

Abstract
Background
Aedes aegypti and Aedes albopictus are invasive mosquito species and significantly impact human health in southern China. Microbiota are confirmed to affect the development and immunity of mosquitoes. However, scientists have focused more on midgut microbiota of female mosquitoes and bacterial differences between female and male Aedes mosquitoes. The relationship between the midgut and entire body microbiota of Aedes is unclear. In this study, we collected mosquito samples reared under the same laboratory conditions and compared the microbial composition of midgut and entire bodies of Aedes aegypti and Aedes albopictus using 16S rRNA gene sequencing.
Methods
In this study, we collected mosquito samples reared under the same laboratory conditions and compared the microbial composition of midgut and entire bodies of Aedes aegypti and Aedes albopictus using 16S rRNA gene sequencing.
Results
A total of 341 OTUs were identified, showing that Proteobacteria was the dominant phylum and Methylobacterium the dominant genus in both Aedes aegypti and Aedes albopictus. The bacterial diversity and community structures of the entire bodies were similar between males and females in both Aedes aegypti and Aedes albopictus. Conversely, the bacterial compositions of male and female Aedes aegypti and Aedes albopictus were significantly different. NMDS analysis, UPGMA analysis, diversity indices and OTU distribution demonstrated that compositions and structures in midgut microbiota were similar but significantly different in the entire bodies of Aedes aegypti and Aedes albopictus. Functional prediction analysis showed that metabolism and environmental information processing were the dominant KEGG pathways at level 1. Our study showed that there were significantly different level 2 and 3 KEGG pathways in the midgut microbiota (16 level 2 and 24 level 3) and the entire bodies (33 level 2 and 248 level 3) between female Aedes albopictus and Aedes Aegypti.
Conclusions
Our findings that Aedes aegypti and Aedes albopictus reared in the same laboratory harbor a similar gut bacterial microbiome but different entire body microbiota imply that the gut microbiota of adult mosquitoes is environmentally determined regardless of the host genotype, but the entire body microbiota is more genetically determined. Our findings improved the understanding of the microbiota in the entire and partial tissues of Aedes mosquitoes.
Graphical abstract
Supplementary Information
The online version contains supplementary material available at 10.1186/s13071-021-05050-4.
Keywords: Mosquito, Vector, High-throughput sequencing, Microbiome, Midgut, Entire body
Introduction
Aedes aegypti and Ae. albopictus, well-known invasive mosquito species, have rapidly expanded and spread to most countries globally in the past 40 years [1–3]. These two mosquitoes are important vectors transmitting dengue virus (DENV) [4], Zika virus (ZIKV) [5], West Nile virus (WNV) [6] and chikungunya virus (CHIKV) [7], which cause enormous damage and public health problems to human and animal organisms [1, 8–11].
Recent studies on the microorganisms of mosquitoes have indicated that the midgut harbors a diverse microbiota, which can significantly affect the reproduction, development, digestion, nutrition and metabolism, immunity, behavior and other physiological functions of their hosts [12–14]. Previous studies have reported that the composition of midgut-derived bacteria is related to the host and parasite interaction, and the midgut microbiota could block the transmission of pathogens such as Zika virus and Plasmodium [5, 15–17]. To investigate the microbial community structure, researchers focused on field-captured and laboratory mosquitoes. They found that the gut bacterial community structures in the adult stage of the field and laboratory mosquitoes were similar [18]. The study revealed that most bacterial genera in wild Culex pipiens and laboratory-reared adult Aedes japonicus could also be found in other mosquito species, such as Ae. aegypti and Ae. albopictus [19], indicating that different mosquitoes may share the same microorganisms. Studies on Ae. aegypti and Anopheles gambiae (An. gambiae) revealed that the development of these mosquitoes depended on gut microbiota [20, 21]. These studies focused on the midgut microbiota and the biological aspects of the hosts.
Previous studies have shown that the entire bodies of insects harbor diverse microbiota [22, 23]. These microbiota can influence insect host vector competence [24, 25] and modulate arbovirus transmission in mosquitoes [24, 25]. However, few studies have focused on the entire body microbiota of Aedes, and the relationship and difference between the entire and midgut microbiota of Aedes mosquitoes are unclear.
To investigate the diversity, composition and network analysis of microbiota in Aedes mosquitoes from South China, we used high-throughput sequencing of the 16S rRNA gene. We revealed the bacterial communities and their associations in the entire bodies and midguts of male and female Aedes mosquitoes reared under laboratory conditions and investigated the relationship between Wolbachia and other microbes of Ae. albopictus.
Methods
Samples collection
Aedes mosquitoes were reared in laboratory conditions. The food consisted of a mixture of lactalbumin hydrolysate, finely ground rat chow and Brewer's yeast (1:1:1). Aedes mosquitoes were maintained at 27 °C, RH 80%, with a photoperiod of 14:10 h (L:D) on 10% sucrose solution ad libitum. For this study, we collected samples of the midgut (from females) and entire body (from both males and females) from adult mosquitoes. Before dissection, the entire mosquito was washed three times with sterile distilled water and surface sterilized using 75% ethanol for 1 min [26, 27]. Each sample consisted of five midguts or one entire mosquito as a pool and then was stored in 95% ethanol at − 80 ˚C until further research.
DNA extraction
Samples collected in a tube were mechanically homogenized using a sterile pestle (an electric homogenizer) in liquid nitrogen. For bacteria community sequencing, total DNA was isolated under a sterile environment according to the protocol of the Hipure Bacterial DNA Kit (Magen, China). Briefly, each sample was put in an Eppendorf tube with 1-mm-diameter inox beads (Qiagen, Germany) and individually crushed using an organization disruptor (Gene Co., Ltd., China). They were removed, and 1 ml extraction STE buffer, 10 μl SDS buffer and 10 μl Proteinase K were added. The samples were heated at 55 ℃ for 1 h. To wipe liquids and proteins, we added 200 μl AL buffer into the tube and then heated it at 70 ℃ for 10 min. Total DNA was precipitated with 250 μl cold absolute ethyl alcohol and centrifugation at 100g for 15 s. After these steps, total nucleic acid was transferred into Hipure DNA mini Column I and washed by GW1 and GW1 buffer step by step. Finally, total DNA was resuspended in 30 μl AE buffer. The DNA quality and quantity examinations were conducted using a Nanodrop (Thermo Scientific, USA).
PCR amplification and sequencing
The 16S rRNA genes of their V3-V4 regions were amplified and sequenced on an Illumina HiSeq 2500 platform. The following primer set was used: forward primer 5'-ACTCCTACGGGAGGCAGCA-3'; reverse primer 5'- GGACTACHVGGGTWTCTAAT-3'. PCR amplification was performed using PrimeStar DNA polymerase (Takara, China). The following PCR cycling conditions were used: denaturation at 95 °C for 5 min, 25 cycles of 95 °C for 30 s, 50 °C for 30 s, 72 °C for 40 s and final extension at 72 °C for 5 min. The PCR products were analyzed by agarose gel electrophoresis. Finally, 16S rRNA gene sequencing was constructed using Biomarker Technologies (Beijing, China).
Analysis of sequencing data
After the base calling analysis, the original data files from the sequencing platform were transformed into the original sequenced Reads Stored in FASTQ format. QIIME (version 1.8.0) was used to cluster reads into operational taxonomic units (OTUs) and identified at 97% or more similarity [28]. To analyze the alpha diversity, we rarified the OTU table and calculated the species abundance based on two metrics: Ace and Shannon [29]. The unweighted pair group method with arithmetic mean analysis (UPGMA) was performed based on the unifrac distance using QIIME (version 1.9.1). As a measure of beta diversity and similarity, the nonmetric multidimensional scaling (NMDS) graph with Bray-Curtis diversity was done in R with the vegan package [30]. Venn diagram analysis was conducted in R statistical software (version 3.0.3) using the vegan packages. The metabolic functions of the bacterial community were inferred by phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt) [31]. The correlation network analysis was conducted using the ClusterMaker app in Cytoscape [32]. For range adjustment, all pairwise comparisons between two groups were tested using Student’s t-test. *P < 0.05 was considered statistically significant.
