

Abbreviations
- GPX4
Glutathione peroxidase 4
- FSP1
Ferroptosis suppressor protein‐1
- DHODH
Dihydroorotate dehydrogenase
- PUFA‐PLs
polyunsaturated‐fatty‐acid–containing phospholipids
- 15‐HpETE‐PE
15‐hydroperoxy‐arachidonoyl‐phosphatidylethanolamine
- PLA2
phospholipase A2
- iPLA2β
Calcium‐independent phospholipase A2β
- PD
Parkinson's disease
- 4‐HNE
4‐hydroxynonenal
Ferroptosis, a form of iron‐dependent regulated cell death caused by excessive accumulation of lipid hydroperoxides, has been associated with various pathological conditions and diseases [1]. Excessive ferroptosis has been causally associated with acute kidney injury, cardiovascular, neurodegenerative and hepatic diseases, whereas impaired ferroptosis in premalignant cells has been shown to contribute to tumor development [2, 3]. To escape from ferroptotic cell death, cells have been equipped with several antioxidant defense systems against lipid peroxidation (Figure 1). Glutathione peroxidase 4 (GPX4) suppresses ferroptosis by converting lipid hydroperoxides into non‐toxic lipid alcohols at the expense of its cofactor glutathione (GSH) [4]. Ferroptosis suppressor protein‐1 (FSP1, also known as AIFM2), a NAD(P)H‐dependent oxidoreductase located on the plasma membrane, catalyzes the reduction of ubiquinone to ubiquinol, a radical trapping antioxidant that suppresses ferroptosis independent of the GSH‐GPX4 axis [5, 6]. In addition, dihydroorotate dehydrogenase (DHODH), an enzyme involved in the de novo pyrimidine biosynthesis pathway, inhibits ferroptosis by reducing ubiquinone to ubiquinol in the inner mitochondrial membrane [7].
FIGURE 1.
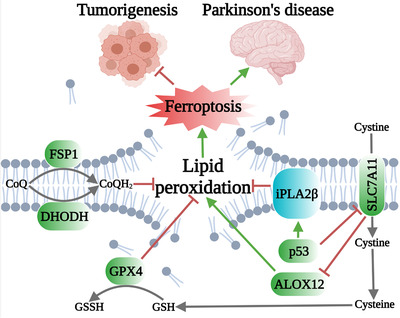
Several cellular defense systems against lipid peroxidation and ferroptosis, and the relevance of ferroptosis to tumorigenesis and Parkinson's disease. GPX4 detoxifies lipid hydroperoxides and represses ferroptosis by converting reduced GSH into GSSG. FSP1 and DHODH catalyze the reduction of CoQ to CoQH2 on the plasma membrane and inner mitochondrial membrane, respectively; CoQH2 acts as a radical trapping antioxidant to suppress lipid peroxidation and ferroptosis. p53 appears to have two opposing functions in ferroptosis. p53 promotes ferroptosis at least partly by suppressing the expression of SLC7A11, which inhibits ferroptosis through both GPX4‐dependent (via GSH) and ‐independent (via ALOX12) pathways. On the other hand, p53 upregulates the expression of iPLA2β, which suppresses ferroptosis. iPLA2β inactivation suppresses tumorigenesis and promotes Parkinson's disease at least partly through promoting ferroptosis. Abbreviations: GPX4, glutathione peroxidase 4; GSH, glutathione; GSSG, oxidized glutathione; FSP1, ferroptosis suppressor protein‐1; DHODH, dihydroorotate dehydrogenase; CoQ, ubiquinone; CoQH2, ubiquinol; SLC7A11, solute carrier family 7 member 11; ALOX12, arachidonate 12‐lipoxygenase; iPLA2β, calcium‐independent phospholipase A2β.
Most lipid peroxidation occurs in polyunsaturated fatty acid‐containing phospholipids (PUFA‐PLs; PUFAs are fatty acids that harbor more than one double bond). Due to the chemical features of double bonds in PUFAs, PUFA‐PLs are exquisitely vulnerable to peroxidation in cellular environments rich in iron and oxygen. One major oxidized PUFA‐PL species that is believed to trigger ferroptosis is 15‐hydroperoxy‐arachidonoyl‐phosphatidylethanolamine (15‐HpETE‐PE) [8]. However, how cells neutralize 15‐HpETE‐PE (and other oxidized PUFA‐PLs) and escape from ferroptotic cell death remains incompletely understood.
