

Abstract
Pre-mRNA processing of the replication-dependent canonical histone mRNAs requires an endonucleolytic cleavage immediately after a conserved stem loop structure which occurs before RNA Pol II encounters any poly(A) signal. Thus, in contrast to all other eukaryotic mRNAs, the canonical histone mRNAs are not polyadenylated in their 3’ ends. The binding of stem-loop binding protein (SLBP) to the stem loop structure of the histone mRNAs is required for this process. SLBP is also involved in regulation of histone mRNA nuclear export, degradation, and translation. Depletion of SLBP has been shown to induce polyadenylation of histone mRNAs and alteration of histone protein levels, which are considered attributable to observed aberrant cell cycle progress and genomic instability by the loss of SLBP function. Recent studies have demonstrated that some heavy metal carcinogens, including arsenic and nickel, can induce the loss of SLBP and the gain of polyadenylation of canonical histone mRNAs. Polyadenylated canonical histone H3 can result in abnormal transcription, cell cycle arrest, genomic instability, and cell transformation, which links SLBP depletion and subsequent histone mRNA misprocessing to cancer. This review seeks to briefly summarize what is known about regulation of SLBP expression, consequence of SLBP depletion, its roles in cancer-related end points, with particular focus on metal-induced SLBP depletion and the potential of SLBP depletion as a new mechanism for metal-induced carcinogenesis.
Keywords: Stem-loop binding protein, Histone, Polyadenylation, Heavy metal, Carcinogenesis
Introduction
Metazoan replication-dependent histone genes are unique–they do not have introns and their mRNAs are not polyadenylated, instead it ends in a conserved stem-loop structure [1, 2]. This hairpin structure is not only involved in processing of the 3’ end of histone mRNAs, but also in cytoplasmic transport, translation, and degradation of canonical histone mRNAs. During attempts to characterize trans-acting factors, the stem-loop binding protein (SLBP), which has sometimes been referred to as hairpin-binding protein (HBP), was identified as the protein that binds to the histone mRNA stem-loop sequence and regulates the 3’ processing of histone mRNAs [3]. The human SLBP gene is located on chromosome 4, encoding a protein with 270 amino acids [4]. A new 75-amino acid RNA-binding domain (RBD) was identified in the SLBP protein [5].
More than two decades ago, SLBP cDNA was separately cloned by two different groups using yeast three-hybrid system [4, 5]. The cloning of SLBP gene accelerated studies on its regulation and biochemical and biological functions. It has been demonstrated that SLBP is cell-cycle dependent with peak levels being found in S phase [6], which is regulated by both translational and post-translational mechanisms [7]. SLBP is considered a key factor for post-transcriptional regulation of canonical histone gene expression. It regulates pre-mRNA processing, nuclear export, stability, and translation of canonical histone mRNAs [2]. Aberrant SLBP expression affects histone homeostasis and is expected to change chromatin functions. Not surprisingly, slbp was found to be essential for embryonic development in Drosophila and C. elegans [8–10]. Moreover, altered SLBP expression and slbp mutations have been observed in several cancers according to the Cancer Genome Atlas (TCGA) dataset (https://www.cancer.gov/tcga). A potential role of SLBP in carcinogenesis is further supported by the finding that exposure to inorganic arsenic induces the loss of SLBP expression and the gain of polyadenylation of canonical histone mRNAs [11–13], leading to disruption of proper histone stoichiometry, genomic instability, and cell transformation.
This review briefly covers what is known about the regulation of SLBP, the consequence of SLBP depletion, and its implication in cancer development. The recent findings about the impact of environmental factors, in particular heavy metals such as arsenic, nickel, cadmium, and chromium on SLBP expression and polyadenylation of canonical histone mRNAs are reviewed. The potential role of SLBP depletion in heavy metal-induced carcinogenesis and the underlying mechanisms as well as the areas where further research is needed are also discussed.
