

Abstract
The aim of this study was to identify RS1 pathogenic variants in Czech patients with X-linked retinoschisis (XLRS) and to describe the associated phenotypes, including natural history, in some cases. Twenty-one affected males from 17 families were included. The coding region of RS1 was directly sequenced and segregation of the identified mutations was performed in available family members. In total, 12 disease-causing variants within RS1 were identified; of these c.20del, c.275G>A, c.[375_379del; 386A>T], c.539C>A and c.575_576insT were novel, all predicted to be null alleles. The c.539C>A mutation occurred de novo. Three patients (aged 8, 11 and 19 years) were misdiagnosed as having intermediate uveitis and treated with systemic steroids. Repeat spectral domain optical coherence tomography examinations in four eyes documented the transition from cystoid macular lesions to macular atrophy in the fourth decade of life. Four individuals were treated with topical dorzolamide and in two of them, complete resolution of the cystic macular lesions bilaterally was achieved, while one patient was noncompliant. Rebound phenomenon after discontinuation of dorzolamide for 7 days was documented in one case. Misdiagnosis of XLRS for uveitis is not uncommon; therefore, identification of disease-causing variants is of considerable benefit to the affected individuals.
Keywords: RS1, X-linked retinoschisis, novel variant, uveitis, steroid treatment
1. Introduction
X-linked retinoschisis (XLRS; OMIM number 312700) is a rare vitreoretinal dystrophy with an estimated prevalence ranging from 1 in 5000 to 20,000 affected inhabitants [1].
Clinically, the disease is characterized in males by early-onset visual loss and bilateral foveal schisis due to splitting of the inner retinal layers; 33–60% of patients with XLRS also have peripheral retinoschisis [2,3,4]. Complications, such as retinal detachment, occur in 5–22% cases, and intraretinal haemorrhage within a schisis cavity or vitreous haemorrhage is present in approximately one third of patients [5,6,7]. Less commonly observed clinical findings include diffuse white retinal flecks [8], Coats disease–like exudative retinopathy, perivascular sheathing, peripheral dendriform lesions and vitreous veils [7,9].
Unless secondary complications occur, XLRS typically shows slow bilateral progression resulting in macular atrophy by the fifth or sixth decade of life [10]. Marked differences, even in members of individual families, can be found [11]. Heterozygous female carriers are typically unaffected [2]. Patients with XLRS may show a benefit from systemic or topical treatment with carbonic anhydrase inhibitors (CAIs) for cystic macular lesions and mid-peripheral retinoschisis [12,13,14].
XLRS is caused by mutations in RS1 encoding retinoschisin [15,16], which plays an important role in maintaining the retinal structure as a cell adhesion protein between photoreceptors and bipolar cells [17,18]. To date, more than 250 pathogenic variants in RS1 have been reported [19,20].
In this study, we have identified disease-causing variants in 17 Czech families with XLRS. We also performed deep phenotyping in our cohort, including natural history, response to topical dorzolamide treatment and optical coherence tomography angiography (OCTA). The study documents the spectrum of genetic and phenotypic variability in XLRS patients resulting in misdiagnosis in three cases for a previously unreported population.
2. Materials and Methods
Affected and unaffected members from 17 Czech families of European descent with XLRS participated in the study. The study was approved by the Ethics Committee of General University Hospital in Prague (reference no. 34/19) and adhered to the tenets set out in the Helsinki Declaration. All participants or their legal guardians signed informed consent prior to the inclusion into the study. Ophthalmic evaluation comprised measurements of best-corrected visual acuity (BCVA) and intraocular pressure, perimetry, fundus photography (Clarus 700 and FF 450 plus IR, Carl Zeiss Meditec AG, Jena, Germany), spectral domain optical coherence tomography (SD-OCT; Spectralis, Heidelberg Engineering GmbH, Heidelberg, Germany and Spectral OCT/SLO, OTI Ophthalmic Technologies Inc., Toronto, Canada), OCTA (Spectralis OCT2, Heidelberg Engineering GmbH), and full-field electroretinography (ERG; RetiPort + mf ERG system, Roland Consult, Brandenburg, Germany) using DTL-electrodes, according to the standards of International Society for Clinical Electrophysiology of Vision [21]. All amplitudes and implicit times were compared with the mean values ± SD of normal age-matched controls.
