

Abstract
Background:
Mitochondrial dysfunction results in an imbalance between energy supply and demand in a failing heart. An innovative therapy that targets the intracellular bioenergetics directly through mitochondria transfer may be necessary.
Objectives:
The purpose of this study was to establish a preclinical proof-of-concept that extracellular vesicles (EVs)-mediated transfer of autologous mitochondria and their related energy source enhance cardiac function through restoration of myocardial bioenergetics.
Methods:
Human-induced pluripotent stem cells (iPSCs)-derived cardiomyocytes (iCMs) were employed. iCMs conditioned-medium were ultracentrifuged to collect mitochondria-rich EVs (M-EVs). Therapeutic effects of M-EVs were investigated using in vivo murine myocardial infarction (MI) model.
Results:
Electron microscopy revealed healthy-shaped mitochondria inside M-EVs. Confocal microscopy showed that M-EV-derived mitochondria were transferred into the recipient iCMs and fused with their endogenous mitochondrial networks. Treatment with 1.0×108/mL M-EVs significantly restored the intracellular ATP production and improved contractile profiles of hypoxia-injured iCMs as early as 3-hours after treatment. In contrast, isolated mitochondria that contained 300 times more mitochondrial proteins than 1.0×108/mL M-EVs showed no effect after 24-hours. M-EVs contained mitochondrial biogenesis-related mRNAs, including PGC-1α, which upon transfer activated mitochondrial biogenesis in the recipient iCMs at 24-hours after treatment. Finally, intramyocardial injection of 1.0×108 M-EVs demonstrated significantly improved post-MI cardiac function through restoration of bioenergetics and mitochondrial biogenesis.
Conclusions:
M-EVs facilitated immediate transfer of their mitochondrial and non-mitochondrial cargos, contributing to improved intracellular energetics in vitro. Intramyocardial injection of M-EVs enhanced post-MI cardiac function in vivo. This therapy can be developed as a novel, precision therapeutic for mitochondria-related diseases including heart failure.
CONDENSED ABSTRACT:
Mitochondrial dysfunction results in an imbalance between energy supply and demand in mitochondria-related diseases, including heart failure (HF). Currently, no therapeutic enhances intracellular energetic supply. The goal of this study is to establish a preclinical proof-of-concept that extracellular vesicles-mediated transfer of autologous mitochondria confers cardioprotection through restoration of cellular bioenergetics. Mitochondria-rich extracellular vesicles (M-EVs) are secreted from induced-pluripotent stem cell-derived cardiomyocytes. Intramyocardial injection of M-EVs facilitate immediate transfer of their mitochondrial and non-mitochondrial cargo into the injured cardiomyocytes, improving cardiac function in mice myocardial infarction model. This novel approach may provide an effective therapeutic for mitochondria-related diseases.
Keywords: human stem cells, mitochondria, myocardial infarction, heart failure, bioenergetics
Introduction
Pharmacologic therapies have improved survival in patients with heart failure (HF) over the past three decades (1). Despite these pioneering medical therapies, the number of HF patients is increasing steadily and represents the top diagnosis of hospital admissions today. This fact highlights the need for a development of innovative treatment strategies (1–2). Heart failure represents bioenergetic imbalance. The disruption of the balance between energy supply and demand is important for the pathogenesis of HF (3). Cardiac tissue from patients with dilated, hypertrophic and ischemic cardiomyopathies exhibits mitochondrial structural abnormalities and diminished ATP production despite increased metabolic energy demands in the failing heart (4–7). Although proliferator-activated receptor γ coactivator-1α (PGC-1α) serves as a master regulator of mitochondrial biogenesis and functional capacity, PGC-1α expression levels are decreased in the myocardium of HF patients (8–9). Insufficient energy generation results in cardiomyocyte contractile abnormality, myocardial dysfunction and, ultimately, decompensated HF. The current standard pharmacologic regimen, including β-blockers (10–11), ivabradine (12–13), and renin–angiotensin-aldosterone antagonism (14–15), attempt to correct this imbalance by reducing cardiac workload, namely, energy demand. These therapeutics are essentially noncurative because they do not target the primary energy source of the failing heart. Therefore, it is essential to develop a novel therapy that targets the intracellular energetic supply directly (16).
Cell-based therapy is reported to improve the cardiac function in animal models of myocardial infarction (MI). Further analysis demonstrated that only a small proportion of the transplanted cells engraft in the recipient heart (17). These data support a ‘paracrine hypothesis’ of stem cell therapy and suggest that the isolation of the secreted factors may have important applications for HF therapy. We and others have reported that extracellular vesicles (EVs) efficiently transfer their cargo into the recipient cells, facilitating various intercellular communications (18–19). Recently, it has been reported that extracellular mitochondria exist inside the EVs (20–22). Astrocytes actively released EVs that contain functional mitochondria via calcium-dependent mechanisms involving CD38 (23). Extracellular-mitochondria were internalized by injured neurons and contributed to better neurologic outcomes in a mice stroke model. These data suggest that EVs facilitate a secure and highly efficient transfer of their mitochondrial cargo into the recipient cells.
Induced pluripotent stem cells (iPSCs) have tremendous therapeutic potential for the treatment of cardiovascular disease (24). The promise of precision medicine can be realized by using patient-specific iPSC derivatives such as iPSC-derived cardiomyocytes (iCMs), which secrete EVs containing functional mitochondria. The goal of this study was to establish a preclinical proof-of-concept that EV-mediated transfer of autologous mitochondria restores myocardial bioenergetics. Moreover, the role of the non-mitochondrial EV cargo in activating PGC-1α-mediated mitochondrial biogenesis in the recipient iCMs was elucidated (Central Illustration).
