

Summary
In this protocol, we describe global proteome profiling for the respiratory specimen of COVID-19 patients, patients suspected with COVID-19, and H1N1 patients. In this protocol, details for identifying host, viral, or bacterial proteome (Meta-proteome) are provided. Major steps of the protocol include virus inactivation, protein quantification and digestion, desalting of peptides, high-resolution mass spectrometry (HRMS)-based analysis, and downstream bioinformatics analysis.
For complete details on the use and execution of this profile, please refer to Maras et al. (2021).
Subject areas: Health Sciences, Proteomics, Mass Spectrometry
Graphical Abstract
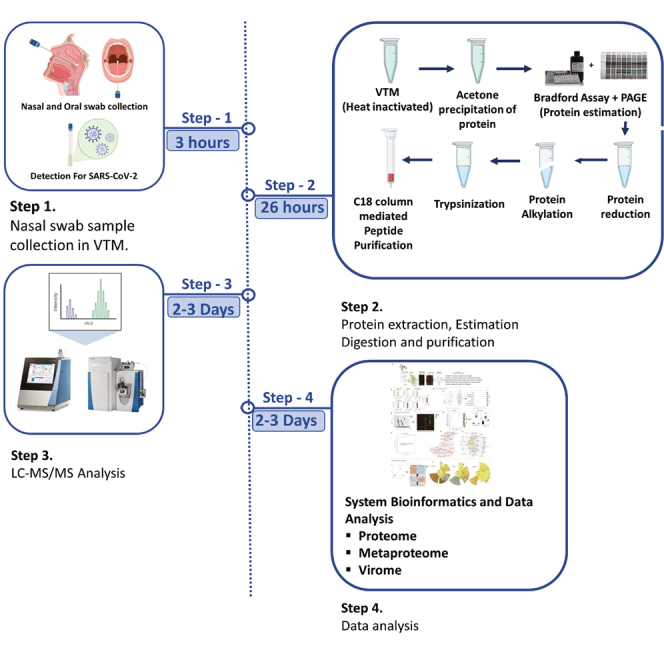
Highlights
-
•
HRMS-based Proteome, Virome, and Metaproteome analysis of COVID 19 patients
-
•
Protocol for Sample preparation and Database curation for HRMS-based analysis
-
•
Downstream bioinformatics data analysis for Proteome, Virome, and Metaproteome dataset
In this protocol, we describe global proteome profiling for the respiratory specimen of COVID-19 patients, patients suspected with COVID-19, and H1N1 patients. In this protocol, details for identifying host, viral, or bacterial proteome (Meta-proteome) are provided. Major steps of the protocol include virus inactivation, protein quantification and digestion, desalting of peptides, high-resolution mass spectrometry (HRMS)-based analysis, and downstream bioinformatics analysis.
Before you begin
This protocol describes specific steps to execute proteome, metaproteome, and virome study in a COVID VTM sample (Maras et al., 2021). However, this protocol can be used for executing proteome, metaproteome, and virome from any sample. In the case of Plasma samples where some proteins are very high in concentration, e.g., albumin and immunoglobulins, you have to specifically deplete them first and then follow the same protocol {Chutipongtanate, 2017 #57}
Nasopharyngeal specimen (NP) collection (performed by a trained healthcare provider, only): (Pondaven-Letourmy et al., 2020)
Tilt head of patient backward around 70 degrees. Then Cautiously insert a mini tip swab through the nostril, parallel to the palate (not upwards) until you feel resistance or the distance is equivalent to that from the ear to the patient's nostril, indicating contact with the nasopharynx. Cautiously rub and roll the mini tip swab and leave the swab in place for several seconds to absorb secretions. Now gently remove the swab while rotating it. Finally, Place the mini tip swab into the transport tube (VTM media) provided.
Note: If a deviated septum or blockage creates difficulty obtaining the specimen from one nostril, use another nostril.
Oropharyngeal (OP) (throat) specimen collection
Insert the mini tip swab into the pharynx and tonsillar areas. Then roll swab over both tonsillar pillars and posterior oropharynx.
Note: Avoid touching the tongue, teeth, and gums.
Finally, Place the swab, tip first, into the viral transport media provided.
Key resource table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Ammonium bicarbonate | Sigma-Aldrich | A6141 |
| Sodium dodecyl sulfate | Sigma-Aldrich | L3771 |
| Dithiothreitol | Sigma-Aldrich | D0632 |
| Iodoacetamide | Acros Organics | 122270050 |
| Trypsin protease, MS grade | Thermo Fisher Scientific | 90305 |
| Trifluoroacetic acid | Sigma-Aldrich | T6508 |
| Acetonitrile | JT Baker | 14650359 |
| Formic acid | Sigma-Aldrich | F0507 |
| Urea | Sigma-Aldrich | U0631 |
| LC-MS grade water | Sigma-Aldrich | 39253 |
| Bovine serum albumin | Thermo Fisher Scientific | 23208 |
| Acetone | JT Baker | 9006–03 |
| Acrylamide | Thermo Fisher Scientific | AM9022 |
| SDS | Thermo Fisher Scientific | 15525017 |
| APS | Thermo Fisher Scientific | HC2005 |
| TEMED | Thermo Fisher Scientific | T9281 |
| Tris-Base | Thermo Fisher Scientific | 17926 |
| HCL | Thermo Fisher Scientific | 24308 |
| Glycine | Thermo Fisher Scientific | A37730IN |
| Glacial acetic acid | Sigma-Aldrich | A0808 |
| Coomassie Brilliant Blue | Thermo Fisher Scientific | 20278 |
| ß-Mercaptoethanol | Sigma-Aldrich | M6250 |
| Bradford reagent | Thermo Fisher Scientific | 22663 |
| Software and algorithms | ||
| Proteome Discoverer 2.2 using Sequest HT | Thermo Fisher Scientific | OPTON-30945 |
| Xcalibur | Thermo Fisher Scientific | OPTON-30965 |
| Uniprot | (https://www.uniprot.org): | |
| Others | ||
| Benchtop centrifuge | Eppendorf | 5427R |
| Sonicator | Helix Biosciences | HBSNII-92 |
| Vortex | Sigma-Aldrich | Z258423 |
| Incubator | Memmert | INB200 |
