

Abstract
G protein–coupled receptor 30 (GPR30) is a membrane receptor reported to bind 17β-estradiol (E2) and mediate rapid nongenomic estrogen responses, hence also named G protein–coupled estrogen receptor. G-1 is a proposed GPR30-specific agonist that has been used to implicate the receptor in several pathophysiological events. However, controversy surrounds the role of GPR30 in G-1 and E2 responses. We investigated GPR30 activity in the absence and presence of G-1 and E2 in several eukaryotic systems ex vivo and in vitro in the absence and presence of the receptor. Ex vivo activity was addressed using the caudal artery from wild-type (WT) and GPR30 knockout (KO) mice, and in vitro activity was addressed using a HeLa cell line stably expressing a synthetic multifunctional promoter (nuclear factor κB, signal transducer and activator of transcription, activator protein 1)–luciferase construct (HFF11 cells) and a human GPR30-inducible T-REx system (T-REx HFF11 cells), HFF11 and human embryonic kidney 293 cells transiently expressing WT GPR30 and GPR30 lacking the C-terminal PDZ (postsynaptic density-95/discs-large /zonula occludens-1 homology) motif SSAV, and yeast Saccharomyces cerevisiae transformed to express GPR30. WT and KO arteries exhibited similar contractile responses to 60 mM KCl and 0.3 μM cirazoline, and G-1 relaxed both arteries with the same potency and efficacy. Furthermore, expression of GPR30 did not introduce any responses to 1 μM G-1 and 0.1 μM E2 in vitro. On the other hand, receptor expression caused considerable ligand-independent activity in vitro, which was receptor PDZ motif-dependent in mammalian cells. We conclude from these results that GPR30 exhibits ligand-independent activity in vitro but no G-1– or E2-stimulated activity in any of the systems used.
SIGNIFICANCE STATEMENT
Much controversy surrounds 17β-estradiol (E2) and G-1 as G protein–coupled receptor 30 (GPR30) agonists. We used several recombinant eukaryotic systems ex vivo and in vitro with and without GPR30 expression to address the role of this receptor in responses to these proposed agonists. Our results show that GPR30 exhibits considerable ligand-independent activity in vitro but no G-1– or E2-stimulated activity in any of the systems used. Thus, classifying GPR30 as an estrogen receptor and G-1 as a specific GPR30 agonist is unfounded.
Introduction
In 2005, two groups reported that 17β-estradiol (E2) binds to and stimulates cAMP production and extracellular-regulated protein kinase (ERK) 1/2 activity through G protein–coupled receptor 30 (GPR30) in recombinant cells ectopically expressing the receptor (Thomas et al., 2005; Revankar et al., 2005), which led the International Union of Pharmacology to rename the receptor G protein–coupled estrogen receptor (GPER) (Alexander et al., 2011). Subsequently, G-1 [(±)-1-[(3aR*,4S*,9bS*)-4-(6-bromo-1,3-benzodioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-ethenone], a substance structurally related to E2, was identified using similar systems and classified as a specific GPR30 agonist (Bologa et al., 2006). Since then, G-1 has been used extensively as a standard of GPR30 agonism to implicate the receptor in a number of pathophysiological systems (Prossnitz and Barton, 2014; Prossnitz and Arterburn, 2015). However, inconsistent results have emerged regarding G-1 and E2 as GPR30 agonists (Levin, 2009; Olde and Leeb-Lundberg, 2009; Langer et al., 2010; Romano and Gorelick, 2018). Indeed, several independent studies done to confirm the original observations in classic recombinant G protein–coupled receptor (GPCR) systems have failed to do so (Pedram et al., 2006; Otto et al., 2008; Kang et al., 2010; Southern et al., 2013; Broselid et al., 2014; Sousa et al., 2017; Gonzalez de Valdivia et al., 2017). Some G-1 and E2 responses are apparently sensitive to knockdown of native GPR30 expression (Prossnitz and Barton, 2014; Prossnitz and Arterburn, 2015), suggesting that these responses depend on receptor expression. However, whether these responses are consequences of a direct interaction with GPR30 or mediated by a distinct target(s) that depends on receptor expression is far from clear. Crosstalk between GPR30 and various ERα isoforms has been reported (Romano and Gorelick, 2018).
Despite conflicting observations, current GPR30 research continues to rely heavily on G-1 responses. The use of pharmacological agents to study receptors requires utmost confidence in receptor specificity. The study of ligand-independent constitutive receptor activity is less common but avoids nonspecific pharmacological effects. All GPCR exhibit ligand-independent activity owing to their nature of existing in an equilibrium between inactive and activated states (Rosenbaum et al., 2009). Assay of such activity often requires well defined recombinant systems where cells expressing the receptor can be compared with those devoid of receptor in the absence of any ligand stimulus. Although this activity is often too low to be detected with many GPCRs, being primarily in the inactive state in the absence of agonist, a number of receptors exhibit considerable constitutive activity, which can be pathophysiologically relevant (Seifert and Wenzel-Seifert, 2002). Evidence for ligand-independent GPR30 activity has been presented in both recombinant (Broselid et al., 2014; Gonzalez de Valdivia et al., 2017) and native systems (Ahola et al., 2002; Ariazi et al., 2010; Broselid et al., 2013; Broselid et al., 2014; Weißenborn et al., 2014).
Considering that G-1 and E2 continue to be used as GPR30 agonists, we felt compelled to further address the involvement of human GPR30 in responses to these agents. Here, we used a number of novel receptor assay systems to monitor several cellular signals in the absence and presence of G-1 and E2, with and without GPR30 expression in several recombinant eukaryotic systems, both ex vivo in transgenic mouse arteries and in vitro in several cell systems including yeast. We report that GPR30 does not show any activity in response G-1 or E2 in any of the investigated systems but exhibits ligand-independent constitutive GPR30 activity in all the in vitro systems used.
Materials and Methods
Mammalian Cell Culturing for Transient Transfection
Human embryonic kidney 293 (HEK293) cells (American Type Culture Collection, Manassas, VA), HeLa cells (American Type Culture Collection), and HFF11 cells were grown in phenol red–free Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS in 5% CO2 at 37°C. HFF11 cells were generated from HeLa cells as previously described (Kotarsky et al., 2003).
cDNA Constructs and Transient Transfection
N-terminally FLAG-tagged human cDNAs of wild-type (WT) GPR30 and GPR30 lacking the C-terminal PSD-95/discs-large/zonula occludens-1 homology (PDZ) motif SSAV (GPR30ΔSSAV) in the pcDNA3.1 plasmid were made as previously described (Broselid et al., 2014). The Rac1Cluc sensor was constructed by replacing the ERK1/2 sensor region of the split click beetle luciferase-based ERK1/2 sensor mitogen-activated protein kinase activity reporter (MAPKAR ) plasmid (Gonzalez-Garcia et al., 2014) with the Rac1 sensor region of Rac1 (EV) from RaichuEV-Rac1 (Komatsu et al., 2011). Briefly, the Rac1 sensor region was amplified using the primers TGGCGAATTCGAGAAAGAGAAAGAGC and TTTAGACTCGAGGCGGACTGCTCGGATC, introducing EcoRI and XhoI sites in the process. The product was then ligated into the EcoRI and XhoI sites of the MAPKAR plasmid. To improve membrane attachment, a CAAX motif was subsequently introduced at the C terminus of the split click beetle luciferase. The MAPKAR plasmid was a kind gift from Dr. Karl Swann (University of Cardiff, UK), the RaichuEV-Rac1(Raichu-2248X) Förster resonance energy transfer plasmid was a kind gift from Dr. Michiyuki Matsuda (Kyoto University, Japan), pGL3–nuclear factor of activated T cells (NFAT) luciferase was a gift from Jerry Crabtree (Addgene plasmid # 17870; http://n2t.net/addgene:17870; Research Resource Identifier: Addgene_17870), and postsynaptic density-95 (PSD-95) FLAG was a gift from Wei-dong Yao (Addgene plasmid # 15463; http://n2t.net/addgene:15463; Research Resource Identifier: Addgene_15463). TransIT-LT1 (Mirus Bio LLC, Madison, WI) was used to transfect plasmid DNA. Cells transiently transfected with plasmid containing receptor constructs were always compared with cells transfected with empty plasmid alone (mock).
Construction and Culturing of Stable T-REx HFF11 Cells
HFF11 cells were transfected with the pcDNA6/TR plasmid (Invitrogen, Carlsbad, CA) using TransIT-LT1 to create an HFF11 cell line expressing the tetracycline (TET) repressor, and stable clones were selected based on blasticidin resistance. FLAG-tagged GPR30 was ligated into the HindIII/XbaI sites of the pcDNA 4T/O plasmid (Invitrogen), the resulting plasmid transfected into HFF11 cells expressing the TET repressor to create T-REx HFF11 cells, and stable clones selected based on blasticidin and zeocin resistance. The cells were then grown in phenol red–free DMEM supplemented with 10% normal or charcoal-treated FBS (growth medium) in 5% CO2 at 37°C.
T-REx HFF11 Promoter-Reporter Assay
T-REx HFF11 and HFF11 cells seeded in white-bottom 96-well plates (20,000 cells/well) in growth medium were incubated without and with TET (Sigma-Aldrich, St. Louis, MO) for 12 or 24 hours. Drug, vehicle (DMSO), or medium was added during the last 12 hours of TET treatment. The promoter-reporter construct was assayed as previously described (Kotarsky et al., 2003). In short, cells were lysed with 20 μl/well reporter lysis buffer (Promega, Madison, WI). After addition of 35 μl/well Luciferin reagent (Biothema, Handen, Sweden) and ATP, promoter-reporter activity was measured as luminescence in a Clariostar luminometer.
ERK Activity
ERK1/2 activity was assayed by immunoblotting as previously described (Gonzalez de Valdivia et al., 2017) using phosphorylated ERK1/2 (pERK) antibody (Santa Cruz Biotechnology, Santa Cruz, CA; 1:1000) for ERK1/2 phosphorylation and ERK1/2 (ERK) antibody (Santa Cruz Biotechnology; 1:1000) for total ERK1/2. Briefly, cells were grown to confluency in 60-mm dishes in phenol red–free DMEM with 10% FBS, washed, incubated without serum for 1 hour, and then incubated without or with vehicle (DMSO) or drug for different times. The cells were then washed, lysed, and subjected to immunoblotting, and immunoreactive bands were visualized as described below. The combined band densities of ERK1 and ERK2 were quantified using ImageJ software, and ERK1/2 activity was expressed as the ratio between the combined pERK band densities and the combined ERK band densities for each condition.