Binding of SLBP to the 3’ End of Canonical Histone mRNAs
The four core histones H2A, H2B, H3, and H4 in duplicate make up the 8-histone core of a nucleosome [1]. There are two types of histones, i.e., canonical and non-canonical histones. The canonical histones, such as the histones H3.1 an H3.2, are also called replication-dependent histones, as their expression peaks during S phase. The non-canonical histones, such as the variant histone H3.3, are expressed throughout the cell cycle and are replication-independent. The mRNAs of canonical histones are unique as instead of having a poly(A) tail like other mRNAs, they have a stem loop structure at the 3’ ends to which SLBP binds [1]. SLBP binding to the stem-loop is needed for proper histone pre-mRNA processing, histone mRNA stabilization and migration, and proper histone expression [1].
The stem loop in the histone mRNA is 26 nucleotides in length [14]. Nucleotides 6 to 11 and 16 to 21 pair to each other forming the 6-nucleotide stem and nucleotides 12–15 form the 4-nucleotide loop of the hairpin structure of the stem loop [14]. The interactions of the stem loop and SLBP are mainly between the RNA back bone and the RNA-binding domain (RBD) of SLBP [15]. SLBP has a 70 amino acid L shaped RBD similar to the high-mobility group proteins (HMG), with 3 alpha helixes through which it interacts with the 5’ flanking region and arm of the stem loop of histone mRNA [16]. Phosphorylated SLBP has been shown to bind the stem loop with a higher affinity [17]. For example, phosphorylation of Thr 171 of SLBP increases its affinity for the stem loop of histone mRNA [18, 19]. A more negative C terminal in SLBP has been shown to lead to a formation of a more compact structure and greater mRNA binding affinity [17]. Thus, structural characteristics and post-translational modifications of SLBP may have impact on its activity through regulating the interaction between SLBP and 3’ end of canonical histone mRNAs.
SLBP Regulation by Translational and Post-translational Mechanisms in Normal Cells
SLBP expression is cell-cycle regulated with peak levels in S phase. SLBP protein level is increased about 20-fold from G1 phase as cells enter S phase and is reduced dramatically in the S/G2 border [7]. By contrast, SLBP mRNA level is constant throughout the cell cycle with less than 2-fold oscillation [7]. This suggested that the amount of SLBP is mostly regulated by the post-transcriptional mechanisms in normal cells.
Translational regulation of SLBP in G1 phase
SLBP is necessary for histone biosynthesis [1]. Accordingly, SLBP level is much higher in S phase as compared to that in outside of S phase. About 10- to 20-fold increase in SLBP amount occurred in the late G1 phase. Elegant work from Marzluff’s group showed that the treatment of G1 cells with MG132, a proteasome inhibitor, had little effect on the SLBP protein levels, which suggested that the change appeared to be regulated at the level of translation [1]. Further analyses of the cell-cycle distribution of SLBP mRNA on polyribosomes showed the presence of SLBP mRNA near the bottom of the sucrose gradient with heavy polyribosomes with G1 cells as seen for GAPDH mRNA. This suggests that the SLBP mRNA is translated as efficiently as GAPDH mRNA and is translationally regulated as cells transition from G1 to S phase [1].
Degradation of SLBP at S/G2 border
After DNA replication is completed at the end of S phase, the SLBP protein levels decrease to the ‘base’ level during G2 phase. When late-S-phase cells were exposed to MG132, SLBP level were returned to the highest level seen in mid-S phase, indicating that SLBP is reduced by proteasome degradation system in S/G2 border to maintain histone homeostasis [1]. The same study found that a phosphorylated form of SLBP accumulates in the presence of MG132 in late-S/G2 cells. It raised the possibility that phosphorylation is required for rapid targeting of SLBP by proteasome. Further studies showed that SLBP is phosphorylated at threonine 62 (Thr62) by CDK1/cyclin A and Thr61 by casein kinase II (CK2), both of which are necessary for the degradation of SLBP [20, 21]. Moreover, phosphorylation of Thr62 primed phosphorylation of Thr61 by CK2. Therefore, activation of CDK1/cyclin A at the end of S phase is considered an initiation step that triggers subsequent SLBP degradation at S/G2 border [20].