| Vacuum evaporator | Genevac | DNA-12060-C00 |
| Microplate reader | Thermo Fisher Scientific | VL0000D0 |
| Nano Reverse-phased capillary HPLC system | Thermo Fisher Scientific | UltiMate 3000 HPLC |
| Q-exactive + orbitrap | Thermo Fisher Scientific | IQLAAEGAAPFADBMBCX |
| Heated electrospray ionization (H-ESI) ion source | Thermo Fisher Scientific | H-ESI probe |
| SDS PAGE Apparatus | Bio-Rad Laboratories | 165-8000 |
| Viral Transfer media | RMBio | VTM-CHT |
| Eppendorf tubes | Eppendorf | 30125150 |
| ELISA plates | Thermo Fisher Scientific | 44-2404-21 |
| Pierce C18 spin columns | Thermo Fisher Scientific | 89870 |
| Water bath | LabcOM | 76308-830 |
| Nano LC Column | Thermo Fisher Scientific | 164570 |
| Calibration solution | Thermo Fisher Scientific | 88323/88324 |
Materials and equipment
Tris's buffer
| Reagent | Final concentration | Volume |
|---|---|---|
| Tris's buffer | 0.1 M Tris, pH 7.8 | 500 mL |
Dissolve 6.05 g tris in LC-MS grade water and makeup to a total volume of 500 mL. Adjust pH to 7.8 with HCl.
Note: Tris buffer solutions can be stored at 25°C or at +4°C for two weeks.
Ammonium bicarbonate buffer
| Reagent | Final concentration | Volume |
|---|---|---|
| Ammonium bicarbonate buffer | 50 mM ammonium bicarbonate in 0.1 M Tris buffer, pH 7.8 | 250 mL |
Dissolve 0.97 g of ammonium bicarbonate in tris buffer and make up a final volume of 250 mL.
Note: Tris's buffer solutions can be stored at 25°C or at +4°C for two weeks.
Urea buffer
| Reagent | Final concentration | Volume |
|---|---|---|
| Urea buffer | 8 M urea in 50 mM ammonium bicarbonate buffer | 125 mL |
Dissolve 110.06 g of urea in 50 mM ammonium bicarbonate buffer and make up a final volume of 125 mL.
Note: Urea solutions should always be freshly prepared and should be stored below 30°C.
Iodoacetamide solution
| Reagent | Final concentration | Volume |
|---|---|---|
| Iodoacetamide solution | 50 mM iodoacetamide in urea buffer | 10 mL |
Dissolve 92 mg of iodoacetamide in urea buffer and makeup to a total volume of 10 mL with 8 M urea buffer.
CRITICAL: Iodoacetamide acts as an acutely toxic, irritant, and health hazard, according to MSDS information. To prevent exposure, wear suitable protective gloves, eye protection, face masks, etc.
Note: Iodoacetamide is unstable and light-sensitive. Prepare solutions immediately before use and perform alkylation in the dark.
Dithiothreitol solution
| Reagent | Final concentration | Volume |
|---|---|---|
| Sample buffer | 1 M DTT in LC-MS grade water | 10 mL |
Dissolve 1.55 gm of DTT in LC-MS grade water and makeup to a total volume of 10 mL.
Note: Dithiothreitol solution is stored at −20°C and should use within 3 months.
LC /MS-MS – Solvent A
| Reagent | Final concentration | Volume |
|---|---|---|
| LC - Solvent A | 0.1% (v/v) formic acid in LC-MS grade water | 1,000 mL |
Gently mix 1 mL formic acid with LC-MS grade water and makeup to a total volume of 1,000 mL with LC-MS grade water.
Note: Always prepare fresh.
LC/MS-MS – Solvent B
| Reagent | Final concentration | Volume |
|---|---|---|
| LC - Solvent B | 95% (v/v) acetonitrile, 0.1% (v/v) formic acid in LC-MS grade water | 1,000 mL |
Gently mix 950 mL of acetonitrile with 0.1 mL of formic acid and makeup to a total volume of 1,000 mL with LC-MS grade water.
Note: Always prepare fresh.
Solution A for peptide purification (Wash buffer)
| Reagent | Final concentration | Volume |
|---|---|---|
| Wash buffer | 50% (v/v) acetonitrile in LC-MS grade water | 5 mL |
In 25 mL of acetonitrile, add 25 mL of LC-MS grade water to make the final volume 50 mL.
Note: Always prepare fresh.
Solution B for peptide purification (Equilibration buffer)
| Reagent | Final concentration | Volume |
|---|---|---|
| Equilibration buffer | 5% (v/v) acetonitrile in LC-MS grade water | 50 mL |
In 2.5 mL of acetonitrile, add 47.5 mL of LC-MS grade water to make the final volume 50 mL.
Note: Always prepare fresh.
Solution C for peptide purification (Elution buffer)
| Reagent | Final concentration | Volume |
|---|---|---|
| Elution buffer | 90% (v/v) acetonitrile in LC-MS grade water | 5 mL |
In 45 mL of acetonitrile, add 5 mL of LC-MS grade water to make the final volume 50 mL.
Note: Always prepare fresh.
Tris's HCL buffer (PH-8.8)
| Reagent | Final concentration | Volume |
|---|---|---|
| Tris's base | 1.5 M Tris's base in deionized water. | 100 mL |
Dissolve 18.15 gm of Tris base in 80 mL of deionized water, now adjust the pH to 8.8 using 6 N HCL and then adjust final volume to 100 mL.
