anton Paar microwave cavity (power = 850 W). The mixture was irradiated at 120 or 150 °C for 20 min under vigorous magnetic stirring. After microwave dielectric heating, the reaction was allowed to cool down to room temperature. To this crude mixture, 2.5 mL of deionized water was added, and the resulting suspension was submitted to ultrasound in a Branson 1510 apparatus at 25 °C for 30 min. Then, the desired insoluble compound 3 was collected by filtration, and 2 mL of absolute ethanol was added to the collected compound 3. The resulting suspension was stirred vigorously under magnetic stirring for 18 h. The desired compound 3 was finally collected by filtration and was dried under high vacuum (10−2 Torr) at 25 °C for 1 h.
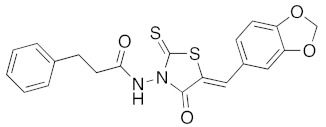
(5Z)-N-(5-Benzo[1,3]dioxol-5-ylmethylene-4-oxo-2-thioxo-thiazolidin-3-yl)-3-phenyl-propionamide (3r). According to the standard procedure, 3r was synthesized at 120 °C from N-(4-oxo-2-thioxo-thiazolidin-3-yl)-3-phenyl-propionamide 1i (50 mg, 0.18 mmol), piperonaldehyde 2a (27 mg, 0.18 mmol), sodium acetate (15 mg, 0.18 mmol, 0.1 eq), and glacial acetic acid (68 μL, 396 mg, 6.6 mmol, 6.6 eq) in 68% yield (50 mg) as light-yellow powder; mp = 226–228 °C. 1H NMR (DMSO-d6) δ: 2.65 (m, 2H, CH2CO); 2.91 (m, 2H, CH2Ph); 6.16 (s, 2H, OCH2O); 7.12–7.32 (m, 8H, Ar); 7.85 (s, 1H, CH=); 11.23 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 30.4 (CH2Ar); 34.5 (CH2CO); 102.3 (OCH2O); 109.4 (Ar); 109.8 (Ar); 116.7 (C=, C-5); 126.1 (Ar); 126.9 (Cipso, Ar); 127.4 (Ar); 128.3–128.4 (Ar); 134.8 (CH=); 140.5 (Cipso, Ar); 148.4 (O-CAr); 150.2 (O-CAr); 163.3 (C=O); 170.0 (C=O, C-4); 190.2 (C=S, C-2). HRMS, m/z: 435.0447 found (calculated for C20H16N2O4NaS2, [M+Na]+. requires 435.0449).
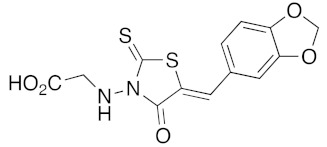
(5Z)-(5-Benzo[1,3]dioxol-5-ylmethylene-4-oxo-2-thioxo-thiazolidin-3-yl)-acetic acid (3u). According to the standard procedure, 3u was synthesized at 120 °C from 2-(4-oxo-2-thioxo-thiazolidin-3-yl)acetic acid 1b (255 mg, 1.33 mmol), piperonaldehyde 2a (200 mg, 1.33 mmol), piperidine (13 μL, 11 mg, 0.13 mmol, 0.1 eq), and glacial acetic acid (8 μL, 7.8 mg, 0.13 mmol, 0.1 eq) in 72% yield (308 mg) as light-yellow powder; mp > 250 °C. 1H NMR (DMSO-d6) δ: 4.81 (s, 2H, CH2COH); 6.26 (s, 2H, OCH2O); 7.23 (d, 1H, J = 8.1 Hz, Ar); 7.31–7.35 (m, 2H, Ar); 7.92 (s, 1H, CH=); 12.37 (br s, 1H, OH). 13C NMR (DMSO-d6) δ: 45.5 (CH2CO2H); 102.0 (OCH2O); 109.1 (Ar); 109.4 (Ar); 118.9 (C=, C-5); 126.7 (Cipso, Ar); 127.0 (Ar); 133.9 (CH=); 148.1 (O-CAr); 149.9 (O-CAr); 166.1 (C=O); 167.1 (C=O, C-4); 192.7 (C=S, C-2). HRMS, m/z: 322.9922 found (calculated for C13H9NO5S2, [M]+. requires 322.9922) and 305.9897 found (calculated for C13H8NO4S2, [M − OH]+ requires 305.9895).
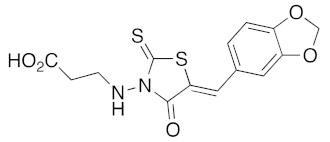
(5Z)-3-(5-Benzo[1,3]dioxol-5-ylmethylene-4-oxo-2-thioxo-thiazolidin-3-yl)-propionic acid (3v). According to the standard procedure, 3v was synthesized at 150°C from 3-(4-oxo-2-thioxo-thiazolidin-3-yl)-propionic acid 1c (50 mg, 0.24 mmol), piperonaldehyde 2a (37 mg, 0.24 mmol), piperidine (2 μL, 2 mg, 0.024 mmol, 0.1 eq), and glacial acetic acid (1.4 μL, 1.4 mg, 0.024 mmol, 0.1 eq) in 62% yield (50 mg) as light-yellow powder; mp = 218–220 °C. 1H NMR (DMSO-d6) δ: 2.58 (m, 2H, CH2CO2H); 4.21 (t, 2H, J = 6.9Hz, NCH2); 6.15 (s, 2H, OCH2O); 7.10–7.24 (m, 3H, Ar); 7.75 (br s, 1H, CH=); 11.95 (br s, 1H, OH). 13C NMR (DMSO-d6) δ: 31.0 (CH2CO2H); 102.2 (OCH2O); 109.3–109.6 (Ar); 119.7 (Cipso, Ar); 126.8 (Ar); 127.2 (C=, C-5); 133.2 (CH=); 148.3 (O-CAr); 149.9 (O-CAr); 166.7 (C=O); 171.8 (C=O, C-4); 193.0 (C=S, C-2). HRMS, m/z: 336.0005 found (calculated for C14H10NO5S2, [M]+. requires 336.0004).
3.1.5. Standard Procedure for the Preparation of (5Z) 2-Amino-5-arylidene-1,3-thiazol-4(5H)-4-one (5a–q) by Sulphur-Nitrogen Displacement in the Synthewave® 402 Microwave Reactor
In a cylindrical quartz reactor (Ø = 1.8 cm), the (5Z)-5-arylidene-2-thioxo-1,3-thiazolidin-4-one 3 (0.5 mmol, 1 eq), and the secondary cyclic amine 4 (1.5 mmol, 3 eq) were successively placed. The reactor was then introduced into the Synthewave® 402 Prolabo microwave cavity (power = 300 W). The stirred mixture was irradiated (after a ramp of 3 min from 20 to the appropriate reaction temperature) at 80–120 °C (power: 80–150 W) for 20–120 min. After microwave dielectric heating, the crude reaction mixture was allowed to cooling down at room temperature, and ethanol (20 mL) was added in the cylindrical quartz reactor. The resulting insoluble product 3 was filtered and recrystallized in absolute EtOH and then dried under high vacuum (10−2 Torr) for 1 h.
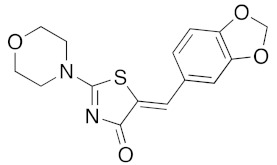
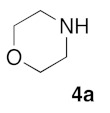
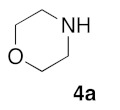
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-(morpholin-1-yl)-1,3-thiazol-4(5H)-one (5a). According to the standard procedure, 5a was synthesized at 80 °C (power: 80 W) after a reaction time of 20 min from (5Z)-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3a (132 mg, 0.5 mmol) and commercial morpholine 4a (131 μL, 131 mg, 1.5 mmol) in 96% yield (153 mg) as light-yellow powder; mp = 250–252 °C. 1H NMR (DMSO-d6) δ: 3.63 (m, 4H, CH2NCH2); 3.90 (m, 4H, CH2OCH2); 6.10 (s, 2H, OCH2O); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7.15 (d, 1H, J = 8 Hz, H-5′); 7.17 (d, 1H, J = 7.9 Hz, H-6′); 7.58 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 66.1 (CH2NCH2); 74.4 (CH2OCH2); 102.2 (OCH2O); 109.1 (C-3′); 109.4 (C-5′); 113.9 (CH=); 125.1 (C-6′); 126.9 (C-1′); 130.6 (C=, C-4); 148.6 (C-4′); 149.2 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 318.0688 found (calculated for C15H14N2O4S, [M]+. requires 318.0674).
(5Z)-5-(3,4-Dimethoxybenzylidene)-2-(morpholin-1-yl)-1,3-thiazol-4(5H)-one (5b). According to the standard procedure, 5b was synthesized at 80 °C (power: 80 W) after a reaction time of 25 min from (5Z)-5-(3,4-dimethoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one 3d (141 mg, 0.5 mmol) and commercial morpholine 4a (131 μL, 131 mg, 1.5 mmol) in 64% yield (107 mg) as light-yellow powder; mp > 260 °C. 1H NMR (DMSO-d6) δ: 3.73 (m, 4H, CH2NCH2); 3.74 (s, 3H, OCH3); 3.81 (s, 3H, OCH3); 3.90 (m, 2H, CH2OCH2); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7.15 (d, 1H, J = 8 Hz, H-5′); 7.17 (d, 1H, J = 7.9 Hz, H-6′); 7.58 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 57.0 (OCH3); 57.1 (OCH3); 66.3 (CH2NCH2); 74.40 (CH2OCH2); 102.2 (OCH2O); 109.1 (C-2′); 109.4 (C-5′); 113.9 (CH=); 125.1 (C-6′); 126.9 (C-1′); 130.6 (C=, C-4); 148.6 (C-4′); 149.2 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 334.0970 found (calculated for C16H18N2O4S, [M]+. requires 334.0987).
