

Abstract
The drugs salmeterol, formoterol, and salbutamol constitute the frontline treatment of asthma and other chronic pulmonary diseases. These drugs activate the β2-adrenergic receptors (β2-AR), a class A G protein–coupled receptor (GPCR), and differ significantly in their clinical onset and duration of actions. According to the microkinetic model, the long duration of action of salmeterol and formoterol compared with salbutamol were attributed, at least in part, to their high lipophilicity and increased local concentrations in the membrane near the receptor. However, the structural and molecular bases of how the lipophilic drugs reach the binding site of the receptor from the surrounding membrane remain unknown. Using a variety of classic and enhanced molecular dynamics simulation techniques, we investigated the membrane partitioning characteristics, binding, and unbinding mechanisms of the ligands. The obtained results offer remarkable insight into the functional role of membrane lipids in the ligand association process. Strikingly, salmeterol entered the binding site from the bilayer through transmembrane helices 1 and 7. The entry was preceded by membrane-facilitated rearrangement and presentation of its phenyl-alkoxy-alkyl tail as a passkey to an access route gated by F193, a residue known to be critical for salmeterol’s affinity. Formoterol’s access is through the aqueous path shared by other β2-AR agents. We observed a novel secondary path for salbutamol that is distinct from its primary route. Our study offers a mechanistic description for the membrane-facilitated access and binding of ligands to a membrane protein and establishes a groundwork for recognizing membrane lipids as an integral component in the molecular recognition process.
SIGNIFICANCE STATEMENT
The cell membrane’s functional role behind the duration of action of long-acting β2-adrenergic receptor (β2-AR) agonists such as salmeterol has been a subject of debate for a long time. This study investigated the binding and unbinding mechanisms of the three commonly used β2-AR agonists, salmeterol, formoterol, and salbutamol, using advanced simulation techniques. The obtained results offer unprecedented insights into the active role of membrane lipids in facilitating access and binding of the ligands, affecting the molecular recognition process and thus their pharmacology.
Introduction
The β2-adrenergic receptors (β2-AR) are class A G proteincoupled receptors (GPCR) and are highly expressed in airway smooth muscle and the alveolar region of lungs. Inhaled selective β2-AR agonists such as salbutamol, salmeterol, and formoterol constitute the first-line treatment of asthma and other pulmonary disorders (Cazzola et al., 2005, 2011; Huggins et al., 2012). By activating β2-AR, these drugs relax constricted airway smooth muscle, cause an immediate reversal of airway obstruction, and prevent bronchoconstriction by multiple mechanisms (Giembycz and Raeburn, 1991; Laurent et al., 1993; Billington et al., 2017). Based on the clinical duration of action, β2-AR agonists are classified into short-acting (e.g., salbutamol), long-acting (salmeterol and formoterol), and ultra-long-acting (indacaterol, olodaterol, and vilanterol) agents. Structurally, despite sharing a similar epinephrine-mimicking saligenin headgroup, salmeterol and formoterol differ from salbutamol by having additional bulky hydrophobic groups (phenyl-alkoxy-alkyl and methoxy-phenyl-alkyl, respectively) at their tail ends (Fig. 1). In the case of formoterol, a formamide group replaces the hydroxymethyl substitution on the saligenin phenyl ring. The apparent differences in the structural features produce markedly different clinical efficacy, onset, and duration of action (Supplemental Table 1). Both formoterol and salbutamol have a quick onset of action (5–10 minutes to produce the bronchial smooth muscle relaxation), whereas salmeterol has a slower onset of action, which is about 15-20 minutes (van Noord et al., 1998; Lötvall, 2001) after inhalation. However, the duration of action of salbutamol is shorter (∼4 hours) compared with that of salmeterol and formoterol, whose actions last for up to 12 hours (Henriksen et al., 1992; Boulet et al., 1997).
Fig. 1.

Membrane partitioning characteristics of salmeterol, formoterol, and salbutamol in the bilayer, made up of POPC and cholesterol. (A) Two-dimensional structures of the studied β2-AR agonists. (B) The PMF curves show the solvation free energies for membrane partitioning and crossing of the ligands, revealing their energetically favorable bilayer locations (of their COMs). Both salmeterol and formoterol show similar free energy profiles, whereas salbutamol has higher energy barriers for partitioning and crossing of the membrane. (C) The time-average preferred orientations of the ligands within the membrane. The nitrogen atom of the choline, phosphorous atom of the phosphate, and oxygen of the glyceryl carbonyl headgroups are represented as balls in blue, olive green, and red colors, respectively. The lipid alkyl chains are represented as lines in gray color. (D) The distinct bilayer orientation of the saligenin head in each ligand is quantified as a tilt angle between the bilayer normal (z-axis) and a vector, connecting O1 and N1 atoms of the ligands. The vertical dashed lines represent the mean values. (E) The percentage (%) H-bond occupancy of the polar atoms of each ligand with lipid headgroups (choline, phosphate, and glyceryl carbonyls) through the simulation time. H-bonds are counted only if the bond distance and angle are within the cutoff values of 3.5 Å and 40 degrees, respectively.
As there is a clear motivation for the clinical relevance of the onset and duration of action of these drugs, there have been many hypotheses to establish the possible mechanism(s) for the long duration of action of salmeterol and formoterol (Coleman, 2009). Many theories, including the existence of putative exosite/exoreceptor binding sites in or near the receptor, rebinding, and plasmalemma diffusion microkinetic models, have been postulated as possible mechanisms for the longer duration of action of salmeterol and formoterol (Anderson et al., 1994; Szczuka et al., 2009; Vauquelin, 2015). According to the exosite theory, salmeterol’s long duration of action and reassertion of its activity after washout are due to its hydrophobic tail anchoring itself in a secondary binding site either on the receptor transmembrane helices (TMHs) or in the surrounding cell membrane (Jack, 1991). A recently published crystal structure of β2-AR bound to salmeterol revealed the ligand’s binding mode and critical residues involved in interactions within and around the binding site (Masureel et al., 2018). The structure reveals that the ether oxygen on the ligand tail forms an H-bond with the main chain amino group of F193ECL2, and the phenyl-alkyl end engages in hydrophobic and π-π interactions with the surrounding residues. The oxygen atom has been proposed to act as the point of support for the pivoting saligenin head to freely reach and leave (reassertion) the central core of the β2-AR by a Charniere (hinge) principle (Ball et al., 1991).
The longer duration of action of salmeterol and formoterol relative to salbutamol is in part attributed to their high lipophilicity and propensity to partition into the membrane surrounding the receptors (Rhodes et al., 1985; Sykes et al., 2014; Dickson et al., 2016). An approximate 5000-fold difference in the membrane partition coefficient of salmeterol compared with salbutamol suggests that the exosite described by Green et al. (1996) likely reflects the phospholipid membrane rather than specific residues on the receptor. According to the plasmalemma diffusion microkinetic model, the lipid bilayer surrounding membrane proteins can affect the molecular recognition (ligand binding) process through multiple mechanisms: 1) by acting as a local reservoir for a drug and thus prolonging the drug exposure of the target; 2) by preorganizing the ligand in orientations and conformations that are thermodynamically favorable for the receptor binding; and/or 3) by altering the time needed for the drug to reach its receptor and thus affecting its binding kinetics without necessarily changing interactions with binding site residues (Swinney, 2006; Vauquelin and Packeu, 2009; Swinney DC, et al., 2015; Schoop and Dey, 2015; Vauquelin, 2015, 2016; Gherbi et al., 2018).
The mechanistic details of how the lipophilic ligands salmeterol and formoterol might access the orthosteric binding site of the receptor from the surrounding membrane remain unknown. Most importantly, it is still unknown how specific ligand-membrane interactions can direct them to a specific bilayer depth and preorganize them in orientations and conformations that affect the kinetics of ligand access to the binding site. In this study, we first determined the membrane partitioning characteristics of salmeterol, formoterol, and salbutamol using steered molecular dynamics and umbrella sampling simulation techniques. Next, we investigated the potential association and dissociation paths for the ligands, elucidating the role of membrane lipid interactions and critical residue interactions along the paths. The differences in the kinetics and thermodynamics of binding and unbinding events among the ligands were studied using enhanced sampling techniques such as well tempered and funnel metadynamics (Tiwary et al., 2015; Tiwary and Parrinello, 2015; Valsson et al., 2016). The obtained results offer useful insights into the functional role of membrane lipids in facilitating ligands’ access to the receptor by serving as a local depot. The critical residue interactions observed in the predominant binding and unbinding paths provide the mechanistic description of the energetic barriers contributing to the observed differences in the binding kinetics and thermodynamics of the studied ligands.
Materials and Methods
Protein and Ligand Structure Preparation
In this study, we used the crystal structure of β2-AR (active state) bound to salmeterol (PDB ID 6MXT) (Masureel et al., 2018). The structure was prepared for simulations using Molecular Operating Environment (MOE) (Molecular Operating Environment (MOE), 2019). The active-state stabilizing Nanobody Nb71 and T4 lysozyme fused to the N-terminal region were removed, and all residue mutations (M96T, M98T, M187E, and N187E) introduced during the crystallization process were reverted to their wild-type counterparts. The QuickPrep tool in MOE was used to assign the protonation states and rotamers, cap the N- and C-terminal ends with acetyl Acetyl and methyl amide groups, respectively, and add the missing hydrogen atoms. Special care was taken to ensure that all the titratable residues were assigned their dominant protonation states at pH 7.4 except for E1223.41, which was kept protonated (neutral) in the simulations as it faces the lipid bilayer (Masureel et al., 2018). The three-dimensional structures of the three studied β2-AR agonists, salmeterol, formoterol, and salbutamol, were built and geometry-optimized in MOE.
Membrane Partitioning Simulations
We used a combination of steered molecular dynamics (SMD) and umbrella sampling simulations to investigate the membrane partitioning characteristics of the studied ligands using the same protocol as published before (Park et al., 2003; Park and Schulten, 2004). For these simulations, the CHARMM-GUI membrane builder module was used to assemble the ligands and lipid bilayer (Brooks et al., 2009; Wu et al., 2014; Lee et al., 2016). The ligands were parameterized by first running geometry optimization in Gaussian 16 (Frisch et al., 2016). The optimization was done in water, using density functional theory and the Becke, 3-parameter, Lee–Yang–Parr B3LYP exchange-correlation functional with basis set 6-31G (Frisch et al., 2016). The geometry-optimized molecules were then submitted to the Paramchem web server, and charges and atom types were assigned (Vanommeslaeghe et al., 2010; Vanommeslaeghe and MacKerell, 2012; Vanommeslaeghe et al., 2012; Yu et al., 2012). The salmeterol simulation system comprised 63 POPC lipids in the upper and lower leaflet along with seven cholesterol molecules in both the upper and lower leaflet. This accounted for 140 lipids total, with dimensions of 67 × 67 × 91 Å3. The salbutamol simulation system comprised 64 POPC lipids in the upper and lower leaflet along with seven cholesterol molecules in the upper and lower leaflet with the dimensions of 68.2 × 68.2 × 93.06 Å3. The formoterol simulation system contained approximately 64 POPC lipids in the upper and lower leaflet, with approximately seven cholesterol molecules in each leaflet with the dimensions of 66.1 × 66.1 × 74.8 Å3. All three systems contained 0.15 M NaCl, and simulations were conducted at 310 K.
The assembled bilayer/ligand systems were subjected to multistage minimization and equilibration protocols. The six-step equilibration protocol recommended by the CHARMM-GUI membrane builder uses canonical ensemble (NVT- Number of atoms, Volume, and Temperature) NVT for the first two equilibration steps, with the last four steps using isothermal-isobaric ensemble (Number of atoms, Pressure, and Temperature) NPT dynamics. The input parameters included a harmonic force constant of 5 kcal/mol restraint on the heavy atoms of the protein, the lipids, the dihedral angles, and a planar restraint to maintain the headgroups in the correct orientation for 50 ps, which was preceded by a 10-ps minimization procedure. This was followed by two steps with a similar 50-ps restraint with a scaling factor on the restraint, slowly releasing the restraints. The final three steps of the equilibration continued with a scaling restraint, but for 200 ps (Lee et al., 2019). For each SMD simulation, the center of mass (COM) of the ligand was translated 30 Å from the center of the bilayer such that the starting position of the ligand is at the aqueous phase above the lipid headgroup stratum of the bilayer. The CHARMM36 Chemistry at HARvard Macromolecular Mechanics (Klauda et al., 2010; Lee et al., 2016) force-field parameters and TIP3P (Jorgensen et al., 1983) water model were used to model lipids and water molecules and their interactions with ions, respectively.
Steered MD simulations were used to pull each ligand from its starting position at the bulk solvent to the bilayer center along the bilayer normal (z-axis) at a speed of 1 Å per ns with a one fs time step. During the pulling simulations, the COM of each ligand was harmonically restrained with a force constant of 5 kcal/mol/Å2. The coordinates of each system located at every 1-Å window over the permeation pathway (a total of 30 windows) were extracted from the simulation trajectories and used as starting structures for subsequent umbrella sampling simulations. These coordinates were equilibrated for an additional 10–20 ns, and umbrella sampling simulations were performed on each window for 50 ns, resulting in a total simulation time of ∼2 μs. The z-component of the distance between the COM of the lipid atoms and the heavy atoms of each ligand was defined as the reaction coordinates. The Colvars module (Fiorin et al., 2013) in NAMD2.12 was used with a biasing harmonic constraint of 1.5 kcal/mol/Å2. The resulting biased probability distributions were reweighted using the weighted histogram analysis method (Kumar et al., 1992) to obtain the unbiased potential of mean force (PMF).
Hydrogen Bond Analysis
We analyzed potential hydrogen bond (H-bond) interactions between the polar functional groups of the ligands and the headgroup of the membrane lipids using the trajectories obtained from the umbrella sampling simulations. For each ligand, the corresponding lowest-energy window ± 2 for a total of five windows was used for H-bond interactions. We calculated the H-bonds between the ligand and cell membrane using the H-bond analysis plugin in VMD (Humphrey et al., 1996) with the donor-H-acceptor distance and angle cutoff values of 3.5 Å and 40°, respectively. Specific hydrogen bonds were grouped together to include bonds made by phosphate, glycerol, and choline groups of the lipids.
