

Abstract
β1 adrenergic receptors (β1ARs) are central regulators of cardiac function and a drug target for cardiac disease. As a member of the G protein–coupled receptor family, β1ARs activate cellular signaling by primarily coupling to Gs proteins to activate adenylyl cyclase, cAMP-dependent pathways, and the multifunctional adaptor-transducer protein β-arrestin. Carvedilol, a traditional β-blocker widely used in treating high blood pressure and heart failure by blocking β adrenergic receptor–mediated G protein activation, can selectively stimulate Gs-independent β-arrestin signaling of β adrenergic receptors, a process known as β-arrestin–biased agonism. Recently, a DNA-encoded small-molecule library screen against agonist-occupied β2 adrenergic receptors (β2ARs) identified Compound-6 (Cmpd-6) to be a positive allosteric modulator for agonists on β2ARs. Intriguingly, it was further discovered that Cmpd-6 is positively cooperative with the β-arrestin–biased ligand carvedilol at β2ARs. Here we describe the surprising finding that at β1ARs unlike β2ARs, Cmpd-6 is cooperative only with carvedilol and not agonists. Cmpd-6 increases the binding affinity of carvedilol for β1ARs and potentiates carvedilol-stimulated, β-arrestin–dependent β1AR signaling, such as epidermal growth factor receptor transactivation and extracellular signal-regulated kinase activation, whereas it does not have an effect on Gs-mediated cAMP generation. In vivo, Cmpd-6 enhances the antiapoptotic, cardioprotective effect of carvedilol in response to myocardial ischemia/reperfusion injury. This antiapoptotic role of carvedilol is dependent on β-arrestins since it is lost in mice with myocyte-specific deletion of β-arrestins. Our findings demonstrate that Cmpd-6 is a selective β-arrestin–biased allosteric modulator of β1ARs and highlight its potential clinical utility in enhancing carvedilol-mediated cardioprotection against ischemic injury.
SIGNIFICANCE STATEMENT
This study demonstrates the positive cooperativity of Cmpd-6 on β1ARs as a β-arrestin–biased positive allosteric modulator. Cmpd-6 selectively enhances the affinity and cellular signaling of carvedilol, a known β-arrestin–biased β-blocker for β1ARs, whereas it has minimal effect on other ligands tested. Importantly, Cmpd-6 enhances the β-arrestin–dependent in vivo cardioprotective effect of carvedilol during ischemia/reperfusion injury–induced apoptosis. The data support the potential therapeutic application of Cmpd-6 to enhance the clinical benefits of carvedilol in the treatment of cardiac disease.
Introduction
β1 adrenergic receptors (β1ARs) are key regulators of heart rate and myocardial contractility and a common therapeutic target for the treatment of cardiac diseases, such as hypertension and heart failure (Wang et al., 2018). As a member of the G protein–coupled receptor (GPCR) family, β1ARs primarily transduce signals through stimulatory guanine nucleotide-binding proteins (Gαs proteins). Upon receptor activation by the endogenous catecholamine epinephrine or norepinephrine, β1ARs couple to Gs proteins to activate the effector enzyme adenylyl cyclase to catalyze the generation of second messenger cAMP, leading to downstream signaling and a diverse array of cellular and physiologic responses (Wang et al., 2018). Sustained Gs signaling activated by chronic neurohumoral stimulation of the β1AR can lead to lethal cardiac arrhythmias and deleterious maladaptive cardiac remodeling (Rockman et al., 2002). Therefore, β-blockers that prevent excessive β1AR activation by blunting catecholamine-stimulated downstream Gs signaling, such as carvedilol, metoprolol, and bisoprolol, are widely used in the treatment of heart failure (Packer et al., 1996, 2001; MERIT-HF-Study-Group, 1999; Bhatt et al., 2017). However, despite the fact that β-blockers effectively decrease morbidity and mortality in heart failure, there is a large variation among patients in the level of responsiveness to these drugs (Bhatt et al., 2017). Moreover, the development of intolerable side effects often leads to difficulty in achieving the targeted drug doses in many patients even though evidence suggests a positive correlation between higher β-blocker doses and improved clinical outcomes (Bhatt et al., 2017). The major side effects of β-blockers involve fatigue because of their potent blockade of exercise-induced heart rate and the nonselective inhibition of both β1 adrenergic receptor (β1AR) and β2 adrenergic receptor (β2AR) subtypes (carvedilol) leading to unwanted effects in other tissues, such as bronchial and blood vessel constriction (Cleland et al., 2000). Therefore, developing new β1AR ligands with improved efficacy, higher receptor subtype specificity, and fewer side effects is a huge unmet need.
It is now appreciated that, in addition to Gs protein coupling, β1ARs also transduce signaling through β-arrestins (Rajagopal et al., 2010), adaptor proteins initially discovered for their roles in interdicting Gs activation and desensitizing GPCRs (Benovic et al., 1989; Lohse et al., 1990). The awareness that a ligand-activated GPCR can selectively couple to different transducers (i.e., G protein or β-arrestin) has led to the current concept of biased agonism (Rajagopal et al., 2010; Wang et al., 2018). The β-blocker carvedilol has been shown to specifically stimulate β adrenergic receptor (βAR) to mediate β-arrestin–dependent extracellular signal-regulated kinase (ERK) signaling (Wisler et al., 2007) and epidermal growth factor receptor (EGFR) transactivation (Kim et al., 2008; Wang et al., 2017) and is therefore termed as a β-arrestin–biased agonist. Moreover, activation of β-arrestin–mediated pathways have been shown to promote cardioprotective effects (Noma et al., 2007; Kim et al., 2008; Wang et al., 2017).
An exciting area of GPCR pharmacology is the emerging field of allosteric modulation. Allosteric modulators bind to receptors in areas that are topographically distinct from the orthosteric site and cooperate with orthosteric ligands to modulate receptor activity (Hauser et al., 2017). Allosteric modulators may revolutionize drug discovery because of their potential advantages over the traditional orthosteric agents, such as greater specificity, precise spatial-temporal regulation of receptor activity, and the possibility to engage or enhance signaling bias by finely modulating potency and efficacy of orthosteric ligands (Hauser et al., 2017; Thal et al., 2018).
Given the cardioprotective effect of β1AR-mediated β-arrestin–biased signaling and the potential therapeutic advantage of allosteric modulators, we set out to identify, characterize, and then translate in vivo novel ligands that function as β-arrestin–biased allosteric modulators on the β1AR. In this study, we investigated the allosteric modulation activity on the β1AR of Compound-6 (Cmpd-6) (Supplemental Fig. 1), a small-molecule agent identified as an unbiased positive allosteric modulator (PAM) for the β2AR through an affinity screen of a DNA-encoded chemical library (Ahn et al., 2018; Liu et al., 2019). Recent work has shown that a unique property of Cmpd-6 is its positive cooperativity with only carvedilol on the β2AR among a diverse panel of β-blockers (Pani et al., 2021). Here, we identify unique allosteric properties of Cmpd-6 on β1AR ligand binding and cellular signaling and demonstrate that Cmpd-6 enhances the in vivo cardioprotective effects of carvedilol in response to cardiac injury. These data suggest that drugs like Cmpd-6 may have therapeutic advantages by enhancing the effectiveness of orthosteric ligands while minimizing unwanted side effects.
Materials and Methods
Reagents
Cmpd-6 was synthesized as previously described (Ahn et al., 2018; Liu et al., 2019). Carvedilol; isoproterenol; epinephrine; norepinephrine; metoprolol; carazolol; atenolol; bisoprolol; ICI-118,551; and epidermal growth factor (EGF) were purchased from Sigma-Aldrich (St. Louis, MO). Alprenolol was from Tocris Bioscience (Minneapolis, MN). Bucindolol was from Santa Cruz Biotechnology (Dallas, TX). Radiolabeled 125I-cyanopindolol was from PerkinElmer Life Sciences (Waltham, MA).
β1AR Expression and Purification
The β1AR was expressed in and purified from Expi293F cells as previously described for the β2AR (Staus et al., 2018) with small modifications. FLAG-tagged β1AR was transfected into Expi293F cells (Invitrogen) with Expifectamine (Invitrogen) as described in the manufacturer’s protocol. Cells were harvested 3 days after transfection and lysed by stirring in lysis buffer (10 mM Tris pH 7.4; 2 mM EDTA; 10 mM MgCl2; 5 units/ml benzonase, benzamidine, and leupeptin; and 1 μM alprenolol) for 30 minutes at room temperature. Cell membrane was pelleted by centrifugation at 32,000 × g for 20 minutes at 4°C, homogenized in solubilization buffer [20 mM HEPES pH 7.4; 100 mM NaCl; 10 mM MgCl2; 1% N-dodecyl-β-D-maltoside (DDM); 0.1% cholesterol hemisuccinate (CHS); 5 units/ml benzonase, benzamidine, and leupeptin; and 1 μM alprenolol], stirred at room temperature and 4°C for 1 hour each, and centrifuged at 32,000 × g for 40 minutes at 4°C. The supernatant was supplemented with 2 mM CaCl2 and loaded on M1-FLAG column at 2–3 ml/min at 4°C. The M1-FLAG column was washed with 5 column volumes each of low-salt (100 mM NaCl) and high-salt (500 mM NaCl) wash buffer (20 mM HEPES pH 7.4; 2 mM CaCl2; 0.1% DDM; 0.01% CHS, benzamidine, and leupeptin; and 1 μM alprenolol). The wash cycle was repeated an additional three times and then completed with 10 column volumes of low-salt wash buffer. Receptor was eluted in elution buffer (20 mM HEPES pH 7.4, 100 mM NaCl, 0.1% DDM, 0.01% CHS, 0.2 mg/ml FLAG-peptide, 5 mM EDTA, and 1 μM alprenolol) and cleaned up by size-exclusion chromatography with Superdex 200 Increase Column (GE Healthcare Life Sciences).