Results
Mosquito colonies study
The Ae. aegypti and Ae. albopictus mosquitoes were captured in Hainan and Guangdong provinces, South China (Additional file 1: Fig. S1). The Ae. albopictus strain was established from 2000 mosquito larvae collected from 20 different districts of the Guangzhou metropolitan area (about 100 larvae collected per district). Then, they were transferred to a laboratory using a special facility for further study. The field-captured mosquitoes were identified under microscope. The Ae. aegypti strain was collected by Guangdong Provincial Center for Disease Control and Prevention, and they offered us this strain. We fed the Aedes under laboratory environmental conditions. After rearing > 30 generations, we collected six groups of Aedes colonies (Additional file 2: Table S1) to investigate the bacterial composition and diversity shaping the environmental factors in 2017.
Composition of microbiota in Ae. aegypti and Ae. albopictus
To analyze the composition and diversity of bacterial communities in both Ae. aegypti and Ae. albopictus from South China, we performed 16S rRNA gene sequencing of their V3-V4 hypervariable regions. In total, 2,686,929 clean tags were retained and classified into operational taxonomic units (OTUs) at a 97% similarity level after rarefaction and quality filtering. An average of 149,274 clean tags were obtained from sequenced samples. The tags were clustered into 341 OTUs representing 14 phyla, 23 classes, 42 orders, 71 families and 123 genera.
We analyzed the differences in the microbial community from both the entire bodies and midguts of Ae. aegypti and Ae. albopictus. Our findings revealed the relative abundances of the predominant taxa in the six groups (Fig. 1). At the phylum level, the Ae. aegypti groups, including the entire bodies of male Ae. Aegypti (AEMW), entire bodies of female Ae. aegypti (AEFW) and midguts of female Ae. aegypti (AEFM), showed close abundances at the main phyla, which were dominated by Proteobacteria (87.6%, 85.2% and 90.6%, respectively) and Bacteroidetes (7.7%, 10.1% and 6.3%, respectively) (Fig. 1a). The total relative abundances of the other phyla were < 5% in each group. The bacterial compositions of the Ae. albopictus groups, including the entire bodies of the male Ae. albopictus (ALMW), entire bodies of the female Ae. albopictus (ALFW) and the midguts of the female Ae. albopictus (ALFM), were dominated by Proteobacteria (71.0%, 98.0% and 93.4%, respectively) (Fig. 1b). The ALFM group showed a higher abundance of Bacteroidetes (4.6%), and the ALMW population harbored the highest relative abundance of Firmicutes (27.2%) compared to those of the other groups. Most microbes in ALFW mosquitoes were identified as the phylum Proteobacteria, the relative abundance of which was higher than those of other groups.
Fig. 1.

Relative abundance of bacteria at phylum and genus level in Ae. aegypti and Ae. albopictus, including males and females. a Phylum, b genus. c Average distribution of top ten genera in Aedes. Only the ten most common bacterial phyla and genera are shown. AEMW: entire body of male Ae. aegypti. AEFW: entire body of female Ae. aegypti. AEFM: midgut of female Ae. aegypti. ALMW: entire body of male Ae. albopictus. ALFW: entire body of female Ae. albopictus. ALFM: midgut of female Ae. albopictus
At the genus level, the top ten genera in the AEMW group were Leptothrix (14.3%), Methylobacterium (8.3%), Enterobacter (6.3%), Methylotenera (6.2%), uncultured bacteria (4.2%), Escherichia-Shigella (3.0%) and Sphingomonas (1.5%). The AEMW group harbored diverse microbiota, including Methylobacterium (12.6%), Asaia (11.4%), Ralstonia (10.8%), Leptothrix (5.6%), Sphingomonas (4.3%), Methylotenera (3.7%), Enterobacter (3.2%), Escherichia-Shigella (3.2%) and uncultured bacteria (3.0%). The relative abundances of the genera Methylobacterium (41.1%), Enterobacter (10.6%) and Sphingomonas (5.9%) in the AEFM group were higher than those in the AEFW group. Among the Ae. albopictus groups, the bacterial composition of ALMW was dominated by the genera Wolbachia (37.7%), Bacillus (22.9%), Enterobacter (14.8%) and Methylobacterium (8.2%), and the ALFW mosquitoes were dominated by Wolbachia (78.7%), Methylobacterium (7.8%) and Enterobacter (4.1%). The ALFM mosquitoes had higher abundances of Methylobacterium (67.4%) and Sphingomonas (7.9%) and lower abundances of Wolbachia (9.5%) and Enterobacter (0.2%) than the ALFW mosquitoes. Our results showed that the dominant microbes colonizing the entire bodies of mosquitoes were distinct and associated with the host’s genetic background. The midguts of female Aedes mosquitoes harbored similarly dominant microbes, which were not dependent on the species or genetic type.
Bacterial community structures of Aedes mosquitoes
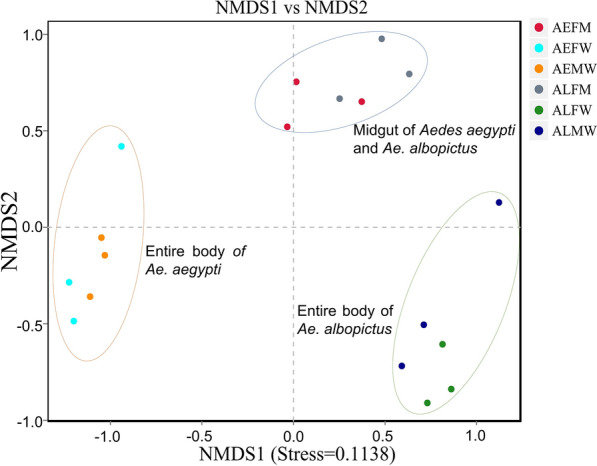
To investigate the bacterial community structures among samples, we performed NMDS analysis (quantified by Bray-Curtis dissimilarity). The NMDS results (Fig. 2) showed that the bacterial communities in the entire bodies of Ae. aegypti and Ae. albopictus were significantly different, while the microbiota compositions of the entire bodies of both the male and female Ae. aegypti and Ae. albopictus were similar. These findings demonstrated that the similarity of the bacterial community structure in the entire body of the Aedes mosquito was associated with the species and genetic type. Interestingly, we found that the bacterial communities in the midguts of Ae. aegypti and Ae. albopictus were highly similar and the bacterial structures in the midgut were significantly different from those in the entire body (Fig. 2). These results showed that the composition and structure of the midgut of Aedes were not dependent on the species or genetic type.
Fig. 2.

NMDS analysis showing microbiome communities of each sample. a Based on the genus level (stress = 0.1138). b Based on the OTU level (stress = 0.1166). The blue ellipse indicates that the bacterial structures in the midgut of both Ae. aegypti and Ae. albopictus are similar to but distinct from those in entire bodies of Aedes mosquitoes. The orange and green ellipses indicated that similar bacterial structures were harbored in the entire bodies of both male and female mosquitoes of Ae. aegypti and Ae. albopictus, respectively. AEMW: entire body of male Ae. aegypti. AEFW: entire body of female Ae. aegypti. AEFM: midgut of female Ae. aegypti. ALMW: entire body of male Ae. albopictus. ALFW: entire body of female Ae. albopictus. ALFM: midgut of female Ae. albopictus
To authenticate the above results, we performed UPGMA analysis. Our findings (Fig. 3) showed that the composition structures in the entire bodies of both males and females of the same Aedes species were clustered in the same branches, while the bacterial communities in the entire bodies of Ae. aegypti and Ae. albopictus were divided into different clusters. Interestingly, the bacterial structures of different samples in the midgut of Aedes mosquitoes were under the same clusters, showing a convergent gut microbiota in Aedes mosquitoes (Fig. 3). These results were similar to the findings shown by NMDS analysis. Our work found a higher similarity of microbiota composition harbored in the midgut compared with that in the entire body of the Aedes mosquito. Our study suggested that the microbiota shaping by the environment in tissue was more than that in the entire body of the Aedes mosquito.
