

Abstract
Microtubule-targeting agents (MTAs), including both microtubule stabilizers and destabilizers are highly effective chemotherapeutic drugs used in the treatment of solid tumors and hematologic malignancies. In addition to the shared ability of all MTAs to block cell cycle progression, growing evidence shows that different agents of this class can also have mechanistically distinct effects on nonmitotic microtubule-dependent cellular processes, including cellular signaling and transport. Herein, we test the biologic hypothesis that MTAs used in the treatment of triple-negative breast cancer (TNBC) can differentially affect innate immune signaling pathways independent of their antimitotic effects. Our data demonstrate that the microtubule destabilizer eribulin, but not the microtubule stabilizer paclitaxel, induces cGAS-STING–dependent expression of interferon-β in both myeloid and TNBC cells. Activation of the cGAS-STING pathway by eribulin was further found to be mediated by the accumulation of cytoplasmic mitochondrial DNA. Together, these findings provide mechanistic insight into how eribulin can induce innate immune signaling independent of its antimitotic or cytotoxic effects.
SIGNIFICANCE STATEMENT
Microtubule-targeting agents (MTAs) are often used in the treatment of breast cancer and have been used in combination with immune checkpoint inhibitors to improve efficacy. Although all clinically approved MTAs share an antimitotic mechanism of action, their distinct effects on interphase microtubules can promote differential downstream signaling consequences. This work shows that the microtubule destabilizer eribulin, but not the microtubule stabilizer paclitaxel, activates the cGAS-STING innate immune signaling pathway through the accumulation of mitochondrial DNA in the cytoplasm.
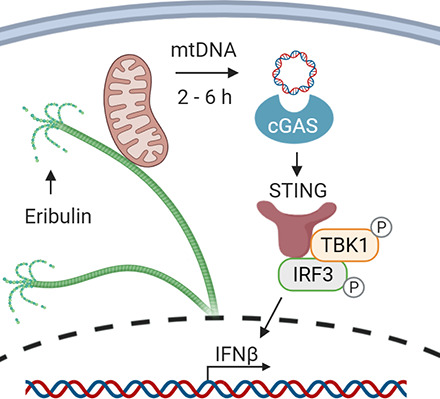
Introduction
Microtubule-targeting agents (MTAs) are a mainstay in the treatment of many malignancies, including breast cancer (Dumontet and Jordan, 2010). MTAs have been historically classified as microtubule stabilizers or destabilizers based on the biochemical mechanism by which they disrupt microtubule dynamics. Microtubule stabilizers, including the taxanes and epothilones, promote the net polymerization of microtubules, whereas microtubule destabilizers, including the vinca alkaloids and eribulin, promote net microtubule depolymerization (Steinmetz and Prota, 2018). Although MTAs have long been thought to exert their anticancer effects primarily through inhibition of mitosis, growing evidence demonstrates that their nonmitotic effects contribute significantly to their anticancer efficacy (Komlodi-Pasztor et al., 2011, 2012; Field et al., 2014; Kaul et al., 2019b). Mechanistically distinct MTAs can also differ from one another in their nonmitotic effects on oncogenic signaling pathways, and such differences could be important factors in their clinical efficacy (Karbowski et al., 2001; Dybdal-Hargreaves et al., 2017; Kaul et al., 2019b).
In addition to their impact directly on cancer cells, MTAs also have differential effects on both innate and adaptive immune cell populations implicated in antitumor immunity (Fong et al., 2019). Although some of the compound or class-specific immunomodulatory effects of MTAs, such as the ability of paclitaxel to directly activate toll-like receptor 4–dependent signaling (Rajput et al., 2013; Wanderley et al., 2018), have been extensively explored, many of the underlying mechanisms of how MTAs elicit distinct immunologic effects are unknown. A more complete interrogation of the effects of MTAs on immune signaling pathways is warranted given the recent clinical evaluations of MTAs, including taxanes and eribulin, in combination with immune checkpoint inhibitors to enhance the therapeutic response in metastatic triple-negative breast cancer (TNBC) (Mittendorf et al., 2020; Schmid et al., 2020; Tolaney et al., 2021). Collectively, these observations prompted us to test the overall biologic hypothesis that MTAs have different effects on innate immune signaling pathways. Herein, we show that eribulin, but not paclitaxel, induces the cGAS-STING–dependent expression of interferon (IFN) β and downstream interferon-stimulated genes (ISGs) in both immune and TNBC cells through the cytoplasmic accumulation of mitochondrial DNA (mtDNA) independent of mitotic arrest or the initiation of apoptosis.
Materials and Methods
Cells and Reagents
BT-549 cells were obtained from the Georgetown University Lombardi Comprehensive Cancer Center, Washington, DC. CAL-51 cells were purchased from Creative Bioarray (Westbury, NY). All other cell lines were purchased from the American Type Culture Collection (Manassas, VA). All TNBC lines were authenticated by short tandem repeat (STR) -based profiling (Genetica DNA Laboratories, Cincinnati OH). Primary wild-type bone marrow–derived macrophages (BMDMs) were generated from BALB/c or C57BL/6 mice purchased from Envigo (Indianapolis, IN) as previously described (Hasan et al., 2013). C57BL/6 Sting gt/gt BMDMs were obtained from animals purchased from Jackson Laboratories (Bar Harbor, ME). BMDMs cells were maintained in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY) supplemented with 10% FBS (Corning, Corning, NY) and 50 μg/ml gentamycin (LifeTechnologies, Carlsbad, CA). THP-1, HCC1937, HCC1806, BT-549, MDA-MB-436, and CAL-51 cells were maintained in RPMI 1640 media (Corning) supplemented with 10% FBS and 50 μg/ml gentamycin. HCC1937 cells depleted of mitochondrial DNA (Rho0) were generated by culturing cells in 150 ng/ml ethidium bromide (EtBr) for 5 days as described (White et al., 2014). All cell lines were routinely checked for mycoplasma contamination. Drugs, ligands, and inhibitors used in this study include eribulin (Eisai Inc. Woodcliff Lake, NJ), paclitaxel (Sigma Aldrich, St Louis, MO), HT-DNA (Sigma Aldrich), ruxolitinib (Invivogen, San Diego, CA), BX795 (Invivogen), TPCA-1 (Sigma Aldrich), SP600125 (Sigma Aldrich), and H-151 (Tocris, Minneapolis, MN), which were all dissolved in DMSO (Fisher, Hampton, NH) and stored at −20°C.
siRNA Transfection, RNA Extraction, and qRT-PCR
Treatment with immune pathway inhibitors was initiated 4 hours before treatment with eribulin or HT-DNA followed by continuous treatment with the inhibitor for the indicated time. For IFNAR knockdown experiments, THP-1 cells were transfected for 48 hours with siRNAs (Sigma Aldrich) against IFNAR1 (SASI_Hs01_00121376, SASI_Hs02_00302973) and IFNAR2 (SASI_Hs01_00208506, SASI_Hs01_00208505) and compared with scramble control (SIC002) using the RNAiMAX reagent (Invitrogen, Carlsbad, CA). Extraction of RNA from cells was done using TRIzol (Ambion, Austin, TX) as indicated by the manufacturer. cDNA was synthesized with iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) and analyzed using a Bio-Rad CFX qRT -PCR using iTaq Universal SYBR Green Supermix (Bio-Rad). mRNA fold change was calculated using the 2−ΔΔCt method, in which GAPDH was used as the control. All treated and stimulated samples were compared with their corresponding vehicle or mock control, which was set to a relative value of 1. All experiments were performed as two technical replicates with a variability cutoff of less than 1 Ct within each of two independent biologic replicates. The range of the biologic replicate values is represented in every bar graph. Data were subject to ordinary one-way ANOVA or two-way ANOVA for statistical analysis as described in figure legends. All primers were ordered from Sigma Aldrich, and sequences can be found in Supplemental Table 1.
