

Abstract
Diet-induced obesity is associated with impaired B-cell-driven humoral immunity, which coincides with chronic inflammation and has consequences for responses to infections and vaccinations. Key nutritional, cellular, and molecular mechanisms by which obesity may impair aspects of humoral immunity such as B cell development, class switch recombination, and formation of long-lived antibody secreting cells are reviewed. A key theme to emerge is the central role of white adipose tissue on the formation and function of pro-inflammatory B cell subsets that exacerbate insulin resistance. The underlying role of select hormones such as leptin is highlighted, which may be driving the formation of pro-inflammatory B cells in the absence of antigen stimulation. This review also extensively covers the regulatory role of lipid metabolites such as prostaglandins and specialized pro-resolving mediators (SPMs) that are synthesized from polyunsaturated fatty acids. Notably, SPM biosynthesis is impaired in obesity and contributes toward impaired antibody production. Future directions for research, including avenues for therapeutic intervention, are included.
Keywords: antibodies, B cells, diet-induced obesity, pro-resolving mediators
Graphical Abstract
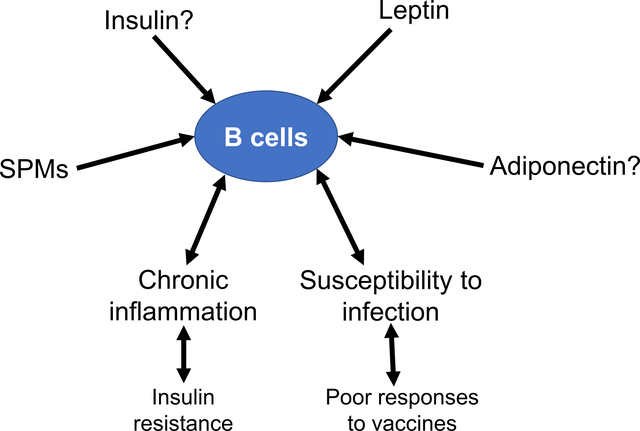
This review article covers experimental data to show that certain hormones and fatty acids have a strong influence on key aspects of inflammation in obesity. We specifically cover results from human and rodent studies that suggest the hormone leptin, which is elevated in obesity, may be driving B lymphocytes to become pro-inflammatory. We also discuss how molecules made from polyunsaturated fatty acids regulate B cell function. Collectively, the review has implications for our understanding of why obese individuals display chronic inflammation and increased susceptibility to infections. The data covered in this review set the basis for the future design of therapeutic strategies for improving immunological outcomes in the obese.
1. Introduction
Dysregulation of innate and adaptive immunity contributes toward the pathogenesis of obesity and its comorbidities. This review focuses on how diet-induced obesity impairs humoral immunity. One of the primary functions of the humoral immune response is to ensure the production of long-lived memory B cells and plasma cells that produce high-affinity, isotype-switched antibodies essential for host defense. Impairments in antibody production in obesity, driven by chronic inflammation, are likely contributing toward increased susceptibility to infection and decreased responses to vaccinations. We specifically review emerging evidence on how specific hormones and nutritional factors dysregulate key aspects of B cell-driven humoral immunity. We also cover recent evidence that metabolites synthesized from polyunsaturated fatty acids (PUFAs) are regulators of the B cell functional response and may have therapeutic potential for improving outcomes related to chronic inflammation and infection.
2. Obesity Impairs B Cell Driven Humoral Immunity in the Context of Inflammation and Infection
2.1. B Cell Development Is Dysregulated in Obesity
Proper B cell development, maturation, and differentiation into long-lived plasma and memory B cells are critical in generating a diverse antibody repertoire that can elicit a strong immune response. Detrimental changes in B cell development can negatively impact humoral immunity. Currently, there is conflicting evidence as to whether obesity enhances or compromises B cell development in the bone marrow. To exemplify, Trottier et al. found that C57BL/6 mice fed a 45% kcal high fat diet for 180 days had a sustained increase in the total number of B cells after 90 days. Furthermore, this study showed that bone marrow cells isolated from mice on a high fat diet for 157 days had increased numbers of pre, immature, and mature B cells compared to their control counterparts.[1] On the contrary, 7-week old C57BL/6 mice that were fed a 60% kcal high fat diet for 2 days, 1 week, and 6 weeks showed B cell numbers were lowered beginning at week 1 of a high fat diet intervention. B cell development markers such as IL-7, EBF1, and Pax5 were notably reduced in mice receiving the high fat diet compared to the control mice.[2] Similarly, long-term high fat administration for 7 months reduced B cell numbers in the bone marrow by 52% and in the blood by 36% compared to control mice.[3] Kosaraju et al. demonstrated that the number of B cells in the bone marrow were lowered in obese male mice although circulating levels of B cells in obese humans were elevated.[4] Conflicting results from these studies could potentially be attributed to differences in experimental diets, the duration of the feeding protocol, and methods used to quantify the number of B cell in the bone marrow. Overall, how obesity dysregulates B cell development is an area of investigation that remains in its infancy but is critical to address.
2.2. B Cells Infiltrate White Adipose Tissue and Contribute toward Chronic Inflammation
In response to excess caloric intake, the adipose tissue undergoes drastic remodeling that is characterized by rapid and massive expansion, changes in extracellular matrix components, secretion of adipokines and hormones, blood vasculature, and infiltration of various immune cells that produce cytokines and chemokines upon activation, which contributes to chronic inflammation.[5–8] B cells traffic to and infiltrate the visceral adipose tissue (VAT) from other tissues during the course of obesity. B cells accumulate in the VAT as early as 3 weeks after initiation of a high fat diet with those numbers remaining constant over the course of the 12-week study.[9] Furthermore, B cells infiltrate the VAT before T cell infiltration, which occurs after 6 weeks of a high fat diet.[9] Total B cell numbers are increased in the VAT of obese mice relative to their control counterparts.[10] Obese mice are reported to have increased follicular B cells, B1a, and B2 cells in the VAT after 6–12 weeks of a high fat diet.[10]
Despite their early recruitment, B cells are not the first immune cells to infiltrate the VAT. Neutrophils have been observed to directly adhere to adipocytes in the VAT of mice 3 days after initiation of a high fat diet.[11] Furthermore, a subpopulation of macrophages (F4/80+CD11b+CD11c+) were detected in the VAT after just 1 week of consuming an obesogenic diet.[12] In addition to their early recruitment to the VAT, B cells can also surround stressed or dying adipocytes in crown-like structures along with other immune cells such as macrophages and T cells.[10] Within these crown-like structures, B cells modulate macrophage and T cell function as well as sample antigens.[10] McDonnell et al. used immunohistochemistry to identify B cells within crown-like structure of subcutaneous adipose tissue and in the perivascular space in both male and female obese subjects. In over 50% of the subjects used in the study, B cells in the subcutaneous crown like structures were relatively more numerous than T cells.[13] Although there is no direct evidence, it is likely that the inflammatory environment of the adipose tissue is contributing toward B cells assuming a pro-inflammatory phenotype, which appears to manifest with a unique metabolic phenotype.[13] Furthermore, it remains to be determined if B cells also accumulate in other depots of fat.