Fig. 3.

UPGMA analysis based on unifrac distance showing the cluster of microbiome communities in Aedes samples. a Based on the genus. b Based on the OTUs. The blue standing string indicates that the bacterial structures in the midgut of both Ae. aegypti and Ae. albopictus are similar and convergent but distinct in relation to that in entire bodies of Aedes mosquitoes. The orange and green standing strings indicate that similar bacterial structures are harbored in the entire bodies of both male and female mosquitoes of Ae. aegypti and Ae. albopictus, respectively. AEMW: entire body of male Ae. aegypti. AEFW: entire body of female Ae. aegypti. AEFM: midgut of female Ae. aegypti. ALMW: entire body of male Ae. albopictus. ALFW: entire body of female Ae. albopictus. ALFM: midgut of female Ae. albopictus. AE: Ae. aegypti. AL: Ae. albopictus
Microbial diversity of Aedes mosquitoes
To examine the alpha diversity between Ae. aegypti and Ae. albopictus, we rarified the OTU table and calculated the richness using two metrics: ACE and Shannon index. Good’s coverage was > 99.98% for each sample. No significant difference was found in the bacterial alpha diversity in the entire body of either the male or female Ae. aegypti or Ae. albopictus. However, the diversities of the bacterial communities in the entire bodies of Ae. albopictus groups (including ALFW and ALMW) were higher than those of Ae. aegypti groups (including AEFW and AEMW) (Table 1). Our findings indicated that there was a significant difference in bacterial diversity between Ae. aegypti and Ae. albopictus, showing that Ae. albopictus harbored more bacterial richness than Ae. aegypti. The diversity of the midgut microbiota was higher than the diversities in the entire bodies of both male and female Ae. aegypti (Table 1). Interestingly, there were significant differences in bacterial diversities between the entire body and midgut of female Ae. albopictus, between the entire body of male Ae. albopictus and midgut of female Ae. albopictus and between the entire body of female Ae. albopictus and midgut of female Ae. aegypti, while no significant difference was found in the midgut microbiota between Ae. aegypti and Ae. albopictus (Table 1). These results demonstrated that higher bacterial diversity existed in the entire body than in the midgut of Aedes, showing a similar bacterial diversity in the midgut. We found similar results on the number of OTUs of microbiomes in Aedes mosquitoes (Fig. 4). Overall, our findings showed that the midgut microbiota was shaped by environmental factors to a greater extent than the entire body of Aedes mosquitoes.
Table 1.
Diversity indices and Good’s coverage of the bacterial composition of Ae. albopictus and Ae. aegypti
| Group | ACE (Mean ± SD) |
Shannon (Mean ± SD) |
Coverage (%) |
|---|---|---|---|
| AEMW | 182.6 ± 5.6 | 4.42 ± 0.18 | 99.98 |
| AEFW | 173.1 ± 3.3 | 4.23 ± 0.29 | 99.98 |
| AEFM | 197.9 ± 10.3 | 3.46 ± 0.28 | 99.99 |
| ALMW# | 266.7 ± 12.9 | 2.81 ± 0.16 | 99.98 |
| ALFW* | 272.0 ± 5.0 | 2.39 ± 0.19 | 99.99 |
| ALFM | 176.6 ± 12.2 | 2.30 ± 0.13 | 99.99 |
#Significant differences (P < 0.05) in microbiota diversities between ALMW and AEMW, between ALMW and AEFW and between ALMW and ALFM
*Significant differences in bacterial diversities between ALFW and AEFW, between ALFW and AEMW and between ALFW and ALFM
Fig. 4.
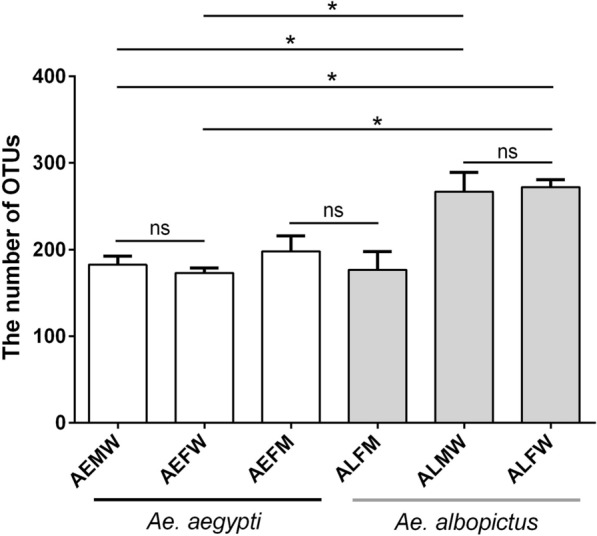
Number of OTUs is shown in each group. * Significant difference determined by the Student’s t-test at P-value < 0.05. AEMW: entire body of male Ae. aegypti. AEFW: entire body of female Ae. aegypti. AEFM: midgut of female Ae. aegypti. ALMW: entire body of male Ae. albopictus. ALFW: entire body of female Ae. albopictus. ALFM: midgut of female Ae. albopictus
In addition, we found that most OTUs were shared in the entire bodies of male and female Aedes mosquitoes, and similar results were observed in the entire body and midgut of Aedes mosquitoes (Fig. 5a–d). However, more unique OTUs existed in the midgut than in the entire body of Ae. aegypti (Fig. 5b). There were more unique OTUs existing in the entire body than in the midgut of Ae. albopictus (Fig. 5d). In addition, the number of unique OTUs harbored by females and males of Ae. albopictus was larger than that of Ae. aegypti, with counts of 114 and 111, respectively (Fig. 5e, f). This finding showed that the number of unique OTUs between different species was larger than that between the same species (Fig. 5a, c, e, f). Interestingly, the numbers of unique OTUs in the midgut microbiota of Ae. albopictus and Ae. aegypti were similar (Fig. 5g) and were smaller than those in the entire bodies of Aedes mosquitoes (Fig. 5e–g). Finally, only 54 OTUs were shared among all samples (Fig. 5h). These findings demonstrated that there was a high similarity in the bacterial composition of the entire body and the midgut of mosquitoes in the same species, regardless of sex. The difference in the compositions of the midgut microbiota between female Ae. albopictus and Ae. aegypti was smaller than that in the entire body.
Fig. 5.

Venn diagrams showing the common and unique OTUs between groups. a Between AEMW and AEFW. b Between AEFW and AEFM. c Between ALMW and ALFW. d Between ALFW and ALFM. e Between AEFW and ALFW. f Between AEMW and ALMW. g Between AEFM and ALFM. h Venn diagrams showing the core OTUs among samples. AEMW: entire body of male Ae. aegypti. AEFW: entire body of female Ae. aegypti. AEFM: midgut of female Ae. aegypti. ALMW: entire body of male Ae. albopictus. ALFW: entire body of female Ae. albopictus. ALFM: midgut of female Ae. albopictus. AE: Ae. aegypti. AL: Ae. albopictus
Functional gene prediction of bacterial composition in Aedes mosquitoes
The functional genes of bacterial communities of the Aedes mosquitoes were explored based on OTUs using PICRUSt software [33, 34]. Functional categories classified into level 1 functional categories were enriched in metabolism, environmental information processing, genetic information processing, cellular processes, human diseases, and organismal systems (Fig. 6). The predicted functions of all groups (AEMW, AEFW, AEFM, ALMW, ALFW and ALFM) were mainly identified in metabolism (72.92%, 73.16%, 72.31%, 66.79%, 63.85% and 70.28%, respectively), the second most common was environmental information processing (13.88%, 13.35%, 13.21%, 16.16%, 17.20% and 13.21%, respectively), and the third most common was genetic information processing (4.96%, 5.10%, 5.15%, 7.86%, 9.26% and 5.77%, respectively).