Cell Cycle and Caspase 3/7 Cleavage Flow Cytometry Assays
For cell cycle analysis, treated cells were harvested on ice by collecting the media and scraping the adhered cells before centrifuging at 500g for 5 minutes at 4°C to pellet both the adherent and nonadherent cells. Cells were mixed with Krishan’s reagent [50 µg/ml propidium iodide, 1 mg/ml sodium citrate, 0.3% (v/v) IGEPAL CA-630, and 20 µg/ml ribonuclease A] and analyzed using a Guava Muse Cell Analyzer (Luminex, Austin, TX) and expressed as percentage of cells in G1, S, and G2/M based on their DNA content, where error bars represent the range among biologic duplicates. For caspase 3/7 cleavage, adherent treated cells were incubated at 37°C for 30 minutes in a 50-μl/ml Caspase 3/7 green reagent (Thermo-Fisher, Waltham, MA) solution. Cells were subsequently rinsed with PBS and analyzed using a Guava Muse Cell Analyzer (Luminex) and expressed as percentage of cells alive and apoptotic, where error bars represent the range in measured values in biologic duplicates.
Interferon-β Flow Cytometry Assay
THP-1 or HCC1937 cells were treated with 1 µl/ml GolgiPlug (555029; BD Biosciences, Franklin Lakes, NJ) with 100 nM eribulin or the DMSO vehicle control for 2–6 hours. Cells were then collected, blocked with TruStain FcX block (BioLegend, San Diego, CA), and stained with Zombie NIR viability dye (BioLegend) for 15 minutes in the dark. After fixation and permeabilization, cells were incubated with anti-IFNβ-fluorescein isothiocyanate(FITC) (PBL Assay Science, Piscataway, NJ) for 20 minutes in the dark. Intracellular IFNβ staining in live cells was determined using the Cytek Aurora at the University of Texas Health Science Center San Antonio Flow Cytometry Facility and analyzed with FlowJo software (Becton, Dickinson and Company, Ashland, OR).
Transient Transfection and Immunoblotting
Ectopic expression of RFP -cGAS in CAL-51 cells was achieved by transfecting 2.5 µg of the pTRIP-CMV-RFP-FLAG-cGAS [Addgene (Watertown, MA) # 86676] plasmid using the LipofectAMINE 3000 (Invitrogen) reagent for a minimum of 16 hours. For immunoblotting, cells were lysed in cell extraction buffer (Life Technologies) containing 1× protease inhibitor cocktail (Sigma Aldrich) and 1 mM phenylmethylsulfonyl fluoride (PMSF) (Sigma Aldrich). Equal amounts of total protein were resolved by SDS-PAGE on a 10% gel and transferred onto a polyvinylidene fluoride (PVDF) Immobilon-FL membrane (Millipore, Burlington, MA), and relative protein levels were evaluated by immunoblotting for cGAS (1:500 dilution, D1D3G; Cell Signaling Technology, Danvers, MA), STING (1:500 dilution, D2P2F; Cell Signaling Technology), TBK1 (1:500 dilution, D1B4; Cell Signaling Technology), P-TBK1 (Ser172) (1:500 dilution, D52C2; Cell Signaling Technology), and β-tubulin (1:10,000 dilution, T4026; Sigma Aldrich) as primary antibodies and species-specific IRDye as secondary antibodies (1:5000 dilution, LI-COR biosciences, Lincoln, NE). Immunoblots were visualized using a LI-COR Biosciences Odyssey Fc Imager.
Intracellular Tubulin Polymerization Assay
Assessment of intracellular tubulin polymerization in nonadherent THP-1 cells in response to MTAs was done as described previously (Giannakakou et al., 1997). In short, THP-1 cells were washed with 37°C PBS and lysed at 37°C for 5 minutes in a hypotonic lysis buffer [1 mM MgCl2, 2 mM EGTA, 0.5% (v/v) Nonidet P-40 (Shell), 50 mM Tris-HCL pH 6.8, and 1× protease inhibitor cocktail]. The lysate was then centrifuged at 14,400g for 10 minutes at 37°C to separate soluble tubulin heterodimers (supernatant) from polymerized microtubules (pellet). The pellet was resuspended in an equal volume to the supernatant, and 10% of each fraction was subjected to SDS-PAGE and immunoblotting using β-tubulin (1:2000 dilution, T4026; Sigma Aldrich) as the primary antibody and anti-mouse IRDye as the secondary antibody (1:5000 dilution; LI-COR biosciences). Immunoblots were visualized using a LI-COR Biosciences Odyssey Fc Imager.
Microtubule Indirect Immunofluorescence and Mitochondrial Staining
BMDM cells grown on coverslips were fixed and permeabilized using cold 100% methanol and immunostained using anti–β-tubulin (1:400 dilution, T4026; Sigma Aldrich) as primary and anti-mouse FITC (1:200 dilution, F-3008; Sigma Aldrich) as a secondary antibody. HCC1937 cells were stained with 50 nM Mitotracker Red CMXRos (Thermo-Fisher) in serum-free RPMI 1640 for 30 minutes at 37°C, fixed with 4% paraformaldehyde, and permeabilized with 0.5% (v/v) Triton X-100 prior to immunostaining with anti–β-tubulin. Samples were imaged on a Nikon (Tokyo, Japan) Eclipse 80i fluorescence microscope with NIS elements using z-stacks at 1000× total magnification.
Subcellular Fractionation and Detection of Mitochondrial DNA
Detection of mtDNA from cytoplasmic and organelle-enriched fractions was performed as described previously (Wolf et al., 2016). Briefly, cells were scraped into 600 µl of cold disruption buffer (20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, and 250 mM sucrose) containing 1× protease inhibitor cocktail (Sigma Aldrich) and placed on ice for 5 minutes. Samples were passed through a 22-gauge syringe 30 times followed by a low spin to remove unbroken cells and subsequently subjected to centrifugation at 10,000g for 10 minutes at 4°C. The pellet was designated the organelle-enriched fraction (containing nucleus and mitochondria) and the supernatant the cleared cytoplasmic fraction as confirmed by the relative enrichment of VDAC (1:1000 dilution, 4866; Cell signaling Technologies), lamin B1 (1:1000 dilution, ab16048; Abcam), and GAPDH (1:1000 dilution, D4C6R; Cell signaling Technologies) by immunoblotting. DNA was isolated from each fraction using a QiaAmp kit (Qiagen, Germantown, MD) and evaluated for genomic or mitochondrial DNA by qRT-PCR. Cell number was used for normalization between samples, and equal percentages of cytoplasmic and organelle-enriched fractions were compared for each sample to evaluate relative distribution. The primers used to assess mitochondrial genes were ordered from Sigma Aldrich and are in Supplemental Table 1.
