

Abstract
Metastasis, tumor progression, and chemoresistance are the major causes of death in patients with pancreatic ductal adenocarcinoma (PDAC). Tumor dissemination is associated with the activation of an epithelial-to-mesenchymal transition (EMT) process, a program by which epithelial cells lose their cell polarity and cell-to-cell adhesion, and acquire migratory and invasive abilities to become mesenchymal stem cells (MSC). These MSCs are multipotent stromal cells capable of differentiating into various cell types and trigger the phenotypic transition from an epithelial to a mesenchymal state. Therefore, EMT promotes migration and survival during cancer metastasis and confers stemness features to particular subsets of cells. Furthermore, a major problem limiting our ability to treat PDAC is the existence of rare populations of pancreatic cancer stem cells (PCSCs) or cancer-initiating cells in pancreatic tumors. PCSCs may represent sub-populations of tumor cells resistant to therapy which are most crucial for driving invasive tumor growth. These cells are capable of regenerating the cellular heterogeneity associated with the primary tumor when xenografted into mice. Therefore, the presence of PCSCs has prognostic relevance and influences the therapeutic response of tumors. PCSCs express markers of cancer stem cells (CSCs) including CD24, CD133, CD44, and epithelial specific antigen as well as the drug transporter ABCG2 grow as spheroids in a defined growth medium. A major difficulty in studying tumor cell dissemination and metastasis has been the identification of markers that distinguish metastatic cancer cells from cells that are normally circulating in the bloodstream or at sites where these cells metastasize. Evidence highlights a linkage between CSC and EMT. In this review, The current understanding of the PCSCs, signaling pathways regulating these cells, PDAC heterogeneity, EMT mechanism, and links between EMT and metastasis in PCSCs are summarised. This information may provide potential therapeutic strategies to prevent EMT and trigger CSC growth inhibition and cell death.
Keywords: Pancreatic cancer, cancer stem cells, epithelial-mesenchymal transition, metastasis, drug resistance
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-related death in the world with a low probability of early diagnosis[1]. KRAS, CDKN2A, TP53, and SMAD4 are frequently mutated genes that define the genetic landscape of PDAC. PDAC is one of the most lethal cancers due to its high metastatic potential and delayed detection[2,3]. The median survival time following diagnosis remains at less than 6 months, with an overall survival rate of less than 4%[1–3]. Gemcitabine (GEM) treatment has only increased the median survival of PDAC patients from 3–4 months to 6–7 months[4–6]. Recent evidence shows that using FOLFIRINOX (leucovorin, fluorouracil, irinotecan, oxaliplatin) in PDAC patients was more effective than GEM as it demonstrated longer survival in pancreatic cancer patients (11.1 months vs. 6.8 months)[6,7]. Resistance to apoptosis is a common feature of PDAC and a major reason why this devastating disease is resistant to various treatment strategies including GEM and FOLFIRINOX[8]. Another major problem limiting our ability to treat PDAC is the existence of rare populations of pancreatic cancer stem cells (PCSCs) or cancer-initiating cells in pancreatic tumors; PCSCs may represent sub-populations of tumor cells resistant to therapy which are most crucial for driving invasive tumor growth[9–13]. The PCSCs express a wide array of markers such as CD44, CD24, epithelial specific antigen (ESA), CD133, c-mesenchymal to epithelial transition (c-MET), CXCR4, PD2/Paf1, and ALDH1[14–17]. These cells are capable of regenerating the cellular heterogeneity associated with the primary tumor when xenografted into mice[13–17]. Therefore, the presence of PCSCs has prognostic relevance and influences the therapeutic response of tumors.
Metastasis is the major cause of high PDAC mortality. As Mu et al.[15] recently described, tumor progression is driven by the cross-interaction between tumor cells, primarily cancer stem cells (CSCs) (or cancer-initiating cells) and surrounding stromal cells as well as distant organs, in which tumor-derived extracellular vesicles (TEX) play a major and important role. Mu et al.[15] report that the PCSC markers Tspan8, alpha6beta4, CD44v6, CXCR4, LRP5/6, LRG5, claudin7, EpCAM, and CD133, participate in a metastatic cascade at various steps, often via PDAC CSC-TEX.
In this review, the models of CSCs in PDAC and their cell-intrinsic and - extrinsic regulatory pathways are described. Insights into the heterogeneity of cell sub-populations of PDAC, plasticity, cancer stemness, and the involvement of epithelial-mesenchymal transition (EMT), which participates in metastasis are highlighted. These properties may account for the unsuccessful clinical trials that test therapeutics designed to directly target CSCs.
PCSCS
CSCs from epithelial tissues were first identified in breast cancer in 2003 by Al-Hajj et al.[18], who found that a distinct sub-population of cancer cells expressing CD44+CD24−/low ESA+ develop into tumors in immunodeficient mice. In PDAC, the presence of CSCs was reported in 2007 by Shah et al.[19] who demonstrated that CD44+CD24+ESA+ cells exhibit high tumorigenic potential.
Two models are proposed to describe the origin of CSCs [Figure 1] and their role in tumorigenesis[20–22]. The first one is the hierarchical model in which the CSCs represent a small distinct sub-population within the tumor with capacity for self-renewals and also the ability to differentiate into progeny cells; the progeny cells then proliferate to initiate and sustain tumor development and progression. Therefore, this model indicates that in the clinical setting, eradication of CSCs may prevent tumor recurrence.
Figure 1.
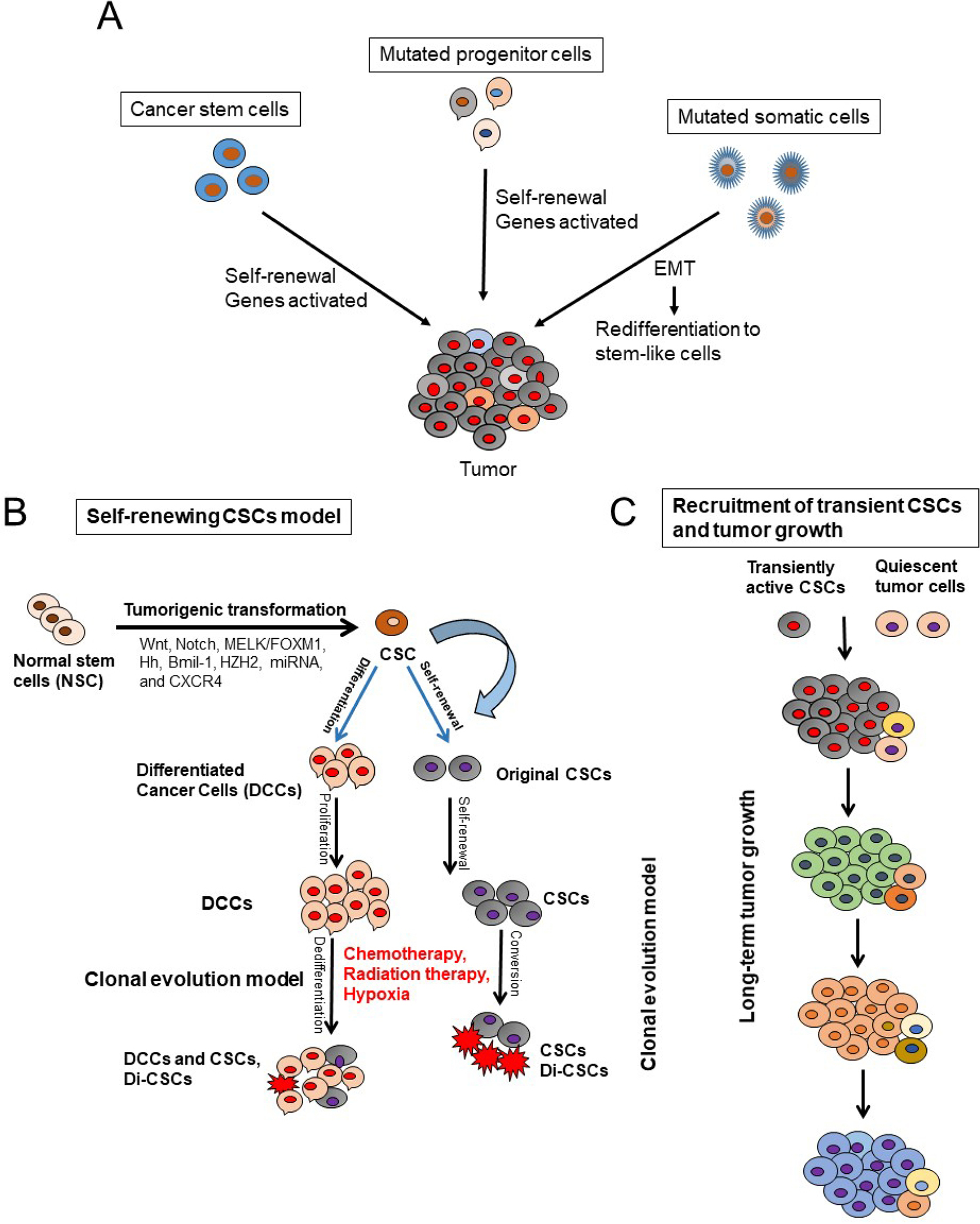
Models that explain pancreatic cancer tumor development and roles of cancer stem cells (CSCs) in tumor initiation and progression. A: stochastic model shows that every cell has the potential to be the tumor initiator; B: CSC model shows that CSCs originate from normal stem cells (NSCs) through mutations and tumorigenic transformation of several potential pathways including Hh: hedgehog, epithelial-to-mesenchymal transition (EMT), and the reverse process mesenchymal-to-epithelial transition (MET). CSCs are also generated by de-differentiation of differentiated malignant cells by chemotherapeutic agents, ionizing radiation, and hypoxia. CSCs and drug-induced CSCs (Di-CSCs) are enriched following conventional chemotherapy treatment; C: the model of long-term tumor growth in PDAC states that a succession of CSC or tumor initiating cells (TIC) clones and drives tumor progression in serial xenotransplantation. Individual TICs contribute to tumor formation transiently and generate mainly non-tumorigenic progeny expressing little or no self-renewal