NFAT Activity
NFAT promoter activity was measured in cells transfected with pGL3-NFAT luciferase plasmid. Transfected cells in white-bottom 96-well plates (20,000 cells/well) were grown in phenol red–free DMEM with 10% FBS overnight and then incubated with vehicle (DMSO) or drug for 12 hours. Cells were then lysed with 20 μl/well reporter lysis buffer (Promega, Madison, WI). After addition of 35 μl/well Luciferin reagent (Biothema, Handen, Sweden) and ATP, NFAT promoter activity was measured as luminescence in a Clariostar luminometer.
Rac1 Activity
Rac1 activity was measured in cells transfected with Rac1Cluc sensor plasmid. Transfected cells were grown in six-well plates for 12 hours and then transferred to white-bottom 96-well plates (20,000 cells/well) and grown in phenol red–free DMEM with 10% FBS for an additional 12 hours. The cells were then incubated with 90 μl/well HEPES-buffered DMEM containing 3% (w/v) Na-luciferin (Promega) for 2–3 hours in the dark at room temperature. Luminescence was then measured with drug for various times in a Clariostar luminometer. The Rac1Cluc sensor was activated by 1 μM bradykinin in HEK293 cells transfected with the B2 bradykinin receptor (Supplemental Fig. 1), as previously described (Wojciak-Stothard and Ridley, 2002).
Flow Cytometry
Cells were incubated with primary mouse M1 FLAG antibody (1:200) or mouse IgG (DAKO, Glostrup, Denmark) for 20 minutes with or without 0.1% saponin/PBS (Sigma-Aldrich) at room temperature, to detect intracellular and cell surface receptors, respectively. Cells were then washed with PBS with Ca2+/Mg2+ and resuspended in PBS with phycoerythrin-labeled goat anti-mouse antibody (DAKO; 1:2000) or Alexa488-labeled donkey anti-goat antibody (ThermoFisher Scientific; 1:1000) as secondary antibody, with or without 0.1% saponin/PBS for 20 minutes at room temperature in the dark. The cells were then washed with PBS, centrifuged at 2000g for 5 minutes and the pellet resuspended in PBS and directly analyzed by flow cytometry. The specificity of the secondary antibody was tested by omitting the primary antibody. The cells were analyzed using a BD FACSCanto Cytometer and FACSDiva Software (Beckton Dickinson Immunocytometry Systems, San Jose, CA). Forward and side scatter measurements were attained with gain settings in linear mode. In all experiments, binding was calculated after subtracting background fluorescence of the control antibody.
Construction and Transformation of CY2797 and EY-1 Yeast Cells
The CY2797 yeast strain was a kind gift from Dr. James Broach (CADUS Corp.) (Manfredi et al., 1996). The EY-1 strain was made from the CY2797 strain by replacing the four C-terminal amino acid residues (KIGII) of with the corresponding residues (DCGLF) of human Gαi2. The targeting was done using the pUG6/pSH47 system, kindly donated by Dr. Johannes Hegemann (University of Düsseldorf, Germany), essentially as described (Güldener et al., 1996). Briefly, the targeting construct was made by polymerase chain reaction (PCR ) using pUG6, containing a loxP-flanked neomycin resistance gene, and the specific primers GPA1 -(AGTGCAGTCACCGATCTAATCATCCAGCAAAACCTTAAAGATTGTGGTCTATTCTGAGCCAGCTGAAGCTTCGTACG) and GPA1 - (ATTTACGTATCTAAACACTACTTTAATTATACAGTTCCTCATAGGCCACTAGTGGATCTG). The product was purified on agarose electrophoresis, electroporated into CY2797 and then selected on G418/ plates. G418-resistant colonies were analyzed for correct construct insertion by colony PCR. The neomycin resistance cassette was removed by loxP recombinase expression using the pSH47 vector. G418-sensitive colonies were selected by replica plating, and correct removal was confirmed using colony PCR. WT GPR30 was cloned into the p426GPD plasmid (Mumberg et al., 1995), containing a URA3 selection marker, and PSD-95 into the p415GPD plasmid, containing a LEU2 selection marker. CY2797 and EY-1 strains were transformed by electroporation with each plasmid individually or together.
Yeast Culturing
Yeast strains were grown and maintained on synthetic complete (SC) agar plates supplemented with 200 mg/l histidine and tryptophan. SC medium was prepared by mixing 1.7 g yeast nitrogen base without amino acids and ammonium sulfate (Sigma-Aldrich), 5 g ammonium sulfate (Prolabo, Paris, France), 1.3 g yeast synthetic dropout medium without histidine, leucine, tryptophan, and uracil (Sigma-Aldrich), 200 mg l-tryptophan (Sigma-Aldrich), and 200 mg l-histidine (Sigma-Aldrich) in 900 ml water. When SC agar plates were prepared, 20 g American bacteriological agar (Pronadisa-Hispanlab, Madrid, Spain) was added. The medium was then autoclaved for 20 minutes at 121°C. After autoclaving, the medium was allowed to cool down to ∼45–50°C before 100 ml sterile-filtered 20% (w/v) d-(+)-glucose (BDH AnalaR, Poole, UK or Sigma-Aldrich) was added.
Precultures were prepared in 250 ml E-flasks (VWR, Randor, PA) the day before the main experiment by inoculating 25 ml SC medium with colonies from SC agar plates, and the cultures were then incubated overnight at 30°C under constant agitation (230 rpm). The following day, precultures were transferred to 50 ml disposable polypropylene tubes (Sarstedt, Nümbrecht, Germany) and harvested by centrifugation (5 minutes at 800g). Supernatants were discarded, and cell pellets were washed twice in 20 ml SC medium before final resuspension in 25 ml. Depending on the application, washed precultures were used to inoculate working cultures in SC medium at different initial values of optical density at 600 nm (OD600 ). All absorbance measurements were performed with the Eppendorf-BioPhotometer (Eppendorf, Hamburg, Germany).
Assay of Yeast Growth
Working cultures were prepared in 50 ml disposable polypropylene tubes by inoculating 8 ml SC medium supplemented with 0.2 mM 3-amino-1,2,4-triazol with CY2797 and EY-1 precultures to an initial OD600 of ∼0.05. Cultures were incubated at 230 rpm for 48 hours at 30°C without or with vehicle (DMSO) or drug, and OD600 measurements were made at 24, 30, and 48 hours.
Cell Lysis
HEK293 cells and HeLa cells were lysed as previously described (Sandén et al., 2011), and yeast cells lysed essentially as described previously (Hoffman et al., 2002). In short, yeast cells corresponding to an OD600=10.0 were centrifuged at 2000g for 10 minutes at 4°C. The supernatant was discarded and the cell pellet resuspended in 1 ml ice-cold H2O, transferred to a 1.5 ml microcentrifuge tube, and centrifuged for 1 minute at 16,000g at room temperature. The supernatant was discarded and the cell pellet resuspended in 200 μl 1× NuPAGE LDS sample buffer (Invitrogen) supplemented with 1× cOmplete protease inhibitor (Sigma-Aldrich). The tube was then incubated at 100°C for 10 minutes, cooled at room temperature for 7 minutes, and supplemented with 200 μl glass beads (Ø=425–600 μm; Sigma-Aldrich). The tube was vortexed for 2 minutes at maximum speed and then inverted two or three times for 1 minute. The glass beads were separated from the cell lysate by introducing a hole at the bottom of each tube. The perforated tube was placed in a fresh tube and centrifuged shortly, separating the cell lysate from the glass beads. The tube was then centrifuged for 2 minutes at 16,000g, and the supernatant was transferred to a fresh 1.5 ml microcentrifuge tube and stored at −20°C.
Immunoprecipitation and Immunoblotting
Immunoprecipitation and immunoblotting were done as described previously (Sandén et al., 2011). Staining was done with goat GPR30 antibody (R&D Systems, Minneapolis, MN; 1:200), mouse FLAG M2 antibody (Sigma-Aldrich; 1:1000), or pan–membrane-associated guanylate kinase antibody (Merck Millipore, Billerica, MA; 1:2000). Immunoreactive bands were visualized with a chemiluminescence immunodetection kit using peroxidase-labeled secondary antibody (Invitrogen) according to the procedure described by the supplier (PerkinElmer Life and Analytical Sciences, Waltham, MA).
Animals
GPR30 knockout (KO) mice were generated as previously described (Mårtensson et al., 2009) and backcrossed 14 generations onto the C57BL/6 background. WT C57BL/6 and GPR30 KO mice were housed in a standard animal facility under controlled temperature (22°C) and photoperiod (12 hours of light, 12 hours of dark), and the mice were fed a standard phytoestrogen-free pellet diet ad libitum. Animal care was in accordance with institutional guidelines. All animal experiments had been approved by the local Ethical Committees for Animal Research (M-416).
Wire Myography
Three-month-old female GPR30 KO mice and age-matched female WT C57BL/6 mice were euthanized using CO2. The tail was marked on the abdominal side, cut off at its base, and then placed in cold HEPES-buffered Krebs solution (135.5 mM NaCl, 11.6 mM HEPES, 11.5 mM glucose, 5.9 mM KCl, 1.2 mM MgCl2, pH 7.4). The caudal artery was exposed by a midline incision (4 cm) through the skin and covering fascia. Two-millimeter segments (1.8–2.1 mm) were cut and mounted in wire myographs (610 M and 620 M, Danish Myotechnology A/S, Aarhus, Denmark) as described (Rippe et al., 2017). All segments were stretched to 5 mN under relaxed conditions. After equilibration for 25 minutes in HEPES-buffered Krebs solution containing 2.5 mM Ca2+, viability was ascertained by depolarization using 60 mM KCl (obtained by exchange of NaCl for KCl). α1-Adrenergic receptors were activated using cirazoline (0.3 µM). G-1 or forskolin was added at increasing concentrations using vehicle (DMSO) as control. At the end of the experiment, 1 μM carbachol was added to cause endothelial- and nitric oxide–dependent dilatation. For the forskolin experiment, the exact length of each preparation was recorded at the end of experiments using a dissection microscope with an ocular scale. This length was then used for normalization of some of the force data. Having established that no differences existed between WT and KO arteries, we instead normalized force to the precontraction level (100%).
