

Abstract
HSD17B13 (17-beta hydroxysteroid dehydrogenase 13) is genetically associated with human non-alcoholic fatty liver disease (NAFLD). Inactivating mutations in HSD17B13 protect humans from NAFLD- and alcohol-associated liver injury, fibrosis, cirrhosis and hepatocellular carcinoma, leading to clinical trials of anti-HSD17B13 therapeutic agents in humans. We aimed to study the in vivo function of HSD17B13 using a mouse model. Single-cell RNAseq and qPCR data revealed that hepatocytes are the main HSD17B13-expressing cells in mice and humans. We compared Hsd17b13 whole-body knockout (KO) mice and wild-type littermate controls (WT) fed regular chow (RC), high fat diet (HFD), Western diet (WD), or the NIAAA model of alcohol exposure. HFD and WD induced significant weight gain, hepatic steatosis and inflammation. However, there was no difference between genotypes with regards to body weight, liver weight, hepatic triglycerides (TG), histological inflammatory scores, expression of inflammatory- and fibrosis-related genes and hepatic retinoid levels. Compared to WT, KO mice on HFD had hepatic enrichment of most cholesterol esters, monoglycerides, and certain sphingolipids species. Extended feeding with WD for 10 months led to extensive liver injury, fibrosis and hepatocellular carcinoma, with no difference between genotypes. Under alcohol exposure, KO and WT mice showed similar hepatic TG and liver enzyme levels. Interestingly, chow-fed KO showed significantly higher body and liver weight compared to WT, while KO mice on obesogenic diets had a shift towards larger lipid droplets. In conclusion, extensive evaluation of Hsd17b13 deficiency in mice under several fatty liver-inducing dietary conditions didn’t reproduce the protective role of HSD17B13 loss-of-function mutants in human NAFLD. Moreover, mouse Hsd17b13 deficiency induces weight gain under RC. It is crucial to understand inter-species differences prior to leveraging HSD17B13 therapies.
Keywords: 17-beta hydroxysteroid dehydrogenase 13, NAFLD, Hsd17b13 deficiency, alcohol-associated liver disease
Introduction
Non-alcoholic fatty liver disease (NAFLD) has become a major global health burden and a leading cause of chronic liver disease (1) with an important heritable component contributing to disease incidence and progression to its severe form, non-alcoholic steatohepatitis (NASH) (2–4). Several genetic loci have been identified and validated to be associated with NAFLD severity, including PNPLA3 (5–10), TM6SF2 (11–13), MBOAT7 (14), and recently, HSD17B13 (15–17).
HSD17B13 is a lipid droplet-associated protein (15, 17–19), expressed predominantly in the liver (18, 19), implying a liver-specific function. Based on structural similarities, HSD17B13 has been assigned to two families: the 17β hydroxysteroid dehydrogenase (HSD) family which includes 14 structurally related enzymes with steroids, bile acids, and fatty acids (20) as potential substrates, and the short-chain dehydrogenase/reductase 16C famiy (SDR16C) which regulates retinoid metabolism (21). We and others (15, 17, 22) identified a single nucleotide polymorphism (SNP) in HSD17B13, rs72613567, that is associated with decreased risk of progression in NASH (15, 17, 23), alcoholic liver disease (15, 17), and lower risk of hepatocellular carcinoma (HCC) (24, 25). The protective allele, rs72615367-TA, leads to the generation of two novel splicing variants, both of which have no enzymatic activity (15, 17). We identified an additional HSD17B13 SNP, rs62305723 (encoding a P260S mutation), that also has no enzymatic activity and protects from injury in NAFLD (15). Cell-based in vitro assays have indicated steroid substrates, bioactive lipids (17), and retinol (15) as potential enzymatic substrates of HSD17B13 and the protein variants generated by the protective SNP alleles have been shown to lose enzymatic activity in these assays (15, 17), confirming the importance of enzymatic activity of HSD17B13. These findings led to ongoing and planned clinical trials, investigating the use of anti-HSD17B13 therapies in humans (26) (https://clinicaltrials.gov/ct2/show/NCT04202354, accessed February 21, 2020).
Despite strong human genetic data linking HSD17B13 to chronic liver disease and in vitro studies, and despite its advancement to clinical stage, the in vivo function of the protein is yet unclear. To study the role of HSD17B13 in vivo, we utilized a Hsd17b13 knock-out mouse model under several dietary conditions to induce fatty liver with short-, medium-, and long-term exposures. Our data suggest that Hsd17b13 deficiency in mice does not reproduce the protective role of HSD17B13 loss-of-function mutants in human fatty liver disease. It is crucial to understand inter-species differences prior to leveraging HSD17B13-based therapies.
Methods
Animals and diets
B6;129S5-Hsd17b13tm1Lex/Mmucd mice (MMRRC, Stock number: 032367-UCD, hereafter Hsd17b13-KO) were back-crossed with C57BL6J mice (The Jackson Laboratory, Stock number: 000664) for a minimum of 8 generations. Exon 1 and 2 of the Hsd17b13 gene were deleted in Hsd17b13-KO mice as described previously (27). Heterzyogote mice were used for breeding, with pups genotyped by PCR (Supplementary Figure 1). Littermate wild type (WT) and Hsd17b13-KO (KO) mice were used for all experiments. Mice were housed in a temperature-controlled environment (22.2 ± 1.6°C) with a 12-hour light/dark cycle and free access to water and feed, unless otherwise indicated. To facilitate food intake measurement, animals in the regular chow (RC, 15% kcal from fat, NIH-07, Envigo, Madison, WI) and high fat diet (HFD, 60% kcal% from fat, D12492, Research Diets, New Brunswick, NJ) experiments were housed separately according to their genotype. In all other experiments, same-gender sibling animals of both genotypes were co-housed. Western Diet (WD, D12079B, 40% Kcal% from fat, 43% kcal carbohydrate; with 0.15% cholesterol, containing 4IU/g vitamin A) was purchased from Research Diets and vitamin A-enriched Western Diet (TD.88137, 42% Kcal% from fat, 43% kcal from carbohydrate; with 0.15% cholesterol, containing 19IU/g vitamin A) was obtained from Envigo. Ten days ad libitum oral feeding with the Lieber-DeCarli ethanol liquid diet plus a single binge ethanol feeding was used to generate alcohol-associated liver disease as decribed previously (28). All animal experiments were approved by the Institutional Animal Care and Use Committees of NIDDK and NIAAA (K060-LDB-18, ASP 141-MMC-18, and LLD-BG-1).
Serum biochemistry
Sera were collected at the end of experiment from each animal cohort after a 5-hour fast. Serum liver enzymes (Catachem. Inc., C164–0B and C154–0A), triglycerides (Pointe Scientific Inc., Canton, MI, #T7532–120), total cholesterol (Thermo Scientific, Middletown, VA, #TR13421), free fatty acids (Fujifilm Waco Diagnostics, Mountain View, CA, reagents # 999–34691, 995–34791, 991–34891, 993–35191) were measured using the indicated colorimetric assays following manufacturers’ instructions. Blood glucose was measured with a Glucometer Contour (Bayer, Mishawaka, IN).
Body composition
Fat mass and lean mass of non-anesthetized mice were measured by time domain Echo MRI 3-in-1 NMR (EchoMRI LLC, Houston, TX). Body composition was measured once before the end of experiments (RC, HFD, WD, and long-term WD), or weekly for energy expenditure calculation purpose.
Food intake and energy expenditure
To assess energy balance on regular chow, food intake and body composition were measured weekly in mice singly-housed in their home cages for 4 weeks, starting form 14 weeks of age. Energy expenditure was estimated by energy balance technique (29). In brief, energy expenditure is calculated from the metabolizable caloric intake, corrected for the change in caloric content of the mouse.
Liver histology
A small piece of freshly dissected liver was fixed in 4% paraformaldehyde or 10% neutral buffered formalin at least overnight. Tissue processing for paraffin block section and H&E staining were performed by American Histolabs (Rockville, MD). Histological evaluation and scoring were performed by a single pathologist (DEK) blinded to mouse genotype and treatment. The degree of steatosis was assessed as the percent of hepatocytes demonstrating fat accumulation. We also recorded separately the percentage of cells with small “foamy” fat accumulation appearance (microsteatosis) and the percentage of cells with large fat vacuoles (macrosteatosis). The degree of inflammation was scored as an average of foci per 20x field (0.933 mm2) and then binned into categories: No foci, <1, 1 to 2, 2 to 4, 4 to 8 and more than 8 foci per 20x field.
Hepatic triglycerides measurement
Small pieces of liver (~100 mg) were used to measure hepatic triglycerides (TG). Hepatic lipids were extracted using chloroform:methanol (1:2) and measured by TG reagents from Pointe Scientific Inc., (Canton, MI, # T7532–120). TG content was normalized to liver weight.
Hepatic collagen quantification
Hepatic collagen content was quantified using colorimetric Total Collagen Assay Kit (BioVision). Briefly, liver tissues were homogenized in ddH2O (m/v, 10 mg/100 μl), to which an equal volume of concentrated HCl was added in a pressure-tight polypropylene screw-capped vial. Samples were hydrolyzed at 120°C for 3 hours, clarified by centrifugation after adding activated charcoal, and dryed in a 96-well plate by heating the plate at 70°C. Collagen content was measured at wavelength of 560 nm after adding the provided reagents following the manufacturer’s instructions.