The second model is stochastic model in which every cell within a tumor is capable of promoting tumor initiation and progression[21]. Furthermore, the heterogeneity within the tumor cell population is due to accumulated genetic mutations[21]. These CSCs models may express plasticity in the presence of tumor microenvironmental stimuli including oxidative and nutritional stress, low oxygen tension, and cytotoxic drugs to which the tumor can be subjected to[16,21–25]. These factors influence the inter-conversion of a non-CSC population to a CSCs[21,22] [Figure 1]. Previous studies have demonstrated that extracellular matrix components like collagen can influence clonogenic tumor growth, tumor initiation, and invasion of PDAC due to activation of the FAK signaling pathway[26,27]. Additionally, a hypoxic tumor microenvironment is known to promote the conversion of non-CSC to CSC populations in PDAC[8]. These data corroborate with observations that the CSCs within the tumor are the cells’ survival advantage, capable of living under stress conditions and to express resistance to cancer therapies.
Accumulated evidence obtained through large-scale genomic studies and single cell RNA sequencing analysis has indicated the existence of CSCs in hematological malignancies and solid tumors including PDAC, breast cancer, malignant glioma, prostate cancer, non-small cell lung cancer, colorectal cancer, and hepatocellular carcinoma[28–37]. Substantial in vitro and in vivo studies have provided convincing evidence supporting CSCs as the critical cause of cancer initiation, growth, metastasis, and treatment resistance in cancers[20–24]. CSCs were also found to exhibit characteristics of an EMT, a process known to enable cancer cell dissemination and metastasis to other organs[35–41]. Additionally, significant and convincing evidence shows that inducing EMT in cancer cells confers cancer stem cell-like characteristics, which promote their metastatic and tumor-initiating abilities[37,38]. While PCSCs self-renewal is essential in the progression, migration, and metastasis of PDAC, clonal evolution and plasticity of PCSCs within PDAC tumors are not well understood[38–46]. In this regard, Ball et al.[47] showed that long-term progression of PDAC in serial xenotransplantation happens by a succession of transiently active PCSCs producing tumor cells in a temporally restricted manner with little overlap between subsequent xenograft generations. Therefore, the clonal PCSC activity in PDAC differs from the continuous activity of limited numbers of self-renewing PCSCs with a defined and rigid cellular hierarchy. Indeed, PCSCs heterogeneity and plasticity make therapeutic targeting of PCSCs very difficult and challenging[24]. Furthermore, several investigations have suggested that CSCs exist in dynamic equilibrium with more differentiated cancer cells via bi-directional regeneration or interconversion of differentiated cancer cells to CSCs, caused by various factors[23,24,48,49,50].
A distinct characteristic of PDAC is its desmoplasia, consisting of a significant amount of cancer-associated fibroblasts (CAFs) and a very dense fibrotic stroma[51] [Figure 2]. The CAFs are pro-inflammatory due to activation of several signaling factors including nuclear factor kappa B (NF-κB), signal transducer and activator of transcription (STAT)-1 and STAT-3, and transforming growth factor (TGF)-β/SMAD[51–54]. These signaling factors cooperate in active cross-talk with cancer cells through paracrine signaling factors including chemokines, insulin-like growth factor, and proteases[52–56]. Furthermore, several pro-stemness paracrine factors are secreted by distinct CAFs[56–62] and support the self-renewal and the stemness properties of initial PCSCs in tumors or promote the conversion of cancer cells into PCSCs[63]. Additionally, chemotherapy (e.g., GEM), can affect CAFs in PDAC, which then acquire a senescence-like secretory phenotype and increase the production of pro-stemness chemokines which enhance tumorstemness and aggressiveness of PDAC after therapy[13]. It is known that CAFs consist of a heterogeneous population, with specific functions within tumors and in the process of metastasis[64].
Figure 2.
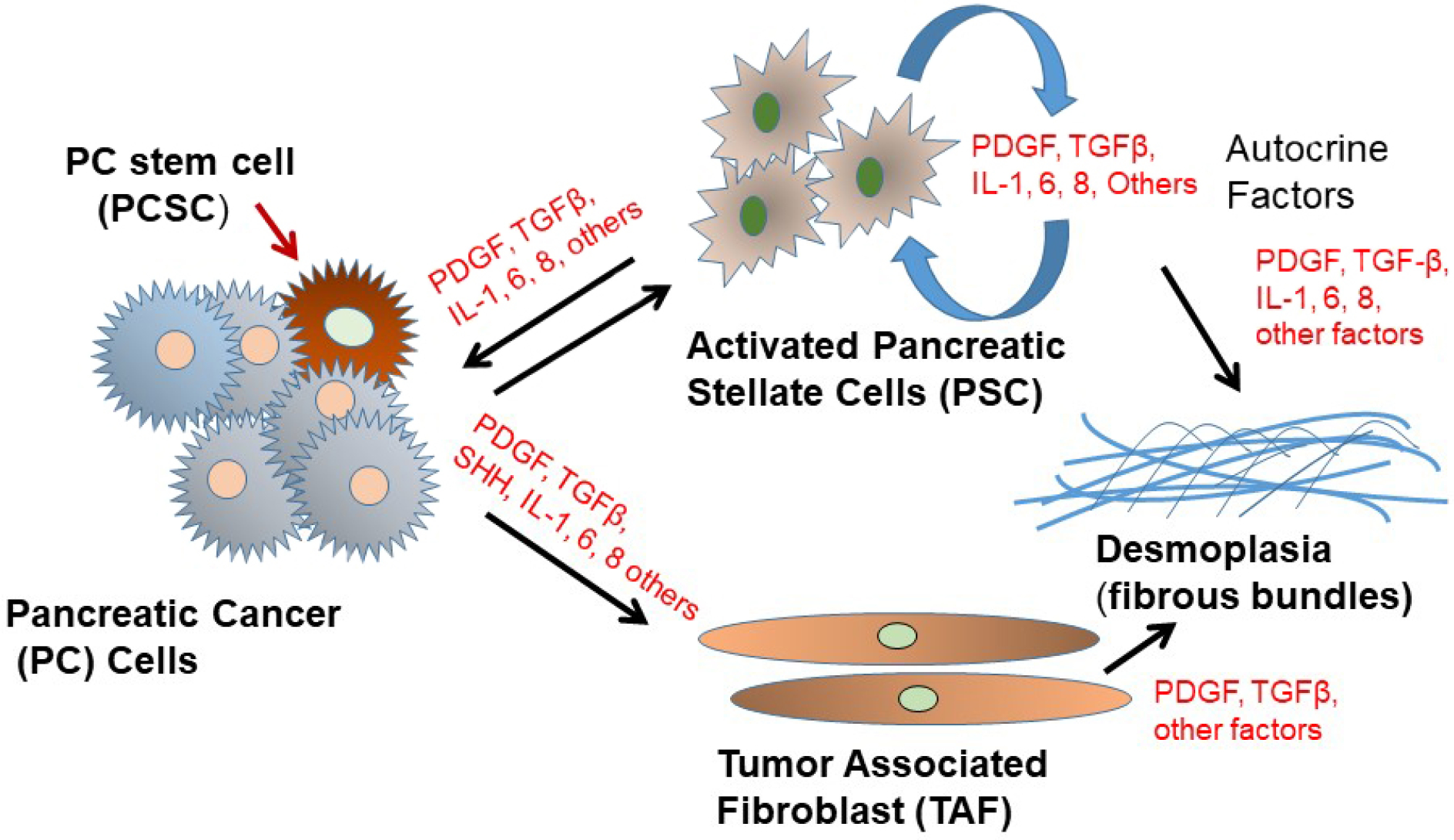
Schematic depiction of pancreatic cancer cell interactions with tumor-associated fibroblasts (TAF) and activated pancreatic stellate cells (PSC) in the tumor microenvironment. PDGF: platelet-derived growth factor; TGFβ: transforming growth factor β
Interestingly, besides CAFs, the PDAC stroma also contains bone marrow-derived mesenchymal stem cells (MSCs)[65]. The MSCs significantly contribute to tumor progression and promote cancer stemness by secreting pro-stemness cytokines, chemokines, and growth factors or by differentiating into pro-stemness CAFs[62,63,66]. MSCs also produce pro-stemness niches in the stroma of PDAC, and infiltrating immune cells produce pro-stemness factors that form pro-PCSC niches[67–70]. Waghray et al.[71] identified and characterized mesenchymal stem cells (MSC) within the human PDAC tumor microenvironment (TME). These cancer-associated MSCs (CA-MSCs) increase the growth, invasion, and metastatic potential of PDAC cancer cells[71], and CA-MSCs secrete the cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF) that is required for tumor cell proliferation, invasion, and trans-endothelial migration. The depletion of GM-CSF in CA-MSCs inhibited the ability of these cells to promote tumor cell growth and metastasis[71]. Therefore, CA-MSCs may provide a potential strategy for a PDAC therapeutic approach. Since the desmoplastic stroma in PDAC is believed to be a major barrier for the efficient penetration of anti-cancer agents into the tumor, efficient anti-PCSC therapeutics plus anti-stromal or stromal remodeling therapeutics may be able to penetrate through the thick layer of stroma to reach PCSCs and the bulk of tumor cells, to trigger their cell death or growth inhibitory effects and effectively inhibit growth and metastasis of PDAC.