Genomic DNA was isolated from peripheral blood samples using standard methods. All six exons of RS1 and intron–exon boundaries were bidirectionally Sanger sequenced. Primer sequences used were as previously reported [22] using NM_000330.3 as the reference sequence. The presence of the identified variants in first-degree relatives was tested also by conventional sequencing. The Genome Aggregation Database v2.1.1 (gnomAD; http://gnomad.broadinstitute.org/, accessed on 17 October 2021) was searched to determine variant frequency [23]. Because of the rarity of XLRS, only variants with minor allele frequency ≤0.005 were further evaluated for potential pathogenicity and cross-referenced with published literature, public archive of interpretations of clinically relevant variants ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, accessed on 9 November 2021) and the RS1 LOVD database (https://databases.lovd.nl/shared/genes/RS1, accessed on 17 October 2021). The pathogenicity of the detected variants was evaluated according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP) recommendations [24].
3. Results
Twenty-one Czech males from 17 not knowingly related families (Supplementary Figure S1) clinically diagnosed with XLRS were included in the study.
Molecular diagnosis was achieved in all families; 12 different RS1 pathogenic variants were identified, of which 5 were novel (Table 1). All novel mutations were predicted to be null alleles, either generating a stop codon or a frameshift with subsequent introduction of a premature or delayed stop codon (Table 1, Supplementary Figure S1). Novel RS1 variants were unique to the families, except for c.20del, which was observed recurrently in families F1 and F2, who interestingly originated in nearby geographical regions (less than 30 km from each other) suggesting a common founder.
Table 1.
RS1 disease-causing mutations identified in Czech families with X-linked retinoschisis.
| Family | DNA Level | Protein Level | ClinVar Interpretation/VCV Accession | References |
|---|---|---|---|---|
| F1, F2 | c.20del | p.(Gly7Alafs*119) | Not present | Novel |
| F3, F4, F15 | c.33_36del | p.(Leu11Phefs*114) | Pathogenic/VCV000098944.7 | [25] |
| F5 | c.187T>C | p.(Cys63Arg) | Not present | [26] |
| F6 | c.275G>A | p.(Trp92*) | Not present | Novel |
| F7, F8 | c.305G>A | p.(Arg102Gln) | Pathogenic/VCV000009896.11 | [11,25,27,28,29] |
| F9 | c.375_379del | p.(Asp126Glufs*16) | Not present | Novel |
| F10 | c.539C>A | p.(Ser180*) | Not present | Novel |
| F16 | c.421C>T | p.(Arg141Cys) | Pathogenic/VCV000098959.9 | [11,25,30] |
| F11, F17 | c.544C>T | p.(Arg182Cys) | Pathogenic/VCV000098986.3 | [25,29,31,32] |
| F12 | c.574C>T | p.(Pro192Ser) | Pathogenic/VCV000098990.4 | [15,25,32] |
| F13 | c.575_576insT | p.(Ile194Hisfs*70) | Not present | Novel # |
| F14 | c.637C>T | p.(Arg213Trp) | Pathogenic/VCV000099009.5 | [25,29,33,34] |
NM_000330.3 was used as the reference sequence; # Novel at DNA level; at protein level p.(Ile194Hisfs*70) has been reported.
Seven mutations, 6 missense and 1 frameshifting, have been previously reported associated with XLRS in multiple families (Table 1). Only one variant c.544C>T had an entry in the gnomAD v2.1.1 population dataset (1 allele out of 182,642—observed in a heterozygous female).
Segregation analysis within the families showed that c.539C>A; p.(Ser180*) occurred de novo in the proband from family F10 (Supplementary Figure S1). All variants were classified as pathogenic according to the ACMG-AMP guidelines as they have been either previously reported to be associated with XLRS and/or lead to a loss of function, and were absent in gnomAD v2.1.1 male samples.
Clinical ophthalmic data of the affected males are summarized in Supplementary Table S1. Sixteen subjects were evaluated at repeated intervals with a mean follow-up of 9.8 years (range 0.6–34 years).