High-Density Lipoprotein Reconstitution
Reconstitution of purified β1ARs into high-density lipoprotein (HDL) particles was performed as previously described for the β2AR (Ahn et al., 2018). In brief, 2 μM purified β1AR was incubated with 100 μM apolipoprotein A1 (Membrane Scaffold Protein 1) and 8 mM phosphatidylcholine/phosphatidylglycerol (3:2 molar ratio mixture) in buffer (20 mM HEPES pH 7.4, 100 mM NaCl, and 100 mM cholate) for 1 hour at 4°C. Biobeads (Bio-Rad) were then added and rotated overnight at 4°C to remove detergent. β1AR-HDL particle was isolated from empty HDL particles using M1-FLAG chromatography.
Competition Binding Assay
Radioligand competition binding assay was performed with the radiolabeled 125I-cyanopindolol (CYP) (PerkinElmer) as previously described for the β2AR (Ahn et al., 2018). The β1AR-HDL particles were incubated with 60 pM 125I-CYP, vehicle (DMSO) or 25 μM Cmpd-6, and a serial dilution of indicated orthosteric ligands in assay buffer (20 mM HEPES pH 7.4, 100 mM NaCl, 0.1% bovine serum albumin, and 1 mM ascorbic acid) at room temperature for 90 minutes. Nonspecific binding was determined in the presence of 20 µM propranolol. The reaction was terminated and harvested by rapid filtration onto 0.3% poly(ethyleneimine)-treated GF/B glass fiber filter paper (Brandel) using a harvester (Brandel). 125I-CYP on the filter paper was measured by a WIZARD2 2-Detector Gamma Counter (PerkinElmer). Binding data were analyzed in GraphPad Prism using a one-site competition-binding logIC50 curve fit with data points weighted equally. Counts per minute values were normalized as percentage of the maximal 125I-CYP binding level and plotted as mean ± S.D.
GloSensor Assay
Analysis of cAMP accumulation was performed in human embryonic kidney (HEK) 293 cells transiently transfected with β1AR and GloSensor constructs. The day before transfection, 5.5 million cells were seeded in one 10-cm tissue culture dish. The cells were transfected with 6 μg GloSensor plasmid DNA and 25 ng β1AR plasmid using FuGene6 (Promega) transfection reagent. After 1 day, 100,000 transfected cells were seeded to each well of 96-well plate in assay media (minimal essential medium with 2% FBS). The next day, the cells were incubated with the GloSensor reagent [Promega; 4% (v/v)] at room temperature for 2 hours; pretreated with 100 nM ICI118,551 to block activation of endogenous β2AR; and then stimulated with indicated ligands for 5 minutes. Luminescence was detected on a Biotek Neo2 microplate reader.
EGFR Transactivation Assay
The level of EGFR transactivation was measured by the endocytosis bioluminescence resonance energy transfer (BRET) assay as previously described (Namkung et al., 2016) with slight modifications. HEK293 cells stably expressing β1ARs were transfected with 250 ng of EGFR-Renilla luciferase variant 2 (RlucII) and 1 μg of recombinant Green Fluorescent Protein (rGFP)-FYVE constructs. The day after transfection, cells were reseeded onto poly(L-ornithine)-coated 96-well plates at ∼25,000 cells per well. The next day, cells were pretreated with vehicle (DMSO), 30 μM Cmpd-6, or 10 μM AG1478 in assay buffer (Hanks’ balanced salt solution, 20 mM HEPES and pH 7.4) for 20 minutes at 37 °C and then stimulated with ligand at indicated concentration for 25 minutes. The cell-permeable substrate coelenterazine 400a (final concentrations of 5 μM) was added ∼3–6 minutes before BRET measurements. The BRET measurements were performed with a Neo2 microplate reader (BioTek) with a filter set (center wavelength/bandwidth) of 410/80 nm (donor) and 515/30 nm (acceptor).
ERK Activation Assay
HEK293 cells with stable overexpression of FLAG-tagged β1AR or β-arrestin knockout were described previously. Cells were periodically treated with BM Cyclin (Roche) to avoid mycoplasma contamination. Cells were incubated for 4 hours in serum-free medium supplemented with 0.1% bovine serum albumin, 10 mM HEPES, and 1% penicillin-streptomycin; pretreated with vehicle (DMSO) or 30 μM Cmpd-6 for 20 minutes; and stimulated with indicated ligands for 5 minutes. After stimulation, cells were lysed in lysis buffer (20 mM Tris pH 7.4, 137 mM NaCl, 20% glycerol, 1% Nonidet P-40, 2 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 10 mM sodium fluoride, 10 μg/ml aprotinin, and 5 μg/ml leupeptin and phosphatase inhibitors) by rotating for 30 minutes at 4°C. Cell lysate samples were separated by SDS-PAGE, transferred to PVDF Membrane (Bio-Rad), and subjected to immunoblotting with anti-MAPK 1/2 (EMD Millipore) and anti-p44/42 MAPK (Cell Signaling) antibodies. Immunoblots were detected using enhanced chemiluminescence (Thermo Fisher Scientific) and analyzed with ImageJ software. The densitometry values from phosphorylated ERK (pERK) blots were normalized to respective total ERK blots and normalized to the maximum response of control group in each experiment.
Isoproterenol-Induced Hemodynamic in Mouse Heart
Eight to 12-week-old C57BL/6J wild-type mice of both sexes were used for this study. Animal experiments carried out for this study were handled according to approved protocols and animal welfare regulations mandated by the Institutional Animal Care and Use Committee of Duke University Medical Center. Mice were treated with vehicle, carvedilol (1, 5, or 20 mg/kg per day), or a combination of carvedilol (1 mg/kg per day) and Cmpd-6 (5 mg/kg per day) for 3 days with Alzet osmotic pump (Durect) implanted into mice subcutaneously. After treatment, mice were anesthetized with ketamine (100 mg/kg) and xylazine (2.5 mg/kg). After bilateral vagotomy, a 1.4 French (0.46 mm) high-fidelity micromanometer catheter (ADInstruments) connected to a pressure transducer (ADInstruments) was inserted into the left ventricle to monitor blood pressure. Basal blood pressure was recorded at steady state after the catheter insertion (2–3 minutes after insertion). Graded doses of isoproterenol were administered at 45-second intervals by intravenous injection through a jugular vein. Blood pressure was monitored continuously and recorded at the steady state (35–45 seconds after each injection). Data analysis was performed using LabChart 8 software (ADInstruments).
Ischemia/Reperfusion-Induced Cell Apoptosis in Mouse Heart
Eight to 12-week-old C57BL/6J wild-type mice or 8- to 27-week-old α-myosin heavy chain (αMyHC)–Cre:Arrb1flox/flox/Arrb2flox/flox mice of both sexes were used for this study. Animals were randomly assigned to and the researchers were masked for treatment groups. Alzet osmotic pumps (Durect) were implanted into mice subcutaneously to deliver vehicle (DMSO), Cmpd-6 (5 mg/kg per day), carvedilol (1 mg/kg per day or 20 mg/kg per day), or a combination of Cmpd-6 (5 mg/kg per day) and carvedilol (1 mg/kg per day). After 3 days of treatment, cardiac ischemia was produced by the ligation of left anterior descending coronary artery. After 45 minutes, the ligation was released to allow the blood flow to be restored for 45 minutes. The mice hearts were perfusion-fixed with 4% paraformaldehyde, excised from the body, and placed in 30% sucrose in PBS for 2–4 hours at 4°C. The hearts were then embedded in optimum cutting temperature compound (Sakura Finetek) and snap-frozen in liquid nitrogen. Terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling (TUNEL) staining on the cryosections were performed with in situ cell death detection kit (Sigma-Aldrich) according to the manufacturer’s protocol. Sections were then mounted in ProLong Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific). Images were recorded with Zeiss Axio Observer Z1 confocal microscope with 20× objective. In each heart, sections from different groups (from base to apex of the tissue, apart by 0.5 mm) were stained, and the group with maximum TUNEL-positive cells were used for quantification. At least nine images from three sections were quantified for each heart. The number of TUNEL-positive cells and the total number of nuclei as determined by 4′,6-diamidino-2-phenylindole staining were counted using ImageJ software.
