

Abstract
Cocaine exerts its stimulant effect by inhibiting dopamine reuptake leading to increased dopamine signaling. This action is thought to reflect binding of cocaine to the dopamine transporter (DAT) to inhibit its function. However, cocaine is a relatively weak inhibitor of DAT, and many DAT inhibitors do not share the behavioral actions of cocaine. We previously showed that toxic levels of cocaine induce autophagic neuronal cell death. Here, we show that subnanomolar concentrations of cocaine elicit neural autophagy in vitro and in vivo. Autophagy inhibitors reduce the locomotor stimulant effect of cocaine in mice. Cocaine-induced autophagy degrades transporters for dopamine but not serotonin in the nucleus accumbens. Autophagy inhibition impairs cocaine conditioned place preference in mice. Our findings indicate that autophagic degradation of DAT modulates behavioral actions of cocaine.
Introduction
The behavioral stimulant actions of cocaine are thought to involve inhibition of the reuptake inactivation of neurotransmitters such as dopamine (DA), norepinephrine, and serotonin, actions that typically occur at micromolar concentrations of the drug [1–3]. Cocaine binds numerous proteins, which may sequester the drug from its targets and influence behavioral [4] or neurotoxic actions [5, 6].
We obtained evidence implicating autophagy in the neurotoxicity of cocaine [5]. First identified by Clark [7] and characterized by Deter and de Duve [8], autophagy is a lysosomal process that recycles proteins and organelles to maintain homeostasis and influence cell signaling by generating autophagosomes [9]. The cytosolic microtubule-associated protein 1A/1B-light chain 3 (LC3-I) is conjugated to phosphatidylethanolamine to form LC3-II, which tags the autophagosomal membranes. Autophagosomes fuse with lysosomes to form autolysosomes, whose hydrolases degrade the autophagic cargo [10].
In the present study, we show that cocaine, at nontoxic subnanomolar concentrations, elicits autophagy in neurons. Autophagy appears to modulate behavioral actions of the drug, which are blocked by autophagy inhibitors. Cocaine degrades the DA transporter (DAT) in the nucleus accumbens (NAc), an action prevented by autophagy inhibitors.
Materials and methods
Reagents
Neurobasal-A medium (no glucose, no sodium pyruvate), B-27 supplement, and B-27 supplement minus antioxidants were purchased from Life Technologies (Grand Island, NY). Bafilomycin A1 and SBI-0206965 were purchased from Cayman Chemical (Ann Arbor, Michigan). Cocaine hydrochloride and hydroxychloroquine (HCQ) sulfate were purchased from Sigma-Aldrich (St. Louis, MO). Vacuolin-1 was purchased from Santa Cruz Biotechnology (Paso Robles, CA). Cocaine hydrochloride (used in the DA microdialysis experiments) was obtained from Mallinckrodt (St. Louis, MO). Deuterated cocaine (cocaine-d3; internal standard) was obtained from Torrent research chemicals (ON, Canada). LC–MS grade water, acetonitrile, and formic acid were obtained from Thermo Fisher Scientific (Waltham, MA, USA).
Antibodies
Anti-beta actin-HRP, catalog number 5125S, and anti-phospho-ULK Ser555, catalog number 5869, were purchased from Cell Signaling Technology (Danvers, MA). Anti-DAT (for western blot analysis), anti-N-terminus, catalog number sc-32258, anti-ULK1 antibody, catalog number sc-33182, and anti-EGFR, catalog number: sc-03-G, were purchased from Santa Cruz Biotechnology (Paso Robles, CA). Anti-serotonin transporter (SERT), catalog number PA1706, was purchased from Boster Bio (Pleasanton, CA). Anti-tyrosine hydroxylase (TH), catalog number 818001, was purchased from BioLegend (San Diego, CA). Anti-DAT (anti-C-terminus), catalog number AB1766, and anti-DAT for immunostaining, catalog number MAB369, were purchased from Millipore Sigma (Burlington, MA). Anti-VPS34, catalog number 3811S, and anti-phospho-VPS34 Ser249, catalog number 13857S, for western blot were purchased from Cell Signaling Technology (Danvers, MA). LAMP2A antibody was purchased from (Thermo Fisher Scientific, USA). GFP antibody, catalog number ab13970 was pruchased from Abcam (USA).
Animals
All experiments involving animals were conducted in accordance with the Johns Hopkins Medical Institutions Animal Care and Use Committee guidelines. C57Bl/6j adult mice were purchased from The Jackson Laboratory (Bar Harbor, ME). CD-1 timed pregnant mice were purchased from Charles River Laboratories. GFP-LC3 mice (No. RBRC00806) were a kind gift from Dr. Mizushima Noboru, provided by the RIKEN BRC through the National Bio-Resource Project of the MEXT, Japan [11]. Animal handling and procedures were conducted in accordance with the National Institutes of Health guidelines for use of experimental animals and the Johns Hopkins animal care and use guidelines. To allocate mice into experimental groups, we used a stratified randomization method. For sample size determination, we used the sample size calculator at ClinCalc. com (http://clincalc.com/Stats/SampleSize.aspx). Continuous endpoint, two independent sample study design was used with the following study parameters: probability of a type-I error = 0.05, probability of a type-II error = 0.1, power = 0.9. Classification and counting of autophagic vacuoles in mice NAc were done by double-blinded independent observers. Also, in primary neurons derived from GFP-LC3 mice, the number of puncta was quantified in a blinded fashion.
Transmission electron microscopy (TEM)
Primary cortical cultures (PCCs) were treated with indicated concentrations of cocaine and bafilomycin. The cells were fixed in 2.5% glutaraldehyde, 3 mM MgCl2, in 0.1 M sodium cacodylate buffer, pH 7.2, for 1 h at room temperature. After buffer rinse, samples were postfixed in 1% osmium tetroxide in buffer (1 h) on ice in the dark. The cells were stained with 2% aqueous uranyl acetate (0.22 μm filtered, 1 h in the dark), dehydrated in a graded series of ethanol solutions and embedded in Eponate 12 (Ted Pella) resin. Samples were polymerized at 37 °C for 2–3 days before moving to 60 °C overnight. Thin sections, 60–90 nm, were cut with a diamond knife on a Reichert-Jung Ultracut E Ultramicrotome and picked up with 2 × 1 mm copper slot grids. Grids were stained with 2% uranyl acetate in 50% methanol and lead citrate at 4 °C and observed with a Hitachi 7600 TEM. Images were captured with an AMT CCD XR50 (2 K × 2 K) camera. Classification and counting of autophagic vacuoles were done by double-blinded independent observers.
Confocal microscopy of primary neural cultures
We used a ZEISS LSM 800 confocal microscopy to image the primary cortical cultures isolated from GFP-LC3 mice. The cells were treated with different concentrations of cocaine. The LC3 GFP puncta were analyzed and counted using Imaris x64 7.7.2 software.
Primary neural cultures
Unless otherwise specified, PCCs were isolated from E15–E18 pregnant CD-1 mice. The pregnant mouse was sacrificed by decapitation, then the uterus was dissected out immediately. Working in sterile conditions, the uterus was opened, the pups were decapitated, and their brains were dissected and placed in dissection media containing DMEM/F12 1:1 supplemented with 10% horse serum. The cerebral cortices were detached from the rest of the brain, then the meninges were removed. The cortices were incubated in 0.025% trypsin for 15 min at 37 °C. The trypsin was washed with dissection media. The cortices were disrupted into single-cell suspension by pipetting up and down ten times, then strained through a 40 μm sterile mesh. The single-cell suspension was cultured in dissection media overnight. Then, the media were replaced by PCCs plating media containing neurobasal-A medium (no glucose, no sodium pyruvate) supplemented with 12.5 mM glucose, 2 mM l-glutamine and 2% B-27. On day 4 in vitro, the media were changed to PCCs maintenance media containing neurobasal-A medium (no glucose, no sodium pyruvate) supplemented with 12.5 mM glucose, 2 mM l-glutamine and 2% B-27 minus antioxidants. Every 3 days thereafter, 50% of the media were replaced with PCCs maintenance media. Postnatal ventral midbrain cultures were prepared as previously described [12] except that the following media were used. Days 1–2: 47.25% MEM, 40% DMEM, 10% F12, 0.25% BSA, 0.5% l-glutamine, and 2% B-27. After day 2: 47.25% MEM, 40% DMEM, 10% F12, 0.25% BSA, 0.5% l-glutamine, 2% B-27 minus antioxidants, and 10 ng/ml GDNF.