Fig. 6.
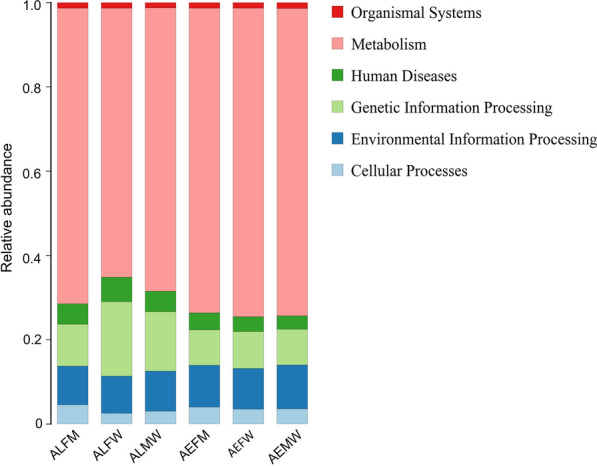
Level 1 of predicted function categories is shown in each group. AEMW: entire body of male Ae. aegypti. AEFW: entire body of female Ae. aegypti. AEFM: midgut of female Ae. aegypti. ALMW: entire body of male Ae. albopictus. ALFW: entire body of female Ae. albopictus. ALFM: midgut of female Ae. Albopictus
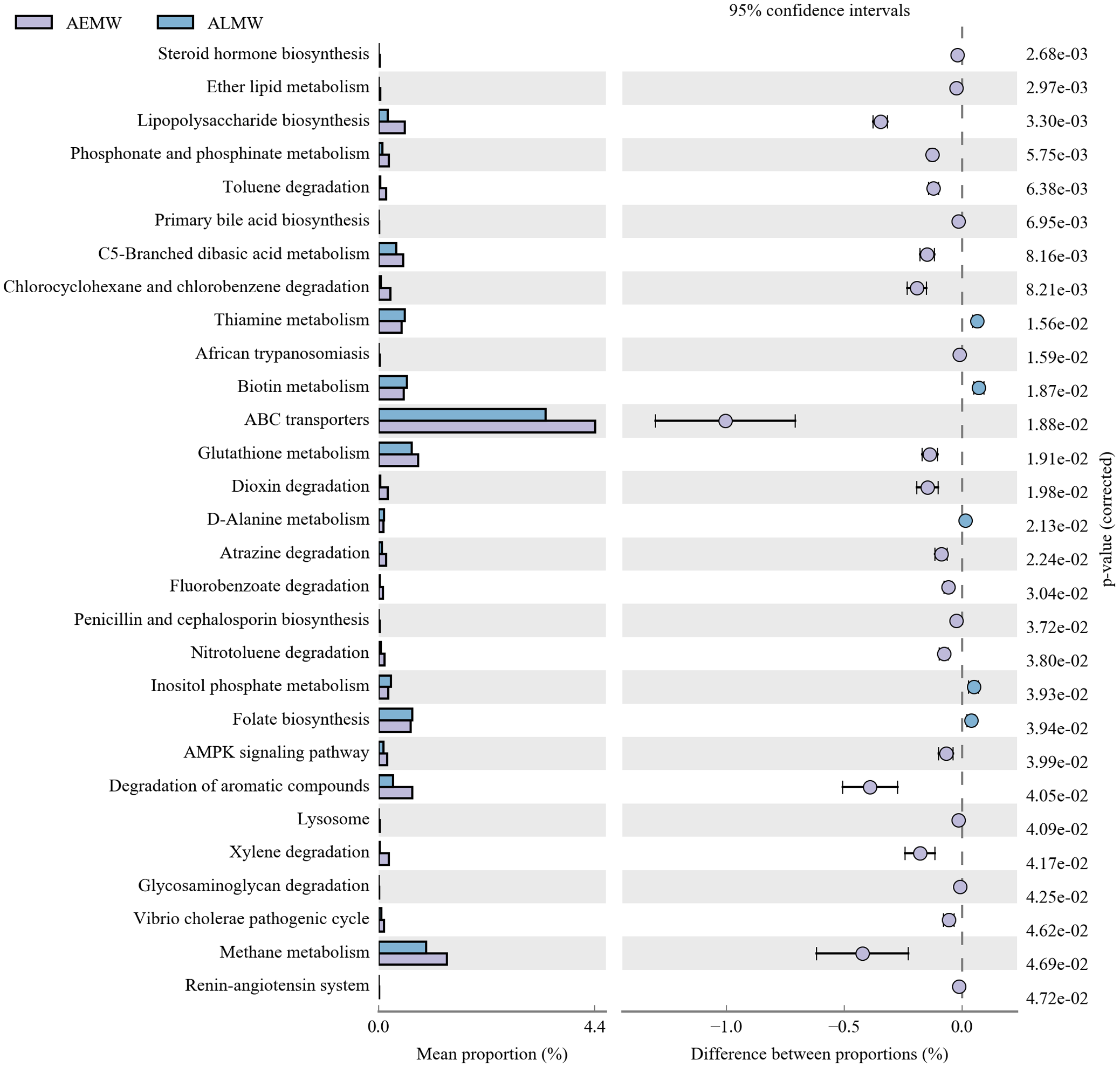
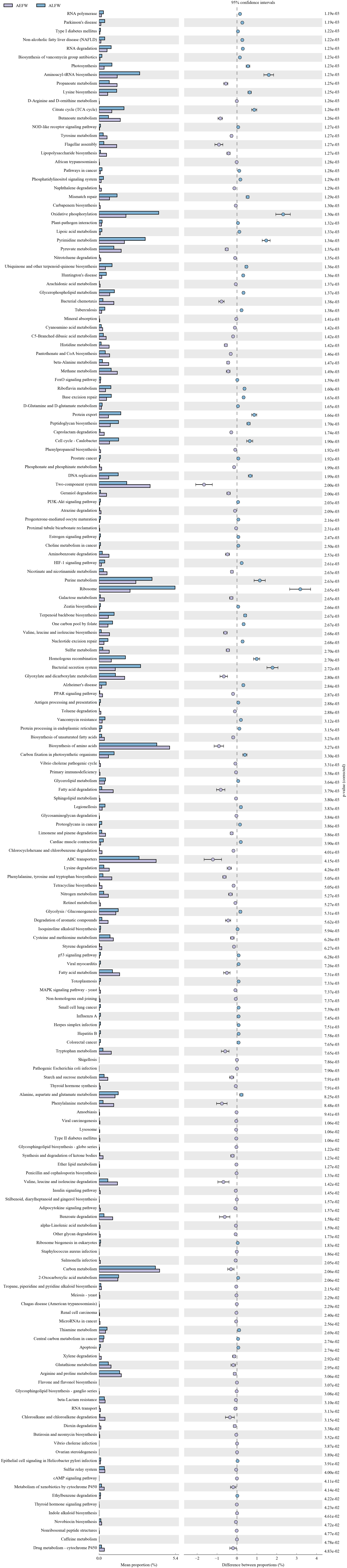
Moreover, the pathways of carbohydrate metabolism, global and overview maps, amino acid metabolism, energy metabolism and metabolism of cofactors and vitamins were among the most abundant functional classes belonging to level 2 (Fig. 7). To further detect the significantly different abundances of metabolic pathways between groups, we used a t-test to compare the functional pathways. We found there were 16 significantly different pathways of Kyoto Encyclopedia of Genes and Genomes (KEGG) categories in the midgut microbiota between Ae. albopictus and Ae. aegypti (Fig. 7a). The abundances of the level 2 KEGG pathways, such as amino acid metabolism, xenobiotic biodegradation and metabolism, global and overview maps, endocrine and metabolic diseases, immune diseases, neurodegenerative diseases and nucleotide metabolism, were significantly different. There were 33 significantly different KEGG pathways of microbiota composition in the entire bodies of female Ae. albopictus and Ae. aegypti (Fig. 7b). Significantly different pathways, such as excretory system, xenobiotic biodegradation and metabolism, neurodegenerative diseases, amino acid metabolism and nucleotide metabolism, were noted. In addition, there were 24, 29 and 248 significantly different level 3 KEGG pathways in the comparisons between ALFM and AEFM (Additional file 3: Figure S2), between ALMW and AEMW (Additional file 4: Figure S3) and between ALFW and AEFW (Additional file 5: Figure S4), respectively. Our findings showed that the difference in predicted functions of midgut microbiota was smaller than that of the entire body microbiota of Aedes mosquitoes under the same rearing conditions.