Statistical Analysis
In figures, data are presented as means ± S.D. except in Fig. 1I, where data are presented as means with whiskers of min-max. In the Results section, effects are presented as fold change ± 95% confidence interval. Data distribution was assessed using Shapiro-Wilk test for normality. To evaluate statistical significance, Student’s two-tailed t test for unpaired or paired data was used for single comparisons of parametrical data, and the Whitney-Mann U test was used for single comparisons of nonparametric data. For multiple comparisons of parametrical data, one-way ANOVA with Kruskal-Wallis test for multiple comparisons, or two-way ANOVA with Tukey’s test of multiple comparisons was used. To test for trends in data, one-way ANOVA with test for trend was used. P values less than 0.05 were regarded as statistically significant. Data analysis was performed using the Prism program version 9.1.0 (GraphPad).
Fig. 1.

Contractility and forskolin- and G-1–driven relaxation in caudal arteries from WT and GPR30 KO mice. Caudal artery tubes were prepared by microdissection and mounted in wire myograph chambers to measure active force development (at a passive force of 5 mN). The length of the arterial tube was measured at the end of the experiment and used for normalization of the force integral over the stimulation periods shown. (A, B) Force in response to depolarization (60 mM K+, n = 17–18 preparations from a minimum of six mice of each genotype) and stimulation with the α1-adrenergic agonist cirazoline (0.3 μM), respectively, statistically analyzed using Mann-Whitney U test. (C) Concentration-dependent relaxation by forskolin in WT and KO arteries (n = 17–18 preparations) after precontraction with cirazoline. Here and in the following panels, force was normalized to the precontraction, but results were the same using absolute force. (D) Original force records from WT arteries stimulated with 0.3 μM cirazoline followed by cumulative addition of G-1 (red trace) or vehicle (DMSO) (blue trace). The muscarinic agonist carbachol (Cch) was given at the end to ascertain endothelium-dependent dilatation. (E) Summarized data for the effect of different concentrations of G-1 in WT (n = 6 preparations receiving G-1, and n = 6 preparations receiving vehicle). (F, G) Effect of G-1 in KO arteries run in parallel with the WT arteries in (D) and (E) [n = 6 preparations receiving G-1, and n = 6 preparations receiving vehicle; six WT and six KO mice were used altogether for the experiments in (D)–(G)]. The data in (E) and (G) were statistically analyzed by paired t test. (H) Concentration-response curves for G-1 in WT and KO arteries are plotted alongside each other. Vehicle (DMSO) controls are pooled for clarity. G-1 relaxation was not different in WT compared with KO arteries but was greater than seen in vehicle-treated arteries in both cases. (I) Area under the curve (AUC) of the responses shown in (H), statistically tested using one-way ANOVA with Tukey’s multiple comparisons test. The results are either representative [(D) and (F)] or means ± S.D. of n = 17–18 measurements from a minimum of 6 mice of each genotype [(A)–(C), (E), (G), (H)], or means with whiskers of min-max (I). *P < 0.05; **P < 0.01; n.s., not significant.
Results
GPR30 Activity in Mouse Caudal Artery Ex Vivo
GPR30 is expressed in mouse peripheral arteries (Isensee et al., 2009), and several studies have used G-1 to conclude that GPR30 mediates relaxation of such arteries from a number of different species (Haas et al., 2009; Broughton et al., 2010, Meyer et al., 2010, Lindsey et al., 2011; Jang et al., 2013; Lindsey et al., 2014; Arefin et al., 2014; Debortoli et al., 2017; Peixoto et al., 2017; Yu et al., 2018). To address the involvement of GPR30 in this response, we used the caudal artery from our GPR30 KO mouse strain (Mårtensson et al., 2009) and compared it with age-matched C57BL/6 WT mice. Fig. 1 shows that 60 mM KCl (Fig. 1A) and 0.1 μM of the α1-adrenergic receptor agonist cirazoline (Fig. 1B) each constricted WT and KO arteries to the same maximal degree. Furthermore, no difference was observed in the dose-response curves of forskolin-promoted relaxation between cirazoline-preconstricted WT and KO arteries (Fig. 1C). G-1 relaxed cirazoline-preconstricted WT arteries dose-dependently and to a maximal degree very similar to that previously reported by other investigators (Fig. 1D and E). An essentially overlapping G-1 dose-response curve was observed with KO arteries (Fig. 1F, G, and H), and the maximal response was not different between the WT and KO arteries (Fig. 1I). We conclude from these results that GPR30 does not influence the contractility of the mouse caudal artery to any statistically significant degree, and that the receptor does not contribute to G-1–promoted relaxation of the artery.
Human WT GPR30 Activity in Mammalian Cells In Vitro
Next, we investigated human GPR30 activity in well defined mammalian recombinant systems in vitro. To capture a broad repertoire of receptor signals, we used HFF11 cells (Kotarsky et al., 2003), a HeLa cell line stably expressing a luciferase-based promoter-reporter construct containing a synthetic multifunctional promoter with several different response motifs [nuclear factor κB (NF -κB), signal transducer and activator of transcription (STAT), and activator protein 1 (AP-1)] upstream from the luciferase gene. In these cells, we also stably expressed a T-REx system, in which the expression of N-terminally FLAG-tagged human WT GPR30 (GPR30) is dependent on TET treatment (T-REx HFF11 cells). Treatment with 1000 ng/ml TET for 12 hours resulted in the appearance of at least three immunoreactive species, at about 35–40 kDa, 70 kDa and 130 kDa, as determined with a polyclonal goat antibody against the human GPR30 N-terminal domain (Fig. 2A). The same species were identified with a monoclonal M2 FLAG antibody, which specifically reacts with the GPR30 FLAG epitope, confirming that the GPR30 antibody recognizes the expressed receptor (Fig. 2A). As expected, removal of TET led to the disappearance of the species (Fig. 2B). Flow cytometry showed that 22% of the induced receptors were localized in the plasma membrane (PM) (Fig. 2C).
Fig. 2.

Expression and ligand-independent activity of human GPR30 in T-REx HFF11 cells. (A, B) T-REx HFF11 cells were treated without (−TET) or with 1000 ng/ml TET (+TET) for 12 hours (A), washed free of TET and incubated in medium for different lengths of time as indicated (B), and then immunoblotted with GPR30 or M2 FLAG antibody. (C) T-REx HFF11 cells were treated without (−TET) or with 1000 ng/ml TET (+TET) for 12 hours, stained live (Cell surface) or after permeabilization (Total) with M1 FLAG , and then subjected to flow cytometry. The data were plotted as GPR30-positive cells as % of total cells. (D) T-REx HFF11 cells were treated without or with increasing concentrations of TET for 12 hours as indicated and then immunoblotted with GPR30 , pERK , and ERK . The combined pERK band intensities were normalized to the combined ERK band intensities for each condition and the ratio graphed and statistically analyzed using a repeated measures one-way ANOVA with test for trend. The intensity of the GPR30 band at approximately 70-kDa was graphed as arbitrary units (AU). The results are either representative or the means ± S.D. of at least three independent experiments. In (A), (B), and (D), molecular mass standards (left side) and receptor species (right side) are indicated.
Treating T-REx HFF11 cells with increasing doses of TET (0–1000 ng/ml) for 12 hours, to dose-dependently increase GPR30 expression, yielded a trend of an increase in basal ERK1/2 activity (trend: slope=0.14 ± 0.076, P = 0.0014) with increased receptor expression (Fig. 2D), as previously reported in transiently transfected HEK293 cells (Gonzalez de Valdivia et al., 2017). To assay ligand-independent constitutive GPR30 activity using the luciferase-based promoter-reporter, luciferase activity was monitored after treatment of cells with 1000 ng/ml TET for 12 and 24 hours and compared with that without TET treatment (Fig. 3A). The reason for the 24-hour time point is that in contrast to receptor stimulation of ERK1/2 activity, which is rapid, the downstream stimulation of luciferase activity is slow, requiring gene transcription to occur. No change in basal reporter activity was observed after 12 hours of TET treatment, whereas a statistically significant increase was observed after 24 hours of treatment both as determined in absolute numbers (Fig. 3B; fold change for vehicle-treated cells: 1.5 ± 0.3) and as a fraction of the response to 0.1 μM phorbol myristate acetate (PMA), a potent stimulator of the promoter-reporter through protein kinase C and used here as a positive control (Fig. 3B and C; fold change for vehicle-treated cells: 2.3 ± 0.4). On the other hand, no increase in reporter activity was observed after TET treatment of HFF11 cells lacking the T-REx system (data not shown). The GPR30-promoted increase in basal reporter activity in T-REx HFF11 cells was similar to that observed in response to 1 mM ATP through native purinergic receptors (fold change 2.7 ± 0.15) (Fig. 3D). Neither 0.1 μM E2 nor 1 μM G-1 had any clear effect on the reporter activity either at low (1 ng/ml TET) or high (1000 ng/ml TET) levels of receptor expression regardless of whether the cells had been grown in normal or charcoal-treated FBS (to remove FBS-derived estrogens) (Fig. 3E). Thus, human GPR30 exhibits ligand-independent effects on reporter activity, whereas no effect is observed after treatment with either 0.1 μM E2 or 1 μM G-1.
Fig. 3.