Brain tissue processing and immunostaining
Mice were anesthetized with intraperitoneal injection of sodium pentobarbital (80 mg/kg), then the heart was exposed, and transcardiac perfusion was performed using phosphate buffered saline (PBS) for 5 min followed by ice-cold 4% paraformaldehyde (PFA) in PBS for 30 min (using 3 ml/min rate of perfusion). The brains were then dissected and cryoprotected with 30% sucrose in PBS for 24 h at 4 °C. Brains were sectioned on a freezing stage sliding microtome into a series of 40 μm sections. Sections were postfixed in 4% PFA for 5 min, permeabilized with 0.5% Triton X-100 in Tris buffered saline, then blocked with 5% normal goat serum. Rat anti-DAT, rabbit anti-TH, and chicken anti-GFP were used as primary antibodies. Anti-rat IgG (H + L) Alexa Fluor 555 conjugate, anti-rabbit IgG (H + L) Alexa Fluor 647 conjugate, and anti-chicken IgG (H + L) Alexa Fluor 488 conjugate were used as secondary antibodies. ZEISS LSM 800 confocal microscopy was used to image immunostained sections. Quantification of the intensity of the fluorescence signal was performed using the NIH ImageJ software. Fluorescence colocalization analysis was performed using Imaris software.
Stereotaxic injection
Stereotaxic injections were performed as previously described with modifications [13]. C57 BL6/j male mice (age range: 10–12 weeks) were used. Mice were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine. Stoelting’s stereotaxic instrument for mice was used to perform the procedure. The skull was exposed, and burr holes were made at the stereotaxic coordinates specified below. Bilateral injections into the NAc were performed using the following stereotaxic coordinates: +1.4 (anterior/ posterior), +/−1.3 (medial/lateral), and −4.3 (dorsal/ventral). One microliter of saline or cocaine (containing 10 fmol) was delivered on each side followed by 5 min pause before removing the needle.
Open field test
Open field activity monitoring was performed in an open field Plexiglas chamber with photocell emitters and receptors to detect locomotor activity using a grid of invisible infrared beams. The chambers were enclosed in illuminated opaque boxes and connected to a computer for beam break data collection. C57 BL6/j male mice (age range:10–16 weeks) were used. The mice were given intraperitoneal injections of HCQ, vacuolin-1, SBI-0206965, or saline 45 min before the test. Then, mice were placed in the chambers for 45 min to detect baseline activity. After baseline monitoring, mice were given intraperitoneal injections of cocaine (20 mg/kg) or saline and returned to the chambers for 75 min to monitor their locomotor behavior. Baseline activity was calculated based on the initial 45 min. The effect of cocaine on locomotor activity was calculated based on the 15 min following cocaine injection (peak activity). Different animals were used for the behavioral and biochemical tests.
Isolation of lysosomes
Lysosomes were isolated as previously described [14] with minor modifications. Briefly, ventral midbrains were homogenized using Potter-Elvehjem tissue grinder with PTFE pestle (Fisher Scientific, Pittsburgh, PA) in homogenization buffer (HB) containing 0.326 M sucrose, 1 mM EDTA, 10 mM HEPES, pH 7.3, 0.5 μg/ml antipain, 1 μg/ ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml chymostatin, and 1 μg/ml pepstatin A. The tissue homogenates were centrifuged at 750 × g for 10 min at 4 °C. The postnuclear supernatant was centrifuged at 20,000 × g for 10 min at 4 °C in Sorvall RC series centrifuge and SS-34 rotor. The pellet was resuspended in HB and centrifuged at 20,000 × g for 10 min at 4 °C in Sorvall RC series centrifuge and SS-34 rotor. The pellet was resuspended in HB and mixed with 50% iodixanol then bottom loaded into a 19–27% linear iodixanol gradient. The gradient was centrifuged at 110,000 × g for 120 min at 4 °C. The lysosomal band at the top of the grad0069ent was collected and diluted with ten volumes of HB then centrifuged at 30,000 × g for 20 min at 4 °C. The pellet was resuspended and used for western blot analysis.
Microdialysis studies
Experimentally naive, male Swiss-Webster mice (Charles River, Wilmington, MA), weighing 25–30 g at the start of the study, were housed four per cage in a temperature and humidity-controlled facility on a 12-h light/dark cycle (lights on from 7:00 a.m. to 7:00 p.m.), where they were kept for at least a week before starting surgical procedures. They had free access to food and water at all times except during microdialysis test sessions. The present study was approved by the Animal Care and Use Committee of the National Institute on Drug Abuse, Intramural Research Program, Baltimore, MD, USA. Care of the subjects was in accordance with the guidelines of the National Institutes of Health and the National Institute on Drug Abuse Intramural Research Program, which is fully accredited by AAALAC International. Mice were anesthetized under a mixture of xylazine (10 mg/kg, i.p.) and ketamine (60 mg/kg, i.p.). They were then placed in a stereotaxic apparatus, the skull was exposed, and a small hole was drilled to expose the dura. Concentric dialysis probes (dialyzing surface of 1.0 mm), prepared with AN69 fibers (Hospal Dasco, Bologna, Italy) as described previously [15–17], were randomly implanted in the right or left NAc shell, uncorrected coordinates [18]: anterior = +1.5, lateral = ±0.6, vertical = −5.1, in millimeters from bregma. After surgery, mice recovered in square Plexiglas cages with bedding on the floor that were equipped with overhead quartz-lined fluid swivels (375/D/22QM; Instech Laboratories, Plymouth Meeting, PA, USA) for connections to the dialysis probes. Microdialysis test sessions in freely moving mice started ~42–47 h after the surgical procedures. Probes were connected to fluid swivels and perfused with Ringer’s solution at a constant flow rate of 1 μl/min as described [15–17]. Collection of dialysate samples (10 μl) started after about 30 min, and samples collected every 10 min were immediately analyzed for DA content. After establishment of a DA baseline, 2–4 samples differing no more than 20%, different groups of naive mice were pretreated with the test compounds or their vehicle and after 90 min all groups received cocaine, 20 mg/kg i.p. Sample collection continued every 10 min up to 2 h after cocaine administration. Dialysate samples (10 μl) were then injected without purification into a high-performance liquid chromatography (HPLC) apparatus in order to detect and quantify extracellular DA levels as detailed previously [15–17]. Assay sensitivity for DA was 2 fmol/sample. After the end of the tests, brains were collected, kept in 4% formalin for at least a week, and then cut on a vibratome in serial coronal slices oriented according to the mice brain atlas [18] to identify the location of the probes. In accordance with previous studies [15–17], only data from animals showing appropriate location of probes inside the NAc shell have been used.
Statistical analysis
Results were expressed as a percentage of basal DA values, calculated as the mean of the last 2–4 consecutive samples immediately preceding the first drug or vehicle injection. All results are presented as group means (±SEM). Differences in basal levels of DA between different experimental groups within the same brain area, or between different brain areas, were analyzed by one-way analysis of variance (ANOVA). Statistical analyses of experimental data were carried out with Statistica 6 software using ANOVA for repeated measures over time, with results from treatments showing overall changes subjected to Duncan’s post hoc test. Results were considered significant at p < 0.05.