Fig. 7.

Significantly different distribution of level 2 of predicted functional categories between groups (P < 0.05). a Sixteen items of functional category between AEFM and ALFM. b Thirty-three items of functional category between AEFW and ALFW. AEFM: midgut of female Ae. aegypti. ALFM: midgut of female Ae. Albopictus. AEFW: entire body of female Ae. aegypti. ALFW: entire body of female Ae. albopictus
Co-occurrence between the genus Wolbachia and other microbes of Ae. albopictus
The genera Bacillus, Methylobacterium and Wolbachia were dominant microbes in Ae. albopictus (Additional file 6: Figure S5). The bacterium Wolbachia plays an important role in blocking the transmission of Zika and dengue virus. Therefore, we built a network analysis to further investigate the co-occurrence networks of the genus Wolbachia and bacterial communities in Ae. albopictus (Fig. 8). The results showed that the bacterial community of the entire body of male Ae. albopictus had more edges than that of female Ae. albopictus. The number of edges observed in the entire body of microbiota of mosquito groups was greater than that in the midgut of Ae. albopictus. We found that the genus Wolbachia was negatively correlated with Solimonas, Rhizobium, Pseudonocardia and Paracoccus (Fig. 8a). Network analysis of the microbiota community of the entire body of female Ae. albopictus showed that the microbe Wolbachia was positively associated with Wautersiella, Undibacterium, Rothia, Rickettsia, Porphyrobacter, Niabella, Methylobacterium, Flavobacterium, Enterococcus, Devosia and Cloacibacterium and negatively associated with Sporocytophaga, Roseomonas, Paracoccus, Microvirga, Microbacterium, Lysinibacillus, Incertae Sedis, Elizabethkingia and Bacillus (Fig. 8b). In addition, the associations of the bacterial community in the male Ae. albopictus between Wolbachia and Sphingobacterium, Rhodovulum, Lactobacillus, Flectobacillus, Delftia, Blastomonas and Acinetobacter were positive, while Wolbachia was negatively associated with Porphyrobacter, Paenibacillus, Microbacterium, Methylobacterium, Kocuria, Incertae Sedis, Asaia, Aeromonas and Aeromicrobium (Fig. 8c). Overall, there was strong and complex connectivity between Wolbachia and other microbes, indicating competing relationships in the bacterial communities of Ae. albopictus.
Fig. 8.

Network analysis between genus Wolbachia and bacterial communities of Ae. albopictus. a In the midgut of female mosquitoes containing 101 nodes and 379 edges. b In the entire female mosquitoes containing 111 nodes and 458 edges. c In the entire male mosquitoes containing 115 nodes and 607 edges. All edges are statistically significant (P < 0.05). The red circle represents these microbiota, which were positively associated with the genus Wolbachia. The gray circle represents a negative correlation between the genus Wolbachia and the microbes
Discussion
Aedes aegypti and Ae. albopictus are among the most unneglected vectors of arboviral diseases and are considered primary public health issues [2, 35]. In addition, Ae. aegypti, as an invasive species, has already spread in Guangdong, Yunnan and Hainan provinces in recent years [36] and may transmit pathogens causing microcephaly and other public health problems [37–39]. As the dominant species of mosquitoes in South China, Ae. albopictus caused outbreaks of dengue fever in tens of thousands of patients in Guangdong Province in 2014 [40–42]. The gut microbiota, which plays an important role in interrupting disease transmission, affects the interaction between the host and pathogens, such as ZIKV and Plasmodium [5, 15–17]. Therefore, it is necessary to investigate the diversity and composition of the microbiota in Aedes species in South China. In the present study, we aimed to detect and compare the bacterial communities and diversity in both the entire body and midgut of Aedes mosquitoes using the V3-V4 variable region of 16S rRNA gene sequencing.
Previous studies showed that the microbiota of mosquitoes were significantly affected by the geographical location [20, 43]. Mosquitoes collected from different geographical regions and habitats harbored significantly diverse microbiota [26, 43, 44]. These previous results indicated that the mosquitoes collected from Hainan and Guangdong provinces may harbor a diverse and distinct composition and diversity of microbiota. The mosquitoes were reared under laboratory conditions for > 30 generations. However, how the midgut and entire body microbiota change when shaped by the laboratory environment is unclear.
We found a lower bacterial diversity of the entire body of Ae. aegypti mosquitoes compared to that of Ae. albopictus. The dominant phyla in both Ae. aegypti and Ae. albopictus were Proteobacteria and Bacteroidetes, the results of which were similar to previous research [45]. The results of similarly dominant phyla of both the entire body and the midgut in Aedes mosquitoes indicated that the main microbiota did not depend on the organ, sex or species.
We cataloged the genus microbiota distribution in each group. Common microbes in the entire bodies of both male and female Ae. aegypti mainly included seven genera: Leptothrix, Methylobacterium, Enterobacter, Methylotenera, Uncultured bacteria, Escherichia Shigella and Sphingomonas. The relative richness of these microbes was > 1%. Meanwhile, the relative abundance of all microbes in Ae. aegypti was < 15%, suggesting that the bacterial composition of Ae. aegypti was rather decentralized, and no microbes were absolutely dominant. In addition, the genera Wolbachia (37.72% and 78.67%, respectively), Methylobacterium and Enterobacter were the common microbes in the entire bodies of both male and female Ae. albopictus, showing higher relative abundances than the genera in Ae. aegypti. These results revealed that the bacterial composition of Ae. albopictus is concentrated in a few genera, such as Wolbachia. Interestingly, the genus Bacillus (phylum: Firmicutes) was the second most dominant bacteria in male Ae. albopictus, while the female mosquito harbored only a few, and this bacterium existed in other species, as reported in previous studies [12, 45].
A previous study revealed that environmental factors dominated host genetics in shaping the host gut microbiota [46]. Recent studies have suggested that a diversity of bacteria-colonizing mosquitoes is acquired from aquatic larval habitats [23, 43]. The environment is an essential factor in researching the bacterial diversity and composition of the host. In addition, the surface of the insect is in contact with the environment and harbors a diverse microbiota [22], and the entire body also houses many microbes [22]. However, the relationship and difference between the entire body and the midgut microbiota of the Aedes mosquito are still unclear. How the entire body and the midgut microbiota change under the same living conditions was not clearly revealed. We found a similar midgut microbiota of Ae. aegypti and Ae. albopictus compared with their entire body microbiota. Our findings revealed that the bacterial diversity and community structures in the midgut of Ae. aegypti and Ae. albopictus were highly similar and significantly different from those in their entire bodies. Our results suggested that the composition and structure of the midgut of Aedes were not dependent on the species or genetic type and that the microbiota shaping in tissue by the environment was more than that in the entire body of the Aedes mosquito. Environmental factors can be neglected in microorganism studies of vector biology in both the tissue and the entire body.