2.3. Hyper-Stimulated B Cells Have Impaired B Cell Cytokine Secretion and Antibody Production
B cells undergo drastic phenotypic and functional changes upon the initiation of high fat diet. B cells from obese mice have increased class switching to IgG+ cells, especially to proinflammatory IgG2c in the VAT as a consequence of increased fat mass, hypoxia, and apoptosis.[10] Furthermore, total B cells from both young and elderly individuals with obesity show higher immune activation as measured by increased intracellular TNFα before stimulation.[14,15]
The percentage of anti-inflammatory B cell subsets (transitional B cells) is lowered and the percentage of proinflammatory late/exhausted memory B cells is increased with obesity.[15] Upon stimulation with CpG, total B cells from these obese individuals have decreased levels of activation-induced cytidine deaminase (AID), a functional measure of class switch recombination. In addition, culture supernatants collected from challenged B cells have increased secretion of IL-6 and decreased secretion of the anti-inflammatory cytokine IL-10 in obese subjects, compared to lean individuals.[15] In another study with obese male subjects, B-cell IL-6 secretion was decreased while IgM, but not IgG, levels were elevated upon ex-vivo challenge with anti-BCR/TLR9 stimulation.[4] Some of these effects may be sex specific. Very recent work by Crouch et al. shows that obese female subjects have elevated levels of ex vivo B cell IL-10 and TNFα upon BCR/TLR9 stimulation with no effect on IgM or IgG levels in culture.[16] Overall, the emerging viewpoint is that hyperstimulated B cells in obese individuals function sub-optimally and perhaps some of these effects are sex-specific, which remains to be investigated in greater depth.[4]
2.4. Obesity-Induced Insulin Resistance and B Cell Function
Obesity is a critical risk factor in the development of insulin resistance.[17] B cells have emerged as mediators of insulin resistance via several mechanisms including modulation of cytokine production, T cells, and antibody production. To elaborate, IL-10 appears to have a protective role in insulin resistance by reducing macrophage and cytokine responses.[18] IL-10 secretion by B cells is decreased in the blood of patients with type 2 diabetes as well as upon challenge with toll-like receptor agonists, which could predispose obese individuals to insulin resistance.[19,20] Furthermore, in diet-induced obesity, B cells interact with T cells to induce IFNγ expression, which contributes to local and systemic inflammation and insulin resistance.[10] Likewise, the VAT of B cell-deficient mice compared to controls displayed reduced T-cell IFNγ and IL-17.[21] Furthermore, the role of B cells in brown adipose tissue is unclear and also likely contributes toward the production of differing cytokines.[22]
Metformin, which has been shown to reduce chronic inflammation through lowering of insulin resistance, hyperglycemia, and atherogenic dyslipidemia in obesity has a strong enhancing effect in B cells.[23] One study shows that B cells from obese patients taking metformin have increased transcript levels of AID in response to stimulation with CpG.[23] Similarly, B cells from obese patients treated in vitro with metformin have increased AID levels compared to those that were not treated with the drug. Obese individuals taking metformin display an increase in the percentage of circulating switched memory B cells, decreased percentage of pro-inflammatory exhausted memory B cells, and reduced B-cell intrinsic inflammation via upregulation of AMPK.[23] Notably, obese individuals on metformin have increased in vitro AID response to the influenza vaccine compared to obese individuals not taking the drug.[23]
Insulin resistance also impacts antibody production and isotype. Winer et al. initially demonstrated that total splenic B cells from mice on a high fat diet have increased IgG secretion and reduced IgM production.[10] Furthermore, overweight children that are associated with an unfavorable metabolic phenotype have increased circulating IgG and IgA, and obese mice have increased class-switched, pro-inflammatory IgG2c antibody.[24] Oral feeding of ovalbumin antigen to obese mice induced the production of IgG2c antibody, whereas in lean mice IgG1 was the predominant antibody isotype produced.[25] While obesity impacts antibody production and isotype, it also appears that the antibody isotype has a direct effect on insulin resistance in obesity. Several studies have demonstrated the transfer of IgG antibodies from insulin-resistant mice on a high fat diet to mice on a normal chow diet increases the polarization of classically activated pro-inflammatory macrophages, increases TNFα production, and enhances targeting of gut-derived antigens.[25–27] Overall, these studies suggest that B cells may be a therapeutic target for improving insulin sensitivity and glucose tolerance.
2.5. Antibody Production Is Impaired in the Context of Infection
Several studies suggest that obesity impairs the B cell response during infection.[28,29,4] Milner et al. reported that obese mice had a diminished concentration of influenza-specific antibodies 35 days post-infection.[28] Furthermore, obese mice had lower influenza-specific antibody production compared to lean mice despite increased production of neutralizing and non-neutralizing influenza-specific antibodies upon adjuvant vaccination.[30] In another study, young and elderly individuals had decreased antibody production to the influenza vaccine.[15] Interestingly, vaccines may fail to provide optimal protection in obese individuals despite sufficient production of neutralizing antibodies. Work from Schultz-Cherry’s lab demonstrates that mice receiving an adjuvanted vaccine have a fourfold increase in neutralizing antibody levels that were considered “protective.” However, despite improved seroconversion with the use of the adjuvant, the mice still succumbed to influenza infection.[30] Similarly, a hemagglutination antibody titer of 40 or higher was not seroprotective against influenza in obese individuals and influenza-like illness.[31] These studies demonstrate that supplementing vaccines with adjuvant and boosting antibody levels fails to diminish an obese individual’s susceptibility to infection. However, improving antibody levels remains an important target as this is critical for the optimal response to differing vaccinations.
The effects of obesity on B cells are not just limited to viral infection. Obese/type 2 diabetic mice infected with Staphylococcus aureus have reduced numbers of germinal centers in the popliteal lymph nodes compared to control mice infected with S. aureus.[29] Despite reduced germinal centers, obese mice have increased total number of B cells at day 0 and day 14 post-infection. Obese mice also have decreased numbers of activated B cells on day 7 and day 14 post-infection as well as reduced germinal center B cells by day 14 post-infection.[29] Furthermore, mice challenged with S. aureus display reduced levels of AID accompanied with decreased IgG and IgE antibody production compared to their control counterparts, further demonstrating obese mice have impaired B cell function upon infection.[29]
3. The Role of Adiponectin and Leptin on B Cell Driven Humoral Immunity
Adiponectin and leptin are fat-derived adipokines that undergo drastic changes during the onset of obesity.[32] Adiponectin exerts anti-inflammatory effects and increases insulin sensitivity via effective disposal of glucose from circulation. Adiponectin is decreased in obesity and circulating levels are inversely correlated with percentage of body fat, increasing after weight reduction. Several studies suggest that this inverse relationship between adiponectin and adiposity phenotypes is solely dependent upon the development of insulin resistance.[33–36] Various studies also support the idea that adiponectin plays a protective role during the development and early stages of obesity and energy balance.[35–37] In particular, C57BL/6J mice on a high fat diet have increased circulating adiponectin concentrations during the initial stages of adiposity development before decreasing after 18 weeks of consuming a high fat diet.[38]
Despite its established role in obesity, adiponectin’s effect on B cell activation, maturation, and expansion through the targeting of adiponectin receptors in the context of obesity is poorly understood. It is suggested that adiponectin promotes myelopoiesis and inhibits B lymphopoiesis in long-term bone marrow cultures when early lymphoid progenitors are in contact with stromal cells. This study also reported that prostaglandin E2 (PGE2) had a direct inhibitory influence on purified hematopoietic cells, suggesting that adiponectin can negatively and selectively influence lymphopoiesis through the induction of prostaglandins.[39] Obeid et al. demonstrated that adiponectin knock-out mice receiving dextran sulfate sodium exhibited severe colitis, increased B cell infiltration in the colon, and upregulated B cell activation, which resulted in the secretion of pro-inflammatory cytokines.[40] Therefore, these findings provide evidence that adiponectin can directly modulate B cell function. However, this relationship has not been established in the context of diet-induced obesity.
