

Abstract
Objective
To evaluate long-term survival in patients with Turner syndrome after congenital heart surgery with a focus on left heart obstructive lesions (LHOLs).
Study design
We queried the Pediatric Cardiac Care Consortium, a US-based registry of congenital heart surgery, for patients with Turner syndrome undergoing congenital heart surgery at <21 years of age between 1982 and 2011. Outcomes were obtained from the Pediatric Cardiac Care Consortium and from national death and transplant registries through 2019. Survival of patients with Turner syndrome and nonsyndromic patients with similar LHOL was compared by Kaplan-Meier survival curves and Cox regression adjusted for age, congenital heart disease, and era.
Results
We identified 179 patients with Turner syndrome operated for LHOL: 161 with 2-ventricle lesions (coarctation n = 149, aortic stenosis n = 12) and 18 with hypoplastic left heart (HLH) variants. There were 157 with 2-ventricle LHOL and 6 with HLH survived to discharge. Among survivors to hospital discharge, the 30-year transplant-free survival was 90.4% for Turner syndrome with 2-ventricle lesions and 90.9% for nonsyndromic comparators (adjusted hazard ratio [aHR] 1.15, 95% CI 0.64–2.04). The postdischarge survival for HLH was 33% for Turner syndrome and 51% for nonsyndromic patients, with these numbers being too small for meaningful comparisons. There was a higher risk for cardiovascular disease events in patients with Turner syndrome vs male (aHR 3.72, 95% CI 1.64–8.39) and female comparators (aHR 4.55, 95% CI 1.87–11.06) excluding heart failure deaths.
Conclusions
The 30-year transplant-free survival is similar for patients with Turner syndrome and nonsyndromic comparators with operated 2-ventricle LHOL without excess congenital heart disease risk. However, patients with Turner Syndrome still face increased cardiovascular disease morbidity, stressing the importance of lifelong comorbidity surveillance in this population.
Turner syndrome, characterized by complete or partial monosomy-X, affects approximately 1 in 2500 live-born female infants.1–3 Congenital heart disease (CHD) is the most serious among the phenotypic features characterizing Turner syndrome, occurring in up to 40% of the cases.4–7 Most of the patients with Turner syndrome and CHD undergo congenital heart surgery at an early age.8,9 These patients do well and have similar mortality after congenital heart surgery with nonsyndromic patients with the exception of certain lesions such as hypoplastic left heart (HLH) and partial anomalous pulmonary venous return (PAPVR).9 Long term, patients with Turner syndrome are at increased risk for the development of other cardiovascular diseases (CVDs) including aortic aneurysms, systemic hypertension, vasculopathy, and ischemic heart disease.7,10–12 Studies have reported a ~40% excess mortality for patients with Turner syndrome because of CVD.1,3,13–15 These reports were based on national registry data and did not include information regarding previous congenital heart surgery and postoperative outcomes for these patients.3,15 It is not clear the exact age of divergence of late mortality, but most known Turner syndrome-related complications and contributing comorbidities start in the third and fourth decade of life.1,5
The most common type of CHDs associated with Turner syndrome are within the spectrum of left heart obstructive lesions (LHOL) such as bicuspid aortic valve, aortic stenosis, coarctation of the aorta, or HLH.11,16,17
In this study, we describe the long-term transplant-free survival of a cohort of patients with Turner syndrome operated on for CHD at <21 years of age using the multi-institutional Pediatric Cardiac Care Consortium (PCCC) registry.18 We examined whether the presence of Turner syndrome and LHOL are risk factors for premature mortality or transplant compared with nonsyndromic male and female patients with similar lesions.
Methods
We conducted a retrospective cohort study using the PCCC registry data, enriched with prospective data linkages from the US National Death Index (NDI) and the Organ Procurement and Transplantation Network (OPTN) through 2019.19,20
PCCC is a large US-based multi-institutional registry tracking interventional outcomes after surgical or transcatheter interventions for children with CHD between 1982 and 2011.18,21 We queried the PCCC for all patients with a diagnosis code for Turner syndrome who were operated for a CHD at <21 years of age and selected a comparison cohort of non-syndromic patients (males and females) operated for similar CHD diagnoses within the same period.
Inclusion criteria were congenital heart surgery in a US PCCC center before the age of 21 years, enrollment in PCCC before April 15, 2003 (implementation date of stricter Health Insurance Portability and Accountability Act privacy rules), and US residence at the time of the first congenital heart surgery. We excluded patients with prior cardiac surgery outside the PCCC, patients with incomplete identifiers, and patients with other chromosome abnormalities (including patients with microduplications or microdeletions) or heterotaxy syndrome.
We abstracted sex, primary CHD diagnosis, age at first surgery, and era of first surgery. Age groups for analyses were divided into 3 subsets: neonates (younger than 28 days), infants (28 days to <1 year), and children (1 year to <21 years old). Era of first surgery was categorized into tertiles using early (1982–1993), middle (1994–1998), and late (1999–2011) groupings.