Characterization of the functional KEGG pathways detected by the PICRUSt tool [33] is a key element in microbiome research. In this study, the functional genes of the microbiomes of Aedes were identified. The level 1 KEGG pathways mainly included metabolism, environmental information processing, genetic information processing, cellular processes, human diseases and organismal systems. These predicted functions were usually detected in the bacterial communities of water and sediments [47]. The study revealed that the metabolism of bacterial communities of fishes is involved in metabolic pathways, environmental information processing and genetic information processing [48]. Previous studies have revealed the importance of microbial communities in nutrient cycling [49] and their metabolites for hosts [50–53]. The microbiota also contributes to the nutrition of insects in different ways [13]. The midgut microbiota can produce compounds such as vitamins, amino acids and sterol, which are directly assimilated by the host [13, 54, 55]. Our results revealed that bacterial symbionts in Aedes were associated with the second level of metabolism, including carbohydrate metabolism, global and overview maps, amino acid metabolism, energy metabolism and metabolism of cofactors and vitamins. These metabolic pathways are involved in essential elements for hosts, such as energy supply, amino acid production, cofactors and vitamins. Interestingly, the metabolism levels of bacterial communities in the entire bodies of both male Ae. aegypti and Ae. albopictus were similar, while they were significantly different in the bacterial compositions of female Ae. aegypti and Ae. albopictus mosquitoes. However, the mechanism of this result needs to be further tested. Our findings revealed that the difference in predicted functions of midgut microbiota was smaller than that of the entire body microbiota of Aedes mosquitoes under the same rearing conditions, showing a similar level on metabolism genes of microbiota. In short, our study suggested that the metabolic functions of microbiota shaped by the environment in tissue were greater than those in the entire body of Aedes mosquitoes. However, functional gene prediction based on the 16S rRNA gene was mainly applied to investigate the metabolic pathways of environmental microbiota communities [47]. In our work, we aimed to explain the relationship and difference between the midgut and the entire body microbiota at the molecular level. More experiments and techniques are needed to fully characterize the bacterial metabolism of Aedes mosquitoes.
Network analysis represents the complexity of ecological relationships among microbial communities [56, 57]. Previous studies have revealed that Wolbachia could block the transmission of Zika and dengue virus [5, 58, 59] and decrease the density of mosquitoes, resulting in disease interruption [60–63], implying the important role of Wolbachia in Aedes. Here, our study also revealed the dominant genus Wolbachia. Our data showed that the relative abundance of Wolbachia in Ae. albopictus was significantly associated with a few microbes. We found that Wolbachia was positively or negatively associated with some microorganisms throughout and in the midgut of Ae. albopictus. These results may help us to promote the design of vectors and disease control by modulating the richness of Wolbachia.
Conclusions
In this study, we revealed the composition and diversity of microbiota of Ae. aegypti and Ae. albopictus from South China. Our finding that Ae. aegypti and Ae. albopictus reared in the same laboratory harbor a similar midgut bacterial microbiome but different entire body microbiota implies that the gut microbiota of adult mosquitoes is environmentally determined regardless of the host genotype, but the entire body microbiota was influenced more by genotype and less by environment. Our study could provide a further understanding of the aspects of the microbiome of Aedes mosquitoes.
Supplementary Information
{kind=link}
Additional file 1: Figure S1. Map showing the sampling site of Ae. aegypti mosquitoes in Hainan province and the sampling site of Ae. albopictus in Guangdong province in 2003. Red triangles represent the sampling sites.
Additional file 2: Table S1. Samples of Aedes colonies reared in laboratory conditions included in this study.
{kind=link}
Additional file 3: Figure S2. Significantly different distribution of level 3 of predicted functional categories between AEFM and ALFM (P < 0.05).
{kind=link}
Additional file 4: Figure S3. Significantly different distribution of level 3 of predicted functional categories between AEMW and ALFW (P < 0.05)
{kind=link}
Additional file 5: Figure S4. Significantly different distribution of level 3 of predicted functional categories between AEFW and ALFW (P < 0.05).
{kind=link}
Additional file 6: Figure S5. Relative abundance of dominant bacteria of Ae. albopictus compared with each group (*P < 0.05, ***P < 0.0001). (A) Phylum Firmicutes and genus Bacillus. (B) Genus Methylobacterium. (C) Genus Wolbachia.
Acknowledgements
We thank Dr. Heinz Mehlhorn, a German scientist, for proofreading this article.
Abbreviations
- WHO
World Health Organization
- AEMW
Entire mosquito of male Ae. aegypti
- AEFW
Entire mosquito of female Ae. aegypti
- AEFM
Midgut of female Ae. aegypti
- ALMW
Entire mosquito of male Ae. albopictus
- ALFW
Entire mosquito of female Ae. albopictus
- ALFM
Midgut of female Ae. albopictus
- AE
Ae. aegypti
- AL
Ae. albopictus
- NMDS
The nonmetric multidimensional scaling
- PICRUSt
Phylogenetic investigation of communities by reconstruction of unobserved states
- OTUs
Operational taxonomic units
- UPGMA
The unweighted pair-group method with arithmetic mean analysis
- KEGG
Kyoto Encyclopedia of Genes and Genomes
Authors' contributions
ZW and XS conceived and designed the study. XZ and TD gave constructive suggestions. DL carried out the experiments, prepared the manuscript and handled statistical analysis and interpretation of the data. BS, TD and DL critically revised the draft version of the paper. All authors read and approved the final manuscript.
Funding
This work was supported by the National Key R&D Program of China (nos. 2020YFC1200100 and 2016YFC1200500), the Natural Science Foundation of Guangdong Province (nos. 2019A1515012068, 2020A1515010896 and 2021A1515010976), the National Natural Science Foundation of China (nos. 82072308 and 82002168), the 6th Nuclear Energy R&D Project (20201192) and the 111 Project (no. B12003). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Availability of data and materials
Please contact the author for additional data requests.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Datao Lin, Email: lindt5@mail.sysu.edu.cn.
Xiaoying Zheng, Email: zhengxy@mail.sysu.edu.cn.
Benjamin Sanogo, Email: 2058548220@qq.com.
Tao Ding, Email: dingt8@mail.sysu.edu.cn.
Xi Sun, Email: sunxi2@mail.sysu.edu.cn.
Zhongdao Wu, Email: wuzhd@mail.sysu.edu.cn.