Conversely, leptin is a pro-inflammatory hormone that is elevated in obesity. Leptin inhibits hunger by physiologically regulating energy balance and is secreted primarily by adipocytes. Circulating levels of leptin are directly correlated with the amount of fat in the body.[41] Leptin signals through the JAK/STAT signaling pathways resulting in translocation of STAT3 into the nucleus and subsequent transcription of leptin-induced genes.[42] Leptin targets the medio-basal nucleus of the hypothalamus, where it inhibits appetite. As a result, leptin deficiency or resistance leads to an energy imbalance, hyperinsulinemia, insulin resistance, uncontrolled food intake, obesity, and type 2 diabetes.[42]
Leptin resistance in obese individuals regulates both the innate and adaptive immune response and contributes to the exacerbated inflammatory environment in obesity.[42] Leptin equally affects immune cells from the innate and adaptive immune system. Almost all immune cells, including B cells, express Ob-R, an isoform of the leptin receptor that is required for leptin signaling.[43–45] Leptin can directly target and modulate B cell responses.[46] For example, leptin signaling promotes B cell survival by inhibiting apoptosis and inducing cell cycle entry through activation of Bcl-2 and cyclin D1.[47] Furthermore, leptin can induce peripheral blood B cells to secrete various cytokines such as IL-6, IL-10, and TNFα.[48]
Leptin also modulates B cell development. Fasted mice, characterized by low leptin levels, displayed decreased numbers of pro-, pre-, and immature B cells and increased numbers of mature B cells in the bone marrow.[49] Continuous leptin administration prevented the reduction in B cells observed in the bone marrow of fasted mice, indicating an important role for leptin in B-cell lymphopoiesis.[49,50] In addition, leptin may be responsible for B cell intrinsic inflammation. B cells from lean individuals treated in vitro with leptin had increased STAT3 phosphorylation (crucial for TNFα production) and decreased phosphorylation of the anti-inflammatory AMPK pathway, which helps mediate an antibody response.[15] As a result, increasing leptin levels associated with obesity could be a potential mechanism responsible for reduced B cell function in obese individuals. An additional avenue for future investigation is the role of insulin on B cell activity as recent data indicate that insulin regulates the CD4+ T cell response to inflammation and infection.[51]
4. The Role of PUFA-Derived Lipid Mediators on the Humoral Immune Response
4.1. Dietary PUFAs Are Precursors to Bioactive Lipid Mediators
Obesity is associated with an increase in free fatty acid levels, which promotes insulin resistance and the development of systemic hyperglycemia.[52–55] Humans consume saturated, monounsaturated, and polyunsaturated fatty acids (PUFA).[56] Some PUFAs cannot be made by humans and must be consumed through the diet, rendering them essential fatty acids.[56] PUFAs are released from membrane phospholipids into the cytosol primarily by the action of the phospholipase A2 and are quickly converted into n-3 and n-6-derived lipid mediator derivatives, fatty acid esters of hydroxy fatty acids, nitroalkenes, and endocannabinoids.[57,58]
Western diets are rich in saturated fats and n-6 PUFAs, and are generally low in n-3 PUFAs.[59,60] Estimated ratios of n-6 PUFAs/n-3 PUFAs in Western diets range from 10:1 to 20:1.[59–61] n-6-derived lipid mediators are often (with exceptions) considered to be pro-inflammatory and pro-thrombotic, whereas n-3-derived lipid mediators display dual anti-inflammatory and pro-resolving actions.[57–59] Thus, diets characterized by lower intake of n-3 PUFAs and higher n-6 PUFA consumption may increase the risk of individuals developing various inflammatory, thrombotic, carbohydrate, and lipid disorders.[58]
Arachidonic acid is the precursor and parent compound of various bioactive signaling lipid mediators that are generated through the actions of cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P-450 (CYP) enzymes.[57,58] The COX pathway results in the production of prostaglandins and thromboxanes, whereas the LOX pathway catalyzes the formation of leukotrienes and hydroxyeicosatetraenoic acids (Figure 1).[57,62,63] Arachidonic acid can also generate bioactive lipid mediators through the CYP epoxygenase.[59,61] Arachidonic acid also has the ability to produce lipoxins (LXs) through the actions of the LOX enzymes. Lipoxins have both anti-inflammatory and pro-resolving activities and are classified as specialized pro-resolving mediators (SPMs).[58,63–65]
Figure 1.