Central Illustration. Extracellular Vesicle-Mediated Autologous Mitochondrial Transplantation.
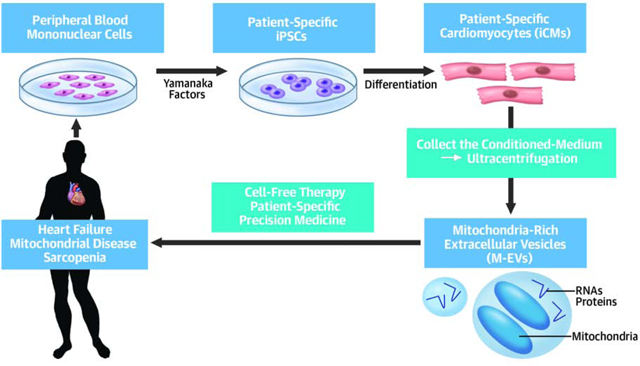
We obtained human-induced pluripotent stem cells (iPSCs)-derived cardiomyocytes (iCMs) and succeeded in collecting mitochondria-rich extracellular vesicles (M-EVs) from iCM conditioned-medium. The autologous M-EVs can be applied to patients with mitochondria-related diseases including heart failure.
Methods
This study has been approved by Stanford University School of Medicine Institutional Review Board committee and Institutional Animal Care and Use Committee. (IRB Approval number: 31517, APLAC Approval number: 33723, and IACUC Assurance Number: A3213–01). All animals received humane care and treatment in accordance with the “Guide for the Care and Use of Laboratory Animals” (www.nap.edu/catalog/5140.html). Detailed methods are described in the Online Appendix.
Preparation of human blood mononuclear cell-derived iPSCs and iCMs
The monoclonal iPSCs lines were generated by transfection of human blood mononuclear cells with non-integrating Sendai virus. iPSCs were chemically differentiated into iCMs.
Isolation of mitochondria-rich extracellular vesicles (M-EVs) and Large vesicle-depleted EVs (Ld-EVs)
The iCM conditioned-medium were collected and M-EVs were obtained based on a differential ultracentrifugation method. Floating cells, cell debris and apoptotic bodies were removed by centrifugation at 1,000g for 10-minutes at 4 °C. Pelleted vesicles were obtained by ultracentrifugation of the supernatant at 10,000g for 30-minutes at 4 °C. Ld-EVs were obtained by filtering of M-EVs through 220-nm polyvinylidene fluoride membrane filter.
Nanoparticle tracking analysis (NTA)
Size and quantity of EVs were evaluated by NTA with a NanoSight LM20 (NanoSight, UK). Samples were loaded into the sample chamber with sterile syringes and imaged using a 40 mW at 640-nm laser.
FCM-based EV analysis
1×108 EVs were washed and suspended in 200 μL of iCM culture medium and stained with 100 nM MTDR (Thermo Fisher Scientific, Waltham, MA) and 100 nM MTG (Thermo Fisher Scientific) for 10-minutes at room temperature (RT) and protected from light. Flow cytometry was conducted at the Stanford Shared FACS Facility (SSFF) on a BD LSRII instrument.
In vitro hypoxia model
iCMs were placed in a Hypoxia Inductor Chamber (Stemcell Technologies, Canada). Oxygen content was reduced to 0.8–1.0% using N2/CO2 gas mix, and monitored continuously. The chamber was placed in a 37°C incubator for 24-hours.
Adenosine Triphosphate (ATP) measurement.
Intracellular ATP was determined by CellTiter-Glo luminescence kit (Promega, G7570). Opaque-walled 96-well plates with iCMs (10,000 cells/well) were prepared. CellTiter-Glo luminescence test solution was added and incubated for 30-minutes at RT. Luminescent signal was determined by a luminescence microplate reader.
Mouse myocardial infarction (MI) model
Adult female CD1 mice (Age: 80–100 days) were purchased from Charles River. Experimental groups were randomized across multiple cages, litters, and location of mouse cages in the husbandry room. Mice were endotracheally intubated. Anesthesia was maintained with inhalational 1.5–2.0 % isoflurane. A thoracotomy was performed, and an 8–0 silk ligature was placed around the left anterior descending artery 1 mm below the atrioventricular border. M-EVs (1 × 108/30 μL PBS/group), Ld-EVs (1 × 108/30 μL PBS/group), Isolated-Mitochondria (1.0 μg protein/30 μL PBS) or 30 μL PBS were injected at 3-sites around infarct regions. The chest was closed, and animals were weaned from the ventilator and extubated.
Statistical Analysis
Data are expressed as the mean ± standard deviations. We analyzed the differences between the two groups using unpaired t-tests; the differences among 3 groups or more were assessed using an ANOVA and post-hoc Tukey’s or Bonferroni’s multiple comparison tests with Prism Software version 4.0 (Graph Pad Software, CA). P values<0.05 were considered statistically significant. Detailed methods and statistical analyses are described in the Online Appendix.
This study has been approved by Stanford University School of Medicine Institutional Review Board committee and Institutional Animal Care and Use Committee.