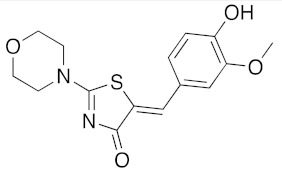
(5Z)-5-(4-Hydroxy-3-methoxybenzylidene)-2-(morpholin-1-yl)-1,3-thiazol-4(5H)-one (5c). According to the standard procedure, 5c was synthesized at 80 °C (power: 80 W) after a reaction time of 20 min from (5Z)-5-(4-hydroxy-3-methoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one 3b (134 mg, 0.5 mmol) and commercial morpholine 4a (131 μL, 131 mg, 1.5 mmol) in 88% yield (141 mg) as light-yellow powder; mp = 251–253 °C. 1H NMR (DMSO-d6) δ: 3.46 (m, 4H, CH2NCH2); 3.81 (s, 3H, OCH3); 3.90 (m, 2H, CH2OCH2); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7,15 (d, 1H, J = 8 Hz, H-5′); 7.17 (d, 1H, J = 7.9 Hz, H-6′); 7.58 (s, 1H, CH=); 9.57 (br s, 1H, OH). 13C NMR (DMSO-d6) δ: 58.1 (OCH3); 66.1 (CH2NCH2); 74.5 (CH2OCH2); 109.1 (C-2′); 109.4 (C-5′); 113.9 (CH=); 125.1 (C-6′); 126.9 (C-1′); 130.6 (C=, C-4); 148.6 (C-4′); 150.4 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 320.0836 found (calculated for C15H16N2O4S, [M]+. requires 320.0831).
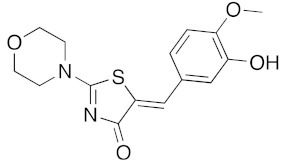
(5Z)-5-(3-Hydroxy-4-methoxybenzylidene)-2-(morpholin-1-yl)-1,3-thiazol-4(5H)-one (5d). According to the standard procedure, 5d was synthesized at 80 °C (power: 80 W) after a reaction time of 20 min from (5Z)-5-(3-hydroxy-4-methoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one 3c (134 mg, 0.5 mmol) and commercial morpholine 4a (131 μL, 131 mg, 1.5 mmol) in 86% yield (138 mg) as light-yellow powder; mp = 256–258 °C. 1H NMR (DMSO-d6) δ: 3.45 (m, 4H, CH2NCH2); 3.91 (s, 3H, OCH3); 4.01 (m, 2H, CH2OCH2); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7.15 (d, 1H, J = 8 Hz, H-5′); 7.17 (d, 1H, J = 7.9 Hz, H-6′); 7.58 (s, 1H, CH=); 9.54 (br s, 1H, OH). 13C NMR (DMSO-d6) δ: 58.1 (OCH3); 66.1 (CH2NCH2); 74.4 (CH2OCH2); 109.1 (C-2′); 109.4 (C-5′); 113.5 (CH=); 125.3 (C-6′); 126.9 (C-1′); 130.3 (C=, C-4); 148.8 (C-4′); 149.2 (C-3′); 175.1 (C=O, C-4). HRMS, m/z: 320.0834 found (calculated for C15H16N2O4S, [M]+. requires 320.0831).
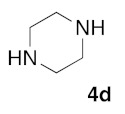
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-(piperazin-1-yl)-1,3-thiazol-4(5H)-one (5e). According to the standard procedure, 5e was synthesized at 80 °C (power: 80 W) after a reaction time of 60 min from (5Z)-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3a (132 mg, 0.5 mmol) and commercial piperazine 4d (129 mg, 1.5 mmol) in 78% yield (124 mg) as light-yellow powder; mp > 260 °C. 1H NMR (DMSO-d6) δ: 3.75 (m, 4H, CH2NCH2); 3.90 (m, 4H, CH2NHCH2); 6.10 (s, 2H, OCH2O); 7.06 (d, 1H, J = 8.1 Hz, H-2′); 7.17 (d, 1H, J = 8 Hz, H-5′); 7.15 (d, 1H, J = 7.9 Hz, H-6′); 7.56 (s, 1H, CH=); 12.58 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 66.2 (CH2NCH2); 74.4 (CH2NHCH2); 102.2 (OCH2O); 109.3 (C-2′); 109.1 (C-5′); 113.9 (CH=); 125.1 (C-6′); 126.9 (C-1′); 130.6 (C=, C-4); 148.6 (C-4′); 149.2 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 317.0831 found (calculated for C15H15N3O3S, [M]+. requires 317.0834).
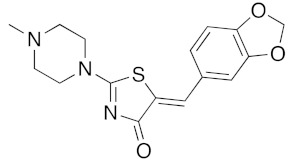
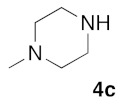
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-(4-N-methylpiperazin-1-yl)-1,3-thiazol-4(5H)-one (5f). According to the standard procedure, 5f was synthesized at 80 °C (power: 80 W) after a reaction time of 60 min from (5Z)-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3a (132 mg, 0.5 mmol) and commercial N-methylpiperazine 4d (166 μL, 150 mg, 1.5 mmol) in 84% yield (139 mg) as light-yellow powder; mp = 202–204 °C. 1H NMR (DMSO-d6) δ: 3.03 (S, 3H, NCH3); 3.73 (m, 4H, CH2NCH2); 3.90 (m, 4H, CH2NMeCH2); 6.10 (s, 2H, OCH2O); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7.15 (d, 1H, J = 8 Hz, H-6′); 7.17 (d, 1H, J = 7.9 Hz, H-6′); 7.57 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 36.1 (NCH3); 65.1 (CH2NCH2); 74.2 (CH2NMeCH2); 102.2 (OCH2O); 109.4 (C-5′); 113.9 (CH=); 125.4 (C-6′); 126.2 (C-1′); 130.6 (C=, C-4); 148.1 (C-4′); 149.6 (C-3′); 173.6 (C=O, C-4). HRMS, m/z: 331.0962 found (calculated for C16H17N3O3S, [M]+. requires 331.0991).
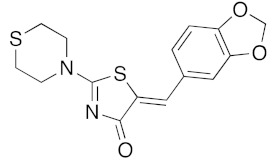
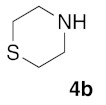
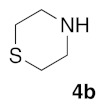
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-(thiomorpholin-1-yl)-1,3-thiazol-4(5H)-one (5g). According to the standard procedure, 5g was synthesized at 80 °C (power: 80 W) after a reaction time of 30 min from (5Z)-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3a (132 mg, 0.5 mmol) and commercial thiomorpholine 4b (150 μL, 154 mg, 1.5 mmol) in 80% yield (134 mg) as light-yellow powder; mp = 204–208 °C. 1H NMR (DMSO-d6) δ: 3.79 (m, 4H, CH2NCH2); 3.90 (m, 4H, CH2SCH2); 6.10 (s, 2H, OCH2O); 7.01 (d, 1H, J = 8.1 Hz, H-2′); 7.16 (d, 1H, J = 8 Hz, H-6′); 7.17 (d, 1H, J = 7.8 Hz, H-6′); 7.57 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 67.0 (CH2NCH2); 74.4 (CH2SCH2); 102.7 (OCH2O); 109.5 (C-5′); 114.1 (CH=); 125.1 (C-6′); 126.9 (C-1′); 130.8 (C=, C-4); 148.6 (C-4′); 149.2 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 334.0441 found (calculated for C15H14N2O3S2, [M]+. requires 334.0449).
(5Z)-5-(3,4-Dimethoxybenzylidene)-2-(thiomorpholin-1-yl)-1,3-thiazol-4(5H)-one (5h). According to the standard procedure, 5h was synthesized at 80 °C (power: 80 W) after a reaction time of 30 min from (5Z)-5-(3,4-dimethoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one 3d (141 mg, 0.5 mmol) and commercial thiomorpholine 4b (150 μL, 154 mg, 1.5 mmol) in 78% yield (137 mg) as light-yellow powder; mp = 240–242 °C. 1H NMR (DMSO-d6) δ: 3.73 (m, 4H, CH2NCH2); 3.74 (s, 3H, OCH3); 3.81 (s, 3H, OCH3); 3.90 (m, 2H, CH2SCH2); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7.15 (d, 1H, J = 8 Hz, H-5′); 7.17 (d, 1H, J = 7.9 Hz, H-6′); 7.60 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 57.0 (OCH3); 57.1 (OCH3); 66.0 (CH2NCH2); 74.4 (CH2SCH2); 109.1 (C-5′); 113.9 (CH=); 125.1 (C-6′); 126.9 (C-1′); 130.6 (C=, C-4); 148.6 (C-4′); 149.2 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 350.0749 found (calculated for C16H18N2O3S2, [M]+. requires 350.0759).