Statistical Analysis
Data are expressed as mean ± S.D. Statistical comparisons were performed using one-way ANOVA with Tukey correction or two-way repeated-measures ANOVA with Sidak correction for multiple comparison in GraphPad Prism. Differences were considered statistically significant at P < 0.05.
Results
Compound-6 Selectively Potentiates the Binding Affinity of Carvedilol to the β1AR
Cmpd-6 is an unbiased positive allosteric modulator for the β2AR that substantially enhances the affinity of agonists and downstream signaling mediated by either G protein or β-arrestin (Ahn et al., 2018). Carvedilol, a traditional β-blocker widely used in the treatment of cardiac diseases, has also been identified as a β-arrestin–biased agonist for both the β1AR and the β2AR (Wisler et al., 2007; Kim et al., 2008). Cmpd-6 potentiates β-arrestin1–induced high-affinity binding of carvedilol to the β2AR as well as carvedilol-stimulated β2AR cellular signaling (Pani et al., 2021). To determine the allosteric modulatory activity of Cmpd-6 on the β1AR, we first tested its effect on the orthosteric binding affinity of a comprehensive ligand panel of agonists and antagonists by performing competition binding assays against radiolabeled antagonist cyanopindolol (125I-CYP) binding to purified β1ARs reconstituted in HDL particles (Whorton et al., 2007). Remarkably, we found that Cmpd-6 leads to ∼1-log leftward shift of the carvedilol competition binding curve [Fig. 1A, vehicle: LogIC50 = −8.89 (−8.98 to −8.80), Cmpd-6: LogIC50 = −9.79 (−9.91 to −9.67), mean (95% confidence intervals)], indicating that Cmpd-6 strongly potentiates the binding affinity of carvedilol to the β1AR. Notably, Cmpd-6 minimally enhanced the binding affinity of the full agonist isoproterenol for β1ARs [Fig. 1B, vehicle: LogIC50 = −7.60 (−7.65 to −7.54), Cmpd-6: LogIC50 = −7.92 (−8.00 to −7.83)], which is in marked contrast to its effect on β2ARs in which it enhanced isoproterenol binding affinity by 50-fold (Ahn et al., 2018). This marginal effect of Cmpd-6 on isoproterenol affinity for β1ARs is consistent with previous studies (Ahn et al., 2018; Liu et al., 2019). When tested against a diverse ligand panel, it showed minimal positive cooperativity with the full agonists epinephrine and norepinephrine and no positive cooperativity with any other antagonist tested (Fig. 1C). Overall, Cmpd-6 showed a subtle increase of agonist binding to the β1AR, as indicated by a slight leftward shift of competition binding curves and change of IC50 value, and no positive modulation of binding affinity for the five other antagonists tested (Fig. 1, C and D). To compare the Cmpd-6 cooperativity with ligands between β1ARs and β2ARs in separate experiments, we determined Cmpd-6–induced affinity shifts of an expanded panel of ligands on the β1AR (Supplemental Fig. 2A) and plotted against the shifts observed for the β2AR shown in Fig. 1B of the accompanying manuscript by Pani et al., 2021 (Supplemental Fig. 2B). Comparison between receptor subtypes highlights that carvedilol is the only ligand among those tested that shows strong cooperativity with Cmpd-6 at both the β1AR and β2AR (Supplemental Fig. 2B). The binding affinity of Cmpd-6 to carvedilol-bound β1ARs can be calculated from the leftward shift in carvedilol binding affinity by Cmpd-6 (ΔLogIC50) by radioligand competition binding for β1ARs and β2ARs. The binding affinity of Cmpd-6 to carvedilol-bound β1ARs is 17 μM and to carvedilol-bound β2ARs is 1.2 μM (Pani et al., 2021).
Fig. 1.

Cmpd-6 selectively potentiates the binding affinity of carvedilol for the β1AR. (A) Cmpd-6 led to a leftward shift of the carvedilol competition binding curve against 125I- CYP to the β1AR, which indicated the increased receptor binding affinity of carvedilol. β1ARs reconstituted in high-density lipoprotein particles were incubated with vehicle (DMSO) or 25 μM Cmpd-6, indicated concentration of carvedilol, and 60 pM 125I-CYP. Values were normalized to the percentage of maximal 125I-CYP binding level. (B) Cmpd-6 modestly enhanced the binding affinity of isoproterenol for the β1AR by 2-fold. (C) Cmpd-6 had minimal effect on the β1AR binding affinity of a panel of agonists and antagonists tested. (D) Cmpd-6–led leftward shift of ligands competition binding curves, shown as the difference of log IC50 values of the vehicle- and Cmpd-6–treated groups for each ligand. Cmpd-6 showed strong cooperativity with carvedilol (red bar), a modest PAM activity on full agonists (black bars), and no positive modulation on other antagonists tested (blue bars). Data represent the mean ± S.D. for three independent experiments. Competition binding curves were generated with GraphPad Prism. The log IC50 values shown in the legends of (A–C) and represented in (D) were calculated from competition binding curves with GraphPad Prism. In (A-C), statistical comparisons were performed using two-way repeated (related)-measures ANOVA with Sidak correction for multiple comparisons. In (D), two-tailed Student’s t tests were performed for each ligand testing the null hypothesis that the shift in LogIC50 is 0 (D). *P < 0.05 and **P < 0.01.
Although traditionally identified and used as a β-blocker, carvedilol has been shown to possess β-arrestin–biased activity by selectively engaging β-arrestins as the transducer in mediating downstream βAR signaling (Wisler et al., 2007; Kim et al., 2008; Wang et al., 2017). Our discovery that Cmpd-6 specifically potentiates the binding affinity of carvedilol to the β1AR (Fig. 1, A and D) suggests that Cmpd-6 may be a β-arrestin–biased allosteric modulator of the β1AR.
Cmpd-6 Shows Positive Cooperativity on β-Arrestin–Mediated but Not Gs Protein–Mediated Signaling Induced by Carvedilol Stimulation of the β1AR
To determine the functional significance of the allosteric modulation activity of Cmpd-6 on β1ARs, we tested the effect of Cmpd-6 on β1AR-mediated cellular signaling in HEK293 cells. We first monitored Gs protein–mediated cAMP generation in the presence or absence of Cmpd-6. HEK293 cells were transfected with β1ARs and the luciferase-based cAMP biosensor GloSensor (Fig. 2A). Upon ligand stimulation of the β1AR, Gs activation of adenylyl cyclase generates cAMP that is detected by a conformational change of GloSensor to produce light (Fig. 2A). We observed that the full agonist isoproterenol robustly induced cAMP production in a dose-dependent manner, whereas the β-arrestin–biased agonist carvedilol induced a low level of cAMP only at high concentrations of ligand (Fig. 2B). This is consistent with a previous study showing that carvedilol displays a very low potency and efficacy with β1AR-mediated Gs activation in β1AR-overexpressed cells (Kim et al., 2008). Importantly, Cmpd-6 showed no enhancement of the dose-response curve for either ligand (Fig. 2B), indicating that Cmpd-6 has little or no PAM activity on the Gs-mediated β1AR signaling.
Fig. 2.

Cmpd-6 potentiates carvedilol-stimulated β-arrestin–mediated β1AR signaling but not Gs protein–mediated β1AR signaling. (A) β1AR-mediated Gs activation was monitored with GloSensor cAMP assay (Promega), a luciferase-based biosensor to monitor cAMP level. (B) Cmpd-6 had no effect on isoproterenol- or carvedilol-induced β1AR-mediated Gs activation. HEK293 cells transiently transfected with β1ARs and GloSensor were pretreated with vehicle (DMSO) or 30 μM Cmpd-6 for 20 minutes with 100 nM ICI118,551, the highly selective β2AR antagonist, to block the activation of endogenous β2ARs. Cells were then stimulated with serial doses of isoproterenol or carvedilol for 5 minutes. The luminescence values were normalized to the percentage of maximal isoproterenol-induced level in the vehicle-treated group. (C) β1AR-mediated EGFR transactivation was determined by monitoring EGFR endocytosis with BRET assay with EGFR_RlucII and endosomal-located rGFP_FYVE. (D) Cmpd-6 potentiated carvedilol-stimulated β1AR-mediated EGFR transactivation, which was indicated by a leftward shift of the dose-response curve. HEK293 cells stably expressing β1ARs were transfected with EGFR-RlucII and rGFP-FYVE constructs. Cells were pretreated with vehicle (DMSO) or 30 μM Cmpd-6 for 20 minutes and then stimulated with indicated concentrations of carvedilol for 25 minutes before BRET measurement. The change in BRET ratio (emission ratio of RlucII to rGFP) is expressed as the difference between ligand-stimulated and unstimulated samples. (E) Carvedilol-induced β1AR activation of ERK is mediated through β-arrestins. Wild-type (WT) or β-arrestin1/2 knockout (β-arr1/2 KO) HEK293 cells transiently transfected with β1ARs were stimulated with serial concentrations of carvedilol for 5 minutes. Data are calculated as the ratio of pERK to total ERK (tERK) and then normalized as percentage of the maximum value in the wide-type group. (F) Cmpd-6 led to a leftward shift of carvedilol dose-response curve on β1AR-mediated ERK activation, indicating its PAM activity on this β-arrestin–dependent signaling. HEK293 cells stably expressing β1ARs were pretreated with vehicle or 30 μM Cmpd-6 for 20 minutes and then stimulated with indicated concentration of carvedilol for 5 minutes. The pERK/tERK ratio is normalized to the maximum value in the vehicle-treated group. Data represent the mean ± S.D. for four to eight independent experiments as marked on the figure. Error bars in some data points in (B) are not visualizable because the error bar is shorter than the size of the symbol. Dose-response curves and log EC50 values shown in legends were generated with GraphPad Prism. Statistical comparisons were performed using two-way repeated (related)-measures ANOVA with Sidak correction for multiple comparisons. *P < 0.05, **P < 0.01, and ****P < 0.0001.