Bilayer Orientations of the Ligands
All three studied ligands differ significantly in size, functional groups, and most importantly, the number of rotatable bonds (see Supplemental Table 1). As ionized species, these molecules interact differently with various bilayer strata, resulting in unique orientations dictated by the constraints posed by the membrane lipids. To characterize the ligands’ orientations within the membrane, we calculated the angle made by the longitudinal vector of the ligand with the bilayer normal vector (z-axis) for each ligand at their energetically favorable locations (|z|energy-minimum ± 2 Å, as described above for H-bond calculations). For each ligand, a straight line connecting atoms O1 and N1 (Fig. 1) represents the longitudinal vector. Calculations were performed using the PLUMED driver to process the trajectory and extract relevant values (Bonomi et al., 2009; Tribello et al., 2014).
Unbiased Flooding Simulation
To choose an appropriate starting point for the association simulations, multiple replicates of unbiased simulations were conducted for each ligand as described below. In this study, we used the crystal structure of the unbound β2-AR protein in an active state, extracted from the β2-AR-salmeterol complex (PDB ID 6MXT) (Masureel et al., 2018). The system was oriented using the Orientations of Proteins in Membrane (OPM) web server for correct orientation in the membrane (Lomize et al., 2012). We placed 7–10 molecules of salmeterol randomly around the receptor in its energetically favorable bilayer location as determined by the PMF calculations. The system simulated consisted of 226 POPC lipids with 10 cholesterol molecules total. The ionic concentration of the system was 0.15 M NaCl. The system was set up using the CHARMM-GUI membrane builder, and the corresponding six steps of equilibration were run before running our production simulation (Lee et al., 2019). The system was then simulated using NPT, with a Nose-Hoover Langevin piston set to 1 atm, at a temperature of 310 K for a total of ∼250 ns. At the end of the simulation, there were multiple molecules in contact with the protein at various positions. However, on multiple attempts, only the system in which salmeterol was located near TMH1 and TMH7 in its energetically favorable bilayer depth and orientation turned out to be a rational starting state. Similarly, for formoterol, the starting position was near the two helices in its energetically favorable bilayer location and orientation. For salbutamol, simulations were run with multiple starting points in and around the receptor, including the aqueous bulk surrounding the receptor’s extracellular vestibule.
Steered MD Simulations to Elucidate Unbinding Pathways
We used SMD simulations to investigate the dissociation mechanism of the ligands from the orthosteric site of β2-AR. Three separate systems were set up for each of the studied ligands. For salmeterol, we used the ligand-bound crystal structure of β2-AR in its active state (PDB ID 6MXT) (Masureel et al., 2018). As there were no experimental structures available for formoterol or salbutamol bound to β2-AR during the beginning of this study, these ligands were docked to the receptor using MOE’s docking module. The saligenin headgroup of each of the three ligands was placed in a similar binding mode, forming conserved interactions with the binding site residues D1133.32, S2035.42, S2075.46, and N3127.39. The binding site was defined by D1133.32, S2035.42, and S2075.46 for both salbutamol and formoterol. Placement of the ligand was done with a triangle matcher, and the scoring was done using London dG scoring. After the ligand placement, an induced-fit refinement strategy was used, and the final poses were scored using the GBVI/WSA dG (Generalized-Born Volume Integral/Weighted Surface area) scoring method. In total, 10 final docked poses were generated for each ligand and then manually inspected for specific interactions and binding mode similarity to the salmeterol-bound crystal structure. The bound poses predicted by the docking program for formoterol and salbutamol were very similar to their crystal structures bound to β1-AR (Warne et al., 2011; Lee et al., 2020) (PDB IDs 6IBL and 2Y04, respectively). Also, a recently published cryo-electron microscopy structure of β2-AR (Zhang et al., 2020) bound to formoterol (PDB ID 7BZ2) revealed a similar binding mode, backing our starting points. The docked complex was submitted to the Orientations of Proteins in Membranes web server for obtaining proper orientation of the receptor in the membrane (Lomize et al., 2012). The previously obtained ligand parameters were used for the ligands. The entire complex was submitted to the CHARMM-GUI membrane builder (Wu et al., 2014; Lee et al., 2016). For each system, ∼90 POPC lipids were chosen for both the lower and upper leaflet, along with ∼10 cholesterol molecules in each leaflet for a total of ∼100 lipids in both leaflets. A water padding with a thickness of 17.5 Å from the lipid bilayer head stratum was added along with 0.15 M of NaCl. The three-site transferrable intermolecular potential (TIP3P) water model model was used for water along with the CHARMM salt ion parameters (Jorgensen et al., 1983). After running the six step equilibration process (Jo et al., 2007) at a temperature of 310 K, each system was simulated for an additional 50-ns production run to ensure the ligands were stable in the binding site. This 50-ns run was an NPT run using periodic boundary conditions, in which the Nose-Hoover Langevin piston was used to maintain a constant pressure of 1 atm. After 50 ns, a constant-velocity SMD simulation began. The phosphorus atoms of the lipid headgroup were harmonically restrained using a spring constant of 0.5 kcal/mol/Å2, and the ligand’s heavy atoms were defined as the SMD atom. The normalized direction between the COM of the binding site residues and the COM of the SMD atom was used to define the initial direction of pulling. A spring constant of 5 kcal/mol/Å2 was used, and the pulling was performed at a constant velocity of 0.000001 Å per time step (1 Å/ns). Each SMD simulation was conducted for 50 ns, in replicates of 4, 4, and 3, for salmeterol, salbutamol, and formoterol, respectively. SMD simulations were run using the GPU version of Nanoscale Molecular Dynamics (NAMD2.12).
PMF Calculation Using Jarzynski Equality
We used SMD, a nonequilibrium enhanced sampling technique, to investigate the unbinding (dissociation) of the studied ligands from the β2-AR binding site. During the SMD simulations, a dummy atom attached to the COM of ligands with force constant (k = 5 kcal/mol) was pulled with a constant velocity (v) of 1 Å/ns in a normalized vector (nx, ny, nz) direction defined by the line connecting COM of ligands and the binding site residues. The total force applied to ligands, f = fxnx + fyny + fznz, where fx, fy, and fz are components of the force along x, y, and z normal vectors. The time-dependent irreversible work done on the system was calculated using the equation
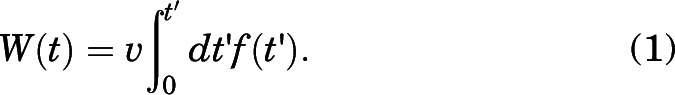 |
Conceptually, SMD is similar to performing an atomic force microscope experiment. The Jarzynski equality (JE) (Jarzynski, 1997) was implemented to extract the equilibrium work done (ΔF) or the PMF from the nonequilibrium SMD simulations, with the following equation:
where ⟨ ⟩ refers to the ensemble average. JE is the expansion of the second law of thermodynamics to nonequilibrium work performed on the system. The right-hand side of eq. 2 can be expanded as a sum of cumulants or ensemble distribution of nonequilibrium work values. If the ensemble distribution of nonequilibrium work values is Gaussian, the free energy profile can be written as follows:
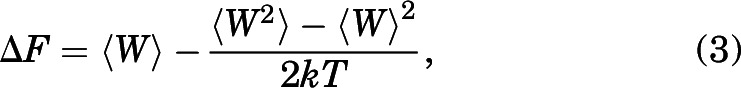 |
where k is the Boltzmann constant, and T is the temperature of the system.
Association Simulation Details
Metadynamics (Laio and Gervasio, 2008; Barducci et al., 2011) and well tempered metadynamics (Barducci et al., 2008) have emerged as excellent simulation techniques to investigate rare and important events within a reasonable timescale. Briefly, metadynamics is a theoretical method in which an external history-dependent potential is added that acts on a few degrees of freedom, called collective variables. This discourages the system from revisiting the sampled configurations. Well tempered metadynamics (WT-MetaD) is a unique scheme of metadynamics in which the height of the bias potential is decreased with the amount of bias already deposited. The free energy surface (FES) for the ligand-binding process can be sampled (Limongelli et al., 2013) by either metadynamics or WT-MetaD.
As we were interested in a membrane-facilitated binding pathway for salmeterol, we conducted a flooding simulation described above with the receptor in an active state. However, we did not observe a spontaneous binding event for salmeterol through the membrane. So, a frame from the end of the simulation was chosen as a starting point for the well tempered metadynamics run. All WT-MetaD runs were set up using PLUMED version 2.3 or PLUMED version 2.4 (Bonomi et al., 2009; Tribello et al., 2014). The simulations were run using GROMACS 2016 (Berendsen et al., 1995; Abraham et al., 2015). The simulations used the six-step equilibration protocol provided by the CHARMM-GUI membrane builder. A minimization step of 1 ps was run using the steepest-descent algorithm, along with the Linear Constraint Solver (LINCS) algorithm constraining H-bonds in the system. The following six-step equilibration process was done using a Berendsen canonical ensemble (NVT) setup for the first two steps, followed by a Berendsen NPT setup. The production run was conducted with an NPT setup and pressure-coupled to a Parrinello-Rahman barostat. The WT-MetaD was set up using two collective variables: 1) the distance between the COM of the ligand and the COM of binding site residues (D1133.32, F193ECL2, F194ECL2, S2035.42, S2075.46) and 2) the angle between three points: the upper saligenin headgroup, represented by the phenyl ring carbon atoms; the center of the molecule, represented by the two carbons attached to the ether oxygen; and the tail end, represented by the phenyl ring carbons. In addition, an upper wall boundary was placed on the distance variable so that the system sampled the region around the binding site and did not drift too far away. This upper wall boundary is activated only when the distance drifts outside of a user-defined distance. The upper wall distance was set to 1.5 nm. A force constant of 200 kJ/mol, an exponent of 2, a rescaling factor of 1, and an offset value of 0 were used for the upper wall. For the WT-MetaD run, the temperature of the system was set to 310 K, the height of the Gaussian hills was 1.5, the width of the hills was 0.05 and 0.35 for distance and angle, respectively, and a bias factor of 15 was used. Each of the systems was simulated for 100 ns. The same parameters were used for salbutamol and formoterol. The angle defined for formoterol was defined in a similar manner to salmeterol, with the COM of the headgroup phenyl carbons used as a point, the nitrogen in the linker region as another point, and the COM of the carbons on the methoxyphenyl group as the third point. Salbutamol’s angle was defined by the COM of the phenyl carbons as a single point, the carbon between the hydroxyl and amine as the second point, and the tert-butyl carbon as the final point. Each system was simulated at least three times.
In addition to the association simulations described above, we wanted to test whether salmeterol or formoterol could enter the binding pocket through salbutamol’s secondary access route. To assess this possibility, we set up SMD simulations similar to previously described dissociation simulations. The starting point for salmeterol and formoterol was chosen near the entry of the access route in the membrane at their preferred low-energy bilayer depths and orientations. After minimization, we defined the normalized direction between the COM of the binding site residues, as described earlier, and the SMD atom as the initial direction of pulling.
Binding Free Energy Calculations Using Funnel Metadynamics
We calculated the absolute protein-ligand binding free energies of the studied β2-AR agonists using funnel metadynamics (FM) (Limongelli et al., 2013; Raniolo and Limongelli, 2020). Simulations were set up using the same receptor-bound structures employed in the steered MD simulations for probing dissociation mechanisms. After a 50-ns equilibration run, the funnel for each ligand was set up. GROMACS 5.1.2 was patched with PLUMED version 2.3b with specific codes for the funnel bias added (Berendsen et al., 1995; Bonomi et al., 2009; Tribello et al., 2014; Abraham et al., 2015). The collective variables used were 1) the position of the COM of the ligand along the funnel axis and 2) the vertical distance of the COM of the ligand to the funnel axis. Gaussian widths of 0.05 nm were applied to the two CVs, and a bias factor of 15 was used. Soft harmonic restraining walls were applied at both ends of the funnel axis to limit the exploration of the ligand inside the region enclosed by the funnel potential. The criteria for the successful simulations included the occurrence of multiple recrossing events between the bound and unbound states as well as the convergence of the free energy around a constant value. Using these criteria, the FM simulations were run until 900 ns for salmeterol, 450 ns for formoterol, and 500 ns for salbutamol in multiple replicates. The funnel for salbutamol was placed to include the binding site region and the extracellular solvent. The Zcc distance, signifying the switching distance between the funnel and cylinder, was kept at 2.5 nm. The α value, which represents the angle of the funnel and the radius of the cylinder (rcyl) were 0.7 and 1 Å, respectively. The funnel for formoterol had a Zcc of 3.0 nm, an α value of 0.7, and rcyl of 1 Å. The salmeterol funnel had a Zcc of 3.0 nm, an α value of 0.6, and an rcyl of 2 Å. The FES was reconstructed using the sum_hills function in PLUMED version 2.3. The binding pathway was determined using the Minimum Energy Path Surface Analysis tool (Marcos-Alcalde et al., 2015). The binding free energy was calculated using eq. 4, which accounts for the funnel restraint applied during FM simulations (Limongelli et al., 2013).
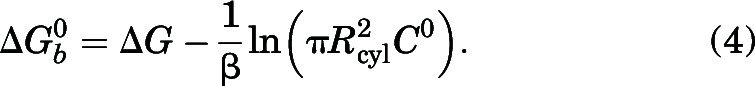 |
The ΔG is the free energy difference between the bound and unbound state. β is a constant and equals 1/kBT, with kB representing the Boltzmann constant, and T is the temperature of the system (310 K). is the surface of the cylinder used for assigning the restraint potential, and C0 is the standard concentration of 1M, which is equal to 1/1,660 Å−3.
Sykes et al. (2014) determined the dissociation constant (Kd) of several β2-AR agonists from their kinetic parameters, koff and kon values, using a competition binding assay with radiolabeled 1-[4,6-propyl-3H]dihydroalprenolol ([3H]DHA) in CHO cell membranes. We calculated the binding free energy (ΔGb0) of the studied ligands using the observed pKd and “corrected” pKd values obtained by decoupling membrane interactions. The calculated binding free energies are summarized in Supplemental Table 1.