RESULTS
Immature iCMs show robust expressions of transcriptional co-activators that regulate mitochondrial biogenesis
iCMs mature in long-term culture (25–26). Based on these findings, we characterized iCMs at 15-days (D15) vs. 50-days (D50) in culture after the observation of initial spontaneous contractions. Consistent with previous reports, D15 showed immature phenotypes in terms of sarcomeric structure, electrical conduction and Ca2+ handling when compared to D50 (Supplemental Figures 1A and 1B). D50 showed higher expression levels of electron transport chain (ETC) proteins/transcripts and mitochondrial DNA (mtDNA) copy numbers (Supplemental Figure 1C to 1E). In contrast, mRNA levels of mitochondrial dynamics-related genes were decreased in D50 (Supplemental Figure 1D). Two types of mitochondria-specific labeling (27) that distinguish functional mitochondria (mitotracker deep red, MTDR) and total mitochondria (mitotracker green, MTG), revealed that D50 showed more functional mitochondria (Supplemental Figure 1F). Intracellular ATP levels were higher in D50 (Supplemental Figure 1G). As D15 continue to differentiate into mature cardiomyocytes, an increase in mitochondria is reported to be orchestrated by PGC-1α-mediated mitochondrial biogenesis (28–29). Consistent with this, the less mature D15 showed higher activity level of mitochondrial biogenesis through increased expression levels of PGC-1α and ERRγ (Supplemental Figures 1C and 1H).
Immature iCMs secrete mitochondria-containing EVs that improve mitochondrial function of hypoxia-injured cardiomyocytes
Flow cytometry (FCM) revealed that D15 conditioned-medium contained more extracellular-mitochondria than D50 (Supplemental Figure 2A). Extracellular-mitochondria from D15 showed greater proportions of MTDR+-functional mitochondria. Filtration of the conditioned-medium with 220-nm filter reduced the quantity of extracellular-mitochondria (Supplemental Figures 2A and 2B), suggesting that extracellular-mitochondria were incorporated in >220 nm EVs. FCM side-scatter-area (SSA) revealed that >240 nm EVs contained 4.7 times more mitochondria than <240 nm EVs (Supplemental Figure 2C). We next assessed the in vitro effects of the conditioned-medium on the mitochondrial function. iCMs were treated with the conditioned-medium for 24-hours and the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction was measured as an indicator of mitochondrial function. The conditioned-medium from D15 or D50 did not change MTT reduction in the iCMs without hypoxic injury (Supplemental Figure 2D). Then iCMs were subjected to hypoxia for 24-hours and treated with the conditioned-medium at the time of reoxygenation. Treatment with the D15 conditioned-medium significantly increased MTT reduction, whereas D50 conditioned-medium showed less effects. 220-nm filtration of the conditioned-medium further compromised the effects, suggesting that the mitochondria-containing EVs (>220 nm) enhanced mitochondrial functions in the hypoxia-injured iCMs.
Characteristics of mitochondria-rich EVs (M-EVs)
To augment the therapeutic effects, we obtained M-EVs by differential ultracentrifugation of the conditioned-medium according to previously published microvesicle-purification (30) and mitochondria isolation methods (Figure 1A). Nanoparticle Tracking Analysis (NTA) revealed that the size of M-EVs from both D15 and D50 ranged from 100 to 600 nm and the mean diameter was 295 and 237 nm, respectively (Supplemental Figure 3A). D15 produced higher numbers of M-EVs that contained more mitochondria than D50 (Figure 1B and Supplemental Figure 3B). 87% of mitochondria inside D15 M-EVs retained their membrane potentials. Double staining with wheat-germ-agglutinin (WGA), a cell membrane dye, and MTG revealed that the majority (81%) of WGA-positive EVs contained mitochondria (Supplemental Figure 3C). Filtration of M-EVs through 220-nm filter resulted in large vesicle-depleted EVs (Ld-EVs) that did not contain mitochondria (Figure1B, Supplemental Figure 3D). NTA confirmed that the filtration technique depleted the EVs >220 nm (Supplemental Figure 3E). 40% and 43% of D15 M-EVs were positive for β1-integrin and CD63, respectively (Supplemental Figure 3F). Transmission electron microscopy of M-EVs revealed round-shaped EVs with a diameter ranging from 98 to 677 nm (mean 204 nm, Figure 1C). Extracellular-mitochondria with preserved structural integrity were observed inside the EVs. ETC proteins were detected in M-EVs, but not in Ld-EVs (Figure 1D). M-EVs contained more mtDNA and ATP than Ld-EVs (Supplemental Figure 3G and Figure 1E).
Figure 1. Characterization of mitochondria-rich extracellular vesicles (M-EVs).

A. Differential ultracentrifugation was employed to isolate M-EVs. B. Representative flow cytometry (FCM) dot plots and histograms of M-EVs and Large vesicle-depleted EVs (Ld-EVs). C. Transmission electron microscopy of D15 M-EVs. D. The protein expression of the electron transport chain (ETC) in D15 M-EVs. E. M-EVs, but not Ld-EVs, produced extracellular ATP (n=4/group). *P<0.0001 by an unpaired t-test. ATP5A=ATP synthase α-subunit; COX II=cytochrome c oxidase subunit-2; MTDR=mitotracker deep red; MTG=mitotracker green; MTT=3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NDUFB8=NADH dehydrogenase-1β subcomplex subunit-8; SDHB=succinate dehydrogenase complex iron sulfur subunit B; SSA=side-scatter-area; UQCRC2=ubiquinol-cytochrome c reductase core protein-2.
M-EVs transfer their mitochondrial cargo into hypoxia-injured iCMs and restore intracellular bioenergetics
We next observed the transfer of M-EV-mitochondria into the recipient cells. We labeled M-EV-mitochondria with red fluorescent proteins (RFP) and native mitochondria in the recipient cells with green fluorescent proteins (GFP) using BacMam technology. FCM revealed that M-EVs containing RFP+-mitochondria were secreted from D15 transduced with BacMam mitochondria-RFP (Supplemental Figure 4). iCMs transduced with BacMam mitochondria-GFP were subjected to hypoxia and treated with 3.0×108/mL RFP+-M-EVs. Confocal microscopy revealed that M-EV-mitochondria (red) were transferred into the recipient iCMs and some of them were fused with the host mitochondrial networks (green) at 3- and 24-hours after treatment (Figure 2A and 2B).