Bioanalysis of cocaine in in microdialysis samples
Quantification of cocaine in dialysate (in vitro and in vivo) was performed by LC–MS/MS. Briefly, standard curves were prepared in ringers buffer ranging from 0.5 to 5000 nM. Prior to extraction, frozen samples were thawed on ice. Twenty microliters of sample or standard was extracted with 150 μl of acetonitrile containing cocaine-d3 (300 nM) as internal standard, followed by vortexing for 30 s and centrifugation at 16,000 × g for 5 min at 4 °C. The resulting supernatants were transferred to a 96-well plate and analyzed on an Accela 1250 HPLC pump and Accela Open Autosampler coupled to TSQ Vantage mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). The mobile phase used for chromatographic separation consisted of 0.1% formic acid in water and 0.1% formic acid in acetonitrile, delivered at a flow rate of 0.3 ml/min using gradient LC method. Separation of analytes was achieved at room temperature with a Newcrom R1 2.1 × 100 mm, 3 μm particle size column (SIELC technologies, Wheeling, IL, USA), in positive mode, using selected reaction monitoring function. The [M + H]+ ion transitions of cocaine at m/z 304.1→182.1 and that of the internal standard at m/z 307.1→185.1 were monitored. Data were acquired and quantified with Xcalibur 2.2.
Calibration curves for cocaine were computed based on peak area ratio (area of analyte/area of IS) using a linear regression analysis with 1/[nominal concentration]2 × weighting factor. The calibration curve correlation coefficients (R2) ≥ 0.9900 were considered acceptable for all analytical runs. The acceptance criteria for each back-calculated standard concentration are ±15% deviation from the nominal value except at the LLOQ which is ±20%. Percent Recovery obtained after in vitro dialysis of cocaine at various concentration levels Following in vitro micro-dialysis, average recovery obtained was 17 ± 4%. The standard concentration was half the nominal concentration and was used for recovery calculations.
Synaptosome fraction preparation and DAT western blot
Crude synaptosome fractions were performed as previously described [19] with minor modifications. Briefly, NAc from mice were dissected and homogenized in a buffer containing 0.32 M sucrose, 20 mM HEPES (pH 7.3), 0.5 μg/ml antipain, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml chymostatin, and 1 μg/ml pepstatin A. The tissue homogenates were centrifuged at 1000 × g for 30 min at 4 °C. The supernatants were centrifuged at 3000 × g for 15 min at 4 °C. The supernatants from the second spin were centrifuged at 10,000 × g for 15 min at 4 °C. The pellets (the crude synaptosome fraction) were resuspended in cell extraction buffer (Thermo Fisher Scientific, USA) containing 10 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM Na4P2O7, 2 mM Na3VO4, 1% TX-100, 105 glycerol, 0.1% SDS, and 0.5% deoxycholate supplemented with 0.5 μg/ml antipain, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml chymostatin, and 1 μg/ml pepstatin A. The crude synaptosome fraction lysates were used for western blot analysis. Western blot was performed following standard procedures except for DAT experiments. For DAT western blot analysis, samples were not boiled before loading into the gels. Instead, samples were heated at 52 °C for 30 min then loaded into the gels. DAT western blots revealed a single band at ~70 KDa that was detected in striatal and midbrain lysates but not detected in lysates prepared from other areas of the brain.
[3H]DA uptake assay
[3H]DA uptake assays were performed as previously described [20] with modifications. Crude synaptosomal fractions were isolated from the NAc of saline- or cocaine-injected mice 30 min after injection, as described above. NAc tissue was combined from five mice per condition. Synaptosomes were incubated with uptake buffer containing 25 mM HEPES, 120 mM NaCl, 5 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 1 μM pargyline, 2 mg/ml glucose, and 0.2 mg/ml sodium ascorbate with or without 1 μM GBR-12935 (Santa Cruz Biotechnology) for 20 min at room temperature to account for nonspecific DA uptake. [3H]DA (Perkin Elmer; final concentration 20 nM) and unlabeled DA (Sigma-Aldrich; concentrations ranging from 0.08 to 2.5 μM) were added to the samples and incubated for 10 min at room temperature. Samples were then passed through polyethyleneimine-coated glass fiber filters and washed four times with ice-cold 50 mM NaCl. The radioactivity retained on filters after washing was determined using a Beckman Coulter LS6500 Liquid Scintillation Counter.
Statistical analysis
Statistical analysis was performed with Prism 7 (GraphPad Software Inc., San Diego, CA) using the alpha power level of 0.05. Two-tailed t-test was used for two group comparisons. One-way ANOVA was used to compare multiple groups followed by Holm–Sidak’s multiple comparisons post hoc test. Data were graphed as means± SEM. Km and Vmax values for [3H]DA uptake were calculated using nonlinear regression analysis, fitting the data to the Michaelis–Menten equation.
Results
Cocaine potently induces autophagy in neurons
In primary cortical and ventral midbrain neuronal cultures, cocaine elicits autophagy, monitored as levels of LC3-II, with extraordinary potency (Fig. 1a; Supplementary Fig. S1a). Microscopic imaging reveals augmentation of neural LC3 puncta at 0.1 nM levels of cocaine (Fig. 1b). Electron microscopy also reveals autophagic influences of 0.1 nM cocaine in primary cortical and ventral midbrain cultures (Fig. 1c, d; Supplementary Fig. S1b). Stereotaxic injection of 10 fmol of cocaine in NAc induces autophagic vacuoles in presynaptic axonal terminals within 15 min (Fig. 1e–g; Supplementary Fig. S1c). Intraperitoneal injection of cocaine also induces autophagic vacuoles at NAc axonal terminals (Supplementary Fig. S1d). Thus, cocaine potently elicits autophagy in neurons. To confirm the selectivity of the influence of cocaine upon autophagy, we monitored other autophagic markers including phospho-ULK1 at Ser555, which is increased fivefold in cortical (Fig. 2a, b) and ventral midbrain cultures (Supplementary Fig. S2a, b) at cocaine concentrations as low as 0.1 nM. Phosphorylation of ULK1 at Ser555 and LC3-II levels are also induced by cocaine in synaptosomes isolated from NAc (Supplementary Fig. S2c).
Fig. 1. Cocaine potently induces autophagy in neurons.