Which ubiquitin ligase is involved in proteasome-mediated SLBP degradation in the end of S and G2 was unknown until Pagano’s group identified the F-box protein cyclin F as the substrate recognition subunit of the SCF (SKP1-CUL1-F-box protein) ubiquitin ligase complex targeting SLBP for degradation [22, 23]. They showed that exogenous cyclin F increased ubiquitylation and degradation of SLBP in G2 cells, which was further enhanced by cyclin A2. Moreover, the SLBP mutants, either TT61/62AA or TT61/62DD, both disrupted the association of SLBP with cyclin F or with SKP1 and CUL1, suggesting that phosphorylation at Thr61 and Thr62 is prerequisite for SCFcyclin F-mediated SLBP degradation in G2. An atypical cyclin-binding (CY) motif, R(97) × L(99), was characterized in SLBP, which was found to be required for binding to cyclin F [22]. Mutated Arg 97 and Leu 99 of SLBP have been shown to lead to a reduction of SLBP binding with cyclin F and decreased SLBP degradation [22]. SLBP is required for 3’ processing of canonical histone mRNAs and H2A.X variant mRNA–the later also ends with a stem loop. Interestingly, the presence of SLBP(RL97/99AA) mutant and depletion of cyclin F, both of which stabilize SLBP, preferentially increased H2A.X levels but other core histones in G2 [22]. High levels of H2A.X increased the susceptibility of cells to apoptosis upon genotoxic stress. Therefore, it was proposed that cyclin F-mediated degradation of SLBP in vertebrate prevents H2A.X accumulation and cell death following genotoxic stress in G2 phase [22].
Degradation of SLBP in G1 phase
Djakbarova et al. reported that SLBP is also regulated by proteasome-mediated degradation in G1 phase, maintaining SLBP protein at low level in early G1 [24]. They found that the SLBP degradation in G1 is independent of the SLBP degradation at S/G2. The work from the Pagano group identified CRL2 (Cullin-RING ligase 2) as the E3 ubiquitin ligase for SLBP degradation in G1 phase [25]. They characterized the sex-determining protein FEM1 as the substrate-recognition subunit of CRL2 for SLBP degradation in G1. SLBP degradation in G1 phase has been shown to result from the binding of three distinct FEM1 isoforms FEM1A, FEM1B, and FEM1C to the distinct motifs within the N-terminus of SLBP [23, 25]. Cyclin F and FEM1 A/B/C all bind to the N terminal region of SLBP, between amino acids 1 to 99 [25]. Depletion of all three FEM1 isoforms resulted in a dramatic increase in SLBP protein levels in G1, while depletion of individual FEM1A, FEM1B, or FEM1C resulted in a modest effect [25]. Thus, timely degradation of SLBP during G1 and S/G2 is orchestrated by two different ubiquitin E3 ligases CRL2FEM1A/B/C and SCFCyclin F to control histone metabolism during the cell cycle progress.
SLBP Regulation by Epigenetic Mechanisms upon Metal Exposure
Epigenetics is changes in gene expression which are heritable and reversible but are not a result of a change in the DNA sequence. Epigenetic mechanisms include, post translational histone modifications, expression of non-coding RNAs, incorporation of histone variants, and DNA and RNA methylation. Depletion of SLBP in the cells exposed to arsenic has been suggested to partially result from transcriptional deregulation of the SLBP gene by epigenetic mechanisms.
Epigenetic regulation of SLBP has been shown to occur possibly through histone deacetylation, DNA methylation, and decreased H3K4me3 in SLBP’s promoter region [13]. Treatment of cells with arsenic leads to depletion of SLBP, however, when these cells were treated with inhibitors of histone deacetylases and DNA methyltransferases, SLBP mRNA levels were recovered, suggesting that downregulation of SLBP transcription is regulated by epigenetic mechanisms [13]. This was further evidenced by the observation that the transcriptional activation mark, H3K4me3, was found to be decreased in the promoter region of SLBP gene after arsenic exposure [13]. This suggests that the amount of SLBP is regulated transactional level at least partially by epigenetic changes in its promoter region.