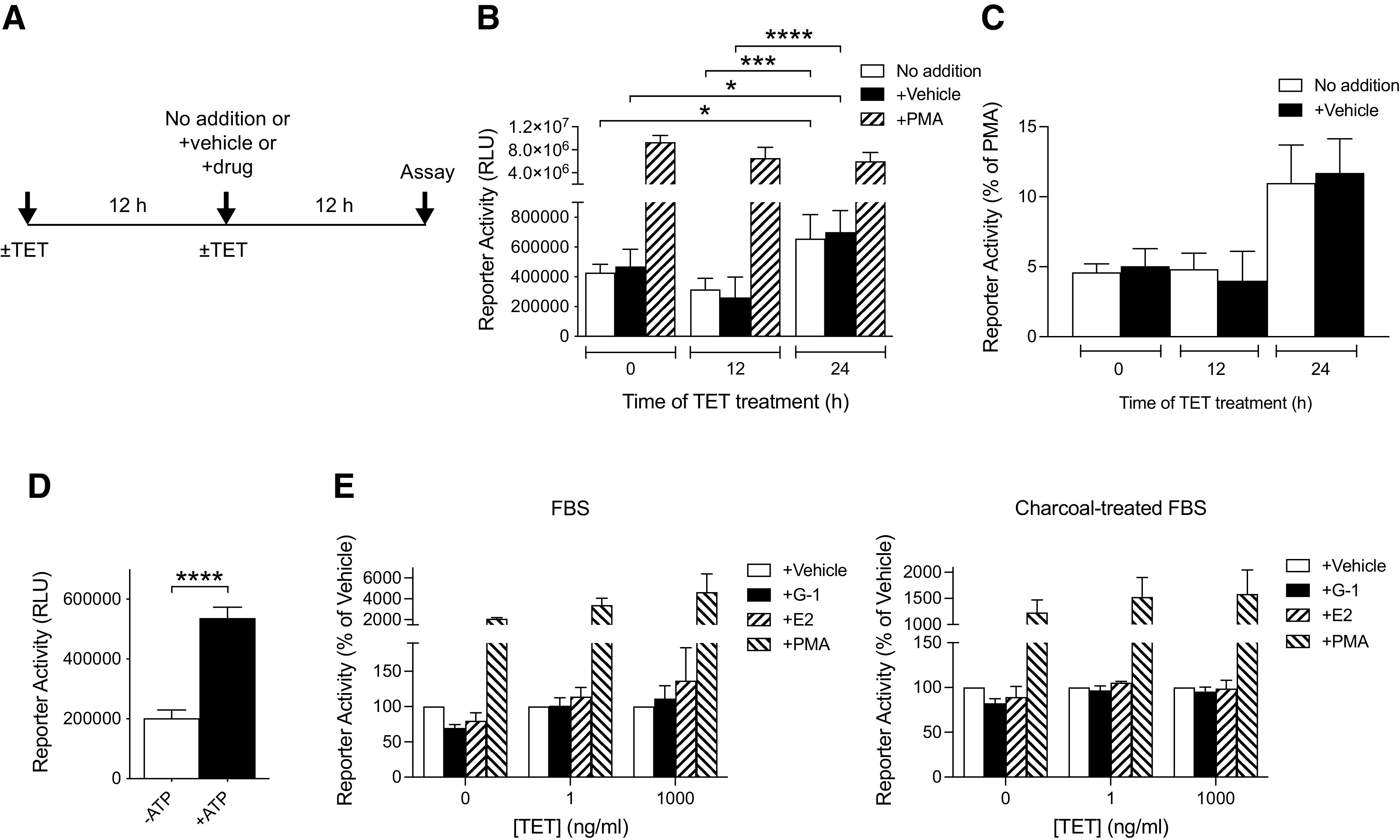
Ligand-dependent and -independent activities of human GPR30 in T-REx HFF11 cells. (A) The treatment protocol of T-REx HFF11 cells for assaying GPR30 activity in (B), (C), and (E) is described. (B, C) T-REx HFF11 cells were treated without or with 1000 ng/ml TET for 12 or 24 hours. During the treatment without TET (−TET) or with TET (+TET), the cells were also treated without (no addition) or with vehicle or 0.1 μM PMA for 12 hours prior to assay for luminescence. The results are expressed as relative light units (RLU) and analyzed statistically using two-way ANOVA with Tukey’s multiple comparisons test (B), or as % of PMA values (C). (D) T-REx HFF11 cells were treated without (-ATP) and with 1 mM ATP (+ATP) for 12 hours prior to assay for luminescence. The results were expressed as RLU and analyzed statistically using an unpaired t test. (E) T-REx HFF11 cells grown in normal FBS (left graph) or in charcoal-treated FBS (right graph) for at least 2 days were treated without or with different concentrations of TET for 24 hours as indicated. During the treatment without or with TET, the cells were also treated with vehicle, 1 μM G-1 (G-1), 0.1 μM E2 (E2), or 0.1 μM PMA for 12 hours prior to assay for luminescence. The results are graphed as % of vehicle. The data are presented as means ± S.D. of at least three independent experiments. *P < 0.05; ***P < 0.001; ****P < 0.0001.
Activity of Human GPR30 With and Without the PSD-95/Discs-Large/Zonula Occludens-1 Homology Motif in Mammalian Cells In Vitro
Next, we evaluated human GPR30 activity in HEK293 cells and HFF11 cells transiently transfected with WT GPR30 or GPR30ΔSSAV, which has increased receptor endocytosis and attenuated ligand-independent receptor activities due to disrupted receptor interaction with the membrane scaffold proteins synapse-associated protein 97 (SAP97) and PSD-95 (Akama et al., 2013; Broselid et al., 2014; Tran et al., 2015; Gonzalez de Valdivia et al., 2017). Both constructs were expressed to approximately the same degree in HEK293 cells (Fig. 4A). Neither 0.1 μM E2 nor 1 μM G-1 had any statistically significant effect on ERK1/2 activity was similar in mock-, GPR30-, or GPR30ΔSSAV-transfected HEK293 cells (Fig. 4B). To address if GPR30 mediates any effect of G-1 or E2 on intracellular free Ca2+, HEK293 cells were transfected with the luciferase promoter-reporter plasmid pGL3-NFAT, which monitors NFAT, a specific transcription factor target for calcineurin, thus showing high sensitivity to Ca2+ (Clipstone and Crabtree, 1992). Although 0.1 μM E2 had a small stimulatory effect on NFAT activity in mock-transfected cells, neither E2 nor 1 μM G-1 had any statistically significant effect on NFAT activity in either GPR30- or GPR30ΔSSAV-transfected cells (Fig. 4C). On the other hand, as previously reported (Gonzalez de Valdivia et al., 2017), GPR30 expression drastically inhibited basal NFAT activity in a PDZ motif–dependent manner, with a fold change compared with mock of 0.29 ± 0.046 for WT GPR30, and 1.1 ± 0.176 for GPR30ΔSSAV (Fig. 4C). To address if GPR30 mediates any effect on Rac1 activity, a downstream effector of phosphoinositide 3-kinase, we constructed a split click beetle luciferase-based Rac1 sensor plasmid (Rac1Cluc). Again, neither 0.1 μM E2 nor 1 μM G-1 had any statistically significant effect on Rac1 activity in either mock-, GPR30-, or GPR30ΔSSAV-transfected cells (Fig. 4E). GPR30 expression inhibited basal Rac1 activity in a PDZ-dependent manner, with fold changes compared with mock-transfected cells of 0.04 ± 0.0049 for WT GPR30, and 1.2 ± 0.81 for GPR30ΔSSAV (Fig. 4D). We also addressed if GPR30 mediates any G-1 or E2 effect on the multifunctional promoter-reporter when GPR30 and GPR30ΔSSAV were transiently transfected in HFF11 cells (Fig. 4F). G-1 (1 μM) stimulated the reporter in mock-transfected cells (fold change of vehicle of 1.5 ± 0.08), whereas 0.1 μM E2 did not (fold change of vehicle of 1.0 ± 0.06). On the other hand, neither GPR30 nor GPR30ΔSSAV expression amplified the response to either G-1 (fold change of vehicle at plasmid concentration 1 µg/1 × 106 cells of 1.4 ± 0.10 for GPR30 and 1.3 ± 0.15 for GPR30ΔSSAV) or E2 (fold change of vehicle at plasmid concentration 1 µg/1 × 106 cells of 1.0 ± 0.05 for GPR30 and 1.1 ± 0.10 for GPR30ΔSSAV (Fig. 4G).
Fig. 4.
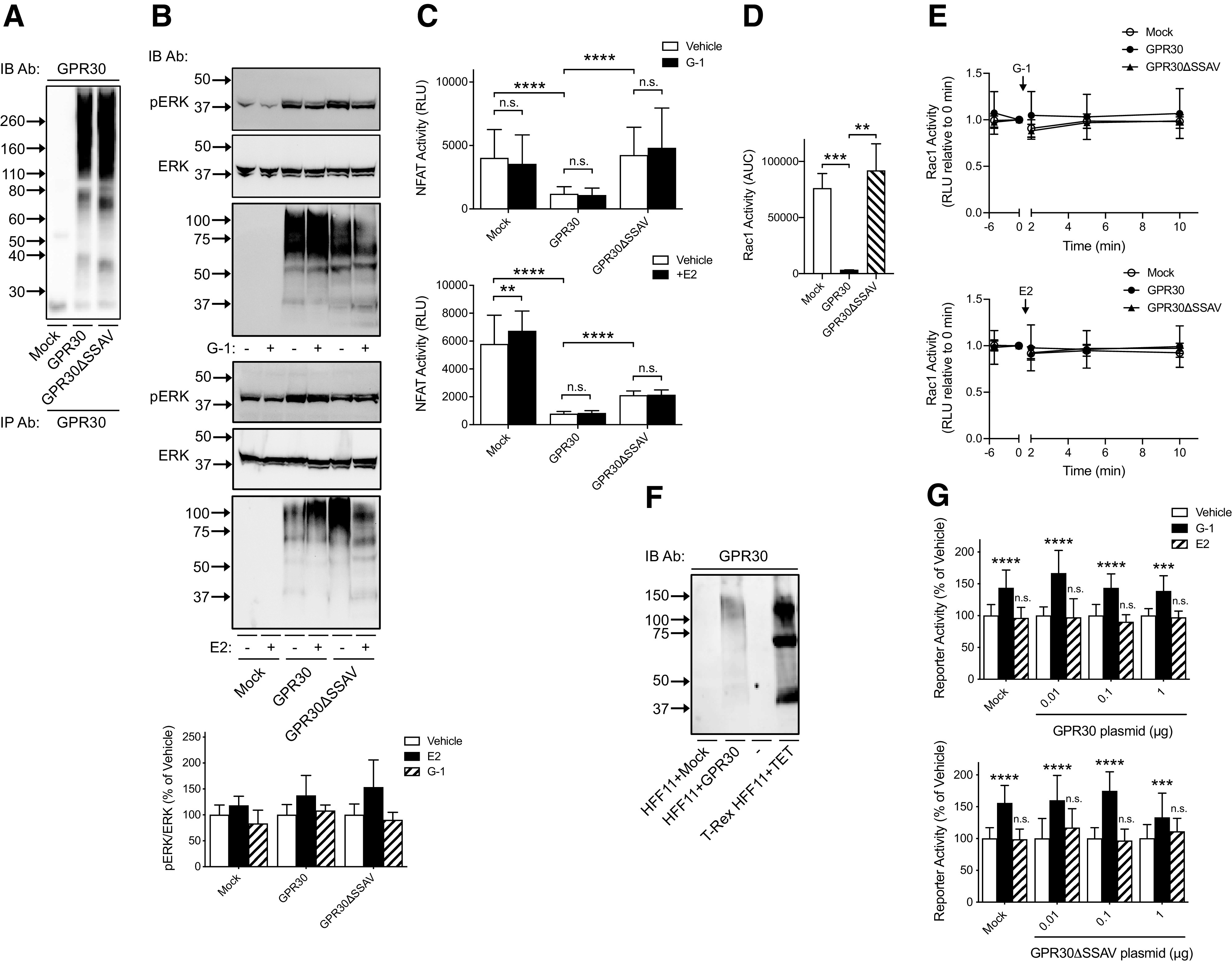
Ligand -dependent and -independent activities of human GPR30 and GPR30ΔSSAV in HEK293 cells and HFF11 cells. (A) HEK293 cells transiently transfected with empty pcDNA3 plasmid (Mock) or plasmid containing GPR30 or GPR30ΔSSAV were immunoprecipitated with GPR30 and then immunoblotted with GPR30 Ab. (B) HEK293 cells transiently transfected with empty pcDNA3 plasmid (Mock) or plasmid containing GPR30 or GPR30ΔSSAV were treated with vehicle (−) or 1 μM G-1 or 0.1 μM E2 (+) for 5 minutes and then immunoblotted with pERK Ab, ERK Ab, or GPR30 Ab. The combined pERK band intensities were normalized to the combined ERK band intensities for each condition and graphed as % of vehicle. (C) HEK293 cells transiently transfected with pGL3-NFAT luciferase plasmid together with either empty pcDNA3 plasmid (Mock) or plasmid containing GPR30 or GPR30ΔSSAV were treated with vehicle, 1 μM G-1, or 0.1 μM E2. NFAT activity was then assayed as luminescence and the results graphed as relative light units (RLU). The data were evaluated statistically using a two-way ANOVA with Tukey’s test for multiple comparisons. (D, E) HEK293 cells transiently transfected with Rac1Cluc plasmid together with either empty pcDNA3 plasmid (Mock) or plasmid containing GPR30 or GPR30ΔSSAV were treated with 1 μM G-1 or 0.1 μM E2 for different times. Rac1 activity was then assayed for luminescence as RLU. In (D), the RLU area under the curve (AUC) in the absence of stimulus was analyzed using an unpaired Student’s t test, and in (E), RLU at different times after addition of stimulus was graphed normalized to time=0. (F) Cells transfected with empty pcDNA3 plasmid (Mock) or 1 μg GPR30 plasmid, or T-REx HFF11 cells treated with 1000 ng/ml TET for 12 hours were with GPR30 Ab. (G) HFF11 cells transiently transfected with either empty pcDNA3 plasmid (Mock) or different amounts of plasmid containing GPR30 or GPR30ΔSSAV were treated with vehicle, 1 μM G-1, or 0.1 μM E2 for 12 hours and luminescence was measured as RLU. Data were analyzed statistically using one-way ANOVA with Kruskal-Wallis test for multiple comparisons. Data are either representative (A), (F) or presented as means ± S.D. of at least three independent experiments. In (A), (B), and (F), molecular mass standards are indicated (left side). **P < 0.01; ***P < 0.001; ****P < 0.0001; n.s., not significant.