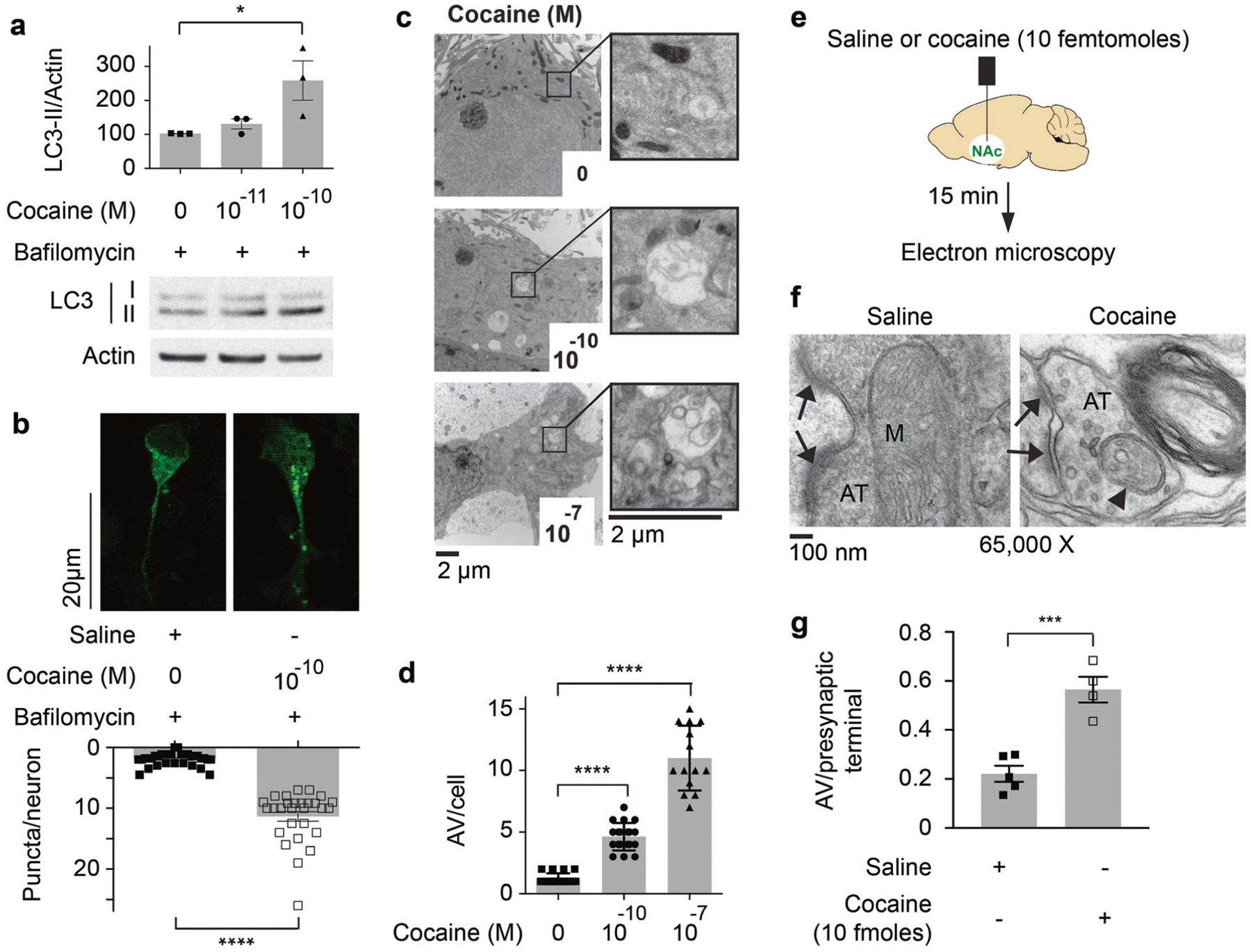
a Primary cortical cultures are treated with indicated concentrations of cocaine with bafilomycin (100 nM) for 30 min. Immunoblot was performed to analyze LC3 lipidation (LC3-II). The densitometric relative value of LC3-II/actin is shown and compared to bafilomycin (100 nM) treated cultures. (n = 3), *P = 0.039, one-way ANOVA, Holm–Sidak’s multiple comparisons test, P = 0.036. b Primary cortical cultures cultured from GFP-LC3 mice were treated with cocaine (100 pM) + bafilomycin (100 nM) and compared with bafilomycin (100 nM) treatment alone for 30 min, number of GFP positive puncta per neuron are shown. The number of puncta was quantified in a blinded fashion. ****P < 0.0001, two-tailed t test. Scale bar: 20 μm. c TEM of primary cortical cultures treated with different concentrations of cocaine and autophagic vacuoles are magnified. d Relative numbers of autophagic vacuoles (AV) per cell are plotted, n > 20, ****P < 0.0001, one-way ANOVA, Holm–Sidak’s multiple comparisons test, P < 0.0001. Scale bar: 2 μm. e A schematic diagram for the stereotaxic injection of cocaine in mice nucleus accumbens (NAc). Ten femtomoles of cocaine were injected. The expected concentration of cocaine in NAc following injection is 1 nM assuming a NAc volume of 10 μl. f TEM of induction of autophagic vacuoles in axonal terminals in NAc after cocaine injection. g The average number of autophagic vacuoles (AV) per presynaptic terminal in NAc is plotted. The number of AV was quantified in a blinded fashion. n = at least 4 mice per group. At least 75 synapses were quantified per mouse. ***P = 0.0006, two-tailed t test. Error bars = ±SEM.
Fig. 2. Autophagy regulates the locomotor stimulant effect of cocaine.
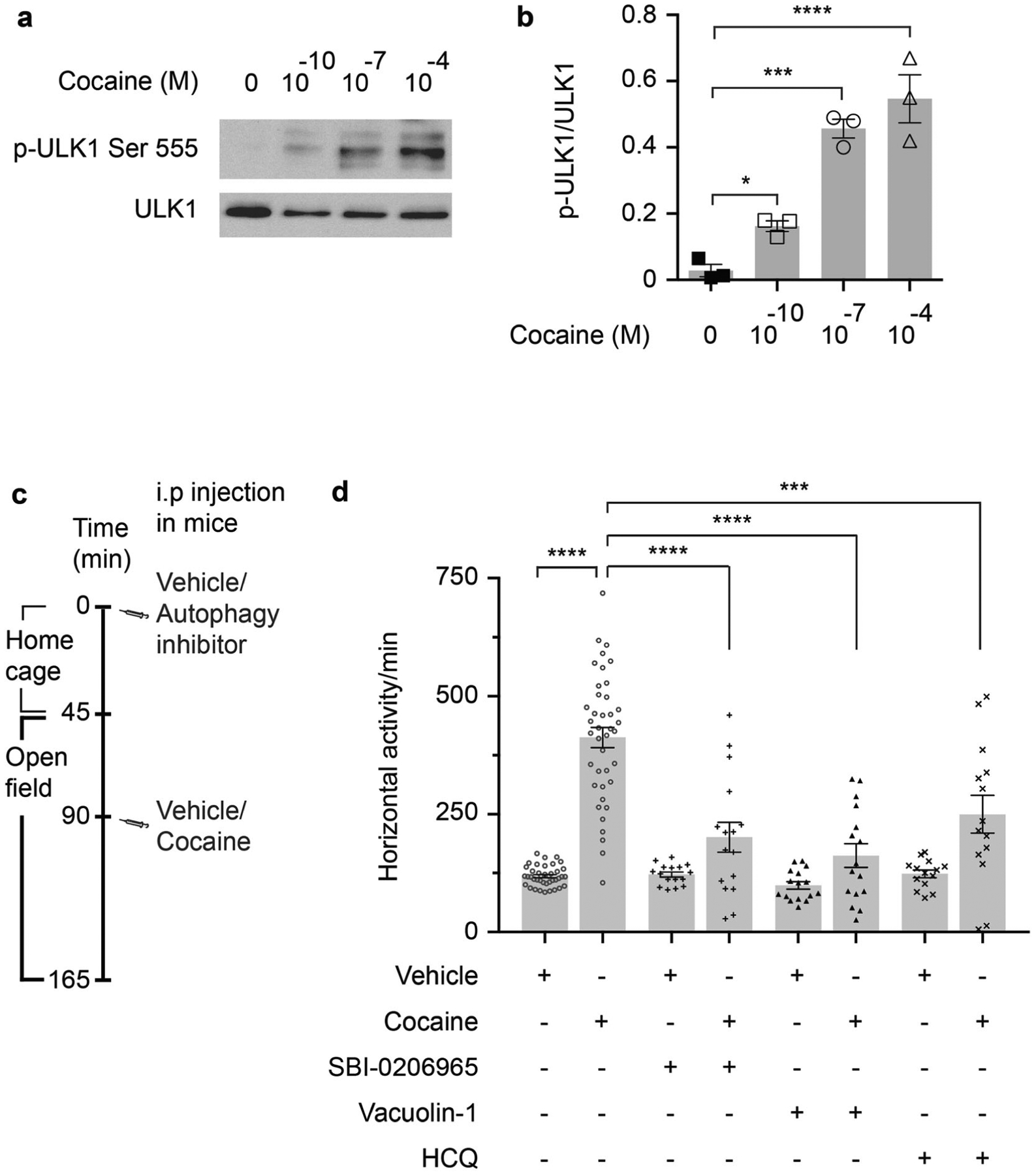
a Western blot of ULK1 phosphorylation at Ser555. b Quantification of western blot bands in a. n = 3, one-way ANOVA, P < 0.0001, Holm–Sidak’s multiple comparisons test, *P < 0.05, ***P < 0.001, ****P < 0.0001. c A schematic diagram of the open field behavior test experimental protocol. d Open field behavioral experiments before and after cocaine injection (20 mg/kg) with and without HCQ (2 mg/kg), vacuolin-1 (2 mg/kg), and SBI-0206965 (2 mg/kg). Vacuolin-1 inhibits the fusion between lysosomes and autophagosomes. SBI-0206965 is an inhibitor of ULK1. HCQ is a lysosomal inhibitor. Vehicle-cocaine n = 40, SBI-0206965 or vacuo-lin-1–cocaine n = 16, HCQ–cocaine n = 14, one-way ANOVA, P < 0.0001, Holm–Sidak’s multiple comparisons test, ****P < 0.0001, ***P = 0.0002. Error bars = ±SEM. Different animals were used for the behavioral and biochemical tests.