Data Analysis
Data are presented with individual data points representing biologic replicates, and errors are shown as a range of these values. Sample sizes were determined in advance based on previous measurements of accuracy and precision for each of the methodologies used in the current study. These parameters were defined by utilizing validated and literature-grounded positive controls for each assay conducted. All statistical analysis was performed on biologic replicates, with technical replicates performed as an internal check for the rigor of each biologic replicate. The statistical analyses presented in this manuscript were conducted using GraphPad Prism and described in the respective figure legends. In summary, we used a one-way ANOVA with Dunnett’s post hoc test compared with the control condition if only one experimental variable was being measured or two-way ANOVA with Tukey’s post hoc test to compare among each condition if two experimental variables were being measured. If only two samples were being compared, we performed an unpaired t test analysis.
Results
Eribulin Stimulates the Expression of IFNβ Independent of Mitotic Arrest or Cytotoxicity
We first determined the time-dependent effects of MTAs on nonadherent THP-1 human monocytic leukemia cells by quantifying the distribution of tubulin in the polymerized microtubule form and soluble tubulin heterodimers by nondenaturing lysis and centrifugation (Giannakakou et al., 1997). When THP-1 cells were treated with 100 nM eribulin or paclitaxel, clinically relevant concentrations of these drugs that promote a rapid microtubule disruption in breast cancer cells (Huizing et al., 1995; Goel et al., 2009; Dybdal-Hargreaves et al., 2017), microtubule depolymerization by eribulin and stabilization by paclitaxel were observed within 2 hours (Fig. 1A). Neither G2/M accumulation nor caspase cleavage were observed in MTA-treated cells at either 2 or 6 hours (Fig. 1, B and C). However, 24 hours after either 100 nM eribulin or paclitaxel treatment to THP-1 cells, there was a significant increase in G2/M accumulation, and approximately 50% of cells had initiated the process of apoptosis as demonstrated by the activation of caspases 3/7 (Fig. 1, B and C).
Fig. 1.

Eribulin but not paclitaxel induces expression of interferon-β and interferon-stimulated genes in THP-1 cells. (A) Soluble (S) or polymerized (P) β-tubulin in THP-1 cells treated with 100 nM eribulin (ERB) or paclitaxel (PTX) for 2, 6, or 24 hours as compared with DMSO (Veh). (B) Cell cycle analysis of THP-1 cells treated with 100 nM eribulin or paclitaxel for 2, 6, or 24 hours compared with DMSO (Veh) as percentage of cells in G1 (black), S (gray), or G2/M (white). (C) Caspase 3/7 cleavage in THP-1 cells treated with 100 nM eribulin or paclitaxel for 2, 6, or 24 hours compared with DMSO as percentage of live (white) or apoptotic (gray) cells from two independent experiments with errors denoting range. (D) IFNβ mRNA in THP-1 cells treated with 100 nM eribulin or paclitaxel for 2, 6, or 24 hours as compared with DMSO. Significance determined by vehicle compared one-way ANOVA with Dunnett’s post hoc test compared with vehicle. (E) IFNβ intracellular protein in live cells treated with DMSO or 100 nM eribulin for 2 or 6 hours. Individual data points represent three independent experiments, and significance was determined by two-way ANOVA (time * drug) with Tukeys’s post hoc test. (F and G) IFIT1 mRNA in THP-1 cells (F) pretreated with 1 µM of ruxolitinib (JAKi) or vehicle for 4 hours and then treated with DMSO or 100 nM eribulin for 24 hours with the inhibitor still present or (G) with siRNA to IFNAR1/2 (siIFNAR1/2) or a scrambled sequence (siCtrl) for 48 hours followed by treatment with DMSO or 100 nM eribulin for 24 hours. Significance was determined by two-way ANOVA (drug * inhibitor/siRNA) with Tukeys’s post hoc test. qRT-PCR data are shown as individual points from two independent biologic replicates with error bars denoting range. *P < 0.05, **P < 0.01, ****P < 0.0001. Veh, vehicle.
At 24 hours, we also evaluated the effects of both drugs on cytokine expression and found that eribulin was distinct from paclitaxel, particularly in its induction of IFNβ and ISGs in THP-1 cells, suggesting mechanistic differences between these MTAs that cannot be strictly attributed to their shared antimitotic and apoptotic effects at this time point (Supplemental Fig. 1A). A more detailed time course demonstrated that eribulin indeed promoted the induction of IFNβ in THP-1 cells within 6 hours (Fig. 1D), a time when interphase microtubules were disrupted, but prior to mitotic arrest or caspase cleavage. Low expression of IFNβ was observed with paclitaxel treatment at each of the time points evaluated with no significant difference from the vehicle control (Fig. 1D). Intracellular flow cytometry further demonstrated an eribulin-mediated increase in IFNβ protein 6 hours after drug treatment in live cells using a dual gating strategy (Fig. 1E). Together, these data demonstrate that eribulin can induce the expression of IFNβ at both the mRNA and protein levels in THP-1 cells prior to mitotic accumulation and apoptosis through a mechanism distinct from paclitaxel.
To determine the downstream functionality of eribulin-induced IFNβ expression, we further evaluated the concomitant expression of the ISG IFIT1 over time after the addition of eribulin to THP-1 cells and found that expression also increased significantly 24 hours after treatment (Supplemental Fig. 1B). Further analysis revealed a similar eribulin-mediated induction of another ISG, OAS1, with a more modest induction of the ISGs IFITm3 and ISG15, which were not observed in paclitaxel-treated cells (Supplemental Fig. 1C). Although ISG induction coincided with the mitotic arrest and apoptotic effects of eribulin, it is important to note that these shared phenotypes were not sufficient to promote ISG induction in paclitaxel-treated cells. To further test whether the eribulin-mediated induction of ISGs was driven by canonical interferon signaling, which is mediated by JAK-STAT signaling downstream of the binding of interferon to the interferon receptor (IFNAR), we took both genetic and pharmacological approaches (Supplemental Fig. 2A). Indeed, inhibition of JAK kinase activity with ruxolitinib or siRNA-mediated knockdown of IFNAR was sufficient to completely suppress eribulin-mediated expression of the ISG IFIT1 (Fig. 1, F and G) without inhibiting induction of IFNβ expression (Supplemental Fig. 2, B–E). Together, these data demonstrate that either pharmacological or genetic inhibition of the canonical interferon receptor signaling pathway was sufficient to decouple the eribulin-mediated induction of IFNβ from downstream ISG induction, further supporting that eribulin induces a functional interferon response.
Previous studies have paradoxically demonstrated a shared upregulation of IFNβ in response to either microtubule stabilization or destabilization driven by cytoplasmic nuclear genomic DNA (gDNA) originating from micronuclei formed after prolonged mitotic arrest and slippage (Harding et al., 2017; Mackenzie et al., 2017; Zierhut et al., 2019; Lohard et al., 2020). However, these findings were distinct from the rapid induction of IFNβ by eribulin, but not paclitaxel, seen in our experiments that occurs prior to mitotic arrest or caspase cleavage. This prompted us to further evaluate the eribulin-mediated IFN response in noncycling primary murine BMDMs. Similar to the results in the THP-1 human cell line, treating murine BMDM with 100 nM eribulin was sufficient to promote depolymerization of the microtubule network within 2 hours (Fig. 2A). However, the primary noncycling BMDMs were refractory to mitotic accumulation or caspase cleavage by eribulin even 24 hours after addition (Fig. 2, B and C). Similar to the THP-1 cells, eribulin-induced IFNβ expression was observed within 2 hours in these noncycling cells with subsequent IFIT1 expression at 24 hours, further demonstrating that eribulin-induced IFNβ production leads to a functional ISG response independent of mitotic arrest or the initiation of apoptosis (Fig. 2, D and E). Altogether, these data demonstrate that acute eribulin-dependent induction of IFNβ and downstream ISG signaling in both primary murine and immortalized human immune cells is independent of antimitotic or apoptotic effects, providing a rationale to further explore the mechanistic underpinnings of this induction.