Death and transplant events were obtained from the PCCC records and by matching to NDI and OPTN files through the end of the study period (December 31, 2019) as previously described.20 The linked dataset with the NDI and the OPTN was shown to have an overall sensitivity of 88.1% for death and 89.7% for transplant events, respectively.
All statistical analyses were completed through Statistical Analysis Software (SAS, v 9.4). Median and IQR were reported for continuous variables with non-normal distribution. Categorical variables were presented as count and percentages.
Kaplan Meier plots were created to visualize transplant-free survival outcomes censored at the date of heart transplant, death, or the end of follow-up (December 31, 2019), whichever occurred first. Follow-up time was calculated from the date of the first surgery in the PCCC to outcome date. In-hospital deaths were accounted for by observing an exit type of “DEATH” at the first surgery in PCCC admission. Wilcoxon log-rank tests were employed to test the difference in survival between cohorts. Statistical significance was assessed at the 0.05 level. Cox proportional hazards models were utilized to assess the association between Turner syndrome and time to death or transplant after adjustment for age at first surgery, diagnosis, and era of surgery. The time to event was censored at the date of heart transplant, death, or end of follow-up, whichever occurred first.
Long-term outcomes were assessed by matching PCCC patients with death events reported in the NDI after discharge. Causes of death were categorized using International Statistical Classification of Diseases and Related Health Problems, Ninth Revision and 10th Revision. Underlying and contributing modes of death were grouped into CHD related, cardiovascular disease related, and non-CHD/non-CVD. CVD-related events included pulmonary hypertension, cerebrovascular disease, endocarditis, heart failure (with transplants counted as heart failure-related events), ischemic heart disease, cardiac arrest, arrhythmias, and miscellaneous causes of CVD.
Results
A total of 636 patients with Turner syndrome were extracted from the PCCC registry, accounting for 0.4% of the registry. Of them, 202 patients were coded as having Turner syndrome (Figure 1), met inclusion criteria, and had available direct identifiers; 179 carried a diagnosis of LHOL or HLH variant of single ventricle. The comparison cohort included 6895 patients (male:female 1.9:1). This is consistent with the sex ratio imbalance reported before for LHOL.22 The percentage of LHOL in patients with Turner syndrome was 88.6% of the PCCC study cohort vs 22.5% in the overall comparator cohort (male 27.3%, female 17.1%).
Figure 1.
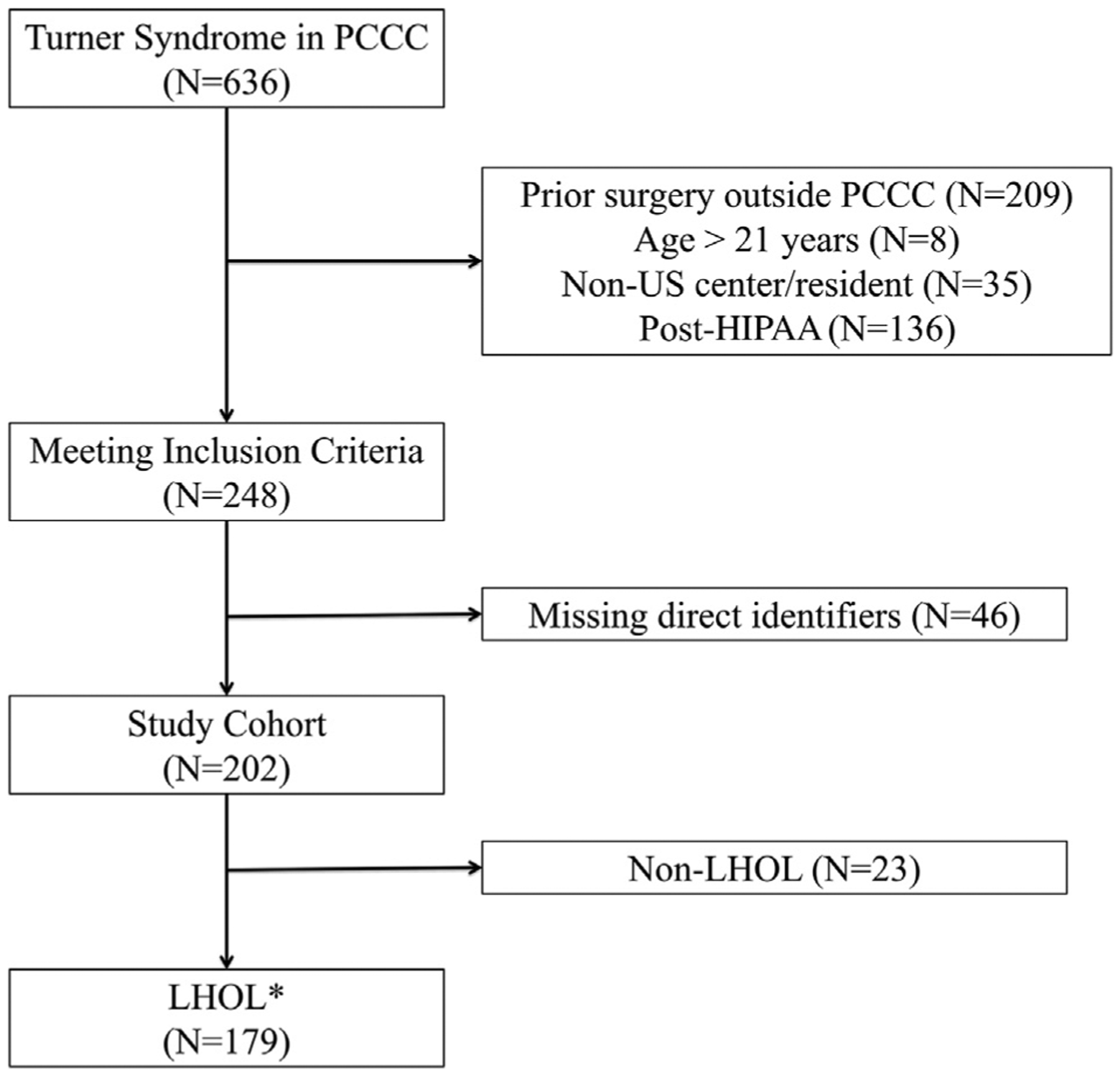
Study cohort flow diagram leading to 179 patients with Turner syndrome and LHOL. *Indicates similar criteria for the selection of the nonsyndromic comparator cohort (n = 6895).