SLBP has also been shown to be regulated by microRNAs in cancer cells. miR-384 levels are altered in a variety of cancer including in osteosarcoma where miR-384 is downregulated [26]. In osteosarcoma cells, overexpression of miR-384 reduced the level of SLBP, while knockdown of miR-384 increased the level of SLBP [26]. Importantly, the luciferase assays, using the vectors containing the wild-type or the mutant miR-384 binding site of SLBP 3’-UTR, showed that overexpression of miR-384 inhibited the luciferase activity of the wild-type reporter but not the mutant reporter. This suggests that miR-384 may directly downregulate SLBP levels through posttranscriptional mechanism by target the 3’-UTR of SLBP [26]. miR-384 overexpression suppressed cell growth and metastasis while silencing of it promoted cell growth and metastasis [26]. Additionally, silencing of miR-384 led to upregulation of SLBP and promotion of ERK and PI3K/AKT pathways [26]. When SLBP was knocked down, it suppressed the effects of silenced miR-384 on promotion of metastasis characteristics [26]. This indicates that SLBP is in the downstream of the miR-384 function, which is critical for miR-384-mediated cell growth and migration of osteosarcoma cells [26].
Together, SLBP expression has been shown to be affected epigenetically. Changes in DNA methylation and histone acetylation are likely to be involved in regulating SLBP transcription while microRNAs have been shown to affect the level of SLBP by post-transcriptional mechanism. Additionally, SLBP is predicted to target 23 miRNAs with variety of functions and effects, which suggests that SLBP expression could be regulated at a post-transcriptional level to a greater extent than we previously thought [6].
Depletion of SLBP, Canonical Histone Polyadenylation, and Histone Homeostasis
SLBP is a key factor for post-transcriptional regulation of canonical histone gene expression. It regulates pre-mRNA processing, nuclear export, stability, and translation of canonical histone mRNAs [2]. Therefore, depletion of SLBP is expected to affect histone homeostasis. Moreover, genome-wide studies have shown that SLBP does not bind non-histone RNAs [27] and it has not been implicated in the direct regulation of any genes other than the histone genes. Thus, the major consequence of SLBP depletion is changes in histone levels.
Depletion of SLBP causes misprocessing of canonical histone mRNAs by inducing polyadenylation at their 3’ ends. In Drosophila SLBP mutant embryos, all five canonical histone mRNAs are polyadenylated. In these embryos, since there is no sufficient SLBP available, RNA polymerase II proceeds past the normal processing site and continues the transcription until a poly(A) signal is encountered. Since each Drosophila canonical histone gene contains at least one cryptic polyadenylation signal, it is not surprising to observe that all five canonical histone genes are polyadenylated in Drosophila SLBP mutant embryos [28].
Knocking down SLBP in human osteosarcoma U2OS cells, however, induced polyadenylation of only H2A and H4 but not H2B and H3 mRNAs at much lower level compared to that in Drosophila [29]. Increased polyadenylation of canonical histone H3.1 mRNAs were observed in a number of different types of human cells and of all other canonical histone mRNAs in BEAS-2B (human bronchial epithelial cells) following arsenic exposure [11]. Moreover, a subset of canonical histone mRNAs, including one of each H2A, H2B, and H3 gene and two of H4 genes are polyadenylated in terminally differentiated tissues [30]. These suggest that all human canonical histone mRNAs could be polyadenylated with the loss of SLBP function, depending on the cell origin and type. In fact, UV irradiation upregulated polyadenylated transcripts of all 29 histone genes in human fibroblasts and keratinocytes with 13 transcripts displaying statistical significance, through slowing down Pol II elongation and inhibition of SLBP binding [31]. The extent of polyadenylation has been shown to be likely depending on the development stage in Drosophila [29].
Importantly, addition of poly(A) to the 3’ end stabilized canonical histone mRNAs in both Drosophila and human cells, resulting in accumulation of canonical histone mRNAs in cells that have already exited S phase [10, 11]. As a result, the synthesis of histone proteins is most likely uncoupled from the cell cycle, which leads to the accumulation of canonical histones outside of S phase where canonical histones are present at very low level, if any, in normal cells [13]. Therefore, while it is not clear how SLBP depletion changes histone protein levels (see below), it appears to be a feasible prediction that there must be an increase in canonical histones proteins in cell cycle phases other than S phase.