In addition to PCSCs and the previously discussed distinct stromal cells, the PDAC immune system also plays a critical and complex role in the development and progression of PDAC. It is well-established that myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) alter the immune environment of the TME and help tumor proliferation as well as metastatic and immunotherapy resistance[72]. Inhibitors of the CSF-1R were shown to reprogram the TME and TAMs and lead to enhanced T-cell-mediated tumor elimination[72]. Furthermore, FAK inhibitors reduced the infiltration of MDSCs, TAMs, and regulatory T-cells[72]. Moreover, C-C motif chemokine receptor (CCR)-2 has been shown to mediate the recruitment of TAMs to the tumor[73]. Bone marrow mesenchymal stem cells (BM‐MPCs) display self‐renewal, differentiation, dormancy, and hematopoiesis properties; BM‐HPCs also secrete cytokines and extracellular matrix for the growth of metastases[74–77]. BM‐MSCs are major players in the tumor microenvironment[78] and affect inflammation, the tumor environment, immunity, and cancer metastasis[79].
Signaling pathways in PCSCs
Various signaling pathways are altered in PCSCs and EMT cells including Hedgehog, Notch, Wnt, NF-κB, and AKT [Figure 3]. Among these, Hedgehog, Notch, and Wnt play particularly important roles in PCSCs[51]. These signaling pathways are critical regulators of PCSC self-renewal, tumor growth, invasion, metastasis, and therapy resistance[16,40,80,81]. Furthermore, miRNAs play a significant role in the regulation of PCSCs[82,83].
Figure 3.
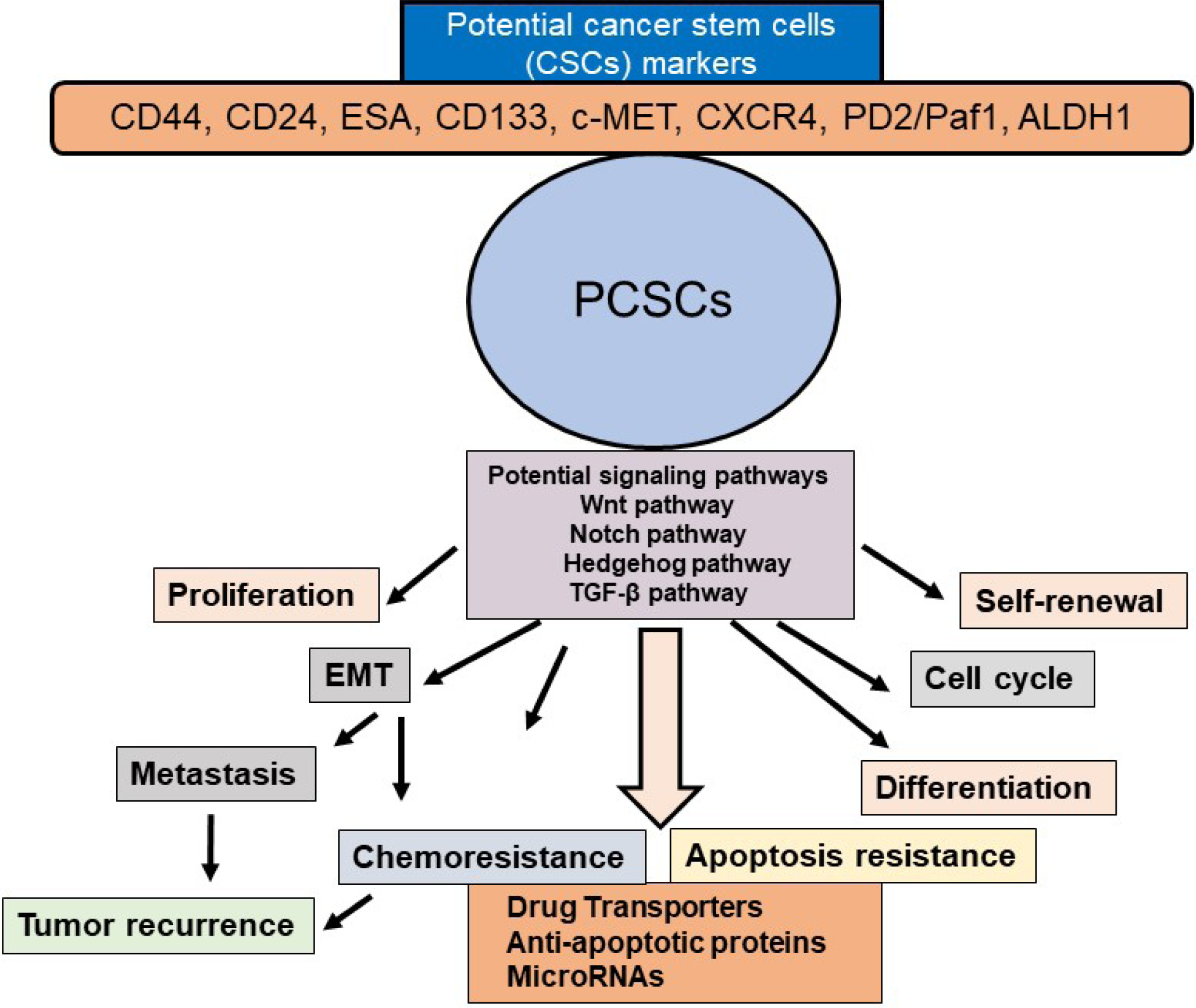
Schematic presentation of PCSC properties and their contribution to EMT, tumor metastasis and recurrence, chemoresistance, and apoptosis resistance. Potential CSC markers and signaling pathways in PCSCs and activation of several signaling pathways in the self-renewal, maintenance, and tumor recurrence in PDAC are also shown. PCSC: pancreatic cancer stem cell; EMT: epithelial-mesenchymal transition
Notch signaling regulates cell proliferation, survival, apoptosis, and the differentiation of various cancers including pancreatic cancer cells and PCSCs as well as promoting EMT by controlling some transcription factors and growth factors including Snail, Slug, and TGF-β. Notch targets many genes which play critical roles in the development and progression of human malignancies[84–86]. Several studies have demonstrated that resistance to chemotherapy in PCSCs is linked to the active Notch signaling pathway[63,87,88].