The average age for the first surgery in patients with Turner syndrome was 1 month of age. The cohort was composed primarily of newborns (<28 days, 47%). The group was further divided into infants (28 days- <1 year; 22%) and children (<1 year- <21 years, 31%). Age ranges for first surgery were similar between patients with Turner syndrome and comparators. Era of surgery was divided into approximate tertiles and grouped into early, middle, and late periods of surgery (Table I). The median age of the Turner syndrome and comparator cohort at the end of the study period (December 31, 2019) was 25.3 years of age (IQR 21.6–32.2).
Table I.
Clinical characteristics of the PCCC long-term study cohort
| LHOLs | n = 179 |
|---|---|
| Median age at surgery (mo) (IQR) | 1.1 (0.3, 33.2) |
| Age group at surgery | |
| Newborns (<28 d) | 84 (46.9) |
| Infants (28d-<1 y) | 40 (22.3) |
| Children (1-<21 y) | 55 (30.7) |
| Era of first surgery | |
| 1982–1993 (Early) | 73 (40.8) |
| 1994–1998 (Middle) | 63 (35.2) |
| 1999–2011 (Late) | 43 (24.0) |
| LHOLs | |
| Coarctation—Simple* | 120 (67.0) |
| Coarctation—Complex* | 29 (16.2) |
| Valvar/subvalvar aortic stenosis | 12 (6.7) |
| Hypoplastic left ventricle | 18 (10.1) |
| Non-LHOL | N = 23 |
| Atrial septal defects | 8 (34.8) |
| Partial anomalous pulmonary venous return | 6 (26.0) |
| Tetralogy of Fallot | 2 (8.7) |
| Ventricular septal defect | 1 (4.3) |
| Valvar/subvalvar pulmonic stenosis | 1 (4.3) |
| Other | 5 (21.7) |
Data is presented as n (%) for categorical variables and median with IQR for continuous variables.
These groups include patients with partial anomalous pulmonary venous return (n = 12).
When focusing on the LHOL, coarctation of the aorta was divided into simple (n = 120, 67%), when coarctation was the only reported CHD, and complex (n = 29, 16.2%), when patients had other coexisting lesions such as ventricular septal defect and aortic valve stenosis or subaortic stenosis. In the complex coarctation group, we also included 1 patient with interrupted aortic arch with ventricular septal defect. Twelve patients had aortic stenosis (6.7%), and 18 patients had HLH variety of single ventricle (10%). Twelve of the patients with coarctation (simple or complex) had PAPVR (6.7%) (Table I).
The median time of follow-up from date of first surgery was 23.2 years (IQR 19.3, 28.0) for those with Turner syndrome, 21.6 years (IQR 17.8, 26.5) for non-syndromic male, and 21.3 years (IQR 17.1, 26.6) for nonsyndromic female patients. There were 16 (8.9%) in-hospital deaths in the patients with Turner syndrome. These patients were more likely to have single ventricle physiology (75%) or be neonates at the time of first surgery (88%). There were 451 (10%) in-hospital deaths in the male comparators and 319 (14%) in the female comparators. Similarly, the majority of these deaths occurred in neonates and among with HLH physiology. The percent survival to discharge was 91% in patients with Turner syndrome, 90% in male comparators, and 86.5 % in female comparators (Table II).
Table II.