Note: Tris buffer solutions can be stored at 25°C or at +4°C for two weeks.
Tris's buffer (pH-6.8)
| Reagent | Final concentration | Volume |
|---|---|---|
| Tris's base | 0.5 M Tris's base in deionized water. | 100 mL |
Dissolve 6 gm of Tris base in 60 mL of deionized water, now adjust the pH to 6.8 using 6 N HCL and then adjust final volume to 100 mL.
Note: Tris's buffer solutions can be stored at 25°C or at +4°C for two weeks.
10% (W/V) SDS
| Reagent | Final concentration | Volume |
|---|---|---|
| SDS | 10% SDS in deionized water. | 100 mL |
Dissolve 10 gm of SDS in 90 mL of deionized water, then adjust the final volume to 100 mL.
Note: Store 10% SDS stock solution at 20°C. Do not store in the fridge, as the SDS will reprecipitate.
10% (W/V) APS
| Reagent | Final concentration | Volume |
|---|---|---|
| APS | 10% APS in deionized water. | 1 mL |
Dissolve 0.1 gm of SDS in 1 mL of deionized water.
Note: Always prepare fresh.
Tris-Glycine buffer (Running buffer)
| Reagent | Final concentration | Volume |
|---|---|---|
| Tris's base | 250 mM | |
| Glycine | 1.92 M | |
| SDS | 1% | |
| water | to 1 L | 1 L |
Dissolve 30.30 gm tris, 144.10 gm Glycine, and SDS 10 gm in 900 mL of water and make final volume 1 L.
Note: don't adjust it will be reached automatically pH 8.3.
Note: Tris buffer solutions can be stored at 25°C or at +4°C for two weeks.
1% Bromophenol Blue
| Reagent | Final concentration | Volume |
|---|---|---|
| Bromophenol blue | 1% bromophenol in water. | 1 mL |
Dissolve 100 mg bromophenol Blue in 10 mL of water.
Note: 1% Bromophenol Blue can be stored at 25°C or at +4°C for two years.
Staining solution
| Reagent | Final concentration | Volume |
|---|---|---|
| Coomassie blue R250 | 0.1% (w/v), | |
| methanol | 20% (v/v) | |
| Acetic acid | 10% (v/v) |
Dissolve 0.4 gm Coomassie blue in 200 mL of 40% (v/v) HPLC grade methanol in water with stirring as required. Filter the solution to remove any insoluble material. Add 200 mL of 20% (v/v) acetic acid in water.
Note: Always prepare fresh.
Destaining solution
| Reagent | Final concentration | Volume |
|---|---|---|
| methanol | 50% (v/v) | |
| Acetic acid | 10% (v/v) | |
| water | to 1 L | 1 L |
Add 500 mL of HPLC- grade methanol to 300 mL of HPLC-grade water, Add 100 mL of reagent grade acetic acid mix and adjust final volume to 1000 mL with water. The final concentrations will be 50% (v/v) methanol in water with 10% (v/v) acetic acid.
Note: Always prepare fresh.
Step-by-step method details
Heat inactivation of virus (Batejat et al., 2021)
Timing: 30 min
Note: By the end of this step, the virus gets inactivated, and now all the remaining sample processing steps can be performed in the BSL-2 facility.
CAUTION: SARS-CoV-2 Samples should be handled in a Biosafety Level 3 (BSL-3) laboratory using BSL-3 practices till virus inactivation is performed and post inactivation, all other steps can be performed on BSL-2 facility.
-
1.
Nasopharyngeal samples (NPS) and oropharyngeal samples together were collected into 1 mL of viral transport media.
-
2.
Around 500 uL of the sample was incubated with a pre-warmed dry heating block maintained at 92°C for 15 min.
Homogenization of respiratory specimen (Dowling et al., 2020)
Note: By the end of this step, all the cell's macromolecules like proteins, metabolites, lipids, etc., will be released in solution.
-
3.
Respiratory specimens may contain host cells, mucus, bacterial and viral components. Thus, it is essential to homogenize the samples.
CAUTION: The high-frequency sound emitted by the Sonicator can damage hearing; therefore, place the sample in a noise isolating chamber and always close the door while operating. Do not grasp an activated horn or touch the tip of a vibrating probe. It can cause severe tissue damage and burns.
-
4.
Take 500 μL of respiratory specimen in a new microcentrifuge tube (MCT) and keep it on the ice during homogenization.
-
5.
Place the sample in a noise isolating chamber and submerge the Sonicator probe into the sample.
Note: The probe should not touch the walls of MCT as it will break the tube and destroy the sample.
-
6.
Close the door of the chamber after properly placing the sample tube.
-
7.
Run the program at Power 20%, Run time 5 min (Cycle – 10 s ON, 10 s OFF) and temperature 22°C.
-
8.
Remove and wipe the probe with ethanol.
Note: Keep the sample on the ice during all the homogenization steps step 2–6.
Pause Point: Respiratory specimen can be stored at −80°C for up to 4 weeks.
Organic solvent-mediated protein precipitation (Bradford, 1976; Simpson and Beynon, 2010)