Autophagy regulates the locomotor stimulant effect of cocaine
We explored a role for autophagy in the behavioral stimulant influences of cocaine by treating mice with autophagy inhibitors such as SBI-0206965 (a highly selective ULK1 kinase inhibitor [21]) (Supplementary Fig. S2c, d), vacuolin-1 (inhibits autophagosome-lysosome fusion [22]) and HCQ (increases lysosomal pH and prevents autophagosomal content degradation [23]). All three drugs reduce the locomotor stimulation elicited by cocaine (Fig. 2c, d; Supplementary Fig. S2e). These results indicate that autophagy regulates the locomotor stimulant effects of cocaine.
Cocaine induces autophagic degradation of DAT
Dopaminergic neurotransmission mediates cocaine-stimulated locomotor activity. Elevated DA levels in NAc are implicated in the stimulant effect of cocaine [24]. We performed DA microdialysis to test whether pharmacologic inhibition of autophagy influences DA levels in NAc following treatment with cocaine. Basal levels of DA (57.6 ± 5.9 fmol/sample, n = 20) in dialysates from the NAc shell are not significantly different between experimental groups (ANOVA, F(3, 16) = 2.03, P = 0.15). Cocaine, 20 mg/kg i.p., enhances extracellular DA levels in dialysates from the NAc shell, when administered 90 min after saline or vehicle pretreatments [data for these effects have been reported as a single group] (Fig. 3a). DA levels rapidly peak (424%) 20 min after cocaine administration, followed by a slower decrease toward basal values. Cocaine administered 90 min after pretreatment with HCQ (2.5 mg/kg i.p.) or SBI-0206965 (2.5 mg/kg i.p.) produces a significantly lower increase in DA levels as compared to the control group (main effect treatment, F(2, 16) = 4.26, P < 0.05; main effect time, F(8, 128) = 20.45, P < 0.05; treatment by time interaction, F(16, 128) = 1.06, P = NS) (Fig. 3a). DA levels peak at 20 min, reaching ~292 and 337% for HCQ and SBI-0206965 treatment groups, respectively. After that, there is a more rapid decrease in stimulation of DA levels produced by cocaine in mice pretreated with HCQ or SBI-0206965 than in the control group (Fig. 3a). These findings indicate that autophagy contributes significantly to cocaine-stimulated DA levels in NAc.
Fig. 3. Cocaine induces autophagic degradation of the dopamine transporter (DAT).
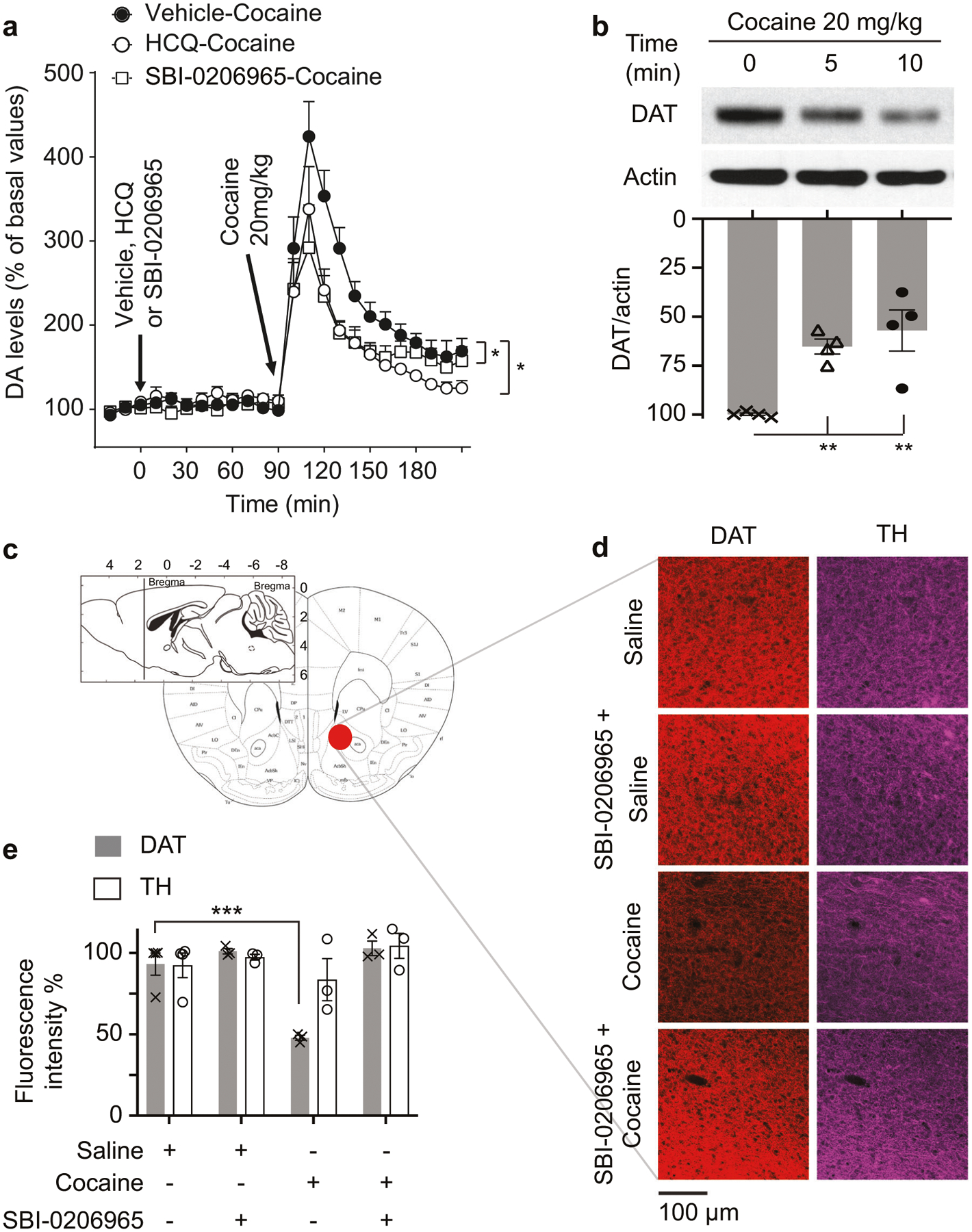
a Time course of dopamine (DA) levels from NAc dialysates before or after intraperitoneal injection of 20 mg/kg cocaine with and without 90 min pre injection with 2.5 mg/kg hydro-xychloroquine (HCQ) or 2.5 mg/kg ULK1 inhibitor SBI-0206965. b Western blot of NAc synaptosomes showing rapid DAT depletion following cocaine 20 mg/kg intraperitoneal injection; quantification of DAT/actin is plotted. n = 4, one-way ANOVA, P = 0.0026, Holm–Sidak’s multiple comparisons test, **P = 0.0043 and 0.0023, respectively. c A schematic diagram illustrating the sectioning and imaging area in NAc. Diagram was modified from Franklin and Paxinos [18]. d Confocal imaging of DAT and tyrosine hydroxylase (TH) in NAc. Mice were treated with saline or cocaine (20 mg/kg) ± SBI-0206965, followed by harvesting tissue and immunostaining for DAT and TH. DAT but not TH is rapidly depleted (30 min) following cocaine injection. Scale bar: 100 μm. e Quantification of immuno-fluorescence staining of DAT and TH in NAc. n = 3, one-way ANOVA, P < 0.0001, Holm–Sidak’s multiple comparisons test, ***P = 0.0003. Error bars = ±SEM.