In-hospital and postdischarge events among patients with Turner syndrome and comparators with LHOL
| Patients with Turner syndrome | Male comparators | Female comparators | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Total | In-hospital events | Postdischarge events | Total | In-hospital events | Postdischarge events | Total | In-hospital events | Postdischarge events | |
| Patient characteristics | n = 179 | n = 16 | n = 17 | n = 4524 | n = 451 | n = 489 | 2371 | n = 319 | n = 236 |
| Age groups | |||||||||
| Newborns (<28 d) | 84 (46.9%) | 14 (87.5%) | 10 | 1940 (42.9%) | 399 (88.5%) | 305 | 1115 (47.0%) | 282 (88.4%) | 157 |
| Infants (28 d -<1 y) | 40 (22.3%) | 2 (12.5%) | 2 | 858 (19.0%) | 48 (10.6%) | 52 | 505 (21.3%) | 31 (9.7%) | 48 |
| Children (1-<21 y) | 55 (30.7%) | 0 (0.0%) | 5 | 1726 (38.2%) | 4 (0.9%) | 132 | 751 (31.7%) | 6 (1.9%) | 31 |
| Era of surgery | |||||||||
| 1982–1993 (Early) | 73 (40.8%) | 4 (25.0%) | 8 | 1556 (34.4%) | 177 (39.3%) | 184 | 861 (36.3%) | 119 (37.3%) | 106 |
| 1994–1998 (Middle) | 63 (35.2%) | 5 (31.3%) | 4 | 1542 (34.1%) | 169 (37.5%) | 167 | 772 (32.6%) | 123 (38.6%) | 77 |
| 1999–2011 (Late) | 43 (24.0%) | 7 (43.8%) | 5 | 1426 (31.5%) | 105 (23.3%) | 138 | 738 (31.1%) | 77 (24.1%) | 53 |
| Type of CHD | |||||||||
| Simple CoA | 120 (67.0%) | 1 (6.3%) | 5 | 1996 (44.1%) | 26 (5.8%) | 100 | 995 (42.0%) | 18 (5.6%) | 32 |
| Complex CoA | 29 (16.2%) | 2 (12.5%) | 6 | 708 (15.7%) | 61 (13.5%) | 76 | 571 (24.1%) | 72 (22.6%) | 70 |
| Valvar, sub-AS | 12 (6.7%) | 1 (6.3%) | 2 | 1048 (23.2%) | 42 (9.3%) | 97 | 400 (16.9%) | 22 (6.9%) | 31 |
| HLH variant | 18 (10.1%) | 12 (75.0%) | 4 | 772 (17.1%) | 322 (71.4%) | 216 | 405 (17.1%) | 207 (64.9%) | 103 |
AS, aortic stenosis; CoA, coarctation of the aorta; SubAS, subaortic stenosis.
Values represent n (% by columns) for each category.
The 30-year transplant-free survival post discharge for patients with Turner syndrome and with 2-ventricle LHOL was 90% (95% CI 85–96.1), compared with 91% for all comparators (95% CI 90–92) (log-rank P = .85) (Figure 2), and without significant differences between male and female patients. Hazard ratios (HRs) adjusted for age at first surgery, CHD diagnosis, and era of surgery, were not statistically different between groups (overall HR 1.15; 95% CI 0.64–2.04, HR 1.18 vs male; 95% CI: 0.66–2.11 and HR 1.13 vs female; 95% CI 0.63–2.07). A total of 157 patients with Turner syndrome and with 2-ventricle LHOL and 6 with HLH survived to discharge. The 30-year postdischarge survival proportion for HLH was 33% for Turner syndrome and 51% for nonsyndromic patients, but the numbers are too small for meaningful comparisons (Table II).