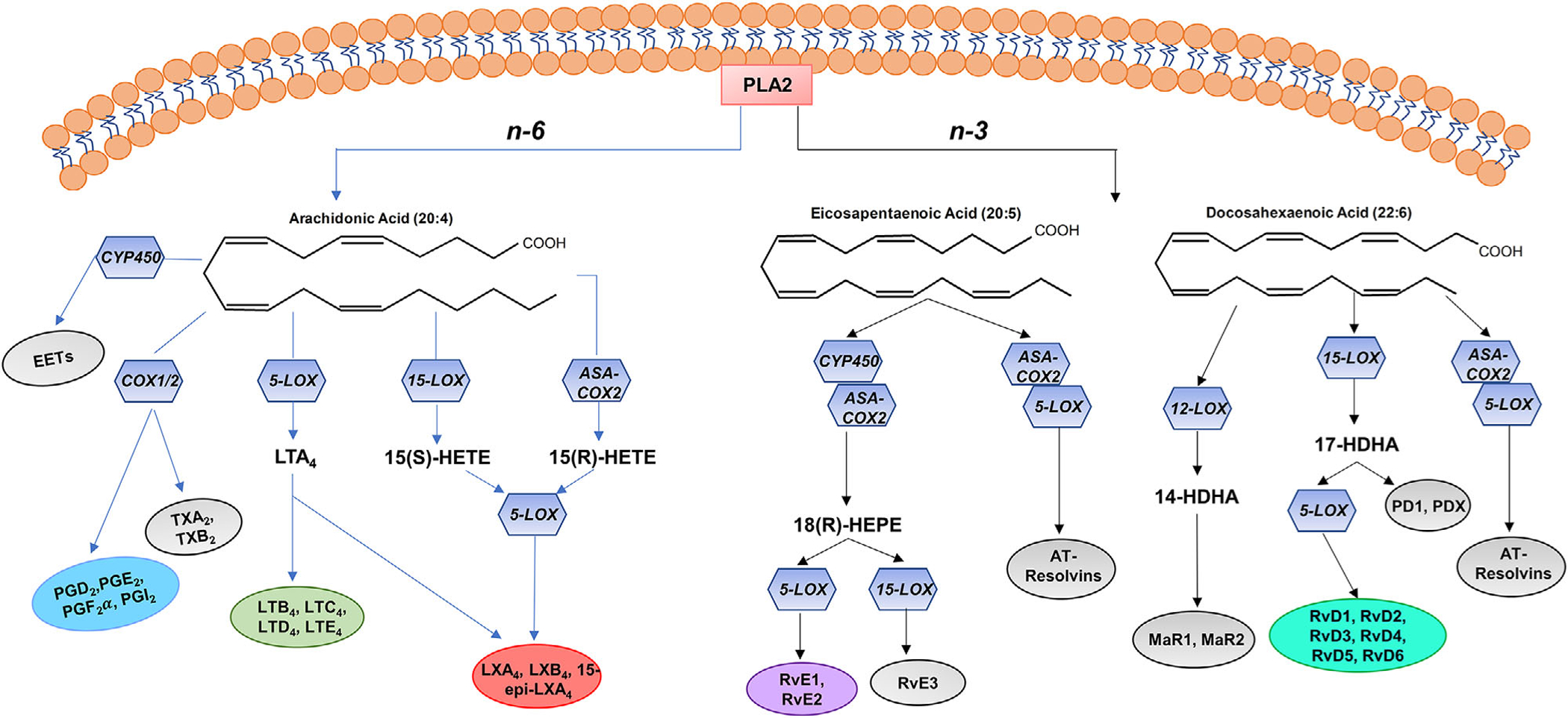
Derivation and biosynthesis of PUFA-derived lipid mediators. PUFAs embedded in membrane phospholipids are released into the cytosol primarily by PLA2 activity. This sets the basis for downstream production of metabolites from both n-6 and n-3 PUFAs.
Arachidonic acid-derived lipid mediators have important homeostatic roles in the acute inflammatory response. The acute inflammatory response is divided into two phases: initiation and resolution. During the initiation phase of the acute inflammatory response, leukocytes traffic from circulation to the damaged tissue or site of injury to form inflammatory exudates.[63] In addition, PUFAs, primarily arachidonic acid, are released from membrane phospholipids or delivered to sites of inflammation by tissue edema and undergo conversion within minutes to specific lipid mediator families via the actions of LOX and COX enzymes.[63] 5-LOX-derived lipid mediator families such as the leukotrienes and hydroxyeicosatetraenoic acids promote bronchoconstriction and regulate vascular permeability and leukocyte recruitment to the sites of tissue damage by promoting transmigration along chemotactic gradients.[57]
The actions of COX-derived prostaglandins and thromboxanes elicit the cardinal signs of inflammation including heat, swelling, redness, pain, and loss of function.[63] Thromboxanes produced by platelets and other cell types induce the platelet response to injury and also have a homeostatic role in platelet aggregation.[66] PGE2 and PGI2 inhibit platelet aggregation and promote vasodilation in endothelial cells.[66,67] Prostaglandins and leukotrienes (i.e., leukotriene B4 - LTB4) along with other chemokines, cytokines, and complement components stimulate chemotaxis of polymorphonuclear neutrophils into the damaged tissue to phagocytize and neutralize invading pathogens.[67–69] These lipid mediator families can bind to and interact with their G-protein coupled receptors (GPCRs) to modulate functions of target tissues and cells (Figure 2).[67,70] Ultimately, the actions of these arachidonic acid-derived lipid mediators are necessary to clear the site of infection and promote wound healing, which occurs during the second phase of the acute inflammatory response, which is resolution.
Figure 2.
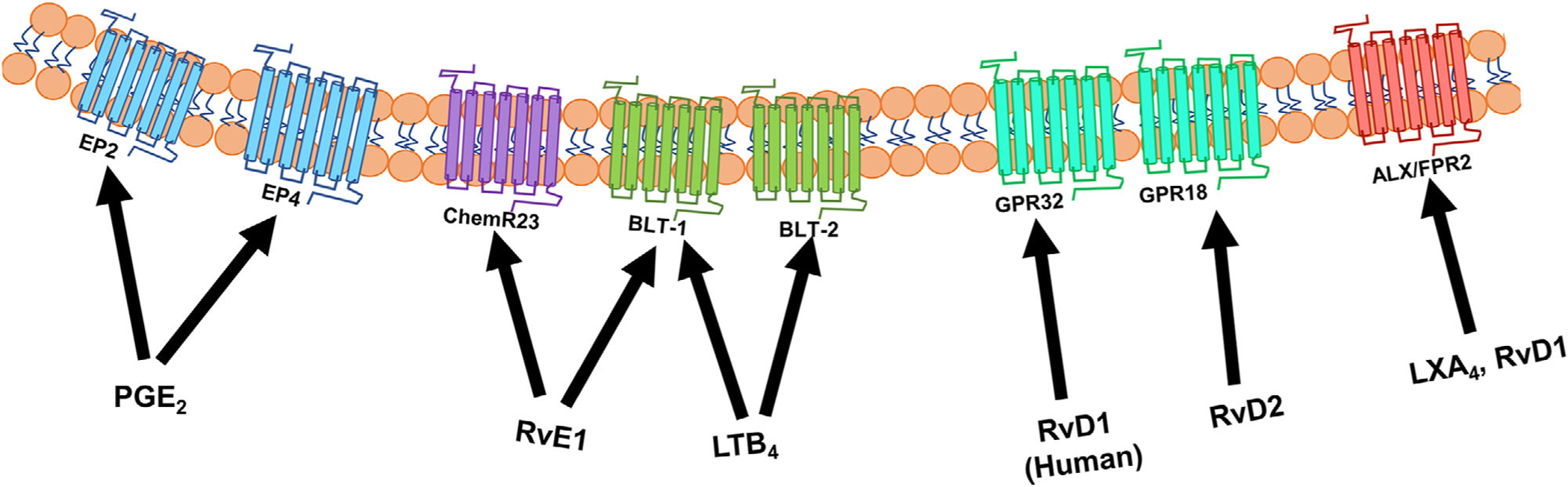
PUFA-derived lipid mediators exert actions by binding to GPCRs. Lipid mediators produced from n-6 and n-3 polyunsaturated fatty acids exert their bioactions through high affinity binding to one or more GPCRs located on the plasma membrane.