Results
Membrane Partitioning Characteristics of Salmeterol, Formoterol, and Salbutamol
To elucidate the differences in membrane partitioning characteristics, we performed steered MD and umbrella sampling simulations using a model membrane containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and cholesterol. The obtained results presented below provide remarkable insights into their distinct membrane interactions in atomistic details.
Salmeterol Has a More Favorable Free Energy of Solvation in the POPC Lipid Bilayer
The PMF curves obtained from the simulations provide the free energy profiles of solvation of each ligand from water to the membrane and along the bilayer normal (z-axis). The bilayer regions at which the solutes exhibit the most and least favorable energies are characterized by energy minima and maxima, respectively (Fig. 1B). Each of the three ligands shows a favorable (negative) free energy of transfer from water to the membrane. The free energy minima of the COM of salbutamol, salmeterol, and formoterol are located at |zmin| ≈ 18, |zmin| ≈ 15, and |zmin| ≈ 14, respectively. The free energy of partitioning for salmeterol was the most favorable (ΔGpartitioning = −3.15 ± 0.02 kcal/mol), followed by formoterol (ΔGpartitioning = −2.53 ± 0.02 kcal/mol) and salbutamol (ΔGpartitioning = −0.98 ± 0.03 kcal/mol). As the COM of each molecule moved across the membrane toward the bilayer center (z = 0), the free energy barrier for crossing the membrane (ΔGcrossing) increased in the reverse order to that of ΔGpartitioning. Salbutamol showed the largest energy barrier (ΔGcrossing = 13.21 ± 0.11 kcal/mol), followed by formoterol (ΔGcrossing = 11.09 ± 0.07 kcal/mol) and salmeterol (ΔGcrossing = 10.39 ± 0.07 kcal/mol). In all cases, the thermally accessible regions (energy barrier of RT = 0.616 kcal/mol at 310 K) extended for 2 Å on both sides around the energy minima (Table 1). It is important to recognize that, unlike salbutamol, both salmeterol and formoterol are lengthy molecules, and their long axes span ∼20 Å and ∼15 Å, respectively, and the time-average bilayer locations of their COMs, i.e., |zmin| values reported above, need to be examined carefully. To obtain precise and detailed bilayer locations and orientations of the molecules, we calculated the mass density profiles for the ligands and various components of the membrane lipids along the bilayer normal (Supplemental Fig. 1). The mass density for salmeterol clearly indicates that the molecule is present in its extended conformation, the long axis of which spans almost half of the entire bilayer. Formoterol’s density profile is similar to that of salmeterol, and it is less spread out due to its smaller size. Salbutamol has the least spread, indicating its preferred location around the lipid headgroup region and the absence of any notable interactions with the lipid core. Whereas the saligenin-ethanolamine head of each ligand is located at the lipid headgroup region (Fig. 1C), the lipophilic tails of salmeterol and formoterol are oriented perpendicular to the membrane plane (i.e., in parallel to the bilayer normal) and buried into the lipid core, clearly displaying their amphiphilic nature.
TABLE 1.
Free energy profiles of membrane partitioning/crossing and time-average bilayer locations of the studied β2-AR agonists
| Ligand Name | Energy-Minimum Position of COM (Bilayer Depth) from the Center | Bilayer Region within Reach of Thermal Motion at 310 K from the Center | ΔGpartitioning | ΔGcrossing | |
|---|---|---|---|---|---|
| Å | kcal/mol | kcal/mol | |||
| Salmeterol | 15 | 13 | 17 | −3.15 0.02 | 10.39 0.07 |
| Formoterol | 14 | 12 | 16 | −2.53 0.02 | 11.09 0.07 |
| Salbutamol | 18 | 16 | 20 | −0.98 0.03 | 13.21 0.11 |
S.E. was calculated using Monte Carlo bootstrap error analysis in weighted histogram analysis method (Kumar et al., 1992).
The Bilayer Orientation and H-Bond Interactions of Saligenin-Ethanolamine Polar Groups Are Distinct among the Ligands
We performed comprehensive analyses of the orientation of the saligenin-ethanolamine head of each ligand and their interactions with various functional groups of the lipids to obtain more insight into the observed differences in the membrane solvation free energies among the ligands. The tilt angle measured between the longitudinal vector of the saligenin-ethanolamine head and the bilayer normal represents the orientational angle and is presented as probability density graphs (Fig. 1D, see Materials and Methods section for details). The H-bond interactions between various polar groups (three -OH groups labeled as O1, O2, and O3, NH3+ group labeled as N1, and -NH group present only in formoterol, which is labeled as N2; Fig. 1A) of the ligands and the lipid headgroups including choline, phosphate, and glyceryl carbonyls were monitored through the simulation time and presented as percent H-bond occupancy (Fig. 1E and Supplemental Fig. 2, see Materials and Methods section for details). In addition, we calculated the atom contacts (as percent occupancy) that give the fraction of total simulation time during which the functional groups from the ligand are located within 4 Å of various lipid functional groups (Supplemental Fig. 3).
Intriguingly, the orientation angles are unique for each ligand, resulting from subtle differences in interaction patterns among their common polar groups as well as specific interactions of additional functional groups that are present only in salmeterol or formoterol. Salmeterol’s saligenin-ethanolamine head orients in two distinct modes, as shown by the bimodal distribution of its orientation angle (Fig. 1D). The major orientation is one in which the head is at an acute angle to the bilayer normal (θ = 40° ± 20°). In the secondary mode, the head orients perpendicular to the bilayer normal (θ = 90° ± 20°). The bimodal distribution of the orientation angle for salmeterol, reflecting its flexible nature, can be attributed to the number of rotatable bonds, which is twice as high as that of formoterol (16 vs. 8; Supplemental Table 1). In the case of formoterol, the saligenin-ethanolamine head is oriented at an acute angle (θ = 60° 30°) that is approximately perpendicular to the bilayer normal. Salbutamol’s orientation is vastly different from salmeterol and formoterol and can be seen mostly at an obtuse angle to the bilayer normal (θ = 140° 20°) in which its phenolic hydroxyl group is oriented toward the lipid alkyl tails.
These unique orientations and conformations are due to the specific interactions between the functional groups located on each ligand and the membrane lipids. The number of H-bonds made by salmeterol (at its low-energy well) is relatively less than the other two ligands (Supplemental Fig. 2, A and B). Whereas the aliphatic ethanolamine hydroxyl group (O3) makes H-bond interactions with oxygen atoms of the lipid phosphate and glyceryl groups in all the ligands, the interactions of both the phenolic OH (O1) and benzyl OH (O2) groups are different. Salmeterol’s O1 is located near the choline group, and its H-bond occupancy with the phosphate oxygen is much less compared with that of formoterol. Formoterol’s O1 makes H-bond interactions with both phosphate and glyceryl oxygens, although the occupancy for phosphate oxygens is higher compared with the glyceryl ones. On the other hand, salbutamol’s O1 is located near the alkyl tail and makes H-bond interaction mainly with glyceryl oxygen atoms (Fig. 1E). The benzyl hydroxy group (O2) in salmeterol and salbutamol follow similar trends to their respective O1 groups but with much higher occupancies. However, the formamide carbonyl oxygen (labeled as O2 in Fig. 1A) of formoterol does not engage in H-bond interactions with the lipid headgroups but rather is oriented toward the aqueous bulk and forms stable H-bonds with the water molecules (Fig. 1C).
The charged ethanolamine amino group (labeled as N1), in all three ligands, is located near the lipid phosphate groups and makes H-bond/electrostatic interactions with the phosphate oxygens. This amino group also makes such interactions with glyceryl oxygens (only in salmeterol and formoterol, but not in salbutamol). The formamide -NH group of formoterol (labeled as N2) is primarily located near the glyceryl/phosphate groups and engages in H-bond interactions with them. The hydrophobic tails of salmeterol and formoterol (including the oxygen atom, labeled as O4 in Fig. 1A) are mostly buried and sandwiched between the alkyl tails of the lipid core region.
The time-average bilayer locations of the ligands at the lipid headgroup/core interface and their distinct orientations clearly indicate that they remain in contact with the aqueous phase while they are partitioned into the membrane. To quantify these critical solvation characteristics, we calculated the number of water molecules within 4 Å of any heavy atom of the ligands and normalized the numbers by their respective heavy atom counts to account for the notable variation in their sizes (Table 1). The numbers are significantly different among the ligands (Supplemental Fig. 2C). Interestingly, salmeterol has significantly fewer water contacts compared with formoterol and salbutamol because only 13 (out of 30) of its heavy atoms are located at the lipid headgroup region, and the majority of them (>56%) are buried deep in the lipid core. In the case of formoterol, 14 (out of 25) of its heavy atoms are in contact with the lipid headgroup, and only 11 (44%) of its heavy atoms are in contact with the lipid core. In contrast, none of the salbutamol atoms are buried in the lipid core region. These unique solvation characteristics of the ligands appear to dictate their association paths (either by aqueous route or by diffusion through the membrane) through which the ligand access and reach the binding site of β2-AR.
Binding Pathways for Salmeterol, Formoterol, and Salbutamol
We examined the plausible receptor access and binding pathways of the three ligands, taking into consideration their membrane partitioning characteristics. All association simulations started with the ligands at their energetically favorable (time-average) locations and orientations within the membrane. For salbutamol, several simulations were run with the ligand in the aqueous phase, above and around the receptor as its starting position. Earlier studies investigating the binding of ligands to orthosteric sites of GPCRs demonstrated that such events occur spontaneously only in microsecond timescales (Dror et al., 2011, 2013). Therefore, to accelerate the ligand access and binding processes, we used WT-MetaD, an enhanced sampling technique that utilizes a biasing force and the specifics of the binding site residues to guide the ligand-receptor association (see Materials and Methods section for details).
Salmeterol Enters the Binding Pocket through TMH1 and 7 from within the Bilayer
Out of 20 WT-MetaD association simulations aimed to characterize the binding mechanism of salmeterol, we observed a complete association of the ligand in 18 simulations. The ligand association process was considered complete only when the distance between the COMs of the ligand and that of the binding site residues is within 4 Å. In 15 of 18 successful simulations, salmeterol reached the binding pocket by entry through its saligenin headfirst. Intriguingly, in the rest of the three simulations, salmeterol entered the pocket with its phenyl tail first. However, in all the cases, salmeterol approached the receptor from within the membrane through TMHs 1, 2, and 7. All the association simulations started with salmeterol partitioned within the membrane in its energetically favorable bilayer depth and orientation, at a minimum distance of ∼20 Å from the receptor binding site. Specifically, salmeterol was placed near TMHs 1 and 7 as several unbiased simulations indicated that salmeterol preferentially interacts with residues from these helices. However, a spontaneous entry of salmeterol into the binding site was not observed in any of these simulations.
The free energy surface, describing the thermodynamics of the ligand access and receptor-binding events, including multiple low-energy intermediate states (A-D), was characterized by two collective variables: the distance between the COM of the ligand (heavy atoms) and the binding site residues (x-axis) and the internal angle of the ligand accounting for the conformational changes (y-axis) (Fig. 2). As salmeterol approached the receptor, its hydrophobic aralkyl-oxy-alkyl tail made contact with several residues from TMHs 1 and 7, including W3137.40, I3097.36, L3107.37, V391.38, W421.41, and I431.42, whereas the saligenin head appeared to explore nearby residues for potential interactions (Fig. 2A). In most of the simulations, E3067.33 made the first contact with the upper end of salmeterol through H-bond interactions with its O1 and O2 groups. These interactions drew the ligand closer to the binding pocket and anchored it in a position favorable for entry.
Fig. 2.

The free energy surface for salmeterol’s access and binding to the β2-adrenergic receptor through the TMHs 1 and 7 from its energetically favorable location in the membrane. The two-dimensional free energy surface is characterized by the distance between the COM of the ligand and binding site residues (x-axis) in nm and the internal angle of the salmeterol molecule (y-axis) in degrees. The minimum energy path, as determined by well tempered metadynamics simulations, is given in bold black line, and several representative intermediate states along the path were labeled (A–D). (A) As salmeterol (licorice representation, magenta color) approaches the receptor from within the membrane, its aryl-alkoxy-alkyl tail comes into contact with the hydrophobic residues, including W3137.40, I3097.36, L3107.37, V391.38, W421.41, and I431.42 from TMHs 1 and 7. (B and C) Salmeterol undergoes a significant conformational change from its extended to bent form and engages in H-bond interactions with D192ECL2 through its O1 and O2 groups, which destabilize the salt bridge between D192ECL2 and K3057.32. The breakage of the salt bridge allows salmeterol to move further, disrupting the hydrophobic lock (between F193ECL2 and Y3087.35) while the tail is still anchored by the hydrophobic residues. (D) Outward movement of the side chain indole ring of W3137.40 releases the tail and facilitates salmeterol’s entry into the pocket, which then assumes its final bound pose, mostly similar to that of the crystal structure pose (PDB ID 6MXT) (Masureel et al., 2018). In its final bound pose, salmeterol engages in polar H-bond and salt bridge interactions with S2035.42, S2075.46, D1133.32, and N3127.39. Salmeterol (crystal pose in dark blue) and the binding site residues (green) are illustrated in licorice representation. The receptor is illustrated in secondary structure representation (light blue).
As salmeterol prepared to enter the binding pocket through the helices, it underwent a significant conformational change from its extended form to a bent one, with the change in the internal angle of the ligand from ∼150° to ∼50° (from A to B) as can be seen in the free energy surface diagram (Fig. 2). The rearrangement caused the saligenin head to proceed toward the site forming H-bonds with D192ECL2 through O1 and O2 groups (Fig. 2B). As the saligenin head of salmeterol moved further toward the pocket, its N1 group formed an H-bond with E3067.33, whereas O1 and O2 groups engaged with the main chain -NH group of F193ECL2, D192ECL2, and K3057.33, causing the breakage of the D192ECL2-K3057.33 salt bridge if the bond was already formed. These interactions drew salmeterol’s head further into the binding site, whereas the tail remained anchored by the residues from TMHs 1 and 7, including V391.38, I431.42, I3097.36, L3107.37, and W3137.40 (Fig. 2C). As the saligenin head entered the binding pocket, the hydrophobic lock between F193ECL2 and Y3087.35 was also broken, which reformed briefly after breaking earlier in the simulation. Eventually, salmeterol slid into the binding pocket and advanced toward its final bound pose with its saligenin head O1 and O2 groups engaging in H-bond interactions to the side chain -OH groups of S2075.46 and S2035.42, respectively (Fig. 2D). The phenyl end was able to reach the pocket only when the side chain indole ring of W3137.40 rotated outward and pushed the phenyl ring away from the anchoring point while closing the transmembrane entry. In its final bound pose, the N1 and O3 groups of the ligand formed polar interactions with D1133.32 and N3127.39, respectively. Interestingly, the phenyl tail adopted a slightly different position relative to the crystal structure pose. The phenyl tail drifts to the other side of the ECL2 region, close to R175ECL2 and Y174ECL2. However, in the crystal structure pose, there is an H-bond between the ether oxygen of salmeterol and the main chain of F193ECL2.