Another self-renewal pathway in PCSCs is Hedgehog signaling, which is involved in tumor initiation, progression, and metastasis[63,89,90]. The three hedgehog genes include Sonic hedgehog (Shh), Indian hedgehog, and the Desert hedgehog homolog[90,91]. It has been shown that one of these three ligands binds to the receptor Patched1 and releases the protein smoothened (Smo)[63,89,90]. Smo triggers the activation of downstream target genes such as the GLI family of transcription factors and PTCH. Yamasaki et al.[92] have reported that a nine-fold increase in Shh mRNA levels has been found in CD44+CD24+ESA+ PCSCs when compared to the bulk of unsorted pancreatic cancer cells. Inhibition of Hedgehog signaling by Smo suppression has been shown to reverse EMT and suppress the invasion of pancreatic cancer cells[93–95].
Substantial evidence has shown that Wnt/β-catenin signaling is involved in cell proliferation, migration, apoptosis, differentiation, and self-renewal of CSCs in several types of cancers[96–98]. Dysregulation of the Wnt/β-catenin signaling pathway is associated with chemotherapy resistance in PDAC, and significant evidence suggests that nuclear β-catenin plays an essential role in EMT[97,99]. The Wnt signaling pathway also plays a significant role in regulating PCSCs[99,100]. Additionally, Wnt signals significantly regulate CSCs in solid tumors including PDAC in the niche environments[98,101,102]. Because of dysregulation in the Wnt signaling pathway, PCSCs are significantly susceptible to Wnt signal inhibitors[98,99,103,104]. Hence, Wnt in the PDAC niche of PCSCs offers a critical therapeutic target in PDAC.
A significantly activated signaling pathway in CSCs is the NF-κB pathway; its inhibition triggers the loss of CSC properties[105,106]. Interestingly, the CCL21/CCR7 axis known to facilitate metastasis to distant organs promoted the metastasis and survival of CD133+ PCSCs and regulated their metastasis by modulating EMT and the Erk/NF-κB pathway[107]. Moreover, NF-κB-mediated invasiveness in CD133+ PCSCs is regulated by autocrine and paracrine activation of IL1 signaling[108]. Furthermore, the crucial role of PCSCs in developing resistance to gemcitabine treatment through the Nox/ROS/NF-κB/STAT3 signaling pathway was demonstrated by Zhang et al.[109]. Significantly, therapeutic targeting of the FGFR1/Src/NF-κB signaling axis has been shown to inhibit PCSCs and oncogenicity[110]. These findings will provide new directions for identifying potential targets that regulate NF-κB-mediated invasiveness of PCSCs and can be used to sensitize pancreatic cells to chemotherapy. In addition to the above major signaling pathways, the mTOR pathway has been shown to be essential for the self-renewal of PCSCs[111,112].
Like other cancers, miRNA expression is dysregulated in PDAC[113,114]. Two classes of miRNAs play crucial roles in cancer cells, oncogenic miRNAs and tumor suppressor miRNAs[23,115,116]. Jung et al.[117] demonstrated that PCSCs exhibit differential expression of miR-99a, miR-100, miR-125b, miR-192, and miR-429 compared with controls. Another study reported the loss of miR-34 in CD44+CD133+ PCSCs, while miR-34 restoration led to the inhibition of spheroidal growth of CSCs and tumor formation[118]. Wellner et al.[119] showed that miR-200c, miR-203, and miR-183 down regulate stem cell factors and described a regulatory feedback loop between miRNAs and CSC in pancreatic cancer. Moreover, they demonstrated that ZEB1 represses expression of stemness-inhibiting miR-203 and that ZEB1 links EMT activation and stemness maintenance by suppressing stemness-inhibiting miRNA expression, and therefore promotes mobile migrating CSCs. These authors concluded that targeting the ZEB1-miR-200 feedback loop may potentially be a valid and promising therapeutic approach for PDAC.
While this review discusses abnormalities in the intracellular signaling pathways known to be involved in carcinogenesis, growth, metastasis, EMT, and chemoresistance in PDAC, the significance of these regulatory pathways to the major risk factors for pancreatic cancer including diabetes, smoking, alcoholism, and psychological stress remains to be discovered.
EPITHELIAL-MESENCHYMAL (EMT) TRANSITION
A small number of cancer cells in the primary tumor are capable of undergoing EMT, which critically promotes tumor invasion and metastatic dissemination[120,121] in human cancer patients including PDAC[40,41,122], as well as in PCSCs and circulating tumor cells[122–124]. Metastatic seeding is frequently initiated before diagnosis of the primary tumor in cancer patients, and disseminated tumor cells can remain dormant in secondary sites before forming metastatic tumors[125–128]. The majority of pancreatic cancer-related mortality is due to metastatic disease.
EMT is critical for this rapid metastatic tumor progression, and it is a multi-stage trans-differentiation cellular process that allows epithelial cells to undergo multiple biochemical changes to gain a mesenchymal phenotype[128,129]. During this process, epithelial cells lose their epithelial markers (such as E-cadherin, occludin, claudin, and laminin 1) and gain mesenchymal markers such as N-cadherin, vimentin, and fibronectin[130]. These changes are caused by the activation of specific EMT transcriptional programs. Some transcriptional regulators such as TWIST, SNAI1, SNAI2, ZEB1, and ZEB2 repress E-cadherin expression, while others play roles in promoting the expression markers of mesenchymal differentiation markers including N- and/or R-cadherin and vimentin, as well as cellular matrix and focal adhesion proteins involved in promoting motility[124,131,132]. Furthermore, EMT is associated with enhanced activity of matrix metalloproteinases[124,131,132].
After the invading cancer cells reach metastatic sites, they undergo the reverse EMT process, MET, and adapt to proliferating in the invaded tissue microenvironment[133–136]. Since metastasis is the major cause of cancer mortality, a detailed understanding of EMT could potentially lead to more effective therapeutic strategies for PDAC.
METASTASIS AND CSC POPULATION
Emerging evidence indicates that as the tumor microenvironment opts for a CSC population, these cells can also metastasize and initiate cells that participate in metastasis[137]. Several cellular signaling pathways regulate the self-renewal capacity of CSCs and control their inter-conversion between the dormantstate and subsequent re-activation upon metastasis[137–140]. These re-activated cells are nurtured by extracellular niches, which crosstalk and support positive cytoprotective signals such as Wnt and Notch[96,141–145]. As Mu et al.[15] described, tumor progression is driven by the cross-interaction between tumor cells, primarily CSCs and surrounding stromal cells as well as distant organs, in which tumor-derived extracellular vesicles (TEX) play a major and important role. Mu et al.[15] report that the PCSC markers Tspan8, alpha6beta4, CD44v6, CXCR4, LRP5/6, LRG5, claudin, EpCAM, and CD133 participate in the metastatic cascade at different stages, often via PDAC TEX. In PDAC tumors, the PCSC population shows over-expression of genes involved in EMT and the distinct ability to metastasize and colonize distant tissues.
CD44 is overexpressed in CSCs, frequently shows alternative spliced variants, and plays a role in cancer development and progression. The CD44 major ligand, hyaluronan, binds to and activates CD44 resulting in stimulation of several cell signaling pathways that trigger cell proliferation, survival, and increased cellular motility[146]. Cancer cells that undergo an EMT acquire properties of CSC and show enhanced CD44 expression[43]. CD44 consists of two isoforms, standard (CD44s) and CD44 variants(CD44v), but the different functional roles of these variants are not well known. CD44v may play a role in regulating EMT and the plasticity of cancer cells[146–148]. Moreover, evidence shows that CD44 activates the MT1-MMP-SNAI1 axis to promote metastasis in PDAC, and CD133 activates the IL1β-NF-κB pathway leading to invasiveness and metastasis[108,149]. Interestingly, Zhang et al.[107] demonstrated that the CCR7–CCL21 axis in PCSCs mediates EMT phenotype via activation of ERK/NF-κB signaling pathways. Additionally, it has been shown that the Hh pathway is implicated in playing a role in EMT in PCSCs. Inhibition of Hh signaling in CSCs reduced self-renewal, EMT tumorgenesis, invasiveness, drug resistance, and metastasis[150]. Activation of the PI3K/AKT/mTOR pathway[151] alone or in combination with the Hh pathway[152] in PCSCs also increased metastasis in these cells.