4.2. Pro-Inflammatory Lipid Mediators and the Humoral Immune Response
Arachidonic acid-derived lipid mediators can directly impact B cell development, activation, and function. Some of the first evidence that lipid mediators could potentially be involved in B cell function was data to show that PGE2 suppressed splenic B cell colony formation due to its ability to directly influence the growth and differentiation of human B cells.[71,72] Prostaglandins, particularly PGE2, are not only produced and secreted by B cells, but can also influence B cell antibody production, cytokine secretion, and class-switching, providing further evidence that lipid mediators can directly target B cells.[71,72] It is intriguing to speculate how lipid mediators such as PGE2 that are made by B cells may be further regulated by adiponectin and leptin. There is evidence that leptin and adiponectin can stimulate release of prostaglandins.[71–73]
In contrast to prostaglandins, leukotriene synthesis is restricted to a few cell types, predominantly myeloid cells and B cells.[67] Interestingly, the p38 mitogen-activated protein kinase seems to be directly involved in leukotriene synthesis in B cells.[67,68] The immunomodulator LTB4 is expressed on B cells and can enhance B cell activation, antibody production, and proliferation in tonsillar B cells.[69,71]
COX-1 is an essential regulator of B cell development in particular from the pro-B to pre-B cell stage, which is dependent upon the activation of the Janus kinase/signal transducer and activator of transcription 5 (JAK/STAT5) signaling.[74] Notably, administration of a thromboxane A2 agonist rescued deficits in B cell development seen in COX-1 deficient mice suggesting that thromboxane A2 plays a crucial role in early B cell development through regulation of JAK/STAT5 signaling.[74] It would be interesting to establish how these pro-inflammatory lipid mediators affect specific B cells subsets and B cell function in the context of obesity.
4.3. SPM Biosynthesis
Resolution of the acute inflammatory response is an active process that inhibits leukocyte infiltration to the inflammatory site, increases apoptosis of PMNs, and promotes clearance of debris and apoptotic cells by macrophages.[75–78] These actions are carried out by bioactive mediators known as the lipoxins, resolvins, protectins, and maresins, which are coined as SPMs.[63,75,76,78] SPMs are derived from PUFAs in an enzyme-dependent manner and are active in the picogram to nanogram range.[63] The first resolution signals and SPM biosynthesis are initiated by lipid mediators of the initiation phase.[63,76,78] For example, PGE2 and PGD2 initiate a temporal lipid mediator class switch, which results in the biosynthesis of lipoxin A4 (LXA4). [63] Lipoxins, derived from the n-6 PUFA arachidonic acid, are formed by transcellular biosynthesis (Figure 1).[63] LXA4 in particular serves as the “stop” signal of the acute inflammatory response resulting in decreased production of pro-inflammatory arachidonic acid-derived lipid mediators and decreased PMN infiltration.[63,78,79] Notably, LXA4 administration to mice consuming a high fat diet improved adipose tissue inflammation, liver triglycerides, and chronic kidney disease in an adiponectin-independent manner.[79]
SPMs enzymatically derived from the n-3 PUFAs are formed from eicosapentaenoic acid (EPA, 20:5) and docosahexaenoic acid (DHA, 22:6). These mediators exert anti-inflammatory and pro-resolving actions.[63,78] EPA can be converted by aspirin-acetylated COX-2 and CYP450 into 18R-hydroxy-EPE (18(R)-HEPE). 18(R)-HEPE is subsequently transformed into resolvin E1 (RvE1) and resolvin E2 (RvE2) by leukocyte 5-LOX (Figure 1).[59] 18(R)-HEPE can also serve as the substrate for 15-LOX to generate RvE3.[59,63] Similarly, DHA can generate resolvins (RvD1-RvD6) and their aspirin-triggered isomers.[59,63] DHA can be converted into 17(S)-HDHA via the actions of 15-LOX. 17(S)-HDHA is subsequently transformed into RvD1-RvD6 by 5-LOX or converted to protectin D1(PD1) and its isomer, PDX, by 15-LOX (Figure 1).[59,63] Furthermore, DHA is unique in that it generates the maresin SPM family in macrophages. DHA is readily converted by 12-LOX into the SPM precursor, 14(S)-HDHA, which undergoes further lipoxygenation to give rise to maresins 1 and 2 (MaR1 and MaR2) in macrophages.[59,63] Maresins stimulate the switch of classically activated, pro-inflammatory macrophages to alternatively activated, anti-inflammatory macrophages.[63]
4.4. Imbalance of SPMs in Obesity
Obesity is associated with a low-grade chronic inflammatory state that results in the overproduction of various pro-inflammatory cytokines and arachidonic acid-derived lipid mediators. Several studies report that levels of LTB4 are increased in the VAT, skeletal muscle, and liver of mice consuming a high fat diet compared to mice consuming a control diet.[80] Increased levels of BLT-1, the receptor for LTB4, also play a negative role in the adipose of obese mice. This was evident in a recent study, which shows that deleting BLT-1 in obese mice protected them from systemic insulin resistance.[81] Furthermore, n-3-derived SPM precursors and SPMs are significantly reduced in the adipose tissue of obese mice. Neuhofer et al. reported that mice consuming a high fat diet had decreased levels of 17-HDHA and PD1 in the gonadal adipose tissue after 4 and 14 days of high fat feeding.[82] In addition, RvD1, PD1, 18-HEPE, 14-HDHA, and 17-HDHA were decreased in the adipose tissue of mice consuming a high fat diet for 18 weeks compared to controls.[82] More recent evidence suggests sex-differences in the loss of SPMs and their precursors in obesity. Obese male but not female mice have lower levels of splenic 14-HDHA, 17-HDHA, and downstream PDX.[16]
VAT from obese patients showed an upregulation of both proinflammatory lipid mediators as well as SPMs. The ratio between SPM production with respect to leukotriene and prostaglandin production was significantly reduced in the VAT from obese patients.[83] Deficits in SPMs in white adipose tissue could be the result of decreased intake of n-3 PUFAs, which drives diminished levels in various tissues.[83,84] Alternatively, obese white adipose tissue could be facilitating the conversion of SPMs to inactive oxidized metabolites (oxo-resolvins) or PUFAs into inactive diols, which would not allow for SPM conversion.[84] However, recent studies suggest that deficits in SPM production could be a result of impaired LOX activity, particularly decreased 15-LOX production rather than altered cellular uptake of DHA.[85] Leukocytes from obese patients had reduced levels of 17-HDHA and unbalanced formation of resolvins derived from DHA accompanied by increased production of LTB4.[85] When the leukocytes were incubated with 17-HDHA in culture, the formation of the resolvins was completely rescued.[85] As DHA serves as the substrate for 15-LOX to yield the production of the SPM precursor, 17-HDHA, these findings highlight 15-LOX as a key player in efficient SPM production at the cellular level and as a potential therapeutic target in diet-induced obesity.