Fig. 2.

Eribulin-mediated interferon-β expression occurs independently of mitotic arrest and caspase cleavage. (A) Immunofluorescence images of microtubules (green) and DNA (blue) in BMDM cells treated with 100 nM eribulin (ERB) for 2, 6, or 24 hours as compared with DMSO (Veh). Scale bar, 10 µm. (B) Cell cycle analysis of BMDM cells treated with 100 nM eribulin for 2, 6, or 24 hours as compared with DMSO as percentage of cells in G1 (black), S (gray), or G2/M (white) from two independent experiments, with error bars denoting range. (C) Caspase 3/7 cleavage in wild-type BMDM cells treated with 100 nM eribulin for 2, 6, or 24 hours compared with DMSO as percentage of live (white) or apoptotic (gray) cells from two independent experiments with errors denoting range. IFNβ (D) and IFIT1 (E) mRNA in BMDM cells treated with 100 nM ERB for 2, 6, or 24 hours compared with DMSO. qRT-PCR data are shown as individual points from two independent biologic replicates with error bars denoting range. Significance was determined by vehicle-compared one-way ANOVA with Dunnett’s post hoc test. **P < 0.01, ****P < 0.0001. Veh, vehicle.
Eribulin Stimulation of IFNβ Is Dependent on the cGAS-STING-TBK1 Pathway
In mammals, the transcription of IFNβ is primarily mediated by interferon regulatory factors 3 and 7, NF-κB, and the AP-1 complex (Honda et al., 2006). To preliminarily investigate the likelihood of involvement of each of these transcription factors in the eribulin-mediated expression of IFNβ, BMDMs were pretreated with pharmacological inhibitors of each of these pathways prior to eribulin treatment at concentrations that did not promote apoptosis within the timeframe evaluated (Supplemental Fig. 3, A–C). The TBK1 inhibitor BX795 was used to suppress the interferon regulatory factors 3 and 7 signaling pathway, the IκB kinase inhibitor TPCA-1 was used to inhibit NF-κB nuclear localization, and the c-Jun N-terminal kinase inhibitor SP600125 was used to inhibit transcription mediated by the AP-1 complex (Bennett et al., 2001; Podolin et al., 2005; Clark et al., 2009). We found that pretreatment of BMDM with the TBK1 inhibitor completely abrogated eribulin-mediated IFNβ and IFIT1 induction and that this response was also partially inhibited by NF-κB, but not AP-1, inhibition (Fig. 3, A and B).
Fig. 3.

Eribulin-dependent upregulation of interferon-β and interferon-stimulated genes is dependent on the STING pathway. IFNβ (A) and IFIT1 (B) mRNA in BMDM cells pretreated with 1 µM of the indicated inhibitor for 4 hours and then treated with 100 nM eribulin (ERB) for 2, 6, or 24 hours as compared with DMSO with the inhibitor still present from two independent experiments with error bars denoting range. IFNβ (C and E) and IFIT1 (D and F) mRNA from BMDM and THP-1 cells pretreated with 1 µM H-151 (STINGi) for 4 hours and then treated with 100 nM eribulin for 2, 6, or 24 hours as compared with DMSO with the inhibitor still present. IFNβ (G) and IFIT1 (H) mRNA in wild-type and Sting gt/gt BMDM cells treated with 100 nM eribulin for 2, 6, or 24 hours. Data are shown as individual points from two independent biologic replicates with error bars denoting range. Significance was determined by two-way ANOVA (time * inhibitor) using a Tukey’s post hoc test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Veh, vehicle.
TBK1 plays a pivotal role in activating the transcriptional expression of IFNβ during the process of intracellular DNA sensing by the cGAS-STING pathway (Supplemental Fig. 4A) (Shang et al., 2019). Therefore, we sought to determine whether the cGAS-STING pathway was involved in the eribulin-mediated stimulation of IFNβ. To test this, we pretreated BMDM and THP-1 cells with a covalent STING inhibitor, H-151 (Haag et al., 2018), prior to eribulin treatment using a concentration that inhibited TBK1 phosphorylation (Supplemental Fig. 4B) but was not toxic to BMDM or THP-1 cells (Supplemental Fig. 4, C and D). Inhibition of STING signaling by H-151 was sufficient to attenuate the induction of IFNβ and IFIT1 by either eribulin (Fig. 3, C–F) or exogenous HT-DNA (Supplemental Fig. 4, E–H) in both primary BMDMs and in THP-1 cells. To further examine the involvement of STING in mediating eribulin-induced expression of IFNβ, we generated BMDMs from Sting gt/gt mice that contain a missense mutant allele that renders them defective for STING-induced INFβ expression (Sauer et al., 2011). Consistent with the results observed with the STING inhibitor, Sting gt/gt derived BMDMs failed to upregulate the expression of IFNβ and concomitant IFIT1 when treated with eribulin (Fig. 3, G and H) or exogenous HT-DNA (Supplemental Fig. 4, I and J). Taken together, these studies demonstrate that the eribulin-mediated induction of IFNβ and concomitant IFIT1 expression occurs through a mechanism dependent on the cGAS-STING-TBK1 pathway.
To further interrogate the importance of an intact DNA sensing pathway for eribulin-mediated expression of IFNβ, we capitalized on the previous finding that CAL-51 TNBC cells are deficient in cytoplasmic DNA sensing because of a lack of cGAS, which is required for the synthesis of the second messenger cyclic GMP-AMP that functions as a STING agonist (Supplemental Fig. 4A) (Zierhut et al., 2019). Immunoblot analysis of a molecularly diverse panel of TNBC cell lines confirmed that CAL-51 cells express STING but do not express cGAS (Fig. 4A). As expected, each of the cGAS-expressing TNBC cell lines evaluated were capable of inducing IFNβ expression when challenged with exogenous HT-DNA, but cGAS-deficient CAL-51 cells were deficient in this response (Fig. 4B). Importantly, ectopic expression of RFP-cGAS (CAL-51-RFP-cGAS) (Fig. 4A) restored the ability of CAL-51 cells to respond to HT-DNA stimulation without introducing gross cytotoxicity (Fig. 4B; Supplemental Fig. 5A). Consistent with the role of the cGAS-STING pathway in the eribulin-mediated induction of IFNβ in immune cells, we found that eribulin also promoted the expression of IFNβ in cGAS- and STING-expressing TNBC cell lines, as well as the CAL-51 line reconstituted with RFP-cGAS, but was unable to promote IFNβ expression in cGAS-deficient CAL-51 cells or low-cGAS-expressing BT-549 cells (Fig. 4, C and D). Consistent with the lack of eribulin-mediated IFNβ induction, cGAS-deficient CAL-51 cells also failed to upregulate IFIT1 when treated with eribulin, although the expression of this ISG was rescued by expression of RFP-cGAS (Fig. 4E). Using flow cytometry, we further demonstrated that live HCC1937 TNBC cells produced IFNβ protein, which was able to promote downstream IFIT1 expression within 6 hours of treatment (Fig. 4, F and G). The finding that IFNβ expression occurs in these TNBC cells within 6 hours, before mitotic accumulation or cytotoxicity are observed (Supplemental Fig. 5, B–D), further demonstrates the independence of eribulin-mediated interferon expression and downstream ISG expression from its antimitotic mechanism of action. These results demonstrate that eribulin-mediated induction of IFNβ and downstream ISGs in immune cells as well as in a molecularly diverse panel of TNBC cells occurs in a cGAS-STING–dependent manner.