Tissue quantitative reverse transcription polymerase chain reaction (qPCR)
Total RNA was extracted from 25–100 mg of liver tissue using Trizol Reagent (Thermo Fisher Scientific). cDNA was synthesized using First-strand cDNA Synthesis Kit (Origene, Rockville, MD) and used as template for qPCR. Predesigned Taq-man probe based primers were purchased from IDT (Supplementary Table) except for G6pc primer (Thermo Fisher, Assay ID: Mm00839363).
Liver lipidomics
Samples were weighed and then soaked in 1:1 dichloromethane:methanol overnight at 4°C. The supernatants were subjected to a modified Bligh-Dyer extraction using methanol/water/dichloromethane in the presence of deuterated internal standards. The extracts were dried under nitrogen and reconstituted in a dichloromethane:methanol solution containing ammonium acetate. The extracts were transferred to vials for infusion-MS analysis, performed on a Shimadzu LC with nano PEEK tubing and the Sciex SelexIon-5500 QTRAP. The samples were analyzed via both positive and negative mode electrospray. The 5500 QTRAP was operated in MRM mode with a total of more than 1,100 MRMs. Individual lipid species were quantified by taking the ratio of the signal intensity of each target compound to that of its assigned internal standard, then multiplying by the concentration of internal standard added to the sample. Lipid class concentrations were calculated from the sum of all molecular species within a class, and fatty acid compositions were determined by calculating the proportion of each class comprised by individual fatty acids.
Statistical analysis
Student’s t test was used for differences between two groups, and one-way analysis of variance followed by Dunnett’s multiple comparison test was used for difference among more than two groups. Two-sided testing was used for all tests. Statistical analyses were performed using Prism V.8 (GraphPad).
Results:
HSD17B13 expression is restricted to hepatocytes
The expression level of HSD17B13 was assessed in 53 normal human tissue types and 33 carcinoma types included in the Genotype-Tissue Expression (GTEx, https://www.gtexportal.org/home/) project and the Cancer Genome Atlas (TCGA, https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga). The highest expression level was found in liver tissue and overall, HSD17B13 has a higher expression level in healthy tissues compared to samples from malignancies (Figure 1A). We tested the tissue distribution of Hsd17b13 in mice and as previously reported (18, 19), we confirmed that Hsd17b13 is predominantly expressed in the liver (Figure 1B), with other digestive tissues showing lower, but non-negligible expression level (duodenum, 26% of liver expression level; jejunum 9.8%; ileum 3.5%; pancreas 5.2%). We further investigated which cell type within the liver drives the high expression level of this gene. Available single cell sequencing data from human livers (30) demonstrates that within the liver, HSD17B13 expression is restricted to hepatocytes (Figure 1C). Similarly, single cell sequencing of mouse tissues (https://tabula-muris.ds.czbiohub.org), shows that among liver cell types, Hsd17b13 is mainly expressed in hepatocytes (Figure 1D). Lower level expression was also detected in pancreatic acinar cells, enterocytes and epithelial cells of intestine (Figure 1D) consistent with our tissue distribution data as determined by qPCR (Figure 1B).
Figure 1. HSD17B13 is expressed in normal hepatocytes.
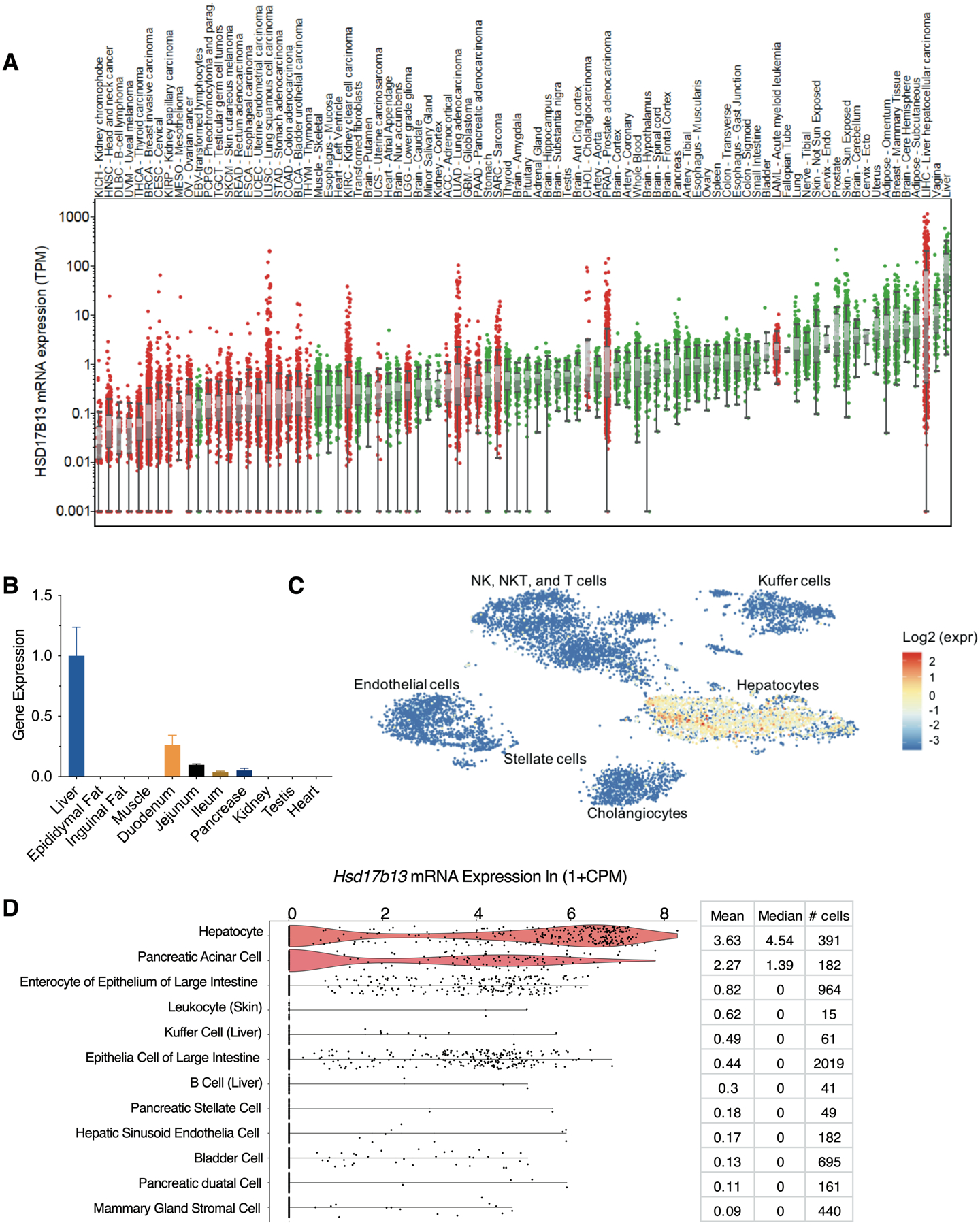
(A) Gene expression of HSD17B13 in 53 healthy and 33 carcinoma tissues from the GTEx and TCGA databases. Cancer tissues are shown in red and normal tissues are shown in green. Data are sorted by median expression. (B) Tissue distribution of Hsd17b13 in mice. Gene expression of Hsd17b13 was determined in the liver, epididymal fat, inguinal fat, muscle, duodenum, jejunum, ileum, pancrase, kidney, testis, and heart tissues of 20 weeks old C57BL/6 mice by qPCR (n=6, Mean±SEM). (C) HSD17B13 is restrictively expressed within the hepatocyte cluster in human liver determined by single cell sequencing (http://human-liver-cell-atlas.ie-freiburg.mpg.de). (D) Single cell transcriptome data (https://tabula-muris.ds.czbiohub.org) showing the top 12 mouse cell types from 20 organs according to expression level of Hsd17b13, sorted by mean value.
Hsd17b13 deficiency induces weight gain
To evaluate the function of Hsd17b13 in vivo, we obtained Hsd17b13 whole-body knock out mice and backcrossed them to the C57BL6 strain (Hsd17b13-KO). When fed regular chow, Hsd17b13-KO mice had increased weight gain starting at age 10 weeks compared to wild type littermate controls (WT) (Figure 2A), reaching a difference of 6.2±2.1 grams (34.9%) by age 22 weeks (p=0.015, Figure 2B). Hsd17b13-KO mice had an increase in fat mass, but not in lean mass percentage (Figure 2C and D). To further understand the mechanism leading to increased body weight in Hsd17b13-KO mice, we measured food intake, body weight, and body mass composition in a separate cohort for 4 consecutive weeks and confirmed an increase in fat mass in Hsd17b13-KO mice (Supplementary Figure 2A–C). There was a mild (8%) increase in food intake in Hsd17b13-KO mice (p=0.04, Supplementary Figure 2D) while the calculated energy expenditure shows a non-significant trend for increase (Supplementary Figure 2E).
Figure 2. Hsd17b13 deficiency induces weight gain in mice fed regular chow diet.