RELEVANCE OF PCSCS IN PDAC DEVELOPMENT
The foregoing discussion provided ample evidence related to the role of PCSCs in carcinogenesis, growth, metastasis, EMT, and chemoresistance in PDAC. The above aberrant signaling pathways govern cancer cell plasticity, which give rise to tumor cellular heterogeneity, EMT, therapeutic resistance, and recurrence through clonal replacement and activation of dormant CSCs in PDAC as well as other cancers[80,88,153,154]. PDAC is characterized by molecular alterations regulating PCSCs, including mutations of K-RAS, TP53, transforming growth factor-β, Hedgehog, WNT and NOTCH signaling pathways. Many genetic alterations were defined in PDAC such as earlier events including K-ras point mutation[155], INK4a/Arf deficiency[156], the epidermal growth factor receptor (EGFR) over-expression, gene amplification and HER2/neu over-expression and TP53, transforming growth factor-β, Hedgehog, WNT and NOTCH signaling pathways maintain PCSCs. Furthermore, the roles of EGFR, Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTORC1/GSK-3 pathways in PCSCs and their relationship to PDAC tumor initiation, EMT, malignant development has been investigated[157]. As discussed above, recent advances in the role of EMT and PCSCs in tumor progression, metastasis, chemo-resistance, and the mechanisms integrated with aberrant biochemical signals and the underlying pathways have been surveyed in this review. Furthermore, WNT signaling cascades cross-interaction with the FGF, Notch, Hedgehog and TGFβ/BMP signaling cascades and regulate expression of functional CSC markers, such as CD44, CD133 (PROM1), EPCAM and LGR5 (GPR49)[40,153–158]. Aberrant canonical and non-canonical WNT signaling in human malignancies, including PDAC are involved in CSC survival, bulk-tumor expansion and invasion/metastasis[158]. Despite these advances, the significance of these regulatory pathways to the major risk factors for pancreatic cancer including diabetes, smoking, alcoholism, and psychological stress remain to be studied in detail in the future.
CONCLUSION
Substantial evidence has demonstrated that CSCs including PCSCs trigger the characteristic hallmarks of various tumors including self-renewal, invasiveness, tumor recurrence, resistance, metastasis, and resistance to chemotherapeutic agents and radiotherapy. Moreover, the bulk of cancer cell population displays plasticity in most tumors including PDAC, which enables them to dynamically inter-convert between non-CSC and CSC states. Another critically important and intriguing characteristic of CSCs including PCSCs is their capacity to disseminate, migrate, and form metastatic lesions expressing resistance to therapies. In some tumors including PDAC, this plasticity has been associated with the EMT process. Furthermore, cytokines and growth factors, provided by the CSC niche containing CAFs, MSCs, endothelial cells and specific immune cells, and hypoxia, trigger transcriptional and epigenetic regulations leading to the induction of plasticity, stemness, EMT, and metastasis. Taken together, the foregoing discussion in this review provides a better understanding of the molecular mechanisms underlying these behaviours in CSCs, including PCSCs, and may lead to the identification of specific therapeutics and novel strategies to prevent EMT and metastasis, trigger CSC growth inhibition and cell death, and increase the sensitivity of tumors including PDAC to cancer therapeutics.
Acknowledgments
I would like to thank Dr. Mary D. Kraeszig for her excellent editorial assistance.
Footnotes
Conflicts of interest
The author declared that there are no conflicts of interest.
Availability of data and materials
Not applicable.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
REFERENCES
- 1.Aier I, Semwal R, Sharma A, Varadwaj PK. A systematic assessment of statistics, risk factors, and underlying features involved in pancreatic cancer. Cancer Epidemiol 2019;58:104–10. [DOI] [PubMed] [Google Scholar]
- 2.Sohal DP, Mangu PB, Khorana AA, Shah MA, Philip PA, et al. Metastatic pancreatic cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 2016;34:2784–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, et al. Pancreatic cancer. Nat Rev Dis Primers 2016;2:16022. [DOI] [PubMed] [Google Scholar]
- 4.Balic A, Dorado J, Alonso-Gómez M, Heeschen C. Stem cells as the root of pancreatic ductal adenocarcinoma. Exp Cell Res 2012;318:691–4. [DOI] [PubMed] [Google Scholar]
- 5.Hassanein MK, Suetsugu A, Saji S, Moriwaki H, Bouvet M, et al. Stem-like and non-stem human pancreatic cancer cells distinguished by morphology and metastatic behavior. J Cell Biochem 2011;112:3549–54. [DOI] [PubMed] [Google Scholar]
- 6.Berlin JD, Catalano P, Thomas JP, Kugler JW, Haller DG, et al. Phase III study of gemcitabine in combination with fluorouracil versus gemcitabine alone in patients with advanced pancreatic carcinoma: Eastern Cooperative Oncology Group Trial E2297. J Clin Oncol 2002;20:3270–5. [DOI] [PubMed] [Google Scholar]
- 7.Singh RR, O’Reilly EM. New treatment strategies for metastatic pancreatic ductal adenocarcinoma. Drugs 2020;80:647–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lambert A, Gavoille C, Conroy T. Current status on the place of FOLFIRINOX in metastatic pancreatic cancer and future directions. Therap Adv Gastroenterol 2017;10:631–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adamska A, Elaskalani O, Emmanouilidi A, Kim M, Abdol Razak NB, et al. Molecular and cellular mechanisms of chemoresistance in pancreatic cancer. Adv Biol Regul 2018;68:77–87. [DOI] [PubMed] [Google Scholar]
- 10.Hermann PC, Sainz B Jr. Pancreatic cancer stem cells: a state or an entity? Semin Cancer Biol 2018;53:223–31. [DOI] [PubMed] [Google Scholar]
- 11.Sergeant G, Vankelecom H, Gremeaux L, Topal B. Role of cancer stem cells in pancreatic ductal adenocarcinoma. Nat Rev Clin Oncol 2009;6:580–6. [DOI] [PubMed] [Google Scholar]
- 12.Bednar F, Simeone DM. Pancreatic cancer stem cells and relevance to cancer treatments. Cell Biochem 2009;107:40–5. [DOI] [PubMed] [Google Scholar]
- 13.Dembinski JL, Krauss S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin Exp Metastasis 2009;26:611–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaz AP, Ponnusamy MP, Seshacharyulu P, Batra SK. A concise review on the current understanding of pancreatic cancer stem cells. J Cancer Stem Cell Res 2014; 2:e1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mu W, Wang Z, Zöller M. Ping-pong-tumor and host in pancreatic cancer progression. Front Oncol 2019;9:1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsai KK, Chan TS, Shaked Y. Next viable routes to targeting pancreatic cancer stemness: learning from clinical setbacks. J Clin Med 2019;8:702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gzil A, Zarębska I, Bursiewicz W, Antosik P, Grzanka D, et al. Markers of pancreatic cancer stem cells and their clinical and therapeutic implications. Mol Biol Rep 2019;46:6629–45. [DOI] [PubMed] [Google Scholar]
- 18.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003;100:3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah AN, Summy JM, Zhang J, Park SI, Parikh NU, et al. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann Surg Oncol 2007;14:3629–37. [DOI] [PubMed] [Google Scholar]
- 20.Hermann PC, Huber SL, Heeschen C. Metastatic cancer stem cells: a new target for anti-cancer therapy? Cell Cycle 2008;7:188–93. [DOI] [PubMed] [Google Scholar]
- 21.Steinbichler TB, Dudás J, Skvortsov S, Ganswindt U, Riechelmann H, et al. Therapy resistance mediated by cancer stem cells. Semin Cancer Biol 2018;53:156–67. [DOI] [PubMed] [Google Scholar]
- 22.Safa AR. Resistance to drugs and cell death in cancer stem cells (CSCs). J Transl Sci 2019;5:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Safa AR, Saadatzadeh MR, Cohen-Gadol AA, Pollok KE, Bijangi-Vishehsaraei K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis 2015;2:152–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chatterjee R, Chatterjee J. ROS and oncogenesis with special reference to EMT and stemness. Eur J Cell Biol 2020;99:151073. [DOI] [PubMed] [Google Scholar]
- 25.Garg M Epithelial plasticity, autophagy and metastasis: potential modifiers of the crosstalk to overcome therapeutic resistance. Stem Cell Rev Rep 2020;16:503–10. [DOI] [PubMed] [Google Scholar]