Note: By the end of this step, all proteins will be precipitated.
-
9.
200 uL of VTM was placed in acetone compatible tube.
-
10.
Add the 6-fold volume of cold acetone to VTM.
-
11.
Vortex and incubate for 1–4 h at −80°C.
-
12.
Centrifuge for 10 min at 18,000 g.
-
13.
Carefully decant the supernatant without disturbing the protein pellet.
-
14.
Allow pellet to air dry for 10–30 min in an uncapped tube. (Do not over dry otherwise, the pellet may not resuspend properly)
-
15.
Resuspend the pellet in 50 uL of Ammonium Bicarbonate Buffer (ABC buffer).
Protein estimation using Bradford assay (Bradford, 1976)
Note: By the end of this step, the protein concentration of all samples can be quantified.
-
16.
To prepare BSA standards and associated calibration curve, make a stock solution of 2 mg/mL of BSA in PBS.
-
17.
Now label vials A to I, and add the stock or the master solution and Buffer as directed in the table below to achieve the desired results concentration of standard.
| Vial | Volume of diluent |
Volume and source vial of BSA |
Final concentration |
|---|---|---|---|
| (μL) | (μL) | (μg/μL) | |
| A | 0 | 300 of stock | 2000 |
| B | 125 | 375 of stock | 1500 |
| C | 325 | 325 of stock | 1000 |
| D | 175 | 175 of vial B dilution | 750 |
| E | 325 | 325 of vial C dilution | 500 |
| F | 325 | 325 of vial E dilution | 250 |
| G | 325 | 325 of vial F dilution | 125 |
| H | 400 | 100 of vial G dilution | 25 |
| I | 400 | 0 | 0 |
-
18.
In a 96 well plate, 5 uL of pre-prepared BSA standards along with blank were added.
-
19.
5 uL of unknown samples was also loaded.
-
20.
Now to each well, 245 uL of Bradford reagent was added.
-
21.
Keep it on a shaker for 15–30 s.
-
22.
Now incubate at 25°C for 10 min in the dark.
-
23.
Take absorbance at 595 nm within 60 min post-incubation.
-
24.
Plot a standard absorbance curve at 595 nm on the "Y" axis versus the protein concentration on the "X" axis.
-
25.
Record the value "x" of unknown samples from the graph corresponding to absorbance reading and determine the protein concentration.
SDS PAGE mediated intra-sample variability detection (Nowakowski et al., 2014)
Note: By the end of this step, the intrasample variability among samples and in their associated group can be detected.
-
26.
In-gel casting apparatus assemble glass plates and spacers.
-
27.
Mix the component of resolving gel (12%).
-
28.
Pour the resolving gel mixture between the gel plates till a level 2 cm below the top of the shorter plate.
-
29.
Pace a layer of Distilled H2O over the top of the resolving gel to prevent meniscus formation in the resolving gel.
-
30.
Incubate resolving gel for 30 min at 25°C
-
31.
Drain the water from the top of the resolving gel and wick any remaining water away with a Kimwipe.
-
32.
Mix stacking gel components and pour into gel plates (on top of resolving gel) to fill the gel plates.
-
33.
Insert comb at the top of the spacers.
-
34.
Remove the comb and assemble cast gel into the SDS apparatus.
-
35.
Add 1× bromophenol blue to each sample.
-
36.
Add freshly prepared 1× running buffer to both chambers of the apparatus
-
37.
Now load DNA ladder in first well, 5 ug BSA in another well, and 30 ug equivalent unknown sample protein in other wells.
-
38.
Run the gel at 100 V till the dye front migrates into the running gel (∼15 min) and then increase to 120 V until the dye front reaches the bottom of the gel (∼1.5 h.).
-
39.
Now Remove the gel from the apparatus and by removing the spacers and glass plates.
-
40.
Place the gel into a small tray.
Note: Never use a metal spatula to separate the glass plates.
-
41.
Add ∼25 mL staining solution and incubate for > 30 min with gentle shaking.
-
42.
Add ∼5 mL destaining solution and destain for ∼1 min with gentle shaking, then Pour off and discard the destain solution.
-
43.
Further, add ∼ 30 mL of destain solution and destain with gentle shaking until the gel is visibly distained (> 2 h).
-
44.
Pour off and discard the destain solution and rewash the gel with distilled water.
-
45.
Visualize on GelDoc.
In-solution protein digestion (Gundry et al., 2009)
Note: By the end of this step, all the proteins will be digested, and tryptic peptides will be generated.
-
46.
50 ug equivalent proteins are diluted using the buffer till the final volume becomes 100 uL.
-
47.
Add 20 uL of 10 mM Dithiothreitol (DTT) and incubate at 60°C for 1 h in a water bath to reduce proteins.
-
48.
Post-reduction, add 15 uL of 10 mM Iodoacetamide and incubate for 30 min in the dark for alkylation of proteins,
-
49.