Once the saligenin head reached the pocket and participated in interactions similar to the crystal structure bound pose, we switched off the bias and continued the simulations unbiased for another 200 ns to observe whether salmeterol’s tail would assume a similar conformation as that of the crystal pose. Although not identical, we observed a binding pose in which the conformation of the tail was similar to that of the crystal pose, with an overall ligand RMSD of 2.56 Å, as shown in Fig. 2D. Another notable binding pose of salmeterol observed during this extended unbiased simulation was one in which the ligand tail adopted an extended conformation with its phenyl ring positioned around K972.63, K3057.32, and H932.64.
Salmeterol’s Lipophilic Tail Acts as a Passkey for the Receptor Entry
Although in the majority of the simulations, salmeterol made entry into the binding pocket with its saligenin head first, in three out of the 18 successful simulations, salmeterol entered in a strikingly different manner (Fig. 3). Initially, in its bent conformation as described above, salmeterol engaged with D192ECL2 and K3057.33 through the saligenin -OH groups O1 and O2, whereas its aryl-alkyl tail was anchored by the residues from TMHs 1 and 7 (W3137.40, V3177.44, I431.42, and V391.38). As the salt bridge interactions were broken (between D192ECL2 and K3057.32), the saligenin head proceeded to pass between the residues, which was prevented by a newly formed bond between the side chain amino group of K3057.33 and the main chain oxygen of F193ECL2. Salmeterol’s attempt to enter the site with its saligenin head was also blocked by the phenyl ring side chains of F193ECL2 and F194ECL2, which appeared to act as a gate (Fig. 3B). In a fascinating manner, salmeterol rearranged, withdrawing its saligenin head from the gate and presenting its aryl-alkyl tail as a passkey instead (Fig. 3C and Supplemental Video 1).
Fig. 3.

Salmeterol enters the β2-AR binding site from within the membrane through the transmembrane helices 1 and 7 using its lipophilic tail as a passkey. (A) Hydrophobic residues from TMHs 1 and 7 anchor the lipophilic tail of salmeterol and facilitate salmeterol’s entry into the pocket from within the membrane. (B) However, in several simulations, salmeterol’s entry by its saligenin head is blocked by the side chain phenyl ring of F193ECL2 that acts as a gate. (C) Fascinatingly, salmeterol flips 180° and presents its aryl-alkyl tail as a passkey to the gate, which immediately opens up the channel by flipping F193ECL2 upward to gain entry into the pocket.
Surprisingly, the gate opened as the phenyl ring of F193ECL2 flipped upward, allowing salmeterol to enter the pocket. It is worth noting that F193ECL2 has been reported to be important for the higher selectivity of salmeterol for the 2-AR over 1-AR relative to other commonly used 2-AR agonists (Carter and Hill, 2005; Baker, 2010). The tail appears to push apart TMHs 7 and 2, breaking the newly formed interactions between K3057.33 and F193ECL2 as well as pushing the hydrophobic lock (Y3087.35-F193ECL2) further apart to facilitate the entry. As the molecule stretched across the binding pocket, the tail end was seen exploring regions between TMH4 and TMH5, which allowed the saligenin head to slide into the pocket to assume its final bound pose identical to other simulations.
Formoterol Preferentially Interacts with Aqueous Bulk before Entering the Binding Pocket
In the case of formoterol, a clearly predominant association path was observed in the majority of the simulations. As the ligand approaches the receptor from within the membrane, it undergoes a slight conformational change such that the methoxyphenyl alkyl tail makes notable hydrophobic contacts with residues I942.65, L3117.38, and W3137.40 of TMHs 2 and 7, respectively (Fig. 4A). After these transient interactions, the ligand moves up to a region just above the transmembrane helices, formed by residues from TMHs 2 and 6 as well as ECL2, and then enters the binding pocket in a manner that is similar to other polar ligands. Intriguingly, the entry of formoterol from this region followed two distinct pathways, remarkably governing the pace at which the ligand reaches the binding pocket.
Fig. 4.

The free energy surface for formoterol’s access and binding to the β2-adrenergic receptor from its energetically favorable location in the membrane. The two-dimensional free energy surface is characterized by the distance between the COM of the ligand and binding site residues (x-axis) and the internal angle of formoterol (y-axis). The minimum energy path, as determined by well tempered metadynamics simulations, is given in bold black line, and several representative low-energy intermediate states along the path were labeled (A–D). (A) As formoterol (licorice representation, yellow color) approaches the receptor from within the membrane, its alkoxy-aryl-alkyl tail comes into contact with TMHs 2 and 7, engaging in hydrophobic contacts with I942.65, L3107.37, and W3137.40. (B and C) Formoterol moves up over the transmembrane helices and remains in a region where it makes extensive H-bond interactions with several polar residues and bulk water molecules. Specifically, the H-bond with D192ECL2 through its O3 appears to destabilize the salt bridge between D192ECL2 and K3057.32. The breakage of the salt bridge allows formoterol to slide down from this region into the binding pocket. As formoterol enters the pocket, the hydrophobic lock (between F193ECL2 and Y3087.35) breaks, resulting in the formation of a π-π interaction between F193’s phenyl ring and the aromatic ring at the tail end of the ligand. (D) Formoterol is seen in its final bound pose, slightly different from its X-ray structure pose (RMSD 2.1Å) bound to turkey β1-AR [PDB ID 6IBL (Lee et al., 2020)]. In its final bound pose, formoterol engages in polar H-bond and salt bridge interactions with S2035.42, S2075.46, D1133.32, and N3127.39. Formoterol (crystal pose in cyan) and the binding site residues (green) are illustrated in licorice representation. The receptor is illustrated in secondary structure representation (light golden yellow).
In 11 out of 15 simulations, prior to entering the binding pocket, formoterol stays in the region for approximately 25 ns, during which it engages in extensive H-bond interactions with many residues. Most importantly, the H-bond with D192ECL2 through its O3 appears to destabilize the salt bridge between D192ECL2 and K3057.32 (Fig. 4B). At the same time, the oxygen (O4) of its methoxyphenyl tail forms another H-bond with Y3167.43. In this extended orientation, formoterol also forms H-bonds through its carbonyl oxygen (O2) and hydroxyl group (O1) of the saligenin head with residues E180ECL2 and N187ECL2, respectively. The breakage of the salt bridge allows formoterol to slide down from this region into the binding pocket while forming a transient interaction between its positively charged amino group (N1) and D192ECL2. As this interaction breaks, formoterol proceeds to the binding site underneath the hydrophobic lock between F193ECL2 and Y3087.35 (Fig. 4C). As the ligand passes down, this lock is broken, resulting in the phenyl ring of F193ECL2 rotating toward the tail end of formoterol, forming a π-π interaction with its aromatic ring enabling the ligand to assume its final pose (Fig. 4D) similar to the crystal bound pose (PDB ID 6IBL) with an RMSD of 2.1Å.
On the other hand, in four out of 15 simulations, formoterol was seen engaging in extensive contacts with water in the region but much fewer interactions with the protein residues before entering the binding pocket. Interestingly, the hydrophobic lock (between F193ECL2 and Y3087.35) was not closed, and the phenyl ring of F193ECL2 was seen rotated back toward Y174ECL2, allowing formoterol to quickly enter into the binding pocket. Remarkably, in these simulations, the entire process of ligand access and reaching the binding pocket to assume its final bound pose took less than 25 ns. Most intriguingly, once formoterol settles into the binding pocket, the residues forming the hydrophobic lock come closer and remain in proximity for the rest of the simulation (see Supplemental Fig. 4).
During association, although both salmeterol and formoterol made initial interactions with residues from TMH1 and 2 before entering the pocket, there were some clear differences. The measured distance between TMH1 and TMH2, using the Cα carbon of Trp321.32 and Met962.66 was much larger for salmeterol than formoterol (Supplemental Fig. 5A), thanks to the bulkier tail group in salmeterol. There was a notable increase in this distance approximately 20 ns into the simulation, which was absent for formoterol (Supplemental Fig. 5A). Also, the change in the backbone RMSD of the entire protein was much larger during salmeterol association than for formoterol. This difference in the RMSD values (calculated using the backbone atoms) is ∼3 Å between the two compounds (Supplemental Fig. 5B). Specifically, the RMSD change comes from ECL2, which underwent significant conformational change (up to 7 Å) upon entry of salmeterol (Supplemental Fig. 6). Salmeterol’s large size and many rotatable bonds allow for the molecule to adopt a multitude of conformations that might not be suitable for entry into the binding site, and both the protein and salmeterol have to undergo conformational changes for the association process.
Salbutamol Enters the Binding Site Primarily through the Canonical Aqueous Path Described for Other Polar β2-AR Ligands
Unlike salmeterol and formoterol, the chemical polarity and hydrophilic nature of salbutamol do not allow the molecule to go deeper into the membrane (Fig. 1, B and C). Instead, salbutamol prefers to locate at the lipid headgroup-water interface, where it remains in contact with a significant number of water molecules (Supplemental Fig. 3). Also, as the solvation free energies for salbutamol in the aqueous bulk and at the membrane are quite comparable (Fig. 1B), spontaneous transfer of the ligand between these two phases can be expected. Therefore, we performed the WT-MetaD simulations for salbutamol in which the molecule was placed randomly around the receptor (a distance of at least 10 Å from any residue), with its starting location either at the aqueous bulk or in the membrane (time-average location and orientation, as determined by PMF; Fig. 1B). The FES for the receptor binding of salbutamol and the minimum energy path (black line) through which it gained access to the binding pocket predominantly is illustrated in Fig. 5.
Fig. 5.

The free energy surface and minimum energy path for salbutamol’s access and binding to the β2-adrenergic receptor. The primary binding path for salbutamol is through the common aqueous route, as determined by well tempered metadynamics simulations (minimum energy path, in bold black line). (A) In each simulation, salbutamol first entered the aqueous bulk before it established its first contact with the receptor. (B) Salbutamol first encountered K972.68 (through its saligenin hydroxyl groups). (C) Salbutamol slid along the extracellular vestibule, establishing interactions with F193ECL2 through its t-butyl end and with D1133.32 and N3127.39 with its saligenin end. (D) Salbutamol is seen in its final bound pose, very similar to its crystal X-ray structure pose (RMSD of 2.25Å) bound to turkey β1-AR (PDB ID 2Y04) (Warne et al., 2011). In its final bound pose, salbutamol engages in polar interactions with S2035.42, S2075.46, D1133.32, and N3127.39. Salbutamol (blue) and the binding site residues (green) are illustrated in licorice representation. The receptor is illustrated in secondary structure representation (light pink).
While approaching the receptor from the aqueous bulk, salbutamol made its first contact with the side chain amino group of K972.68 and the carboxyl group of D300ECL3 through its saligenin hydroxyl group and protonated ethanolamine amino group, respectively (Fig. 5B). These polar interactions drew salbutamol near the salt bridge between D192ECL2 and K3057.32 and enabled further contacts with D192ECL2, F193ECL2, and T195ECL2. This was followed by destabilization and breaking of the salt bridge, allowing further entry of salbutamol into the binding pocket. The two hydroxyl groups of the saligenin head were seen engaging in H-bond interactions with D1133.32 and N3127.39 as the molecule glided down into the binding site (Fig. 5C). Subsequently, salbutamol assumed its final bound pose in which its two hydroxyl groups of saligenin head made H-bond interactions with side chain -OH groups of S2035.42 and S2075.46. Also, the -OH and -NH3+ groups of the ethanolamine chain were engaged in H-bond/electrostatic interactions with the side chain amino group of N3127.39 and the carboxyl group of D1133.32, respectively (Fig. 5D).
Salbutamol Accesses the Binding Site through a Novel Polar Channel as a Secondary Route
Most surprisingly, in nearly half of the simulations (5 out of 12), salbutamol entered the binding pocket through a novel polar channel that opens up on the distal and opposite side of the common access route (Fig. 6, A and B). The entrance of this channel is located at the extracellular ends of TMH3/TMH4 and ECL2 and is lined by several polar residues, including N1033.22, Y185ECL2, R175ECL2, E1073.26, and Y174ECL2. As soon as salbutamol approaches the channel opening, it quickly engages in polar interactions with these residues through its two hydroxyl groups of the saligenin head first. In addition, brief interactions were observed with ethanolamine and t-butyl groups of salbutamol with F1043.23, S1113.30, and Q1704.62. Specifically, N1033.22, R175ECL2, and E1073.26 make initial contacts with the ligand that draws the molecule inside the channel, during which Y174ECL2 and Y185ECL2 together act as an arm, re-engaging the two hydroxyl groups and pulling the ligand deeper into the pocket (Fig. 6, C and D). Subsequently, salbutamol interacts with T1103.29, T195ECL2, and Y1995.38 as it slides deeper into the pocket before assuming its final pose akin to the crystal structure bound pose (PDB ID 2Y04) (Warne et al., 2011). Salbutamol also made a π-π interaction with the aromatic ring of Y1995.38, the orientation of which seems to push salbutamol further into the pocket and prevent its exit back through the hole. In its final pose, the two hydroxyl groups of saligenin head made H-bond interactions with side chain -OH groups of S2035.42 and S2075.46. Also, the -OH and -NH3+ groups of the ethanolamine chain were engaged in H-bond/electrostatic interactions with the side chain amino group of N3127.39 and the carboxyl group of D1133.32, respectively. To examine whether salmeterol or formoterol could also enter the binding pocket through this polar channel, we ran multiple SMD simulations (association) in which the ligands were kept near (within 5 Å) the channel entrance to facilitate the entry. Neither of the ligands entered the channel in any of the simulations, suggesting that this polar channel is unlikely to accommodate these larger lipophilic β2-AR agonists and thus signifies a unique access path for the small and polar salbutamol.