Figure 2. M-EVs facilitate transfer of their mitochondrial cargo and restore intracellular bioenergetics in hypoxia-injured iCMs.

A and B. Representative microscopic image of iCMs treated with M-EVs containing RFP+-mitochondria. Transferred M-EV-mitochondria (red) were observed in the recipient iCMs harboring GFP+-mitochondria (green). Some of the transferred mitochondria were fused with the native mitochondrial networks (white arrows). Scale bar: 50 and 20 μm. C to F. ATP levels in iCMs treated with M-EVs or Ld-EVs. (0, 1.0×107, 3.0×107, 1.0×108 or 3.0×108/mL: wedge). Left, 3-hours after treatment. Right, 24-hours after treatment (n=4–6/group). *P<0.05 and **P<0.001 by a one-way ANOVA followed by Tukey’s test.
We next examined the effects of M-EVs on intracellular bioenergetics. When iCMs were subjected to hypoxia, ATP levels fell as expected (Figure 2C). Treatment with D15 M-EVs at the time of reoxygenation increased ATP levels in a dose dependent manner and 1.0×108/mL or 3.0×108/mL M-EVs showed significant effects at 3- and 24-hours after treatment (Figure 2C). D50 M-EVs also showed significant effects, albeit to a lesser extent (Figure 2D). We and others reported that EVs from cardiomyocytes deliver their RNA or protein cargos into the recipient cells, providing various therapeutic effects (19, 31). Therefore, we next examined whether the non-mitochondrial cargo in M-EVs had significant effects on the intracellular bioenergetics. Although Ld-EVs showed no significant effect at 3-hours after treatment, 3.0×108/mL D15 Ld-EVs significantly improved intracellular ATP levels at 24-hours after treatment (Figure 2E). In contrast, D50 Ld-EVs showed no effects within 24-hours (Figure 2F). To confirm the effects of non-mitochondrial cargo, D15 were treated with oligomycin A (32), a specific ATP synthase inhibitor, and collected M-EVs (Oligo-EVs). The size distribution of Oligo-EVs were similar to M-EV (Supplemental Figure 5A). Mitochondria inside Oligo-EVs were dysfunctional (Supplemental Figure 5B and C). However, 3.0×108/mL Oligo-EVs significantly improved ATP levels at 24-hours after treatment (Supplemental Figure 5D). These findings suggest that D15 M-EVs contain non-mitochondrial cargos that enhance bioenergetics in the hypoxia-injured iCMs.
Non-mitochondrial cargos in D15 M-EVs enhance mitochondrial biogenesis and bioenergetics in the recipient iCMs
The RNA/protein profiles of EVs are reported to mirror those of their parent cells (33). D15 showed on-going mitochondrial development and higher expressions of mRNA that regulate mitochondrial biogenesis. We therefore hypothesized that D15 M-EVs contain more mRNAs that facilitate mitochondrial biogenesis. D15 Ld-EVs showed higher expression levels of PGC-1α and ERRγ mRNA than D50 Ld-EVs (Figure 3A). These mRNAs were uniformly distributed in the D15 M-EVs, D15 Ld-EVs and Mitotracker-positive EVs that were purified by fluorescence-activated cell sorter (Supplemental Figure 6A and 6B). Treatment with D15 M-EVs, but not D50 M-EVs, induced upregulation of mitochondrial biogenesis-related mRNAs in the hypoxia-injured iCMs at 24-hours after treatment (Figure 3B). The protein expressions of PGC-1α and ETC complexes were upregulated in the iCMs treated with D15 M-EVs, whereas D50 M-EVs showed no effect (Supplemental Figure6C). D15 Ld-EVs treatment showed a trend toward an increase in PGC-1α and ETC protein expressions. We measured the mitochondrial respiratory function with a mitochondrial stress test using the Seahorse XF96 extracellular-flux analyzer. D15 M-EVs treatment significantly increased basal, ATP-linked and maximal respiration in hypoxia-injured iCMs (Figure 3C). D15 Ld-EVs also restored basal and maximal respiration. These data suggested that non-mitochondrial cargo in D15 M-EVs enhance the intracellular bioenergetics through activation of mitochondrial biogenesis.
Figure 3. Non-mitochondrial cargo in D15 M-EVs restored bioenergetics through activation of PGC-1α-mediated mitochondrial biogenesis in the recipient iCMs.

A. PGC-1α and ERRγ mRNA, normalized to U6, were upregulated in D15 Ld-EVs (n=4/group). *P<0.05 by an unpaired t-test. B. Quantitative gene expression in iCMs, normalized to ACTB shown as fold change relative to normoxic iCMs (n=4–5/group). C. Seahorse extracellular-flux assays measuring oxygen-consumption rate (OCR) (n=5–6/group). *P<0.01, **P<0.001 and ***P<0.0001 by a one-way ANOVA followed by Tukey’s test. ATP5A1=ATP synthase F1 α-subunit; COX6A1=cytochrome c oxidase subunit-6A1; Cs=citrate synthase; CYC1=cytochrome c1; ERRγ=estrogen-related receptor γ; FCCP=carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone; NRF1=Nuclear respiratory factor-1; NDUFA1=NADH dehydrogenase-1α subcomplex subunit-1; PGC-1α=peroxisome proliferator-activated receptor γ coactivator 1α; PKM=pyruvate kinase M1/2; SDHA=succinate dehydrogenase complex flavoprotein subunit-A; TFAM=mitochondrial transcription factor-A.