Human WT GPR30 Activity in Yeast
We also addressed GPR30 activity in the yeast Saccharomyces cerevisiae, a system previously used to study GPCR activity and pharmacology by monitoring G protein–mediated coupling to the pheromone response pathway (Liu et al., 2016). To this end, we used the S. cerevisiae strain CY2797, in which the endogenous α-factor GPCR gene STE2 is disrupted and expresses the native yeast G protein Gpa1. Furthermore, the strain is auxotrophic for histidine unless activated by the pheromone pathway, and activation of the pathway leads to increased growth (Manfredi et al., 1996). From CY2797, we made EY-1, in which we expressed a chimeric Gα protein where the five C-terminal residues of Gpa1 (KIGII) were replaced with those of human Gαi2 (DCGLF).
GPR30 was readily expressed in both CY2797 and EY-1, migrating as a single species at about 35–40 kDa in both strains as determined using the GPR30 antibody (Fig. 5A). GPR30 expression led to a clear increase in the basal growth of both strains, with a fold change at 48 hours compared with 0 hours of 2.3 ± 0.25 and 2.0 ± 0.32 for GPR30-expressing CY2797 and EY-1, respectively, and a fold change of 1.7 ± 0.38 and 1.5 ± 0.34 for CY2797 and EY-1, respectively, without GPR30 expression (Fig. 5B and C). On the other hand, incubation with 1 μM G-1 for 30 hours had no effect on CY2797 growth either with or without GPR30 expression (Fig. 5D). Thus, GPR30 constitutively couples to the pheromone response pathway but does not respond to G-1 in this system.
Fig. 5.
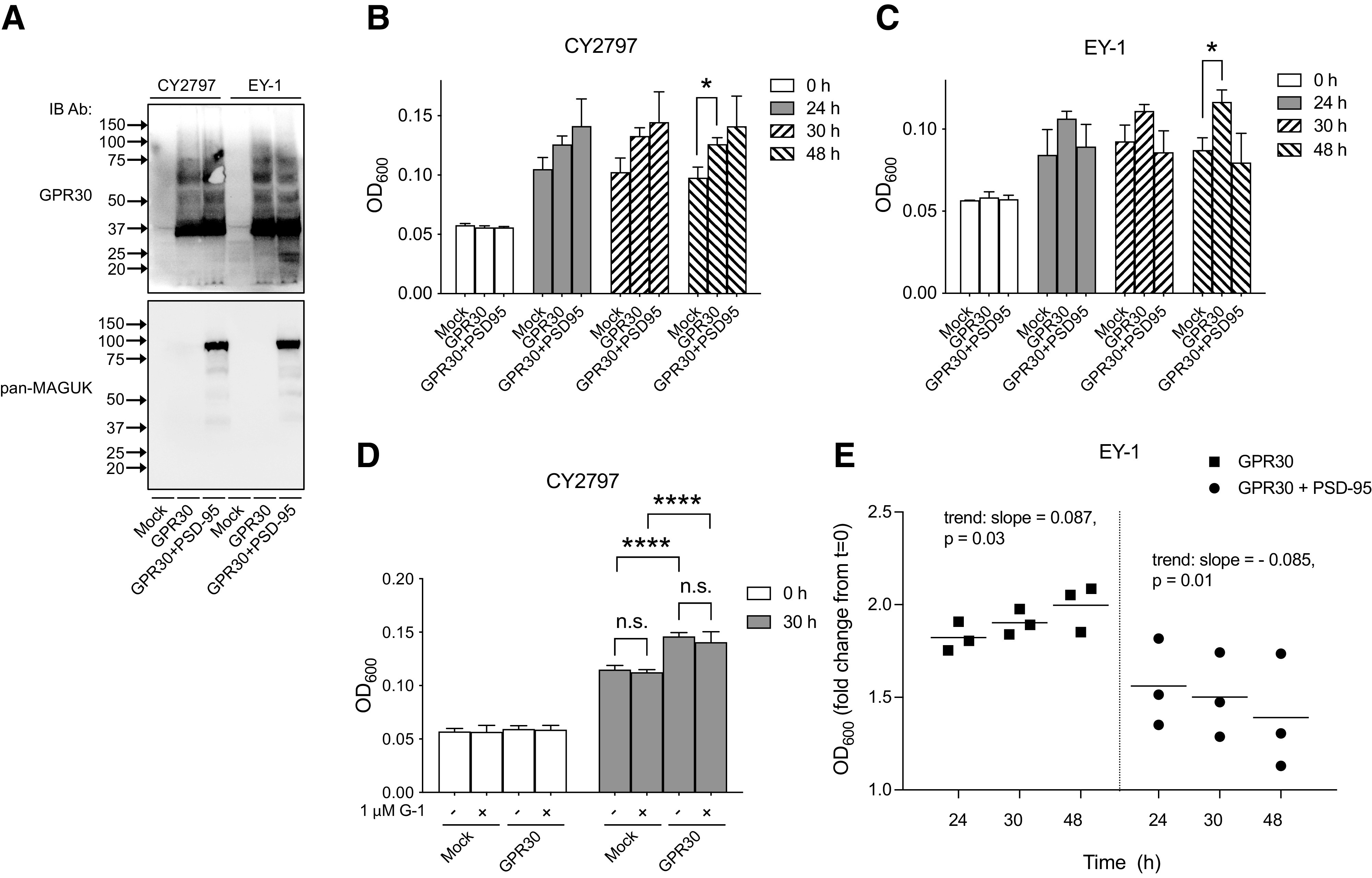
Expression and ligand-independent activity human GPR30 activity with and without PSD-95 in yeast S. cerevisiae. (A) CY2797 and EY-1 yeast strains transformed with empty plasmid (Mock), plasmid containing GPR30, or plasmids containing GPR30 and PSD-95 were immunoblotted with GPR30 and pan–membrane-associated guanylate kinase (MAGUK) Ab. (B, C) Cell growth was followed for various times and graphed as OD600. Two-way ANOVA with Tukey’s multiple comparisons test was used to statistically analyze the relationship between all groups. (D) Cells were treated with vehicle (−) or 1 μM G-1 (+), and OD600 was determined and graphed for time 0 hours and 30 hours of treatment. Statistical analysis was performed using two-way ANOVA with Tukey’s multiple comparisons test. (E) Data from (C) were normalized to the OD600 at t=0 and analyzed using repeated measures one-way ANOVA with test for trend to assess time-dependent linear relationships for each condition. The results are either representative (A) or the means ± S.D. (C)–(E) of at least three independent experiments. In (A), molecular mass standards are indicated (left side). *P < 0.05; ****P < 0.0001; n.s., not significant.
We also evaluated the effect of PSD-95, a GPR30 PDZ domain partner (Broselid et al., 2014), on the ligand-independent GPR30 activity. Figure 5A shows that human FLAG-tagged PSD-95 (Zhang et al., 2007) was readily expressed in both CY2797 and EY-1 cells. PSD-95 expression had no effect on the GPR30 response in CY2797 cells (Fig. 5B). On the other hand, PSD-95 inhibited the GPR30 response when the chimeric Gpa1/Gαi2 protein was expressed in EY-1 cells, with fold change in growth at 48 hours of 1.4 ± 0.77 in yeast expressing both GPR30 and PSD-95, as compared with 1.5 ± 0.34 in mock, and 2.0 ± 0.32 when GPR30 was expressed without PSD-95 (Fig. 5C). Indeed, although there was a positive linear relationship between time and growth in EY-1 expressing only GPR30 (trend: slope = 0.087 ± 0.07, P = 0.03), growth of EY-1 expressing both GPR30 and PSD-95 had a negative linear relationship with time (trend: slope = −0.085 ± 0.056, P = 0.01) (Fig. 5E).