Fig. 6.

Salbutamol enters the β2-AR binding site through a novel polar channel. (A) From within the membrane view of the channel entrance (in yellow color and surface representation) displaying critical polar residues (in green color and licorice representation) that make immediate interactions with salbutamol (in blue color and licorice representation) as it enters. (B) The top view of the polar channel illustrating its position that is distal to the common access path lined by F193ECL2. (C and D) The snapshots show the intermediate states of salbutamol before reaching the binding pocket in its final bound pose, as presented in Fig. 6D. The receptor is illustrated in secondary structure representation (white).
The Distances between the D192-K305 Salt Bridge and the Hydrophobic F193-Y308
Salmeterol seems to break the salt bridge interaction before entering the binding pocket in the majority of simulations. During association, salmeterol forms interactions with D192ECL2, E3067.33, and K3057.32 during association that likely help break this interaction prior to entering between the transmembrane helices. As salmeterol slips into the binding pocket, it pushes aside the F193ECL2-Y3087.35 residues and breaks the hydrophobic lock to facilitate its entry. However, the rift of hydrophobic lock does not seem to follow a particular trend, as in many simulations, the residues are already far apart. For formoterol, a similar trend is seen with the salt bridge, in that the D192ECL2-K3057.33 is intact prior to association, and as formoterol gets closer to the binding site, it breaks these contacts (almost like a knife slicing through the bond). Formoterol forms contacts with D192ECL2 during the association in various simulations and might attribute to the salt bridge breaking open. The F193ECL2-Y3087.35 interaction is similar for formoterol in its distance, and as formoterol settles in the binding site, it pushes apart the F193-Y308 interaction. Salbutamol also appears to break the D192ECL2-K3057.33 interaction in the aqueous pathway, but for the polar channel access, it slips underneath the F193ECL2-Y3087.35, and the salt bridge, for the most part, seems to be unaffected until salbutamol is in the binding site. Salbutamol tends to explore where it can enter easiest, whereas salmeterol and formoterol seem to need to break the salt bridge to enter because of their size and location.
Unbinding Pathways for the β2-AR Agonists: Salmeterol Prefers to Localize into the Membrane after Dissociation from the Receptor
To elucidate the unbinding mechanisms of the studied ligands, including the paths and critical residue interactions affecting the kinetics of the dissociation processes, we performed SMD simulations using the β2-AR-ligand complexes in their native membrane environment. For salmeterol, the ligand-bound crystal structure of β2-AR (PDB ID 6MXT) (Masureel et al., 2018) was used as the starting point. For both formoterol and salbutamol, the ligands were docked to the same β2-AR crystal structure, replacing salmeterol and using their respective β1-AR-bound crystal poses [PDB IDs 6IBL (Lee et al., 2020) and 2Y04 (Warne et al., 2011), respectively] as guidance for the docking simulations. After a 50-ns equilibration run, in which all ligands maintained stable contacts and remained in the binding pocket, for each ligand, three SMD runs of 50 ns were performed. The ligands were pulled out of the binding pocket with a minimal force in the direction of least resistance (see Materials and Methods section for details). The SMD simulations revealed unique dissociation paths from the binding site for each ligand (Fig. 7, A–C). Most interestingly, in two of three simulations, salmeterol dissociated from the binding pocket and eventually localized into the membrane (Fig. 7A). The bilayer depth and orientation of salmeterol were identical to those determined by the membrane partitioning simulations carried out without the receptor (Fig. 1, B and C). Unlike salmeterol, both formoterol and salbutamol dissociated toward the region around ECL2/TMH4/TMH5 and remained in the aqueous bulk above the receptor (Fig. 7, B and C).
Fig. 7.

Unbinding paths and critical residue interactions along the dissociation pathways for salmeterol, formoterol, and salbutamol. (A–C) Dissociation paths for salmeterol, formoterol, and salbutamol, respectively. Salmeterol dissociates into the aqueous bulk and spends a transient period there before partitioning into the membrane near the receptor. Formoterol and salbutamol dissociate into the aqueous bulk surrounding the extracellular loop region. The start, intermediate, and final poses of the molecules are represented as density maps in red, gray, and blue colors, respectively. (D–F) Critical residues in contact with the ligands at their bound states and along the dissociation path. (G–I) Pie charts representing residues from various TMHs and ECL2 and the fraction of simulation time (percent occupancy) during which they are within 4 Å of the ligands.
When bound, all three ligands make conserved polar interactions (H-bonds) with the binding site residues S2035.43, S2075.46, D1133.32, and N3127.39 through their two hydroxyl groups of saligenin ring (one hydroxyl and one formyl amino group in the case of formoterol) and a hydroxyl and positively charged amino group of ethanolamine, respectively (Fig. 1A). As the ligands dissociated from the pocket, these interactions were broken at various time points for each ligand, indicating the specific strengths of those interactions attributed to the differences in their structures. Also, as the ligands drifted away from the site, they were in contact with a different set of residues from various TMHs and ECL2 along the way. The fraction of the total simulation time (50 ns) during which each residue was in contact with a ligand was given as percent occupancy in Fig. 7, G–I. For each ligand, as the simulation started, the amount of force used for pulling the ligand out of the pocket increased gradually as a function of time (Fig. 8A). It reached a maximum at which critical bonds were broken successively, leading to a decrease until the ligands were entirely dissociated. Ligands formed new interactions as they drifted away from the pocket, which caused momentary fluctuations in the required force, which was more pronounced for formoterol. The time points at which the individual polar interactions severed (bond length > 4 Å) were slightly different for the ligands.
Fig. 8.

Force profiles, critical electrostatic/H-bond interactions, and PMF profiles of salmeterol, formoterol, and salbutamol while unbinding from the β2-adrenergic receptor revealed by steered MD simulations. (A) Average force profile for each ligand calculated using three independent SMD simulations. The cumulative force applied increases until critical interactions get disrupted and then decreases to zero as bonds are broken successively. (B) The time evolution of the H-bond distances between the ligands and the binding site residues, indicating the strength and longevity of the interactions contributing to binding affinities of the ligands. (C) Salmeterol bound to the receptor in its crystal structure pose, showing all the critical residues involved in its dissociation path. (D) The PMF profile for each ligand was calculated by Jarzynski equality.
For salmeterol, the H-bond with S2075.46 was the first one to break at approximately 8 ns, followed by S2035.43, D1133.32, and F193ECL2 at around 12 ns. The H-bond between the positively charged ethanolamine amino group of salmeterol with N3127.39 lasted until approximately 15 ns, at which point all the critical interactions with the binding site residues were broken. A similar trend was observed for formoterol, except that it made additional interactions with several other residues (Fig. 7, E and H) along the way. Interestingly, for salbutamol, the bonds with S2035.43 and S2075.46 broke at the same time (∼13 ns), followed by D1133.32 and N3127.39 at approximately 15 ns.
Notably, formoterol remained in contact with binding site residues for a longer time relative to the other two ligands. Specifically, the H-bond between N3127.39 and the positively charged amine group on formoterol was intact until after 16 ns (Fig. 8, B and D). All the other H-bonds between binding site residues and formoterol begin breaking at 10 ns, starting with S2035.42 and S2075.46, and followed by D1133.32 around 12 ns. Formoterol also made contacts with many other residues, including H2966.58, Y3087.35, A2005.39, and Q1975.36, during dissociation (Fig. 7, E and H). Similarly, salbutamol also interacted with residues T195ECL2, A2005.39, Y3087.35, and F193ECL2 during dissociation (Fig. 7, F and I). Furthermore, unbinding of the ligands disrupted the salt bridge between D192ECL2 and K3057.33 and the hydrophobic lock between F193ECL2 and Y3087.35 along the way, which occurred mostly after 10 ns into the simulations (Supplemental Fig. 10).
The free energy profile along the reaction coordinate for each ligand was constructed as the PMF from multiple dissociation SMD simulations using the JE (Jarzynski, 1997), as described in the Materials and Methods section. The PMF values obtained for salmeterol, formoterol, and salbutamol are 35.266 ± 0.796 kcal/mol, 39.859 ± 0.29462 kcal/mol, and 34.621±0.495 kcal/mol, respectively (Fig. 8D). For salmeterol, all the binding site interactions are broken after 15 ns, and the dip in the PMF curve is due to its interactions with the ECL2 residues (D192, F193, F194, and T195) and surrounding water molecules before completely dissociating from the receptor and eventually settling in the membrane environment near the receptor. The PMF profile for salbutamol shows a similar dip as salmeterol after all the binding site interactions were broken due to its interactions with ECL2/TMH5 residues (F193, T195, and A200) along the dissociation path. Once these transient interactions were broken, salbutamol entered the aqueous phase and remained there for the rest of the simulation. The PMF profile for formoterol was unique, which clearly revealed strong interactions with other residues after leaving the main binding site cleft. In summary, all the three studied ligands dissociated from the binding site around the same time. However, the PMF curves indicate formoterol as having a slightly higher binding free energy compared with salmeterol and salbutamol, which have approximately the same binding free energies (Fig. 8D).
Hydration of Ligands and Binding Site Residues during Dissociation Simulations
Desolvation (removal of water or dewetting) of a ligand and its binding site is an integral step before the receptor-ligand binding process (Abel et al., 2008). The opposite process, meaning solvation of the ligand and binding site, occurs when the ligand dissociates from the binding site. To assess the extent of solvation along the dissociation paths, we calculated the number of water contacts (number of water molecules that are within 3.5 Å from any heavy atom) for the studied ligands and the binding site residues (D1133.32, S2035.42, S2075.46, F193ECL2, F194ECL2, T195ECL2, D192ECL2, N3127.39). The plots of water contacts for the ligands and binding site against the simulation time showed unique characteristics for each ligand (Fig. 9A). As expected, during dissociation, each ligand’s water contacts increased as the ligand drifted away from the binding site. However, interestingly, the water contacts for salmeterol increased until 30 ns (from 6 to 17) but decreased afterward (from 16 to 10) as the ligand partitioned into the membrane. Formoterol and salbutamol had a similar trend in that the number of water contacts increased steadily as the ligands dissociated (from 4 to 15 for formoterol and from 5 to 10 for salbutamol) but attained steady levels afterward. However, the decrease was less pronounced for these ligands.
Fig. 9.

The number of water contacts as a measure of solvation (wetting or hydration) of the ligands (A) and binding site residues (B) during the dissociation of salmeterol, formoterol, and salbutamol. The water contacts were at a minimum at the start of the simulation (t = 0 ns) when the ligands were bound. The contacts increased gradually as the ligands drifted away from the binding site and the degree of increase was directly proportional to the ligand size in the order of salmeterol > formoterol > salbutamol. Notably, the water contacts decreased for salmeterol, indicating its relocation into the surrounding membrane. Similarly, the number of water contacts with the binding site residues increased gradually, converging to approximately the same number of contacts (∼28).
The water contacts of the binding site residues exhibited similar differences among the three ligands (Fig. 9B), clearly reflecting the ligand sizes. While bound, salbutamol had more water in the binding site than formoterol and salmeterol (∼22, ∼20, and ∼18, respectively). The smaller size of salbutamol compared with the other bulky ligands can explain this difference. Once the ligands drifted away from the binding site, the number of water contacts converged to similar values (26–28 water molecules) for all the ligands, indicating an absence of significant conformational change in the receptor after ligand dissociation (Mason et al., 2012).
Binding Free Energies Calculated by Funnel Metadynamics
We used FM (Limongelli et al., 2013; Raniolo and Limongelli, 2020) to elucidate all possible binding modes and calculate the absolute binding free energies of the three β2-AR agonists. For integral membrane proteins such as β2-AR, the molecular recognition process occurs at the interface between protein, membrane, and solvent molecules, including ions. It is critical to employ an appropriate method for large and flexible ligands such as salmeterol, explicit solvent molecules, membrane lipids surrounding the receptor with a buried binding site, and regions that undergo notable conformational changes (for example, ECL2). Although a priori knowledge of the ligand-binding mode is not required for FM simulations, we used the crystal bound pose as starting conformations as used in the dissociation SMD simulations described earlier. Also, the results obtained from the WT-MetaD simulations for elucidating the association mechanisms provided useful information on the proper placement of the funnel in FM simulations (see the Materials and Methods section).
For salmeterol, the funnel was placed such that the binding and unbinding states incorporate the ligand access through TMH1 and TMH7 from within the membrane. This step is critical, as salmeterol has been found to enter the binding pocket from within the membrane through the transmembrane helices in the association simulations. Figure 10 illustrates the binding free energy surface labeled A–F with the bound and multiple unbound states of the ligand (clustered) and placement of the funnel (G). The binding free energy was calculated from the energy difference between the unbound and bound region characterized by two collective variables (lp, the position of the ligand along the funnel axis, and ld, the orthogonal distance of the ligand from the funnel axis) with a correction for the restraining potential of the cylinder portion of the funnel. The bound state was characterized by lp and ld values in the range of 0.5–0.7 nm and 0.1–0.25 nm, respectively. The lp and ld values for the unbound state are 2.5–3.0 nm and 0.1 nm, respectively. The Hills potential deposited along the simulation time was monitored, and the near absence of any depositions indicated convergence of the free energy (Supplemental Fig. 9A). During the 900-ns simulation time, there were at least five crossing events between bound and unbound states (Supplemental Fig. 9B). After applying the correction for the restraining potential of the cylinder portion of the funnel, the calculated binding free energy (ΔGb0) was −14.0 kcal/mol, which is reasonably in agreement with the published experimental value of −11.59 kcal/mol.
Fig. 10.