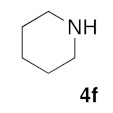
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-(piperidin-1-yl)-1,3-thiazol-4(5H)-one (5i). According to the standard procedure, 5i was synthesized at 80 °C (power: 80 W) after a reaction time of 20 min from (5Z)-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3a (132 mg, 0.5 mmol) and commercial piperidine 4f (149 μL, 128 mg, 1.5 mmol) in 80% yield (127 mg) as light-yellow powder; mp = 201–203 °C. 1H NMR (DMSO-d6) δ: 3.79 (m, 4H, CH2NCH2); 3.90 (m, 4H, CH2SCH2); 6.10 (s, 2H, OCH2O); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7.15 (d, 1H, J = 8 Hz, H-5′; 7.17 (d, 1H, J = 7.8 Hz, H-6′); 7.58 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 60.0 (CH2CH2CH2); 65.9 (CH2CH2CH2); 74.4 (CH2NCH2); 102.3 (OCH2O); 109.1 (C-5′); 109.4 (C-5′); 113.7 (CH=); 125.1 (C-6′); 126.6 (C-1′); 130.5 (C=, C-4); 148.8 (C-4′); 149.4 (C-3′); 174.7 (C=O, C-4). HRMS, m/z: 316.0860 found (calculated for C16H16N2O3S, [M]+. requires 316.0882).
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-[1-(1,3-benzodioxol-5-ylmethyl)piperazin-1-yl]-1,3-thiazol-4(5H)-one (5j). According to the standard procedure, 5j was synthesized at 100 °C (power: 80 W) after a reaction time of 30 min from (5Z)-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3a (132 mg, 0.5 mmol) and commercial 1-(1,3-benzodioxol-5-ylmethyl)piperazine 4e (330 mg, 1.5 mmol) in 75% yield (169 mg) as light-yellow powder; mp > 260 °C. 1H NMR (DMSO-d6) δ: 3.24 (s, 2H, CH2N); 3.73 (m, 4H, CH2NCH2); 3.92 (m, 4H, CH2NCH2); 6.12 (s, 2H, OCH2O); 6.15 (s, 2H, OCH2O); 7.06 (d, 1H, J = 8.1 Hz, H-2′); 7.09 (d, 1H, J = 8.1 Hz, H-2″); 7.15 (d, 1H, J = 8 Hz, H-5′); 7.16 (d, 1H, J = 8 Hz, H-3″); 7.18 (d, 1H, J = 7.9 Hz, H-6′); 7.19 (d, 1H, J = 7.9 Hz, H-2″); 7.59 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 66.1 (CH2NCH2); 74.6 (CH2NCH2); 102.4 (OCH2O); 103.2 (OCH2O); 109.1 (C-2′); 109.2 (Ar); 109.4 (C-5′); 110.1 (Ar); 113.9 (CH=); 125.3 (C-6′); 125.6 (Ar); 126.9 (C-1′); 127.3 (Cipso Ar); 130.6 (C=, C-4); 148.6 (C-4′); 148.8 (Cipso Ar); 149.2 (C-3′); 149.8 (Ar); 174.9 (C=O, C-4). HRMS, m/z: 451.1191 found (calculated for C21H21N3O5S, [M]+. requires 451.1202).
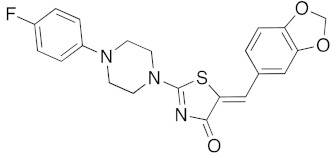
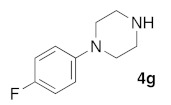
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-[1-(4-fluorophenyl)piperazin-1-yl]-1,3-thiazol-4(5H)-one (5k). According to the standard procedure, 5k was synthesized at 120 °C (power: 150 W) after a reaction time of 30 min from (5Z)-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3a (132 mg, 0.5 mmol) and commercial 1-(4-fluorophenyl)piperazine 4g (270 mg, 1.5 mmol) in 72% yield (148 mg) as light-yellow powder; mp > 260 °C. 1H NMR (DMSO-d6) δ: 3.71 (m, 4H, CH2NCH2); 3.90 (m, 4H, CH2NCH2); 6.10 (s, 2H, OCH2O); 7.04 (d, 1H, J = 8 Hz, H-2′); 7.12 (d, 2H, J = 8.1 Hz, H-2″); 7.15 (d, 1H, J = 8 Hz, H-5′); 7.16 (d, 1H, J = 7.9 Hz, H-6); 7.18 (d, 1H, J = 7.9 Hz, H-3″); 7.56 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 65.9 (CH2NCH2); 74.4 (CH2NCH2); 102.7 (OCH2O); 109.4 (C-2′); 109.2 (Cortho Ar); 109.4 (C-5′); 110.1 (Cmeta Ar); 113.7 (CH=); 125.1 (C-6′); 125.6 (C-4″); 126.9 (C-1′); 127.3 (Ar); 130.6 (C=, C-4); 148.4 (C-4′); 148.7 (Cipso Ar); 149.5 (C-3′); 174.5 (C=O, C-4). HRMS, m/z: 411.1021 found (calculated for C21H18N3O3S19F, [M]+. requires 411.1053).
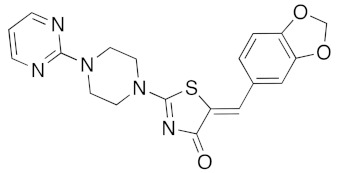
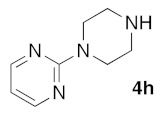
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-[4-(pyrimidin-2-yl)piperazin-1-yl]-1,3-thiazol-4(5H)-one (5l). According to the standard procedure, 5l was synthesized at 100 °C (power: 150 W) after a reaction time of 25 min from (5Z)-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3a (132 mg, 0.5 mmol) and commercial 2-(1-piperazin-1-yl)pyrimidine 4h (212 μL, 246 mg, 1.5 mmol) in 80% yield (158 mg) as light-yellow powder; mp > 260 °C. 1H NMR (DMSO-d6) δ: 3.75 (m, 4H, CH2NCH2); 3.87 (m, 4H, CH2NCH2); 6.14 (s, 2H, OCH2O); 7.06 (d, 1H, J = 7.9 Hz, H-2′); 7.13 (d, 1H, J = 8 Hz, H-5′); 7.16 (d, 1H, J = 7.9 Hz, H-6′); 7.18 (m, 3H, Ar); 7.61 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 66.0 (CH2NCH2); 74.2 (CH2NCH2); 102.5 (OCH2O); 109.2 (C-2′); 109.4 (C-5′); 110.1 (Ar); 113.9 (CH=); 125.2 (C-6′); 125.6 (Cipso Ar); 126.9 (C-1′); 130.5 (C=, C-4); 148.6 (C-4′); 148.8 (Cipso Ar); 149.2 (C-3′); 174.7 (C=O, C-4). HRMS, m/z: 395.1006 found (calculated for C19H17N5O3S, [M]+. requires 395.1052).
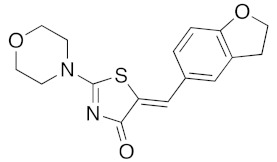
(5Z)-5-(2,3-Dihydro-benzofuran-5-ylmethylene)-2-(morpholin-4-yl)-1,3-thiazol-4(5H)-one(5m). According to the standard procedure, 5m was synthesized at 80 °C (power: 90 W) after a reaction time of 20 min from (5Z)-5-(2,3-dihydro-benzofuran-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3g (131 mg, 0.5 mmol) and commercial morpholine 4a (131 μL, 131 mg, 1.5 mmol) in 80% yield (127 mg) as light-yellow powder; mp = 222–224 °C. 1H NMR (DMSO-d6) δ: 3.70 (t, 2H, J = 6.8 Hz; CH2CH2O); 3.73 (m, 4H, CH2NCH2); 3.90 (t, 2H, J = 6.9 Hz; CH2CH2O); 5.20 (m, 2H, CH2OCH2); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7.15 (d, 1H, J = 8 Hz, H-5′); 7.17 (d, 1H, J = 7.9 Hz, H-6′); 7.58 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 29.2 (CH2CH2O); 65.9 (CH2NCH2); 71.7 (CH2CH2O); 74.4 (CH2OCH2); 109.1 (C-2′); 109.5 (C-5′); 113.9 (CH=); 125.1 (C-6′); 126.9 (C-1′); 130.6 (C=, C-4); 148.6 (C-4′); 149.4 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 316.0864 found (calculated for C16H16N2O3S, [M]+. requires 316.0882).
(5Z)-5-(2,3-Dihydro-benzofuran-5-ylmethylene)-2-(thiomorpholin-4-yl)-1,3-thiazol-4(5H)-one(5n). According to the standard procedure, 5n was synthesized at 80 °C (power: 90 W) after a reaction time of 20 min from (5Z)-5-(2,3-dihydro-benzofuran-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3g (131 mg, 0.5 mmol) and commercial thiomorpholine 4b (150 μL, 154 mg, 1.5 mmol) in 84% yield (140 mg) as light-yellow powder; mp = 230–232 °C. 1H NMR (DMSO-d6) δ: 3.73 (t, 2H, J = 6.8 Hz; CH2CH2O); 4.02 (m, 4H, CH2NCH2); 4.09 (m, 4H, CH2SCH2); 5.22 (t, 2H, J = 6.9 Hz; CH2CH2O); 7.02 (d, 1H, J = 8.1 Hz, H-2′); 7.13 (d, 1H, J = 8 Hz, H-5′); 7.19 (d, 1H, J = 7.9 Hz, H-6′); 7.60 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 29.3 (CH2CH2O); 66.1 (CH2NCH2); 71.7 (CH2SCH2); 74.4 (CH2CH2O); 108.9 (C-2′); 109.4 (C-5′); 113.7 (CH=); 125.2 (C-6′); 126.9 (C-1′); 130.7 (C=, C-4); 148.4 (C-4′); 149.2 (C-3′); 174.6 (C=O, C-4). HRMS, m/z: 332.0643 found (calculated for C16H16N2O2S2, [M]+. requires 332.0653).