Though cocaine impairs DA transport, heretofore there has been no evidence for a direct action of cocaine upon DAT. We observe a substantial depletion of DAT in synaptosomal fractions isolated from NAc in mice treated with 20 mg/kg cocaine (Fig. 3b; Supplementary Fig. S3a,b). This action of cocaine is highly potent, with cocaine at 0.1 mg/kg significantly depleting DAT in NAc synaptosomal fractions (Supplementary Fig. S3c, d). We confirm depletion of DAT in NAc synaptosomal fractions following cocaine injection using both anti-N-terminal and anti-C-terminal antibodies against DAT (Supplementary Fig. S3e). The depletion of DAT by cocaine is selective, as the drug does not alter the serotonin transporter (SERT) or tyrosine hydroxylase (TH) (Supplementary Fig. S3b, f, g). The autophagy inhibitors SBI-0206965 and HCQ, administered 90 min before cocaine treatment, rescue DAT in NAc (Fig. 3c–e; Supplementary Fig. S3f, g). The bulk of neural autophagy is initiated at the axonal terminals and completed via retrograde transport and fusion with lysosomes at the soma [25–27]. We explored the influence of cocaine on the accumulation of DAT in the lysosomes of the ventral midbrain after inhibiting lysosomal degradation. DAT levels are increased in the lysosomal fraction 6 h following i.p. cocaine injection (Supplementary Fig. S3h). The influence of cocaine on DAT is not due to a generalized effect upon endocytosis. Thus, endocytic downregulation of the epidermal growth factor (EGF) receptor in response to EGF is not altered by the autophagy inhibitor SBI-0206965, suggesting that SBI-0206965 does not impair endocytosis (Supplementary Fig. S3i). We examined the actions of another DAT inhibitor; bupropion on DAT depletion in NAc synaptosomal fractions. A behaviorally active dose of bupropion does not affect DAT protein levels in NAc synaptosomes (Supplementary Fig. S3j, k). Collectively, these data demonstrate that autophagy mediates cocaine-induced degradation of DAT.
Inhibiting autophagy impairs cocaine conditioned place preference (CPP)
Intraperitoneal administration of cocaine reduces the Vmax of [3H]DA uptake in striatal synaptosome fractions (Fig. 4a), suggesting a decrease in the number of DAT molecules. The Kd of DA binding to DAT increases somewhat following cocaine injection (Fig. 4b). Western blot analysis of synaptosomal fractions reveals diminished DAT protein levels (Fig. 4c). We measured cocaine by in vivo microdialysis in NAc. Cocaine levels peak at about 1 μM in NAc following 20 mg/kg drug doses (Supplementary Fig. S4a, b). Our data suggest that autophagy degrades DAT in response to cocaine (Fig. 3b–e; Supplementary Fig. S3b–i). We examined whether autophagy is induced in dopaminergic nerve terminals in the NAc. In saline-injected GFP-LC3 mice, there is no colocalization between GFPLC3 and TH reflected by a negative Pearson’s correlation coefficient (Supplementary Fig. S5a, b). In contrast, GFPLC3 and TH colocalize in NAc 3 min after intraperitoneal injection of 20 mg/kg cocaine reflected by a positive Pearson’s correlation coefficient (Supplementary Fig. S5a, b). We confirmed the purity of cocaine used in our studies using proton nuclear magnetic resonance (Supplementary Fig. S6a).
Fig. 4. The autophagy inhibitor hydroxychloroquine impairs cocaine conditioned place preference.
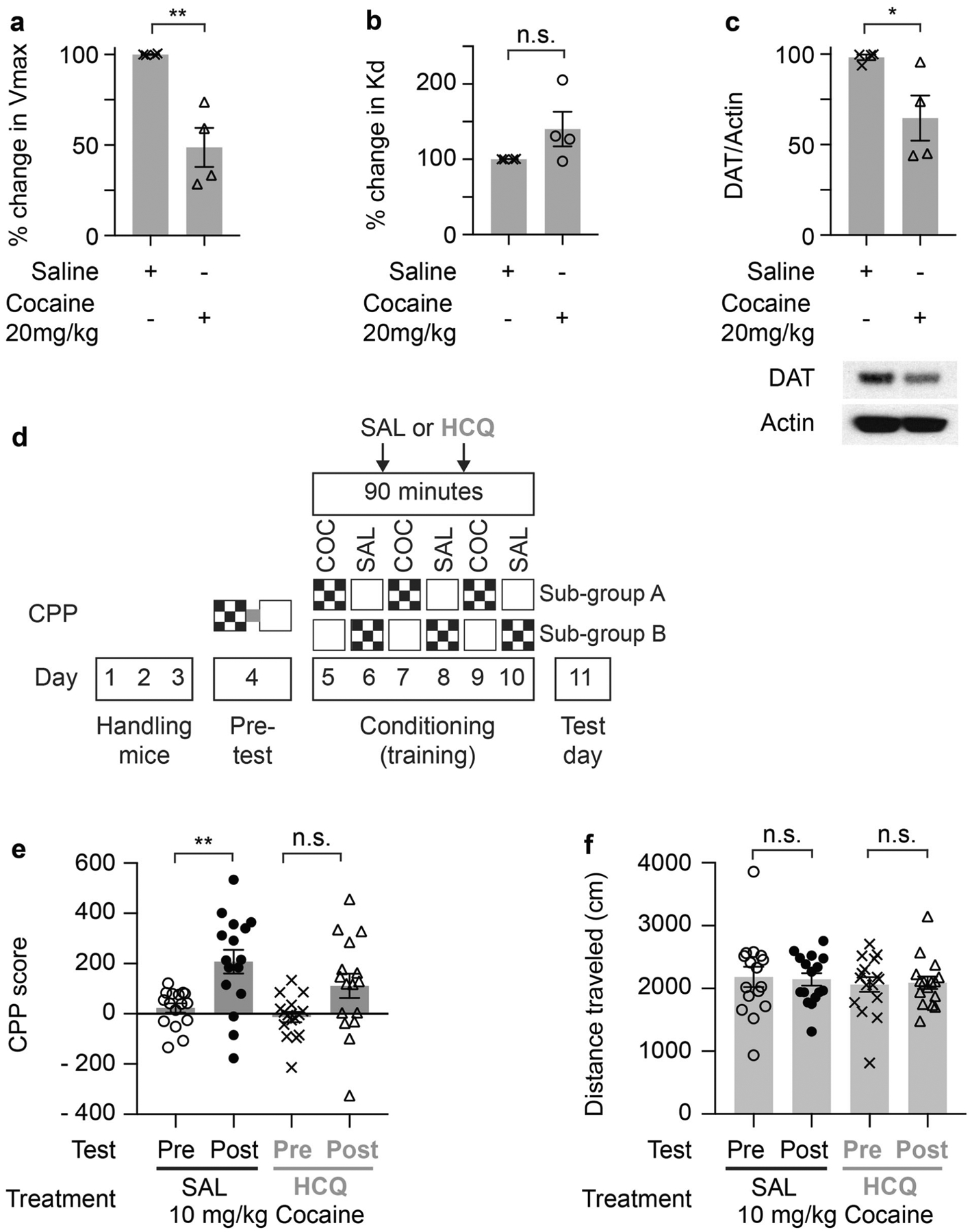
Change in Vmax of [3H]DA uptake (a) or Kd (b) by mouse striatal synaptosome fraction isolated 30 min after intraperitoneal injection of saline or 20 mg/kg cocaine. Error bars = ±SEM, n = 4, **P = 0.003, two-tailed t-test. c Western blot and quantification of DAT in striatal synaptosome fraction isolated 30 min after intraperitoneal injection of saline or 20 mg/kg cocaine. The westerns are from the same samples used in a and b. n = 4, P = 0.036, two-tailed t-test. Error bars = ±SEM. d A schematic diagram illustrating the conditioned place preference (CPP) experimental design. We used an unbiased, counterbalanced protocol. Each experimental group, saline (SAL) or hydroxychloroquine (HCQ), were randomly divided into two subgroups (A and B). For subgroup A, we paired cocaine with one chamber and paired saline with the alternate chamber. For subgroup B, we paired cocaine with the alternate chamber and vice versa. COC = 10 mg/kg cocaine. e Cocaine conditioned place preference expression indicated by mean CPP score (time (s) spent in drug paired compartment minus time (s) spent in saline paired compartment pre and post conditioning). The saline treated group showed a significant increase in CPP score, but the autophagy inhibitor (HCQ) treatment impaired CPP expression. n = 16/group, one-way ANOVA, P < 0.001, Tukey’s multiple comparisons test, **P = 0.0038. Error bars = ±SEM. f Autophagy inhibition by HCQ did not affect the distance traveled during the CPP post test vs pre test compared to the saline treated control group. n = 16/group, Error bars = ±SEM.