The free energy surface of salmeterol binding to β2-AR obtained from the funnel metadynamics simulation using lp, the position of the ligand along the funnel axis, and ld, the distance of the ligand from this axis, as collective variables. Two minimum energy paths, one passing through the aqueous bulk (green) and another through the transmembrane helices (red), reaching the membrane, were determined by the Minimum Energy Path Surface Analysis tool. (A–F) Multiple clusters representing the fully bound state (A) to several intermediate conformational states (B–F) were sampled by several recrossing between bound and unbound states during the entire 900-ns simulation. (G) The funnel placement was based on several association/dissociation simulations during which salmeterol was seen leaving the pocket either by aqueous routes or by a transmembrane route through TMH 1 and 7. The cone region of the funnel was defined by a vertex height Zcc of 3.0 nm from the origin and an α angle of 0.6 rad. The radius of the cylindrical portion of the funnel rcyl was 2 Å.
For formoterol, the FM simulation successfully captured the bound pose (similar to the docked pose) and unbound pose multiple times during the 450-ns simulation time (Supplemental Fig. 9B). The funnel was placed such that the simulation captures the ligand’s association through the aqueous access route. The bound region was characterized by the collective variables in the ranges of 0–0.4 nm for lp and 0–0.3 nm for ld. The values for lp and ld in the unbound region were 3 nm and 0 nm, respectively. The final calculated (see the Materials and Methods section for details) binding free energy (ΔGb0) for formoterol was −10.14 kcal/mol, which is in good agreement with the published experimental value of −11.7 kcal/mol. Supplemental Fig. 7 illustrates the binding free energy surface, labeled A–F, with the bound and multiple unbound states of the ligand (clustered) and placement of the funnel (G).
For salbutamol, the placement of the funnel was to incorporate the dissociated state in the aqueous solvent. Over the course of 500 ns, there were only two recrossing events capturing bound and unbound states (Supplemental Fig. 9B). Supplemental Fig. 8 illustrates the binding free energy surface, labeled A–E, with the bound and multiple unbound states of the ligand (clustered). Considering a correction for the restraining potential of the funnel, the final calculated binding free energy (ΔGb0) was −12.5 kcal mol−1, which is comparable to the experimental data [ Gb_exp = −9.78 kcal mol−1 (Baur et al., 2010), see detailed information in Materials and Methods]. The small discrepancy in binding free energy can be explained by fewer recrossing events captured during the simulation relative to formoterol and salmeterol. It is important to note that even though MD-based FM protocol has been shown to predict absolute binding free energies in good agreement with the experimental values, one should be cautious about inaccuracy in force-field parameters that may introduce error. Our simulations not only resulted in predicting the binding free energy in good agreement with the experimental values but also provided useful insights into the binding poses and conformations of the ligands in the unbound states.
Discussion
The molecular mechanism responsible for the persistent action of the long-acting β2-AR agonists has been a matter of debate for a long time (Coleman, 2009). However, recent studies provide evidence to support the plasmalemma diffusion microkinetic model, which suggests that the duration of action of long-acting β2-AR agonists such as salmeterol and formoterol are, at least in part, due to their lipophilicity and distinct interactions with the membrane lipids surrounding the receptor (Sykes and Charlton, 2012; Sykes et al., 2014; Dickson et al., 2016).
Our results offer valuable insights into the differences in the membrane partitioning characteristics of salmeterol and formoterol in comparison with a short-acting drug, salbutamol. The presence of additional phenyl-alkoxy-alkyl and alkoxy-phenyl-alkyl tails in salmeterol and formoterol, respectively, contributes to the differential partitioning of the ligands at varied bilayer depths (Fig. 1A; Supplemental Table 1). Specifically, the lipid tails contribute to preferential accumulation of salmeterol and formoterol at bilayer depths much deeper than for salbutamol, as evidenced by time-average mass density profiles (Supplemental Fig. 1). The specific locations and orientations of salmeterol and formoterol are in good agreement with a magic-angle spinning NMR study that showed a similar depth and orientation for both the molecules (Yan et al., 2017). The difference in bilayer depths resulted in distinct orientations of their saligenin heads and markedly different interactions with the polar and alkyl lipid functional groups (Fig. 1, C–E). The characteristic bilayer depths, orientations, and interactions of individual ligands dictate the solvation characteristics of the ligands and, in turn, affect their access paths to the receptor. The low number of water contacts per heavy atom (Supplemental Fig. 2C) and a high degree of burial deep into the lipid alkyl region while partitioned in the bilayer (Supplemental Fig. 1 and 3) seem to favor salmeterol’s access to the receptor through a lipid path. Interestingly, for formoterol, higher water contact per heavy atom and less burial into the alkyl region while partitioned in the bilayer seem to reduce the energetic barrier associated with desolvation from the bilayer, favoring the receptor entry through the aqueous path. Salbutamol’s unique orientation (Fig. 1C) results in the highest number of water contacts per heavy atom (Supplemental Fig. 2B) and seems to entirely discourage its interaction with the alkyl tail region of the bilayer. For lipophilic compounds such as salmeterol and formoterol, there seem to exist subtle differences in the thermodynamics of desolvation from the aqueous phase, solvation into the membrane, desolvation from the membrane, and interactions at the lipid-water interface before sliding into the binding pocket, determining the magnitude of the energetic barriers involved in the binding process. Although the lipophilic “tail” of salmeterol serves to anchor the molecule in the surrounding membrane after dissociation and be available for rebinding, there seems to be a trade-off on the onset of action. Therefore, achieving such a fine balance between preferential partitioning into the membrane and the ability to access the receptor through a route involving a combination of membrane and aqueous routes will likely contribute, at least in part, to the fast onset and slow offset for β2-agonists administered by inhalation. The various pharmacokinetic processes, such as the rate of adsorption and solubility in the lung fluids of the inhaled drugs, metabolism (in lung tissues and by the liver after systemic absorption), and excretion by various organs, also contribute to the biologic half-life of drugs and need attention during structural optimization.
Drugs that enter (wash-in) rapidly into membranes achieve relatively low concentrations in the membrane and leave (washout) rapidly. These drugs remain within the membrane compartment for a short period of time relative to slowly accumulating drugs that “load” into the membrane at higher concentrations and washout slowly due to their favorable membrane interactions (Teschemacher and Lemoine, 1999). The three parameters, wash-in and washout rates and equilibrium concentration in the membranes, have a combined effect of keeping the ligands surrounding membrane-embedded targets for longer periods, thus affecting the therapeutic window of activity. Thus, lipophilic/amphiphilic drugs such as salmeterol and formoterol are available to interact with their membrane-associated receptor over a longer period. A similar membrane-involved molecular mechanism has been previously reported for 1,4-dihydropyridine calcium channel blockers targeting voltage-gated channels in cardiac and smooth muscle sarcolemma (Herbette et al., 1993a,b, 1994). Specifically, the unique clinical pharmacokinetic properties of lacidipine (short plasma half-life, gradual onset of action, coupled with longer duration of action) are mainly due to its interactions with the membrane of the arteriole wall, where it is stored over a long period. Unlike amlodipine and nimodipine, which are primarily located at the headgroup/core interface (12–16 Å from the center of the membrane) of the bilayer, lacidipine is located much deeper (∼7 Å from the center of the membrane) in the membrane because of a unique tert-butyl hydrophobic side chain on the phenyl moiety that makes lacidipine both highly lipophilic and sterically bulky. Thus, lacidipine diffuses very slowly into and out (low wash-in and washout rates) of the lipid bilayer and accumulates to a high concentration in this compartment. The high concentration of lacidipine combined with slow washout ensures relatively long residence time. Also, the slow loading into the membrane compartment compared with other drugs in this class avoids a “burst” effect and offers a reduced side effect profile.
In a membrane-facilitated ligand-binding process, the ligand molecule first partitions to a specific, energetically favorable location and adopts an appropriate orientation and conformation within the bilayer before laterally diffusing to its transmembrane binding site (Szlenk et al., 2019; Yuan et al., 2018). The specific makeup and organization of the bilayer restrict lipophilic or amphiphilic ligands at a specific depth, limiting the translational and rotational freedom, resulting in a certain orientation and conformation relative to its binding site (Sargent and Schwyzer, 1986; Schwyzer, 1991, 1995a,b). Most importantly, for ionized ligand molecules, the strong interactions of ionized species (bases and acids) with various phospholipid functional groups dictate their membrane partitioning characteristics and thus affect their access and binding to the receptor (Burges et al., 1987; Jeevan B. GC et al., 2020; Mason et al., 1989). In a series of studies (Sykes and Charlton, 2012; Sykes et al., 2014; Dickson et al., 2016), researchers from Novartis showed that for several clinically relevant β2-AR agonists, the degree of phospholipid interaction affects the drug’s binding kinetics profile and thus the observed affinity. Notably, the measured affinity of salmeterol was three orders of magnitude higher than salbutamol, driven by the large difference in their association rates, which appears to be mainly due to higher membrane partitioning of salmeterol (Szczuka et al., 2009). Upon correcting for this increased membrane affinity, both agonists have comparable intrinsic binding affinity (corrected pKd; Supplemental Table 1) values for β2-AR (Mason et al., 1991; Vauquelin and Packeu, 2009).
Our simulations offer unprecedented details on the access and binding mechanisms of the three ligands from their energetically favorable locations within the membrane-aqueous environment surrounding the receptor. Salmeterol took a consistent lipid path to access the binding site through transmembrane helices 1 and 7. Earlier mutagenesis studies reported the importance of these residues for salmeterol’s efficacy and affinity (Isogaya et al., 1998; Baker et al., 2015). Salmeterol entered the binding pocket by two distinct ways in which the ligand breached the salt bridge and hydrophobic lock either through its saligenin headfirst or by its tail. However, in both modes, the lipid molecules surrounding the ligand appear to play an active role in its access and entry into the binding site. During the initial steps, salmeterol continued to interact with three POPC molecules while it made contact with TMHs 1 and 7. Most intriguingly, the hydrophobic interactions between the aryl-alkoxy-alkyl tail of the ligand with the alkyl tails of the lipids seem to facilitate the conformational rearrangement of the ligand. This near 180° flip of the ligand is critical to present the phenyl end as the passkey to F193ECL2, a gatekeeper residue, shown to be critical for salmeterol’s affinity and efficacy (Isogaya et al., 1998; Baker et al., 2015). In addition, salmeterol continued to interact with both glyceryl and phosphate oxygens until it engaged with the receptor residues. Such membrane-facilitated mechanisms have been demonstrated for ligands of multiple GPCRs, including the cannabinoid receptor CB2 (Hurst et al., 2010; Jakowiecki and Filipek, 2016), the sphingosine-1 phosphate-1 receptor (Hanson et al., 2012), the rhodopsin receptor (Hildebrand et al., 2009), the P2Y purinoceptor (P2Y1) (Yuan et al., 2018), and proteinase-activated receptor 1 (Bokoch et al., 2018). Also, it appears that topological arrangement of TMH1 with respect to TMH7 in β2-AR is critical for such entry through the transmembrane helices from within the membrane. The distance measured between two residues (W32 in TMH1 and E306 in TMH7) is approximately 14.5 Å. Interestingly, this distance is largely conserved among many class A GPCRs with the α-helix ECL2 region (see Supplemental Table 2). In contrast, the distance is much shorter in class A GPCRs with the β-sheet ECL2 region indicating tighter assembly of the two helices. To exploit these larger clefts between TM1 and TM7 helices (for GPCRs with the α-helix ECL2 region), a designing strategy must involve additional consideration for ligand flexibility to navigate through the cleft and energetically favorable residue interactions that effectively draw the ligand from within the membrane and let the molecule slide into the binding pocket.
Intriguingly, our results show that formoterol’s access and binding mechanisms are different from that of salmeterol and follow the patterns reported for other polar β2-AR agents (Dror et al., 2011). The subtle structural differences at the saligenin head and tail regions (Fig. 1A) among the ligands clearly affect their partitioning into and out of the membrane and subsequently affect their access to the receptor. Specifically, the strong electrostatic interactions of formoterol with the polar residues and aqueous region around the receptor seem to provide sufficient force to overcome the favorable interactions of its tail region with the membrane (Fig. 4, A–C). In contrast, the prerequisites, such as the release of its anchored tail from the hydrophobic interactions with the lipids and residues of TMHs 1 and 7, breaking of the D192-K305 salt bridge and F193-Y308 hydrophobic lock, together appear to be the rate-limiting factor for salmeterol’s entry into the pocket (the energy barrier between C and D in the FES of Fig. 2), possibly contributing to its slower onset of action. As anticipated, in the majority of the simulations, salbutamol entered the receptor binding site through the common aqueous route, as reported for other polar β2-AR agents (Dror et al., 2011). However, unexpectedly, we found another novel polar channel that is located on the other side of the route, distal to the ECL2. Salbutamol’s entry through this novel polar channel did not require the breaking of the hydrophobic lock (Y3087.35-F193ECL2), unlike the other primary access pathway, as it remained intact throughout the entire simulation time. The ability of salbutamol to enter the binding site through multiple paths with minimal energy barriers, thanks to its small size, may explain to a certain degree its quicker onset of action.
Our dissociation simulations offer useful insights into the force profiles, critical residue interactions along the dissociation paths, and accompanying PMF profiles, illustrating the strength of interactions that were broken along the way. It is important to note that these results should not be interpreted in the context of the nature of the biological response (formoterol, a full agonist, salbutamol, and salmeterol, partial agonists), which is independent of ligand affinity. The unbinding of the ligands from the binding pocket caused disruptions of the salt bridge (between D192ECL2-K3057.33) and hydrophobic lock (between residues F193ECL2 and Y3087.35) along the way, as previously reported by similar computational studies (Wang and Duan, 2009; González et al., 2011) performed using β2-AR antagonists such as carazolol. Interestingly, in one of these studies (Wang and Duan, 2009), based on the dissociation results, the authors proposed a putative ligand entry pathway in which the ligand first dives into the cleft between TM2, TM3, and TM7 at the extracellular opening, interacts with ECL2 residues (D192 and F193), and then reaches the other end of the binding pocket (the cleft between TM4, TM5, and TM6). Our association simulations seem to support their hypothesis and captures the events in the proposed sequence. The number of water contacts around the ligands as well as the binding site residues during the dissociation events clearly capture the distinction between the ligands, in terms of their size, unique interactions with the residues and aqueous bulk around the region, and eventual re-entry into the membrane, in the case of salmeterol. Interestingly, we did not observe any notable lasting interactions of salmeterol’s phenyl tail with the ECL2 during dissociation. This observation supports the results from earlier experiments involving reassertion of salmeterol’s activity after treatment with an antagonist, followed by washout, indicating that the exosite represents the membrane (Green et al., 1996).