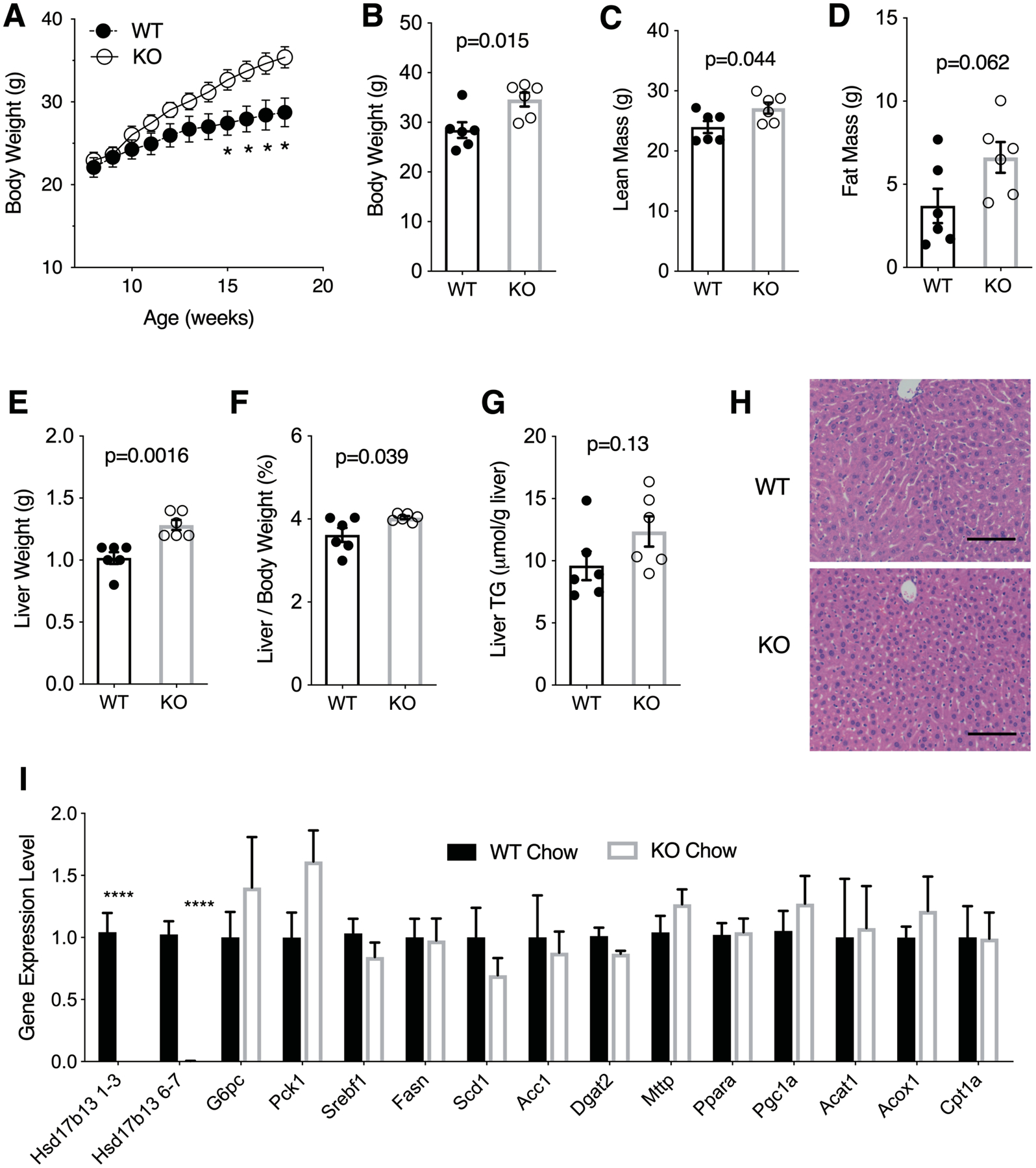
Eight week-old male wild type (WT) and Hsd17b13-knockout (KO) mice were fed regular chow for 12 weeks and sacrified at age 20 weeks, after a 5 hours fast. (A) Body weight of WT and KO mice was measured weekly. (B) Body weight, (C) Lean mass relative to body weight, and (D) Fat mass relative to body weight. (E) Liver weight, (F) Liver-to-body weight ratio, and (G) Liver triglycerides. (H) Hematoxylin & eosin staining of liver sections. Bar indicates 100 μM. (I) Hepatic gene expression, normalized to WT level. Hsd17b13 1–3 and 6–7 denotes two separate regions in the gene. N=6 male mice per group. B-I assessed at sacrifice at age 20 weeks. Mean±SEM; *p<0.05, ****p<0.0001
Concomitantly with their increased weight, Hsd17b13-KO mice had significantly higher liver weights (Figure 2E), and liver/body weight ratios (Figure 2F), as well as a trend for increased liver triglyceride content (Figure 2G). No histological difference was observed (Figure 2H). Hepatic expression of genes associated with lipogenesis (srebf1, Fasn, Scd, and Acc1), gluconeogenesis (Pck1 and G6pc), or lipid/energy metabolism (Dgat2, Mttp, Ppara, Pgc1a, Acat1, Acox1, and Cpt1a) were unaffected (Figure 2I), consistent with a previous report (27).
Limited impact of Hsd17b13 deficiency on high fat diet liver injury
To assess the response of Hsd17b13-KO mice to an obesogenic diet, we challenged the mice with 12 weeks of high fat diet (HFD). Interestingly, despite the mildly obese phenotype of KO mice on chow diet, no difference in body weight was noted between Hsd17b13-KO and WT mice when fed HFD (Figure 3A). There was also no difference between genotypes in liver weight, liver triglyceride content and serum liver enzyme levels (Figure 3B–D). Histologically, there was no difference in overall steatosis scores between genotypes (Figure 3E and F); however, the morphology of lipid vacuoles appeared different in Hsd17b13-KO mice which had a higher proportion of macro-steatotic hepatocytes compared to controls (Figure 3G). The magnitude of histological inflammation was mild and did not differ between genotypes (Figure 3H). Collagen content was not significantly different between groups (Figure 3I). Serum cholesterol (Figure 3J), triglycerides (Figure 3K), free fatty acids (Figure 3L), and blood glucose levels (Figure 3M) were also similar between genotypes. Hepatic expression of inflammatory and fibrogenic genes demonstrated a modest increase in three inflammatory genes (Itgax, Tnfa, and Il-1b) in Hsd17b13-KO mice with no significant difference otherwise (Figure 3N).
Figure 3. Limited impact of Hsd17b13 deficiency on high fat diet (HFD) liver injury.