No surprisingly, how the loss of SLBP changes the protein levels for canonical histones is complicated. In addition to polyadenylation of canonical histone mRNAs due to the depletion of SLBP, SLBP is also required for histone mRNA degradation during the end of the S phase or when DNA synthesis is arrested. Both mechanisms are likely involved in leading to the increase of canonical histone protein levels. On the other hand, SLBP is required for nuclear export and translation of canonical histones. Thus, the loss of SLBP function could compromise these processes, thereby downregulating histone protein levels. This complexity may explain partially inconsistencies among different studies. Coomassie blue staining of nuclear proteins prepared from SLBP knockdown human U2OS cells showed a slight decrease in protein levels for all four core histones, while Western blotting of whole cell lysates showed about 50% reduction of H3 in the SLBP knockdown cells compared to the control cells [29]. In another study, moderate SLBP depletion reduced protein levels for H3 and H2B in HCT116 cells [32]. Knocking down of SLBP by siRNA reduced histone protein synthesis in HeLa cells [33]. Also, similar results were obtained in C. elegans study where reduction of both H3 and H4 proteins were observed by depletion of SLBP function [9].
Salzler et al. observed genomic instability in Slbp mutant Drosophila. When they measured histone protein levels using western blot analysis, no changes in protein levels for H3 and H2b were found in homozygous Slbp15 and Slbp10 mutants compared to that in wild-type counterparts. Thus, they concluded that the observed genomic instability in Slbp mutants cannot be the results of significant alterations in the level of canonical histone proteins [34]. Moreover, it was reported that arsenic exposure induced SLBP depletion in vitro and in vivo, which was accompanied with the increase in the levels of polyadenylated mRNAs and proteins for all four core histones [11, 13]. Given that overexpression of SLBP suppressed arsenic-induced polyadenylation of H3.1 mRNA [13], it is possible that arsenic-mediated upregulation of histone proteins might resulting from SLBP depletion. Differential effects of SLBP depletion on changes in histone mRNA and protein levels are likely affected by the extent and length of SLBP knockdown [29]. Moreover, the cell origin, type, and differentiation state, as well as cell cycle status, all might contribute to the direction and level of changes of the core histone mRNAs and proteins due to SLBP depletion. Nonetheless, SLBP depletion can have two major consequences. First, polyadenylation of histone mRNAs, which induces accumulation of canonical histones outside S phase, leading to an imbalance between canonical and variant histones. Second, either reduction or upregulation of total canonical histones. Both may disrupt stoichiometry of histones, causing abnormal chromatin assembly and genomic instability.
SLBP and Carcinogenesis
SLBP and cell cycle progress and genomic instability
Salzler et al. reported that the loss of SLBP in Drosophila causes genomic instability and impaired cellular proliferation [34]. Reduction of SLBP increased the frequency of loss of heterozygosity (LOH), which is a common occurrence in cancer development. Slbp mutants also displayed increased frequency of DNA double-strand breaks and were tetraploid. Moreover, Slbp mutants showed enhancement of position-effect variegation, suggesting increased heterochromatin formation by Slbp mutations [34]. Therefore, it was proposed that genomic instability in Slbp mutants results from alterations of chromatin structure.
Slowed chromatin assembly behind the replication fork is believed to cause collapsed forks, double-strand breaks, and genomic instability. SLBP is required for coupling rates of histone protein synthesis with DNA replication, thus Slbp mutantions may induce defective chromatin, causing prolonged S-phase. In fact, cell cycle analysis showed that cell cycle phasing in slbp mutant cells was significantly different than wild-type cells, with a significantly increased cell population in S phase of the mutant cells as compared to wild-type cells. This supports the idea that inhibited chromatin assembly due to slbp mutations can impair S-phase progression, thereby generating DNA damage and genomic instability.
Moreover, it has been shown that SLBP is required for chromosome condensation, progression of cell death and morphogenesis in C. elegans [8]. Knockdown of slbp by RNA interference caused chromosome condensation and segregation in C. elegans embryos. Downregulation of SLBP expression by RNAi also resulted in the inhibition of cell cycle progression through S phase and DNA synthesis in several human cell lines [35, 36]. Knocking down SLBP in U2OS cells resulted in a reduction in the rate of cell growth and an accumulation of cells in S-phase. Reducing the levels of SLBP decreased the rate of DNA synthesis and activated an S-phase cell cycle checkpoint [29].