Figure 2.
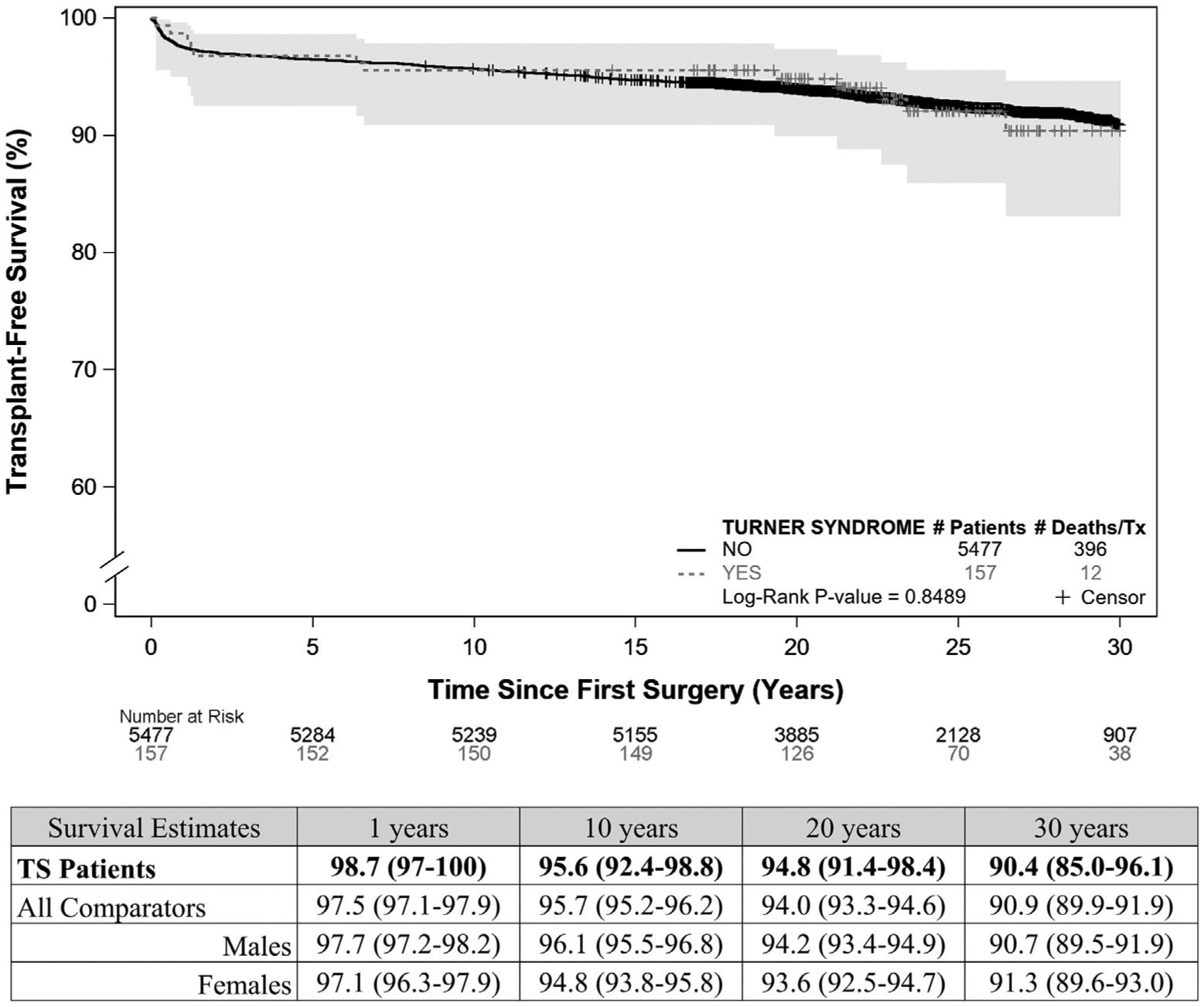
Kaplan-Meier survival plots for patients with Turner syndrome and comparators after congenital heart surgery for 2-ventricle LHOL. Thirty-year transplant-free survival of patients with Turner syndrome with 2-ventricle LHOL Compared with nonsyndromic comparators: Kaplan-Meier curves for patients with Turner syndrome and nonsyndromic comparators with 2 ventricles and LHOL, conditioned on being discharged alive after first congenital heart surgery. Survival estimates given in % with 95% CI in parentheses for all groups.
The distribution and comparison of underlying and contributing causes of death between patients with Turner syndrome and nonsyndromic female and male comparators are presented in Table III (available at www.jpeds.com).
Table III.
Causes of death in patients with Turner syndrome and non-syndromic male and female comparators with LHOL
| Underlying causes of death | Any cause of death (contributing and underlying) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Turner syndrome | Male | P value | Female | P value | Turner syndrome | Male | P value | Female | P value | |
| Causes | n = 17 | n = 489 | n = 236 | n = 17 | n = 489 | n = 236 | ||||
| CHD | 6 (35%) | 227 (46%) | .366 | 141 (60%) | .048 | 10 (59%) | 257 (53%) | .611 | 153 (65%) | .617 |
| CVD | 7 (41%) | 139 (28%) | .279 | 48 (20%) | .063 | 13 (76%) | 296 (61%) | .185 | 132 (56%) | .098 |
| PHTN | 2 (12%) | 3 (1%) | .01 | 3 (1%) | .038 | 3 (18%) | 18 (4%) | .029 | 11 (5%) | .058 |
| Cerebrovascular disease | 0 | 8 (2%) | 1 | 3 (1%) | 1 | 0 | 24 (5%) | 1 | 8 (3%) | 1 |
| Endocarditis | 0 | 3 (1%) | 1 | 1 (0%) | 1 | 0 | 10 (2%) | 1 | 2 (1%) | 1 |
| Heart failure | 0 | 78 (16%) | .089 | 27 (11%) | .23 | 0 | 136 (28%) | .009 | 57 (24%) | .016 |
| Ischemic heart disease | 0 | 6 (1%) | 1 | 3 (1%) | 1 | 0 | 18 (4%) | 1 | 9 (4%) | 1 |
| Cardiac arrest | 0 | 4 (1%) | 1 | 2 (1%) | 1 | 1 (6%) | 65 (13%) | .711 | 36 (15%) | .481 |
| Arrhythmia | 0 | 4 (1%) | 1 | 2 (1%) | 1 | 1 (6%) | 30 (6%) | 1 | 19 (8%) | 1 |
| Miscellaneous CVD | 5 (29%) | 39 (8%) | .011 | 8 (3%) | <.001 | 8 (47%) | 49 (10%) | <.001 | 16 (7%) | <.001 |
| Non-CHD/CVD | 4 (24%) | 123 (25%) | 1 | 47 (20%) | .755 | 1 (6%) | 76 (16%) | .49 | 31 (13%) | .704 |
PHTN, pulmonary hypertension.