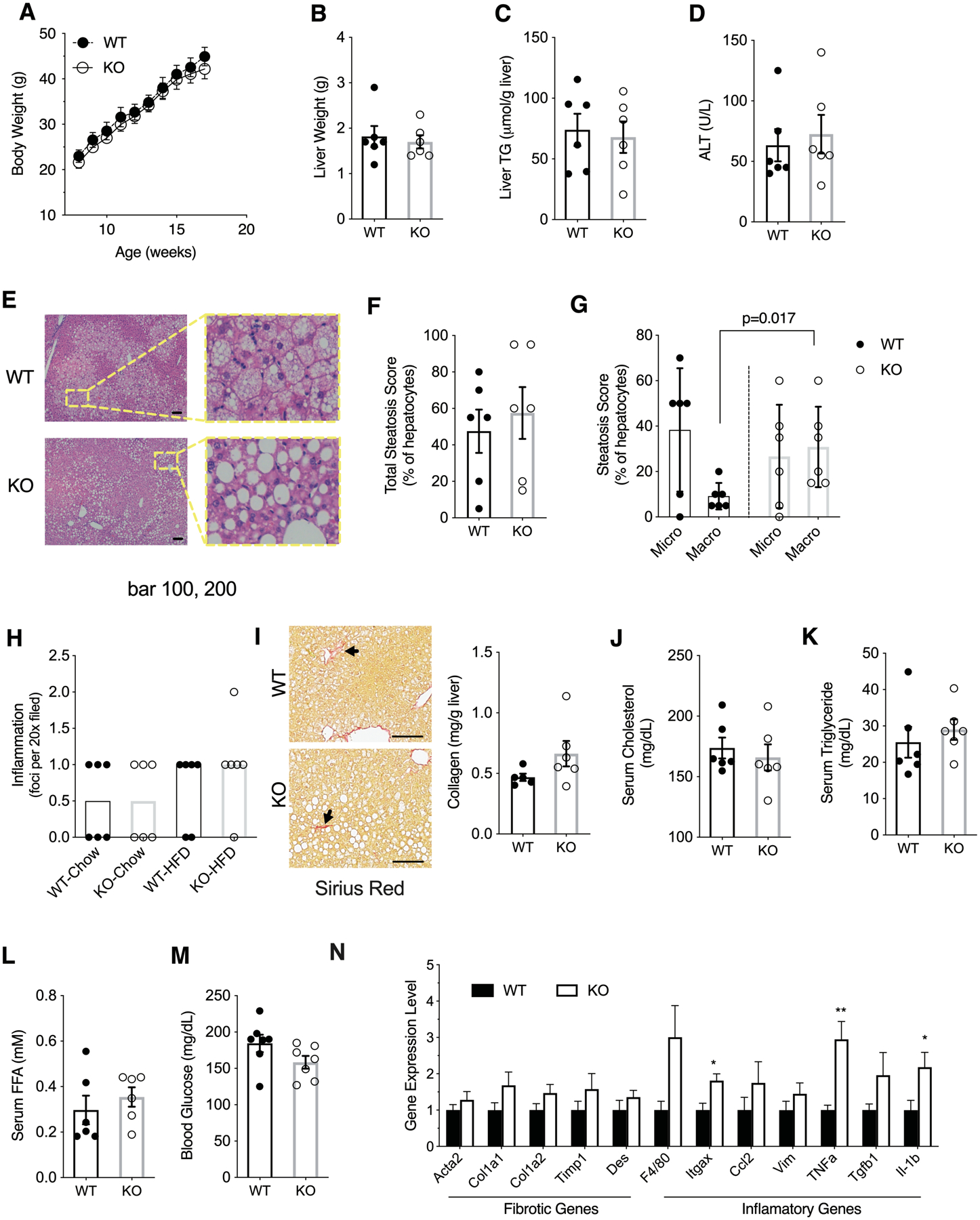
Eight week-old male wild type (WT) and Hsd17b13-knockout (KO) mice were fed with HFD for 12 weeks and sacrified at age 20 weeks after a 5 hours fast. (A) Weekly body weight measurements, (B) Liver weights, (C) Liver triglycerides, and (D) Serum alanine aminotransferase (ALT). (E) Hematoxylin & eosin staining of liver sections. Bar indicates 100 μM. (F) Percentage of steatotic hepatocytes. (G) Percentage of macro- and microtsteatotic hepatocytes. (H) Histological inflammation score. (I) Hepatic collagen content by Sirus Red staining (left) or colorimetric assay (right). Arrow indicates representive positive staining. Bar indicates 100 μM. (J) Serum cholesterol, (K) Serum triglycerides, (L) Serum free fatty acids, and (M) Blood glucose. (N) Hepatic gene expression level of fibrosis- and inflammation-related genes. N=6 male mice per group. B-N assessed at sacrifice at age 20 weeks. Mean±SEM; *p<0.05, **p<0.01
Hsd17b13 deficiency does not impact liver injury with Western diet
To generate liver fibrosis and injury resembling human NASH, mice were challenged for 16 weeks with Western Diet (WD), shown to induce more liver injury compared to HFD (31, 32). Similarly to our findings with HFD, there were no differences in body weight (Figure 4A), liver weight (Figure 4B), liver triglyceride content (Figure 4C), or serum liver enzyme levels (Figure 4D) between genotypes. WD induced a greater degree of steatosis and inflammation compared to HFD. However, Hsd17b13 genotype did not affect overall steatosis (Figure 4E–F) and inflammation (Figure 4H) scores, nor did it affect the collagen content (Figure 4I). Similarly to HFD, we noted a marked shift towards macro-steatosis in Hsd17b13-KO mice (Figure 4G). There were no difference in the expression level of fibrosis- or inflammation-related genes (Figure 4J).
Figure 4. Hsd17b13 deficiency does not protect mice from Western diet (WD) liver injury.
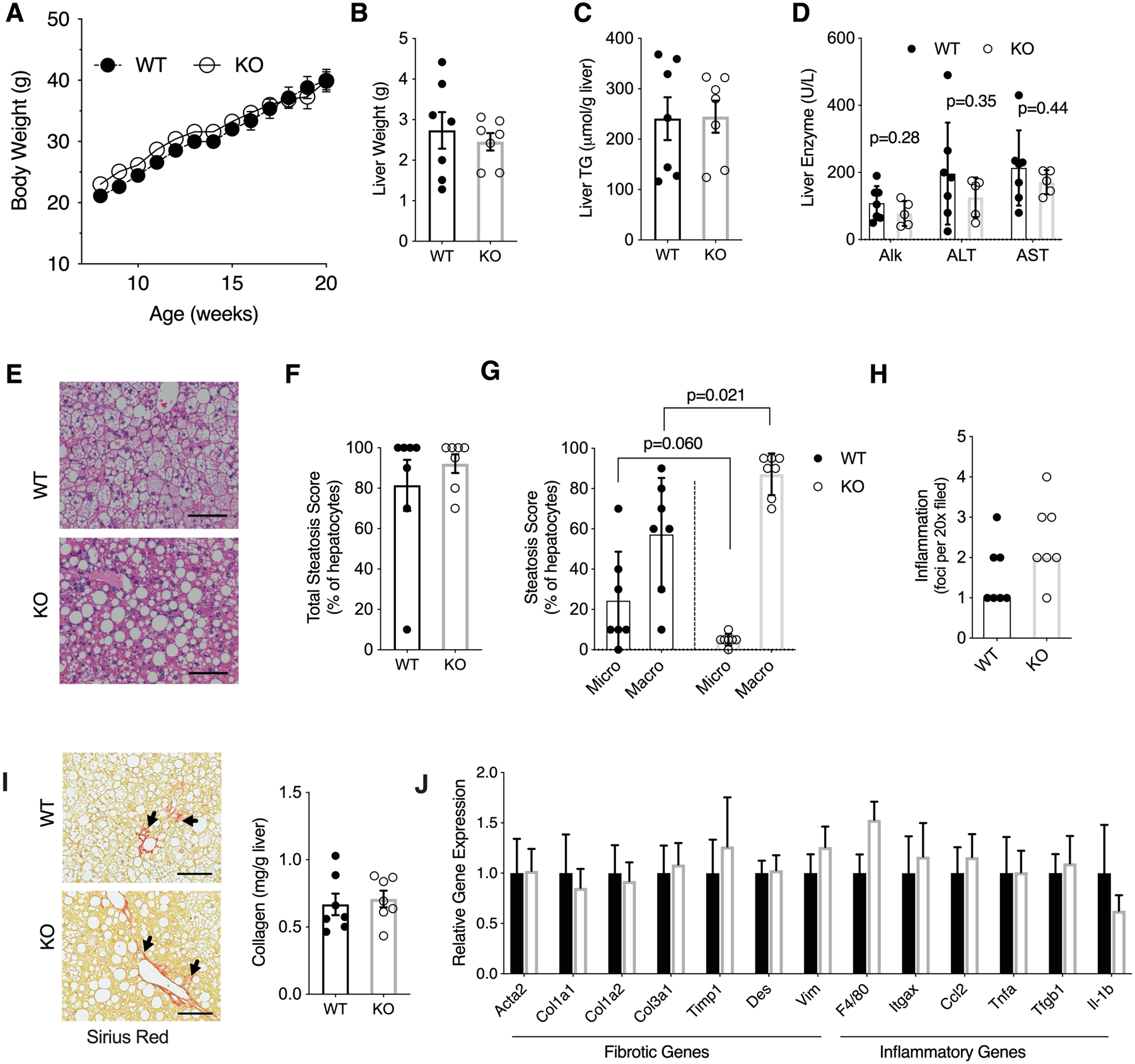
Eight week-old male wild type (WT) and Hsd17b13-knockout (KO) mice were fed with WD for 16 weeks and sacrified at age 24 weeks after a 5 hours fast. (A) Weekly body weight, (B) Liver weight, (C) Liver triglycerides, and (D) Serum liver enzymes. (E) H&E staining of liver sections. Bar indicates 100 μM. (F) Percentage of steatotic hepatocytes. (G) Percentage of hepatocytes with macro- and micro-steatosis. (H) Histological inflammation score. (I) Hepatic collagen content by Sirius Red staining (left) or colorimetric assay (right). Arrow indicates representive positive staining. Bar indicates 100 μM. (J) Hepatic expression level of fibrosis- and inflammation-related genes. N=7 male mice per group. B-J assessed at sacrifice at age 24 weeks. Mean±SEM
We previously identified HSD17B13 as a retinol dehydrogenase but noted that commercial HFD or WD preparations are low (4 IU/g) in vitamin A. To ensure that the lack of difference between genotypes is not due to the low level of putative HSD17B13 substrate, we assessed the response of Hsd17b13-KO mice and littermate controls to a WD enriched with vitamin A (19 IU/g) but again, found no difference between genotypes in obesity or features of fatty liver disease (Supplementary Figure 3).
Hsd17b13 deficiency does not protect mice from injury induced by long-term Western diet
To determine the effects of long-term dietary exposure, we challenged Hsd17b13-KO and WT mice with WD for 10 months. There was no difference between genotypes in body weight (Figure 5A), liver weight (Figure 5B), liver triglyceride content (Figure 5C), serum liver enzyme levels (Figure 5D–E), or liver histology (Figure 5F–I), though a trend for increased macrosteatosis was seen again (Figure 5H). Long-term WD induced more collagen formation compared with the 16 week diet treatment, however, there was no difference in collagen content between genotypes (Figure 5J). We also did not note a difference in fibrosis- and inflammation-related gene expression between genotypes (Figure 5K). Three of six animals in WT group and four of six animals in Hsd17b13-KO group had visible tumors in their livers (Figure 5L) with no difference in surface tumor number (Figure 5M).