Add 5 uL of modified sequencing grade trypsin (1 ug/5 uL) reconstituted in 100 uL ABC buffer.
-
50.
Incubate at 37°C for 20–24 h in a water bath.
-
51.
After incubation, add 5 uL of 0.1% (v/v) Formic acid to the reaction mixture to inhibit trypsin activity.
C18 column mediated desaltation of peptides (Dowling et al., 2020)
Note: By the end of this step, all the contaminants, e.g., salt, will be removed.
-
52.
Please open a new Pierce C18 column, place it in an empty 1.5 mL Eppendorf tube,
-
53.
Incubate it with 250 uL of Buffer C for 5 min (Column Washing).
-
54.
Centrifuge for 2 min on a tabletop centrifuge and discard the flowthrough.
-
55.
Add 200 uL of Buffer A in the column, then incubate for 2 min (Column Washing).
-
56.
Centrifuge for 2 min on a tabletop centrifuge and discard the flowthrough.
-
57.
Add 200 uL of Buffer B in the column,
-
58.
Incubate for 2 min (Column equilibration).
-
59.
Discard the flowthrough.
-
60.
Now load the digested sample.
-
61.
Incubate it for 5 min.
-
62.
Centrifuge for 2 min on a tabletop centrifuge and then reload the flowthrough (peptide-binding).
-
63.
Incubate again for 5 min and repeat step 49.
-
64.
Repeat steps 49 and 50 two more times.
-
65.
Centrifuge for 2 min on a tabletop centrifuge and discard the flowthrough.
-
66.
Add 200 uL of Buffer B in the column, then incubate for 2 min (Washing).
-
67.
Centrifuge for 2 min on a tabletop centrifuge and discard the flowthrough.
-
68.
Add 30 uL of Buffer C and Centrifuge for 2 min on a tabletop centrifuge.
-
69.
Repeat step 55.
-
70.
Finally, add 40 uL Buffer C and Centrifuge for 2 min on a tabletop centrifuge.
-
71.
Take the flowthrough and discard the used column.
-
72.
Lyophilize the samples at 4°C and 60 millibar pressure till the sample is dry (do not over dry sample; otherwise, they will be tough to reconstitute).
-
73.
Reconstitute in 40 uL of 0.1% (v/v) Formic acid.
-
74.
Centrifuge at 15,000 g for and load the supernatant in an HPLC vial.
LC-MS/MS analysis (Bhat et al., 2020; Das et al., 2017, 2019; Dowling et al., 2020)
Note: By the end of this step, the MS.raw files would be curated.
-
75.
The peptides were ionized by nano-electrospray and subsequent tandem mass spectrometry (MS/MS) on a Q-ExactiveTM Plus (Thermo Fisher Scientific, San Jose, CA, United States).
-
76.
The peptides were eluted by a 3–95% gradient of buffer B (aqueous 80% acetonitrile in 0.1% formic acid) with a continuous flow rate of 300 nL/min for about 60 min on a 25-cm analytical C18 column (C18, 3 mm, 100 A).
-
77.
The peptides were analyzed using a mass spectrometer with the collision-induced dissociation mode with the electrospray voltage 2.3 kV.
-
78.
Analysis on the orbitrap was performed with full scan MS spectra with a resolution of 70,000 from m/z 350 to 1800. Mascot algorithm (Mascot 2.4, Matrix Science).
-
79.
Significant proteins were identified at (p < 0.05) and q values (p < 0.05). The threshold of false discovery rate was kept at 0.01.
Database curation for host proteome, metaproteome, and virome (Maras et al., 2021)
Note: By the end of this step, the database would be curated on which MS.raw files would be mapped.
-
80.
The raw files generated post-LC-MS/MS analysis were analyzed using Proteome discoverer 2.2 for protein identification and quantification.
-
81.
One of the most crucial parts is database curation, against which the mass spectrometric files need to be searched. So, in our case, we created three databases: a Virome (COVID) database, another for humans, and one for metaproteome.
-
82.
To curate the database, visit https://www.uniprot.org/proteomes/.
-
83.
Find proteome for your organism of interest by searching for them from proteome ID, e.g., UP000005640 – Homo sapiens) or taxonomic name or ID.
-
84.
Now click on the download button> Select all protein entries > Fasta (canonical, compressed, and isoform). (Figure 1)
Note: the databases in Uniprot are of three categories reviewed (Uniprot/Swissport), Unreviewed (Uniprot/TREMBL), or both (UniprotKB) options. Always prefer the reviewed databases because reviewed databases are carefully annotated with minimal redundancy and can integrate into other databases efficiently.
-
85.
We used the following databases
| Application | Organism | Source |
|---|---|---|
| Host Proteome | Homo sapiens | Uniprot homo sapiens (Human) database (UP000005640; reviewed) |
| Virome | COVID-19 | https://covid-19.uniprot.org/uniprote |
| Metaproteome | Amalgamation | bacterial/fungal sequence (UniprotSwP_20170609, with sequences 467231 and MG_BG_UPSP with sequences2019194) |
Figure 1.