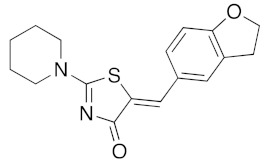
(5Z)-5-(2,3-Dihydro-benzofuran-5-ylmethylene)-2-(piperidin-1-yl)-1,3-thiazol-4(5H)-one(5o). According to the standard procedure, 5o was synthesized at 80 °C (power: 90 W) after a reaction time of 20 min from (5Z)-5-(2,3-dihydro-benzofuran-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3g (131 mg, 0.5 mmol) and commercial piperidine 4f (149 μL, 128 mg, 1.5 mmol) in 86% yield (135 mg) as light-yellow powder; mp = 252–254 °C. 1H NMR (DMSO-d6) δ: 3.71 (m, 6H, CH2); 3.73 (t, 2H, J = 6.8 Hz; CH2CH2O); 4.02 (m, 4H, CH2NCH2); 5.22 (t, 2H, J = 6.9 Hz; CH2CH2O); 7.06 (d, 1H, J = 8 Hz, H-2′); 7.15 (d, 1H, J = 8.2 Hz, H-5′); 7.17 (d, 1H, J = 7.9 Hz, H-6′); 7.56 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 24.1 (CH2CH2CH2); 29.1 (CH2CH2CH2); 31.2 (CH2CH2O); 66.2 (CH2NCH2); 74.3 (CH2CH2O); 109.1 (C-2′); 109.3 (C-5′); 113.9 (CH=); 125.1 (C-6′); 126.8 (C-1′); 130.6 (C=, C-4); 148.6 (C-4′); 149.3 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 314.1075 found (calculated for C17H18N2O2S, [M]+. requires 314.1089).
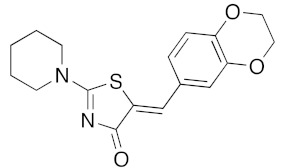
(5Z)-5-(2,3-Dihydro-1,4-benzodioxin-6-ylmethylene)-2-(piperidin-1-yl)-1,3-thiazol-4(5H)-one (5p). According to the standard procedure, 5p was synthesized at 80 °C (power: 90 W) after a reaction time of 20 min from (5Z)-5-(2,3-dihydro-1,4-benzodioxin-6-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3f (140 mg, 0.5 mmol) and commercial piperidine 4f (149 μL, 128 mg, 1.5 mmol) in 88% yield (145 mg) as light-yellow powder; mp = 257–259 °C. 1H NMR (DMSO-d6) δ: 2.93 (m, 6H, CH2); 3.72 (m, 4H, CH2NCH2); 4.28 (t, 2H, OCH2CH2O); 4.30 (t, 2H, OCH2CH2O); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7,14 (d, 1H, J = 8 Hz, H-5′); 7.18 (d, 1H, J = 7.9 Hz, H-6′); 7.60 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 24.0 (CH2CH2CH2); 29.0 (CH2CH2CH2); 63.1 (CH2NCH2); 64.8 (OCH2CH2O); 66.4 (OCH2CH2O); 109.2 (C-2′); 109.4 (C-5′); 113.7 (CH=); 125.2 (C-6′); 126.9 (C-1′); 130.7 (C=, C-4); 148.7 (C-4′); 149.2 (C-3′); 175.1 (C=O, C-4). HRMS, m/z: 330.1081 found (calculated for C17H18N2O3S, [M]+. requires 330.1038).
(5Z)-5-(2,3-Dihydro-1,4-benzodioxin-6-ylmethylene)-2-(thiomorpholin-4-yl)-1,3-thiazol-4(5H)-one (5q). According to the standard procedure, 5q was synthesized at 80 °C (power: 90 W) after a reaction time of 20 min from (5Z)-5-(2,3-dihydro-1,4-benzodioxin-6-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3f (140 mg, 0.5 mmol) and commercial thiomorpholine 4b (150 μL, 154 mg, 1.5 mmol) in 80% yield (139 mg) as light-yellow powder; mp > 260 °C. 1H NMR (DMSO-d6) δ: 3.71 (m, 4H, CH2NCH2); 3.90 (m, 2H, CH2SCH2); 4.27 (t, 2H, OCH2CH2O); 4.30 (t, 2H, OCH2CH2O); 5.10 (m, 2H, CH2OCH2); 7.01 (d, 1H, J = 8.2 Hz, H-2′); 7.15 (d, 1H, J = 8 Hz, H-5′); 7.19 (d, 1H, J = 7.9 Hz, H-6′); 7.58 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 64.8 (OCH2CH2O); 66.2 (CH2NCH2O); 66.6 (OCH2CH2O); 74.40 (CH2SCH2); 102.2 (OCH2O); 109.0 (C-2′); 109.4 (C-5′); 113.9 (CH=); 125.1 (C-6′); 126.9 (C-1′); 130.8 (C=, C-4); 148.6 (C-4′); 149.4 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 348.0603 found (calculated for C16H16N2O3S2, [M]+. requires 348.0602).
3.1.6. Standard Procedure for the Preparation of (5Z) 2-N-Heteroarylamino-5-arylidene-1,3-thiazol-4(5H)-one Derivatives (5r–x) by Solution Phase Condensation under Microwave in the Explorer® 24 Reactor
In a 10 mL glass tube, the 2-N-heteroarylamino-thiazolidin-4-one 8 (1 eq), aromatic aldehyde 2 (1 eq), piperidine (0.1 eq) or sodium acetate (0.1 eq), and glacial acetic acid (0.1–6.6 eq) were successively placed. The glass tube was placed in the Explorer® 24 CEM microwave cavity (power = 300 W). The mixture was irradiated at 120 or 150 °C (with a power of 100 or 200 W) for 20 or 40 min under vigorous magnetic stirring. After microwave dielectric heating, the reaction was allowed to cool down to room temperature. To this crude mixture, 1–2 mL of deionized water was added, and the resulting suspension was submitted to ultrasound in a Branson 1510 apparatus at 25 °C for 30 min. Then, the desired insoluble compound 3 was collected by filtration, and 2 mL of absolute ethanol was added to this collected compound 5. The resulting suspension was stirred vigorously under magnetic stirring for 18 h. The desired compound 3 was finally collected by filtration and was dried under high vacuum (10−2 Torr) at 25 °C for 1 h.
(5Z)-5-Benzo[1,3]dioxol-5-ylmethylene-2-phenylamino-1,3-thiazol-4(5H)-one (5r). According to the standard procedure, 5r was synthesized at 150 °C with a power of 200 W for 20 min from 2-phenylamino-1,3-thiazolidin-4-one 8a (192 mg, 1 mmol), piperonaldehyde 2a (150 mg, 1 mmol), piperidine (10 μL, 9 mg, 0.1 mmol, 0.1 eq), and glacial acetic acid (6 μL, 6 mg, 0.1 mmol, 0.1 eq) in 34% yield (100 mg) as reddish powder; mp > 250 °C. 1H NMR (DMSO-d6) δ: 6.08 (s, 2H, OCH2O); 7.03–7.22 (m, 5H, Ar + CH=); 7.42 (q, J = 7.4Hz, 2H, Ar); 7.56–7.78 (m, 2H, Ar); 12.21 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 101.8 (OCH2O); 108.7 (C-4′); 109.0 (C-7′); 120.4 (C-6′); 121.0 (Cipso, C-5′); 121.4 (C-2″); 124.8 (C-2″); 125.2 (C-4″); 127.5 (C=, C-5); 129.1 (C-3″); 129.4 (C-3″); 130.6 (CH=); 147.9 (Cipso, C-1″); 148.1 (Cipso, C-3a′); 148.7 (Cipso, C-7a′); 170.4 (C=N, C-2); 180.5 (C=O, C-4). HRMS, m/z: 325.0646 found (calculated for C17H13N2O3S, [M+H]+. requires 325.0647).
(5Z)-5-Benzo[1,3]dioxol-5-ylmethylene-2-(pyridin-2-yl)amino-1,3-thiazol-4(5H)-one (5s). According to the standard procedure, 5s was synthesized at 120 °C with a power of 100 W for 20 min from 2-(pyridin-2-yl)amino-1,3-thiazolidin-4-one 8b (50 mg, 0.26 mmol), piperonaldehyde 2a (39 mg, 0.26 mmol), sodium acetate (21 mg, 0.26 mmol, 1 eq), and glacial acetic acid (0.1 mL, 102 mg, 1.7 mmol, 6.6 eq) in 90% yield (76 mg) as green-yellowish powder; mp > 250 °C. 1H NMR (DMSO-d6) δ: 6.13 (s, 2H, OCH2O); 7.12 (d, J = 7.9 Hz, 1H, CH=); 7.17–7.23 (m, 4H, H-7′, H-4′, H-3″, H-5″); 7.58 (m, 1H, H-6′); 7.85 (t, J = 7.6 Hz, 1H, H-4″); 8.52 (d, J = 4.1 Hz, 1H, H-6″); 12.39 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 101.9 (OCH2O); 109.0 (C-4′); 120.1 (Ar); 127.8 (C=, C-5); 130.2 (Ar); 131.1 (CH=); 133.1 (Ar); 138.9 (Ar); 145.6 (Cipso, C-2″); 148.0 (Cipso, C-3a′); 148.8 (Cipso, C-7a′); 163.6 (C=N, C-2); 187.4 (C=O, C-4). HRMS, m/z: 348.0418 found (calculated for C16H11N3O3SNa, [M+Na]+. requires 348.0419).