Figure 5. Hsd17b13 deficiency does not protect mice from long-term Western diet induced injury.
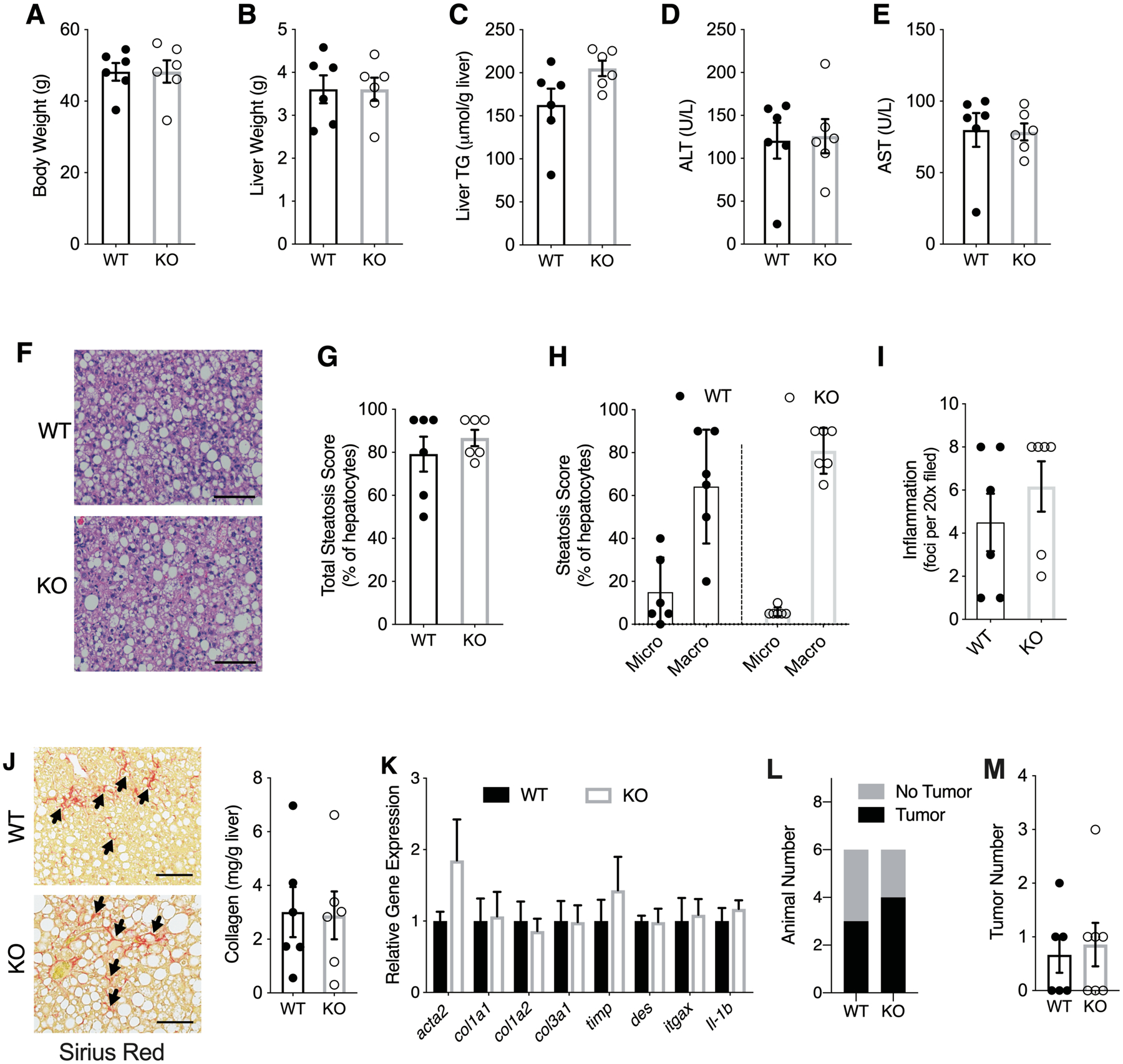
Eight week-old male wild type (WT) and Hsd17b13-KO (KO) mice were fed Western diet (WD) for 10 months and sacrified after a 5 hour fast. (A) Body weight, (B) liver weight, (C) Liver triglycerides, and (D-E) serum liver enzymes. (F) Hematoxyline & eosin staining of liver sections. Bar indicates 100 μM. (G) Percent of steatotic hepatocytes. (H) Percent of hepatocytes with micro- and macro-steatosis. (I) Histological inflammation score. (J) Hepatic collagen content by Sirius Red staining (left) or colorimetric assay (right). Arrow indicates representive positive staining. Bar indicates 100 μM. (K) Hepatic expression level of fibrosis- and inflammation-related genes. (L) Liver tumor incidence and (M) Liver surface tumor number. N=6 male mice per group. All assessments at sacrifice, after 10 months of WD. Mean±SEM
Hsd17b13 deficiency in alcohol-induced liver disease
The HSD17B13 loss of function SNP rs7261356 is associated with protection from human alcohol-associated liver disease (15, 17). To determine whether this can be replicated in a murine model, we applied the NIAAA model of chronic plus one binge ethanol exposure (28). Compared to WT controls, Hsd17b13-KO mice showed no difference in ALT level (Figure 6A), hepatic triglyceride content (Figure 6B) or hepatic cholesterol level (Figure 6C) when exposed to alcohol containing diet. Interestingly, despite no difference in biochemically-measured hepatic triglyceride content, Hsd17b13-KO mice were noted to have higher histological steatosis scores, reflecting more hepatocytes with visible lipid droplets (Figure 6D–E) and suggesting Hsd17b13 may affect lipid droplet morphology in vivo. The Hsd17b13-KO mice had a non-significant trend for higher hepatic inflammation scores (Figure 6F). Again, no significant changes in gene expression were observed (Figure 6G).
Figure 6. Effect of Hsd17b13 deficiency on alcohol-induced liver disease.
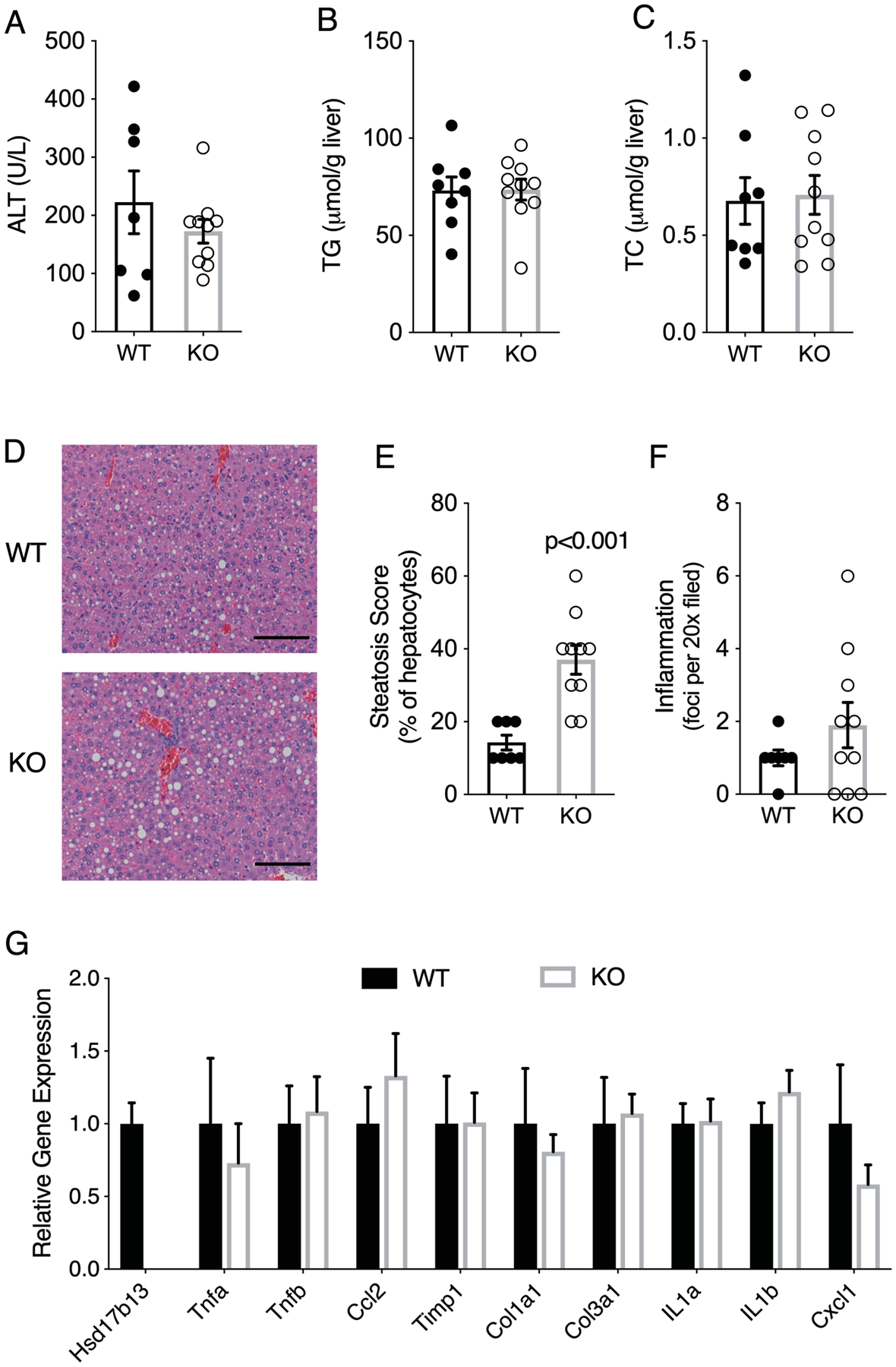
Female wild type (WT, n=8) and Hsd17b13-KO (KO, n=10) mice were given 10 days of Lieber-DeCarli ethanol diet followed by one binge dose of alcohol (ETOH) at age of 18 weeks. (A) Serum ALT level of WT (n=7) and KO (n=10) mice. (B) Liver triglycerides. (C) Liver total cholesterol. (D) Hematoxyline & eosin staining of liver sections. Bar indicates 100 μM. (E) Percentage of steatotic hepatocytes. (F) Histological inflammation score. WT, n=8; KO, n=10 for all experiments unless stated otherwise. Mean±SEM
Effect of Hsd17b13 deficiency on liver lipids
It was recently reported that phospholipids are enriched in the livers of morbidly obese human carriers of the HSD17B13 rs72613567 minor allele (33). We compared the liver lipidome of Hsd17b13-KO and WT controls fed HFD and, in contrast to the human data, found a non-significant trend for decreased levels of lysophospatidylethanolamines (p=0.08) and phosphatidylethanolamines (p=0.09) in the knockouts and no difference in the total abundance of other lipid species (Figure 7A). Few individual lipid species were significantly changed, most notably cholesterol esters with very long chain polyunsaturated fatty acids (Figure 7B). However, despite the changes for most individual lipid species not meeting statistical significance, we noted an effect by class, where the vast majority of cholesterol esters (89%) and monoacylglyecerols (85%) as well as sphingolipids with very long chain fatty acids were increased in Hsd17b13-KO, while the majority of TG (77%) and lysophosphatidylethanolamines (93%) were decreased (Figure 7C–D). Phosphatidylcholine, phosphatidylethanolamines, and ceramide analogue have been reported to regulate LD size but whether the subtle changes we observed in lipid species are sufficient to induce the increased macro-steatosis in Hsd17b13-KO mice requires further investigation (34, 35).