Transplants included as heart failure.
Bold fonts indicate stastistical significance.
CHD was less likely to be identified as the underlying cause of death among the patients with Turner syndrome vs the female comparators (35% vs 60%, P = .05), but this was not the case vs the male comparators (46%, P = .37). There was no statistical difference in the number of CVD events as underlying cause of death among patients with Turner syndrome vs male or female comparators; however, subanalysis revealed increased number of deaths in patients with Turner syndrome because of pulmonary hypertension (12%) vs nonsyndromic comparators (female 1%, P = .038, male 1%, P = .01) as well as because of miscellaneous CVD (Turner syndrome 29% vs female 3%, P < .001, male 8% P < .011); 4 out of the 5 miscellaneous CVD deaths in the patients with Turner syndrome were attributed to aortic dissection. Heart failure-related events (deaths or transplants) did not occur among patients with Turner syndrome, albeit the relatively small number of events among comparators did not reach statistical significance. There were no significant differences between the groups in regards to non-CHD/non-CVD-related deaths.
When analyzing cause-specific adjusted hazard ratios (aHR), patients with Turner syndrome had a higher risk for a CVD event as underlying cause of death vs the female comparators (aHR 2.29; 95% CI 1.02–5.13), but not vs male comparators (aHR 1.71; 95% CI 0.79–3.70). After excluding transplant and heart failure as underlying cause of death because they did not occur in the Turner syndrome group, patients with Turner syndrome had an even higher risk for nonheart failure CVD events overall (aHR 3.91; 95% CI 1.77–8.63) and in comparison with both male (aHR 3.72; 95% 1.64–8.39) and female (aHR 4.55; 95% 1.87–11.06) patients. There were no differences in aHR for CHD alone or non-CHD/non-CVD as an underlying cause of death.
Additional analysis after incorporating the multiple causes of death information, where death was attributed directly or indirectly to the cause of interest, revealed the same pattern for events between the groups. More specifically the distribution of CHD, overall CVD, and non-CHD/non-CVD events was not different between groups. However, subanalysis of CVD events showed again more pulmonary hypertension related events among patients with Turner syndrome vs male patients (18% vs 4%, P = .029). Although there was a higher proportion of pulmonary hypertension events in patients with Turner syndrome vs female patients, this did not reach statistical significance (females 5%, P = .058). There were also more miscellaneous CVD-related events including aortic dissection (Turner syndrome 47% vs female 7%, P < .001 and male 10%, P < .001). Heart failure-related events were not encountered among patients with Turner syndrome and this reached statistical significance vs the non-syndromic comparators (female 25%, P = .016 and male 28%, P = .009).
Discussion
A previous report from the PCCC identified increased risk for perioperative adverse outcomes after congenital heart interventions in patients with Turner syndrome. There was a significant increase in mean length of stay in patients with Turner syndrome after several congenital heart interventions, albeit there was no significant difference in mortality at the time of discharge except patients with HLH and PAPVR.9 This is further supported by another study in which patients with Turner syndrome and with CHD were more likely to have a prolonged length of stay but no difference in increased mortality.23 Improved early postoperative outcomes were reported by a study from the Society of Thoracic Surgeons, but the prolonged hospitalization remained an issue for the Turner syndrome cohort.24 Because prolonged hospitalization post cardiac surgery may be linked with increased risk for post-discharge adverse outcomes and patients with Turner syndrome are at risk for late cardiovascular events,5,25 we analyzed the long-term outcomes of the PCCC cohort of patients with Turner syndrome and with congenital heart surgery. Our data demonstrate that children surviving congenital heart surgery with 2-ventricle LHOL have excellent long-term (30-year) transplant-free survival and are comparable with their nonsyndromic counterparts.
Although the overall 30-year survival was similar between the 2 groups, patients with Turner syndrome had a higher early risk for certain cardiovascular nonheart failure-related deaths in this relatively young cohort. This increased mortality related to aortopathy and pulmonary hypertension does not appear to be due to the underlying CHD disease itself, because the CHD-related mortality was similar or lower between Turner syndrome and nonsyndromic comparators with LHOL. Regarding the subgroup of patients with Turner syndrome and with HLH variants, very few survived to discharge and of those discharged, only 33.3% survived.