(5Z)-5-Benzo[1,3]dioxol-5-ylmethylene-2-(pyridin-4-yl)amino-1,3-thiazol-4(5H)-one (5t). According to the standard procedure, 5t was synthesized at 120 °C with a power of 100 W for 40 min from 2-(pyridin-4-yl)amino-1,3-thiazolidin-4-one 8c (150 mg, 0.78 mmol), piperonaldehyde 2a (117 mg, 0.78 mmol), sodium acetate (64 mg, 0.78 mmol, 1 eq), and glacial acetic acid (295 μL, 307 mg, 5.11 mmol, 6.6 eq) in 43% yield (109 mg) as orange-yellowish powder; mp > 250 °C. 1H NMR (DMSO-d6) δ: 6.11 (s, 2H, OCH2O); 7.11 (m, 2H, H-3″); 7.13 (s, 1H, H-7′); 7.72–7.67 (m, 3H, CH=, H-6′, H-4′); 8.72 (m, 2H, H-2″). 13C NMR (DMSO-d6) δ: 102.4 (OCH2O); 109.6–109.6 (C-7′, C-3″); 117.9 (C-4′); 126.1 (C-6′); 127.6 (C-5′); 132.4 (CH=); 146.3–146.3 (C-2″); 149.2–148.1 (C-3a′, C-7a′); 170.4 (C=N, C-2); 180.5 (C=O, C-4). HRMS, m/z: 370.0243 found (calculated for C16H10N3O3Na2S, [M-H+2Na]+. requires 370.0238).
(5Z)-5-Benzo[1,3]dioxol-5-ylmethylene-2-(4-methoxy-phenylamino)-1,3-thiazol-4(5H)-one (5u). According to the standard procedure, 5u was synthesized at 120 °C with a power of 100 W for 20 min from 2-(4-methoxyphenyl)amino-1,3-thiazolidin-4-one 8d (50 mg, 0.22 mmol), piperonaldehyde 2a (34 mg, 0.22 mmol), sodium acetate (18 mg, 0.22 mmol, 1 eq), and glacial acetic acid (80 μL, 87 mg, 1.45 mmol, 6.6 eq) in 90% yield (72 mg) as light-yellow powder; mp > 250 °C. 1H NMR (DMSO-d6) δ: 3.76 (s, 3H, CH3O); 6.13 (s, 2H, OCH2O); 7.04–7.16 (m, 5H, CH=, Ar); 7.55–7.70 (m, 3H, Ar); 11.56 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 55.3 (CH3O); 101.8 (OCH2O); 108.7 (C-5′); 109.0 (C-4′); 114.2 (C-3″); 114.6 (C-3″); 122.0 (C-7′); 123.1 (C=, C-5); 124.8 (C-2″); 125.1 (C-2″); 127.7 (Cipso, C-1″); 128.2 (C-4″); 129.4 (C-6′); 130.0 (CH=); 147.9 (C-3a′); 148.1 (C-7a′); 148.7 (C=O, C-4); 156.5 (C=N, C-2). HRMS, m/z: 377.0574 found (calculated for C18H14N2O4NaS, [M+Na]+. requires 377.0572).
(5Z)-5-Benzo[1,3]dioxol-5-ylmethylene-2-(4-chloro-phenylamino)-1,3-thiazol-4(5H)-one (5v). According to the standard procedure, 5v was synthesized at 120 °C with a power of 100 W for 20 min from 2-(4-chlorophenyl)amino-1,3-thiazolidin-4-one 8e (50 mg, 0.22 mmol), piperonaldehyde 2a (34 mg, 0.22 mmol), sodium acetate (18 mg, 0.22 mmol, 1 eq), and glacial acetic acid (80 μL, 87 mg, 1.45 mmol, 6.6 eq) in 79% yield (62 mg) as light-yellow powder; mp > 250 °C. 1H NMR (DMSO-d6) δ: 6.08 (s, 2H, OCH2O); 7.04–7.17 (m, 4H, Ar); 7.43–7.51 (m, 2H, Ar); 7.57–7.68 (m, 2H, Ar); 7.81 (d, J = 7.2 Hz, 1H, CH=); 11.65 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 101.8 (OCH2O); 108.8 (C-4″); 109.1 (C-5′); 109.3 (C-4′); 122.0 (C-7′); 123.1 (C-2″); 124.9 (C-2″); 125.3 (C-5′); 127.4 (C=, C-5); 129.0 (C-1″); 129.4 (C-4″); 129.8 (C-6′); 131.0 (CH=); 148.0 (C-3a′); 148.1 (C-7a′); 148.8 (C=O, C-4); 156.7 (C=N, C-2). HRMS, m/z: 381.0079 found (calculated for C17H11N2O335ClSNa, [M+Na]+. requires 381.0077).
(5Z)-5-(2,3-Dihydro-benzofuran-5-ylmethylene)-2-phenylamino-1,3-thiazol-4(5H)-one (5w). According to the standard procedure, 5w was synthesized at 150 °C with a power of 200 W for 20 min from 2-phenylamino-1,3-thiazolidin-4-one 8a (100 mg, 0.22 mmol), 2,3-dihydro-1-benzofuran-5-carbaldehyde 2b (77 mg, 0.52 mmol), piperidine (5 mL, 4 mg, 0.052 mmol, 0.1 eq), and glacial acetic acid (3 μL, 3 mg, 0.052 mmol, 0.1 eq) in 64% yield (107 mg) as light-yellow powder; mp > 250 °C. 1H NMR (DMSO-d6) δ: 3.21 (m, 2H, CH2); 4.54–4.65 (m, 2H, CH2O); 6.90 (m, 1H, H-2″); 7.03 (m, 1H, H-4″); 7.19 (t, J = 7.4 Hz, 1H, H-7′); 7.27–7.66 (m, 5H, H-4′, H-6′, H-3″); 7.77 (d, J = 7.7 Hz, 1H, CH=); 12.25 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 28.5 (CH2); 71.8 (OCH2); 108.7 (C-7′); 120.4 (C-2″); 121.4 (C-2″); 124.8 (C-4″); 125.9 (C-3a′); 126.5 (C-6′); 128.9 (C=, C-5); 129.0 (C-1″); 129.2 (C-4′); 129.4 (C-3″); 129.9 (C-3″); 131.0 (CH=); 148.0 (C-3a′); 148.1 (C-7a′); 161.4 (C=O, C-4); 206.7 (C=N, C-2). HRMS, m/z: 345.0676 found (calculated for C18H14N2O2SNa, [M+Na]+. requires 345.0674).
(5Z)-5-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethylene)-2-phenylamino-1,3-thiazol-4(5H)-one (5x). According to the standard procedure, 5x was synthesized at 120 °C with a power of 100 W for 20 min from 2-phenylamino-1,3-thiazolidin-4-one 8a (100 mg, 0.52 mmol), 2,3-dihydro-1,4-benzodioxine-6-carbaldehyde 2c (85 mg, 0.52 mmol), sodium acetate (43 mg, 0.52 mmol, 1 eq), and glacial acetic acid (0.2 μL, 205 mg, 3.42 mmol, 6.6 eq) in 98% yield (175 mg) as light-yellow powder; mp > 250 °C. 1H NMR (DMSO-d6) δ: 4.24–4.33 (m, 4H, OCH2CH2O); 6.93–7.06 (m, 2H, H-2″); 7.12 (m, 1H, H-8′); 7.20 (t, J = 7.4 Hz, 1H, H-4″); 7.42 (m, 2H, H-3″); 7.52 (m, 1H, H-5′); 7.62 (m, 1H, H-7′); 7.77 (m, 1H, CH=); 12.26 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 63.9–64.4 (CH2CH2O); 117.7 (C-5′); 117.8 (C-8′); 120.5 (C-2″); 121.0 (C-1′); 121.4 (C-2″); 123.3 (C-7′); 124.9 (C-4″); 126.7 (C=, C-5); 127.1 (C-1″); 129.1 (C-3″); 129.4 (C-3″); 130.4 (CH=); 143.5 (C-4a′); 145.1 (C-8a′); 170.5 (C=O, C-4); 117.3 (C=N, C-2). HRMS, m/z: 361.0623 found (calculated for C18H14N2O3SNa, [M+Na]+. requires 361.0623).
3.2. Biochemistry Part
3.2.1. Protein Kinase Assay Buffers
Buffer A: 10 mM MgCl2, 1 mM EGTA, 1 mM DTT, 25 mM Tris-HCl pH 7.5, 50 µg heparin/mL.
Buffer C: 60 mM β-glycerophosphate, 15 mM p-nitrophenyl-phosphate, 25 mM Mops (pH 7.2), 5 mM EGTA, 15 mM MgCl2, 1 mM DTT, 1 mM sodium vanadate, 1 mM phenylphosphate.
3.2.2. Kinase Preparations and Assays
Kinase activities for each enzyme were assayed in buffer A or C, with their corresponding substrates, in the presence of 15 µM ATP in a final volume of 30 µL. After 30-min incubation at 30 °C, the reaction was stopped by harvesting, using a FilterMate harvester (Packard), onto P81 phospho-cellulose papers (GE Healthcare, Chicago, IL, USA) which were washed in 1% phosphoric acid. Scintillation fluid was added and the radioactivity measured in a Packard counter. Blank values were subtracted and activities calculated as pmoles of phosphate incorporated for the 30-min incubation. The activities were expressed in % of the maximal activity, i.e., in the absence of inhibitors. Controls were performed with appropriate dilutions of DMSO.