Overview of data curation process at UNIPROT website
Proteome discoverer 2.2 mediated data analysis (Maras et al., 2021)
Note: By the end of this step, the excel data files would be curated.
Note: The steps for data curation from raw files for Proteome, Metaproteome, and virome are the same, and the same raw files can be used for data curation analysis of all three.
The only difference among the three is that each need to be run individually against their respective database, e.g., Host proteome should be mapped against Human Uniprot database)
-
86.
Open Proteome discoverer 2.2.
-
87.
Click New Study. (Figure 2)
-
88.
Name your study > Decide the root directory > Add your raw files > Press ok. (Figure 3)
-
89.
Click on New Analysis. (Figure 4)
-
90.
Click on Input files > Select all files and drag it below the processing workflow window > Click on the icon of the processing step. (Figure 5)
-
91.
Click on Open common > Select your processing workflow and press enter. (Figure 6)
-
92.
Click on Spectrum files and > go to Search settings > Select the Protein database, e.g., for COVID virome, select the already curated COVID database> Select the type of digestion we performed, e.g., tryptic peptides. (Figure 7)
Note: This is the place where you will select the database for proteome, metaproteome, and virome or any other organism associated.
-
93.
Now, select each step of the Processing workflow.
Note: Setting for each parameter group is given in the image below. (Figure 8)
Figure 2.