Many studies have shown overall decreased long-term survival in patients with Turner syndrome.3,5,15 There are many reasons for this including increased risk for diabetes, elevated triglycerides, obesity, systemic hypertension, and ischemic heart disease.11 In a study by Fuchs et al, evaluating long-term mortality up to 68 years in 318 patients with Turner syndrome with an average age of diagnosis of 9 years, there was mortality of 14%. They showed that the most common cause of death was CVD (22%), which was attributed to late postoperative care past the age of 30 years, late end-stage heart failure, and aortic dissection.5 The study suggested that most cardiovascular complications were late and related to comorbidities such as systemic hypertension, hyperlipidemia, aortopathy, and coronary artery disease. Other comorbidities among patients with Turner syndrome include sleep apnea, diabetes mellitus, thyroid dysfunction, and ovarian failure.26
Our cohort, including only pediatric cases with early surgery for CHD, provides evidence of increased early CVD-related mortality in patients with Turner syndrome. The specific CVD mortality observed in our cohort, though, appears to be related to the increased risk factors (ie, vasculopathy) in this population, rather than consequences of the CHD itself, such as heart failure or arrhythmias. It remains to be seen whether at a later age the interaction of the Turner syndrome-associated comorbidities will disproportionately affect the survival of these patients compared with nonsyndromic comparators. The pattern of increased CVD morbidity in patients with Turner syndrome stresses the importance of following surveillance guidelines for Turner syndrome including monitoring for known comorbidities, and providing anticipatory guidance11,26; however, undergoing congenital heart surgery as a child does not appear to predispose the patients with Turner syndrome to advanced heart failure or worse outcomes, at least up to 30 years of age.
There are some limitations to the study. We did not have access to individual karyotypes for patients with Turner syndrome and, thus, cannot conclude whether they had monosomy X or mosaic Turner syndrome. In addition, over the years the type of genetic testing for Turner syndrome has evolved from karyotype to microarray; however, microarray has not been proven to be more reliable for identifying Turner syndrome than karyotype.27 Nevertheless, it is likely that genetic testing, regardless of method, was not performed in all female patients with left ventricular outflow tract obstruction, so some patients with Turner syndrome may have been misclassified as non-Turner syndrome and, thus, be included in the female comparator group. As a result, such misclassification could blunt the outcome differences between the Turner syndrome and non-Turner syndrome female comparators. Although using a large multicenter database allows for heterogeneity and a larger sample population with linkage to national death and transplant databases, there are inherent limitations of the database itself, including record availability and verification of specific clinical characteristics. Our findings are limited to the extracted data and it is not possible to query for additional clinical information. It is also possible that some deaths can be missed because of errors in matched variables, so the reported events represent a low boundary for this cohort. Despite the relatively large size of this cohort of patients with Turner syndrome, death events from certain causes such as pulmonary hypertension are still too rare to draw definite conclusions.
Further monitoring of this cohort via updated linkage with national databases such as the NDI and OPTN will allow the assessment of the burden of cardiovascular morbidity in these patients over time.
Supplementary Material
Acknowledgments
We thank PCCC directors, coordinators, and the Pediatric Heart Disease Data Registry Core at Emory University for their help with data analysis.
Supported in part by grants from the NIH/The National Heart, Lung, and Blood Institute (R01 HL122392) and the Department of Defense (PR180683). The authors declare no conflicts of interest.
Glossary
- aHR
Adjusted hazard ratio
- CHD
Congenital heart disease
- CVD
Cardiovascular disease
- HLH
Hypoplastic left heart
- HRs
Hazard ratios
- LHOL
Left heart obstructive lesions
- NDI
National Death Index
- OPTN
Organ Procurement and Transplantation Network
- PAPVR
Partial anomalous pulmonary venous return
- PCCC
Pediatric Cardiac Care Consortium
References
- 1.Bondy CA. Congenital cardiovascular disease in Turner syndrome. Congenit Heart Dis 2008;3:2–15. [DOI] [PubMed] [Google Scholar]
- 2.Nielsen J, Wohlert M. Chromosome abnormalities found among 34 910 newborn children: results from a 13-year incidence study in Arhus, Denmark. Hum Genet 1991;87:81–3. [DOI] [PubMed] [Google Scholar]
- 3.Stochholm K, Juul S, Juel K, Naeraa RW, Gravholt CH. Prevalence, incidence, diagnostic delay, and mortality in Turner syndrome. J Clin Endocrinol Metab 2006;91:3897–902. [DOI] [PubMed] [Google Scholar]
- 4.Dulac Y, Pienkowski C, Abadir S, Tauber M, Acar P. Cardiovascular abnormalities in Turner’s syndrome: what prevention? Arch Cardiovasc Dis 2008;101:485–90. [DOI] [PubMed] [Google Scholar]