- 26.Valle S, Martin-Hijano L, Alcalá S, Alonso-Nocelo M, Sainz B Jr. The ever-evolving concept of the cancer stem cell in pancreatic cancer. Cancers (Basel) 2018;10:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Begum A, Ewachiw T, Jung C, Huang A, Norberg KJ, et al. The extracellular matrix and focal adhesion kinase signaling regulate cancer stem cell function in pancreatic ductal adenocarcinoma. PLoS One 2017;12:e0180181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu H, Wang D, Liu Y, Su Z, Zhang L, et al. Role of the hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer Cell Int 2013;13:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santamaria-Martinez A Huelsken J The niche under siege: Novel targets for metastasis therapy. J Intern Med 2013;274:127136. [DOI] [PubMed] [Google Scholar]
- 30.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J. Identification of human brain tumour initiating cells. Nature 2004;432:396–401. [DOI] [PubMed] [Google Scholar]
- 31.Ginestier C, Hur MH, Charafe-Jau EC, Monville F, Dutcher J. et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007;1:555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balic A, Dorado J, Alonso-Gómez M, Heeschen C. Stem cells as the root of pancreatic ductal adenocarcinoma. Exp Cell Res 2012;318:691–4. [DOI] [PubMed] [Google Scholar]
- 33.Van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Lippitt JM, et al. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Res 2010;70:5163–73. [DOI] [PubMed] [Google Scholar]
- 34.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007;445:106–10. [DOI] [PubMed] [Google Scholar]
- 35.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008;8:755–68. [DOI] [PubMed] [Google Scholar]
- 36.Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 2018;173:338–54.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014;344:1396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson MM, Weinberg RA, Lees JA, Guen VJ. Emerging mechanisms by which EMT programs control stemness. Trends Cancer 2020;6:775–80. [DOI] [PubMed] [Google Scholar]
- 39.Castellanos JA, Merchant NB, Nagathihalli NS. Emerging targets in pancreatic cancer: epithelial-mesenchymal transition and cancer stem cells. Onco Targets Ther 2013;6:1261–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou P, Li B, Liu F, Zhang M, Wang Q, et al. The epithelial to mesenchymal transition (EMT) and cancer stem cells: implication for treatment resistance in pancreatic cancer. Mol Cancer 2017;16:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodriguez-Aznar E, Wiesmüller L, Sainz B Jr, Hermann PC. EMT and stemness-Key players in pancreatic cancer stem cells. Cancers (Basel) 2019;11:1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brabletz T To differentiate or not-Routes towards metastasis. Nat Rev Cancer 2012;12:425–36. [DOI] [PubMed] [Google Scholar]
- 43.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008;133:704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pattabiraman DR. Weinberg RA. Tackling the cancer stem cells-What challenges do they pose? Nat Rev Drug Discov 2014;13:497–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012;148:349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011;145:926–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ball CR, Oppel F, Ehrenberg KR, Dubash TD, Dieter M, et al. Succession of transiently active tumor-initiating cell clones in human pancreatic cancer xenografts. EMBO Mol Med 2017;9:918–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Batlle E, Clevers H. Cancer stem cells revisited. Nat Med 2017;23:1124–34. [DOI] [PubMed] [Google Scholar]
- 49.Yang G, Quan Y, Wang W, Fu Q, Wu J, et al. Dynamic equilibrium between cancer stem cells and non-stem cancer cells in human SW620 and MCF-7 cancer cell populations. Br J Cancer 2012;106:1512–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dalla Pozza E, Dando I, Biondani G, Brandi J, Costanzo C, et al. , Pancreatic ductal adenocarcinoma cell lines display a plastic ability to bi directionally convert into cancer stem cells. Int J Oncol 2015;46:1099–108. [DOI] [PubMed] [Google Scholar]
- 51.Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-κB-dependent manner. Cancer Cell 2010;17:135–47. [DOI] [PubMed] [Google Scholar]
- 52.Bazzichetto C, Conciatori F, Falcone I, Cognetti F, Milella M, et al. Advances in tumor-stroma interactions: Emerging role of cytokine network in colorectal and pancreatic cancer. J Oncol 2019;2019:5373580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geismann C, Schäfer H, Gundlach JP, Hauser C, Egberts JH, et al. NF-κB dependent chemokine signaling in pancreatic cancer. Cancers (Basel) 2019;11:1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Knapinska AM, Estrada CA, Fields GB. The roles of matrix metalloproteinases in pancreatic cancer. Prog Mol Biol Transl Sci 2017;148:339–54. [DOI] [PubMed] [Google Scholar]
- 55.Mutgan AC, Besikcioglu HE, Wang S, Friess H, Ceyhan GO, et al. Insulin/IGF-driven cancer cell-stroma crosstalk as a novel therapeutic target in pancreatic cancer. Mol Cancer 2018;17:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee NH, Nikfarjam M, He H. Functions of the CXC ligand family in the pancreatic tumor microenvironment. Pancreatology 2018;18:705–16. [DOI] [PubMed] [Google Scholar]
- 57.Chen WJ, Ho CC, Chang YL, Chen HY, Lin CA, et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat Commun 2014;5:3472. [DOI] [PubMed] [Google Scholar]
- 58.Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Investig 2011;121:3804–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsuyada A, Chow A, Wu J, Somlo G, Chu P, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res 2012;72:2768–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lonardo E, Hermann PC, Mueller MT, Huber S, Balic A, et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 2011;9:433–46. [DOI] [PubMed] [Google Scholar]
- 61.Shi Y, Gao W, Lytle NK, Huang P, Yuan X, et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019;569:131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chan TS, Shaked Y, Tsai KK. Targeting the interplay between cancer fibroblasts, mesenchymal stem cells, and cancer stem cells in desmoplastic cancers. Front Oncol 2019;9:688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hwang HJ, Oh MS, Lee DW, Kuh HJ. Multiplex quantitative analysis of stroma-mediated cancer cell invasion, matrix remodeling, and drug response in a 3D co-culture model of pancreatic tumor spheroids and stellate cells. J Exp Clin Cancer Res 2019;38:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gieniec KA, Butler LM, Worthley DL, Woods SL. Cancer-associated fibroblasts-heroes or villains? Br J Cancer 2019;121:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu T, Zhou L, Li D, Andl T, Zhang Y. Cancer-associated fibroblasts build and secure the tumor microenvironment. Front Cell Dev Biol 2019;7:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yin Z, Jiang K, Li R, Dong C, Wang L. Multipotent mesenchymal stromal cells play critical roles in hepatocellular carcinoma initiation, progression and therapy. Mol Cancer 2018;17:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sainz B Jr, Alcala S, Garcia E, Sanchez-Ripoll Y, Azevedo MM, et al. Microenvironmental hCAP-18/LL-37 promotes pancreatic ductal adenocarcinoma by activating its cancer stem cell compartment. Gut 2015;64:1921–35. [DOI] [PubMed] [Google Scholar]
- 68.Iovanna JL, Closa D. Factors released by the tumor far microenvironment are decisive for pancreatic adenocarcinoma development and progression. Oncoimmunology 2017;6:e1358840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sainz B Jr, Martin B, Tatari M, Heeschen C, Guerra S. ISG15 is a critical microenvironmental factor for pancreatic cancer stem cells. Cancer Res 2014;74:7309–20. [DOI] [PubMed] [Google Scholar]
- 70.Benson DD, Meng X, Fullerton DA, Moore EE, Lee JH, et al. Activation state of stromal inflammatory cells in murine metastatic pancreatic adenocarcinoma. Am J Physiol Regul Integr Comp Physiol 2012;302:1067–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Waghray M, Yalamanchili M, Dziubinski M, Zeinali M, Erkkinen M, et al. GM-CSF mediates mesenchymal-epithelial cross-talk in pancreatic cancer. Cancer Discov 2016;6:886–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Osipov A, Saung MT, Zheng L, Murphy AG. Small molecule immunomodulation: the tumor microenvironment and overcoming immune escape. J Immunother Cancer 2019;7:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: A role for targeting the CCL2/CCR2 axis. Clin Cancer Res 2013.19:3404–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mitchem JB, Brennan DJ, Knolho BL, Belt BA, Zhu Y, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 2013;73:1128–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jones EA, Kinsey SE, English A, Jones RA, Straszynski L, et al. Isolation and characterization of bone marrow multipotential mesenchymal progenitor cells. Arthritis Rheum 2002;46:3349–60. [DOI] [PubMed] [Google Scholar]