SLBP and cellular transformation
To investigate the role of SLBP in carcinogenesis, SLBP gene expression was knocked down by shRNA in BEAS-2B cells followed by soft-agar assays. About 50% of knockdown of SLBP with stable expression of shRNA vector for SLBP promoted anchorage-independent growth of BEAS-2B cells compared to the control cells [12], suggesting that depletion of SLBP alone is sufficient to induce cell transformation. The importance of SLBP in carcinogenesis was further supported by the observation that overexpression of SLBP was able to prevent arsenic-induced cell transformation [11]. In the study, BEAS-2B cells were stably transfected with SLBP-expressing plasmid, resulting in the increase of SLBP levels by about three-folds. Overexpression of SLBP attenuated arsenic-induced polyadenylation of H3.1 mRNA. More importantly, arsenic-induced colony formation in soft agars was significantly reduced in the cells that expressed SLBP [11], indicating an essential role of SLBP depletion and subsequent polyadenylation of canonical histone mRNAs in arsenic-induced cell transformation.
Association of SLBP with Cancer
SLBP is considered to be a prognostic factor in liver cancer and high expression is associated with a worse prognosis of a shorter survival [37]. 51 cases had SLBP mutations and 798 cases had copy number variation (CNV) in the Cancer Genome Atlas (TCGA) projects data set (https://www.cancer.gov/tcga). This is interesting as 2 cases with mutations and 13 with CNV in TCGA were liver cancer. Interestingly, uterine cancer in TCGA database had high numbers for mutations and CNV for SLBP gene, which would present a possible area to look into whether the SLBP expression levels are altered and for if the levels have an effect on outcome. Additionally, from TCGA, with SLBP, CNV predominately lead to gain of function rather than loss of function.
Potential mechanisms of SLBP in carcinogenesis
SLBP regulates biosynthesis of canonical histones from their pre-mRNA processing, stability, and nuclear export to translation [2]. Altered histone levels resulting from altered SLBP levels and subsequent disruption of proper histone stoichiometry and genome integrity appeared to be the major mechanism for SLBP-induced carcinogenesis. Exposure to arsenic induced polyadenylation of canonical histone H3.1 mRNA both in vitro and in vivo, which stabilized H3.1 mRNA, leading to the increase in H3.1 protein level [11]. Notably, overexpression of polyadenylated H3.1 mRNA resulted in aberrant expression of cancer-associated genes, mitotic arrest, chromosome aneuploidy, and aberrations [11]. Moreover, polyadenylation of H3.1 mRNA lead to cell transformation of BEAS-2B cells and enhanced anchorage-independent growth of human embryonic kidney HEK293 and human papillomavirus transformed prostate epithelial PZ-HPV-7 cells. Importantly, the transfection of a H3.1Loop plasmid, which contains a stem-loop sequence in front of the poly(A) signal and thus generates non-polyadenylated H3.1 mRNA, attenuated H3.1poly(A)-induced cell transformation. This indicated that acquisition of a poly(A) tail is the determining factor for cell transformation induced by transfection of H3.1poly(A) plasmid [11].
Additionally, overexpression of polyadenylated H3,1 mRNA resulted in genome-wide reduction of histone variant H3.3 at critical gene regulatory elements, including active promoters, enhancers, and insulator regions by not-yet known mechanisms [11]. Given that canonical and variant histones possess distinctive properties [38], this substitution could disrupt appropriate chromatin structure and affect functions relevant to H3.3 including transcription, cell cycle progress, DNA replication and repair, and eventually cause genomic instability [39, 40]. In fact, the transcriptome was affected by loss of H3f3b, one of two H3.3-encoding genes, in mouse embryonic fibroblasts (MEFs). H3f3b knockout mice exhibited defective cell division and chromosome segregation. In another study, complete depletion of H3.3 in mice caused dysfunction of heterochromatin structures and triggered cell cycle suppression during mitosis. Moreover, H3.3 knockout mice displayed severe chromosomal aberrations [41]. These results support the hypothesis that polyadenylation of H3.1 mRNA may cause abnormal gene expression, cell cycle defect, genomic instability, and cell transformation through displacement of H3.3 at critical genomic loci. Taken together, aberrant biosynthesis of canonical histones along with subsequent disruption of histone stoichiometry between canonical and variant histones are likely the mechanism that underlies SLBP-induced carcinogenesis.