At the most recent examinations, typical macular retinoschisis was detected in 15 individuals (29 eyes), atrophic changes in the macula in 6 individuals (11 eyes), and peripheral retinoschisis in 7 (13 eyes). Additional rarer phenotypes included vitreous veils in 3 (4 eyes), sheathing of retinal vessels, retinal holes in 2 patients (3 eyes) and vitreous haemorrhage observed in 1 patient (2 eyes) (Figure 1). Progression of macular retinoschisis to macular atrophy was documented using SD-OCT in 4 eyes of 4 individuals; none had been treated with topical dorzolamide (Figure 2).
Figure 1.
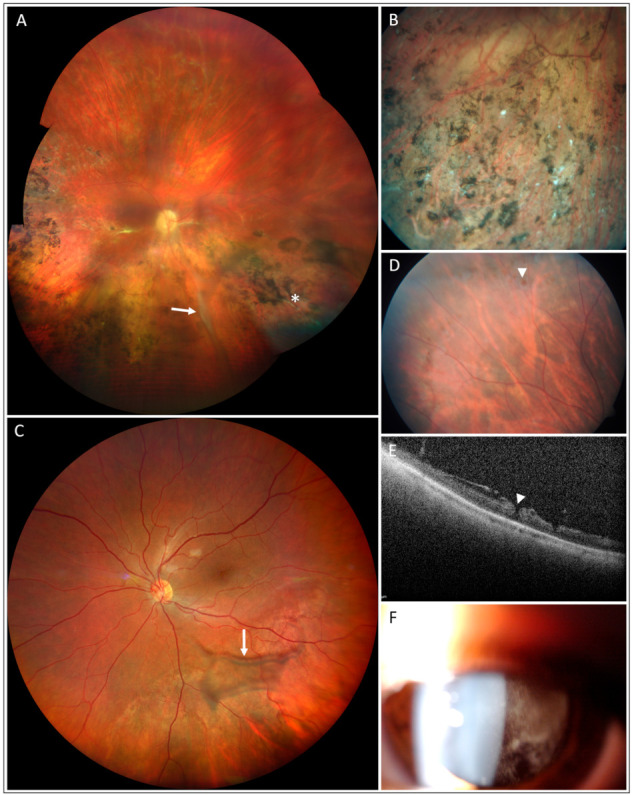
Less frequent clinical findings in patients with XLRS. Vitreous veil (arrow), vascular sheathing (asterisk) and pigmentary changes in the right eye of individual F1-III:1 aged 39 years (A,B). Vitreous veil of the left eye (arrow) in individual F17-II:1 aged 35 years (C). Retinal hole in the right eye (arrowhead) of the individual F5-II:1 aged 11 years (D,E). Vitreous haemorrhage in the left eye of the individual F13-II:1 aged 9 years (F).
Figure 2.

Repeated imaging with optical coherence tomography in individuals with XLRS documenting natural history disease course from cystic changes to macular atrophy. y = years, LE = left eye, RE = right eye.
Notably, findings in four patients revealed atypical clinical phenotypes, which led to diagnostic difficulties. Individual III:1 from family F1 presented at the age of 30 years with subcapsular cataract, dense vitreous opacities, migration and clumping of retinal pigment epithelium (RPE) in the macula and the periphery, perivascular sheathing and white spicules in the right eye (Figure 1A,B), while in the left eye only macular retinoschisis was documented. Fluorescein angiography however excluded vasculitis. Complete ophthalmic imaging of the proband is shown in Supplementary Figure S2. A diagnosis of XLRS was subsequently made using ERG, which demonstrated a typical selective reduction of the b-wave of the standard scotopic response, with preservation of the a-wave resulting in a reduction of the b/a quotient, and subsequently confirmed by genetic testing. Both the proband, and his male cousin with a typical XLRS phenotype (Figure 2, Supplementary Table S1), were found to possess the novel variant c.20del in RS1.