Discussion
Here, we used several recombinant eukaryotic systems with and without GPR30 expression to rigorously address the ability of G-1 and E2 to influence GPR30 activity and the ligand-independent GPR30 activity ex vivo and in vitro. No receptor-dependent activity was observed in response to 1 μM G-1 ex vivo and in vitro or 0.1 μM E2 in vitro. On the other hand, ligand-independent receptor activity was observed in all the in vitro mammalian cell systems and in yeast. Thus, classifying GPR30 as an estrogen receptor sensitive to G-1 and E2 is unfounded.
Transgenic KO mouse models with and without receptor expression offer an excellent opportunity to address in a physiologic setting if the effect of a pharmacological agent is receptor-dependent. GPR30 is expressed in several peripheral arteries in mouse (Isensee et al., 2009), and numerous studies have used G-1 to conclude that GPR30 mediates relaxation of WT arteries from a number of species (Haas et al., 2009; Broughton et al., 2010, Meyer et al., 2010, Lindsey et al., 2011; Jang et al., 2013; Lindsey et al., 2014; Arefin et al., 2014; Debortoli et al., 2017; Peixoto et al., 2017; Yu et al., 2018). Considering that this conclusion is based only on one GPR30 KO mouse strain (Haas et al., 2009), and that phenotypic variations exist between different GPR30-deficient strains (Olde and Leeb-Lundberg, 2009; Langer et al., 2010), we were compelled to readdress the specificity of this response in a unique mouse KO strain (Mårtensson et al., 2009). G-1 relaxed the mouse caudal artery with a potency and to a maximal degree virtually identical to that previously reported by other investigators using other peripheral arteries. An essentially overlapping G-1 dose-response curve was obtained with the KO mouse caudal artery. Thus, GPR30 apparently does not contribute to G-1–promoted vasorelaxation, at least not of the mouse caudal artery.
At the cellular level, G-1 was previously reported to cause an antiproliferative effect through GPR30 in human vascular smooth muscle cells (Haas et al., 2009). We subsequently showed that G-1 is antiproliferative in mouse aortic endothelial and smooth muscle cells, which correlates with microtubule disruption (Holm et al., 2012). This response was identical in cells isolated from WT and GPR30 KO mice, again showing in a physiologically relevant system that G-1 elicits effects independently of native GPR30.
Native systems are complex, rendering them difficult to use to more specifically determine if an agonist response is a consequence of a direct interaction with a receptor or mediated by a distinct target(s) that depends on receptor expression. To attempt to reduce the complexity of the system, we used T-REx HFF11 cells, which were designed to capture a multitude of receptor signals, including mitogen-activated protein kinase activity and Ca2+, through a synthetic multifunctional promoter (NF-κB, STAT, AP-1) at different receptor levels. Receptor species were identified at about 35–40 kDa, about 70 kDa, and about 130 kDa, consistent with that observed by a number of investigators in several native systems (Maiti et al., 2011; Jala et al., 2012; Akama et al., 2013; Scaling et al., 2014; Waters et al., 2015). The species at 35–40 kDa, the theoretical mass of the receptor, most likely represents an immature, unglycosylated form of the receptor. On the other hand, the 70 kDa species represents a mature plasma membrane–localized receptor form (Cheng et al., 2011; Gonzalez de Valdivia et al., 2019), a form that has been reported to be functionally active (Filardo et al., 2007; Broselid et al., 2014; Gonzalez de Valdivia et al., 2017), capable of coupling to G proteins (Filardo et al., 2007; Gonzalez de Valdivia et al., 2017), and undergo receptor-mediated endocytosis (Cheng et al., 2011; Sandén et al., 2011). At least 22% of the expressed receptor resided at the plasma membrane in the T-REx HFF11 cells. Nevertheless, no GPR30-dependent reporter activity was observed in response to either G-1 or E2 at any receptor level in these cells, or in HFF11 cells transiently expressing the receptor, regardless of whether the cells had been grown in normal or charcoal-treated FBS, to remove serum-derived estrogens. Transient expression of GPR30 in HEK293 cells also did not reveal any receptor-dependent G-1 or E2 effects on intracellular free Ca2+, as determined with an NFAT luciferase reporter, Rac1, as determined with a Rac1Cluc reporter, or ERK1/2 activity, as determined with pERK immunoblotting.
To further reduce system complexity, and to exclude cell- or medium-derived estrogens as a reason for the lack of ligand-stimulated GPR30 activity, we also addressed receptor activity in response G-1 in the yeast S. cerevisiae, a system previously used to study GPCR activity and pharmacology by monitoring G protein–mediated coupling to the pheromone response pathway (Liu et al., 2016). Even in this system did G-1 lack the ability to trigger GPR30 activity. Together, these results are in line with those of several earlier studies (Pedram et al., 2006; Otto et al., 2008; Kang et al., 2010; Southern et al., 2013; Broselid et al., 2014; Sousa et al., 2017; Gonzalez de Valdivia et al., 2017) failing to reproduce the initial observations that G-1 and E2 act as agonists at GPR30 (Thomas et al., 2005 ; Revankar et al., 2005; Bologa et al., 2006). We conclude from these results that G-1 and E2 either do not interact directly with GPR30, or that some critical information is missing in our understanding of this interaction. Unfortunately, only limited attention has been paid to resolve this disagreement, which likely has discouraged independent efforts in both academia and industry to further unravel the molecular details of this pathophysiologically interesting receptor, and to develop high-throughput screening systems to identify specific pharmacological tools.
Ligand-independent constitutive GPR30 activity is less investigated but has the advantage of being independent of any off-target pharmacological effects. Such activity can normally be observed only in well defined recombinant systems, where receptor expression can be compared with a receptor-negative control. We showed previously that GPR30 constitutively decreases cAMP production and NFAT activity and increases ERK1/2 activity in transiently transfected HEK293 or CHO cells (Broselid et al., 2014; Gonzalez de Valdivia et al., 2017). Here, we show that GPR30 also constitutively decreases Rac1 activity in HEK293 cells and increases the activity of a multifunctional promoter (NF-κB, STAT, AP-1) reporter in stable T-REx HFF11 cells. An argument can be made that ligand-independent activity is only apparent, instead being the consequence of a cell- or medium-derived ligand. However, the fact that GPR30 expression also increased yeast growth, an entirely heterologous system without any human growth factors or steroids produced by the cells or in the medium, argues against this explanation.
Evidence that native GPR30 exhibits ligand-independent activity was suggested already in 2002, when progestin treatment of estrogen receptor–positive MCF7 cells was shown to yield both increased GPR30 expression and growth inhibition in the absence of E2, and GPR30 knockdown increased growth whereas GPR30 over-expression decreased growth (Ahola et al., 2002). That receptor knockdown increased growth of MDA-MB-231, MDA-MB-468 cells (Weißenborn et al., 2014), and MCF7 cells (Ariazi et al., 2010; Broselid et al., 2013), and receptor overexpression decreased growth of HEK293 cells (Broselid et al., 2013) provided further evidence for such activity.
All ligand-independent GPR30 activities so far identified in recombinant mammalian systems depend on the receptor C-terminal PDZ motif (Broselid et al., 2014; Gonzalez de Valdivia et al., 2017, Tran et al., 2015). Removing the PDZ motif also increases GPR30 endocytosis, suggesting that these activities require PM localization (Broselid et al., 2014). Interestingly, we report here that ectopic GPR30 couples to the pheromone pathway–mediated yeast growth, which is also a PM-dependent event (Alvaro and Thorner, 2016). Furthermore, PM-localized GPR30 staining of breast cancer patient biopsies has a stronger prognostic significance than total cellular staining ( Sjöström et al., 2014 ; Tutzauer et al., 2020 ). Thus, PM localization appears to be critical for GPR30 function and rely on the PDZ motif.
PDZ-dependent GPR30 anchoring in the PM has in some systems been found to involve a direct interaction between the receptor C-terminal PDZ motif and the PDZ domain membrane scaffold proteins PSD-95 (Akama et al., 2013; Broselid et al., 2014; Tran et al., 2015) or SAP97 (Broselid et al., 2014; Waters et al., 2015). Contributing to receptor PM anchoring may also be the direct interaction of the receptor with the PDZ domain protein , which was reported to increase GPR30 stability (Meng et al., 2016). GPR30 also interacts with and favors PM localization of the chaperone protein receptor activity–modifying protein 3 (Lenhart et al., 2013), which contains a PDZ motif capable of interacting with the PDZ domain of NHERF-1 (Bomberger et al., 2005). Through PSD-95 and/or SAP97, GPR30 in turn interacts with A-kinase anchoring protein 5 (Broselid et al., 2014). Thus, GPR30 appears to be part of a larger PM complex with several partner proteins that may allosterically enable the receptor to elicit ligand-independent activity.
GPR30-promoted ERK1/2 activity is dependent on both pertussis toxin and PDZ (Gonzalez de Valdivia et al., 2017), suggesting that a Gi/o protein is also part of the PM GPR30 complex. Interestingly, PSD-95 expression inhibited the growth-stimulatory effect of GPR30 when expressed in a yeast strain harboring a humanized chimeric yeast Gα protein with the five C-terminal residues from human Gαi2 (DCGLF). These residues directly interact with critical functional residues in GPCR (Oldham and Hamm, 2008; Hilger et al., 2018), with the cysteine in this sequence ADP-ribosylated by pertussis toxin (West et al., 1985). Although yeast expresses very few PDZ domain proteins only distantly related to the mammalian proteins (Harris and Lim, 2001), and does not provide any direct information about complex mammalian protein networks, it has been very useful in identifying physiologically relevant binary protein-protein interactions, including PDZ-dependent interactions with GPCR (Bockaert et al., 2003). Thus, it is tempting to speculate that the binding epitopes for a PDZ domain scaffold protein and Gαi in GPR30 partly overlap.