- 5.Fuchs MM, Attenhofer Jost C, Babovic-Vuksanovic D, Connolly HM, Egbe A. long-term outcomes in patients with turner syndrome: a 68-year follow-Up. J Am Heart Assoc 2019;8:e011501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Volkl TM, Degenhardt K, Koch A, Simm D, Dorr HG, Singer H. Cardiovascular anomalies in children and young adults with Ullrich-Turner syndrome: the Erlangen experience. Clin Cardiol 2005;28:88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pierpont ME, Brueckner M, Chung WK, Garg V, Lacro RV, McGuire AL, et al. Genetic basis for congenital heart disease: revisited: a scientific statement from the American Heart Association. Circulation 2018;138: e653–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cramer JW, Bartz PJ, Simpson PM, Zangwill SD. The spectrum of congenital heart disease and outcomes after surgical repair among children with Turner syndrome: a single-center review. Pediatr Cardiol 2014;35:253–60. [DOI] [PubMed] [Google Scholar]
- 9.Madriago E, Nguyen T, McFerson M, Larson EV, Airhart N, Moller JH, et al. Frequency and outcomes of cardiac operations and catheter interventions in Turner syndrome. Am J Cardiol 2012;110:580–5. [DOI] [PubMed] [Google Scholar]
- 10.Elsheikh M, Dunger DB, Conway GS, Wass JA. Turner’s syndrome in adulthood. Endocr Rev 2002;23:120–40. [DOI] [PubMed] [Google Scholar]
- 11.Silberbach M, Roos-Hesselink JW, Andersen NH, Braverman AC, Brown N, Collins RT, et al. Cardiovascular health in Turner syndrome: a scientific statement from the American Heart Association. Circ Genom Precis Med 2018;11:e000048. [DOI] [PubMed] [Google Scholar]
- 12.Sybert VP. Cardiovascular malformations and complications in Turner syndrome. Pediatrics 1998;101:E11. [DOI] [PubMed] [Google Scholar]
- 13.Price WH, Clayton JF, Collyer S, De Mey R, Wilson J. Mortality ratios, life expectancy, and causes of death in patients with Turner’s syndrome. J Epidemiol Community Health 1986;40:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schoemaker MJ, Swerdlow AJ, Higgins CD, Wright AF, Jacobs PA, United Kingdom Clinical Cytogenetics G. Mortality in women with turner syndrome in Great Britain: a national cohort study. J Clin Endocrinol Metab 2008;93:4735–42. [DOI] [PubMed] [Google Scholar]
- 15.Swerdlow AJ, Hermon C, Jacobs PA, Alberman E, Beral V, Daker M, et al. Mortality and cancer incidence in persons with numerical sex chromosome abnormalities: a cohort study. Ann Hum Genet 2001;65:177–88. [DOI] [PubMed] [Google Scholar]
- 16.Chou YY, Wang CJ, Lin CH, Chung HT, Lo FS. Association between cardiovascular anomalies and karyotypes in Turner syndrome patients in Taiwan: a local cohort study. Pediatr Neonatol 2020;61:188–94. [DOI] [PubMed] [Google Scholar]
- 17.Morales-Demori R Congenital heart disease and cardiac procedural outcomes in patients with trisomy 21 and Turner syndrome. Congenit Heart Dis 2017;12:820–7. [DOI] [PubMed] [Google Scholar]
- 18.Vinocur JM, Moller JH, Kochilas LK. Putting the Pediatric Cardiac Care Consortium in context: evaluation of scope and case mix compared with other reported surgical datasets. Circ Cardiovasc Qual Outcomes 2012;5: 577–9. [DOI] [PubMed] [Google Scholar]
- 19.Spector LG, Menk JS, Knight JH, McCracken C, Thomas AS, Vinocur JM, et al. Trends in long-term mortality after congenital heart surgery. J Am Coll Cardiol 2018;71:2434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spector LG, Menk JS, Vinocur JM, Oster ME, Harvey BA, St Louis JD, et al. In-hospital vital status and heart transplants after intervention for congenital heart disease in the Pediatric Cardiac Care Consortium: completeness of ascertainment using the National Death Index and United Network for Organ Sharing Datasets. J Am Heart Assoc 2016;5:e003783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moller JH, Powell CB, Joransen JA, Borbas C. The pediatric cardiac care consortium–revisited. Jt Comm J Qual Improv 1994;20:661–8. [DOI] [PubMed] [Google Scholar]
- 22.Aboulhosn J, Child JS. Left ventricular outflow obstruction: subaortic stenosis, bicuspid aortic valve, supravalvar aortic stenosis, and coarctation of the aorta. Circulation 2006;114:2412–22. [DOI] [PubMed] [Google Scholar]
- 23.Singh I, Duca LM, Kao D, Chatfield KC, Khanna AD. Outcomes in hospitalisations of women with Turner syndrome compared to women without Turner syndrome. Cardiol Young 2021: 1–8. 10.1017/S1047951121000858 [DOI] [PubMed] [Google Scholar]
- 24.Chew JD, Hill KD, Jacobs ML, Jacobs JP, Killen SAS, Godown J, et al. Congenital heart surgery outcomes in Turner syndrome: the Society of Thoracic Surgeons database analysis. Ann Thorac Surg 2019;108: 1430–7. [DOI] [PubMed] [Google Scholar]
- 25.Namachivayam SP, d’Udekem Y, Millar J, Cheung MM, Butt W. Survival status and functional outcome of children who required prolonged intensive care after cardiac surgery. J Thorac Cardiovasc Surg 2016;152:1104–12.e3. [DOI] [PubMed] [Google Scholar]
- 26.Gravholt CH, Andersen NH, Conway GS, Dekkers OM, Geffner ME, Klein KO, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol 2017;177: G1–70. [DOI] [PubMed] [Google Scholar]
- 27.Wapner RJ, Martin CL, Levy B, Ballif BC, Eng CM, Zachary JM, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med 2012;367:2175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.