4.5. SPMs Up- and Down-Regulate B Cell Activity
There is compelling evidence that SPMs can regulate B cell cytokine secretion and antibody production. Initial evidence emerged from studies with dietary EPA and DHA, the parent compounds of SPMs, which showed differential effects on B cell cytokines and antibody production in lean and obese mice.[86] Notably, DHA rescued impairments in antibody production in response to a T-independent antigen in obese mice.[87] More recently, a pilot study also shows EPA and DHA differentially influence B cell cytokines of obese humans.[88]
Ramon et al. demonstrated that RvD1 and 17-HDHA but not PD1 enhanced B cell antibody production.[89] Treatment of human B cells with 17-HDHA without antigen stimulation did not increase antibody production. However, when these B cells were challenged with CpG plus anti-IgM, 17-HDHA increased antibody production without enhancing cell death or proliferation.[89] Challenged B cells treated with 17-HDHA also increased the number of IgM and IgG producing, CD27+CD38+ plasma cells, and increased IL-10 production.[89] These results suggest that the SPM precursor 17-HDHA could promote B cell differentiation towards an antibody-secreting phenotype.[89] This same group also reported that 17-HDHA enhanced the antigen-specific antibody response in a pre-clinical influenza immunization mouse model. Mice vaccinated with hemagglutinin plus 17-HDHA had a twofold increase in the number of bone marrow CD138+ B cells and hemagglutinin-specific antibody secreting cells, which was confirmed by an increase in the transcript levels of BLIMP-1.[90] Upon influenza infection, mice vaccinated with hemagglutinin plus 17-HDHA had increased survival and higher antibody titers. These results suggest that 17-HDHA might be a useful adjuvant to improve vaccine efficiency.[90] Furthermore, several SPMs, particularly RvD1, can boost the antibody response upon murine influenza vaccination.[4]
SPMs differentially affect B cell subsets and class-switching. One study demonstrated that treatment of human memory B cells with LXA4 decreased antibody production and cell proliferation via an ALX/FPR2-dependent mechanism. Interestingly, they reported that human memory B cells had increased expression of ALX/FPR2 compared to human naïve B cells.[91] SPMs seem to differentially affect Ig classes. 17-HDHA and RvD1 promote the differentiation of IgG-secreting B cells but inhibit IgE production in human B cells. This inhibition is due to a decrease in the expression of the epsilon germline transcript, which is essential for IgE class switching.[92] Overall, this further supports the idea that SPMs can differentially affect B cell subsets and potentially alter B cell class switching by targeting specific Ig classes. More work is clearly needed in this area as the effects of SPMs are likely not just limited to B cells in humoral immunity.
5. Potential Therapeutic Approaches for Improving Outcomes to Impaired B Cell Responses
Lifestyle and pharmacological approaches have been examined to identify therapeutic approaches to modulate B cell function. One example is metformin. It is a widely prescribed medication to reduce hyperglycemia and has been shown to increase the B-cell antibody response to influenza vaccination. Flow cytometry analysis of patients on metformin treatment shows an increased percentage of class-switched memory B cells potentially leading to the improved antibody response.[93] Metformin also affected late/exhausted memory B cells. Exhausted memory B cells, prevalent in elderly individuals, express proinflammatory cytokines and are correlated with an impaired vaccination response.[94] However, metformin treated patients had a reduction in exhausted memory B cells.[93] Mechanistically, these studies are intriguing as they suggest that B cell metabolism is likely a target for intervention.
As described above, dietary fat quality also has a role and perhaps small changes in dietary intake of n-3 PUFAs may influence B cell responses in the context of inflammation and infection. There is no clear indication on how much increased n-3 PUFA intake or a lowering of n-6 PUFAs would impact humoral immunity and remains an unanswered question. Aside from pharmaceutical and dietary induced B-cell changes, it has been hypothesized that intense exercise aids in hastening B cell antigen detection and response. Intense exercise induces a two-fold increase in PBMC B cells.[95] Specifically, immature B cells were the most elevated in circulation and seemed to traffic from the bone marrow to the spleen. This suggests that exercise-induced immature B cell mobilization may help in faster detection of circulating antigens. The trafficking of immature B cells to the spleen after exercise-induced stress poises the humoral immune system for a prepared response to an injury or pathogenic infection.[95] In all, multiple therapeutic approaches have been and continue to be explored to enhance impaired B cell activity in a multifaceted manner. An important avenue of research will be to study the combination of exercise, diet, and pharmaceuticals to establish improvements in B cell driven humoral immunity in obese rodents and humans.
6. Conclusion
B cells have a critical role in inflammation and infection, yet our understanding of how obesity impairs B cell activity is just emerging. A range of nutritional and hormonal factors such as leptin, adiponectin, and PUFA-derived mediators including prostaglandins and SPMs regulate differing aspects of B cell development, activation, and antibody production. Establishing the mechanisms by which obesity impairs B cell mediated responses will set the basis for future strategies for therapeutic intervention for chronic inflammation, insulin resistance, and the response to differing infections and vaccinations.
Acknowledgements
This work was supported by NIH R01AT008375 and P30DK05635. This material is also based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. 1650116 to AEA. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Trottier MD, Naaz A, Li Y, Fraker PJ, Proc. Natl. Acad. Sci. USA 2012, 109, 7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Adler BJ, Green DE, Pagnotti GM, Chan ME, Rubin CT, PLoS One 2014, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chan ME, Adler BJ, Green DE, Rubin CT, FASEB J 2012, 26, 4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kosaraju R, Guesdon W, Crouch MJ, Teague HL, Sullivan EM, Karlsson EA, Schultz-Cherry S, Gowdy K, Bridges LC, Reese LR, Neufer PD, Armstrong M, Reisdorph N, Milner JJ, Beck M, Shaikh SR, J. Immunol 2017, 198, 4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lazar MA, Science 2005, 307, 373. [DOI] [PubMed] [Google Scholar]