To determine the activity of Cmpd-6 on carvedilol-stimulated β-arrestin–mediated β1AR signaling, we tested the effect of Cmpd-6 on β1AR-stimulated EGFR transactivation. Previous studies showed that ligand stimulation of the β1AR induces G protein–coupled receptor kinase–mediated receptor phosphorylation and subsequent β-arrestin and Src recruitment (Kim et al., 2008). This in turn leads to the activation of matrix metalloproteinases and shedding of heparin-binding EGF that binds to EGFRs promoting its activation and internalization, a process known as EGFR transactivation (Noma et al., 2007; Kim et al., 2008). To allow sensitive and quantitative detection of β-arrestin–mediated EGFR transactivation, we developed a BRET sensor using EGFR-RlucII and the early endosome-targeted FYVE-rGFP (Namkung et al., 2016) to monitor the internalization of EGFRs (Fig. 2C). To assess the fidelity of this biosensor assay system we show that EGF ligand induced a robust change in BRET ratio, which was blocked by the EGFR inhibitor AG1478 (Supplemental Fig. 3A). In β1AR stably expressing HEK293 cells transfected with EGFR-RlucII and FYVE-rGFP, both the full agonist isoproterenol and the β-arrestin–biased agonist carvedilol dose-dependently induce EGFR internalization, and the response is diminished by the small interfering RNA knockdown of β-arrestin1/2 (Supplemental Fig. 3B). Cmpd-6 potentiated the carvedilol-induced response as indicated by the leftward shift of carvedilol dose-response curve [Fig. 2D, vehicle: logEC50 = −5.93 (−6.28 to −5.61), Cmpd-6: logEC50 = −6.86 (−7.19 to −6.46)], whereas it showed no PAM activity on the isoproterenol response (Supplemental Fig. 3C).
We next tested Cmpd-6 in β1AR-mediated ERK activation induced by carvedilol. We have previously shown that carvedilol induces β1AR-mediated ERK signaling in a β-arrestin–dependent manner (Kim et al., 2008; Wang et al., 2017; Luttrell et al., 2018). Consistent with these earlier studies, we show that carvedilol-induced ERK activation is largely lost in the HEK293 cells in which the genes for both β-arrestin1 and 2 are deleted through CRISPR gene editing (Fig. 2E). The low level of ERK activation in β-arrestin1/2 knockout cells is likely the result of some activation of Gs at high concentrations of carvedilol (Fig. 2B). In marked contrast to the effect observed on Gs activation, Cmpd-6 substantially potentiates carvedilol-induced ERK activation, as indicated by an 11-fold leftward shift in the ERK dose-response curve along with a small increase in the maximal response [Fig. 2F, vehicle: logEC50 = −7.20 (−7.44 to −6.98), Cmpd-6: logEC50 = −8.25 (−8.68 to −7.85)]. Additionally, isoproterenol stimulation showed no cooperativity by Cmpd-6 (Fig. 3A). When expanding our testing to a broad panel of ligands, we found that the effect of Cmpd-6 is highly selective to carvedilol with minimal or no augmentation of ERK phosphorylation in response to stimulation by a comprehensive panel of βAR ligands (Fig. 3). Data of the ERK activation assay are also presented as fold over nonstimulated samples of each treatment (Supplemental Fig. 4).
Fig. 3.
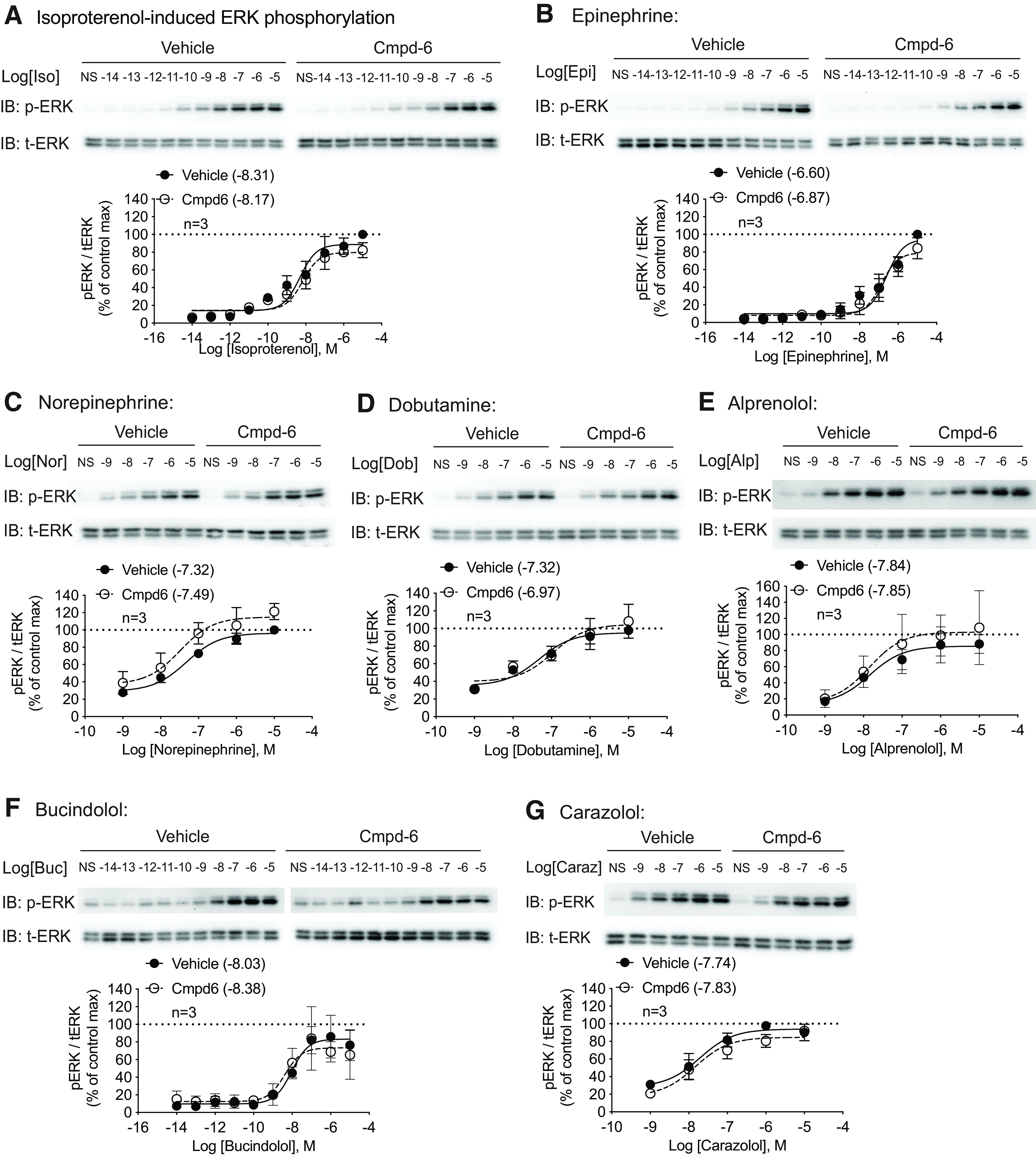
Cmpd-6 shows no PAM activity on β1AR-mediated ERK stimulated by a comprehensive panel of ligands. HEK293 cells stably expressing β1ARs were pretreated with vehicle or 30 μM Cmpd-6 for 20 minutes and then stimulated with serial concentrations of indicated ligands for 5 minutes. A panel of agonists (A–D) and β-blockers (E–G) dose-dependently induced β1AR-mediated ERK phosphorylation. Cmpd-6 pretreatment showed no effect on these ligands. Data are presented as pERK/total ERK (tERK) ratios normalized to the maximum value in the vehicle group. Data represent the mean ± S.D. for three independent experiments. Dose-response curves and log EC50 values shown in legends were generated with GraphPad Prism. Statistical comparisons were performed using two-way repeated (related)-measures ANOVA with Sidak correction for multiple comparisons and showed no difference between vehicle- and Cmpd-6–treated groups for each ligand. IB, immunoblotting.