PGC-1α knockdown compromises the restoration of bioenergetics and mitochondrial biogenesis in the hypoxia-injured iCMs
We evaluated the role of PGC-1α in the effects of M-EV therapy. D15 were transduced with lentivirus harboring anti-PGC-1α short hairpin RNA (shPGC-1α). We confirmed that shPGC-1α decreased the expression levels of PGC-1α mRNA and protein compared with lentivirus harboring non-targeted shRNA (Supplemental Figure 7A and 7B). Ld-EVs from D15 transduced with shPGC-1α showed marked reduction in PGC-1α and ERRγ mRNA levels (Figure 4A). Importantly, PGC-1α knockdown significantly reduced the effects of Ld-EVs treatment on the restoration of ATP levels (Figure 4B) and PGC-1α/ETC protein levels in hypoxia-injured iCMs at 24-hours after treatment (Figure 4C). These data suggest the significant role of PGC-1α-related mRNA and protein in the M-EVs therapy.
Figure 4. PGC-1α knockdown compromised the restoration of mitochondrial bioenergetics and biogenesis in the hypoxia-injured iCMs.

A. Relative PGC-1α and ERRγ mRNA expression, normalized to U6, in Ld-EVs from D15 transduced with pLKO.1 expressing control shRNA (shCTRL) or anti-PGC-1α shRNA (shPGC-1α) (n=3/group). *P<0.0001 by an unpaired t-test. B. PGC-1α knockdown reduced restoration of ATP levels in hypoxia-injured iCMs at 24hous (n=5–8/group). C. Representative immunoblots and quantitative analyses of PGC-1α, ETC complexes and β-actin (n=4–6/group). *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001 by a one-way ANOVA followed by Tukey’s test. Abbreviations as in Figure 1.
Isolated mitochondria did not restore bioenergetics in hypoxia-injured iCMs within 24-hours after treatments
We next obtained isolated mitochondria from homogenized D15 (Isolated-Mito) and confirmed that the Isolated-Mito (3.0 μg/mL protein) harbored the same amounts of mitochondrial proteins (VDAC and COX IV) in approximately 3.0×108/mL D15 M-EVs (Supplemental Figure 8A). Hypoxia-injured iCMs were treated with RFP-labeled Isolated-Mito (3.0 μg/mL protein) and observed by confocal microscopy. Isolated-Mito (red) were rarely detected inside the recipient iCMs within 24-hours after treatment (Supplemental Figure 8B). Isolated-Mito (300 μg/mL) that contain 100 times more mitochondrial proteins than 3.0×108/mL M-EVs had no effect on the recovery of ATP levels within 24-hours (Supplemental Figure 8C). These data suggested that the EVs facilitated the transfer of their mitochondrial cargo into the recipient iCMs.
Mitochondria inside EVs are more resistant to Ca2+ overload and oxidative stress than isolated mitochondria
Extracellular environment with high Ca2+ concentration (~2.0 mM) and oxidative stress may compromise the viability of exogenous mitochondria injected into the injured heart tissues. Therefore, we investigated whether M-EV-mitochondria vs. Isolated-Mito maintain function after incubation in buffers with various concentrations of Ca2+ or hydrogen peroxide (H2O2). After 3- or 24-hours incubation, D15 M-EVs or Isolated-Mito were labeled with MTDR and analyzed by FCM. M-EVs retained functional mitochondria up to concentration of 2 mM Ca2+ within 24-hours, whereas 24-hours incubation with Ca2+ at 0.2 and 2 mM significantly reduced MTDR signals in Isolated-Mito (Supplemental Figure 9A). EV-mitochondria retained MTDR signals up to concentration of 100 mM H2O2 within 24-hours, whereas MTDR signals were decreased in Isolated-Mito incubated with buffers containing 100 mM H2O2 at 3- and 24-hours after treatment (Supplemental Figure 9B). These data suggested that M-EV-mitochondria were more resistant to Ca2+ overload and oxidative stress than Isolated-Mito.
M-EVs improve mitochondrial function, contractile profile and cell survival of hypoxia-injured iCMs
Appropriate level of intracellular bioenergetic is essential for mitochondrial quality, contractile activity and, ultimately, cell survival. iCMs were exposed to hypoxia for 24-hours and treated with 3.0×108/mL D15 M-EVs at the time of reoxygenation. Double-labeling of injured iCMs with MTG and MTDR revealed that M-EV treatment significantly increased the proportion of functional mitochondria (MTG+MTDR+) at 24-hours after treatment (Figure 5A). 3.0×108/mL M-EVs significantly improved the contractile profiles, including D peak and max contractile rate at 24-hours after treatment (Figure 5B). M-EVs treatment increased viable cells by reducing early phase apoptotic cell death at 24-hours after treatment (Figure 5C). In contrast, Isolated-Mito that contained the same amount of mitochondrial proteins as 3.0×108/mL M-EVs showed no effect on the mitochondrial and cellular phenotypes.
Figure 5. D15 M-EVs improved mitochondrial function, contractile property, and cell survival in hypoxia-injured iCMs.

A. Representative FCM dotplots of iCMs labeled with MTG/MTDR (n=5–6/group). B. D peak and maximum contractile rate in iCMs (n=5–6/group). C. Representative FCM dotplots of iCMs stained with Annexin V/Propidium iodide (n=4–5/group). *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001 by a one-way ANOVA followed by Tukey’s test.