The binding free energies calculated by funnel metadynamics, taking into account the membrane-facilitated access and dissociation paths observed in SMD and WT-MetaD simulations, are in good agreement with the experimental data and appear to support the described mechanisms (Kästner, 2011; Bochicchio et al., 2015; Schneider et al., 2015; Troussicot et al., 2015).
As with any study, there may be some limitations associated with our simulations. It is possible that the obtained results from the membrane partitioning, association, and dissociation simulations might be different if we have used a heterogeneous lipid composition to mimic the asymmetric nature of the biologically relevant plasma membrane of lung smooth muscle cells. Lipidomic analyses (Zemski Berry et al., 2017; Kyle et al., 2018) of major lung cells (alveolar macrophages, bronchial epithelial cells, and alveolar type II cells) isolated from human subjects revealed that these cell types have varying distributions of lipid species, with glycerophospholipids being the most abundant of all. In addition, the alveolar type II cells produce pulmonary surfactant, which contains 90% lipid and 10% protein. Phosphatidylcholines are the most abundant class of phospholipids (∼80%), present in the pulmonary surfactant. It is noteworthy to recognize that for orally inhaled β2-AR agonists, such as salmeterol, the drug molecules often encounter the lung surfactant lipids (∼80% phosphatidylcholines) first and then reach alveolar epithelial cells, each layer of which is lined by capillaries. So, unlike other organs, drug molecules do not need to cross multiple cell layers to penetrate lung tissues. Considering all these facts, we chose to keep the membrane composition relatively simple and to reflect the most commonly occurring 16:0–18:1 PC (POPC) lipids and cholesterol (10%–30%). We believe that the obtained results, to a large extent, are representative of the mechanisms occurring in the human lungs.
Also, the choice of force field for simulating partitioning of solutes in the bilayer has been shown to affect the outcome (Paloncyova et al., 2014). This is critical, especially given the complex environment (the interface between membrane, protein, and solvent molecules) in which the ligands’ access and binding phenomena were studied. In addition, the conformational changes associated with the different functional states of the receptor (active, inactive, and intermediate transition states) as well as the recruitment of signaling partners (G proteins) at the intracellular end may affect the observed results.
In summary, our results offer valuable insights into the functional role of membrane lipids in the molecular recognition of ligands to β2-AR and lay a groundwork for incorporating membrane interactions of drugs as an integral component in rational drug design (Payandeh and Volgraf, 2021). The ability of the membrane to preferentially accumulate and facilitate the receptor access of ligands opens an exciting new avenue of study for better understanding of structure-membrane interactions relationships of new drug candidates. Although it is important not to overlook the inherent side effects associated with excessive accumulation in membranes, recent structural studies revealing an increasing number of lipid-facing binding sites at the extrahelical surfaces of GPCRs (Shao et al., 2019; Bueno et al., 2020; Xiao et al., 2021) warrant comprehensive and systematic investigation of membrane interactions of drugs. Understanding the specific contribution of functional groups in small-molecule drugs toward target membrane partitioning characteristics will significantly assist in designing new drug candidates with desirable pharmacology.
Conclusions
GPCRs are the largest family of integral membrane proteins that are responsible for the therapeutic effects of more than one-third (∼33%) of all Food and Drug Administration–approved small-molecule drugs. An increasing body of evidence suggests that the cell membrane surrounding these receptors may actively participate in the molecular recognition processes. Most importantly, for lipophilic and amphiphilic ligands, specific interactions with the membrane lipids can affect access and binding mechanisms, ultimately influencing their pharmacology. In this study, we present a mechanistic description of how differences in the membrane partitioning characteristics of three commonly used asthma drugs, salmeterol, formoterol, and salbutamol, affect their mode of access and binding to the membrane-embedded β2-adrenergic receptor. The distinction in the structural features in these drugs produces specific interactions with lipid bilayer strata, resulting in varying bilayer depth, orientations, and conformations, and results in markedly different receptor access paths. Most notably, salmeterol’s phenyl-alkoxy-alkyl tail has been observed to play a novel role as a passcode to gain access into the binding site through transmembrane helices from within the membrane. Despite containing a hydrophobic tail and being moderately lipophilic, formoterol prefers to access the binding site using a path that is comparable to ones used by other known hydrophilic ligands, possibly explaining its quicker onset of action compared with salmeterol. In addition to the common aqueous path, salbutamol has been observed to access the binding site using a novel path that is located at the distant side of the ECL2 loop, and this narrow access route is impermeable to bulkier salmeterol and formoterol. The structural details reported here provide useful insights into the clear and active role played by membrane lipids in preferential accumulation, access, and binding of ligands binding to the membrane-embedded protein. Future investigations will help us discern how to exploit membrane interactions to avail desired pharmacology while designing drugs targeting membrane-embedded proteins such as GPCRs.
Acknowledgments
We thank Dr. Stefano Raniolo and Dr. Vittorio Limongelli for providing the PLUMED version 2.3 patch code for performing funnel metadynamics simulations.
Abbreviations
- β2-AR
β2-adrenergic receptor
- COM
center of mass
- ECL
extracellular loop
- FES
free energy surface
- FM
funnel metadynamics
- GPCR
G protein–coupled receptor
- MD
molecular dynamics
- MOE
Molecular Operating Environment
- PDB
Protein Data Bank
- PMF
potential of mean force
- SMD
steered molecular dynamics
- TMH
transmembrane helix
- WT-MetaD
well tempered metadynamics
- CHARMM-GUI
Chemistry at HARvard Molecular Mechanics – graphical user interface
- POPC
– 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine
- PLUMED
PLUgin for MolEcular Dynamics
- RMSD
root mean square deviation
Authorship Contributions
Participated in research design: Szlenk, GC, Natesan.
Conducted experiments: Szlenk, GC.
Performed data analysis: Szlenk, GC, Natesan.
Wrote or contributed to the writing of the manuscript: Szlenk, GC, Natesan.
Footnotes
This work was supported by National Institutes of Health National Institute of General Medical Sciences [Grant R15 GM131293-01].
The authors declare no competing interests.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- (2019)Molecular Operating Environment (MOE), 2013.08, Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada. [Google Scholar]
- Abel R, Young T, Farid R, Berne BJ, Friesner RA (2008) Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J Am Chem Soc 130:2817–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2:19–25. [Google Scholar]
- Anderson GP, Lindén A, Rabe KF (1994) Why are long-acting beta-adrenoceptor agonists long-acting? Eur Respir J 7:569–578. [DOI] [PubMed] [Google Scholar]
- Baker JG (2010) The selectivity of beta-adrenoceptor agonists at human beta1-, beta2- and beta3-adrenoceptors. Br J Pharmacol 160:1048–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG, Proudman RGW, Hill SJ (2015) Salmeterol’s extreme β2 selectivity is due to residues in both extracellular loops and transmembrane domains. Mol Pharmacol 87:103–120. [DOI] [PubMed] [Google Scholar]
- Ball DI, Brittain RT, Coleman RA, Denyer LH, Jack D, Johnson M, Lunts LHC, Nials AT, Sheldrick KE, Skidmore IF (1991) Salmeterol, a novel, long-acting β 2-adrenoceptor agonist: characterization of pharmacological activity in vitro and in vivo. Br J Pharmacol 104:665–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barducci A, Bonomi M, Parrinello M (2011) Metadynamics. Wiley Interdiscip Rev Comput Mol Sci 1:826–843. [Google Scholar]
- Barducci A, Bussi G, Parrinello M (2008) Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys Rev Lett 100:020603. [DOI] [PubMed] [Google Scholar]
- Baur FBeattie DBeer DBentley DBradley MBruce ICharlton SJCuenoud BErnst RFairhurst RA, et al. (2010) The identification of indacaterol as an ultralong-acting inhaled β2-adrenoceptor agonist. J Med Chem 53:3675–3684. [DOI] [PubMed] [Google Scholar]
- Berendsen HJC, van der Spoel D, van Drunen R (1995) GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun 91:43–56. [Google Scholar]
- Billington CK, Penn RB, Hall IP (2017) β2 Agonists. Handb Exp Pharmacol 237:23–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochicchio D, Panizon E, Ferrando R, Monticelli L, Rossi G (2015) Calculating the free energy of transfer of small solutes into a model lipid membrane: comparison between metadynamics and umbrella sampling. J Chem Phys 143:144108. [DOI] [PubMed] [Google Scholar]
- Bokoch MPJo HValcourt JRSrinivasan YPan ACCapponi SGrabe MDror ROShaw DEDeGrado WF, et al. (2018) Entry from the lipid bilayer: a possible pathway for inhibition of a peptide G protein-coupled receptor by a lipophilic small molecule. Biochemistry 57:5748–5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomi MBranduardi DBussi GCamilloni CProvasi DRaiteri PDonadio DMarinelli FPietrucci FBroglia RA, et al. (2009) PLUMED: a portable plugin for free-energy calculations with molecular dynamics. Comput Phys Commun 180:1961–1972. [Google Scholar]
- Boulet L-P, Laviolette M, Boucher S, Knight A, Hébert J, Chapman KR (1997) A twelve-week comparison of salmeterol and salbutamol in the treatment of mild-to-moderate asthma: a Canadian multicenter study. J Allergy Clin Immunol 99:13–21. [DOI] [PubMed] [Google Scholar]
- Brooks BRBrooks CL 3rdMackerell AD JrNilsson LPetrella RJRoux BWon YArchontis GBartels CBoresch S, et al. (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30:1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno ABSun BWillard FSFeng DHo JDWainscott DBShowalter ADVieth MChen QStutsman C, et al. (2020) Structural insights into probe-dependent positive allosterism of the GLP-1 receptor. Nat Chem Biol 16:1105–1110. [DOI] [PubMed] [Google Scholar]
- Burges RA, Gardiner DG, Gwilt M, Higgins AJ, Blackburn KJ, Campbell SF, Cross PE, Stubbs JK (1987) Calcium channel blocking properties of amlodipine in vascular smooth muscle and cardiac muscle in vitro: evidence for voltage modulation of vascular dihydropyridine receptors. J Cardiovasc Pharmacol 9:110–119. [PubMed] [Google Scholar]
- Carter AA, Hill SJ (2005) Characterization of isoprenaline- and salmeterol-stimulated interactions between β2-adrenoceptors and β-arrestin 2 using β-galactosidase complementation in C2C12 cells. J Pharmacol Exp Ther 315:839–848. [DOI] [PubMed] [Google Scholar]
- Cazzola M, Calzetta L, Matera MG (2011) β(2) -adrenoceptor agonists: current and future direction. Br J Pharmacol 163:4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzola M, Matera MG, Lötvall J (2005) Ultra long-acting β 2-agonists in development for asthma and chronic obstructive pulmonary disease. Expert Opin Investig Drugs 14:775–783. [DOI] [PubMed] [Google Scholar]
- Coleman RA (2009) On the mechanism of the persistent action of salmeterol: what is the current position? Br J Pharmacol 158:180–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CSBrad AHIsabelle Van LGeorges V, (2015) The role of binding kinetics in GPCR drug discovery. Curr Top Med Chem 15(24):2504–2522. [DOI] [PubMed] [Google Scholar]
- Dickson CJHornak VVelez-Vega CMcKay DJJReilly JSandham DAShaw DFairhurst RACharlton SJSykes DA, et al. (2016) Uncoupling the structure–activity relationships of β2 adrenergic receptor ligands from membrane binding. J Med Chem 59:5780–5789. [DOI] [PubMed] [Google Scholar]
- Dror ROGreen HFValant CBorhani DWValcourt JRPan ACArlow DHCanals MLane JRRahmani R, et al. (2013) Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503:295–299. [DOI] [PubMed] [Google Scholar]
- Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, Shaw DE (2011) Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc Natl Acad Sci USA 108:13118–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorin G, Klein ML, Hénin J (2013) Using collective variables to drive molecular dynamics simulations. Mol Phys 111:3345–3362. [Google Scholar]
- Frisch MJTrucks GWSchlegel HBScuseria GERobb MACheeseman JRScalmani GBarone VPetersson GANakatsuji H, et al. (2016) Gaussian 16 Rev. B.01, Gaussian, Inc., Wallingford, CT. [Google Scholar]
- Gherbi K, Briddon SJ, Charlton SJ (2018) Micro-pharmacokinetics: quantifying local drug concentration at live cell membranes. Sci Rep 8:3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giembycz MA, Raeburn D (1991) Putative substrates for cyclic nucleotide-dependent protein kinases and the control of airway smooth muscle tone. J Auton Pharmacol 11:365–398. [DOI] [PubMed] [Google Scholar]
- Green SA, Spasoff AP, Coleman RA, Johnson M, Liggett SB (1996) Sustained activation of a G protein-coupled receptor via “anchored” agonist binding. Molecular localization of the salmeterol exosite within the 2-adrenergic receptor. J Biol Chem 271:24029–24035. [DOI] [PubMed] [Google Scholar]
- Hanson MARoth CBJo EGriffith MTScott FLReinhart GDesale HClemons BCahalan SMSchuerer SC, et al. (2012) Crystal structure of a lipid G protein-coupled receptor. Science 335:851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen JM, Agertoft L, Pedersen S (1992) Protective effect and duration of action of inhaled formoterol and salbutamol on exercise-induced asthma in children. J Allergy Clin Immunol 89:1176–1182. [DOI] [PubMed] [Google Scholar]
- Herbette LG, Gaviraghi G, Dring RJ, Briggs LJ, Mason RP (1993a) Interactions of lacidipine and other calcium channel drugs with biological membranes: a structural model for receptor/drug binding utilizing the membrane bilayer, in Calcium Antagonists: Pharmacology and Clinical Research (Godfraind T, Paoletti R, Govoni S, Vanhoutte PM, eds) pp 15–23, Springer Netherlands, Dordrecht. [Google Scholar]