To determine the effect of autophagy on the rewarding actions of cocaine, we performed the CPP behavioral assay (Fig. 4d). Inhibiting autophagy by HCQ impairs the expression of cocaine CPP compared to saline treatment (Fig. 4e), while the distance traveled is not affected (Fig. 4f).
In summary, we have established that cocaine enhances autophagic degradation of DAT with extraordinary potency, an action that may mediate behavioral actions of cocaine.
Discussion
In 1960, Whitby et al. [28] demonstrated that cocaine inhibits norepinephrine transport. Cocaine also impairs transport of DA [29–31] and serotonin [32–34], actions restricted to neuronal tissues.
The generally accepted model of cocaine action involves competitive inhibition of DA uptake, thereby elevating synaptic levels of the transmitter. However, many DA reuptake inhibitors with comparable or greater potency than cocaine such as sibutramine [35, 36], bupropion [37], mazindol [38, 39], and tesofensine [40] do not elicit cocaine-like actions in human volunteers. Moreover, most antidepressant drugs, like cocaine, are monoamine reuptake inhibitors. However, cocaine does not act as an anti-depressant in patients [41]. Here, we show that behaviorally active concentrations of cocaine induce autophagic degradation of DAT. Hence, cocaine may inhibit DA reuptake through autophagic degradation of the transporter in addition to directly blocking DAT activity. DAT regulates basal as well as cocaine-induced locomotor activity, both of which are linked to DAT expression levels [42]. Complete depletion of DAT leads to hyperlocomotion, since mice with genetic deletion of DAT (slc6a3) display markedly elevated spontaneous locomotor activity. In DAT-deficient mice, cocaine fails to enhance locomotor activity, demonstrating that DAT is required for cocaine-induced locomotor stimulation [43–45]. Furthermore, partial depletion of DAT results in hyperlocomotion in the open field test. Mice with lower levels of DAT display increased rather than reduced locomotor stimulation following cocaine injection [46, 47]. Hence, diminished DAT levels lead to enhanced basal and cocaine-stimulated locomotor activity. These studies along with our data suggest that partial depletion of DAT by autophagy contributes substantially to cocaine‐stimulated locomotor activity and to cocaine-induced elevations in DA levels, as evidenced by the effects of HCQ and SBI-0206965 treatments in DA microdialysis studies. Tilley et al. generated a knock-in (KI) mouse carrying a triple mutant DAT that retains >50% of transport activity but is insensitive to cocaine. While DAT levels in the triple mutant DAT KI mice are similar to WT mice, DA transport activity is decreased at lower DA concentrations because of the lower affinity for DA of the mutant DAT. The triple mutant DAT KI mice display enhanced basal locomotor activity but decreased locomotor stimulation by cocaine. While the interpretation of these findings is complicated by the reduced affinity of the triple mutant DAT for DA, the study demonstrates the importance of cocaine binding to DAT in mediating the locomotor stimulant effect of cocaine [48]. It remains to be determined whether the triple mutant DAT of these KI mice is recognized by selective autophagic machinery similar to WT DAT. Also, it is unclear whether autophagy is dysregulated in the triple mutant DAT KI mice.
Based on our findings, we propose that at lower cocaine concentrations that do not inhibit the DA transport activity, autophagic degradation of DAT is the main driver of DA reuptake inhibition, while at higher cocaine concentrations both autophagic degradation of DAT and direct transport inhibition by cocaine binding to the transporter might underlie DA reuptake inhibition.
Cocaine induces autophagy with very great potency (0.1 nM) both in vitro and in vivo. Pharmacologic inhibition of autophagy diminishes the locomotor stimulant effect of cocaine. Acute inhibition of autophagy using pharmacological blockers is not likely to affect neuronal viability contrasted to chronic inhibition achieved by deleting genes, which leads to neurodegeneration [49, 50]. Inhibitors of autophagy are likely to have off-target effects. We used three different inhibitors of autophagy, all of which block the locomotor stimulant effect of cocaine. It is unlikely that all three inhibitors, SBI-0206965, vacuolin-1, and HCQ, reduce the locomotor stimulant action of cocaine through off-target effects.
Many psychostimulants, such as methamphetamine, 3,4-methylenedioxymethcathinone (methylone), 3,4-methylenedioxypyrovalerone, 3,4-methylenedioxymethamphetamine, and cocaine, induce neurotoxicity that is mediated by autophagy [5, 51–53]. Here, we demonstrate that low/nontoxic levels of cocaine induce neural autophagy, which modulates the drug’s psychotropic actions. Pharmacologic inhibition of autophagy reduces the locomotor stimulant effect of cocaine and impairs cocaine CPP. The extraordinary potency of cocaine in eliciting autophagy implies that minute doses of the drug influence physiologic functions of diverse organs. Conceivably such low doses may exert beneficial effects that warrant exploration for therapeutic application.
Supplementary Material
Acknowledgements
We thank L. Hester, R. Barrow, A. Snowman, S. McTeer, and L. Albacarys from the SHS laboratory for their assistance. We thank B. Smith and the Microscope Core Facility at the Institute for Basic Biomedical Sciences, Johns Hopkins School of Medicine, for helping in preparation of the TEM samples. We are also grateful for fruitful discussions with members of the SHS Laboratory.
Funding
This work was supported by U.S. Public Health Service Grants DA00266 and DA044123 to SHS and a NARSAD Young Investigator Grant (# 25360) from the Brain & Behavior Foundation to MMH. Support for this research was provided in part by the National Institute on Drug Abuse—Intramural Research Program, NIH/DHHS (Z1A DA000611).
Footnotes
Supplementary information The online version of this article (https://doi.org/10.1038/s41380-020-00978-y) contains supplementary material, which is available to authorized users.
Conflict of interest The authors declare that they have no conflict of interest.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Data files).