M-EVs therapy prevents post-MI cardiac remodeling
In an in vivo mouse model of MI, intramyocardial injection of D15 M-EVs (1.0×108) into the peri-infarct region significantly improved left ventricular ejection fraction (LVEF) as compared with control group (Figure 6A, 6B and Online Table. 3). Furthermore, MRI measurements, including LV end-diastolic volume (LVEDV), LV end-systolic volume (LVESV) and LV mass demonstrated significantly reduced LV remodeling in D15 M-EV treated animals (Figure 6C to 6E). Manganese-enhanced MRI (MEMRI) detected significantly greater viable myocardium in D15 M-EVs (Figure 6F). D15 Ld-EVs improved LV-size and LVEF significantly less when compared to D15 M-EVs. D50 M-EVs or D50 Ld-EVs prevented post-MI LV-dilatation, while no effect was observed in LVEF (Supplemental Figures 10A to 10E). Isolated-Mito that contained the same amount of mitochondrial proteins as D15 M-EVs (1.0×108) showed no effect. At week-4, we measured lung weight and LV weight, showing that the treatment with D15 M-EVs had trends toward reduced lung and LV weight when indexed by tibia-length (Supplemental Figure 10F and 10G). These data suggested the superiority of D15 M-EV therapy in preventing post-MI LV remodeling.
Figure 6. The effects of intramyocardial injection of D15 M-EVs on post-myocardial infarction (MI) cardiac remodeling.

A. Representative images of short-axis acquisitions at mid-left ventricle (LV). B to E. LV ejection fraction (LVEF), LV end-diastolic volume (LVEDV), LV end-systolic volume (LVESV) and LV mass at weeks-2 and −4 after MI, evaluated by MRI. F. Representative images and quantitative analyses of myocardial viability as visualized by manganese-enhanced MRI (MEMRI). Red arrows: non-viable regions (n=8/group). *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001 by a two-way ANOVA followed by Tukey’s test.
M-EV therapy restores bioenergetics and activates PGC-1α-mediated mitochondrial biogenesis in the failing heart.\
We next examined the mechanisms by which D15 M-EVs prevented progression of post-MI HF. MTDR-labeled M-EVs (1.0×108) were injected into myocardium in the peri-infarct region. Immunohistochemistry at 8-hours after injection demonstrated that MTDR+-functional mitochondria were detected inside the troponin I+-cardiomyocytes in the peri-infarct region (Figure 7A). To trace the transferred mitochondria, we measured human mtDNA in the peri-infarct myocardium. D15 M-EVs injected myocardium showed 62 times higher human mtDNA when compared to Isolated-Mito group at 3-days after MI (Supplemental Figure 11A). However, human mtDNA was not detected in either group at day-28. Intramyocardial injections of D15 M-EVs, but not Isolated-Mito, significantly increased tissue ATP levels at day-3 and −28 (Figure 7B). M-EVs treatment induced upregulation of mitochondrial biogenesis-related mRNAs, including PGC-1α, ERRγ, SDHB, CYTC and COX4I1 at day-3 (Figure 7C). The protein expressions of PGC-1α and COX IV were up-regulated at day-28 (Figure 7D). Finally, D15 M-EV injection increased mtDNA copy numbers at day-28 (Supplemental Figure11B). These data suggested that M-EV therapy restored bioenergetics and facilitated mitochondrial biogenesis in the peri-infarct region.
Figure 7. D15 M-EV therapy enhances bioenergetics and PGC-1α-mediated mitochondrial biogenesis in the peri-infarct region.

A. MTDR+-mitochondria were detected inside the Troponin I+-cardiomyocytes, highlighted with white arrows. Scale bar: 50 μm. B. Tissue ATP levels, normalized by tissue protein levels (n=4–6/group). C. Mitochondrial biogenesis-related gene expression levels in the peri-infarct region, normalized to GAPDH, measured by RT-PCR (n=4–5/group). D. Representative immunoblots and quantitative analyses of PGC-1α, VDAC and COX IV, normalized to GAPDH (n=4–5/group). *P<0.05 by a one-way ANOVA followed by Tukey’s test. ATP5ME=ATP synthase subunit-e; COX IV=cytochrome c oxidase subunit-4; COX4I1=cytochrome c oxidase subunit-4 isoform-1; NDUFAB1=NADH ubiquinone oxidoreductase subunit-AB1; VDAC=voltage-dependent anion channels; other abbreviations as in Figure 3.
Discussion
The novel findings of the present study are the following: 1) D15 M-EVs contained more functional mitochondria than D50 M-EVs; 2) EV-mediated transfer of mitochondrial and non-mitochondrial cargo restore intracellular bioenergetics and contractile property in hypoxia-injured iCMs; 3) Therapeutic benefit from non-mitochondrial cargos in D15 M-EVs contents depends on PGC-1α-related mRNA/proteins; and 4) intramyocardial injection of 1.0×108 D15 M-EVs improves post-MI cardiac function in mice.
The effectiveness of iCMs transplantation has been reported in preclinical MI models (34). Our data suggest that immature iCMs can be a promising candidate for cell therapy because they may supply mitochondria and/or activate mitochondrial biogenesis and enhance dynamics. However, immature iCMs exhibit less developed conduction and Ca2+ handling (Supplemental Figure1), which may cause automaticity-induced arrhythmias in the transplanted myocardium, as previously reported (34). To address this, we explored the therapeutic effects of paracrine factors. Although the arrhythmogenicity of M-EVs injection needs to be examined using large animal models, this cell-free approach is expected to confer cardioprotection without inducing ventricular arrhythmia.