Figure 7. Effect of Hsd17b13 deficiency on the liver lipidome.
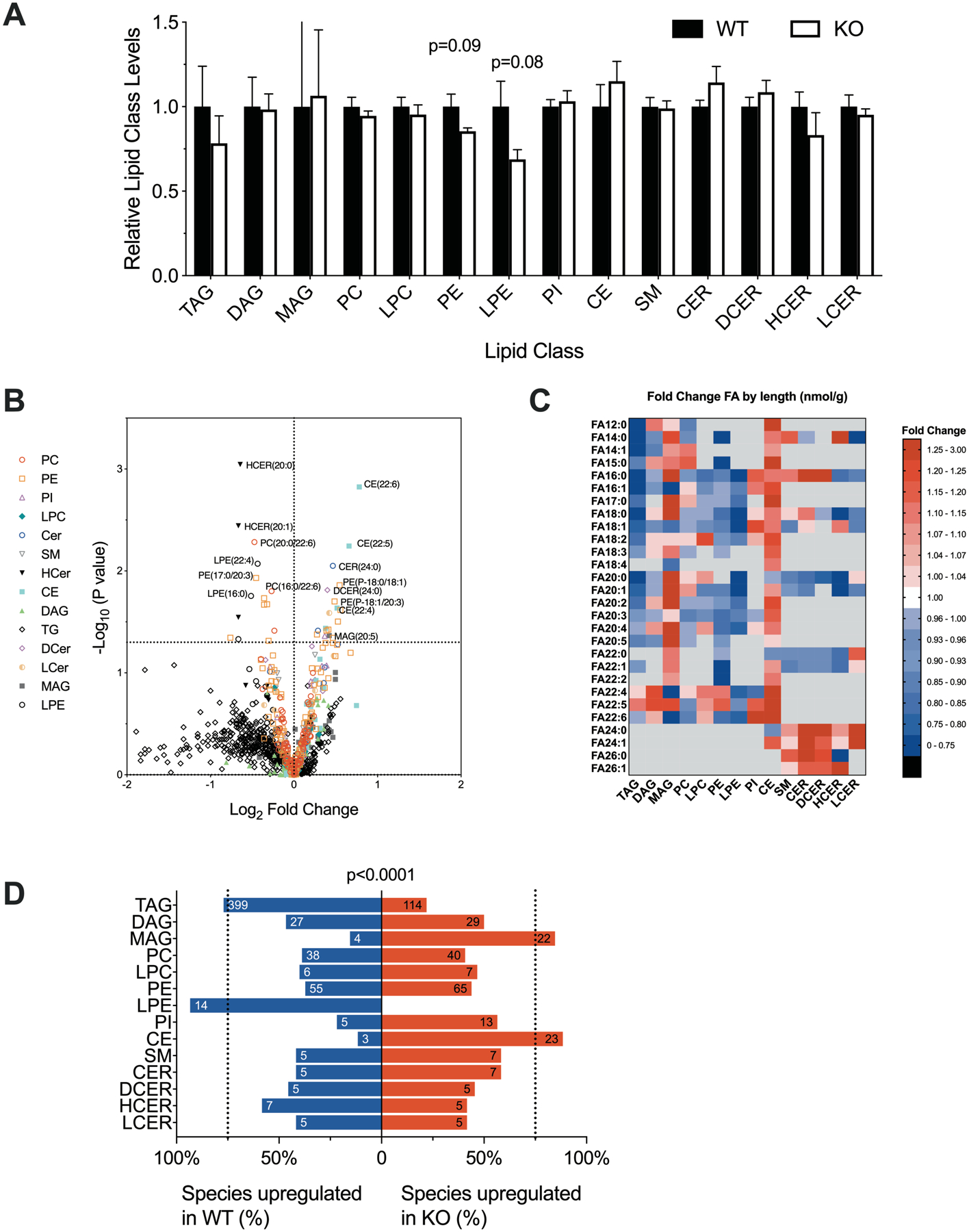
Eight week-old male wild type (WT) and Hsd17b-13-KO (KO) were fed high-fat diet for 12 weeks and sacrified at age 20 weeks after a 5 hours fast. Liver samples were analyzed for lipid composition. (A) Total amounts of liver lipids by lipid class, relative to controls. Mean±SEM. P-values from Student’s t-test. (B) Volcano plot of 986 individual liver lipid species. The x-axis denotes the log2 fold change in concentration in KO compared to WT and the y-axis the −log10 of the unadjusted p-value. (C) Heatmap of individual hepatic lipid species by class and fatty acid composition. Color reflects fold-change in concentrations between KO and WT. Red denotes species increased in KO and blue, in WT. (D)Percent of all species within a class that are upregulated in KO (red) or WT (blue). Numbers denote the absolute number of species in each category. Significance calculated by Γ2. TAG, triacylglycerol; DAG, diacylglycerol; MAG, monoacylglycerol; PC, phosphatidylcholine; LPC, lysophosphatidylcholine; PE, phosphatidylethanolamine; LPE, lysophosphatidylethanolamine; PI, phosphatidylinositol; CE, cholesterol ester; SM, sphingomyelin; CER, ceramide; DCER, dihydroceramide; HCER, hexosylceramide; LCER, lactosylceramide. N=6 male mice per group. Animals are the same as depicted in Figure 3.
Effect of Hsd17b13 deficiency on liver retinoids
We have previously characterized human HSD17B13 as a retinol dehydrogenase (RDH) (15). To determine whether retinol metabolism is affected in Hsd17b13 KO mice, ex-vivo RDH activity of liver extracts (Supplementary Figure 4A) and levels of hepatic retinoids (retinol, retinyl ester, retinoic acid) (Supplementary Figure 4B–D) were determined. We did not find differences in RDH activity or retinoid levels between Hsd17b13-KO and controls. To determine whether mouse Hsd17b13 protein has RDH activity, we performed an in vitro assay as previously described (15). In contrast to human HSD17B13, the mouse protein demonstrated no RDH activity (Supplementary Figure 4E–F).
Finally, we previously reported that HSD17B13 is upregulated in NASH patients (15). We confirmed that human HSD17B13 is indeed upregulated by comparing absolute transcript count in normal controls and NASH patients determined by RNA-seq (Supplementary Figure 5A). However, the expression of Hsd17b13 in mice was not affected by HFD, WD, or long term WD feeding, despite the resultant fatty liver (Supplementary Figure 5B–D), suggesting a differenct regulation mechanism between the species.
Discussion
Genetic studies identified a strong association between variants in HSD17B13 and serum liver enzyme levels (15–17), nonalcoholic (15, 17, 23) and alcoholic liver disease (15, 17), and hepatocellular carcinoma (24, 25), suggesting this gene has a role in regulating hepatic response to metabolic injury. In this study, we sought to characterize the impact of knocking out the mouse orthologue, Hsd17b13, to help understand its mechanism of action. We found that Hsd17b13 deficiency in mice does not affect liver injury, fibrosis and fat content under several fatty liver inducing-dietary conditions indicating that Hsd17b13 deficiency may not have a protective role in these models.