Metals and SLBP Depletion
Heavy metals can be found in the environment often at trace levels, however, environmental contamination from anthropocentric sources can lead to elevated environmental and everyday exposure to them. These elevated exposure levels can amount to levels above those biologically necessary or safe for these metals. Metal exposure can also occur through occupational exposure besides ingestion and inhalation from which most everyday life exposure is derived. Heavy metal exposure has been linked to a variety of toxic effects and outcomes, however, the mechanisms underlying metals effects are under investigation as they are not fully understood. Three heavy metals whose effects on SLBP have been studied are arsenic, nickel, and cadmium [13, 42]. Interestingly, depletion of SLBP has been shown to result from exposure of cells to heavy metals. Nickel, cadmium and arsenic exposure have all been shown to affect cellular levels of SLBP [13, 42].
Nickel and SLBP depletion
Nickel exposure is mainly through ingestion and then dermal exposure through skin contact with products with nickel or nickel alloys [43]. Dermal contact with nickel is known to be able to lead to allergy in the form of contact dermatitis, along with inhalation negatively effecting the lung and nasal passageway resulting in respiratory effects and it can lead to lung cancer [43]. Exposure to nickel has been shown to affect SLBP levels leading to depletion of SLBP protein and mRNA levels in both BEAS-2B and BL41 (Burkitt lymphoma cells) [42]. The depletion of SLBP levels from nickel exposure was suggested to be result of proteasome degradation of the protein or altered transcription of the mRNA [42]. Interestingly, nickel-induced SLBP depletion was still apparent 7 days post nickel exposure, suggesting that this change is relatively stable and can be inherited through multiple cellular divisions [42]. Moreover, nickel exposure led to significantly increased amount of polyadenylated H3.1 mRNA in BL41 cells. By contrast, the levels of polyadenylated H3.1 mRNAs were slightly increased only at lower doses but it was not significant in BEAS-2B cells [42].
Cadmium and SLBP depletion
Cadmium exposure is mainly through inhalation which is mainly in workers or people who smoke and ingestion of contaminated foods [44]. Cadmium is known to accumulate in the kidneys high levels and can consequently lead to kidney damage, while inhalation of it has been shown to lead to lung damage and lung cancer [44]. Cadmium exposure of BL41 cells has been shown to affect SLBP levels and exposure lead to significant depletion of SLBP protein and mRNA levels. However, a less pronounced reduction of SLBP mRNA was observed in BEAS-2B cells [42], indicating a possible tissue specificity of cadmium effect on SLBP regulation. Like seen with nickel, cadmium-induced SLBP depletion exhibited the same reduction pattern at 4 days post exposure. However, the SLBP levels were restored to above control levels by 6 days post cadmium exposure [42], suggesting that the changes of SLBP depletion by nickel are more stable than those of cadmium exposure. Unexpectedly, cadmium exposure did not induce an increase in polyadenylated H3.1 mRNA as seen with nickel and arsenic [42]. Further study is needed to explore the nature of this observation.
Chromium and SLBP depletion
Hexavalent chromium Cr(VI) is an established carcinogen that induces human lung cancer and is associated with increased risks of cancer of the paranasal sinuses and nasal cavity [45]. Inhalation of Cr(VI) has been shown to induce lung cancer, while Cr(VI) ingestion has been linked to gastrointestinal (GI) cancer [46]. Moreover, ingestion of Cr(VI) in their drinking water facilitated UV-induced skin cancer in mice [45]. A study has shown an increased level of polyadenylated H3.1 mRNA in arsenic-transformed BEAS-2B cell clones, whereas the increase in polyadenylation of H3.1 mRNA was not observed in Cr(VI)-transformed BEAS-2B cell clones [13]. Although changes in SLBP ware not tested directly, given the essential role for SLBP in 3’ processing of H3.1 mRNA, these results suggest that the Cr(VI) exposure may not induce depletion of SLBP as seen for other metals.
Arsenic and SLBP depletion
Arsenic contamination of drinking water has been found worldwide, and at levels above which has been set by the WHO of 10 μg/L [47]. Arsenic exposure has been correlated to a variety of human health effects including skin lesions, increased risk of cardiovascular disease and diabetes, and increased risk of skin, bladder, lung, liver, and kidney cancer [47]. Sodium arenite exposure of BEAS-2B and BL41 cells has been shown to lead to the dose-dependent depletion of SLBP protein levels [13]. SLBP protein levels in arsenic-transformed BEAS-2B clones were also significantly lower than that in spontaneous clones. The decrease in SLBP levels was attributed to an observed increase proteasome degradation of the SLBP protein and inhibition of transcription lead to decreased SLBP mRNA levels [13]. The downregulation of SLBP transcription appeared to be regulated by epigenetic mechanisms, since arsenic-induced reduction of SLBP mRNA was reversed by inhibitors of histone deacetylases or DNA methyltransferases.