Investigation of Individual II:1 from family F5 was undertaken after a bilateral BCVA decrease was noted at a routine paediatric check-up at the age of 10 years. Due to the presence of cystoid macular edema with vascular sheathing, a diagnosis of intermediate uveitis was made, and systemic steroids were introduced when he was aged 11 years. Full thickness retinal holes and retinoschisis in the fundus periphery were also documented in both eyes (Figure 1D,E, Supplementary Figure S3). A range of tests were performed to identify the underlying cause of the observed ocular pathology, including MRI and laboratory examination of cerebrospinal fluid. The patient was treated with systemic methylprednisolone for 3.5 years until the patient was 14 years old, at which point ERG was performed, demonstrating a negative b-wave suggesting a diagnosis of XLRS. Subsequent molecular genetic testing identified hemizygosity for a known c.187T>C mutation in RS1. After obtaining a molecular diagnosis, steroid treatment was gradually tapered down and discontinued.
Individual II:1 from family F10, aged 19 years, was treated elsewhere with systemic methylprednisolone for 4 months because of bilateral cystoid macular edema, until referred to our tertiary care setting where an XLRS diagnosis was made, subsequently confirmed by the identification of a novel pathogenic variant c.539C>A in RS1. Based on the positive genetic test, steroid administration was gradually decreased and stopped.
Individual II:1 from family F13 was treated elsewhere for cystoid macular oedema and intermediate uveitis with systemic steroids from 9 years of age. He also received a dexamethasone intravitreal implant bilaterally. When examined by the authors at the age of 10 years, a typical star-shaped macular lesion was found in the right eye and resolving vitreous haemorrhage in the left eye (Figure 1F). No signs of inflammation were present. SD-OCT showed bilateral typical schitic cavities (Supplementary Figure S4). Molecular genetic analysis identified the novel RS1 variant c.575_576insT predicted to be pathogenic.
Available clinical documentation in individuals F4-II:1 and F14-III:3 permitted comparison of BCVA over more than three decades. No significant decrease was noted.
Therapeutic intervention for the schitic macular lesions with CAIs (dorzolamide, 2% drops three times a day) was started in four individuals (F8-II:1, F12-II:1, F14-IV:1, F15-II:1). Decrease of retinal thickness was observed on SD-OCT in all eyes. Patient F14-IV:1, aged 7 years at the start of the treatment, had the longest follow-up (37 months) during which we were able to document complete resolution of the schitic cavities; however, after changing the treatment regimen to twice a day, rebound occurred. After returning to dosing three times a day improvement was observed (Figure 3). Individuals F12-II:1 and F15-II:1 responded well, with none or very few cystic changes noted 3 months after the start of the treatment; however, discontinuation of the treatment for only 7 days led to a marked rebound phenomena (Figure 3). Two patients (F8-II:1, F14-IV:1) noticed subjective improvement of visual functions, which was also documented on BCVA measurements (Supplementary Table S1).
Figure 3.
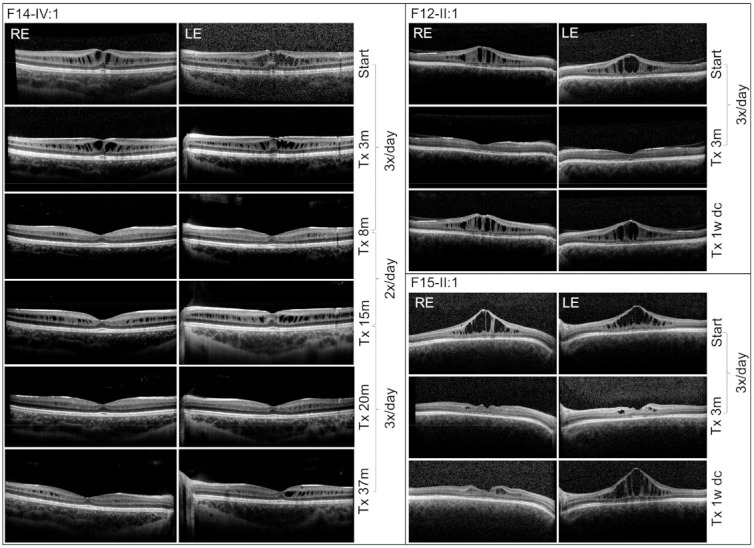
Effects of dorzolamide treatment in three XLRS patients as documented by SD-OCT. Results of individual treatment dosing as well as the length of the therapy are shown. Note rebound phenomena in patient F15-II:1 only after one week of discontinuation of drop administration. dc = discontinued, m = months, Tx = treatment, w = week.