The phospholipase A2 (PLA2) gene family encodes protein enzymes that specifically hydrolyze the sn‐2 ester bond on PLs, yielding free fatty acids and lysophosphatidic acids. Calcium‐independent phospholipase A2β (iPLA2β), a member of the PLA2 family, is a cytosolic protein that is catalytically active in the absence of calcium and can be stimulated by ATP. In response to stress or injury, iPLA2β preferentially releases free fatty acids (particularly PUFAs, such as arachidonic acid, eicosapentaenoic acid, and docosahexaenoic acid) from the sn‐2 position of PLs. While iPLA2β was initially implicated as a housekeeping enzyme in membrane remodeling, recent studies have suggested an engaged role of iPLA2β in a variety of cellular processes, including calcium homeostasis, apoptosis, and inflammation, and its dysregulation has been genetically linked to diverse human diseases, such as Parkinson's disease (PD), male infertility, cardiovascular abnormalities, and cancer [9]. However, the underlying mechanisms by which iPLA2β dysregulation is linked to these human diseases or pathological conditions still remain largely unknown. Two recent studies revealed that iPLA2β preferentially hydrolyzes peroxidized PLs, such as 15‐HpETE‐PE, and acts as a GPX4‐independent repressor of p53‐driven ferroptosis [10, 11]. These findings further suggest that iPLA2β is a promising therapeutic target for cancer therapy and that its mutation may be relevant to PD.
Sun et al. [10] examined the specific hydrolytic activity of iPLA2β, revealing that both 1‐stearoyl (SA)‐2‐ETE‐PE and 1‐SA‐2‐15‐HpETE‐PE (one specific 15‐HpETE‐PE) were highly hydrolyzed by iPLA2β. Further studies from both experimental analyses and computational modeling suggested that 1‐SA‐2‐15‐HpETE‐PE is likely the preferred substrate for iPLA2β. R747W is a loss‐of‐function mutant in iPLA2β that is associated with infantile neuroaxonal dystrophy and adult‐onset dystonia‐parkinsonism. Notably, the R747W mutant was shown to exhibit lower hydrolytic activity toward 1‐SA‐2‐15‐HpETE‐PE than did the wild‐type iPLA2β protein. Likewise, the phospholipase activity of iPLA2β toward 1‐SA‐2‐15‐HpETE‐PE was dramatically lower in cells harboring PD‐associated mutation (fPDR747W) than in wild‐type counterparts. Mechanistically, it was proposed that R747W iPLA2β mutant protein has less accessible catalytic site and weaker membrane association as compared with the wild‐type iPLA2β protein, which likely explains its lower hydrolytic activity toward its substrate 1‐SA‐2‐15‐HpETE‐PE. Given the important role of 15‐HpETE‐PE in driving ferroptosis, these studies further suggest that iPLA2β might suppress ferroptosis by hydrolyzing 15‐HpETE‐PE. In support of this hypothesis, Sun et al. [10] showed that fPDR747W cells contained more oxidized PEs and exhibited more sensitivities to GPX4 inhibitor RSL3‐induced ferroptosis than did wild‐type cells.