To date three independent variants in HSD17B13 were associated with protection from liver injury. A splice site SNP, rs72613567 (linked to rs6834314), which generates two novel splicing variants (HSD17B13-G insertion and HSD17B13-Exon 6 deletion) (15, 17); rs62305723 which encodes a P260S mutation (15); and rs143404524 which generates a frame-shift and premature truncation (36). Importantly, in all 3 variants, the allele associated with protection from liver injury generates a loss-of-function protein. Thus, disruption of the enzymatic function is associated with a decrease in liver injury and justifies exploring HSD17B13 as a therapeutic target.
Based on these data, we expected Hsd17b13-KO mice to display a phenotype similar to the human loss-of-function mutants and be protected from fatty liver-associated injury. We treated mice with an obesity-inducing diet (HFD), NASH-inducing diet (WD, 16 weeks), NASH and HCC inducing diet (WD, 10 months), and a diet modeling alcohol-associated liver injury. In all of these models, the expected liver phenotype was observed (e.g. steatosis, injury or HCC) but Hsd17b13-KO did not lead to decreased injury in any of the models. Specifically, we did not see an impact of the knock-out on histological liver steatosis or inflammation scores, serum liver enzymes, liver fat content, HCC incidence, and induction of inflammatory or fibrogenic genes. Furthermore, even if not significant, many of these features were numerically worse, not better, in the Hsd17b13-KO animals. Thus, we conclude that loss of Hsd17b13 does not affect diet-induced fatty liver disease in mice.
There could be several explanations for the discrepancy between mouse and human phenotypes. First, it is possible that the dietary models we employed did not produce sufficient injury to allow for a genetic effect to be noticeable. No one mouse model fully captures the human phenotype of NASH (32). The models we used are characterized by the development of obesity, metabolic syndrome and inflammation but do not develop marked ballooning degeneration (which is generally uncommon in mouse models) or fibrosis. However, beyond fibrosis, the human loss-of-function variants of HSD17B13 are also associated with inflammation and injury (15) thought to be precursor to, and drivers of, fibrosis, and these were not affected by the KO in our models. In addition, a difference between genotypes was not seen in either the alcohol induced liver disease which induces marked injury, or the long term WD model which induced HCC and is more advanced than fibrosis only.
Second, it is possible that in mice, the loss of Hsd17b13 enzymatic activity is compensated for by another enzyme. Two paralogues of HSD17B13, HSD17B11 and RDH10, share conserved cofactor binding, homodimer interaction, substrate binding and catalytic sites, as well as the critical P260 site of unknown function (15), which could indicate a similar substrate preference. In fact, steroids were proposed as substrates for HSD17B13 (17) and HSD17B11 (37, 38), and retinol was found to be a substrate for both HSD17B13 (15) and RDH10 (39). Moreover, all three proteins localize to lipid droplets (15, 17–19, 40). We did not observe a compensatory upregulation of Hsd17b11 and Rdh10 in Hsd17b13-KO mice livers (Supplementary Figure 6A). However, in WT mice, the hepatic expression level of Hsd17b11 is about half of that of Hsd17b13 (Supplementary Figure 6B) while in human liver HSD17B13 is expressed at 3–4 fold higher levels compared to HSD17B11, and 16 fold higher than RDH10 (Supplementary Figure 6C). Considering the naturally high expression level of Hsd17b11 in mouse liver (175.5 TPM vs 57.98 TPM in human), it is plausible that this highly-expressed paralogous protein compensates for the loss of Hsd17b13 activity in mice but not in humans.
Third, it is possible that the mouse and human HSD17B13 orthologues have different functions or substrate specificity. In contrast to our previous characterization of human HSD17B13 as a retinol dehydrogenase, we did not find in vivo or in vitro evidence of RDH activity of mice Hsd17b13. In addition to retinol, human HSD17B13 was found to be active toward steroid substrates and bioactive lipids (17). We found increased cholesterol ester species in Hsd17b13-KO mice livers (Figure 7); whether this reflects a cholesterol ester hydrolase activity remains to be seen. It is possible that Hsd17b13 prefers steroid substrates over retinol in mouse in vivo, and Hsd17b13-KO leads to accumulated cholesterol caused by decreased usage.
Finally, it is possible that the loss-of-function human HSD17B13 mutants exhibit a protective role in a mechanism independent of their enzymatic activity, for example if they also function in a dominat negative manner to affect other proteins. This brings to mind the initial mouse studies on Patatin-like phospholipase domain-containing protein 3 (PNPLA3). A common variant in PNPLA3 (rs738409, encoding I148M mutation) was identified in several genetic studies as a modulator of human NAFLD (5–10), but despite the strong human evidence, no clear phenotype was seen after Pnpla3 deletion in mice (41, 42), likely because I148M leads to a dominant negative effect. Hepatic steatosis was only observed when PNPLA3-I148M (but not wildtype PNPLA3) (43) was overexpressed or PNPLA3-I148M was knocked-in (44, 45) in mice. The discrepancy in phenotype between Hsd17b13-KO in mice and human variants could thus point at a mechanism separate from loss of function.
Although we did not find an effect of Hsd17b13-KO on overall hepatic TG content under the various dietary conditions, we observed a consistent change in lipid droplet morphology, with a shift towards macrosteatosis occurring in Hsd17b13-KO mice at an earlier stage. This may explain a discrepance in in vivo human findings where the inactivating variants in rs72613567 and rs6834314 were found to be associated with steatosis when assessed by histological scores (reflecting visible lipid vacuoles) (15) but not when assessed by MRI-based measurement of hepatic TG content (17). In vitro, we and Abul-Husn et al found that intracellular TG content is not affected by HSD17B13 overexpression (15, 17). In contrast, Su et al reported that HSD17B13 overexpression in human hepatocytes cell lines increased lipid droplets number and size (19). In that context, Luukkonen et al recently described an increase in liver phospholipids in morbidly obese carriers of the rs72613567 minor allele undergoing bariatric surgery (33), but we found no evidence for a similar effect in mice fed HFD, instead seeing an increase in cholesterol esters and monoacylglycerols. Whether and how Hsd17b13 affects lipid droplet dynamics in vivo is intriguing and requires further studies.
Another intriguing finding is the development of increased weight gain and hepatic steatosis in Hsd17b13 KO mice fed regular chow diet. The genetic effect size was small and was completely overcome when exposed to nutrient overload. Consistent with our findings, Adam et al recently reported that Hsd17b13 deficiency leads to hepatic steatosis and inflammation with prolonged (9 months) regular chow diet (27), but did not find a difference in body weight. Interestingly, they also noted a greater prevalence of macrovesicular steatosis in their Hsd17b13-KO model. In contrast to the findings associated with Hsd17b13 deficiency, Su et al noted an increase in steatosis in mouse livers 4 days after a single injection of adenovirus-encoded HSD17B13 (19). More studies are needed to understand the discrepancy between the different reports.
We have previously characterized HSD17B13 as a retinol dehydrogenase (15). Yang et al described that mice heterozygous for the paralogue Rdh10 display mild obesity and increased adiposity (46), and a similar phenotype was observed in Rdh1-null mice, which also have disrupted retinoid metabolism (47). It is unclear whether the similar phenotypes of Hsd17b13 KO, Rdh10 heterozygotes, and Rdh1-null mice are caused by retinol metabolism since Rdh10 heterozygouss mice only have very mild decrease (≤25%) in the hepatic all-trans-retinoic acid concentration.
In conclusion, our data shows for the first time that, although Hsd17b13 clearly modulates hepatic lipid droplet formation, its deficiency in mice does not protect from injury associated with nonalcoholic and alcoholic fatty liver disease, nor from diet-induced HCC development. Understanding inter-species differences may be key to unraveling the mechanism of action of human HSD17B13 and advance its role as a therapeutic target for fatty liver disease.
Supplementary Material
Acknowledgement:
The authors would like to thank Suman Karki for his comments on this paper (University of Alabama - Birmingham). The results shown here are partly based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga and the Genotype-Tissue Expression-GTEx Portal: https://gtexportal.org/home/.
Source of funding
This study was supported by the Intramural Research Programs of the National Institutes of Health (NIH), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and NIH, National Cancer Institute, NIH Office of Dietary Supplements (ODS) Research Scholars Program to YM, and by National Institute of Alcohol Abuse and Alcoholism (AA012153, to N.Y.K.).