Patient F16-II:1 underwent OCTA in addition to other examinations. An irregular foveal avascular zone and flow loss within the deep capillary plexus corresponding to the distribution of the schisis were noted (Supplementary Figure S5).
We also comprehensively examined one heterozygous carrier (F14-III:2). No fundus pathology was noted both on wide-field imaging nor SD-OCT.
4. Discussion
In this study, we report the clinical and molecular genetic findings in 17 families with XLRS ascertained in the Czech Republic. In addition to the identification of novel pathogenic variants, our study provides further data on XLRS responsivity to topical dorzolamide treatment and the natural history of the disease.
Five novel disease-causing mutations in RS1 were identified, all predicted to be loss of function alleles. Four pathogenic variants, c.20del, c.33_36del, c.305G>A and c.544C>T, were observed in more than one family, although haplotyping to either confirm or disprove a founder effect was not undertaken. Given the close geographical origin, we hypothesise that at least families F1 and F2 are likely to share a common ancestor.
In family F10, the c.539C>A variant in RS1 was present only in the proband and not in the genomic DNA of his mother, indicating a likely de novo origin, albeit mosaicism cannot be entirely excluded. Interestingly, the rate of de novo RS1 mutations is low, and to the best of our knowledge, previously only reported in seven cases with XLRS [16,25,29,30,35].
Three probands were misdiagnosed with uveitis and treated with steroids, administered systemically in two patients, and bilateral dexamethasone intravitreal implants were inserted in one individual.
The progressive nature of XLRS has been debated [10,36,37,38]. Available clinical documentation suggested that the disease is relatively stable with no major deterioration unless proliferative vitreoretinopathy develops.
In this study, we have also documented on SD-OCT, in five eyes of three individuals, the transition from cystic changes to macular atrophy, interestingly without any major effect on BCVA. OCTA imaging, which is a relatively new method, was similar to previously described cases and documented perifoveal microvascular changes. The usefulness of this method for monitoring disease progression or treatment response is yet to be established [39,40,41,42].
To conclude, the diagnosis of XLRS can be challenging due to phenotypic variability. XLRS may be misdiagnosed as uveitis, and in these cases in particular, the combined modalities of electrophysiology and molecular genetic testing can be crucial in confirming a diagnosis. Longitudinal records of BCVA in XLRS seem to be stable, even in older individuals who are documented to develop atrophic changes in the macula.
Acknowledgments
We thank Martin Meliska for SD-OCT and OCTA examination.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/genes12111816/s1, Figure S1: Pedigrees of 17 Czech families with X-linked retinoschisis and segregation of the identified RS1 mutations, Figure S2: Clinical findings in individual F1-III:1 aged 39 years, Figure S3: Clinical findings in individual F5-II:1 aged 15 years, Figure S4: Clinical findings in individual F13-II:1 aged 9 years, Figure S5: Clinical findings in individual F16-II:1 aged 40 years, Table S1: Summary of clinical data in 21 male individuals with X linked retinoschisis.