Interestingly, in our expanded ligand panel on ERK activation, we noticed that a number of other β-blockers robustly induced β1AR-mediated ERK activation (Fig. 3, E–G). To dissect the mechanism for the various β-blockers on β1AR signaling, we determined the level of β-arrestin dependence on ERK signaling and Gs activation. In contrast to carvedilol, which activates ERK mainly through β-arrestin (Fig. 2F), the activation induced by alprenolol and carazolol is partially (∼50%) dependent on β-arrestin, whereas bucindolol activates ERK independent of β-arrestin (Supplemental Fig. 5, A–C). We then performed GloSensor assays of Gs-mediated cAMP generation, which showed that bucindolol and carazolol induced robust activation of cAMP (40% of the maximal isoproterenol response), whereas alprenolol only modestly induced a response (20% of the maximal isoproterenol response), and metoprolol induced no response (Supplemental Fig. 5D). Importantly, none of the β-blockers tested had positive cooperativity to Cmpd-6 (Supplemental Fig. 5D). These data show that although a number of β-blockers can activate β1AR signaling, they differentially engage signal transducers to activate downstream pathways as indicated by their differential cooperativity with Cmpd-6, dependence on β-arrestin, and activity on Gs-mediated cAMP production.
Taken together, we show the selective positive cooperativity of Cmpd-6 for the β1AR with carvedilol with respect to binding affinity and the selective enhancement of carvedilol-stimulated β-arrestin–mediated signaling but not Gs-mediated cAMP production. These data support our notion that Cmpd-6 is a β-arrestin–biased PAM of the carvedilol-occupied β1ARs.
Cmpd-6 Potentiates β-Arrestin–Dependent Cell Survival by Carvedilol in Response to Ischemia/Reperfusion Myocardial Injury
Our data from in vitro and cellular assays strongly support that Cmpd-6 selectively cooperates with carvedilol to enhance β-arrestin–biased β1AR signaling. Previous studies have shown that sustained G protein activation by β1ARs is associated with deleterious cardiac remodeling, whereas β-arrestin signaling is potentially cardioprotective (Noma et al., 2007; Wang et al., 2018). Although carvedilol, among other β-blockers, is considered a standard therapy in heart failure, a major limitation is that it is often difficult to achieve the maximally tolerated dose because of the development of adverse effects (Bhatt et al., 2017). Carvedilol may lead to fatigue and impair functional capacity by virtue of its blockade of the catecholamine-stimulated heart rate response during exercise. We therefore reasoned that a β-arrestin–biased PAM for the β1AR, such as Cmpd-6, as we identify in this study, could potentially enhance the cardioprotective effect of carvedilol in vivo while minimizing the hemodynamic perturbation.
To test this hypothesis, we first determined the in vivo β-blockade effect of carvedilol on catecholamine-induced cardiac hemodynamics by assessing the response to isoproterenol on heart rate and contractility in mice pretreated with increasing doses of carvedilol. We treated mice with 1, 5, or 20 mg/kg per day carvedilol with Alzet osmotic minipump for 3 days and then intravenously injected boluses of increasing doses of isoproterenol while monitoring the hemodynamics with a high-fidelity micromanometer catheter inserted retrograde into the left ventricle (Fig. 4A). Isoproterenol dose-dependently increased cardiac contractility, as indicated by an increase in dP/dt max as well as heart rate (Fig. 4, A and B). Low dose of carvedilol at 1 mg/kg per day had minimal effect on isoproterenol-induced hemodynamics, whereas higher doses of carvedilol blocked both the heart rate and contractility response (Fig. 4B). Coadministration of Cmpd-6 (5 mg/kg per day) in a ∼3:1 molar ratio to carvedilol (1 mg/kg per day) showed only a modest potentiation of the inhibitory effect on heart rate and dP/dt max (Fig. 4C). Importantly, the β-blocker effect of low-dose carvedilol (1 mg/kg per day) with Cmpd-6 is substantially smaller than that of higher doses of carvedilol alone (Fig. 4, B and C), despite the 7.9-fold enhancement on receptor binding affinity of carvedilol in the presence of Cmpd-6 as shown in Fig. 1A. The lack of a rightward shift of the dose-response curve by Cmpd-6 suggests that it has minimal regulatory effects in vivo on β1AR-mediated Gs signaling.
Fig. 4.
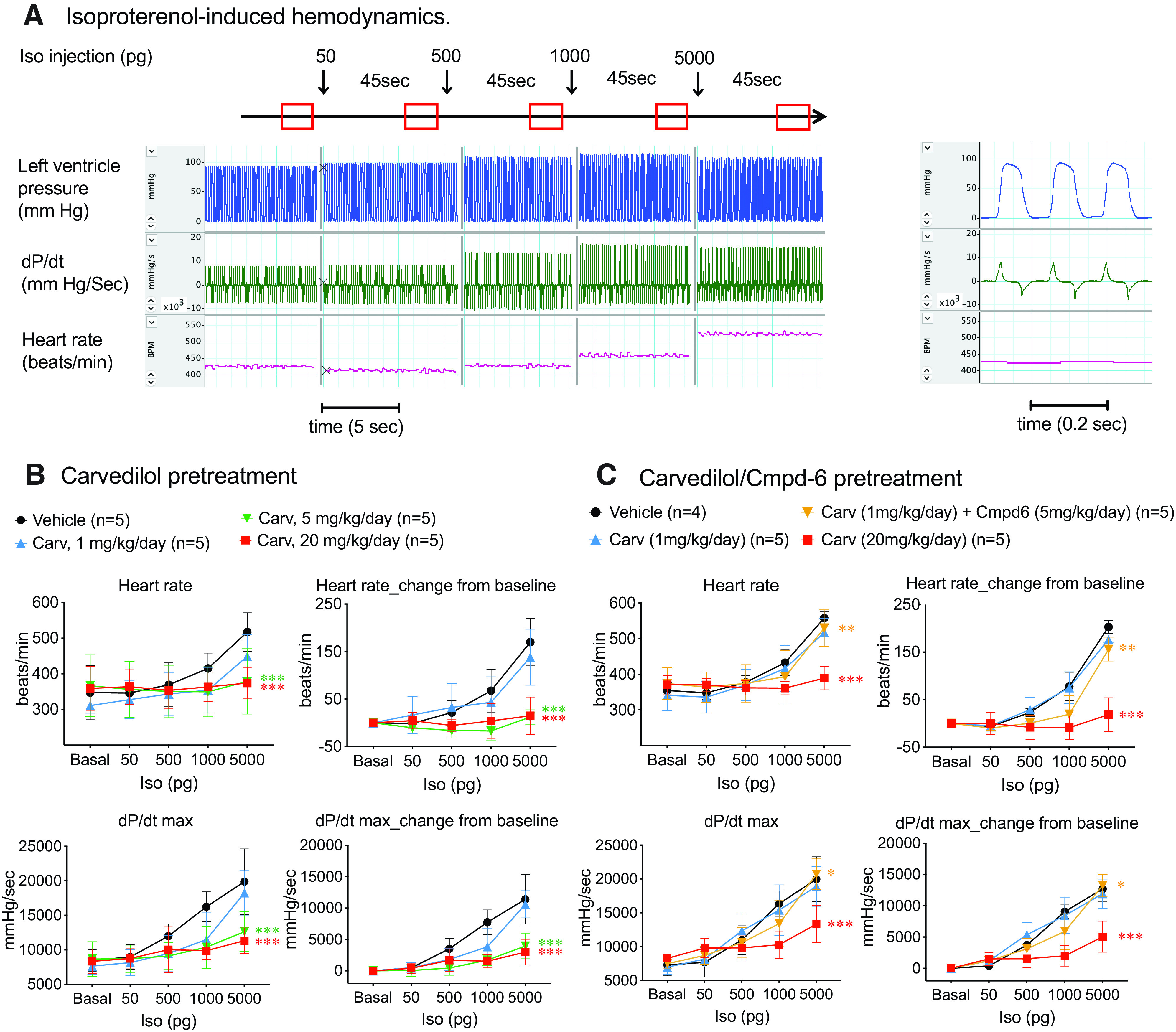
Cmpd-6 modestly enhances the inhibitory effect of carvedilol on catecholamine-induced hemodynamics in mouse hearts. (A) Representative traces of hemodynamic measurements in mouse. A high-fidelity micromanometer catheter connected with a pressure transducer was inserted retrograde into the left ventricle to monitor blood pressure. Serial doses of isoproteronol (50, 500, 1000, and 5000 pg) were injected intravenously at 45-second intervals. Left: hemodynamics were monitored continuously and recorded between 35 and 45 seconds after each injection when steady state was reached. Right: expanded view of cardiac cycles enlarged on x-axis (time). (B) Carvedilol dose-dependently blocks isopreteronol-induced increase in heart rate and contractility (indicated by dP/dt max). Wild-type C57BL/6J mice were treated with vehicle or indicated doses of carvedilol (1, 5, and 20 mg/kg per day) for 3 days with Alzet osmotic pumps before the isoproterenol-induced hemodynamic study. (C) Cmpd-6 led to a modest enhancement of the inhibitory effect of carvedilol. Mice were treated with vehicle, carvedilol (1 or 20 mg/kg per day), or a combination of carvedilol (1 mg/kg per day) and Cmpd-6 (5 mg/kg per day) for 3 days with Alzet osmotic pump. Hemodynamics were monitored and calculated using LabChart8 software. Data represent the mean ± S.D. for four or five animals as marked on the figure. Data of each drug-treated group were compared with the vehicle-treated group using two-way repeated (related)-measures ANOVA with Sidak correction for multiple comparisons. *P < 0.05, **P < 0.01, and ***P < 0.0001, interaction vs. vehicle group.