Abbreviations:
- HSD17B13
17-beta hydroxysteroid dehydrogenase 13
- GWAS
Genome wide association studies
- NAFLD
Non-alcoholic fatty liver disease
- NASH
Non-alcoholic steatohepatitis
- HCC
hepatocellular carcinoma
- SNP
Single nucleotide polymorphism
- ALT
Asparate aminotransferase
- AST
Alanine aminotransferase
- RC
Regular Chow
- HFD
High fat diet
- WD
Western diet
- TG
Triglycerides
References
- 1.Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018;15:11–20. [DOI] [PubMed] [Google Scholar]
- 2.Loomba R, Schork N, Chen CH, Bettencourt R, Bhatt A, Ang B, Nguyen P, et al. Heritability of Hepatic Fibrosis and Steatosis Based on a Prospective Twin Study. Gastroenterology 2015;149:1784–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwimmer JB, Celedon MA, Lavine JE, Salem R, Campbell N, Schork NJ, Shiehmorteza M, et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology 2009;136:1585–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 2004;40:1387–1395. [DOI] [PubMed] [Google Scholar]
- 5.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuan X, Waterworth D, Perry JR, Lim N, Song K, Chambers JC, Zhang W, et al. Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am J Hum Genet 2008;83:520–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sookoian S, Castano GO, Burgueno AL, Gianotti TF, Rosselli MS, Pirola CJ. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J Lipid Res 2009;50:2111–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rotman Y, Koh C, Zmuda JM, Kleiner DE, Liang TJ, Nash CRN. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology 2010;52:894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Speliotes EK, Butler JL, Palmer CD, Voight BF, Consortium G, Consortium MI, Nash CRN, et al. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology 2010;52:904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valenti L, Al-Serri A, Daly AK, Galmozzi E, Rametta R, Dongiovanni P, Nobili V, et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010;51:1209–1217. [DOI] [PubMed] [Google Scholar]
- 11.Holmen OL, Zhang H, Fan Y, Hovelson DH, Schmidt EM, Zhou W, Guo Y, et al. Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat Genet 2014;46:345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjaerg-Hansen A, Vogt TF, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahdessian H, Taxiarchis A, Popov S, Silveira A, Franco-Cereceda A, Hamsten A, Eriksson P, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci U S A 2014;111:8913–8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buch S, Stickel F, Trepo E, Way M, Herrmann A, Nischalke HD, Brosch M, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet 2015;47:1443–1448. [DOI] [PubMed] [Google Scholar]
- 15.Ma Y, Belyaeva OV, Brown PM, Fujita K, Valles K, Karki S, de Boer YS, et al. 17-Beta Hydroxysteroid Dehydrogenase 13 Is a Hepatic Retinol Dehydrogenase Associated With Histological Features of Nonalcoholic Fatty Liver Disease. Hepatology 2019;69:1504–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, Van der Harst P, Holm H, et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet 2011;43:1131–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abul-Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, Liu Y, et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N Engl J Med 2018;378:1096–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horiguchi Y, Araki M, Motojima K. 17beta-Hydroxysteroid dehydrogenase type 13 is a liver-specific lipid droplet-associated protein. Biochemical and biophysical research communications 2008;370:235–238. [DOI] [PubMed] [Google Scholar]
- 19.Su W, Wang Y, Jia X, Wu W, Li L, Tian X, Li S, et al. Comparative proteomic study reveals 17beta-HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 2014;111:11437–11442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marchais-Oberwinkler S, Henn C, Moller G, Klein T, Negri M, Oster A, Spadaro A, et al. 17beta-Hydroxysteroid dehydrogenases (17beta-HSDs) as therapeutic targets: protein structures, functions, and recent progress in inhibitor development. The Journal of steroid biochemistry and molecular biology 2011;125:66–82. [DOI] [PubMed] [Google Scholar]
- 21.Belyaeva OV, Chang C, Berlett MC, Kedishvili NY. Evolutionary origins of retinoid active short-chain dehydrogenases/reductases of SDR16C family. Chem Biol Interact 2015;234:135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma Y, Brown PM, Rotman Y. Reply to “Does the HSD17B13 rs72613567 splice variant actually yield a new type of alternative splicing”. Hepatology 2019. [DOI] [PubMed] [Google Scholar]
- 23.Pirola CJ, Garaycoechea M, Flichman D, Arrese M, San Martino J, Gazzi C, Castano GO, et al. Splice variant rs72613567 prevents worst histologic outcomes in patients with nonalcoholic fatty liver disease. J Lipid Res 2019;60:176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stickel F, Lutz P, Buch S, Nischalke HD, Silva I, Rausch V, Fischer J, et al. Genetic variation in HSD17B13 reduces the risk of developing cirrhosis and hepatocellular carcinoma in alcohol misusers. Hepatology 2019. [DOI] [PubMed] [Google Scholar]
- 25.Yang J, Trepo E, Nahon P, Cao Q, Moreno C, Letouze E, Imbeaud S, et al. A 17-Beta-Hydroxysteroid Dehydrogenase 13 Variant Protects From Hepatocellular Carcinoma Development in Alcoholic Liver Disease. Hepatology 2019;70:231–240. [DOI] [PubMed] [Google Scholar]
- 26.ClinicalTrials.gov. Study of ARO-HSD in Healthy Volunteers and Patients With Non-Alcoholic Steatohepatitis (NASH) or Suspected NASH. In; 2019. December 17.
- 27.Adam M, Heikela H, Sobolewski C, Portius D, Maki-Jouppila J, Mehmood A, Adhikari P, et al. Hydroxysteroid (17beta) dehydrogenase 13 deficiency triggers hepatic steatosis and inflammation in mice. FASEB J 2018:fj201700914R. [DOI] [PubMed] [Google Scholar]
- 28.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc 2013;8:627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ravussin Y, Gutman R, LeDuc CA, Leibel RL. Estimating energy expenditure in mice using an energy balance technique. Int J Obes (Lond) 2013;37:399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aizarani N, Saviano A, Sagar, Mailly L, Durand S, Herman JS, Pessaux P, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019;572:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanuri G, Bergheim I. In vitro and in vivo models of non-alcoholic fatty liver disease (NAFLD). Int J Mol Sci 2013;14:11963–11980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willebrords J, Pereira IV, Maes M, Crespo Yanguas S, Colle I, Van Den Bossche B, Da Silva TC, et al. Strategies, models and biomarkers in experimental non-alcoholic fatty liver disease research. Prog Lipid Res 2015;59:106–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luukkonen PK, Tukiainen T, Juuti A, Sammalkorpi H, Haridas PAN, Niemela O, Arola J, et al. Hydroxysteroid 17-beta dehydrogenase 13 variant increases phospholipids and protects against fibrosis in nonalcoholic fatty liver disease. JCI Insight 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kato Y, Arakawa S, Terasawa K, Inokuchi JI, Iwata T, Shimizu S, Watabe T, et al. The ceramide analogue N-(1-hydroxy-3-morpholino-1-phenylpropan-2-yl)decanamide induces large lipid droplet accumulation and highlights the effect of LAMP-2 deficiency on lipid droplet degradation. Bioorg Med Chem Lett 2020;30:126891. [DOI] [PubMed] [Google Scholar]
- 35.van der Veen JN, Kennelly JP, Wan S, Vance JE, Vance DE, Jacobs RL. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim Biophys Acta Biomembr 2017;1859:1558–1572. [DOI] [PubMed] [Google Scholar]
- 36.Kozlitina J, Stender S, Hobbs HH, Cohen JC. HSD17B13 and Chronic Liver Disease in Blacks and Hispanics. N Engl J Med 2018;379:1876–1877. [DOI] [PubMed] [Google Scholar]
- 37.Li KX, Smith RE, Krozowski ZS. Cloning and expression of a novel tissue specific 17beta-hydroxysteroid dehydrogenase. Endocr Res 1998;24:663–667. [DOI] [PubMed] [Google Scholar]
- 38.Brereton P, Suzuki T, Sasano H, Li K, Duarte C, Obeyesekere V, Haeseleer F, et al. Pan1b (17betaHSD11)-enzymatic activity and distribution in the lung. Mol Cell Endocrinol 2001;171:111–117. [DOI] [PubMed] [Google Scholar]
- 39.Belyaeva OV, Johnson MP, Kedishvili NY. Kinetic analysis of human enzyme RDH10 defines the characteristics of a physiologically relevant retinol dehydrogenase. J Biol Chem 2008;283:20299–20308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang W, Napoli JL. The retinol dehydrogenase Rdh10 localizes to lipid droplets during acyl ester biosynthesis. J Biol Chem 2013;288:589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen W, Chang B, Li L, Chan L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology 2010;52:1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Basantani MK, Sitnick MT, Cai L, Brenner DS, Gardner NP, Li JZ, Schoiswohl G, et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J Lipid Res 2011;52:318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, Castro-Perez J, et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest 2012;122:4130–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smagris E, BasuRay S, Li J, Huang Y, Lai KM, Gromada J, Cohen JC, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015;61:108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Linden D, Ahnmark A, Pingitore P, Ciociola E, Ahlstedt I, Andreasson AC, Sasidharan K, et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol Metab 2019;22:49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang D, Vuckovic MG, Smullin CP, Kim M, Lo CP, Devericks E, Yoo HS, et al. Modest Decreases in Endogenous All-trans-Retinoic Acid Produced by a Mouse Rdh10 Heterozygote Provoke Major Abnormalities in Adipogenesis and Lipid Metabolism. Diabetes 2018;67:662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang M, Hu P, Krois CR, Kane MA, Napoli JL. Altered vitamin A homeostasis and increased size and adiposity in the rdh1-null mouse. FASEB J 2007;21:2886–2896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.