References
- 1.Bonizzoni M, Gasperi G, Chen X, James AA. The invasive mosquito species Aedes albopictus: current knowledge and future perspectives. Trends Parasitol. 2013;29(9):460–468. doi: 10.1016/j.pt.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ducheyne E, Tran MN, Haddad N, Bryssinckx W, Buliva E, Simard F, et al. Current and future distribution of Aedes aegypti and Aedes albopictus (Diptera: Culicidae) in WHO Eastern Mediterranean Region. Int J Health Geogr. 2018;17(1):4. doi: 10.1186/s12942-018-0125-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen H, Li K, Wang X, Yang X, Lin Y, Cai F, et al. First identification of kdr allele F1534S in VGSC gene and its association with resistance to pyrethroid insecticides in Aedes albopictus populations from Haikou City, Hainan Island, China. Infect Dis Poverty. 2016;5:31. doi: 10.1186/s40249-016-0125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Callender DM. Factors contributing to and strategies to combat emerging arboviruses. Glob Public Health. 2018;13:1–7. doi: 10.1080/17441692.2018.1464588. [DOI] [PubMed] [Google Scholar]
- 5.Dutra HLC, Rocha MN, Dias FBS, Mansur SB, Caragata EP, Moreira LA. Wolbachia blocks currently circulating Zika virus isolates in Brazilian Aedes aegypti mosquitoes. Cell Host Microbe. 2016;19(6):771–774. doi: 10.1016/j.chom.2016.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suthar MS, Diamond MS, Gale MJ. West Nile virus infection and immunity. Nat Rev Microbiol. 2013;11(2):115–128. doi: 10.1038/nrmicro2950. [DOI] [PubMed] [Google Scholar]
- 7.Melan A, Aung MS, Khanam F, Paul SK, Riaz BK, Tahmina S, et al. Molecular characterization of chikungunya virus causing the 2017 outbreak in Dhaka, Bangladesh. New Microbes New Infect. 2018;24:14–16. doi: 10.1016/j.nmni.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leta S, Beyene TJ, De Clercq EM, Amenu K, Kraemer M, Revie CW. Global risk mapping for major diseases transmitted by Aedes aegypti and Aedes albopictus. Int J Infect Dis. 2018;67:25–35. doi: 10.1016/j.ijid.2017.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khan A, Hassan M, Imran M. Estimating the basic reproduction number for single-strain dengue fever epidemics. Infect Dis Poverty. 2014;3:12. doi: 10.1186/2049-9957-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo L, Jiang LY, Xiao XC, Di B, Jing QL, Wang SY, et al. The dengue preface to endemic in mainland China: the historical largest outbreak by Aedes albopictus in Guangzhou, 2014. Infect Dis Poverty. 2017;6(1):148. doi: 10.1186/s40249-017-0352-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ridde V, Agier I, Bonnet E, Carabali M, Dabire KR, Fournet F, et al. Presence of three dengue serotypes in Ouagadougou (Burkina Faso): research and public health implications. Infect Dis Poverty. 2016;5:23. doi: 10.1186/s40249-016-0120-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yadav KK, Datta S, Naglot A, Bora A, Hmuaka V, Bhagyawant S, et al. Diversity of cultivable midgut microbiota at different stages of the Asian tiger mosquito, Aedes albopictus from Tezpur, India. PLoS ONE. 2016;11(12):e167409. doi: 10.1371/journal.pone.0167409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Minard G, Mavingui P, Moro CV. Diversity and function of bacterial microbiota in the mosquito holobiont. Parasite Vector. 2013;6(1):146. doi: 10.1186/1756-3305-6-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ponnusamy L, Boroczky K, Wesson DM, Schal C, Apperson CS. Bacteria stimulate hatching of yellow fever mosquito eggs. PLoS ONE. 2011;6(9):e24409. doi: 10.1371/journal.pone.0024409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang S, Dos-Santos A, Huang W, Liu KC, Oshaghi MA, Wei G, et al. Driving mosquito refractoriness to Plasmodium falciparum with engineered symbiotic bacteria. Science. 2017;357(6358):1399–1402. doi: 10.1126/science.aan5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong Y, Manfredini F, Dimopoulos G. Implication of the mosquito midgut microbiota in the defense against malaria parasites. PLoS Pathog. 2009;5(5):e1000423. doi: 10.1371/journal.ppat.1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang S, Ghosh AK, Bongio N, Stebbings KA, Lampe DJ, Jacobs-Lorena M. Fighting malaria with engineered symbiotic bacteria from vector mosquitoes. Proc Natl Acad Sci USA. 2012;109(31):12734–12739. doi: 10.1073/pnas.1204158109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Gilbreath TR, Kukutla P, Yan G, Xu J. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS ONE. 2011;6(9):e24767. doi: 10.1371/journal.pone.0024767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zotzmann S, Steinbrink A, Schleich K, Frantzmann F, Xoumpholphakdy C, Spaeth M, et al. Bacterial diversity of cosmopolitan Culex pipiens and invasive Aedes japonicus from Germany. Parasitol Res. 2017;116(7):1899–1906. doi: 10.1007/s00436-017-5466-2. [DOI] [PubMed] [Google Scholar]
- 20.Coon KL, Brown MR, Strand MR. Mosquitoes host communities of bacteria that are essential for development but vary greatly between local habitats. Mol Ecol. 2016;25(22):5806–5826. doi: 10.1111/mec.13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coon KL, Vogel KJ, Brown MR, Strand MR. Mosquitoes rely on their gut microbiota for development. Mol Ecol. 2014;23(11):2727–2739. doi: 10.1111/mec.12771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park R, Dzialo MC, Spaepen S, Nsabimana D, Gielens K, Devriese H, et al. Microbial communities of the house fly Musca domestica vary with geographical location and habitat. Microbiome. 2019;7(1):147. doi: 10.1186/s40168-019-0748-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guegan M, Zouache K, Demichel C, Minard G, Tran VV, Potier P, et al. The mosquito holobiont: fresh insight into mosquito-microbiota interactions. Microbiome. 2018;6(1):49. doi: 10.1186/s40168-018-0435-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiss B, Aksoy S. Microbiome influences on insect host vector competence. Trends Parasitol. 2011;27(11):514–522. doi: 10.1016/j.pt.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jupatanakul N, Sim S, Dimopoulos G. The insect microbiome modulates vector competence for arboviruses. Viruses. 2014;6(11):4294–4313. doi: 10.3390/v6114294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muturi EJ, Kim CH, Bara J, Bach EM, Siddappaji MH. Culex pipiens and Culex restuans mosquitoes harbor distinct microbiota dominated by few bacterial taxa. Parasit Vectors. 2016;9:18. doi: 10.1186/s13071-016-1299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma P, Sharma S, Maurya RK, Das De T, Thomas T, Lata S, et al. Salivary glands harbor more diverse microbial communities than gut in Anopheles culicifacies. Parasite Vector. 2014;7(1):235. doi: 10.1186/1756-3305-7-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324(5931):1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kambura AK, Mwirichia RK, Kasili RW, Karanja EN, Makonde HM, Boga HI. Bacteria and archaea diversity within the hot springs of Lake Magadi and Little Magadi in Kenya. BMC Microbiol. 2016;16(1):136. doi: 10.1186/s12866-016-0748-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langille MG, Zaneveld J, Caporaso JG, Mcdonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friedman J, Alm EJ. Inferring correlation networks from genomic survey data[J] PLoS Comput Biol. 2012;8(9):e1002687. doi: 10.1371/journal.pcbi.1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asshauer KP, Wemheuer B, Daniel R, Meinicke P. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics. 2015;31(17):2882–2884. doi: 10.1093/bioinformatics/btv287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014;30(21):3123–3124. doi: 10.1093/bioinformatics/btu494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kraemer MU, Sinka ME, Duda KA, Mylne AQ, Shearer FM, Barker CM, et al. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. Elife. 2015;4:e8347. doi: 10.7554/eLife.08347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi QM, Zhang HD, Wang G, Guo XX, Xing D, Dong YD, et al. The genetic diversity and population structure of domestic Aedes aegypti (Diptera: Culicidae) in Yunnan Province, southwestern China. Parasit Vectors. 2017;10(1):292. doi: 10.1186/s13071-017-2213-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yuan L, Huang XY, Liu ZY, Zhang F, Zhu XL, Yu JY, et al. A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science. 2017;358(6365):933–936. doi: 10.1126/science.aam7120. [DOI] [PubMed] [Google Scholar]