Based on the above data, we tested whether the positive cooperative effect of Cmpd-6 on low-dose carvedilol could potentiate its action on cell survival in response to ischemia/reperfusion (I/R) cardiac injury. Wild-type mice were treated with vehicle, Cmpd-6 (5 mg/kg per day), carvedilol (1 mg/kg per day), or coadministration of both compounds through Alzet osmotic pump for 3 days to reach the steady state. The left anterior descending coronary artery was occluded for 45 minutes to cause myocardial ischemia and then released for 45 minutes of tissue reperfusion (Fig. 5A). We chose the 1 mg/kg per day dose of carvedilol since this dose was sufficient to provide partial cardioprotection in response to I/R injury (Supplemental Fig. 6A), whereas it only minimally enooohanced the β-blockade effect on cardiac hemodynamics (Fig. 4B). In contrast, although higher doses of carvedilol (5 or 20 mg/kg per day) inhibited I/R-induced apoptosis (Supplemental Fig. 6), carvedilol also showed a high level of competitive antagonism (i.e., β-blocker activity) to isoproterenol-stimulated, Gs protein–mediated hemodynamics (Fig. 4B).
Fig. 5.

Cmpd-6 potentiates the β-arrestin–dependent in vivo protective effect of carvedilol against ischemia/reperfusion injury–induced myocardium apoptosis. (A) Scheme of the study. Mice were treated with carvedilol and Cmpd-6 individually or in combination for 3 days with Alzet osmotic pumps. The left anterior descending (LAD) coronary artery was ligated for 45 minutes to produce cardiac ischemia and then released to allow blood flow restoration for 45 minutes. Level of apoptosis was assessed with TUNEL staining. (B) Carvedilol diminishes the ischemia/reperfusion-induced apoptosis in hearts, and coadministration of Cmpd-6 enhances this protective effect. Wild-type C57BL/6J mice were treated with vehicle, Cmpd-6 (5 mg/kg per day), carvedilol (1 mg/kg per day), or a combination of both compounds for 3 days with Alzet osmotic pumps before the ischemia/reperfusion procedure was performed. (C) The antiapoptotic effect of carvedilol is abolished in mice with cardiomyocyte-specific deletion of β-arrestins. The β-arrestin1/2 cardiac knockout animals (αMyHC-Cre:Arrb1flox/flox/Arrb2flox/flox) were treated with vehicle or carvedilol (1 or 20 mg/kg per day) for 3 days before the ischemia/reperfusion procedure was performed. Data represent the mean ± S.D. for 5–10 animals as marked on the figure. Statistical comparisons were performed using one-way ANOVA with Tukey correction for multiple comparisons. DAPI, 4',6-diamidino-2-phenylindole.
Cmpd-6 alone has no intrinsic activity on I/R-induced apoptosis as shown by the ∼10% TUNEL-positive cells in the ischemic zone in mice pretreated with either vehicle or Cmpd-6 (Fig. 5B). Carvedilol decreased the level of I/R-induced apoptosis compared with vehicle-treated animals to ∼5% albeit with considerable variability. In marked contrast, the addition of Cmpd-6 to the same dose of carvedilol substantially and more consistently enhanced the antiapoptotic effect, as indicated by the reduction in the level of TUNEL-positive cells to ∼2% with many hearts showing very low levels of injury (Fig. 5B).
To determine whether the antiapoptotic effect of carvedilol is mediated through β-arrestins, we tested carvedilol in mice with cardiomyocyte-specific deletion of β-arrestin1/2 achieved by αMyHC promoter-driven Cre recombinase (αMyHC-Cre:Arrb1flox/flox/Arrb2flox/flox, Supplemental Fig. 6B). Pretreatment with either low- or high-dose carvedilol did not reduce I/R-induced apoptosis in the β-arrestin1/2 knockout animals (Fig. 5C), indicating that the cardioprotective action of carvedilol in vivo is mediated by signals downstream of β-arrestin.
Taken together, these data support our contention that the β-arrestin–biased β1AR PAM Cmpd-6 enhances the in vivo cardioprotective effects of carvedilol against cardiac injury–induced cell apoptosis and further supports Cmpd-6 as having a potential therapeutic benefit in enhancing the effectiveness of carvedilol under conditions of cardiac injury.
Discussion
In this study, we show Cmpd-6 is a β-arrestin–biased positive allosteric modulator for β1ARs occupied with carvedilol. Cmpd-6 was identified as a PAM for the β2AR that enhances agonist binding affinity and potentiates both the Gs protein– and β-arrestin–dependent signaling. Cmpd-6 was initially shown to cooperate with carvedilol but not other β-blockers on the β2AR (Pani et al., 2021). This prompted us to assess its properties at the β1AR. Here we identify that Cmpd-6 selectively enhances the binding affinity of the β-arrestin–biased agonist carvedilol to the β1AR, whereas it has minimal effects on the affinity of other agonists and antagonists tested. Cmpd-6 also potentiates carvedilol-induced β-arrestin–dependent β1AR signaling, including EGFR transactivation and ERK activation, whereas it has no effect on Gs protein–activated cAMP generation. In contrast to its minimal effect on the in vivo β-blockade function of carvedilol on catecholamine-induced cardiac hemodynamics, Cmpd-6 augments the cardioprotective roles of carvedilol against myocardial ischemia/reperfusion-induced apoptosis, which we demonstrate is a β-arrestin–dependent process since the antiapoptotic effect of carvedilol is abolished in mice with cardiac-specific deletion of β-arrestin1/2. The highly selective cooperativity of Cmpd-6 with carvedilol on ligand binding affinity to the β1AR, β-arrestin–biased cellular signaling, and the β-arrestin–dependent antiapoptotic role in vivo suggest that Cmpd-6 is a β-arrestin–biased PAM for the β1AR and may have therapeutic potential to enhance the clinical effects of carvedilol, which is widely used in the treatment of cardiac diseases.
β1ARs and β2ARs are the most abundant GPCRs expressed in mammalian hearts and are principal regulators of cardiac pathophysiology (Woo and Xiao, 2012). Prolonged catecholamine stimulation leads to cardiac injury (Haft, 1974) and cardiomyocyte apoptosis (Communal et al., 1999; Noma et al., 2007), and mice with cardiac-specific β1AR overexpression develop myocyte hypertrophy and cardiac dysfunction (Engelhardt et al., 1999), which indicates that excessive catecholamine activation of β1ARs is pathogenic to the heart (Yoo et al., 2009). In contrast, β-arrestin–mediated β1AR signaling appears to provide cardioprotection (Noma et al., 2007). In this study, we show that carvedilol protects hearts from ischemia/reperfusion-induced apoptosis, and Cmpd-6 enhances this antiapoptotic effect. Importantly, we demonstrated that the antiapoptotic effect of carvedilol in cardiomyocyte is at least in part mediated through β-arrestin–driven signaling pathways.
In contrast to the predominant myocyte expression and the proapoptotic effect of the β1ARs, β2ARs are primarily expressed on fibroblasts and endothelial cells (Myagmar et al., 2017) and have been shown to be cardioprotective from apoptosis (Communal et al., 1999; Devic et al., 2001; Wang et al., 2018). Based on the predominant expression of β1ARs in myocytes and the opposing effect of agonist-activated β1AR and β2AR on apoptosis, we reason that the protective effects of carvedilol and Cmpd-6 against ischemia/reperfusion-induced apoptosis are most likely to be mediated by the β1AR. However, a previous study showed that the β2AR modulator pepducin ICL1-9 reduces I/R-induced cardiomyocyte death in a β2AR- and β-arrestin–dependent manner (Grisanti et al., 2018). Therefore, it is possible that the enhanced antiapoptotic effect of carvedilol by Cmpd-6 also involves the β2AR. The precise dissection of the contribution of receptor subtypes would require testing carvedilol and Cmpd-6 in β1AR- or β2AR-knockout animals.