Fig. 4.

Expression of interferon-β by eribulin in TNBC cell lines requires the DNA sensor cGAS. (A) Immunoblot analysis of cGAS, STING, and β-tubulin expression in THP-1 and TNBC cells. (B) IFNβ mRNA in TNBC cells transfected with 1 µg of HT-DNA for 24 hours or mock-transfected. (C and D) IFNβ mRNA in TNBC cells treated with 100 nM eribulin (ERB) for 2 hours (C) or 6 hours (D) as compared with DMSO controls. Significance was determined by two-way ANOVA (cell line * drug) with Tukey’s post hoc test. (E) IFIT1 mRNA in CAL-51 cells transfected with RFP-cGAS or mock-transfected and treated with DMSO (Veh) or 100 nM ERB for 24 hours. Significance was determined by two-way ANOVA (cGAS * drug) with Tukey’s post hoc test. (F) HCC1937 IFNβ intracellular protein in live cells treated with DMSO or 100 nM eribulin for 6 hours. Significance determined by an unpaired two-tailed t test. (G) Human IFIT1 mRNA in HCC1937 cells treated with 100 nM eribulin for 2, 6, or 24 hours compared with DMSO. Significance determined by vehicle-compared one-way ANOVA with Dunnett’s post hoc test. Data are shown as individual points from two independent biologic replicates with error bars denoting range. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Veh, vehicle.
Eribulin Activates the cGAS-STING Pathway by Inducing the Accumulation of Cytoplasmic Mitochondrial DNA
The dependence of cells on the cytoplasmic cGAS-STING pathway for the induction of IFNβ in response to eribulin suggests that eribulin could be promoting the accumulation of DNA in the cytoplasm. Since mammalian gDNA and mtDNA serve as the major endogenous cGAS-STING DNA ligands (Härtlova et al., 2015; West et al., 2015), we hypothesized that gDNA and/or mtDNA sequences were enriched in the cytoplasm of cells after treatment with eribulin. To test this hypothesis, we performed differential centrifugation in HCC1937 cells to generate an organelle-enriched (OE) fraction that contained mitochondria and nuclei as evidenced by the enrichment in VDAC and lamin B1, respectively, as well as a cytoplasmic fraction that was enriched for GAPDH (Supplemental Fig. 6A). We then quantified distinct mtDNA and gDNA sequences (six each) that were enriched in the cytoplasmic fraction of HCC1937 cells after a 6-hour treatment with eribulin by qRT-PCR and found a significant increase in the enrichment of mtDNA sequences in the cytoplasm (Fig. 5A). Furthermore, we found that the cytoplasmic enrichment of the COX-1 mtDNA sequence specifically occurred in eribulin-treated HCC1937 and BMDM cells as compared with vehicle or paclitaxel treatment (Fig. 5, B and C). These results suggest that the release of mtDNA into the cytoplasmic space could initiate eribulin-mediated induction of the cGAS-STING DNA sensing pathway.
Fig. 5.

Eribulin promotes cytoplasmic accumulation of mtDNA. (A) qRT-PCR analysis of relative abundance of genomic DNA (gDNA) (ACTb, GAPDH, HPRT, PGK, RPS18, and TBP) and mitochondrial DNA (mtDNA) (ATP6, ATP8, COX-1, ND1, ND4, and ND6) sequences present in the cytoplasm of HCC1937 cells treated with 100 nM eribulin (ERB) for 6 hours. Data are shown as the ratio of gDNA or mtDNA in the cytoplasmic fraction as compared with the organelle-enriched fraction and normalized to the vehicle control as fold change. Significance was determined by one-way ANOVA with Dunnett’s post hoc test. (B and C) COX-1 mtDNA present in the cytoplasm of HCC1937 (B) or BMDM (C) cells treated with 100 nM eribulin or paclitaxel (PTX) for 6 hours as compared with vehicle. Significance determined by vehicle-compared one-way ANOVA with Dunnett’s post hoc test compared with vehicle. (D) IFNβ mRNA in THP-1 cells treated with 10, 100, or 1000 nM ERB, vinorelbine (VNR), ixabepilone (IXA), PTX, or docetaxel (DTX) for 24 hours. Significance determined by vehicle-compared two-way ANOVA (drug * concentration) with Tukey’s post hoc test compared with vehicle. (E) IFIT1 mRNA in THP-1 cells treated with 10, 100, or 1000 nM ERB, VNR, or PTX for 24 hours. Significance determined by vehicle-compared two-way ANOVA (drug * concentration) with Tukey’s post hoc test compared with vehicle. (F) COX-1 mRNA in control and ethidium bromide cultured (Rho0) HCC1937 cells treated with 100 nM eribulin for 2 or 6 hours as compared with DMSO. Significance was determined by two-way ANOVA (Rho status * drug) with Tukey’s post hoc test. (G) IFNβ mRNA in control and Rho0 HCC1937 cells treated with 100 nM eribulin for 2 or 6 hours as compared with DMSO. Significance was determined by two-way ANOVA (Rho status * drug) with Tukey’s post hoc test. (H) Cytoplasmic COX-1 DNA present in control and Rho0 HCC1937 cells treated with 100 nM eribulin or paclitaxel for 6 hours as compared with DMSO. Significance was determined by two-way ANOVA (Rho status * drug) with Tukey’s post hoc test. Data are shown as individual points from two independent biologic replicates with error bars denoting range. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Veh, vehicle; ctrl, control.
We hypothesized that microtubule destabilization could lead to the release of mitochondrial DNA into the cytoplasm because of the critical role microtubules play in maintaining mitochondrial homeostasis. Intriguingly, microtubule stabilizing and destabilizing agents have previously been shown to elicit differential effects on mitochondrial biogenesis that increase or reduce mitochondrial mass, respectively (Karbowski et al., 2001), and eribulin can effectively disrupt mitochondrial membrane potential within 30 minutes of treatment (Sampson et al., 2016). Therefore, we speculated that eribulin could disrupt mitochondrial homeostasis in a manner that could lead to the cytosolic accumulation of mtDNA. We thus examined the morphology of both microtubules and mitochondria in HCC1937 cells treated for 4 hours with eribulin or paclitaxel. These cells were chosen because of their large size and fine resolution of the microtubule and mitochondrial network. As previously shown for other microtubule destabilizers (Okatsu et al., 2010), we found that eribulin-treated cells acquired a somewhat fragmented mitochondrial phenotype, a feature not observed in paclitaxel-treated cells (Supplemental Fig. 6B). Based on these findings, we hypothesized that other drugs that destabilized microtubules could also promote IFNβ and ISG expression. Indeed, when we treated THP-1 cells with increasing concentrations (10–1000 nM) of five different MTAs used in the treatment of breast cancer, we found that only the microtubule destabilizers (eribulin and vinorelbine) but not stabilizers (ixabepilone, paclitaxel, and docetaxel) upregulated IFNβ expression (Fig. 5D). Additionally, this IFNβ induction by vinorelbine was associated with downstream IFIT1 expression (Fig. 5E). These data support our hypothesis that destabilization of the interphase microtubule network leads to disruption of mitochondrial homeostasis and subsequent release of mtDNA to drive innate immune activation by these agents.