CDK5/p25 (human, recombinant) was prepared as previously described [57]. Its kinase activity was assayed in buffer C, with 1 mg histone H1/mL, in the presence of 15 μM [γ-33P] ATP (3000 Ci/mmol; 10 mCi/mL) in a final volume of 30 μL. After 30-min incubation at 30 °C, 25 μL aliquots of supernatant were spotted onto 2.5 × 3 cm pieces of Whatman P81 phospho-cellulose paper, and, 20 s later, the filters were washed five times (for at least 5 min each time) in a solution of 10 mL phosphoric acid/litter of water. The wet filters were counted in the presence of 1 mL ACS (Amersham, UK) scintillation fluid.
DYRK1A (rat, recombinant, expressed in Escherichia coli as a GST fusion protein, provided by Dr. W. Becker) was purified by affinity chromatography on glutathione–agarose and assayed as described for CDK5/p25 using myelin basic protein (1 mg/mL) as a substrate.
Casein kinase 1 (CK1δ/ε) (porcine brain, native) was assayed with 0.67 µg of CKS peptide (RRKHAAIGpSAYSITA), a CK1 specific substrate obtained from Millegen (Labege, France) [58].
GSK-3α/β (porcine brain, native) was assayed, as described for CDK5/p25 but in Buffer A and using a GSK-3 specific substrate (GS-1: YRRAAVPPSPSLSRHSSPHQSpEDEEE) (pS stands for phosphorylated serine) [59]. GS-1 was synthesized by Millegen (Labege, France).
3.2.3. Cell Culture and Survival Assays
Skin diploid fibroblastic cells were provided by BIOPREDIC International Company (Rennes, France). Caco2 (Ref ECACC: 86010202), Huh-7D12 (Ref ECACC: 01042712), MDA-MB-231 (Ref ECACC: 92020424), HCT-116 (Ref ECACC: 91091005), PC3 (Ref ECACC: 90112714), and NCI-H727 (Ref ECACC: 94060303) cell lines were obtained from the ECACC collection. Cells were grown according to ECACC recommendations [60]. The toxicity test of the compounds on these cells was as follows: 2 × 103 cells for HCT-116 cells or 4 × 103 for the other cells were seeded in 96-multi-well plates in triplicate and left for 24 h for attachment, spreading and growing. Then, cells were exposed for 48 h to increasing concentrations of the compounds, ranging from 0.1 to 25 mM, in a final volume of 120 μL of culture medium. Cells were fixed in cooled 90% ethanol/5% acetic acid solution, and nuclei were stained with Hoechst 3342 (Sigma) and counted using automated imaging analysis (Cellomics Arrayscan VTI/HCS Reader, Thermo/Scientific, Waltham, MA, USA). The IC50 were graphically determined.
4. Conclusions
All the data presented in this study demonstrate the interest and the potential of the 1,3-thiazolidin-4-one core in inhibition of the protein kinase DYRK1A. In this preliminary project of pharmacology, we have developed two series of new compounds under microwave irradiation. In the first library, the twenty-two 5-arylidene-2-thioxo-1,3-thiazolidin-4-ones 3a–v were synthesized in yields ranging from 26 to 98% by microwave irradiation assisted Knoevenagel condensation using an “open or closed vessel” approach using organic bases (propylamine, TEA, and AcONa). Next, most of the compounds 3 were transformed into 2-amino-5-arylidene-1,3-thiazol-4(5H)-ones 5a–q under microwave irradiation by sulphur/nitrogen displacement with appropriate secondary cyclic amines 4a–h. This second family was completed by the preparation of seven new 2-N-heteroarylamino-5-arylidene-1,3-thiazol-4(5H)-ones 5r–x from the 2-N-heteroarylamino-1,3-thiazolidin-4-one building blocks 8a–e under microwave irradiation (34–98% yield). In the two libraries, all the 46 compounds 3 and 5 have been built with a Z-geometry and were evaluated against four protein kinases. Among all these compounds, nine of them turned out to be very interesting because they presented sub-micromolar inhibition activity on DYRK1A (0.090 μM > IC50 > 0.028 μM). The most effective compounds in these two libraries are the molecules 3e and 5s, as new nano-molar DYRK1A inhibitors (3e: IC50 0.028 μM and 5s: IC50 0.033 μM). The current results are the starting point of a new larger program within our group to investigate the biological properties of these new inhibitors with potential application in Alzheimer’s disease or in cancer.
Acknowledgments
The authors are grateful to the assistance of the staff (N. le Yondre, P. Jéhan, F. Lambert) of CMRPO analytical chemistry core facility for HRMS analysis (CRMPO platform UMS 2001 CNRS, Université de Rennes 1, Bât. 11A, Campus de Beaulieu, Rennes, France). We are thankful to Merck Eurolab Prolabo (France) for providing the Synthewave® 402 microwave apparatus.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/ph14111086/s1, Supporting information.
Author Contributions
Organic synthesis under microwave, K.B., S.G. and E.L.; supervision for organic synthesis under microwave, L.P. and M.R.; tumoral cell lines assays, R.L.G.; supervision for tumoral cell lines assays, T.C.; protein kinase screening, E.D. and O.L.; supervision for protein kinase screening, L.M.; supervision for organic synthesis, F.C.; supervision of project, writing-review, editing and funding acquisition of NCI contracts, J.-P.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research work was funded by the “Ministère de l’Enseignement Supérieur et de la Recherche de la République Algérienne Démocratique et Populaire” (PhD fellowship for K.B.) and by the “Ministère de l’Enseignement Supérieur et de la Recherche de la République Française” (PhD fellowship for S.G.). This research was supported by the “Cancéropôle Grand Ouest” of French National Cancer Institute (contracts PRIR 04-8390 and ACI 04-2254). This research was supported by grants from the “Fondation Jérôme Lejeune” (L.M.), the “Agence Nationale pour la Recherche (ANR)” (DYRK-DOWN) (L.M.), the “Fonds Unique Interministériel” (FUI) PHARMASEA and TRIAD (L.M., J.-P.B., F.C.) projects, an FP7-KBBE-2012 grant (BlueGenics) (L.M.). This project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement No 848077. This reflects only the authors’ view and the European Commission is not responsible for any use that may be made of the information it contains.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest regarding the publication of this paper.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fedorov O., Müller S., Knapp S. The (un)targeted cancer kinome. Nat. Chem. Biol. 2010;6:166–169. doi: 10.1038/nchembio.297. [DOI] [PubMed] [Google Scholar]
- 2.Dar A.C., Shokat K.M. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Ann. Rev. Biochem. 2011;80:769–795. doi: 10.1146/annurev-biochem-090308-173656. [DOI] [PubMed] [Google Scholar]
- 3.Kimura R., Kamino K., Yamamoto M., Nuripa A., Kida T., Kazui H., Hashimoto R., Tanaka T., Kudo T., Yamagata H., et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between beta-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 2007;16:15–23. doi: 10.1093/hmg/ddl437. [DOI] [PubMed] [Google Scholar]
- 4.Smith B., Medda F., Gokhale V., Dunckley T., Hulme C. Recent Advances in the Design, Synthesis, and Biological Evaluation of Selective DYRK1A Inhibitors: A New Avenue for a Disease Modifying Treatment of Alzheimer’s? ACS Chem. Neurosci. 2012;3:857–872. doi: 10.1021/cn300094k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ling Y., Wang Z.-Q., Xiao Y.-A., Zhu C., Shen L., Wang X.-M., Hui Y., Wang X.-Y. Benzylidene 2-aminoimidazolones derivatives: Synthesis and in vitro evaluation of anti-tumor carcinoma activity. Chem. Pharm. Bull. 2013;61:1081–1084. doi: 10.1248/cpb.c13-00340. [DOI] [PubMed] [Google Scholar]
- 6.Ionescu A., Dufrasne F., Gelbcke M., Jabin I., Kiss R., Lamoral-Theys D. DYRK1A Kinase inhibitors with emphasis on cancer. Mini Rev. Med. Chem. 2012;12:1315–1329. doi: 10.2174/13895575112091315. [DOI] [PubMed] [Google Scholar]
- 7.Xiao Y.-A., Wang Z.-Q., Wang X.-M., Hui Y., Ling Y., Wang X.-Y., He L.-Q. Synthesis and in-vitro biological evaluation of novel 2-aminoimidazolinone derivatives as anti-tumor agents. Chin. Chem. Lett. 2013;24:727–730. doi: 10.1016/j.cclet.2013.05.009. [DOI] [Google Scholar]
- 8.Lindberg M., Meijer L. Dual-specificity, tyrosine phosphorylation-regulated kinases (DYRKs) and cdc2-like kinases (CLKs) in human disease, an overview. Int. J. Mol. Sci. 2021;22:6047. doi: 10.3390/ijms22116047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen T.L., Fruit C., Hérault Y., Meijer L., Besson T. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) inhibitors: A survey of recent patent literature. Expert Opin. Ther. Pat. 2017;27:1183–1199. doi: 10.1080/13543776.2017.1360285. [DOI] [PubMed] [Google Scholar]