In regard to the mechanism of action for the in vivo cardioprotection by carvedilol, we speculate that the enhancement by Cmpd-6 on carvedilol-stimulated βAR–β-arrestin signaling may play an important role. β1AR-mediated EGFR transactivation is β-arrestin–dependent and confers cardioprotection against myocardial apoptosis induced by chronic catecholamine stimulation (Noma et al., 2007). β1AR-mediated EGFR transactivation stimulates differential subcellular activation of ERK and Akt (protein kinase B) (Grisanti et al., 2014; Tilley et al., 2009) to modulate caspase 3 activity and apoptotic gene expression (Grisanti et al., 2014). It has also been shown that β-arrestin mediates angiotensin II type 1 receptor–induced EGFR/ERK transactivation and protects mouse hearts against mechanical stress–induced apoptosis (Rakesh et al., 2010). Angiotensin II type 1 receptor stimulation with the β-arrestin–biased agonist SII activates both ERK/p90 ribosomal S6 kinase and phosphoinositide 3-kinase/AKT pathways leading to inactivation of the proapoptotic protein B-cell lymphoma 2–associated agonist of cell death to protect cells from oxidative stress–induced apoptosis (Ahn et al., 2009). Furthermore, the antiapoptotic effects of Cmpd-6 and carvedilol may also be mediated by regulation of microRNA (miR) processing. Carvedilol-stimulated β1AR-activated β-arrestin1 promotes the processing of a subset of miRs in the mouse heart (Kim et al., 2014), among which miR-125b-5p reduced expression of proapoptotic genes BCL2 antagonist/killer 1 and Kruppel-like factor 13 in cardiomyocytes to protect the mouse heart from ischemic injury (Bayoumi et al., 2018). Finally, the β-arrestin–biased β2AR modulator pepducin ICL1-9 protects against I/R-induced cardiac injury and cardiomyocyte death through activating the Ras homolog family member A/rhodopsin-associated protein kinase pathway and reducing mitochondrial oxidative stress (Grisanti et al., 2018).
Based on the central role of βARs in the heart, β-blockers are first-line agents for the treatment of cardiac diseases, such as heart failure (Yancy et al., 2017). However, different β-blockers have variable therapeutic effectiveness in treating heart failure (Bhatt et al., 2017). Advances in understanding the complexities of GPCR biology and detailed dissections of pharmacological actions of β-blockers may assist in understanding their differential efficacy. In this study, we show that the β-blockers alprenolol, carvedilol, bucindolol, and carazolol differentially engage Gs and β-arrestin to activate downstream signaling. We postulate that the unique properties of carvedilol in stimulating β-arrestin–dependent β1AR signaling may contribute to its potential clinical superiority as suggested by meta-analyses showing that carvedilol has the lowest all-cause mortality among different β-blockers tested in heart failure (Chatterjee et al., 2013; DiNicolantonio et al., 2013).
Although β-blockers are considered standard of therapy in heart failure, the level of patient unresponsiveness to β-blocker treatment and intolerable adverse effects (Bhatt et al., 2017) support a quest to develop novel βAR ligands with improved drug efficacy and safety. Recent developments in identifying allosteric modulators for GPCRs may offer great potential as novel therapeutics since they have the potential for greater receptor subtype specificity, fewer off-target side effects, and better drug safety profiles than orthosteric ligands (Lane et al., 2017; Wisler et al., 2018).
Cmpd-6 was identified by a DNA-encoded small-molecule library affinity screen for agonist-bound β2ARs (Ahn et al., 2018). Cmpd-6 potentiates both Gs-mediated cAMP generation and β-arrestin recruitment to β2ARs with comparative efficacy in HEK293 cells (Ahn et al., 2018), indicating its unbiased PAM activity on β2ARs. Cmpd-6 also cooperates with the β-arrestin–biased agonist carvedilol but not other β-blockers on β2ARs (Pani et al., 2021). In contrast to its ∼50-fold enhancement of isoproterenol binding affinity for β2ARs, Cmpd-6 enhances isoproterenol binding affinity for β1ARs by only 2-fold as shown in this study as well as previous ones (Ahn et al., 2018; Liu et al., 2019). The selectivity of Cmpd-6 on agonist binding affinity for β2ARs over β1ARs might be because of the structural differences between the two receptor subtypes in the allosteric binding pocket for Cmpd-6. Mutational studies introducing key β2AR residues interacting with Cmpd-6 into the corresponding β1AR sites created gain-of-function mutant β1ARs, to which Cmpd-6 salvaged its PAM behavior for agonist binding (Liu et al., 2019). Interestingly, in contrast to its marginal effect on agonist binding to β1ARs, Cmpd-6 enhances the affinity of the antagonist carvedilol to β1ARs by ∼8-fold, leading to a potentiation of β-arrestin signaling. We speculate that this unique property of Cmpd-6 on carvedilol and not any other antagonist tested is likely based on structural determinants whereby Cmpd-6 binding to carvedilol-occupied β1ARs or β2ARs stabilizes the receptor to adopt a “β-arrestin” conformation, thereby engaging β-arrestin and not Gs as its preferred signaling transducer.
Our data demonstrate that Cmpd-6 potentiates the β-arrestin–dependent antiapoptotic effects of carvedilol in vivo, whereas it has minimal effect on blocking the physiologic response to catecholamines. Major side effects of carvedilol treatment in patients with heart failure are exertional intolerance, dizziness, and advanced heart block (Krum et al., 1995) due to sympathetic activity attenuation. Accordingly, allosteric modulators, such as Cpmd-6, could provide important therapeutic advantages by enhancing the cardioprotective action of carvedilol but minimizing the adverse effects often found with high-dose carvedilol. In the future, determining the effects of Cmpd-6 in chronic left ventricle dysfunction will be of great interest to further evaluate the therapeutic potential of this compound. Additionally, the structure-activity relationship study of Cmpd-6 may lead to identification of new compounds with desirable receptor subtype selectivity or signaling modulation effects. For instance, a chemically modified analog of Cmpd-6 selectively cooperates with carvedilol on the β2AR while showing no PAM activity on agonists (Pani et al., 2021), which is similar to the effects of Cmpd-6 on the β1AR. Based on the differential functional consequences of β1AR and β2AR activation in the heart (Wang et al., 2018), further development of Cmpd-6 analogs with high receptor selectivity and selective carvedilol cooperativity may provide additional therapeutic benefits.
In conclusion, we show that Cmpd-6 is a β-arrestin–biased PAM for β1ARs. It selectively increases the affinity of the β-arrestin–biased agonist carvedilol for β1ARs and potentiates β-arrestin–mediated signaling stimulated with carvedilol. Importantly, Cmpd-6 enhances the β-arrestin–dependent in vivo effects of carvedilol to protect hearts from ischemia/reperfusion injury–induced apoptosis. Our data suggest that the development of β-arrestin–biased βAR allosteric modulators may provide a new direction for improving current treatments or developing novel agents for cardiac diseases.
Acknowledgments
The authors thank the Duke Cardiovascular Research Center Small Animal Physiology Core for performing the in vivo mouse experiments and Weili Zou for her expert technical assistance.
Abbreviations
- AR
β adrenergic receptor
- AR
β1 adrenergic receptor
- AR
β2 adrenergic receptor
- BRET
bioluminescence resonance energy transfer
- CHS
cholesterol hemisuccinate
- Cmpd-6
Compound-6
- CYP
cyanopindolol
- DDM
N-dodecyl-β-D-maltoside
- EGF
epidermal growth factor
- EGFR
EGF receptor
- ERK
extracellular signal-regulated kinase
- GPCR
G protein–coupled receptor
- HDL
high-density lipoprotein
- HEK
human embryonic kidney
- I/R
ischemia/reperfusion
- miR
microRNA
- MyHC
α-myosin heavy chain
- PAM
positive allosteric modulator
- pERK
phosphorylated ERK
- rGFP
recombinant Green Fluorescent Protein; RlucII, Renilla luciferase variant 2
- TUNEL
terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling
Authorship Contributions
Participated in research design: Wang, Pani, Gokhan, Xiong, Kahsai, Jiang, Rockman.
Conducted experiments: Wang, Pani, Gokhan, Xiong, Kahsai, Jiang.
Contributed new reagents or analytic tools: Wang, Pani, Gokhan, Xiong, Kahsai, Jiang.
Performed data analysis: Wang, Pani, Gokhan, Xiong, Kahsai, Jiang.
Wrote or contributed to the writing of the manuscript: Wang, Pani, Gokhan, Xiong, Kahsai, Jiang, Ahn, Lefkowitz, Rockman.
Footnotes
This work was supported by National Institutes of Health National Heart, Lung, and Blood Institute [Grants 5R01-HL056687 and 5P01-HL075443] (to H.A.R.) and [Grant 5R01-HL016037] (to R.J.L.). R.J.L. is an investigator of the Howard Hughes Medical Institute.
No author has any actual or perceived conflicts of interest with the contents of this article.