Intramyocardial injection of isolated mitochondria reduced infarct size in animal models of ischemia/reperfusion injury (35). However, clinical application of this technique is extremely challenging (36) due to the following constraints of isolated mitochondria: (1) effective function in the extracellular environment, (2) transmembrane transfer into the recipient cardiomyocytes, and (3) sufficient and sustainable ATP generation for contractile performance in the cardiomyocytes. M-EV therapy addresses these challenges. M-EV-mitochondria were more resistant to high extracellular Ca2+ concentrations than isolated-mitochondria (Supplemental Figure 9), suggesting that the lipid bilayer of the EVs prevented Ca2+ diffusion to damage the mitochondria and the related cargo. EVs facilitated quick transfer of their mitochondrial cargo into the recipient iCMs in contrast to the Isolated-Mito that are rarely internalized by the iCMs (Figure 2, Supplemental Figure 8). These findings suggested that the lipid bilayer of EVs is able to fuse with the cell membrane, resulting in direct release of their cargo into the cytoplasm of the recipient cells. In addition, the non-mitochondrial cargo of D15 M-EVs facilitated PGC-1α-mediated mitochondrial biogenesis in the recipient iCMs (Figure 3B), suggesting that mitochondrial and non-mitochondrial cargos may contribute to sufficient and persistent restoration of intracellular energetics in hypoxia-injured iCMs and post-MI murine heart.
M-EVs are smaller than the expected size range of intracellular mitochondria, suggesting that the fragmented mitochondria via mitochondrial fission machinery are engulfed into EVs. Consistent with this finding, D15 that exhibited higher expression level of mitochondrial dynamics-related genes secreted more mitochondria-containing EVs than D50 (Figure 1B). These mitochondria are not “dysfunctional mitochondria” that are destined to be eliminated by autophagy because they retain structural and functional integrity (Figures 1B –1E).
Our data showed that human-derived M-EVs restored bioenergetics in mouse heart tissues. It is reported that mitochondrial fusion proteins such as Mfn1/2 and Opa1 in mouse and human cells have high degree of homologies. Human-derived mitochondria efficiently fuse to the mitochondrial networks in mice cells and mixing of the matrix protein content is completed within 4-hours (37). These data suggest that human-derived mitochondria inside the M-EVs were transferred into mouse cardiomyocytes and fuse to native mitochondrial networks, resulting in enhanced mitochondrial function.
Study limitation
We acknowledge that this study has several experimental limitations. First, we have only examined the EVs secreted from iCMs on the assumption that the cardiomyocytes harbor the most active mitochondria and produce the most effective M-EVs. However, we cannot exclude other cell-types, which could yield equally effective M-EVs. Second, we used differential ultracentrifugation to isolate the M-EVs. However, it is unknown what other methods may isolate the M-EVs more efficiently. Future studies are warranted to clarify the most effective EV cell source and optimal purification methods. Finally, we have only investigated mitochondrial biogenesis-related mRNA profiles as non-mitochondrial therapeutic factors. It is, however, reported that transfer of protein clusters or non-coding RNAs inside the EVs also initiates various intracellular signaling. Comprehensive analysis of EV-related proteins/RNAs is required to identify non-mitochondrial factors that activate mitochondrial biogenesis.
Conclusion
This study demonstrated the feasibility of M-EV-mediated transfer of mitochondria and their related bioenergetic cargo for the treatment of failing hearts. Our data provide an important proof-of-concept of successful in vitro augmentation of cellular bioenergetics and effective in vivo intramyocardial delivery of M-EVs in a murine model of MI. This iCM-based autologous source of bioenergetics may provide an effective therapy for mitochondria-related diseases including advanced HF patients.
Supplementary Material
Clinical Perspectives.
Competency in Medical Knowledge:
Mitochondria-rich extracellular vesicles (M-EV) transfer mitochondrial and non-mitochondrial substances into cardiomyocytes, stimulating mitochondrial biogenesis and enhancing energy metabolism in failing myocardium.
Translational Outlook:
Studies of transendocardial administration of M-EV in larger animal models would facilitate assessment of the therapeutic potential of this strategy for patients with heart failure.
Acknowledgments:
We thank Meredith Weglarz (Stanford Shared FACS Facility) and John Perrino (Stanford Cell Science Imaging Facility).