How then may G-1 and E2 depend on GPR30 in some systems? Several modes of crosstalk between GPR30 and various ERα isoforms have been proposed in recent years (Romano and Gorelick, 2018). GPR30 was reported to interact with the 66-kDa main isoform of ERα (ERα66) (Vivacqua et al., 2009). GPR30 also upregulates and interacts with the 36-kDa ERα isoform (ERα36), mediating effects overlapping with those of GPR30 (Kang et al., 2010; Pelekanou et al., 2016), and both G-1 and E2 activate ERα36 (Kang et al., 2010). Furthermore, the gene promoters of receptor activity–modifying protein 3 and NHERF-1 contain ERE elements through which E2, via ERα66, upregulates these proteins (Ediger et al., 1999; Watanabe et al., 2006). In addition, G-1 increases PSD-95 expression in mouse brain (Waters et al., 2015), a membrane scaffold protein that interacts with the GPR30 PDZ motif, a motif necessary to retain the receptor in the plasma membrane (Akama et al., 2013; Broselid et al., 2013). Given this background, it is tempting to propose that E2 and G-1 influence the ligand-independent activity of GPR30 by interacting with and/or regulating the expression of one or more partners in a GPR30 PM complex.
In this study, we provide ample evidence both ex vivo and in vitro that G-1 and E2 do not act as agonists directly at GPR30. Instead, we propose that this receptor forms a PM complex with partner proteins through which it harbors ligand-independent activity. Considering that the expression or signaling of some components of this complex is sensitive to G-1 and/or E2 stimulation, this could be the reason why some responses to these agents are GPR30-dependent.
Acknowledgments
The authors thank Joanna Daszkiewicz-Nilsson for expert technical.
Abbreviations
- AP-1
activator protein 1
- DMEM
Dulbecco’s modified Eagle’s medium
- E2
17β-estradiol
- ERK
extracellular-regulated protein kinase
- G-1
(±)-1-[(3aR*,4S*,9bS*)-4-(6-bromo-1,3-benzodioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-ethenone
- GPCR
G protein–coupled receptor
- GPR30
G protein–coupled receptor 30
- GPR30ΔSSAV
GPR30 lacking the C-terminal PDZ motif SSAV
- HEK293
human embryonic kidney 293
- KO
knockout
- MAPKAR
mitogen-activated protein kinase activity reporter
- NF-κB
nuclear factor κB
- NFAT
nuclear factor of activated T cells
- OD600
optical density at 600 nm
- PCR
polymerase chain reaction
- PDZ
PSD-95/discs-large/zonula occludens-1 homology
- pERK
phosphorylated ERK1/2
- PM
plasma membrane
- PMA
phorbol myristate acetate
- PSD-95
postsynaptic density-95
- SAP97
synapse-associated protein 97
- SC
synthetic complete
- STAT
signal transducer and activator of transcription
- TET
tetracycline
- WT
wild type
Authorship Contributions
Participated in research design: Olde, Leeb-Lundberg.
Conducted experiments: Tutzauer, Gonzalez de Valdivia, Swärd, Alexandrakis Eilard, Broselid, Kahn, Olde.
Contributed new reagents or analytic tools: Gonzalez de Valdivia, Olde.
Performed data analysis: Tutzauer, Gonzalez de Valdivia, Swärd, Alexandrakis Eilard, Broselid, Kahn, Leeb-Lundberg, Olde.
Wrote or contributed to the writing of the manuscript: Tutzauer, Olde, Leeb-Lundberg.
Footnotes
This work was supported by the Swedish Cancer Foundation (CAN 2016/423, 19 0479 Pj) and the Swedish Research Council (2016-02427) (to L.M.F.L.-L.).
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Ahola TM, Manninen T, Alkio N, Ylikomi T (2002) G protein-coupled receptor 30 is critical for a progestin-induced growth inhibition in MCF-7 breast cancer cells. Endocrinology 143:3376–3384. [DOI] [PubMed] [Google Scholar]
- Akama KT, Thompson LI, Milner TA, McEwen BS (2013) Post-synaptic density-95 (PSD-95) binding capacity of G-protein-coupled receptor 30 (GPR30), an estrogen receptor that can be identified in hippocampal dendritic spines. J Biol Chem 288:6438–6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA (2011) Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol 164:S1–S324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvaro CG, Thorner J (2016) Heterotrimeric G protein-coupled receptor signaling in yeast mating pheromone response. J Biol Chem 291:7788–7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arefin S, Simoncini T, Wieland R, Hammarqvist F, Spina S, Goglia L, Kublickiene K (2014) Vasodilatory effects of the selective GPER agonist G-1 is maximal in arteries of postmenopausal women. Maturitas 78:123–130. [DOI] [PubMed] [Google Scholar]
- Ariazi EA, Brailoiu E, Yerrum S, Shupp HA, Slifker MJ, Cunliffe HE, Black MA, Donato AL, Arterburn JB, Oprea TI, et al. (2010) The G protein-coupled receptor GPR30 inhibits proliferation of estrogen receptor-positive breast cancer cells. Cancer Res 70:1184–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockaert J, Marin P, Dumuis A, Fagni L (2003) The ‘magic tail’ of G protein-coupled receptors: an anchorage for functional protein networks. FEBS Lett 546:65–72. [DOI] [PubMed] [Google Scholar]
- Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, et al. (2006) Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol 2:207–212. [DOI] [PubMed] [Google Scholar]
- Bomberger JM, Spielman WS, Hall CS, Weinman EJ, Parameswaran N (2005) Receptor activity-modifying protein (RAMP) isoform-specific regulation of adrenomedullin receptor trafficking by NHERF-1. J Biol Chem 280:23926–23935. [DOI] [PubMed] [Google Scholar]
- Broselid S, Cheng B, Sjöström M, Lövgren K, Klug-De Santiago HL, Belting M, Jirström K, Malmström P, Olde B, Bendahl PO, et al. (2013) G protein-coupled estrogen receptor is apoptotic and correlates with increased distant disease-free survival of estrogen receptor-positive breast cancer patients. Clin Cancer Res 19:1681–1692. [DOI] [PubMed] [Google Scholar]
- Broselid S, Berg KA, Chavera TA, Kahn R, Clarke WP, Olde B, Leeb-Lundberg LMF (2014) G protein-coupled receptor 30 (GPR30) forms a plasma membrane complex with membrane-associated guanylate kinases (MAGUKs) and protein kinase A-anchoring protein 5 (AKAP5) that constitutively inhibits cAMP production. J Biol Chem 289:22117–22127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton BR, Miller AA, Sobey CG (2010) Endothelium-dependent relaxation by G protein-coupled receptor 30 agonists in rat carotid arteries. Am J Physiol Heart Circ Physiol 298:H1055–H1061. [DOI] [PubMed] [Google Scholar]
- Cheng SB, Quinn JA, Graeber CT, Filardo EJ (2011) Down-modulation of the G-protein-coupled estrogen receptor, GPER, from the cell surface occurs via a trans-Golgi-proteasome pathway. J Biol Chem 286:22441–22455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clipstone NA, Crabtree GR (1992) Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature 357:695–697. [DOI] [PubMed] [Google Scholar]
- Debortoli AR, Rouver WDN, Delgado NTB, Mengal V, Claudio ERG, Pernomian L, Bendhack LM, Moysés MR, Santos RLD (2017) GPER modulates tone and coronary vascular reactivity in male and female rats. J Mol Endocrinol 59:171–180. [DOI] [PubMed] [Google Scholar]
- Ediger TR, Kraus WL, Weinman EJ, Katzenellenbogen BS (1999) Estrogen receptor regulation of the Na+/H+ exchange regulatory factor. Endocrinology 140:2976–2982. [DOI] [PubMed] [Google Scholar]
- Filardo E, Quinn J, Pang Y, Graeber C, Shaw S, Dong J, Thomas P (2007) Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology 148:3236–3245. [DOI] [PubMed] [Google Scholar]
- Gonzalez de Valdivia E, Broselid S, Kahn R, Olde B, Leeb-Lundberg LMF (2017) G protein-coupled estrogen receptor 1 (GPER1)/GPR30 increases ERK1/2 activity through PDZ motif-dependent and -independent mechanisms. J Biol Chem 292:9932–9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez de Valdivia E, Sandén C, Kahn R, Olde B, Leeb-Lundberg LMF (2019) Human G protein-coupled receptor 30 is N-glycosylated and N-terminal domain asparagine 44 is required for receptor structure and activity. Biosci Rep 39:BSR20182436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Garcia JR, Bradley J, Nomikos M, Paul L, Machaty Z, Lai FA, Swann K (2014) The dynamics of MAPK inactivation at fertilization in mouse eggs. J Cell Sci 127:2749–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güldener U, Heck S, Fielder T, Beinhauer J, Hegemann JH (1996) A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res 24:2519–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas E, Bhattacharya I, Brailoiu E, Damjanović M, Brailoiu GC, Gao X, Mueller-Guerre L, Marjon NA, Gut A, Minotti R, et al. (2009) Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ Res 104:288–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris BZ, Lim WA (2001) Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci 114:3219–3231. [DOI] [PubMed] [Google Scholar]
- Hilger D, Masureel M, Kobilka BK (2018) Structure and dynamics of GPCR signaling complexes. Nat Struct Mol Biol 25:4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman GA, Garrison TR, Dohlman HG (2002) Analysis of RGS proteins in Saccharomyces cerevisiae. Methods Enzymol 344:617–631. [DOI] [PubMed] [Google Scholar]
- Holm A, Grände PO, Ludueña RF, Olde B, Prasad V, Leeb-Lundberg LMF, Nilsson BO (2012) The G protein-coupled oestrogen receptor 1 agonist G-1 disrupts endothelial cell microtubule structure in a receptor-independent manner. Mol Cell Biochem 366:239–249. [DOI] [PubMed] [Google Scholar]
- Isensee J, Meoli L, Zazzu V, Nabzdyk C, Witt H, Soewarto D, Effertz K, Fuchs H, Gailus-Durner V, Busch D, et al. (2009) Expression pattern of G protein-coupled receptor 30 in LacZ reporter mice. Endocrinology 150:1722–1730. [DOI] [PubMed] [Google Scholar]
- Jala VR, Radde BN, Haribabu B, Klinge CM (2012) Enhanced expression of G-protein coupled estrogen receptor (GPER/GPR30) in lung cancer. BMC Cancer 12:624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang EJ, Seok YM, Arterburn JB, Olatunji LA, Kim IK (2013) GPER-1 agonist G1 induces vasorelaxation through activation of epidermal growth factor receptor-dependent signalling pathway. J Pharm Pharmacol 65:1488–1499. [DOI] [PubMed] [Google Scholar]