Drs. Lefkowitz and Rockman are scientific cofounders of Trevena, Inc.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Ahn S, Kim J, Hara MR, Ren XR, Lefkowitz RJ (2009) beta-Arrestin-2 mediates anti-apoptotic signaling through regulation of BAD phosphorylation. J Biol Chem 284:8855–8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn SPani BKahsai AWOlsen EKHusemoen GVestergaard MJin LZhao SWingler LMRambarat PK, et al. (2018) Small-molecule positive allosteric modulators of the β2-adrenoceptor isolated from DNA-encoded libraries. Mol Pharmacol 94:850–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayoumi AS, Park KM, Wang Y, Teoh JP, Aonuma T, Tang Y, Su H, Weintraub NL, Kim IM (2018) A carvedilol-responsive microRNA, miR-125b-5p protects the heart from acute myocardial infarction by repressing pro-apoptotic bak1 and klf13 in cardiomyocytes. J Mol Cell Cardiol 114:72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benovic JL, DeBlasi A, Stone WC, Caron MG, Lefkowitz RJ (1989) Beta-adrenergic receptor kinase: primary structure delineates a multigene family. Science 246: 235–240. [DOI] [PubMed] [Google Scholar]
- Bhatt AS, DeVore AD, DeWald TA, Swedberg K, Mentz RJ (2017) Achieving a maximally tolerated β-blocker dose in heart failure patients: is there room for improvement? J Am Coll Cardiol 69:2542–2550. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Biondi-Zoccai G, Abbate A, D’Ascenzo F, Castagno D, Van Tassell B, Mukherjee D, Lichstein E (2013) Benefits of β blockers in patients with heart failure and reduced ejection fraction: network meta-analysis. BMJ 346:f55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland JG, Freemantle N, Eastaugh J, Young PJ, Harrison J (2000) Beta-blockers for heart failure. Cochrane Database Syst Rev (2):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Communal C, Singh K, Sawyer DB, Colucci WS (1999) Opposing effects of beta(1)- and beta(2)-adrenergic receptors on cardiac myocyte apoptosis: role of a pertussis toxin-sensitive G protein. Circulation 100:2210–2212. [DOI] [PubMed] [Google Scholar]
- Devic E, Xiang Y, Gould D, Kobilka B (2001) Beta-adrenergic receptor subtype-specific signaling in cardiac myocytes from beta(1) and beta(2) adrenoceptor knockout mice. Mol Pharmacol 60:577–583. [PubMed] [Google Scholar]
- DiNicolantonio JJ, Lavie CJ, Fares H, Menezes AR, O’Keefe JH (2013) Meta-analysis of carvedilol versus beta 1 selective beta-blockers (atenolol, bisoprolol, metoprolol, and nebivolol). Am J Cardiol 111:765–769. [DOI] [PubMed] [Google Scholar]
- Engelhardt S, Hein L, Wiesmann F, Lohse MJ (1999) Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc Natl Acad Sci USA 96:7059–7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisanti LA, Talarico JA, Carter RL, Yu JE, Repas AA, Radcliffe SW, Tang HA, Makarewich CA, Houser SR, Tilley DG (2014) β-Adrenergic receptor-mediated transactivation of epidermal growth factor receptor decreases cardiomyocyte apoptosis through differential subcellular activation of ERK1/2 and Akt. J Mol Cell Cardiol 72:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisanti LA, Thomas TP, Carter RL, de Lucia C, Gao E, Koch WJ, Benovic JL, Tilley DG (2018) Pepducin-mediated cardioprotection via β-arrestin-biased β2-adrenergic receptor-specific signaling. Theranostics 8:4664–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft JI (1974) Cardiovascular injury induced by sympathetic catecholamines. Prog Cardiovasc Dis 17:73–86. [DOI] [PubMed] [Google Scholar]
- Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, Gloriam DE (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov 16:829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IM, Tilley DG, Chen J, Salazar NC, Whalen EJ, Violin JD, Rockman HA (2008) Beta-blockers alprenolol and carvedilol stimulate beta-arrestin-mediated EGFR transactivation. Proc Natl Acad Sci USA 105:14555–14560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IMWang YPark KMTang YTeoh JPVinson JTraynham CJPironti GMao LSu H, et al. (2014) β-arrestin1-biased β1-adrenergic receptor signaling regulates microRNA processing. Circ Res 114:833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krum HSackner-Bernstein JDGoldsmith RLKukin MLSchwartz BPenn JMedina NYushak MHorn EKatz SD, et al. (1995) Double-blind, placebo-controlled study of the long-term efficacy of carvedilol in patients with severe chronic heart failure. Circulation 92:1499–1506. [DOI] [PubMed] [Google Scholar]
- Lane JR, May LT, Parton RG, Sexton PM, Christopoulos A (2017) A kinetic view of GPCR allostery and biased agonism. Nat Chem Biol 13:929–937. [DOI] [PubMed] [Google Scholar]
- Liu XMasoudi AKahsai AWHuang LYPani BStaus DPShim PJHirata KSimhal RKSchwalb AM, et al. (2019) Mechanism of β2AR regulation by an intracellular positive allosteric modulator. Science 364:1283–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ (1990) beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science 248:1547–1550. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Wang J, Plouffe B, Smith JS, Yamani L, Kaur S, Jean-Charles PY, Gauthier C, Lee MH, Pani B, et al. (2018) Manifold roles of β-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci Signal 11:eaat7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MERIT-HF-Study-Group (1999) Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 353:2001–2007. [PubMed] [Google Scholar]
- Myagmar BEFlynn JMCowley PMSwigart PMMontgomery MDThai KNair DGupta RDeng DXHosoda C, et al. (2017) Adrenergic receptors in individual ventricular myocytes: the beta-1 and alpha-1B are in all cells, the alpha-1A is in a subpopulation, and the beta-2 and beta-3 are mostly absent. Circ Res 120:1103–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung Y, Le Gouill C, Lukashova V, Kobayashi H, Hogue M, Khoury E, Song M, Bouvier M, Laporte SA (2016) Monitoring G protein-coupled receptor and β-arrestin trafficking in live cells using enhanced bystander BRET. Nat Commun 7:12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma TLemaire ANaga Prasad SVBarki-Harrington LTilley DGChen JLe Corvoisier PViolin JDWei HLefkowitz RJ, et al. (2007) Beta-arrestin-mediated beta1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Invest 117:2445–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM, Shusterman NH; U.S. Carvedilol Heart Failure Study Group (1996) The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. N Engl J Med 334:1349–1355. [DOI] [PubMed] [Google Scholar]
- Packer MCoats AJFowler MBKatus HAKrum HMohacsi PRouleau JLTendera MCastaigne ARoecker EB, et al. ; Carvedilol Prospective Randomized Cumulative Survival Study Group (2001) Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med 344:1651–1658. [DOI] [PubMed] [Google Scholar]
- Pani B, Ahn S, Rambarat PK, Vege S, Kahsai AW, Liu A, Valan BN, Staus DP, Costa T, Lefkowitz RJ(2021) Unique positive cooperativity between the β-arrestin-biased β-blocker carvedilol and a small molecule positive allosteric modulator of the β2-adrenergic receptor. Mol Pharmacol 100:513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S, Rajagopal K, Lefkowitz RJ (2010) Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov 9:373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA (2010) beta-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal 3:ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman HA, Koch WJ, Lefkowitz RJ (2002) Seven-transmembrane-spanning receptors and heart function. Nature 415:206–212. [DOI] [PubMed] [Google Scholar]
- Staus DP, Wingler LM, Choi M, Pani B, Manglik A, Kruse AC, Lefkowitz RJ (2018) Sortase ligation enables homogeneous GPCR phosphorylation to reveal diversity in β-arrestin coupling. Proc Natl Acad Sci USA 115:3834–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DM, Glukhova A, Sexton PM, Christopoulos A (2018) Structural insights into G-protein-coupled receptor allostery. Nature 559:45–53. [DOI] [PubMed] [Google Scholar]
- Tilley DG, Kim IM, Patel PA, Violin JD, Rockman HA (2009) beta-Arrestin mediates beta1-adrenergic receptor-epidermal growth factor receptor interaction and downstream signaling. J Biol Chem 284:20375–20386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Gareri C, Rockman HA (2018) G-protein-coupled receptors in heart disease. Circ Res 123:716–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Hanada K, Staus DP, Makara MA, Dahal GR, Chen Q, Ahles A, Engelhardt S, Rockman HA (2017) Gαi is required for carvedilol-induced β1 adrenergic receptor β-arrestin biased signaling. Nat Commun 8:1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, Sunahara RK (2007) A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci USA 104:7682–7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, Shenoy SK, Lefkowitz RJ (2007) A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci USA 104:16657–16662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisler JW, Rockman HA, Lefkowitz RJ (2018) Biased G protein-coupled receptor signaling: changing the paradigm of drug discovery. Circulation 137:2315–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo AY, Xiao RP (2012) β-Adrenergic receptor subtype signaling in heart: from bench to bedside. Acta Pharmacol Sin 33:335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancy CWJessup MBozkurt BButler JCasey DE JrColvin MMDrazner MHFilippatos GSFonarow GCGivertz MM, et al. (2017) 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines and the Heart Failure Society of America. J Card Fail 23:628–651. [DOI] [PubMed] [Google Scholar]
- Yoo B, Lemaire A, Mangmool S, Wolf MJ, Curcio A, Mao L, Rockman HA (2009) Beta1-adrenergic receptors stimulate cardiac contractility and CaMKII activation in vivo and enhance cardiac dysfunction following myocardial infarction. Am J Physiol Heart Circ Physiol 297:H1377–H1386. [DOI] [PMC free article] [PubMed] [Google Scholar]