Author Contributions
Conceptualization, B.K., L.H. and P.L.; writing—original draft preparation, B.K., L.H. and P.L.; writing—review and editing, B.K., L.H., H.V., L.D. (Lenka Dvorakova), M.B., Z.D., H.L., A.L.V., L.D. (Lubica Dudakova) and P.L.; visualization, B.K., L.H., L.D. (Lubica Dudakova) and P.L.; supervision, P.L.; project administration, P.L.; funding acquisition, P.L. and B.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the AZV NU20-07-00182 research grant from the Ministry of Health of the Czech Republic.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Molday R.S., Kellner U., Weber B.H. X-linked juvenile retinoschisis: Clinical diagnosis, genetic analysis, and molecular mechanisms. Prog. Retin. Eye Res. 2012;31:195–212. doi: 10.1016/j.preteyeres.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.George N.D., Yates J.R., Moore A.T. X linked retinoschisis. Br. J. Ophthalmol. 1995;79:697–702. doi: 10.1136/bjo.79.7.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu Q.R., Huang L.Z., Chen X.L., Xia H.K., Li T.Q., Li X.X. Genetic analysis and clinical features of X-linked retinoschisis in Chinese patients. Sci. Rep. 2017;7:44060. doi: 10.1038/srep44060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hinds A.M., Fahim A., Moore A.T., Wong S.C., Michaelides M. Bullous X linked retinoschisis: Clinical features and prognosis. Br. J. Ophthalmol. 2018;102:622–624. doi: 10.1136/bjophthalmol-2017-310593. [DOI] [PubMed] [Google Scholar]
- 5.George N.D., Yates J.R., Moore A.T. Clinical features in affected males with X-linked retinoschisis. Arch. Ophthalmol. 1996;114:274–280. doi: 10.1001/archopht.1996.01100130270007. [DOI] [PubMed] [Google Scholar]
- 6.Kellner U., Brümmer S., Foerster M.H., Wessing A. X-linked congenital retinoschisis. Graefe’s Arch. Clin. Exp. Ophthalmol. 1990;228:432–437. doi: 10.1007/BF00927256. [DOI] [PubMed] [Google Scholar]
- 7.Wang N.K., Liu L., Chen H.M., Tsai S., Chang T.C., Tsai T.H., Yang C.M., Chao A.N., Chen K.J., Kao L.Y., et al. Clinical presentations of X-linked retinoschisis in Taiwanese patients confirmed with genetic sequencing. Mol. Vis. 2015;21:487–501. [PMC free article] [PubMed] [Google Scholar]
- 8.Hotta Y., Nakamura M., Okamoto Y., Nomura R., Terasaki H., Miyake Y. Different mutation of the XLRS1 gene causes juvenile retinoschisis with retinal white flecks. Br. J. Ophthalmol. 2001;85:238–239. doi: 10.1136/bjo.85.2.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fahim A.T., Ali N., Blachley T., Michaelides M. Peripheral fundus findings in X-linked retinoschisis. Br. J. Ophthalmol. 2017;101:1555–1559. doi: 10.1136/bjophthalmol-2016-310110. [DOI] [PubMed] [Google Scholar]
- 10.Apushkin M.A., Fishman G.A., Rajagopalan A.S. Fundus findings and longitudinal study of visual acuity loss in patients with X-linked retinoschisis. Retina. 2005;25:612–618. doi: 10.1097/00006982-200507000-00012. [DOI] [PubMed] [Google Scholar]
- 11.Pimenides D., George N.D., Yates J.R., Bradshaw K., Roberts S.A., Moore A.T., Trump D. X-linked retinoschisis: Clinical phenotype and RS1 genotype in 86 UK patients. J. Med. Genet. 2005;42:e35. doi: 10.1136/jmg.2004.029769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khandhadia S., Trump D., Menon G., Lotery A.J. X-linked retinoschisis maculopathy treated with topical dorzolamide, and relationship to genotype. Eye. 2011;25:922–928. doi: 10.1038/eye.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collison F.T., Genead M.A., Fishman G.A., Stone E.M. Resolution of mid-peripheral schisis in x-linked retinoschisis with the use of dorzolamide. Ophthalmic Genet. 2014;35:125–127. doi: 10.3109/13816810.2013.779383. [DOI] [PubMed] [Google Scholar]