Pictographic representation of step 87 associated to proteome discoverer 2.2 mediated data analysis section
Figure 3.

Pictographic representation of step 88 associated to proteome discoverer 2.2 mediated data analysis section
Figure 4.

Pictographic representation of step 89 associated to proteome discoverer 2.2 mediated data analysis section
Figure 5.

Pictographic representation of step 90 associated to Proteome discoverer 2.2 mediated data analysis section
Figure 6.

Pictographic representation of step 91 associated to proteome discoverer 2.2 mediated data analysis section
Figure 7.

Pictographic representation of step 92 associated to proteome discoverer 2.2 mediated data analysis section
Figure 8.

Pictographic representation of step 93 associated to proteome discoverer 2.2 mediated data analysis section
-
94.
Now click on consensus workflow > then Open common > and select your workflow. (Figure 9) Step 10-Now select each step of the processing workflow.
Note: Setting for each parameter group is given in the image below. (Figure 10)
Figure 9.

Pictographic representation of step 94 associated to proteome discoverer 2.2 mediated data analysis section
Figure 10.

Pictographic representation of step 94 associated to proteome discoverer 2.2 mediated data analysis section
-
95.
Click on grouping and quantification> Then select Sample type. (Figure 11)
-
96.
Click on Run. (Figure 12)
-
97.Wait for 24–72 h. for data analysis to happen, and finally, you will get a file like below. (Figure 13)Note: You have to repeat Proteome discoverer 2.2 mediated data analysis five times.
-
a.For proteome run all raw files of COVID positive, COVID negative, and H1N1 samples together.
-
b.For virome, similarly run all raw files of COVID positive, COVID negative, and H1N1 samples together.
-
c.For the metaproteome, run three times for COVID positive, once for COVID negative, and then finally once for H1N1.)Note: From here, the further downstream data processing is the same for Host proteome and virome and different for metaproteome.
-
a.
Figure 11.

Pictographic representation of step 95 associated to proteome discoverer 2.2 mediated data analysis section
Figure 12.

Pictographic representation of step 96 associated to proteome discoverer 2.2 mediated data analysis section
Figure 13.

Final output result file from proteome discoverer (step 105)
Downstream data Processing for Metaproteome analysis (Maras et al., 2021)
Note: By the end of this step final metaproteome excel file will be curated.
-
98.
By the end of step 13, you will have three files for the metaproteome, one for each group.
-
99.
Open the excel file of one group. (Figure 14)
-
100.
Copy all the tryptic peptides
-
101.
Visit the Unipept website https://unipept.ugent.be/. (Figure 15)
-
102.
Enter the list of tryptic peptides here > Click search (Figure 16).
-
103.
You will get a sunburst plot from Unipept (Figure 17).
-
104.
Now download the data from Unipept by clicking on the download tab. (Figure 18).
-
105.
You will get a file like this (Figure 19).
-
106.
Repeat these steps for the other two groups.
-
107.
Now, you will have this excel file for all groups.
-
108.
Now, compile all three files and master peptide files to get the abundance of each bacterium (Figure 20).
-
109.
During this process, you can identify each disease group-specific metaproteome and compile the relative abundance of metaproteome.
-
110.
Now use this file for statistical analysis
Note: the files generated at step 13 of proteome and virome can also be used for statistical analysis.
Figure 14.

Pictographic representation of step 99 associated to downstream data processing for metaproteome analysis
Figure 15.

Pictographic representation of step 101 associated to downstream data processing for metaproteome analysis
Figure 16.