- 10.Hagiwara M., Shibuya H., Onishi E., Ogawa Y., Hosoya T., Hiramatsu T., Yoshida M. Pharmaceutical Composition Comprising DYRK-Inhibiting Compound. WO 2010/010797 A1. Patent. 2010 January 28;
- 11.Fedorov O., Huber K., Eisenreich A., Filippakopoulos P., King O., Bullock A.-N., Szklarczyk D., Jensen L.-J., Fabbro D., Trappe J., et al. Specific CLK Inhibitors from a Novel Chemotype for Regulation of Alternative Splicing. Chem. Biol. 2011;18:67–76. doi: 10.1016/j.chembiol.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Göckler N., Jofre G., Papadopoulous C., Soppa U., Tejedor F.-J., Becker W. Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. FEBS J. 2009;276:6324–6337. doi: 10.1111/j.1742-4658.2009.07346.x. [DOI] [PubMed] [Google Scholar]
- 13.Wang S., Wood G., Duncan K.W., Meades C., Gibson D., McLachlan J.C., Perry A., Blake D., Zheleva D.L., Fischer P.M. Pyrimidin-4-yl-3,4-thione Compounds and their Use in Therapy. WO 2005/042525 A1. Patent. 2005 May 12;
- 14.Mott B.T., Tanega C., Shen M., Maloney D.J., Shinn P., Leister W., Marugan J.J., Inglese J., Austin C.P., Misteli T., et al. Evaluation of substituted 6-arylquinazolin-4-amines as potent and selective inhibitors of cdc2-like kinases (Clk) Bioorg. Med. Chem. Lett. 2009;19:6700–6705. doi: 10.1016/j.bmcl.2009.09.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosenthal A.S., Tanega C., Shen M., Mott B.T., Bougie J.M., Nguyen D.-T., Misteli T., Auld D.S., Maloney D.J., Thomas C.J. Potent and selective small molecule inhibitors of specific isoforms of Cdc2-like kinases (Clk) and dual specificity tyrosine-phosphorylation-regulated kinases (Dyrk) Bioorg. Med. Chem. Lett. 2011;21:3152–3158. doi: 10.1016/j.bmcl.2011.02.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leblond B., Casagrande A.-S., Désiré L., Foucourt A., Besson T. Tricyclic Pyrimidines as Inhibitors of DYRK1A/DYRK1B as Potential Treatment for Down’s Syndrome or Alzheimer’s Disease. WO/026806 A1. Patent. 2013 February 28;
- 17.Giraud F., Alves G., Debiton E., Nauton L., Thiéry V., Durieu E., Ferandin Y., Lozach O., Meijer L., Anizon F., et al. Synthesis, protein kinase inhibitory potencies, and in vitro antiproliferative activities of Meridianin derivatives. ACS J. Med. Chem. 2011;54:4474–4489. doi: 10.1021/jm200464w. [DOI] [PubMed] [Google Scholar]
- 18.Echalier A., Bettayeb K., Ferandin Y., Lozach O., Clement M., Valette A., Liger F., Marquet B., Morris J.C., Endicott J.A., et al. Meriolins (3-(pyrimidin-4-yl)-7-azaindoles): Synthesis, Kinase Inhibitory Activity, Cellular Effects, and Structure of a CDK2/CyclinA/Meriolin Complex. ACS J. Med. Chem. 2008;51:737–751. doi: 10.1021/jm700940h. [DOI] [PubMed] [Google Scholar]
- 19.Kassis P., Brzeszcz J., Bénéteau V., Lozach O., Meijer L., Le Guével R., Guillouzo C., Lewinski K., Bourg S., Colliandre L., et al. Synthesis and biological evaluation of new 3-(6-hydroxyindol-2-yl)-5-(Phenyl) pyridine or pyrazine V-Shaped molecules as kinase inhibitors and cytotoxic agents. Eur. J. Med. Chem. 2011;46:5416–5434. doi: 10.1016/j.ejmech.2011.08.048. [DOI] [PubMed] [Google Scholar]
- 20.Neagoie C., Vedrenne E., Buron F., Mérour J.-Y., Rosca S., Bourg S., Lozach O., Meijer L., Baldeyrou B., Lansiaux A., et al. Synthesis of chromeno[3,4-b]indoles as Lamellarin D analogues: A novel DYRK1A inhibitor class. Eur. J. Med. Chem. 2012;49:379–396. doi: 10.1016/j.ejmech.2012.01.040. [DOI] [PubMed] [Google Scholar]
- 21.Yoshida K., Itoyama R., Yamahira M., Tanaka J., Loaëc N., Lozach O., Durieu E., Fukuda T., Ishibashi F., Meijer L., et al. Synthesis, resolution, and biological evaluation of atropisomeric (aR)- and (aS)-16-methyllamellarins N: Unique effects of the axial chirality on the selectivity of protein kinases inhibition. ACS J. Med. Chem. 2013;56:7289–7301. doi: 10.1021/jm400719y. [DOI] [PubMed] [Google Scholar]
- 22.Davis R.A., Aalbersberg W., Meo S., da Rocha R.M., Ireland C.M. The isolation and synthesis of polyandrocarpamines A and B. Two new 2-aminoimidazolone compounds from the Fijian ascidian, Polyandrocarpa sp. Tetrahedron Lett. 2002;58:3263–3269. doi: 10.1016/S0040-4020(02)00228-4. [DOI] [Google Scholar]
- 23.Loaëc N., Attanasio E., Villiers B., Durieu E., Tahtouh T., Cam M., Davis R.A., Alencar A., Roué M., Bourguet-Kondracki M.L., et al. Marine derived 2-aminoimidazolone alkaloids. Leucettamine B -related polyandrocarpamines inhibit mammalian and protozoan DYRK & CLK kinases. Mar. Drugs. 2017;15:316. doi: 10.3390/md15100316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis R.A., Baron P.S., Neve J.E., Cullinane C. A microwave-assisted stereoselective synthesis of polyandrocarpamines A and B. Tetrahedron Lett. 2009;50:880–882. doi: 10.1016/j.tetlet.2008.12.010. [DOI] [Google Scholar]
- 25.Lindel T., Hoffmann H. Synthesis of dispacamide from the marine sponge Agelas dispar. Tetrahedron Lett. 1997;38:8935–8938. doi: 10.1016/S0040-4039(97)10387-2. [DOI] [Google Scholar]
- 26.Fresneda P.M., Molina P., Sanz M.A. A convergent approach to midpacamide and dispacamide pyrrole-imidazole marine alkaloids. Tetrahedron Lett. 2001;42:851–854. doi: 10.1016/S0040-4039(00)02120-1. [DOI] [Google Scholar]
- 27.Roué M., Domart-Coulon I., Ereskovsky A., Djediat C., Perez T., Bourguet-Kondracki M.-L. Cellular localization of clathridimine, an antimicrobial 2-aminoimidazole alkaloid produced by the Mediterranean calcareous sponge Clathrina clathrus. J. Nat. Prod. 2010;73:1277–1282. doi: 10.1021/np100175x. [DOI] [PubMed] [Google Scholar]
- 28.Chan G.W., Mong S., Hemling M.E., Freyer A.J., Offen P.H., De Brosse C.W., Sarau H.M., Westley J.W. New leukotriene B4 receptor antagonist: Leucettamine A and related imidazole alkaloids from the marine sponge Leucetta microraphis. J. Nat. Prod. 1993;56:116–121. doi: 10.1021/np50091a016. [DOI] [PubMed] [Google Scholar]
- 29.Cimino G., de Rosa S., de Stefano S., Mazzarella L., Puliti R., Sodano G. Isolation and X-ray crystal structure of a novel bromo-compound from two marine sponges. Tetrahedron Lett. 1982;23:767–768. doi: 10.1016/S0040-4039(00)86943-9. [DOI] [Google Scholar]
- 30.Sharma G.M., Buyer J.S., Pomerantz M.W. Characterization of a yellow compound isolated from the marine sponge Phakellia flabellata. J. Chem. Soc. Chem. Commun. 1980;10:435–436. doi: 10.1039/c39800000435. [DOI] [Google Scholar]
- 31.Williams D.H., Faulkner J. Isomers and tautomers of hymenialdisine and debromohymenialdisine. Nat. Prod. Lett. 1996;9:57–64. doi: 10.1080/10575639608043579. [DOI] [Google Scholar]
- 32.Xu Y.Y., Yakushijin K., Horne D.A. Synthesis of C(11)N(5) Marine sponge alkaloids: (±)-hymenin, stevensine, hymenialdisine, and debromohymenialdisine. J. Org. Chem. 1997;62:456–464. doi: 10.1021/jo9619746. [DOI] [PubMed] [Google Scholar]
- 33.Papeo G., Posteri H., Borghi D., Varasi M. A new glycociamidine ring precursor: Syntheses of (Z)-hymenialdisine, (Z)-2-debromohymenialdisine, and (±)-endo-2-debromohymenialdisine. Org. Lett. 2005;7:5641–5644. doi: 10.1021/ol052266m. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen T.N., Tepe J.J. Preparation of hymenialdisine, analogues and their evaluation as kinase inhibitors. Curr. Med. Chem. 2009;16:3122–3143. doi: 10.2174/092986709788803015. [DOI] [PubMed] [Google Scholar]
- 35.Meijer L., Thunnissen A.M.W.H., White A., Garnier M., Nikolic M., Tsai L.H., Walter J., Cleverley K.E., Salinas P.C., Wu Y.Z., et al. Inhibition of cyclin-dependent kinases, GSK-3 and casein kinase 1 by hymenialdisine, a marine sponge constituent. Chem. Biol. 2000;7:51–63. doi: 10.1016/S1074-5521(00)00063-6. [DOI] [PubMed] [Google Scholar]
- 36.Wan Y., Hur W., Cho C.Y., Liu Y., Adrian F.J., Lozach O., Bach S., Mayer T., Fabbro D., Meijer L., et al. Synthesis and target identification of hymenialdisine analogs. Chem. Biol. 2004;11:247–259. doi: 10.1016/j.chembiol.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 37.Roué N., Bergman J. Synthesis of the marine alkaloid leucettamine B. Tetrahedron. 1999;55:14729–14738. doi: 10.1016/S0040-4020(99)00918-7. [DOI] [Google Scholar]
- 38.Bazureau J.-P., Carreaux F., Renault S., Meijer L., Lozach O. Imidazolone Derivatives, Preparation Method Thereof and Biological Use of Same. WO 2009/05032 A2. Patent. 2009 April 23; Demande PCT/FR 2008/001152, 1 October 2008.