References
- 1.Hall FS, Sora I, Drgonova J, Li X-F, Goeb M, Uhl GR. Molecular mechanisms underlying the rewarding effects of cocaine. Ann N Y Acad Sci. 2004;1025:47–56. [DOI] [PubMed] [Google Scholar]
- 2.Rothman RB, Baumann MH. Monoamine transporters and psychostimulant drugs. Eur J Pharm. 2003;479:23–40. [DOI] [PubMed] [Google Scholar]
- 3.Nestler EJ, Malenka RC. The addicted brain. Sci Am. 2004;290:78–85. [DOI] [PubMed] [Google Scholar]
- 4.Edwards DJ, Bowles SK. Protein binding of cocaine in human serum. Pharm Res. 1988;5:440–2. [DOI] [PubMed] [Google Scholar]
- 5.Guha P, Harraz MM, Snyder SH. Cocaine elicits autophagic cytotoxicity via a nitric oxide-GAPDH signaling cascade. Proc Natl Acad Sci USA. 2016;113:1417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Majewska MD. Neurotoxicity and neuropathology associated with chronic cocaine abuse. NIDA Res Monogr. 1996;162:70–72. [PubMed] [Google Scholar]
- 7.CLARK SL. Cellular differentiation in the kidneys of newborn mice studies with the electron microscope. J Biophys Biochem Cytol. 1957;3:349–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deter RL, de Duve C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol. 1967;33:437–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dent P, Booth L, Poklepovic A, Hancock JF. Signaling alterations caused by drugs and autophagy. Cell Signal. 2019;64:109416. [DOI] [PubMed] [Google Scholar]
- 10.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rayport S, Sulzer D, Shi WX, Sawasdikosol S, Monaco J, Batson D, et al. Identified postnatal mesolimbic dopamine neurons in culture: morphology and electrophysiology. J Neurosci. 1992;12:4264–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harraz MM, Eacker SM, Wang X, Dawson TM, Dawson VL. MicroRNA-223 is neuroprotective by targeting glutamate receptors. Proc Natl Acad Sci USA. 2012;109:18962–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graham JM. Isolation of lysosomes from tissues and cells by differential and density gradient centrifugation. Curr Protoc Cell Biol. 2001;Chapter 3:Unit 3.6. [DOI] [PubMed] [Google Scholar]
- 15.Tanda G, Newman AH, Ebbs AL, Tronci V, Green JL, Tallarida RJ, et al. Combinations of cocaine with other dopamine uptake inhibitors: assessment of additivity. J Pharm Exp Ther. 2009;330:802–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mereu M, Tronci V, Chun LE, Thomas AM, Green JL, Katz JL, et al. Cocaine-induced endocannabinoid release modulates behavioral and neurochemical sensitization in mice. Addict Biol. 2015;20:91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keighron JD, Quarterman JC, Cao J, DeMarco EM, Coggiano MA, Gleaves A, et al. Effects of (R)-modafinil and modafinil analogues on dopamine dynamics assessed by voltammetry and microdialysis in the mouse nucleus accumbens shell. ACS Chem Neurosci. 2019;10:2012–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Compact third eddition. San Diego, CA: Academic Press; 2008. [Google Scholar]
- 19.Li N, Lee B, Liu R-J, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid anti-depressant effects of NMDA antagonists. Science. 2010;329:959–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janowsky A, Neve K, Eshleman AJ. Uptake and release of neurotransmitters. Curr Protoc Neurosci 2001;Chapter 7: Unit7.9–7.9.22. [DOI] [PubMed] [Google Scholar]
- 21.Egan DF, Chun MGH, Vamos M, Zou H, Rong J, Miller CJ, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015;59:285–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu Y, Dong S, Hao B, Li C, Zhu K, Guo W, et al. Vacuolin-1 potently and reversibly inhibits autophagosome-lysosome fusion by activating RAB5A. Autophagy. 2014;10:1895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Degtyarev M, De Mazière A, Orr C, Lin J, Lee BB, Tien JY, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol. 2008;183:101–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uhl GR, Hall FS, Sora I. Cocaine, reward, movement and monoamine transporters. Mol Psychiatry. 2002;7:21–26. [DOI] [PubMed] [Google Scholar]
- 25.Maday S, Wallace KE, Holzbaur ELF. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol. 2012;196:407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maday S, Holzbaur ELF. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell. 2014;30:71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hill SE, Kauffman KJ, Krout M, Richmond JE, Melia TJ, Colón-Ramos DA. Maturation and clearance of autophagosomes in neurons depends on a specific cysteine protease isoform, ATG-4.2. Dev Cell. 2019;49:251–266.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.WHITBY LG HERTTING G, AXELROD J. Effect of cocaine on the disposition of noradrenaline labelled with tritium. Nature. 1960;187:604–5. [DOI] [PubMed] [Google Scholar]
- 29.Kilty JE, Lorang D, Amara SG. Cloning and expression of a cocaine-sensitive rat dopamine transporter. Science. 1991;254:578–9. [DOI] [PubMed] [Google Scholar]
- 30.Shimada S, Kitayama S, Lin CL, Patel A, Nanthakumar E, Gregor P, et al. Cloning and expression of a cocaine-sensitive dopamine transporter complementary DNA. Science. 1991;254:576–8. [DOI] [PubMed] [Google Scholar]
- 31.Amara SG, Sonders MS. Neurotransmitter transporters as molecular targets for addictive drugs. Drug Alcohol Depend. 1998;51:87–96. [DOI] [PubMed] [Google Scholar]
- 32.Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–23. [DOI] [PubMed] [Google Scholar]
- 33.Ramamoorthy S, Bauman AL, Moore KR, Han H, Yang-Feng T, Chang AS, et al. Antidepressant- and cocaine-sensitive human serotonin transporter: molecular cloning, expression, and chromosomal localization. Proc Natl Acad Sci USA. 1993;90:2542–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blakely RD, Bauman AL. Biogenic amine transporters: regulation in flux. Curr Opin Neurobiol. 2000;10:328–36. [DOI] [PubMed] [Google Scholar]
- 35.Cole JO, Levin A, Beake B, Kaiser PE, Scheinbaum ML. Sibutramine: a new weight loss agent without evidence of the abuse potential associated with amphetamines. J Clin Psychopharmacol. 1998;18:231–6. [DOI] [PubMed] [Google Scholar]
- 36.Schuh LM, Schuster CR, Hopper JA, Mendel CM. Abuse liability assessment of sibutramine, a novel weight control agent. Psychopharmacology. 2000;147:339–46. [DOI] [PubMed] [Google Scholar]
- 37.Peck AW, Bye CE, Clubley M, Henson T, Riddington C. A comparison of bupropion hydrochloride with dexamphetamine and amitriptyline in healthy subjects. Br J Clin Pharm. 1979;7:469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chait LD, Uhlenhuth EH, Johanson CE. Reinforcing and subjective effects of several anorectics in normal human volunteers. J Pharm Exp Ther. 1987;242:777–83. [PubMed] [Google Scholar]
- 39.Chait LD, Uhlenhuth EH, Johanson CE. The discriminative stimulus and subjective effects of phenylpropanolamine, mazindol and d-amphetamine in humans. Pharm Biochem Behav. 1986;24:1665–72. [DOI] [PubMed] [Google Scholar]
- 40.Schoedel KA, Meier D, Chakraborty B, Manniche PM, Sellers EM. Subjective and objective effects of the novel triple reuptake inhibitor tesofensine in recreational stimulant users. Clin Pharm Ther. 2010;88:69–78. [DOI] [PubMed] [Google Scholar]
- 41.Post RM, Kotin J, Goodwin FK. The effects of cocaine on depressed patients. Am J Psychiatry. 1974;131:511–7. [PubMed] [Google Scholar]
- 42.Uhl GR. Dopamine transporter: basic science and human variation of a key molecule for dopaminergic function, locomotion, and parkinsonism. Mov Disord. 2003;18:S71–80. [DOI] [PubMed] [Google Scholar]
- 43.Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–12. [DOI] [PubMed] [Google Scholar]
- 44.Sora I, Wichems C, Takahashi N, Li XF, Zeng Z, Revay R, et al. Cocaine reward models: conditioned place preference can be established in dopamine- and in serotonin-transporter knockout mice. Proc Natl Acad Sci USA. 1998;95:7699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sora I, Hall FS, Andrews AM, Itokawa M, Li XF, Wei HB, et al. Molecular mechanisms of cocaine reward: combined dopamine and serotonin transporter knockouts eliminate cocaine place preference. Proc Natl Acad Sci USA. 2001;98:5300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rao A, Sorkin A, Zahniser NR. Mice expressing markedly reduced striatal dopamine transporters exhibit increased locomotor activity, dopamine uptake turnover rate, and cocaine responsiveness. Synapse. 2013;67:668–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tilley MR, Cagniard B, Zhuang X, Han DD, Tiao N, Gu HH. Cocaine reward and locomotion stimulation in mice with reduced dopamine transporter expression. BMC Neurosci. 2007;8:42–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen R, Tilley MR, Wei H, Zhou F, Zhou F-M, Ching S, et al. Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter. Proc Natl Acad Sci USA. 2006;103:9333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. [DOI] [PubMed] [Google Scholar]
- 50.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J-I, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. [DOI] [PubMed] [Google Scholar]
- 51.Valente MJ, Amaral C, Correia-da-Silva G, Duarte JA, Bastos M, de L, et al. Methylone and MDPV activate autophagy in human dopaminergic SH-SY5Y cells: a new insight into the context of β-keto amphetamines-related neurotoxicity. Arch Toxicol. 2017;91:3663–76. [DOI] [PubMed] [Google Scholar]
- 52.Mercer LD, Higgins GC, Lau CL, Lawrence AJ, Beart PM. MDMA-induced neurotoxicity of serotonin neurons involves autophagy and rilmenidine is protective against its pathobiology. Neurochem Int. 2017;105:80–90. [DOI] [PubMed] [Google Scholar]
- 53.Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci. 2002;22:8951–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its Supplementary Data files).