- 38.Christian KA, Iuliano AD, Uyeki TM, Mintz ED, Nichol ST, Rollin P, et al. What we are watching-top global infectious disease threats, 2013–2016: an update from CDC's global disease detection operations center. Health Secur. 2017;15(5):453–462. doi: 10.1089/hs.2017.0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monath TP. Yellow fever: an update. Lancet Infect Dis. 2001;1(1):11–20. doi: 10.1016/S1473-3099(01)00016-0. [DOI] [PubMed] [Google Scholar]
- 40.Yiguan W, Xin L, Chengling L, Su T, Jianchao J, Yuhong G, et al. A survey of insecticide resistance in Aedes albopictus (Diptera: Culicidae) during a 2014 dengue fever outbreak in Guangzhou, China. J Econ Entomol. 2017;110(1):239–244. doi: 10.1093/jee/tow254. [DOI] [PubMed] [Google Scholar]
- 41.Shen JC, Luo L, Li L, Jing QL, Ou CQ, Yang ZC, et al. The impacts of mosquito density and meteorological factors on dengue fever epidemics in Guangzhou, China, 2006–2014: a time-series analysis. Biomed Environ Sci. 2015;28(5):321–329. doi: 10.3967/bes2015.046. [DOI] [PubMed] [Google Scholar]
- 42.Li MT, Sun GQ, Yakob L, Zhu HP, Jin Z, Zhang WY. The driving force for 2014 dengue outbreak in Guangdong, China. PLoS ONE. 2016;11(11):e166211. doi: 10.1371/journal.pone.0166211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bascunan P, Nino-Garcia JP, Galeano-Castaneda Y, Serre D, Correa MM. Factors shaping the gut bacterial community assembly in two main Colombian malaria vectors. Microbiome. 2018;6(1):148. doi: 10.1186/s40168-018-0528-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Osei-Poku J, Mbogo CM, Palmer WJ, Jiggins FM. Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Mol Ecol. 2012;21(20):5138–5150. doi: 10.1111/j.1365-294X.2012.05759.x. [DOI] [PubMed] [Google Scholar]
- 45.Yadav KK, Bora A, Datta S, Chandel K, Gogoi HK, Prasad GBKS, et al. Molecular characterization of midgut microbiota of Aedes albopictus and Aedes aegypti from Arunachal Pradesh, India. Parasites Vectors. 2015;8(1):1. doi: 10.1186/s13071-015-1252-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555(7695):210–215. doi: 10.1038/nature25973. [DOI] [PubMed] [Google Scholar]
- 47.Abia A, Alisoltani A, Keshri J, Ubomba-Jaswa E. Metagenomic analysis of the bacterial communities and their functional profiles in water and sediments of the Apies River, South Africa, as a function of land use. Sci Total Environ. 2018;616–617:326–334. doi: 10.1016/j.scitotenv.2017.10.322. [DOI] [PubMed] [Google Scholar]
- 48.Baldo L, Pretus JL, Riera JL, Musilova Z, Bitja NA, Salzburger W. Convergence of gut microbiotas in the adaptive radiations of African cichlid fishes. ISME J. 2017;11(9):1975–1987. doi: 10.1038/ismej.2017.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wobus A, Bleul C, Maassen S, Scheerer C, Schuppler M, Jacobs E, et al. Microbial diversity and functional characterization of sediments from reservoirs of different trophic state. FEMS Microbiol Ecol. 2003;46(3):331–347. doi: 10.1016/S0168-6496(03)00249-6. [DOI] [PubMed] [Google Scholar]
- 50.Shoaie S, Ghaffari P, Kovatcheva-Datchary P, Mardinoglu A, Sen P, Pujos-Guillot E, et al. Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 2015;22(2):320–331. doi: 10.1016/j.cmet.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 51.Xu J, Galley JD, Bailey MT, Thomas-Ahner JM, Clinton SK, Olivo-Marston SE. The impact of dietary energy intake early in life on the colonic microbiota of adult mice. Sci Rep. 2016;6:19083. doi: 10.1038/srep19083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500(7464):541–546. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 53.Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, Le Chatelier E, et al. Dietary intervention impact on gut microbial gene richness. Nature. 2013;500(7464):585–588. doi: 10.1038/nature12480. [DOI] [PubMed] [Google Scholar]
- 54.Aksoy S. Tsetse - A Haven for Microorganisms. Parasitol Today. 2000;16(3):114–118. doi: 10.1016/s0169-4758(99)01606-3. [DOI] [PubMed] [Google Scholar]
- 55.Douglas AE. Nutritional interactions in insect-microbial symbioses: aphids and their symbiotic bacteria Buchnera. Ann Rev Entomol. 1998;43(1):17–37. doi: 10.1146/annurev.ento.43.1.17. [DOI] [PubMed] [Google Scholar]
- 56.Leite D, Salles JF, Calderon EN, Castro CB, Bianchini A, Marques JA, et al. Coral bacterial-core abundance and network complexity as proxies for anthropogenic pollution. Front Microbiol. 2018;9:833. doi: 10.3389/fmicb.2018.00833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benjamino J, Lincoln S, Srivastava R, Graf J. Low-abundant bacteria drive compositional changes in the gut microbiota after dietary alteration. Microbiome. 2018;6(1):86. doi: 10.1186/s40168-018-0469-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Caragata E, Dutra H, Moreira L. Inhibition of Zika virus by Wolbachia in Aedes aegypti. Microbial Cell. 2016;3(7):293–295. doi: 10.15698/mic2016.07.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walker T, Johnson PH, Moreira LA, Iturbe-Ormaetxe I, Frentiu FD, Mcmeniman CJ, et al. The wMel Wolbachia strain blocks dengue and invades caged Aedes aegypti populations. Nature. 2011;476(7361):450–453. doi: 10.1038/nature10355. [DOI] [PubMed] [Google Scholar]
- 60.Baldini F, Segata N, Pompon J, Marcenac P, Robert Shaw W, Dabiré RK, et al. Evidence of natural Wolbachia infections in field populations of Anopheles gambiae. Nat Commun. 2014;5:1. doi: 10.1038/ncomms4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xi Z, Khoo CC, Dobson SL. Wolbachia establishment and invasion in an Aedes aegypti laboratory population. Science. 2005;310(5746):326–328. doi: 10.1126/science.1117607. [DOI] [PubMed] [Google Scholar]
- 62.Bian G, Joshi D, Dong Y, Lu P, Zhou G, Pan X, et al. Wolbachia invades Anopheles stephensi populations and induces refractoriness to Plasmodium infection. Science. 2013;340(6133):748–751. doi: 10.1126/science.1236192. [DOI] [PubMed] [Google Scholar]
- 63.Zheng X, Zhang D, Li Y, Yang C, Wu Y, Liang X, et al. Incompatible and sterile insect techniques combined eliminate mosquitoes. Nature. 2019;572(7767):56–61. doi: 10.1038/s41586-019-1407-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Map showing the sampling site of Ae. aegypti mosquitoes in Hainan province and the sampling site of Ae. albopictus in Guangdong province in 2003. Red triangles represent the sampling sites.
Additional file 2: Table S1. Samples of Aedes colonies reared in laboratory conditions included in this study.
Additional file 3: Figure S2. Significantly different distribution of level 3 of predicted functional categories between AEFM and ALFM (P < 0.05).
Additional file 4: Figure S3. Significantly different distribution of level 3 of predicted functional categories between AEMW and ALFW (P < 0.05)
Additional file 5: Figure S4. Significantly different distribution of level 3 of predicted functional categories between AEFW and ALFW (P < 0.05).
Additional file 6: Figure S5. Relative abundance of dominant bacteria of Ae. albopictus compared with each group (*P < 0.05, ***P < 0.0001). (A) Phylum Firmicutes and genus Bacillus. (B) Genus Methylobacterium. (C) Genus Wolbachia.
Data Availability Statement
Please contact the author for additional data requests.