- Herbette LG, Gaviraghi G, Tulenko T, Mason RP (1993b) Molecular interaction between lacidipine and biological membranes. J Hypertens Suppl 11:S13–S19. [DOI] [PubMed] [Google Scholar]
- Herbette LG, Mason PE, Gaviraghi G, Tulenko TN, Mason RP (1994) The molecular basis for lacidipine’s unique pharmacokinetics: optimal hydrophobicity results in membrane interactions that may facilitate the treatment of atherosclerosis. J Cardiovasc Pharmacol 23 (Suppl 5):S16–S25. [PubMed] [Google Scholar]
- Hildebrand PW, Scheerer P, Park JH, Choe H-W, Piechnick R, Ernst OP, Hofmann KP, Heck M (2009) A ligand channel through the G protein coupled receptor opsin. PLoS One 4:e4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins DJ, Sherman W, Tidor B (2012) Rational approaches to improving selectivity in drug design. J Med Chem 55:1424–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38, 27–28. [DOI] [PubMed] [Google Scholar]
- Hurst DP, Grossfield A, Lynch DL, Feller S, Romo TD, Gawrisch K, Pitman MC, Reggio PH (2010) A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J Biol Chem 285:17954–17964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogaya M, Yamagiwa Y, Fujita S, Sugimoto Y, Nagao T, Kurose H (1998) Identification of a key amino acid of the β2-adrenergic receptor for high affinity binding of salmeterol. Mol Pharmacol 54:616–622. [PubMed] [Google Scholar]
- Jack D (1991) The 1990 Lilly Prize Lecture. A way of looking at agonism and antagonism: lessons from salbutamol, salmeterol and other beta-adrenoceptor agonists. Br J Clin Pharmacol 31:501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakowiecki J, Filipek S (2016) Hydrophobic ligand entry and exit pathways of the CB1 cannabinoid receptor. J Chem Inf Model 56:2457–2466. [DOI] [PubMed] [Google Scholar]
- Jarzynski C (1997) Nonequilibrium equality for free energy differences. Phys Rev Lett 78:2690–2693. [Google Scholar]
- JB GC, Szlenk C, Jin G, Xinyue D, Zhenjia W, Natesan S (2020) Molecular Dynamics Simulations Provide Insight into the Loading Efficiency of Pro-Resolving Lipid Mediators Resolvin D1 and D2 in Cell Membrane-Derived Nanovesicles. Molecular Pharmaceutics 17 (6) 2155–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Im W (2007) Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS One 2:e880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935. [Google Scholar]
- Kästner J (2011) Umbrella sampling. Wiley Interdiscip Rev Comput Mol Sci 1:932–942. [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr, Pastor RW (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B 114:7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA (1992) THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J Comput Chem 13:1011–1021. [Google Scholar]
- Kyle JEClair GBandyopadhyay GMisra RSZink EMBloodsworth KJShukla AKDu YLillis JMyers JR, et al. (2018) Cell type-resolved human lung lipidome reveals cellular cooperation in lung function. Sci Rep 8:13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laio A, Gervasio FL (2008) Metadynamics: a method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep Prog Phys 71:126601. [Google Scholar]
- Laurent F, Michel A, Bonnet PA, Chapat JP, Boucard M (1993) Evaluation of the relaxant effects of SCA40, a novel charybdotoxin-sensitive potassium channel opener, in guinea-pig isolated trachealis. Br J Pharmacol 108:622–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JCheng XSwails JMYeom MSEastman PKLemkul JAWei SBuckner JJeong JCQi Y, et al. (2016) CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J Chem Theory Comput 12:405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JPatel DStahle JPark S-JKern NKim SLee JCheng XValvano MHolst O, et al. (2019) CHARMM-GUI Membrane Builder for Complex Biological Membrane Simulations with Glycolipids and Lipoglycans. J Chem Theory Comput 15(1):775–786. [DOI] [PubMed] [Google Scholar]
- Lee Y, Warne T, Nehmé R, Pandey S, Dwivedi-Agnihotri H, Chaturvedi M, Edwards PC, García-Nafría J, Leslie AGW, Shukla AK, et al. (2020) Molecular basis of β-arrestin coupling to formoterol-bound β1-adrenoceptor. Nature 583:862–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limongelli V, Bonomi M, Parrinello M (2013) Funnel metadynamics as accurate binding free-energy method. Proc Natl Acad Sci USA 110:6358–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL (2012) OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res 40:D370–D376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lötvall J (2001) Pharmacological similarities and differences between β2-agonists. Respir Med 95 (Suppl B):S7–S11. [DOI] [PubMed] [Google Scholar]
- Marcos-Alcalde I, Setoain J, Mendieta-Moreno JI, Mendieta J, Gómez-Puertas P (2015) MEPSA: minimum energy pathway analysis for energy landscapes. Bioinformatics 31:3853–3855. [DOI] [PubMed] [Google Scholar]
- Mason JS, Bortolato A, Congreve M, Marshall FH (2012) New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol Sci 33:249–260. [DOI] [PubMed] [Google Scholar]
- Mason RP, Campbell SF, Wang SD, Herbette LG (1989) Comparison of location and binding for the positively charged 1,4-dihydropyridine calcium channel antagonist amlodipine with uncharged drugs of this class in cardiac membranes. Mol Pharmacol 36:634–640. [PubMed] [Google Scholar]
- Mason RP, Rhodes DG, Herbette LG (1991) Reevaluating equilibrium and kinetic binding parameters for lipophilic drugs based on a structural model for drug interaction with biological membranes. J Med Chem 34:869–877. [DOI] [PubMed] [Google Scholar]
- Masureel MZou YPicard L-Pvan der Westhuizen EMahoney JPRodrigues JPGLMMildorf TJDror ROShaw DEBouvier M, et al. (2018) Structural insights into binding specificity, efficacy and bias of a β2AR partial agonist. Nat Chem Biol 14:1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paloncýová M, Fabre G, DeVane RH, Trouillas P, Berka K, Otyepka M (2014) Benchmarking of force fields for molecule-membrane interactions. J Chem Theory Comput 10:4143–4151. [DOI] [PubMed] [Google Scholar]
- Park S, Khalili-Araghi F, Tajkhorshid E, Schulten K (2003) Free energy calculation from steered molecular dynamics simulations using Jarzynski’s equality. J Chem Phys 119:3559–3566. [Google Scholar]
- Park S, Schulten K (2004) Calculating potentials of mean force from steered molecular dynamics simulations. J Chem Phys 120:5946–5961. [DOI] [PubMed] [Google Scholar]
- Payandeh J, Volgraf M (2021) Ligand binding at the protein-lipid interface: Strategic considerations for drug design. Nat Rev Drug Discov. 10.1038/s41573-021-00240-2. [DOI] [PubMed] [Google Scholar]
- Raniolo S, Limongelli V (2020) Ligand binding free-energy calculations with funnel metadynamics. Nat Protoc 15:2837–2866. [DOI] [PubMed] [Google Scholar]
- Rhodes DG, Sarmiento JG, Herbette LG (1985) Kinetics of binding of membrane-active drugs to receptor sites. Diffusion-limited rates for a membrane bilayer approach of 1,4-dihydropyridine calcium channel antagonists to their active site. Mol Pharmacol 27:612–623. [PubMed] [Google Scholar]
- Sargent DF and Schwyzer R (1986) Membrane lipid phase as catalyst for peptide-receptor interactions. Proc Natl Acad Sci USA 83:5774–5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider S, Provasi D, Filizola M(2015) The dynamic process of drug–GPCR binding at either orthosteric or allosteric sites evaluated by metadynamics, in G Protein-Coupled Receptors in Drug Discovery: Methods and Protocols (Filizola M ed) pp 277–294, Springer, New York. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoop A, Dey F (2015) On-rate based optimization of structure-kinetic relationship--surfing the kinetic map. Drug Discov Today Technol 17 (Supplement C):9–15. [DOI] [PubMed] [Google Scholar]
- Schwyzer R (1991) New principle in QSAR: membrane requirements. J Recept Res 11:45–57. [DOI] [PubMed] [Google Scholar]
- Schwyzer R (1995a) 100 years lock-and-key concept: are peptide keys shaped and guided to their receptors by the target cell membrane? Biopolymers 37:5–16. [DOI] [PubMed] [Google Scholar]
- Schwyzer R (1995b) In search of the ‘bio-active conformation’--is it induced by the target cell membrane? J Mol Recognit 8:3–8. [DOI] [PubMed] [Google Scholar]
- Shao Z, Yan W, Chapman K, Ramesh K, Ferrell AJ, Yin J, Wang X, Xu Q, Rosenbaum DM (2019) Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat Chem Biol 15:1199–1205. [DOI] [PubMed] [Google Scholar]
- Szlenk CJB GCNatesan S, (2019) Does the Lipid Bilayer Orchestrate Access and Binding of Ligands to Transmembrane Orthosteric/Allosteric Sites of GPCRs?. Molecular Pharmacology 96(5) 527–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinney DC (2006) Can binding kinetics translate to a clinically differentiated drug? From theory to practice. Lett Drug Des Discov 3:569–574. [Google Scholar]
- Swinney DC, Haubrich BA, Van Liefde I, Vauquelin G (2015) The role of binding kinetics in GPCR drug discovery. Curr Top Med Chem 15:2504–2522. [DOI] [PubMed] [Google Scholar]
- Sykes DA and Charlton SJ (2012) Slow receptor dissociation is not a key factor in the duration of action of inhaled long-acting β2-adrenoceptor agonists. Br J Pharmacol 165:2672–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes DA, Parry C, Reilly J, Wright P, Fairhurst RA, Charlton SJ (2014) Observed drug-receptor association rates are governed by membrane affinity: the importance of establishing “micro-pharmacokinetic/pharmacodynamic relationships” at the β2-adrenoceptor. Mol Pharmacol 85:608–617. [DOI] [PubMed] [Google Scholar]
- Szczuka A, Wennerberg M, Packeu A, Vauquelin G (2009) Molecular mechanisms for the persistent bronchodilatory effect of the β 2-adrenoceptor agonist salmeterol. Br J Pharmacol 158:183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschemacher A, Lemoine H (1999) Kinetic analysis of drug-receptor interactions of long-acting β2 sympathomimetics in isolated receptor membranes: evidence against prolonged effects of salmeterol and formoterol on receptor-coupled adenylyl cyclase. J Pharmacol Exp Ther 288:1084–1092. [PubMed] [Google Scholar]
- Tiwary P, Limongelli V, Salvalaglio M, Parrinello M (2015) Kinetics of protein-ligand unbinding: predicting pathways, rates, and rate-limiting steps. Proc Natl Acad Sci USA 112:E386–E391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwary P, Parrinello M (2015) A time-independent free energy estimator for metadynamics. J Phys Chem B 119:736–742. [DOI] [PubMed] [Google Scholar]
- Tribello GA, Bonomi M, Branduardi D, Camilloni C, Bussi G (2014) PLUMED 2: new feathers for an old bird. Comput Phys Commun 185:604–613. [Google Scholar]
- Troussicot L, Guillière F, Limongelli V, Walker O, Lancelin J-M (2015) Funnel-metadynamics and solution NMR to estimate protein-ligand affinities. J Am Chem Soc 137:1273–1281. [DOI] [PubMed] [Google Scholar]
- Valsson O, Tiwary P, Parrinello M (2016) Enhancing important fluctuations: rare events and metadynamics from a conceptual viewpoint. Annu Rev Phys Chem 67:159–184. [DOI] [PubMed] [Google Scholar]
- van Noord JA, Smeets JJ, Maesen FPV (1998) A comparison of the onset of action of salbutamol and formoterol in reversing methacholine-induced bronchoconstriction. Respir Med 92:1346–1351. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe KHatcher EAcharya CKundu SZhong SShim JDarian EGuvench OLopes PVorobyov I, et al. (2010) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31:671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K, MacKerell AD Jr (2012) Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J Chem Inf Model 52:3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K, Raman EP, MacKerell AD Jr (2012) Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J Chem Inf Model 52:3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vauquelin G (2015) On the ‘micro’-pharmacodynamic and pharmacokinetic mechanisms that contribute to long-lasting drug action. Expert Opin Drug Discov 10:1085–1098. [DOI] [PubMed] [Google Scholar]
- Vauquelin G (2016) Cell membranes… and how long drugs may exert beneficial pharmacological activity in vivo. Br J Clin Pharmacol 82:673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vauquelin G, Packeu A (2009) Ligands, their receptors and ... plasma membranes. Mol Cell Endocrinol 311:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Duan Y (2009) Ligand entry and exit pathways in the β2-adrenergic receptor. J Mol Biol 392:1102–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Moukhametzianov R, Baker JG, Nehmé R, Edwards PC, Leslie AGW, Schertler GFX, Tate CG (2011) The structural basis for agonist and partial agonist action on a β(1)-adrenergic receptor. Nature 469:241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ELCheng XJo SRui HSong KCDávila-Contreras EMQi YLee JMonje-Galvan VVenable RM, et al. (2014) CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J Comput Chem 35:1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao PYan WGou LZhong Y-NKong LWu CWen XYuan YCao SQu C, et al. (2021) Ligand recognition and allosteric regulation of DRD1-Gs signaling complexes. Cell 184:943–956.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S, Shaw DE, Yang L, Sandham DA, Healy MP, Reilly J, Wang B (2017) Interactions between β2-adrenoceptor ligands and membrane: atomic-level insights from magic-angle spinning NMR. J Med Chem 60:6867–6879. [DOI] [PubMed] [Google Scholar]
- Yu W, He X, Vanommeslaeghe K, MacKerell AD Jr (2012) Extension of the CHARMM General Force Field to sulfonyl-containing compounds and its utility in biomolecular simulations. J Comput Chem 33:2451–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Raniolo S, Limongelli V, Xu Y (2018) The molecular mechanism underlying ligand binding to the membrane-embedded site of a G-protein-coupled receptor. J Chem Theory Comput 14:2761–2770. [DOI] [PubMed] [Google Scholar]
- Zemski Berry KA, Murphy RC, Kosmider B, Mason RJ (2017) Lipidomic characterization and localization of phospholipids in the human lung. J Lipid Res 58:926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yang F, Ling S, Lv P, Zhou Y, Fang W, Sun W, Zhang L, Shi P, Tian C (2020) Single-particle cryo-EM structural studies of the β2AR–Gs complex bound with a full agonist formoterol. Cell Discov 6:45. [DOI] [PMC free article] [PubMed] [Google Scholar]