In another study, Chen et al. [11] uncovered a role of iPLA2β in ferroptosis governance through their efforts to understand tumor suppressor p53's function in ferroptosis. Combined treatment of tert‐butyl hydroperoxide (a ferroptosis inducer) and Nutlin (a p53 activator) was found to induce high levels of lipid peroxidation and to result in potent p53‐dependent ferroptotic cell death; notably, p53‐driven ferroptosis appeared to be independent of GPX4. This prompted the authors to identify additional downstream targets of p53 to control ferroptosis. Through mining RNA‐sequencing data, Chen et al. [11] revealed that iPLA2β is a direct transcriptional target of p53; both pharmacological and genetic activation of p53 significantly upregulated iPLA2β mRNA levels, whereas p53 deficiency dampened Nutlin‐induced iPLA2β expression. They further showed that iPLA2β depletion promoted ferroptosis in p53 wild‐type cells but not in p53‐null counterparts. In addition, overexpression of iPLA2β, but not its enzymatic inactive mutant, was found to reduce the levels of oxidized PEs and to inhibit p53‐driven ferroptosis. Collectively, these data suggest that iPLA2β functions to suppress p53‐induced ferroptosis likely through hydrolyzing oxidized PUFA‐PLs. It should be noted that although p53 promotes iPLA2β expression, p53 and iPLA2β have opposite functions in regulating ferroptosis: p53 promotes whereas iPLA2β suppresses ferroptosis. Therefore, iPLA2β is unlikely the p53 transcriptional target that mediates p53's function in promoting ferroptosis; rather, it seems that iPLA2β as a p53's target serves to buffer p53's ability to induce ferroptosis (Figure 1).
The above two studies also examined the disease relevance of iPLA2β’s function in ferroptosis governance. Chen et al. [11] showed that iPLA2β deficiency suppressed xenograft tumor growth and increased ferroptosis marker expression in p53 wild‐type but not in p53‐null tumors, suggesting that iPLA2β could promote tumor growth in a p53‐dependent manner and possibly through suppressing ferroptosis in vivo. It should be noted that while iPLA2β is a transcriptional target of p53, the dependency of iPLA2β‐mediated regulation of ferroptosis and tumor growth on p53 suggests that somehow p53 can also operate downstream of iPLA2β, although the underlying mechanism remains unclear. Sun et al. [10] studied iPLA2β in the context of PD. iPLA2β is encoded by the PNPLA9 gene. Since mouse Pnpla9 R748W mutant corresponds to human PNPLA9 R747W mutant (which is associated with PD), they developed Pnpla9 R748W /R748W mouse models and found that Pnpla9 R748W /R748W mice indeed developed behavioral defects related to PD pathogenesis, with increased levels of 15‐HpETE‐PE and 4‐hydroxynonenal (4‐HNE, a lipid peroxidation marker) in midbrains of these mice compared with those in wild‐type mice. In addition, the levels of tyrosine hydroxylase, a biomarker for dopaminergic neurons in the central nervous system and its product dopamine were found to be significantly lower in the midbrain of Pnpla9 R748W/R748W mutant mice than in control mice. These analyses suggest a causal effect of ferroptosis in mediating iPLA2β mutation's function in PD pathogenesis.
Together, these two intriguing studies [10, 11] reveal that iPLA2β acts as a guardian against ferroptosis by hydrolyzing and thereby eliminating pro‐ferroptotic oxidized PUFA‐PLs, and further suggest that iPLA2β deficiency or its loss‐of‐function mutation can suppress tumor growth or promote PD pathogenesis, potentially by inducing aberrant ferroptosis (Figure 1). These studies also raised several outstanding questions. Since the PLA2 gene family encodes multiple phospholipases, whether other PLA2 members are also involved in ferroptosis governance remains to be investigated. iPLA2β normally localizes in the cytosol, but upon stimulation it can mobilize to organelles such as the endoplasmic reticulum, mitochondria, and nucleus. For example, iPLA2β has been implicated in repairing mitochondrial phospholipids [12]. In light of recent findings suggesting separate ferroptosis defense systems on the plasma membrane and in the mitochondria in cells [7, 13], whether iPLA2β participates in ferroptosis defense in different subcellular compartments under diverse cellular conditions remains a fascinating area for future studies. In addition, although these two studies have causally linked ferroptosis to iPLA2β’s functions in tumor biology and PD pathogenesis, the extent to which ferroptosis contributes to tumor suppression or PD‐associated phenotypes caused by iPLA2β deletion or mutation remains unclear. Considering the multifaceted roles of iPLA2β in cellular signaling, other biological functions of iPLA2β might also be involved. Rescue analyses by ferroptosis inhibitors such as liproxstatin‐1 or ferrostatin‐1 will help address this important question. Further, as a key transcription factor involved in ferroptosis regulation, p53's activity is tightly regulated by post‐translational modifications such as phosphorylation and acetylation. For example, the suppressor of cytokine signaling 1 (SOCS1) reduces the expression of SLC7A11 through promoting p53 serine‐15 phosphorylation and thereby sensitizes cells to ferroptosis [14]. In addition, p53 acetylation at lysine K98 is essential for its ability to inhibit SLC7A11 expression and to promote ferroptosis in mouse xenograft models [15]. Whether these post‐translational modifications of p53 are also involved in regulating iPLA2β expression remains to be tested. Likewise, it will be interesting to identify other transcription factors that regulate iPLA2β expression and ferroptosis in future investigations. Finally, from a therapeutic perspective, the study by Chen et al. [11] suggests iPLA2β inhibitors as potential therapeutic strategies in treating p53 wild‐type tumors. However, the study by Sun et al. [10] would argue that such therapeutic interventions might cause other complications, including promoting PDs. It remains to be established whether there exist appropriate therapeutic windows for iPLA2β inhibitors to selectively kill tumors without inducing obvious toxicities in the brain and other organs. Future investigations will be directed to understanding these important questions.