Pictographic representation of step 102 associated to downstream data processing for metaproteome analysis
Figure 17.

Pictographic representation of step 102 associated to downstream data processing for metaproteome analysis
Figure 18.

Pictographic representation of step 104 associated to downstream data processing for metaproteome analysis
Figure 19.

Pictographic representation of step 105 associated to downstream data processing for metaproteome analysis
Figure 20.

Pictographic representation of step 108 associated to downstream data processing for metaproteome analysis
Expected outcomes
The importance of Global Proteomics in SARS-CoV-2 biology can help us understand the virus-host protein interaction and provide the index of its variability depending on the cell type used or sample type studied. Together, this can help us understand the mechanism of virus infection and provide us with potential biomarkers. On the contrary SARS-CoV-2 infection often precedes with bacterial co-infection and is linked with longer duration and more severe infection (van den Brand et al., 2014) thus metaproteome studies can give us insights into how the COVID-19 infection changes the microbiome of the human body and is this change in microflora can be associated to the severity of the disease. This protocol describes how to execute global proteomics to identify host proteome, virus-linked proteome (Virome), and bacteria-linked proteome (metaproteome) in the respiratory specimen of the study group. This protocol describes an approach that combines the Sample collection, virus inactivation, protein isolation, alkylation, digestion, desalination, and peptide sequencing using a Q-exactive + mass spectrometer bioinformatics approaches for data analysis like database curation, using proteome discoverer 2.2 and metaproteome analysis.
The expected outcome of these studies would be a list of a few hundred to several thousands of proteins and their expression pattern. Now via the proteomics approach, you can understand the change in host proteome pattern, virome can reveal the insights of virus proteins in the host, and finally, the metaproteome study can reveal the change in it and intensity of change. For example, in our COVID study (Maras et al., 2021), we identified 6 significantly differentially expressed COVID-19 viral proteins; around 1256 host proteins were identified. We also found an increase in respiratory pathogens (e.g., Burkholderiales, Klebsiella pneumonia) and decreased lactobacillus salivary (FDR<0.05) in the COVID-19 specimen. Finally, an increase in the basal level of WARS (tryptophan-tRNA ligase) and MX1 (MX dynamin-like GTPase 1) was correlated with SARS-CoV-2 infection outcome. Further, these were validated in a separate cohort of 200 patients.
Quantification and statistical analysis
Systems bioinformatics
For further downstream analysis of proteome, virome, and Metaproteome data is performed using the following software's-
-
a.
Uniprot (https://www.uniprot.org): A comprehensive database of protein sequences. (The usage is explained in detail in the bioinformatics section, especially database curation).
-
b.
Enricher (https://maayanlab.cloud/Enrichr/): Program for the functional enrichment analysis of proteins.
(Open the site > Copy the list of Gene symbols and paste in the Query section > Press Submit > Open the Reactome database > Download)
-
c.
PANTHER (http://www.pantherdb.org): Program for the functional enrichment analysis of proteins.
(Open the site > Copy the list of Gene symbols and paste in Query section > Select ID type > Select organism > Select statistical overrepresentation test > Choose Reactome annotation > Submit > Select reference genome and run.
-
d.
Metaboanalyst 4.0 (https://www.metaboanalyst.ca/): A web server used for all the statistical analysis associated with the proteome, virome, and Metaproteome data.
(Open site > Click on start > Click on statistical analysis > select data type, file and format > Submit and select preferred normalization > Perform desired statistics)
-
e.
Cytoscape (https://cytoscape.org/): a global cross-correlation map between the virome, metaproteome, and the pathways linked to the proteins and metabolites using Cytoscape.
-
f.
Unipept (https://unipept.ugent.be/): Database that's designed for metaproteomic data analysis with a focus on interactive data visualizations.
(The usage is explained in detail in the bioinformatics section, especially database curation)
Limitations
Although mass spectrometry-based proteomics and viromics are non-replaceable techniques in terms of how much they are essential in giving the snapshot of Protein expression of whole-cell, unlike genome, the proteome of cells is highly dynamic, and thus, multiple samples and time points need to be considered if study demands temporal profiling (Betzen et al., 2015). The starting sample should be fresh as degradation in the proteins would result in lower identification. The extensive protein and peptide cleanup procedure used in the protocol tends to lose some proteins and peptides, resulting in lower identification.
Troubleshooting
Problem 1
Interference of chemicals such as polymers in LC-MS/MS analysis (steps 83–87).
Potential solution
In such a scenario, the prepared peptides samples need to be reprecipitated. Add 6× acetone to your peptide samples and incubate them at −20°C for 1 h > centrifuge at maximum g possible and discard the supernatant. Dissolve the pellet again and reanalyze in mass-spectrometer.
If still the problem persists, reprepare your samples, and this time before using any plastic ware, soak them in any organic solvent (e.g., ethanol or acetone) for 24–72 h to remove any contaminant polymer.
Problem 2
The trap column of Nano-LC gets blocked (steps 83–87).
Potential solution
In such a scenario, first, you disconnect the column from source > then reverse the direction of flow in the column by reversing the direction of column > then start the isocratic flow and wait till water starts to appear or flow from the open end of the column once it happens. Rearrange the column and start.
Problem 3
The air bubble got trapped in the nano-LC pump (steps 83–87).
Potential solution
In such a scenario, first, you disconnect the column from source > then join the column end with waste outlet> then start purge > the flush finally performs a leak test if passed. Rearrange the column and start.
Problem 4
No Peaks/Very Small Peaks (steps 83–87).
Potential solution
In such a scenario, check that the lamp is on and the cables are well connected. Next, ensure that the flow is normal and the automatic sampler is working properly, not blocked or jammed.
Problem 5
No or low protein detection (steps 60–82).
Potential solution
In such a scenario take your flowthrough and repurify peptide from it again. Never discard the flowthrough.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Jaswinder Singh Maras (jassi2param@gmail.com).
Materials availability
The study did not generate any materials.
Acknowledgments
We want to acknowledge the Science and Engineering Research Board Department of Science and Technology Government of INDIA for providing funds to build the manuscript. The work was supported by project DST (DST-SERB) (EMR/2016/004829).
Author contributions
J.S.M. conceptualized the work. The manuscript was written by G.T. and J.S.M., with help from M.Y., N.S., B.M., V.B., S.S., and S.K.S. The manuscript was read and approved by all authors.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Jaswinder Singh Maras, Email: jassi2param@gmail.com.
Shiv Kumar Sarin, Email: shivsarin@gmail.com.
Data and code availability
No data is provided, and the raw data for the manuscript is available on request to the lead contact Dr. Jaswinder Singh Maras (jassi2param@gmail.com).
References
- Batejat C., Grassin Q., Manuguerra J.C., Leclercq I. Heat inactivation of the severe acute respiratory syndrome coronavirus 2. J. Biosaf Biosecur. 2021;3:1–3. doi: 10.1016/j.jobb.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzen C., Alhamdani M.S., Lueong S., Schroder C., Stang A., Hoheisel J.D. Clinical proteomics: promises, challenges and limitations of affinity arrays. Proteomics Clin. Appl. 2015;9:342–347. doi: 10.1002/prca.201400156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat A., Das S., Yadav G., Chaudhary S., Vyas A., Islam M., Gupta A.C., Bajpai M., Maiwall R., Maras J.S., et al. Hyperoxidized albumin modulates platelets and promotes inflammation through CD36 receptor in severe alcoholic hepatitis. Hepatol. Commun. 2020;4:50–65. doi: 10.1002/hep4.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Das S., Hussain M.S., Maras J.S., Kumar J., Shasthry S.M., Nayak S., Arora V., Vijayaraghavan R., Sharma S., Maiwall R., et al. Modification patterns of urinary albumin correlates with serum albumin and outcome in severe alcoholic hepatitis. J. Clin. Gastroenterol. 2019;53:e243–e252. doi: 10.1097/MCG.0000000000000990. [DOI] [PubMed] [Google Scholar]
- Das S., Maras J.S., Hussain M.S., Sharma S., David P., Sukriti S., Shasthry S.M., Maiwall R., Trehanpati N., Singh T.P., et al. Hyperoxidized albumin modulates neutrophils to induce oxidative stress and inflammation in severe alcoholic hepatitis. Hepatology. 2017;65:631–646. doi: 10.1002/hep.28897. [DOI] [PubMed] [Google Scholar]
- Dowling P., Gargan S., Zweyer M., Henry M., Meleady P., Swandulla D., Ohlendieck K. Protocol for the bottom-up proteomic analysis of mouse spleen. STAR Protoc. 2020;1:100196. doi: 10.1016/j.xpro.2020.100196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundry R.L., White M.Y., Murray C.I., Kane L.A., Fu Q., Stanley B.A., Van Eyk J.E. Preparation of proteins and peptides for mass spectrometry analysis in a bottom-up proteomics workflow. Curr. Protoc. Mol. Biol. 2009;Chapter 10:Unit10 25. doi: 10.1002/0471142727.mb1025s88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maras J.S., Sharma S., Bhat A., Rooge S., Aggrawal R., Gupta E., Sarin S.K. Multi-omics analysis of respiratory specimen characterizes baseline molecular determinants associated with SARS-CoV-2 outcome. iScience. 2021;24:102823. doi: 10.1016/j.isci.2021.102823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski A.B., Wobig W.J., Petering D.H. Native SDS-PAGE: high resolution electrophoretic separation of proteins with retention of native properties including bound metal ions. Metallomics. 2014;6:1068–1078. doi: 10.1039/c4mt00033a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pondaven-Letourmy S., Alvin F., Boumghit Y., Simon F. How to perform a nasopharyngeal swab in adults and children in the COVID-19 era. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2020;137:325–327. doi: 10.1016/j.anorl.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson D.M., Beynon R.J. Acetone precipitation of proteins and the modification of peptides. J. Proteome Res. 2010;9:444–450. doi: 10.1021/pr900806x. [DOI] [PubMed] [Google Scholar]
- van den Brand J.M., Haagmans B.L., van Riel D., Osterhaus A.D., Kuiken T. The pathology and pathogenesis of experimental severe acute respiratory syndrome and influenza in animal models. J. Comp. Pathol. 2014;151:83–112. doi: 10.1016/j.jcpa.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data is provided, and the raw data for the manuscript is available on request to the lead contact Dr. Jaswinder Singh Maras (jassi2param@gmail.com).