- 39.Debdab M., Carreaux F., Renault S., Soundararajan M., Fedorov O., Filippakopoulos P., Lozach O., Babault L., Tahtouh T., Baratte B., et al. Design, synthesis and biological evaluation of leucettines, a class of potent CLK and DYRK kinases inhibitors derived from the marine sponge leucettamine B. Modulation of alternative RNA splicing. ACS J. Med. Chem. 2011;54:4172–4186. doi: 10.1021/jm200274d. [DOI] [PubMed] [Google Scholar]
- 40.Tahtouh T., Elkins J.M., Filippakopoulos P., Soundararajan M., Burgy G., Durieu E., Cochet C., Schmid R.S., Lo D.C., Delhommel F., et al. Selectivity, cocrystal structures, and neuroprotective properties of Leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid Leucettamine B. ACS J. Med. Chem. 2012;55:9312–9330. doi: 10.1021/jm301034u. [DOI] [PubMed] [Google Scholar]
- 41.Mendgen T., Steuer C., Klein C.D. Privileged Scaffolds or Promiscuous Binders: A comparative Study on Rhodanines and Related Heterocycles in Medicinal Chemistry. ACS J. Med. Chem. 2012;55:743–753. doi: 10.1021/jm201243p. [DOI] [PubMed] [Google Scholar]
- 42.Nasr M.N.A., Said S.-A. Novel 3, 3a, 4, 5, 6, 7-Hexahydroindazole and arylthiazolylpyrazoline derivatives as anti-inflammatory agents. Arch. Pharm. 2003;336:551–559. doi: 10.1002/ardp.200300796. [DOI] [PubMed] [Google Scholar]
- 43.Martin L., Rabasseda X., Castaner J. Darbufelone Mesilate. Drugs Futur. 1999;24:853–857. doi: 10.1358/dof.1999.024.08.534526. [DOI] [Google Scholar]
- 44.Cutshall N.-S., O’Day C., Prezhdo M. Rhodanine derivatives as inhibitors of JSP-1. Bioorg. Med. Chem. Lett. 2005;15:3374–3382. doi: 10.1016/j.bmcl.2005.05.034. [DOI] [PubMed] [Google Scholar]
- 45.Radi M., Botta M., Falchi F., Maga G., Baldanti F., Paolucci S. Compounds with ddx3 Inhibitory Activity and Uses Thereof. WO 2011/039735 A2. Patent. 2011 April 7; Demande PCT/IT 2010/054475, 4 October 2010.
- 46.Powers J.-P., Piper D.-E., Li Y., Mayorga V., Anzola J., Chen J.-M., Jaen J.-C., Lee G., Liu J., Peterson M.G., et al. SAR and Mode of Action of Novel Non-Nucleoside Inhibitors of Hepatitis C NS5b RNA Polymerase. ACS J. Med. Chem. 2006;49:1034–1046. doi: 10.1021/jm050859x. [DOI] [PubMed] [Google Scholar]
- 47.Guihéneuf S., Paquin L., Carreaux F., Durieu E., Roisnel T., Meijer L., Bazureau J.-P. New 5-ylidene rhodanine derivatives based on the Dispacamide A model. Mol. Divers. 2014;18:375–388. doi: 10.1007/s11030-014-9509-7. [DOI] [PubMed] [Google Scholar]
- 48.Song Y., Connor D.T., Doubleday R., Sorenson R.J., Sercel A., Unangst P.C., Roth B.D., Gilbertsen R.B., Chan K., Schrier D.J., et al. Synthesis, structure-activity relationships, and in vivo evaluations of substituted di-tert-butylphenols as a novel class of potent, selective, and orally active cyclooxygenase-2 inhibitors. 1. Thiazolone and oxazolone series. ACS J. Med. Chem. 1999;42:1151–1160. doi: 10.1021/jm9805081. [DOI] [PubMed] [Google Scholar]
- 49.Lohray B.B., Bhushan V., Rao P.B., Madhavan G.R., Murali N., Rao K.N., Reddy K.A., Rajesh B.M., Reddy P.G., Chakrabarti R., et al. Novel indole containing thiazolidinedione derivatives as potent euglycemic and hypolipidaemic agents. Bioorg. Med. Chem. Lett. 1997;7:785–788. doi: 10.1016/S0960-894X(97)00118-2. [DOI] [Google Scholar]
- 50.Zhou J.F., Zhu F.X., Song Y.Z., Zhu Y.L. Synthesis of 5-arylalkylidenerhodanines catalyzed by tetrabutylammonium bromine in water under microwave irradiation. Arkivoc. 2006;14:175–180. doi: 10.3998/ark.5550190.0007.e18. [DOI] [Google Scholar]
- 51.Safonov I.G., Heerding D.A., Keenan R.M., Price A.T., Erickson-Miller C.L., Hopson C.B., Levin J.L., Lord K.A., Tapley P.M. New benzimidazoles as thrombopoietin receptor agonists. Bioorg. Med. Chem. Lett. 2006;16:1212–1216. doi: 10.1016/j.bmcl.2005.11.096. [DOI] [PubMed] [Google Scholar]
- 52.Bourahla K., Derdour A., Rahmouni M., Carreaux F., Bazureau J.-P. A practical access to novel 2-amino-5-arylidene-thiazol-4-ones via sulfur/nitrogen displacement under solvent-free microwave irradiations. Tetrahedron Lett. 2007;48:5785–5789. doi: 10.1016/j.tetlet.2007.06.078. [DOI] [Google Scholar]
- 53.Xia Z., Knaak C., Ma J., Beharry Z.M., McInnes C., Wang W., Kraft A.S., Smith C.D. Synthesis and evaluation of novel inhibitors of Pim-1 and Pim-2 protein kinases. ACS J. Med. Chem. 2009;52:74–86. doi: 10.1021/jm800937p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Unangst P.C., Connor D.T., Cetenko W.A., Sorenson R.J., Kostlan C.R., Sircar J.C., Wright C.D., Schrier D.J., Dyer R.D. Synthesis and biological evaluation of 5-[[3,5-bis(1,1-dimethylethyl)-4-hydroxyphenyl]methylene]oxazoles, -thiazoles, and -imidazoles: Novel dual 5-lipoxygenase and cyclooxygenase inhibitors with antiinflammatory activity. ACS J. Med. Chem. 1994;37:322–328. doi: 10.1021/jm00028a017. [DOI] [PubMed] [Google Scholar]
- 55.Guihéneuf S., Paquin L., Carreaux F., Durieu E., Bénédetti H., Le Guével R., Corlu A., Meijer L., Bazureau J.-P. Microwave Assisted Organic Synthesis (MAOS) of new Dispacamide A derivatives bearing a thiazolinone platform, biological assays on inhibition of protein kinases and cell effects. Curr. Microw. Chem. 2014;1:33–40. doi: 10.2174/22133356114019990002. [DOI] [Google Scholar]
- 56.Pulici M., Quartieri F. Traceless solid-phase synthesis of 2-amino-5-alkylidene-thiazol-4-ones. Tetrahedron Lett. 2005;46:2387–2391. doi: 10.1016/j.tetlet.2005.02.059. [DOI] [Google Scholar]
- 57.Leclerc S., Garnier M., Hoessel R., Marko D., Bibb J.-A., Snyder G.L., Greengard P., Biernat J., Mandelkow E.M., Eisenbrand G., et al. Indirubins inhibit glycogen synthase kinase-3 beta and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors? J. Biol. Chem. 2001;276:251–260. doi: 10.1074/jbc.M002466200. [DOI] [PubMed] [Google Scholar]
- 58.Reinhardt J., Ferandin Y., Meijer L. Purification of CK1 by affinity chromatography on immobilised axin. Protein Expr. Purif. 2007;54:101–109. doi: 10.1016/j.pep.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 59.Primot A., Baratte B., Gompel M., Borgne A., Liabeuf S., Romette J.L., Costantini F., Meijer L. Purification of GSK-3 by affinity chromatography on immobilised axin. Protein Expr. Purif. 2000;20:394–404. doi: 10.1006/prep.2000.1321. [DOI] [PubMed] [Google Scholar]
- 60.Nakabayashi H., Taketa K., Miyano K., Yamane T., Sato J. Growth of human hepatoma cell lines with differentiated functions in chemically defined medium. Cancer Res. 1982;42:3858–3863. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in this study are available on request from the corresponding author.