Funding
Stanford Dean’s Postdoctoral Fellowship (GI)
Japan Heart Foundation/Bayer Yakuhin (GI)
AHA postdoctoral fellowship (GI)
Alpha Omega Alpha Carolyn B. Kuckein Student Research Fellowship (MS)
NIH/NHLBI 1 K24 HL130553K (PY)
NIH/NHLBI UM1 L12026 (PY)
SPARK Stanford University (PY)
ABBREVIATIONS
- D15
iCMs cultured for shorter than 15 days
- D50
iCMs cultured for longer than 50 days
- EV
Extracellular vesicle
- HF
heart failure
- iPSC
Induced pluripotent stem cell
- iCM
iPSC-derived cardiomyocyte
- Isolated-Mito
Isolated mitochondria from homogenized D15
- Ld-EV
Large vesicle-depleted extracellular vesicle
- M-EV
Mitochondria-rich extracellular vesicle
- MI
Myocardial infarction
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. J Am Coll Cardiol. 2013. October 15;62(16):e147–239. [DOI] [PubMed] [Google Scholar]
- 2.Taylor CJ, Ordóñez-Mena JM, Roalfe AK, et al. Trends in survival after a diagnosis of heart failure in the United Kingdom 2000–2017:population based cohort study. BMJ. 2019;364:1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neubauer S. The failing heart-an engine out of fuel. N Engl J Med. 2007;356:1140–1151. [DOI] [PubMed] [Google Scholar]
- 4.Sekiguchi M. Electron microscopical observations of the myocardium in patients with idiopathic cardiomyopathy using endomyocardial biopsy. J Mol Cell Cardiol. 1974;6:111–122. [DOI] [PubMed] [Google Scholar]
- 5.Starling RC, Hammer DF, Altschuld RA. Human myocardial ATP content and in vivo contractile function. Mol Cell Biochem. 1998;180:171–177. [PubMed] [Google Scholar]
- 6.Beer M, Seyfarth T, Sandstede J, et al. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol. 2002;40:1267–1274. [DOI] [PubMed] [Google Scholar]
- 7.Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN. Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol. 2000;32:2361–2367. [DOI] [PubMed] [Google Scholar]
- 8.Sebastiani M, Giordano C, Nediani C, et al. Induction of mitochondrial biogenesis is a maladaptive mechanism in mitochondrial cardiomyopathies. J Am Coll Cardiol. 2007;50:1362–1369. [DOI] [PubMed] [Google Scholar]
- 9.Garnier A, Zoll J, Fortin D, et al. Control by circulating factors of mitochondrial function and transcription cascade in heart failure:a role for endothelin-1 and angiotensin-II. Circ Heart Fail. 2009;2:342–350. [DOI] [PubMed] [Google Scholar]
- 10.Waagstein F, Hjalmarson A, Varnauskas E, Wallentin I. Effect of chronic beta-adrenergic receptor blockade in congestive cardiomyopathy. Br Heart J. 1975;37:1022–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Packer M, Coats AJ, Fowler MB, et al. Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med. 2001;344:1651–1658. [DOI] [PubMed] [Google Scholar]
- 12.Fox K, Ford I, Steg PG, Tendera M, Ferrari R. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL):a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:807–816. [DOI] [PubMed] [Google Scholar]
- 13.Swedberg K, Komajda M, Böhm M, et al. Ivabradine and outcomes in chronic heart failure (SHIFT):a randomised placebo-controlled study. Lancet. 2010;376:875–885. [DOI] [PubMed] [Google Scholar]
- 14.CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med. 1987;316:1429–1435. [DOI] [PubMed] [Google Scholar]
- 15.Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–717. [DOI] [PubMed] [Google Scholar]
- 16.Brown DA, Perry JB, Allen ME, et al. Expert consensus document:Mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. 2017;14:238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murry CE, Soonpaa MH, Reinecke H, et al. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature. 2004;428:664–668. [DOI] [PubMed] [Google Scholar]
- 18.Jung J-H, Fu X, Yang PC. Exosomes Generated From iPSC-Derivatives:New Direction for Stem Cell Therapy in Human Heart Diseases. Circ Res. 2017;120:407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Couto G, Gallet R, Cambier L, et al. Exosomal MicroRNA Transfer into Macrophages Mediates Cellular Postconditioning. Circulation. 2017;136:200–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jang SC, Crescitelli R, Cvjetkovic A, et al. Mitochondrial protein enriched extracellular vesicles discovered in human melanoma tissues can be detected in patient plasma. J Extracell Vesicles. 2019. August 27;8(1):1635420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puhm F, Afonyushkin T, Resch U, et al. Mitochondria Are a Subset of Extracellular Vesicles Released by Activated Monocytes and Induce Type I IFN and TNF Responses in Endothelial Cells. Circ Res. 2019. June 21;125(1):43–52. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Martinez I, Santoro N, Chen Y, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest. 2016. March 1;126(3):859–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayakawa K, Esposito E, Wang X, et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 2016;535:551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell. 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- 25.Yang X, Pabon L, Murry CE. Engineering adolescence:maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Res. 2014;114:511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lundy SD, Zhu W-Z, Regnier M, Laflamme MA. Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev. 2013;22:1991–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. [DOI] [PubMed] [Google Scholar]
- 28.Vega RB, Horton JL, Kelly DP. Maintaining Ancient Organelles:Mitochondrial Biogenesis and Maturation. Circ Res. 2015;116:1820–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu R, Greening DW, Rai A, Ji H, Simpson RJ. Highly-purified exosomes and shed microvesicles isolated from the human colon cancer cell line LIM1863 by sequential centrifugal ultrafiltration are biochemically and functionally distinct. Methods. 2015;87:11–25. [DOI] [PubMed] [Google Scholar]
- 31.Tachibana A, Santoso MR, Mahmoudi M, et al. Paracrine Effects of the pluripotent stem cell-derived cardiac myocytes salvage the injured myocardium. Circ Res. 2017;121:e22–e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim MJ, Hwang JW, Yun C-K, Lee Y, Choi Y-S. Delivery of exogenous mitochondria via centrifugation enhances cellular metabolic function. Sci Rep. 2018;8:3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adamiak M, Cheng G, Bobis-Wozowicz S, et al. Induced Pluripotent Stem Cell (iPSC)-Derived Extracellular Vesicles Are Safer and More Effective for Cardiac Repair than iPSCs. Circ Res. 2018;122(2);296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shiba Y, Gomibuchi T, Seto T, et al. Allogeneic transplantation of iPS cell-derived cardiomyocytes regenerates primate hearts. Nature. 2016. October 20;538(7625):388–391. [DOI] [PubMed] [Google Scholar]
- 35.Kaza AK, Wamala I, Friehs I, et al. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J Thorac Cardiovasc Surg. 2017;153:934–943. [DOI] [PubMed] [Google Scholar]
- 36.Bertero E, Maack C, O’Rourke B. Mitochondrial transplantation in humans:“magical” cure or cause for concern? J Clin Invest. 2018;128:5191–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoon YG, Haug CL, Koob MD. Interspecies mitochondrial fusion between mouse and human mitochondria is rapid and efficient. Mitochondrion. 2007;7:223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.