- [6].Ang XM, Lee MHC, Blocki A, Chen C, Ong LL, Asada HH, Sheppard A, Raghunath M, Tissue Eng., Part A 2014, 20, 966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].McCulloch LJ, Rawling TJ, Sjöholm K, Franck N, Dankel SN, Price EJ, Knight B, Liversedge NH, Mellgren G, Nystrom F, Carlsson LM, Kos K, Endocrinology 2015, 156, 134. [DOI] [PubMed] [Google Scholar]
- [8].Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, Brekken RA, Scherer PE, Mol. Cell. Biol 2009, 29, 4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Duffaut C, Galitzky J, Lafontan M, Bouloumié A, Biochem. Biophys. Res. Commun 2009, 384, 482. [DOI] [PubMed] [Google Scholar]
- [10].Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, Tsui H, Wu P, Davidson MG, Alonso MN, Leong HX, Glassford A, Caimol M, Kenkel JA, Tedder TF, McLaughlin T, Miklos DB, Dosch HM, Engleman EG, Nat. Med 2011, 17, 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Elgazar-Carmon V, Rudich A, Hadad N, Levy R, J. Lipid Res 2008, 49, 1894. [DOI] [PubMed] [Google Scholar]
- [12].Nguyen MTA, Favelyukis S, Nguyen A-K, Reichart D, Scott PA, Jenn A, Liu-Bryan R, Glass CK, Neels JG, Olefsky JM, J. Biol. Chem 2007, 282, 35279. [DOI] [PubMed] [Google Scholar]
- [13].McDonnell ME, Ganley-Leal LM, Mehta A, Bigornia SJ, Mott M, Rehman Q, Farb MG, Hess DT, Joseph L, Gokce N, Apovian CM, Obesity 2012, 20, 1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Frasca D, Diaz A, Romero M, Thaller S, Blomberg BB, PLoS One 2019, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Frasca D, Ferracci F, Diaz A, Romero M, Lechner S, Blomberg BB, Obesity (Silver Spring) 2016, 24, 615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Crouch MJ, Kosaraju R, Guesdon W, Armstrong M, Reisdorph N, Jain R, Fenton J, Shaikh SR, J. Leukocyte Biol 2019, 106, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sabio G, Davis RJ, Trends Biochem. Sci 2010, 35, 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hong E-G, Ko HJ, Cho Y−R, Kim HJ, Ma Z, Yu TY, Friedline RH, Kurt-Jones E, Finberg R, Fischer MA, Granger EL, Norbury CC, Hauschka SD, Philbrick WM, Lee CG, Elias JA, Kim JK, Diabetes 2009, 58, 2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].van Exel E, Gussekloo J, de Craen AJM, Frölich M, Bootsma-Van Der Wiel A, Westendorp RG, Diabetes 2002, 51,1088. [DOI] [PubMed] [Google Scholar]
- [20].Kowalski GM, Nicholls HT, Risis S, Watson NK, Kanellakis P, Bruce CR, Bobik A, Lancaster GI, Febbraio MA, Diabetologia 2011, 54, 888. [DOI] [PubMed] [Google Scholar]
- [21].DeFuria J, Belkina AC, Jagannathan-Bogdan M, Snyder-Cappione J, Carr JD, Nersesova YR, Markham D, Strissel KJ, Watkins AA, Zhu M, Allen J, Bouchard J, Toraldo G, Jasuja R, Obin MS, McDonnell ME, Apovian C, Denis GV, Nikolajczyk BS, Proc. Natl. Acad. Sci. USA 2013, 110, 5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Peterson KR, Flaherty DK, Hasty AH, Obesity (Silver Spring) 2017,25, 1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Diaz A, Romero M, Vazquez T, Lechner S, Blomberg BB, Frasca D, Vaccine 2017, 35, 2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bassols J, Prats-Puig A, Gispert-Saüch M, Crehuet-Almirall M, Carreras-Badosa G, Díaz-Roldán F, Montesinos-Costa M, de Zegher F, Ibáñez L, López-Bermejo A, Pediatric Obesity 2014, 9, 232. [DOI] [PubMed] [Google Scholar]
- [25].Mito N, Kaburagi T, Yoshino H, Imai A, Sato K, Life Sci 2006, 79, 1056. [DOI] [PubMed] [Google Scholar]
- [26].Palming J, Gabrielsson BG, Jennische E, Smith U, Carlsson B, Carlsson LM, Lönn M, Biochem. Biophys. Res. Commun 2006, 343, 43. [DOI] [PubMed] [Google Scholar]
- [27].Shulzhenko N, Morgun A, Hsiao W, Battle M, Yao M, Gavrilova O, Orandle M, Mayer L, Macpherson AJ, McCoy KD, Fraser-Liggett C, Matzinger P, Nat. Med 2011, 17, 1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Milner JJ, Sheridan PA, Karlsson EA, Schultz-Cherry S, Shi Q, Beck MA, J. Immunol 2013, 191, 2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Farnsworth CW, Schott EM, Benvie A, Kates SL, Schwarz EM, Gill SR, Zuscik MJ, Mooney RA, J. Immunol 2018, 201, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Karlsson EA, Hertz T, Johnson C, Mehle A, Krammer F, Schultz-Cherry S, mBio 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Neidich SD, Green WD, Rebeles J, Karlsson EA, Schultz-Cherry S, Noah TL, Chakladar S, Hudgens MG, Weir SS, Beck MA, Int J Obes 2017, 41, 1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Agrawal M, Kern PA, Nikolajczyk BS, Curr Diab Rep 2017, 17, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hotta K, Funahashi T, Bodkin NL, Ortmeyer HK, Arita Y, Hansen BC, Matsuzawa Y, Diabetes 2001, 50, 1126. [DOI] [PubMed] [Google Scholar]
- [34].Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, Tataranni PA, J Clin Endocrinol Metab 2001, 86, 1930. [DOI] [PubMed] [Google Scholar]
- [35].Pellmé F, Smith U, Funahashi T, Matsuzawa Y, Brekke H, Wiklund O, Taskinen MR, Jansson PA, Diabetes 2003, 52, 1182. [DOI] [PubMed] [Google Scholar]
- [36].Côté M, Mauriège P, Bergeron J, Alméras N, Tremblay A, Lemieux I, Després JP, J Clin Endocrinol Metab 2005, 90, 1434. [DOI] [PubMed] [Google Scholar]
- [37].Havel PJ, Curr. Opin. Lipidol 2002, 13, 51. [DOI] [PubMed] [Google Scholar]
- [38].Anderson NJ, King MR, Delbruck L, Jolivalt CG, Dis. Models Mech 2014, 7, 625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yokota T, Meka CSR, Kouro T, Medina KL, Igarashi H, Takahashi M, Oritani K, Funahashi T, Tomiyama Y, Matsuzawa Y, Kincade PW, J. Immunol 2003, 171, 5091. [DOI] [PubMed] [Google Scholar]
- [40].Obeid S, Wankell M, Charrez B, Sternberg J, Kreuter R, Esmaili S, Ramezani-Moghadam M, Devine C, Read S, Bhathal P, Lopata A, Ahlensteil G, Qiao L, George J, Hebbard L, J. Biol. Chem 2017, 292, 6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].German JP, Wisse BE, Thaler JP, Oh-I S, Sarruf DA, Ogimoto K, Kaiyala KJ, Fischer JD, Matsen ME, Taborsky GJ Jr, Schwartz MW, Morton GJ, Diabetes 2010, 59, 1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Francisco V, Pino J, Campos-Cabaleiro V, Ruiz-Fernández C, Mera A, Gonzalez-Gay MA, Gómez R, Gualillo O, Front Physiol 2018, 9, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI, Nature 1998, 394, 897. [DOI] [PubMed] [Google Scholar]
- [44].Zarkesh-Esfahani H, Pockley G, Metcalfe RA, Bidlingmaier M, Wu Z, Ajami A, Weetman AP, Strasburger CJ, Ross RJ, J. Immunol 2001, 167, 4593. [DOI] [PubMed] [Google Scholar]