- Kang L, Zhang X, Xie Y, Tu Y, Wang D, Liu Z, Wang ZY (2010) Involvement of estrogen receptor variant ER-alpha36, not GPR30, in nongenomic estrogen signaling. Mol Endocrinol 24:709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu N, Aoki K, Yamada M, Yukinaga H, Fujita Y, Kamioka Y, Matsuda M (2011) Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol Biol Cell 22:4647–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotarsky K, Antonsson L, Owman C, Olde B (2003) Optimized reporter gene assays based on a synthetic multifunctional promoter and a secreted luciferase. Anal Biochem 316:208–215. [DOI] [PubMed] [Google Scholar]
- Langer G, Bader B, Meoli L, Isensee J, Delbeck M, Noppinger PR, Otto C (2010) A critical review of fundamental controversies in the field of GPR30 research. Steroids 75:603–610. [DOI] [PubMed] [Google Scholar]
- Lenhart PM, Broselid S, Barrick CJ, Leeb-Lundberg LMF, Caron KM (2013) G-protein-coupled receptor 30 interacts with receptor activity-modifying protein 3 and confers sex-dependent cardioprotection. J Mol Endocrinol 51:191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ER (2009) G protein-coupled receptor 30: estrogen receptor or collaborator? Endocrinology 150:1563–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey SH, Carver KA, Prossnitz ER, Chappell MC (2011) Vasodilation in response to the GPR30 agonist G-1 is not different from estradiol in the mRen2.Lewis female rat. J Cardiovasc Pharmacol 57:598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey SH, Liu L, Chappell MC (2014) Vasodilation by GPER in mesenteric arteries involves both endothelial nitric oxide and smooth muscle cAMP signaling. Steroids 81:99–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Wong W, IJzerman AP (2016) Human G protein-coupled receptor studies in Saccharomyces cerevisiae. Biochem Pharmacol 114:103–115. [DOI] [PubMed] [Google Scholar]
- Maiti K, Paul JW, Read M, Chan EC, Riley SC, Nahar P, Smith R (2011) G-1-activated membrane estrogen receptors mediate increased contractility of the human myometrium. Endocrinology 152:2448–2455. [DOI] [PubMed] [Google Scholar]
- Manfredi JP, Klein C, Herrero JJ, Byrd DR, Trueheart J, Wiesler WT, Fowlkes DM, Broach JR (1996) Yeast alpha mating factor structure-activity relationship derived from genetically selected peptide agonists and antagonists of Ste2p. Mol Cell Biol 16:4700–4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mårtensson UEA, Salehi SA, Windahl S, Gomez MF, Swärd K, Daszkiewicz-Nilsson J, Wendt A, Andersson N, Hellstrand P, Grände PO, et al. (2009) Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology 150:687–698. [DOI] [PubMed] [Google Scholar]
- Meng R, Qin Q, Xiong Y, Wang Y, Zheng J, Zhao Y, Tao T, Wang Q, Liu H, Wang S, et al. (2016) NHERF1, a novel GPER associated protein, increases stability and activation of GPER in ER-positive breast cancer. Oncotarget 7:54983–54997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MR, Baretella O, Prossnitz ER, Barton M (2010) Dilation of epicardial coronary arteries by the G protein-coupled estrogen receptor agonists G-1 and ICI 182,780. Pharmacology 86:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Müller R, Funk M (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156:119–122. [DOI] [PubMed] [Google Scholar]
- Olde B, Leeb-Lundberg LMF (2009) GPR30/GPER1: searching for a role in estrogen physiology. Trends Endocrinol Metab 20:409–416. [DOI] [PubMed] [Google Scholar]
- Oldham WM, Hamm HE (2008) Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol 9:60–71. [DOI] [PubMed] [Google Scholar]
- Otto C, Rohde-Schulz B, Schwarz G, Fuchs I, Klewer M, Brittain D, Langer G, Bader B, Prelle K, Nubbemeyer R, et al. (2008) G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol. Endocrinology 149:4846–4856. [DOI] [PubMed] [Google Scholar]
- Pedram A, Razandi M, Levin ER (2006) Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol 20:1996–2009. [DOI] [PubMed] [Google Scholar]
- Peixoto P, Aires RD, Lemos VS, Bissoli NS, Santos RLD (2017) GPER agonist dilates mesenteric arteries via PI3K-Akt-eNOS and potassium channels in both sexes. Life Sci 183:21–27. [DOI] [PubMed] [Google Scholar]
- Pelekanou V, Kampa M, Kiagiadaki F, Deli A, Theodoropoulos P, Agrogiannis G, Patsouris E, Tsapis A, Castanas E, Notas G (2016) Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERα36 and GPR30/GPER1. J Leukoc Biol 99:333–347. [DOI] [PubMed] [Google Scholar]
- Prossnitz ER, Arterburn JB (2015) International Union of Basic and Clinical Pharmacology. XCVII. G protein-coupled estrogen receptor and its pharmacologic modulators. Pharmacol Rev 67:505–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz ER, Barton M (2014) Estrogen biology: new insights into GPER function and clinical opportunities. Mol Cell Endocrinol 389:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER (2005) A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307:1625–1630. [DOI] [PubMed] [Google Scholar]
- Rippe C, Zhu B, Krawczyk KK, Bavel EV, Albinsson S, Sjölund J, Bakker ENTP, Swärd K (2017) Hypertension reduces soluble guanylyl cyclase expression in the mouse aorta via the Notch signaling pathway. Sci Rep 7:1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano SN, Gorelick DA (2018) Crosstalk between nuclear and G protein-coupled estrogen receptors. Gen Comp Endocrinol 261:190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Rasmussen SG, Kobilka BK (2009) The structure and function of G-protein-coupled receptors. Nature 459:356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandén C, Broselid S, Cornmark L, Andersson K, Daszkiewicz-Nilsson J, Mårtensson UE, Olde B, Leeb-Lundberg LMF (2011) G protein-coupled estrogen receptor 1/G protein-coupled receptor 30 localizes in the plasma membrane and traffics intracellularly on cytokeratin intermediate filaments. Mol Pharmacol 79:400–410. [DOI] [PubMed] [Google Scholar]
- Scaling AL, Prossnitz ER, Hathaway HJ (2014) GPER mediates estrogen-induced signaling and proliferation in human breast epithelial cells and normal and malignant breast. Horm Cancer 5:146–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K (2002) Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch Pharmacol 366:381–416. [DOI] [PubMed] [Google Scholar]
- Sjöström MHartman LGrabau DFornander TMalmström PNordenskjöld BSgroi DCSkoog LStål OLeeb-Lundberg LMF, et al. (2014) Lack of G protein-coupled estrogen receptor (GPER) in the plasma membrane is associated with excellent long-term prognosis in breast cancer. Breast Cancer Res Treat 145:61–71. [DOI] [PubMed] [Google Scholar]
- Sousa C, Ribeiro M, Rufino AT, Leitão AJ, Mendes AF (2017) Assessment of cell line competence for studies of pharmacological GPR30 modulation. J Recept Signal Transduct Res 37:181–188. [DOI] [PubMed] [Google Scholar]
- Southern C, Cook JM, Neetoo-Isseljee Z, Taylor DL, Kettleborough CA, Merritt A, Bassoni DL, Raab WJ, Quinn E, Wehrman TS, et al. (2013) Screening β-arrestin recruitment for the identification of natural ligands for orphan G-protein-coupled receptors. J Biomol Screen 18:599–609. [DOI] [PubMed] [Google Scholar]
- Thomas P, Pang Y, Filardo EJ, Dong J (2005) Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 146:624–632. [DOI] [PubMed] [Google Scholar]
- Tran QK, VerMeer M, Burgard MA, Hassan AB, Giles J (2015) Hetero-oligomeric complex between the G protein-coupled estrogen receptor 1 and the plasma membrane Ca2+-ATPase 4b. J Biol Chem 290:13293–13307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tutzauer J, Sjöström M, Bendahl P-O, Rydén L, Fernö M, Leeb-Lundberg LMF, Alkner S(2020) Plasma membrane expression of G protein-coupled estrogen receptor (GPER)/G protein-coupled receptor 30 (GPR30) is associated with worse outcome in metachronous contralateral breast cancer. PLoS ONE 15:e0231786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivacqua A, Lappano R, De Marco P, Sisci D, Aquila S, De Amicis F, Fuqua SA, Andò S, Maggiolini M (2009) G protein-coupled receptor 30 expression is up-regulated by EGF and TGF alpha in estrogen receptor alpha-positive cancer cells. Mol Endocrinol 23:1815–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Takahashi E, Kobayashi M, Goto M, Krust A, Chambon P, Iguchi T (2006) The estrogen-responsive adrenomedullin and receptor-modifying protein 3 gene identified by DNA microarray analysis are directly regulated by estrogen receptor. J Mol Endocrinol 36:81–89. [DOI] [PubMed] [Google Scholar]
- Waters EM, Thompson LI, Patel P, Gonzales AD, Ye HZ, Filardo EJ, Clegg DJ, Gorecka J, Akama KT, McEwen BS, et al. (2015) G-protein-coupled estrogen receptor 1 is anatomically positioned to modulate synaptic plasticity in the mouse hippocampus. J Neurosci 35:2384–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weißenborn C, Ignatov T, Ochel HJ, Costa SD, Zenclussen AC, Ignatova Z, Ignatov A (2014) GPER functions as a tumor suppressor in triple-negative breast cancer cells. J Cancer Res Clin Oncol 140:713–723. [DOI] [PubMed] [Google Scholar]
- West RE Jr, Moss J, Vaughan M, Liu T, Liu TY (1985) Pertussis toxin-catalyzed ADP-ribosylation of transducin. Cysteine 347 is the ADP-ribose acceptor site. J Biol Chem 260:14428–14430. [PubMed] [Google Scholar]
- Wojciak-Stothard B, Ridley AJ (2002) Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol 39:187–199. [DOI] [PubMed] [Google Scholar]
- Yu X, Stallone JN, Heaps CL, Han G (2018) The activation of G protein-coupled estrogen receptor induces relaxation via cAMP as well as potentiates contraction via EGFR transactivation in porcine coronary arteries. PLoS One 13:e0191418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Vinuela A, Neely MH, Hallett PJ, Grant SG, Miller GM, Isacson O, Caron MG, Yao WD (2007) Inhibition of the dopamine D1 receptor signaling by PSD-95. J Biol Chem 282:15778–15789. [DOI] [PMC free article] [PubMed] [Google Scholar]