To directly test the hypothesis of whether the eribulin-mediated release of mtDNA was indeed responsible for the increase in IFNβ expression, we took advantage of the fact that EtBr depletes mtDNA while preserving mitochondria themselves (Armand et al., 2004). This method has been used by others to evaluate mtDNA-mediated activation of the cGAS-STING pathway (White et al., 2014; Yamazaki et al., 2020; Hu et al., 2021). We therefore cultured HCC1937 cells in EtBr for 5 days to generate mtDNA-depleted (rho0) HCC1937 cells as noted by the depletion of mitochondrial genes in these cells as compared with the untreated controls (Fig. 5F). Strikingly, when mitochondria-deficient HCC1937 rho0 cells were treated with eribulin, there was a complete loss of eribulin-mediated IFNβ induction (Fig. 5G) that coincided with the absence of eribulin-induced accumulation of mtDNA in the cytoplasm (Fig. 5H). Importantly, HCC1937 rho0 cells retained the ability to induce IFNβ expression in response to exogenously added HT-DNA (Supplemental Fig. 6C), indicating that these cells retain a functional cGAS-STING pathway. Collectively, these studies demonstrate that cellular mitochondria are critical for mediating the eribulin-dependent accumulation of cytoplasmic mtDNA, which promotes innate immune activation via the cGAS-STING pathway to drive expression of IFNβ and ISGs.
Discussion
MTAs, including the microtubule stabilizer paclitaxel and the microtubule destabilizer eribulin, are some of the most effective agents used in the treatment of metastatic TNBC (Dumontet and Jordan, 2010). Historically, their clinical success has been attributed to their shared ability to suppress mitosis, leading to the apoptosis of rapidly dividing cancer cells. However, accumulating evidence in both patients and preclinical models demonstrates that the anticancer effects of MTAs cannot solely be explained by their shared antimitotic effects and that inhibition of interphase microtubule dynamics significantly contributes to their antitumor properties (Komlodi-Pasztor et al., 2012; Field et al., 2014; Bates and Eastman, 2017). Moreover, different MTAs can elicit distinct effects on cellular oncogenic signaling pathways as well as on mitochondrial homeostasis that may underlie unappreciated clinical effects between these drugs (Karbowski et al., 2001; Dybdal-Hargreaves et al., 2017; Kaul et al., 2019a). Herein, we show that the microtubule destabilizer eribulin induces the cGAS-STING pathway–mediated expression of IFNβ in innate immune cells as well as in TNBC cells within 2–6 hours by promoting the accumulation of cytoplasmic mtDNA.
One of our most surprising and important findings is that eribulin is distinct from paclitaxel in its ability to promote cytoplasmic accumulation of mtDNA leading to cGAS-STING–dependent interferon and downstream ISG induction. Our results demonstrate that the activation of this immune pathway occurs within 2–6 hours of drug addition and is not dependent on mitotic arrest or the initiation of apoptosis. This is different from studies demonstrating that both microtubule stabilizers and destabilizers can promote activation of the cGAS-STING pathway, specifically during extended periods of mitotic arrest, when genomic DNA is released into the cytoplasm (Mackenzie et al., 2017; Zierhut et al., 2019). Importantly, this previously reported genomic DNA release was dependent on mitosis and not observed in noncycling cells. Our current finding that eribulin rapidly and specifically activates cGAS-STING through mtDNA release in both TNBC and immune cells including those that are terminally differentiated is significant particularly in solid tumors that have a much lower mitotic index than cancer cells in culture (Komlodi-Pasztor et al., 2011).
These findings are timely, as they may contribute to a mechanistic understanding behind the efficacy of combinations of molecularly distinct classes of MTAs, including eribulin, with immune checkpoint inhibition in patients with TNBC (Schmid et al., 2020; Tolaney et al., 2021). The cGAS-STING pathway has been shown to be indispensable for immune checkpoint inhibitors to exert their antitumor effects (Wang et al., 2017), and agents that activate the cGAS-STING pathway have been shown to enhance the efficacy of immune checkpoint therapy in metastatic breast cancer models primarily by priming the immune system to acquire an antitumor phenotype (Chandra et al., 2014; Cheng et al., 2018). These findings have prompted the discovery and development of pharmacological STING agonists; however, our data demonstrate that eribulin and likely a subset of currently approved microtubule destabilizers that innately possess this activity are already in clinical use. Consistent with our findings that the microtubule destabilizer vinorelbine promotes STING activation, preclinical studies have shown an increased response to immune checkpoint inhibitors when combined with vinorelbine (Orecchioni et al., 2018). We speculate that the mitotic-independent activation of the cGAS-STING pathway by eribulin and other microtubule destabilizers could provide an advantage over stabilizers in activating this antitumor-associated immune signaling pathway.
Although our studies demonstrate that the interphase effects of eribulin disrupt mitochondrial localization and promote the release of mtDNA into the cytoplasm and that mtDNA is required for eribulin-mediated activation of the cGAS-STING pathway, the mechanism by which eribulin promotes these mitochondrial effects is implicated from previous studies. The cytoplasmic release of mtDNA under cellular stress can be mediated by the formation of macropores in the mitochondrial outer membrane either through oligomerization of the Bcl-2–associated proteins Bax and Bak (Galluzzi and Vanpouille-Box, 2018) or the formation of VDAC oligomers (Kim et al., 2019). Previous studies have demonstrated that increases in cytoplasmic free tubulin heterodimers, similar to those we observe within 2 hours of eribulin treatment (Figs. 1A and 2A), can directly interact with VDAC channels and disrupt mitochondrial membrane potential (Carre et al., 2002; Maldonado et al., 2010; Rovini, 2019). Additionally, other microtubule destabilizers, including the vinca alkaloids and combretastatin A-4, induce the expression of NOXA, which promotes the release and activation of Bax and Bak to form mitochondrial pores in noncycling cells within the same timeframe in which we observe an increase of cytoplasmic mtDNA with eribulin (Bates et al., 2013). Therefore, there is a strong precedent and rationale for how microtubule destabilization can alter mitochondrial permeability through multiple pathways to lead to the innate immune signaling observed in the current study. A rigorous determination of the relative impact of these mechanisms on the release of mtDNA by eribulin will require further investigation.
Together, this work sets the stage for investigation of other innate immune pathways that are specifically modulated by MTAs downstream of their effects on microtubule dynamics and structure. It also specifically prompts interrogation of the adaptive immunologic events downstream of the activation of these innate immune sensing pathways, including whether activation of the cGAS-STING pathway within the tumor microenvironment can serve as a predictive biomarker of response to eribulin, particularly in combination with immunotherapy in TNBC.