- 14.Thobani A., Fishman G.A. The use of carbonic anhydrase inhibitors in the retreatment of cystic macular lesions in retinitis pigmentosa and X-linked retinoschisis. Retina. 2011;31:312–315. doi: 10.1097/IAE.0b013e3181e587f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sauer C.G., Gehrig A., Warneke-Wittstock R., Marquardt A., Ewing C.C., Gibson A., Lorenz B., Jurklies B., Weber B.H. Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nat. Genet. 1997;17:164–170. doi: 10.1038/ng1097-164. [DOI] [PubMed] [Google Scholar]
- 16.Gehrig A., Weber B.H., Lorenz B., Andrassi M. First molecular evidence for a de novo mutation in RS1 (XLRS1) associated with X linked juvenile retinoschisis. J. Med. Genet. 1999;36:932–934. [PMC free article] [PubMed] [Google Scholar]
- 17.Khan N.W., Jamison J.A., Kemp J.A., Sieving P.A. Analysis of photoreceptor function and inner retinal activity in juvenile X-linked retinoschisis. Vision Res. 2001;41:3931–3942. doi: 10.1016/S0042-6989(01)00188-2. [DOI] [PubMed] [Google Scholar]
- 18.Molday L.L., Hicks D., Sauer C.G., Weber B.H., Molday R.S. Expression of X-linked retinoschisis protein RS1 in photoreceptor and bipolar cells. Investig. Ophthalmol. Vis. Sci. 2001;42:816–825. [PubMed] [Google Scholar]
- 19.Xiao S., Sun W., Xiao X., Li S., Luo H., Jia X., Ouyang J., Li X., Wang Y., Jiang Y., et al. Clinical and genetic features of retinoschisis in 120 families with RS1 mutations. Br. J. Ophthalmol. 2021 doi: 10.1136/bjophthalmol-2021-319668. [DOI] [PubMed] [Google Scholar]
- 20.Kim D.Y., Mukai S. X-linked juvenile retinoschisis (XLRS): A review of genotype-phenotype relationships. Semin. Ophthalmol. 2013;28:392–396. doi: 10.3109/08820538.2013.825299. [DOI] [PubMed] [Google Scholar]
- 21.McCulloch D.L., Marmor M.F., Brigell M.G., Hamilton R., Holder G.E., Tzekov R., Bach M. ISCEV Standard for full-field clinical electroretinography (2015 update) Doc. Ophthalmol. 2015;130:1–12. doi: 10.1007/s10633-014-9473-7. [DOI] [PubMed] [Google Scholar]
- 22.Yi J., Li S., Jia X., Xiao X., Wang P., Guo X., Zhang Q. Novel RS1 mutations associated with X-linked juvenile retinoschisis. Int. J. Mol. Med. 2012;29:644–648. doi: 10.3892/ijmm.2012.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The Retinoschisis Consortium Functional implications of the spectrum of mutations found in 234 cases with X-linked juvenile retinoschisis. Hum. Mol. Genet. 1998;7:1185–1192. doi: 10.1093/hmg/7.7.1185. [DOI] [PubMed] [Google Scholar]
- 26.Huang L., Sun L., Wang Z., Chen C., Wang P., Sun W., Luo X., Ding X. Clinical manifestation and genetic analysis in Chinese early onset X-linked retinoschisis. Mol. Genet. Genomic Med. 2020;8:e1421. doi: 10.1002/mgg3.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hotta Y., Fujiki K., Hayakawa M., Ohta T., Fujimaki T., Tamaki K., Yokoyama T., Kanai A., Hirakata A., Hida T., et al. Japanese juvenile retinoschisis is caused by mutations of the XLRS1 gene. Hum. Genet. 1998;103:142–144. doi: 10.1007/PL00008705. [DOI] [PubMed] [Google Scholar]
- 28.Hiriyanna K.T., Bingham E.L., Yashar B.M., Ayyagari R., Fishman G., Small K.W., Weinberg D.V., Weleber R.G., Lewis R.A., Andreasson S., et al. Novel mutations in XLRS1 causing retinoschisis, including first evidence of putative leader sequence change. Hum. Mutat. 1999;14:423–427. doi: 10.1002/(SICI)1098-1004(199911)14:5<423::AID-HUMU8>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 29.Kim S.Y., Ko H.S., Yu Y.S., Hwang J.M., Lee J.J., Kim S.Y., Kim J.Y., Seong M.W., Park K.H., Park S.S. Molecular genetic characteristics of X-linked retinoschisis in Koreans. Mol. Vis. 2009;15:833–843. [PMC free article] [PubMed] [Google Scholar]
- 30.Riveiro-Alvarez R., Trujillo-Tiebas M.J., Gimenez-Pardo A., Garcia-Hoyos M., Lopez-Martinez M.A., Aguirre-Lamban J., Garcia-Sandoval B., Vazquez-Fernandez del Pozo S., Cantalapiedra D., Avila-Fernandez A., et al. Correlation of genetic and clinical findings in Spanish patients with X-linked juvenile retinoschisis. Investig. Ophthalmol. Vis. Sci. 2009;50:4342–4350. doi: 10.1167/iovs.09-3418. [DOI] [PubMed] [Google Scholar]