DECLARATIONS
AUTHORS’ CONTRIBUTIONS
CM drafted the manuscript with additional support from GL for checking relevant literature. BG provided critical revision of the manuscript with additional manuscript editing support from LZ. All authors read and approved the final manuscript.
COMPETING INTERESTS
BG is an inventor of patent applications involving targeting ferroptosis in cancer therapy. Other authors have no conflicts of interest to declare.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
Research in the authors’ lab has been supported by The University of Texas MD Anderson Cancer Center, National Institutes of Health grants R01CA181196, R01CA244144, and R01CA247992 (to BG) and by Cancer Center Support (Core) Grant P30 CA016672 from the National Cancer Institute (to The University of Texas MD Anderson Cancer Center).
ACKNOWLEDGMENTS
Not applicable.
REFERENCES
- 1. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An iron‐dependent form of nonapoptotic cell death. Cell. 2012;149:1060‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jiang L, Kon N, Li T, Wang S‐J, Su T, Hibshoosh H, et al. Ferroptosis as a p53‐mediated activity during tumour suppression. Nature. 2015;520:57‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. Bap1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20:1181‐1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020;32:920‐937. [DOI] [PubMed] [Google Scholar]
- 5. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. Fsp1 is a glutathione‐independent ferroptosis suppressor. Nature. 2019;575:693‐698. [DOI] [PubMed] [Google Scholar]
- 6. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The coq oxidoreductase fsp1 acts parallel to gpx4 to inhibit ferroptosis. Nature. 2019;575:688‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. Dhodh‐mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586‐590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, St Croix C, et al. Oxidized arachidonic and adrenic pes navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Malley KR, Koroleva O, Miller I, Sanishvili R, Jenkins CM, Gross RW, et al. The structure of ipla2beta reveals dimeric active sites and suggests mechanisms of regulation and localization. Nat Commun. 2018;9:765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun WY, Tyurin VA, Mikulska‐Ruminska K, Shrivastava IH, Anthonymuthu TS, Zhai Y‐J, et al. Phospholipase ipla2beta averts ferroptosis by eliminating a redox lipid death signal. Nat Chem Biol. 2021;17:465‐476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen D, Chu B, Yang X, Liu Z, Jin Y, Kon N, et al. Ipla2beta‐mediated lipid detoxification controls p53‐driven ferroptosis independent of gpx4. Nat Commun. 2021;12:3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ma ZA. The role of peroxidation of mitochondrial membrane phospholipids in pancreatic beta ‐cell failure. Curr Diabetes Rev. 2012;8:69‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gan B. Mitochondrial regulation of ferroptosis. J Cell Biol. 2021;220:e202105043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saint‐Germain E, Mignacca L, Vernier M, Bobbala D, Ilangumaran S, Ferbeyre G. Socs1 regulates senescence and ferroptosis by modulating the expression of p53 target genes. Aging (Albany NY). 2017;9:2137‐2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, et al. Acetylation is crucial for p53‐mediated ferroptosis and tumor suppression. Cell Rep. 2016;17:366‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.