Acknowledgments
The authors thank Dr. Bruce A. Littlefield, Dr. Taro Semba, and members of the Risinger and Mooberry laboratories for their valuable comments and suggestions. The authors are grateful to Dr. Juli Bai and Dr. Feng Liu for providing them with Sting gt/gt mice. The authors also thank Dr. Nan Yan for kindly sharing the THP-1 cells and are very appreciative of Dr. Wouter Koek for his assistance in the statistical analysis of the data presented. Flow Cytometry data were generated in the University of Texas Health Science Center San Antonio Flow Cytometry Shared Resource Facility, which is supported by the University of Texas Health Science Center San Antonio Mays Cancer Center P30 Cancer Center Support Grant [NIH-NCI P30 CA054174] and the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant UL1 TR002645.
Abbreviations
- ACTb
beta actin
- AP-1
activator protein 1
- Bak
bcl-2 homologous antagonist/killer protein
- Bax
bcl-2-associated x protein
- BMDM
bone marrow–derived macrophage
- Bcl-2
b-cell lymphoma 2
- cGAS
cyclic GMP-AMP synthase
- COX1
cytochrome c oxidase I
- Ctrl
control
- EtBr
ethidium bromide
- FITC
fluorescein isothiocyanate
- GAPDH
glycerol-3-phosphate dehydrogenase
- gDNA
genomic DNA
- gt
golden ticket
- HPRT
hypoxanthine-guanine phosphoribosyltransferase
- HT-DNA
herring testes DNA
- IFIT
interferon-induced protein with tetratricopeptide repeats
- IFN
interferon
- IFNAR
interferon receptor
- IkB
I kappa B
- ISG
interferon-stimulated gene
- JAK
Janus kinase
- Mt
mitochondrial
- MTA
microtubule-targeting agent
- mtDNA
mitochondrial DNA
- ND
NADH-ubiquinone oxidoreductase chain
- NF-κB
nuclear factor κ-light-chain-enhancer of activated B cell
- NOXA (PMAIP1)
phorbol-12-myristate-13-acetate-induced protein 1
- OAS1
2’-5’-Oligoadenylate synthetase 1
- PGK
phosphoglycerate kinase 1
- PMSF
phenylmethylsulfonyl fluroride
- PVDF
polyvinylidene fluoride
- qRT-PCR
quantitative real time polymerase chain reaction
- RFP
red fluorescent protein
- Rho0
cells depleted of mitochondrial DNA
- RPS18
ribosomal protein S18
- siRNA
small interfering RNA
- STAT
signal transducer and activator of transcription
- STING
stimulator of interferon genes
- STR
short tandem repeat
- TBK1
tank binding kinase
- TBP
TATA-binding protein
- TNBC
triple-negative breast cancer
- VDAC
voltage-dependent anion channel
Authorship Contributions
Participated in research design: Fermaintt, Takahashi-Ruiz, Risinger.
Conducted experiments: Fermaintt, Takahashi-Ruiz, Liang.
Performed data analysis: Fermaintt, Takahashi-Ruiz, Risinger.
Wrote or contributed to the writing of the manuscript: Fermaintt, Takahashi-Ruiz, Liang, Mooberry, Risinger.
Footnotes
This work was funded by a Sponsored Research Agreement with Eisai Inc. (S.L.M. and A.L.R.). Postdoctoral training is supported by the US National Institutes of Health National Institute of General Medical Sciences K12 [Grant GM111726 (to C.S.F.)]. The flow cytometry core facility is supported by the University of Texas Health Science Center San Antonio Mays Cancer Center P30 Cancer Center Support Grant [NIH-NCI P30 CA054174] and the National Center for Advancing Translational Sciences, National Institutes of Health [Grant UL1 TR002645].
Dr. Risinger and Dr. Mooberry are funded by Eisai Inc. through a Sponsored Research Agreement. Eisai Inc. holds the intellectual property of eribulin mesylate.
Primary laboratory of origin: Risinger.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Armand R, Channon JY, Kintner J, White KA, Miselis KA, Perez RP, Lewis LD (2004) The effects of ethidium bromide induced loss of mitochondrial DNA on mitochondrial phenotype and ultrastructure in a human leukemia T-cell line (MOLT-4 cells). Toxicol Appl Pharmacol 196:68–79. [DOI] [PubMed] [Google Scholar]
- Bates DJ, Danilov AV, Lowrey CH, Eastman A (2013) Vinblastine rapidly induces NOXA and acutely sensitizes primary chronic lymphocytic leukemia cells to ABT-737. Mol Cancer Ther 12:1504–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D, Eastman A (2017) Microtubule destabilising agents: far more than just antimitotic anticancer drugs. Br J Clin Pharmacol 83:255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BLSasaki DTMurray BWO’Leary ECSakata STXu WLeisten JCMotiwala APierce SSatoh Y, et al. (2001) SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA 98:13681–13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carré M, André N, Carles G, Borghi H, Brichese L, Briand C, Braguer D (2002) Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J Biol Chem 277:33664–33669. [DOI] [PubMed] [Google Scholar]
- Chandra D, Quispe-Tintaya W, Jahangir A, Asafu-Adjei D, Ramos I, Sintim HO, Zhou J, Hayakawa Y, Karaolis DK, Gravekamp C (2014) STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer Immunol Res 2:901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng NWatkins-Schulz RJunkins RDDavid CNJohnson BMMontgomery SAPeine KJDarr DBYuan HMcKinnon KP, et al. (2018) A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1-insensitive models of triple-negative breast cancer. JCI Insight 3:120638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K, Plater L, Peggie M, Cohen P (2009) Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase ϵ: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J Biol Chem 284:14136–14146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumontet C, Jordan MA (2010) Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov 9:790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dybdal-Hargreaves NF, Risinger AL, Mooberry SL (2017) Regulation of E-cadherin localization by microtubule targeting agents: rapid promotion of cortical E-cadherin through p130Cas/Src inhibition by eribulin. Oncotarget 9:5545–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field JJ, Kanakkanthara A, Miller JH (2014) Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg Med Chem 22:5050–5059. [DOI] [PubMed] [Google Scholar]
- Fong A, Durkin A, Lee H (2019) The potential of combining tubulin-targeting anticancer therapeutics and immune therapy. Int J Mol Sci 20:586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vanpouille-Box C (2018) BAX and BAK at the gates of innate immunity. Trends Cell Biol 28:343–345. [DOI] [PubMed] [Google Scholar]
- Giannakakou P, Sackett DL, Kang Y-K, Zhan Z, Buters JTM, Fojo T, Poruchynsky MS (1997) Paclitaxel-resistant human ovarian cancer cells have mutant β-tubulins that exhibit impaired paclitaxel-driven polymerization. J Biol Chem 272:17118–17125. [DOI] [PubMed] [Google Scholar]
- Goel SMita ACMita MRowinsky EKChu QSWong NDesjardins CFang FJansen MShuster DE, et al. (2009) A phase I study of eribulin mesylate (E7389), a mechanistically novel inhibitor of microtubule dynamics, in patients with advanced solid malignancies. Clin Cancer Res 15:4207–4212. [DOI] [PubMed] [Google Scholar]
- Haag SMGulen MFReymond LGibelin AAbrami LDecout AHeymann Mvan der Goot FGTurcatti GBehrendt R, et al. (2018) Targeting STING with covalent small-molecule inhibitors. Nature 559:269–273. [DOI] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA (2017) Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548:466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Härtlova AErttmann SFRaffi FAMSchmalz AMResch UAnugula SLienenklaus SNilsson LMKröger ANilsson JA, et al. (2015) DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 42:332–343. [DOI] [PubMed] [Google Scholar]