- 76.Kum JJ, Khan ZA. Propranolol inhibits growth of hemangioma-initiating cells but does not induce apoptosis. Pediatr Res 2014;75:381–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kaplan RN, Psaila B, Lyden D. Bone marrow cells in the ‘pre-metastatic niche’: within bone and beyond. Cancer Metastasis Rev 2006;25:521–9. [DOI] [PubMed] [Google Scholar]
- 78.Shao H, Moller M, Wang D, Ting A, Boulina M, et al. A novel stromal fibroblast-modulated 3D tumor spheroid model for studying tumor-stroma interaction and drug discovery. J Vis Exp 2020; doi: 10.3791/60660. [DOI] [PubMed] [Google Scholar]
- 79.Wang CC, Zhao YM, Wang HY, Zhao YP. New insight into the role of exosomes in pancreatic cancer. Ann Clin Lab Sci 2019;49:385–392. [PubMed] [Google Scholar]
- 80.Ercan G, Karlitepe A, Ozpolat B. Pancreatic cancer stem cells and therapeutic approaches. Anticancer Res 2017;37:2761–75. [DOI] [PubMed] [Google Scholar]
- 81.Venkatesh V, Nataraj R, Thangaraj GS, Karthikeyan M, Gnanasekaran A, et al. Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig 2018;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xu YF, Hannafon BN, Ding WQ. MicroRNA regulation of human pancreatic cancer stem cells. Stem Cell Investig 2017;4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bimonte S, Barbieri A, Leongito M, Palma G, Del Vecchio V, et al. The role of miRNAs in the regulation of pancreatic cancer stem cells. Stem Cells Int 2016;2016:8352684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee HM, Hwang KA, Choi KC. Diverse pathways of epithelial mesenchymal transition related with cancer progression and metastasis and potential effects of endocrine disrupting chemicals on epithelial mesenchymal transition process. Mol Cell Endocrinol 2017;457:103–13. [DOI] [PubMed] [Google Scholar]
- 85.Chen X, Xiao W, Liu X, Zeng M, Luo L, et al. Blockade of Jagged/Notch pathway abrogates transforming growth factor β2-induced epithelial-mesenchymal transition in human retinal pigment epithelium cells. Curr Mol Med 2014;14:523–34. [DOI] [PubMed] [Google Scholar]
- 86.Wang Y, Shi J, Chai K, Ying X, Zhou BP. The role of Snail in EMT and tumorigenesis. Curr Cancer Drug Targets 2013;13:963–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jiang X, Hao HX, Growney JD, Woolfenden S, Bottiglio C, et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci USA 2013;110:12649–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity. Int J Oncol 2017;51:1357–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Onishi H, Katano M. Hedgehog signaling pathway as a new therapeutic target in pancreatic cancer. World J Gastroenterol 201;20:2335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ma Y, Yu W, Shrivastava A, Alemi F, Lankachandra K, et al. Sanguinarine inhibits pancreatic cancer stem cell characteristics by inducing oxidative stress and suppressing sonic hedgehog-Gli-Nanog pathway. Carcinogenesis 2017;38:1047–56. [DOI] [PubMed] [Google Scholar]
- 91.Hebrok M Hedgehog signaling in pancreas development. Mech Dev 2003;120:45–57. [DOI] [PubMed] [Google Scholar]
- 92.Yamasaki A, Onishi H, Imaizumi A, Kawamoto M, Fujimura A, et al. Protein-bound polysaccharide-K inhibits Hedgehog signaling through down-regulation of MAML3 and RBPJ transcription under hypoxia, suppressing the malignant phenotype in pancreatic cancer. Anticancer Res 2016;36:3945–52. [PubMed] [Google Scholar]
- 93.Chiorean EG, Coveler AL. Pancreatic cancer: optimizing treatment options, new, and emerging targeted therapies. Drug Des Devel Ther 2015;9:3529–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lei J, Ma J, Ma Q, Li X, Liu H, et al. Hedgehog signaling regulates hypoxia induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand-independent manner. Mol Cancer 2013;12:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang F, Ma L, Zhang Z, Liu X, Gao H, et al. Hedgehog signaling regulates epithelial-mesenchymal transition in pancreatic cancer stem-like cells. J Cancer 2016;7:408–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Crawford HC, Pasca di Magliano M, Banerjee S. Signaling networks that control cellular plasticity in pancreatic tumorigenesis, progression, and metastasis. Gastroenterology 2019;156:2073–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cui J, Jiang W, Wang S, Wang L, Xie K. Role of Wnt/β-catenin signaling in drug resistance of pancreatic cancer. Curr Pharm Des 2012;18:2464–71. [DOI] [PubMed] [Google Scholar]
- 98.Tanase CP, Neagu AI, Necula LG, Mambet C, Enciu AM, Cancer stem cells: involvement in pancreatic cancer pathogenesis and perspectives on cancer therapeutics. World J Gastroenterol 2014;20:10790–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yao H, Ashihara E, Maekawa T. Targeting the Wnt/β-catenin signaling pathway in human cancers. Expert Opin Ther Targets 2011;15:873–87. [DOI] [PubMed] [Google Scholar]
- 100.Bailey JM, Alsina J, Rasheed ZA, McAllister FM, Fu YY, et al. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology 2014;146:245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang Z, Ahmad A, Li Y, Azmi AS, Miele L, et al. Targeting notch to eradicate pancreatic cancer stem cells for cancer therapy. Anticancer Res 2011;31:1105–13. [PubMed] [Google Scholar]
- 102.Ma J, Xia J, Miele L, Sarkar FH, Wang Z. Notch signaling pathway in pancreatic cancer progression. Pancreat Disord Ther 2013;3:1000114. [PMC free article] [PubMed] [Google Scholar]
- 103.Güngör C, Hofmann BT, Wolters-Eisfeld G, Bockhorn M. Pancreatic cancer. Br J Pharmacol 2014;171:849–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yabuuchi S, Pai SG, Campbell NR, de Wilde RF, De Oliveira E, et al. Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett 2013;335:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang Y, Jiang F, Jiao K, Ju L, Liu Q, et al. De-methylation of miR-148a by arsenic trioxide enhances sensitivity to chemotherapy via inhibiting the NF-κB pathway and CSC like properties. Exp Cell Res 2020;386:111739. [DOI] [PubMed] [Google Scholar]
- 106.Zakaria N, Mohd Yusoff N, Zakaria Z, Widera D, Yahaya BH, et al. Inhibition of NF-κB signaling reduces the stemness characteristics of lung cancer stem cells. Front Oncol 2018;8:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang L, Wang D, Li Y, Liu Y, Xie X, et al. CCL21/CCR7 axis contributed to CD133+ pancreatic cancer stem-like cell metastasis via EMT and Erk/NF-κB pathway. PLoS One 2016;11:e0158529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nomura A, Gupta VK, Dauer P, Sharma NS, Dudeja V, et al. NFκB-mediated Invasiveness in CD133+ pancreatic TICs is regulated by autocrine and paracrine activation of IL1 signaling. Mol Cancer Res 2018;16:162–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang Z, Duan Q, Zhao H, Liu T, Wu H, et al. Gemcitabine treatment promotes pancreatic cancer stemness through the Nox/ROS/NF-κB/STAT3 signaling cascade. Cancer Lett 2016;382:53–63. [DOI] [PubMed] [Google Scholar]
- 110.Lai SW, Bamodu OA, Tsai WC, Chang YM, Lee WH, et al. The therapeutic targeting of the FGFR1/Src/NF-κB signaling axis inhibits pancreatic ductal adenocarcinoma stemness and oncogenicity. Clin Exp Metastasis 2018;35:663–77. [DOI] [PubMed] [Google Scholar]
- 111.Xu H, Zhang L, Qian X, Zhou X, Yan Y, et al. GSK343 induces autophagy and downregulates the AKT/mTOR signaling pathway in pancreatic cancer cells. Exp Ther Med 2019;18:2608–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhou HY, Yao XM, Chen XD, Tang JM, Qiao ZG, et al. Mechanism of metformin enhancing the sensitivity of human pancreatic cancer cells to gemcitabine by regulating the PI3K/Akt/mTOR signaling pathway. Eur Rev Med Pharmacol Sci 2019;23:10283–9. [DOI] [PubMed] [Google Scholar]