- 31.Mashima Y., Shinoda K., Ishida S., Ozawa Y., Kudoh J., Iwata T., Oguchi Y., Shimizu N. Identification of four novel mutations of the XLRS1 gene in Japanese patients with X-linked juvenile retinoschisis. Mutation in brief no. 234. Online. Hum. Mutat. 1999;13:338. doi: 10.1002/(SICI)1098-1004(1999)13:4<338::AID-HUMU16>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 32.Inoue Y., Yamamoto S., Okada M., Tsujikawa M., Inoue T., Okada A.A., Kusaka S., Saito Y., Wakabayashi K., Miyake Y., et al. X-linked retinoschisis with point mutations in the XLRS1 gene. Arch. Ophthalmol. 2000;118:93–96. doi: 10.1001/archopht.118.1.93. [DOI] [PubMed] [Google Scholar]
- 33.Curat C.A., Eck M., Dervillez X., Vogel W.F. Mapping of epitopes in discoidin domain receptor 1 critical for collagen binding. J. Biol. Chem. 2001;276:45952–45958. doi: 10.1074/jbc.M104360200. [DOI] [PubMed] [Google Scholar]
- 34.Wang T., Waters C.T., Rothman A.M., Jakins T.J., Römisch K., Trump D. Intracellular retention of mutant retinoschisin is the pathological mechanism underlying X-linked retinoschisis. Hum. Mol. Genet. 2002;11:3097–3105. doi: 10.1093/hmg/11.24.3097. [DOI] [PubMed] [Google Scholar]
- 35.Robson A.G., Mengher L.S., Tan M.H., Moore A.T. An unusual fundus phenotype of inner retinal sheen in X-linked retinoschisis. Eye. 2009;23:1876–1878. doi: 10.1038/eye.2008.358. [DOI] [PubMed] [Google Scholar]
- 36.Vijayasarathy C., Ziccardi L., Zeng Y., Smaoui N., Caruso R.C., Sieving P.A. Null retinoschisin-protein expression from an RS1 c354del1-ins18 mutation causing progressive and severe XLRS in a cross-sectional family study. Investig. Ophthalmol. Vis. Sci. 2009;50:5375–5383. doi: 10.1167/iovs.09-3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kjellström S., Vijayasarathy C., Ponjavic V., Sieving P.A., Andréasson S. Long-term 12 year follow-up of X-linked congenital retinoschisis. Ophthalmic. Genet. 2010;31:114–125. doi: 10.3109/13816810.2010.482555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bennett L.D., Wang Y.Z., Klein M., Pennesi M.E., Jayasundera T., Birch D.G. Structure/Psychophysical Relationships in X-Linked Retinoschisis. Investig. Ophthalmol. Vis. Sci. 2016;57:332–337. doi: 10.1167/iovs.15-18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stringa F., Tsamis E., Papayannis A., Chwiejczak K., Jalil A., Biswas S., Ahmad H., Stanga P.E. Segmented swept source optical coherence tomography angiography assessment of the perifoveal vasculature in patients with X-linked juvenile retinoschisis: A serial case report. Int. Med. Case. Rep. J. 2017;10:329–335. doi: 10.2147/IMCRJ.S136310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Padrón-Pérez N., Català-Mora J., Díaz J., Arias L., Prat J., Caminal J.M. Swept-source and optical coherence tomography angiography in patients with X-linked retinoschisis. Eye. 2018;32:707–715. doi: 10.1038/eye.2017.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mastropasqua R., Toto L., Di Antonio L., Parodi M.B., Sorino L., Antonucci I., Stuppia L., Di Nicola M., Mariotti C. Optical Coherence Tomography Angiography Findings in X-Linked Retinoschisis. Ophthalmic Surg. Lasers Imaging Retina. 2018;49:e20–e31. doi: 10.3928/23258160-20180907-03. [DOI] [PubMed] [Google Scholar]
- 42.Han I.C., Whitmore S.S., Critser D.B., Lee S.Y., DeLuca A.P., Daggett H.T., Affatigato L.M., Mullins R.F., Tucker B.A., Drack A.V., et al. Wide-Field Swept-Source OCT and Angiography in X-Linked Retinoschisis. Ophthalmol. Retina. 2019;3:178–185. doi: 10.1016/j.oret.2018.09.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.