- Hasan M, Koch J, Rakheja D, Pattnaik AK, Brugarolas J, Dozmorov I, Levine B, Wakeland EK, Lee-Kirsch MA, Yan N (2013) Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat Immunol 14:61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Takaoka A, Taniguchi T (2006) Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360. [DOI] [PubMed] [Google Scholar]
- Hu M, Zhou M, Bao X, Pan D, Jiao M, Liu X, Li F, Li C-Y (2021) ATM inhibition enhances cancer immunotherapy by promoting mtDNA leakage and cGAS/STING activation. J Clin Invest 131:139333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizing MT, Vermorken JB, Rosing H, ten Bokkel Huinink WW, Mandjes I, Pinedo HM, Beijnen JH (1995) Pharmacokinetics of paclitaxel and three major metabolites in patients with advanced breast carcinoma refractory to anthracycline therapy treated with a 3-hour paclitaxel infusion: a European Cancer Centre (ECC) trial. Ann Oncol 6:699–704. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Spodnik JH, Teranishi M, Wozniak M, Nishizawa Y, Usukura J, Wakabayashi T (2001) Opposite effects of microtubule-stabilizing and microtubule-destabilizing drugs on biogenesis of mitochondria in mammalian cells. J Cell Sci 114:281–291. [DOI] [PubMed] [Google Scholar]
- Kaul R, Risinger AL, Mooberry SL (2019a) Eribulin rapidly inhibits TGF-β-induced Snail expression and can induce Slug expression in a Smad4-dependent manner. Br J Cancer 121:611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul R, Risinger AL, Mooberry SL (2019b) Microtubule-targeting drugs: more than antimitotics. J Nat Prod 82:680–685. [DOI] [PubMed] [Google Scholar]
- Kim JGupta RBlanco LPYang SShteinfer-Kuzmine AWang KZhu JYoon HEWang XKerkhofs M, et al. (2019) VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 366:1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komlodi-Pasztor E, Sackett D, Wilkerson J, Fojo T (2011) Mitosis is not a key target of microtubule agents in patient tumors. Nat Rev Clin Oncol 8:244–250. [DOI] [PubMed] [Google Scholar]
- Komlodi-Pasztor E, Sackett DL, Fojo AT (2012) Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin Cancer Res 18:51–63. [DOI] [PubMed] [Google Scholar]
- Lohard SBourgeois NMaillet LGautier FFétiveau ALasla HNguyen FVuillier CDumont AMoreau-Aubry A, et al. (2020) STING-dependent paracriny shapes apoptotic priming of breast tumors in response to anti-mitotic treatment. Nat Commun 11:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJCarroll PMartin C-AMurina OFluteau ASimpson DJOlova NSutcliffe HRainger JKLeitch A, et al. (2017) cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548:461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado EN, Patnaik J, Mullins MR, Lemasters JJ (2010) Free tubulin modulates mitochondrial membrane potential in cancer cells. Cancer Res 70:10192–10201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittendorf EAZhang HBarrios CHSaji SJung KHHegg RKoehler ASohn JIwata HTelli ML, et al. (2020) Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): a randomised, double-blind, phase 3 trial. Lancet 396:1090–1100. [DOI] [PubMed] [Google Scholar]
- Okatsu KSaisho KShimanuki MNakada KShitara HSou YSKimura MSato SHattori NKomatsu M, et al. (2010) p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 15:887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orecchioni S, Talarico G, Labanca V, Calleri A, Mancuso P, Bertolini F (2018) Vinorelbine, cyclophosphamide and 5-FU effects on the circulating and intratumoural landscape of immune cells improve anti-PD-L1 efficacy in preclinical models of breast cancer and lymphoma. Br J Cancer 118:1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podolin PL, Callahan JF, Bolognese BJ, Li YH, Carlson K, Davis TG, Mellor GW, Evans C, Roshak AK (2005) Attenuation of murine collagen-induced arthritis by a novel, potent, selective small molecule inhibitor of IkappaB Kinase 2, TPCA-1 (2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide), occurs via reduction of proinflammatory cytokines and antigen-induced T cell Proliferation. J Pharmacol Exp Ther 312:373–381. [DOI] [PubMed] [Google Scholar]
- Rajput S, Volk-Draper LD, Ran S (2013) TLR4 is a novel determinant of the response to paclitaxel in breast cancer. Mol Cancer Ther 12:1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovini A (2019) Tubulin-VDAC interaction: molecular basis for mitochondrial dysfunction in chemotherapy-induced peripheral neuropathy. Front Physiol 10:671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson VB, Vetter NS, Zhang W, Patil PU, Mason RW, George E, Gorlick R, Kolb EA (2016) Integrating mechanisms of response and resistance against the tubulin binding agent Eribulin in preclinical models of osteosarcoma. Oncotarget 7:86594–86607. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sauer JDSotelo-Troha Kvon Moltke JMonroe KMRae CSBrubaker SWHyodo MHayakawa YWoodward JJPortnoy DA, et al. (2011) The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun 79:688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid PRugo HSAdams SSchneeweiss ABarrios CHIwata HDiéras VHenschel VMolinero LChui SY, et al. ; IMpassion130 Investigators (2020) Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 21:44–59. [DOI] [PubMed] [Google Scholar]
- Shang G, Zhang C, Chen ZJ, Bai XC, Zhang X (2019) Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 567:389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz MO, Prota AE (2018) Microtubule-targeting agents: strategies to hijack the cytoskeleton. Trends Cell Biol 28:776–792. [DOI] [PubMed] [Google Scholar]
- Tolaney SM, Kalinsky K, Kaklamani V, D'Adamo DR, Aktan G, Tsai ML, O'Regan RM, Kaufman PA, Wilks ST, Andreopoulou E, Patt DA, Yuan Y, Wang G, Savulsky C, Xing D, Kleynerman E, Karantza V, Diab S (2021) Eribulin plus pembrolizumab in patients with metastatic triple-negative breast cancer (ENHANCE 1): a phase 1b/2 study. Clin Cancer Res 27:3061–3068. [DOI] [PubMed] [Google Scholar]
- Wanderley CWColón DFLuiz JPMOliveira FFViacava PRLeite CAPereira JASilva CMSilva CRSilva RL, et al. (2018) Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1- profile in a TLR4-dependent manner. Cancer Res 78:5891–5900. [DOI] [PubMed] [Google Scholar]
- Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, Chen ZJ (2017) cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci USA 114:1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West APKhoury-Hanold WStaron MTal MCPineda CMLang SMBestwick MDuguay BARaimundo NMacDuff DA, et al. (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520:553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MJMcArthur KMetcalf DLane RMCambier JCHerold MJvan Delft MFBedoui SLessene GRitchie ME, et al. (2014) Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159:1549–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AJReyes CNLiang WBecker CShimada KWheeler MLCho HCPopescu NICoggeshall KMArditi M, et al. (2016) Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 166:624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki TKirchmair ASato ABuqué ARybstein MPetroni GBloy NFinotello FStafford LNavarro Manzano E, et al. (2020) Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol 21:1160–1171. [DOI] [PubMed] [Google Scholar]
- Zierhut C, Yamaguchi N, Paredes M, Luo J-D, Carroll T, Funabiki H (2019) The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell 178:302–315.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]