- 113.Meng Q, Liang C, Hua J, Zhang B, Liu J, et al. A miR-146a-5p/TRAF6/NF-kB p65 axis regulates pancreatic cancer chemoresistance: functional validation and clinical significance. Theranostics 2020;10:3967–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yoshizawa N, Sugimoto K, Tameda M, Inagaki Y, Ikejiri M, et al. miR-3940–5p/miR-8069 ratio in urine exosomes is a novel diagnostic biomarker for pancreaticductal adenocarcinoma. Oncol Lett. 2020;19:2677–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shams R, Asadzadeh Aghdaei H, Behmanesh A, Sadeghi A, Zali M, et al. MicroRNAs targeting MYC expression: trace of hope for pancreatic cancer therapy. A systematic review. Cancer Manag Res 2020;12:2393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Han W, Cui H, Liang J, Su X. Role of MicroRNA-30c in cancer progression. J Cancer 2020;11:2593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jung DE, Wen J, Oh T, Song SY. Differentially expressed microRNAs in pancreatic cancer stem cells. Pancreas 2011;40:1180–7. [DOI] [PubMed] [Google Scholar]
- 118.Ji Q, Hao X, Zhang M, Tang W, Yang M, et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One. 2009;4:e6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol 2009;11:1487–95. [DOI] [PubMed] [Google Scholar]
- 120.Smith BN, Bhowmick NA. Role of EMT in metastasis and therapy resistance. J Clin Med 2016;5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Beuran M, Negoi I, Paun S, Ion AD, Bleotu C, et al. The epithelial to mesenchymal transition in pancreatic cancer: a systematic review. Pancreatol 2015;15:217–25. [DOI] [PubMed] [Google Scholar]
- 122.Edderkaoui M, Chheda C, Soufi B, Zayou F, Hu RW, et al. An inhibitor of GSK3B and HDACs kills pancreatic cancer cells and slows pancreatic tumor growth and metastasis in mice. Gastroenterology 2018;155:1985–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li K, Chen Y, Li A, Tan C, Liu X. Exosomes play roles in sequential processes of tumor metastasis. Int J Cancer 2019;144:1486–95. [DOI] [PubMed] [Google Scholar]
- 124.Rodriguez-Aznar E, Wiesmüller L, Sainz B Jr, Hermann PC. EMT and stemness-Key players in pancreatic cancer stem cells. Cancers (Basel) 2019;11:1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Banys-Paluchowski M, Reinhardt F, Fehm T. Disseminated tumor cells and dormancy in breast cancer progression. Adv Exp Med Biol 2020;1220:35–43. [DOI] [PubMed] [Google Scholar]
- 126.Dianat-Moghadam H, Azizi M, Eslami-S Z, Cortés-Hernández LE, Heidarifard M, et al. The role of circulating tumor cells in the metastatic cascade: biology, technical challenges, and clinical relevance. Cancers (Basel) 2020;12:867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Aiello NM, Bajor DL, Norgard RJ, Sahmoud A, Bhagwat N, et al. Metastatic progression is associated with dynamic changes in the local microenvironment. Nat Commun 2016;7:12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014;15:178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012;148:349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009;119:1420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Forte E, Chimenti I, Rosa P, Angelini F, Pagano F, et al. EMT/MET at the crossroad of stemness, regeneration and oncogenesis: the ying-yang equilibrium recapitulated in cell spheroids. Cancers (Basel) 2017;9;98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Procacci P, Moscheni C, Sartori P, Sommariva M, Gagliano N. Tumor-stroma cross-talk in human pancreatic ductal adenocarcinoma: a focus on the effect of the extracellular matrix on tumor cell phenotype and invasive potential. Cells 2018;7;158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Begum A, Ewachiw T, Jung C, Huang A, Norberg KJ, et al. The extracellular matrix and focal adhesion kinase signaling regulate cancer stem cell function in pancreatic ductal adenocarcinoma. PLoS One 2017;12:e0180181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pitarresi JR, Rustgi AK. Mechanisms underlying metastatic pancreatic cancer. Adv Exp Med Biol 2019;1164:3–10. [DOI] [PubMed] [Google Scholar]
- 135.Lawlor RT, Veronese N, Nottegar A, Malleo G, Smith L, et al. Prognostic role of high-grade tumor budding in pancreatic ductal adenocarcinoma: a systematic review and meta-analysis with a focus on epithelial to mesenchymal transition. Cancers (Basel) 2019;11;113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pelosi E, Castelli G, Testa U. Pancreatic cancer: molecular characterization, clonal evolution and cancer stem cells biomedicines. 2017;5;65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kai F, Drain AP, Weaver VM. The extracellular matrix modulates the metastatic journey, Dev Cell 2019;49:332–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Najafi M, Farhood B, Mortezaee K. Cancer stem cells (CSCs) in cancer progression and therapy. J Cell Physiol 2019;234:8381–95. [DOI] [PubMed] [Google Scholar]
- 139.Celià-Terrassa T, Kang Y. Distinctive properties of metastasis-initiating cells Genes Dev 2016;30:892–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Agnoletto C, Corrà F, Minotti L, Baldassari F, Crudele F, et al. Heterogeneity in circulating tumor cells: the relevance of the stem-cell subset. Cancers (Basel) 2019;11:483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Giancotti FG. Mechanisms governing metastatic dormancy and reactivation. Cell 2013;155:750–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 2014;14:611–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yang L, Shi P, Zhao G, Xu J, Feng W, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Crawford HC, Pasca di Magliano M, Banerjee S. Signaling networks that control cellular plasticity in pancreatic tumorigenesis, progression, and metastasis gastroenterology. Gastroenterology 2019;156:2073–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hindriksen S, Bijlsma MF. Cancer stem cells, EMT, and developmental pathway activation in pancreatic tumors cancers (Basel) 2012;4:989–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen C, Zhao S, Karnad A, Freeman JW. The biology and role of CD44 in cancer progression: therapeutic implications J Hematol Oncol 2018;11:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhao S, Chen C, Chang K, Karnad A, Jagirdar J, et al. CD44 expression level and isoform contributes to pancreatic cancer cell plasticity, invasiveness and response to therapy. Clin Cancer Res 2016;22:5592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nielsen MFB, Mortensen MB, Detlefsen S. Typing of pancreatic cancer-associated fibroblasts identifies different subpopulations. World J Gastroenterol 2018;24:4663–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Stokes JB1, Adair SJ, Slack-Davis JK, Walters DM, Tilghman RW, et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther 2011;10:2135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Miyazaki K, Oyanagi J, Hoshino D, Togo S, Kumagai H, et al. Cancer cell migration on elongate protrusions of fibroblasts in collagen matrix. Sci Rep 2019;9:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sharma N, Nanta R, Sharma J, Gunewardena S, Singh KP. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget 2015;6:32039–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang F, Li H, Yan XG, Zhou ZW, Yi ZG, et al. Alisertib induces cell cycle arrest and autophagy and suppresses epithelial-to-mesenchymal transition involving PI3K/Akt/mTOR and sirtuin 1-mediated signaling pathways in human pancreatic cancer cells. Drug Des Devel Ther 2015;9:575–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Makena DP, Gatla H, Verlekar D, Sukhavasi S, Pandey MK, et al. Wnt/β-Catenin signaling: the culprit in pancreatic carcinogenesis and therapeutic resistance. Int J Mol Sci 2019;20:4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Razi E, Radak M, Mahjoubin-Tehran M, Talebi S, Shafiee A, et al. Cancer stem cells as therapeutic targets of pancreatic cancer. Fundam Clin Pharmacol. 2020;34:202–12. [DOI] [PubMed] [Google Scholar]
- 155.Okada M, Shibuya K, Sato A, Seino S, Suzuki S, et al. Targeting the K-Ras--JNK axis eliminates cancer stem-like cells and prevents pancreatic tumor formation. Oncotarget 2014;5:5100–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wang Z, Ali S, Banerjee S, Bao B, Li Y, et al. Activated K-Ras and INK4a/Arf deficiency promote aggressiveness of pancreatic cancer by induction of EMT consistent with cancer stem cell phenotype. J Cell Physiol 2016;231:2304. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 157.Fitzgerald TL, Lertpiriyapong K, Cocco L, Martelli AM, Libra M, et al. Roles of EGFR and KRAS and their downstream signaling pathways in pancreatic cancer and pancreatic cancer stem cells. Adv Biol Regul 2015;59:65–81. [DOI] [PubMed] [Google Scholar]
- 158.Zhang B, Ye H, Ren X, Zheng S, Zhou Q, et al. Macrophage-expressed CD51 promotes cancer stem cell properties via the TGF-β1/smad2/3 axis in pancreatic cancer. Cancer Lett 2019;10;459:204–15. [DOI] [PubMed] [Google Scholar]