- [45].Papathanassoglou E, El-Haschimi K, Li XC, Matarese G, Strom T, Mantzoros C, J. Immunol 2006, 176, 7745. [DOI] [PubMed] [Google Scholar]
- [46].Frasca D, Diaz A, Romero M, Blomberg BB, Cell Immunol 2019, 103994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lam QLK, Wang S, Ko OKH, Kincade PW, Lu L, Proc. Natl. Acad. Sci. USA 2010, 107, 13812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Agrawal S, Gollapudi S, Su H, Gupta S, J Clin Immunol 2011, 31, 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tanaka M, Suganami T, Kim-Saijo M, Toda C, Tsuiji M, Ochi K, Kamei Y, Minokoshi Y, Ogawa Y, J. Neurosci 2011, 31, 8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fujita Y, Yanagida H, Mimori T, Jin ZX, Sakai T, Kawanami T, Sawaki T, Masaki Y, Fukushima T, Okazaki T, Umehara H, Cell. Immunol 2012, 273, 52. [DOI] [PubMed] [Google Scholar]
- [51].Tsai S, Clemente-Casares X, Zhou AC, Lei H, Ahn JJ, Chan YT, Choi O, Luck H, Woo M, Dunn SE, Engleman EG, Watts TH, Winer S, Winer DA, Cell Metab 2018, 28, 922. [DOI] [PubMed] [Google Scholar]
- [52].Björntorp P, Bergman H, Varnauskas E, Acta Med Scand 1969, 185, 351. [DOI] [PubMed] [Google Scholar]
- [53].Boden G, Endocrinol Metab Clin North Am 2008, 37, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Boden G, Shulman GI, Eur J Clin Invest 2002, 32, 14. [DOI] [PubMed] [Google Scholar]
- [55].Boden G, Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 545. [DOI] [PubMed] [Google Scholar]
- [56].Surette ME, Can. Med. Assoc. J 2008, 178, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Calder PC, Biochim Biophys Acta 2015, 1851, 469. [DOI] [PubMed] [Google Scholar]
- [58].Calder PC, Mol. Nutr. Food Res 2012, 56, 1073. [DOI] [PubMed] [Google Scholar]
- [59].Lopategi A, López-Vicario C, Alcaraz-Quiles J, García-Alonso V, Rius B, Titos E, Clària J, Mol. Cell. Endocrinol 2016, 419, 44. [DOI] [PubMed] [Google Scholar]
- [60].Zeldin DC, J. Biol. Chem 2001, 276, 36059. [DOI] [PubMed] [Google Scholar]
- [61].Calder PC, Br. J. Clin. Pharmacol 2013, 75, 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Patterson E, Wall R, Fitzgerald GF, Ross RP, Stanton C, J. Nutr. Metab 2012, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Serhan CN, Nature 2014, 510, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Schmitz G, Ecker J, Prog. Lipid Res 2008, 47, 147. [DOI] [PubMed] [Google Scholar]
- [65].Samuelsson B, Dahlén SE, Lindgren JA, Rouzer CA, Serhan CN, Science 1987, 237, 1171. [DOI] [PubMed] [Google Scholar]
- [66].Astudillo AM, Balgoma D, Balboa MA, Balsinde J, Biochim. Biophys. Acta 2012, 1821, 249. [DOI] [PubMed] [Google Scholar]
- [67].Romano M, Claria J, FASEB J 2003, 17, 1986. [DOI] [PubMed] [Google Scholar]
- [68].Werz O, Klemm J, Rådmark O, Samuelsson B, J. Leukoc. Biol 2001, 70, 830. [PubMed] [Google Scholar]
- [69].Odlander B, Jakobsson PJ, Medina JF, Rådmark O, Yamaoka KA, Rosén A, Claesson HE, Int. J. Tissue React 1989, 11, 277. [PubMed] [Google Scholar]
- [70].Im D-S, Prostaglandins Other Lipid Mediat 2009, 89, 53. [DOI] [PubMed] [Google Scholar]
- [71].Simkin NJ, Jelinek DF, Lipsky PE, J. Immunol 1987, 15, 138. [PubMed] [Google Scholar]
- [72].Roper RL, Brown DM, Phipps RP, J. Immunol 1995, 154, 162. [PubMed] [Google Scholar]
- [73].Lappas M, Permezel M, Rice GE, Endocrinology 2005, 146, 3334. [DOI] [PubMed] [Google Scholar]
- [74].Yang Q, Shi M, Shen Y, Cao Y, Zuo S, Zuo C, Zhang H, Gabrilovich DI, Yu Y, Zhou J, Blood 2014, 124, 1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Basil MC, Levy BD, Nat. Rev. Immunol 2016, 16, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Serhan CN, Savill J, Nat. Immunol 2005, 6, 1191. [DOI] [PubMed] [Google Scholar]
- [77].Nathan C, Nature 2002, 420, 846. [DOI] [PubMed] [Google Scholar]
- [78].Lawrence T, Willoughby DA, Gilroy DW, Nat. Rev. Immunol 2002, 2, 787. [DOI] [PubMed] [Google Scholar]
- [79].Börgeson E, Johnson AMF, Lee YK, Till A, Syed GH, Ali-Shah ST, Guiry PJ, Dalli J, Colas RA, Serhan CN, Sharma K, Godson C, Cell Metab 2016, 22, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Li P, Oh DY, Bandyopadhyay G, Lagakos W, Talukdar S, Osborn O, Johnson A, Chung H, Maris M, Ofrecio JM, Taguchi S, Lu M, Olefsky JM, Nat. Med 2015, 21, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Spite M, Hellmann J, Tang Y, Mathis SP, Kosuri M, Bhatnagar A, Jala V, Haribabu B, J. Immunol 2011, 187, 1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Neuhofer A, Zeyda M, Mascher D, Itariu B, Murano I, Leitner L, Hochbrugger E, Fraisl P, Cinti S, Serhan CN, Stulnig TM, Diabetes 2013, 62, 1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Titos E, Rius B, López-Vicario C, Alcaraz-Quiles J, García-Alonso V, Lopategi A, Dalli J, Lozano JJ, Arroyo V, Delgado S, Serhan CN, Clària J, J. Immunol 2016, 197, 3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Clària J, López-Vicario C, Rius B, Titos E, Mol. Aspects Med 2017, 58, 83. [DOI] [PubMed] [Google Scholar]
- [85].López-Vicario C, Titos E, Walker ME, Alcaraz-Quiles J, Casulleras M, Durán-Güell M, Flores-Costa R, Pérez-Romero N, Forné M, Dalli J, Clària J, FASEB J 2019, 33, 7072. [DOI] [PubMed] [Google Scholar]
- [86].Teague H, Harris M, Fenton J, Lallemand P, Shewchuk BM, Shaikh SR, J. Lipid Res 2014, 55, 1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Teague H, Fhaner CJ, Harris M, Duriancik DM, Reid GE, Shaikh SR, J. Lipid Res 2013, 54, 3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Guesdon W, Kosaraju R, Brophy P, Clark A, Dillingham S, Aziz S, Moyer F, Wilson K, Dick J, Patil SP, Balestrieri N, Armstrong M, Reisdroph N, Shaikh SR, J Nutr Biochem 2018, 53, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Ramon S, Gao F, Serhan CN, Phipps RP, J. Immunol 2012, 189, 1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ramon S, Baker SF, Sahler JM, Kim N, Feldsott E, Serhan C, Martínez-Sobrido L, Topham D, Phipps R, J. Immunol 2014, 193, 6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Ramon S, Bancos S, Serhan CN, Phipps RP, Eur. J. Immunol 2014, 44, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Kim N, Ramon S, Thatcher TH, Woeller CF, Sime PJ, Phipps RP, Eur. J. Immunol 2016, 46, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Diaz A, Romero M, Vazquez T, Lechner S, Blomberg BB, Frasca D, Vaccine 2017, 35, 2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Frasca D, Diaz A, Romero M, Thaller S, Blomberg BB, PLoS One 2019, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Turner JE, Spielmann G, Wadley AJ, Aldred S, Simpson RJ, Campbell JP, Physiol. Behav 2016,164, 376. [DOI] [PubMed] [Google Scholar]