As we described earlier, polyadenylation of canonical histone mRNAs causes abnormal transcription, mitotic arrest, chromosome instability, and cell transformation, indicating that arsenic-induced polyadenylation of canonical histone mRNAs may be critical for arsenic carcinogenesis. SLBP depletion is likely to play a key role in the arsenic-induced polyadenylation of H3.1 mRNA. This is supported by the observation that overexpression of SLBP decreased the level of polyadenylated H3.1 mRNA even in the absence of arsenic treatment. More importantly that arsenic exposure was not able to increase polyadenylated H3.1 mRNA in the cells overexpressing SLBP [11].
Possible mechanism of metals’ effect on SLBP
Nickel, Cadmium and Arsenic have all been shown to reduce SLBP protein and mRNA levels, which suggests that heavy metal exposure could lead to SLBP depletion. While the exact mechanism by which these metals lead to SLBP depletion is yet to be discovered, it was shown that these metals could possibly be leading to the depletion of SLBP through proteasomal degradation of SLBP. When BEAS-2B cells were exposed to arsenic, which has been shown to downregulate SLBP expression, a reduced depletion of SLBP was observed in the presence of the proteasomal inhibitor MG-132 [13]. Also, arsenic was suggested to deplete SLBP by epigenetic regulation by leading to histone deacetylation and DNA methylation, and decreased H3K4me3 in SLBP’s promoter region [13]. Further research is thus necessary to fully understand the complete mechanisms by which metals affect SLBP expression.
SLBP depletion and metal carcinogenesis
A number of heavy metals have been identified as carcinogens. The mechanisms for metal-induced carcinogenesis include genotoxic effect, oxidative stress, and epigenetic regulation among others. Given the carcinogenic effects of polyadenylation of canonical histone mRNAs, metal-induced SLBP depletion and subsequent polyadenylation of canonical histones mRNAs, presents a possible new mechanism for carcinogenesis induced by certain types of heavy metals, such as arsenic and nickel. Both arsenic and nickel induced the loss of SLBP and the gain of polyadenylated canonical H3.1 mRNA [11, 42]. Arsenic exposure also increased the levels of polyadenylated canonical histone mRNAs for all other canonical histones including H3.2, H2A, H2B, and H4 [11]. Although cadmium also reduced the level of SLBP, it did not change significantly the level of polyadenylated H3.1 mRNA [42]. Careful and comprehensive experiments are needed to be done to determine if cadmium induces polyadenylation of H3.1 or other canonical histone mRNAs. Moreover, the consequence of H3.1 polyadenylation is the displacement of histone variant H3.3 at critical gene regulatory elements including active promoters, enhancers, and insulator regions. This supports the idea that H3.3 displacement might be central to carcinogenesis induced by polyadenylated H3.1 mRNA upon arsenic exposure [11]. It is important to examine if other metals, such as nickel or cadmium, also lead to change in H3.3 enrichment at these critical genomic loci.
Discussion and Conclusions
Degradation of SLBP is essential for keeping it at correct levels throughout the cell cycle (Fig. 1). Majority of SLBP degradation occurs when cells enter G2, limiting the peak levels to S phases. This degradation is due to the phosphorylation of Thr 62 by CDK1/cyclin A followed by phosphorylation of Thr 61 by CK2, and subsequent ubiquitination by E3 ligase SCFCyclin F, leading to proteasomal degradation of SLBP. Degradation in G1 occurs through the binding of FEM 1A/B/C, the substrate recognition subunit of another E3 ligase CRL2, while in G2 it involves cyclin F. While the mechanisms of degradation vary between the point in the cell cycle where the degradation of SLBP is occurring, all mechanisms are necessary to ensure proper levels of SLBP are maintained.
Figure 1. A schematic summarizing the regulation and functions of SLBP in normal cells and in the cells exposed to heavy metals.
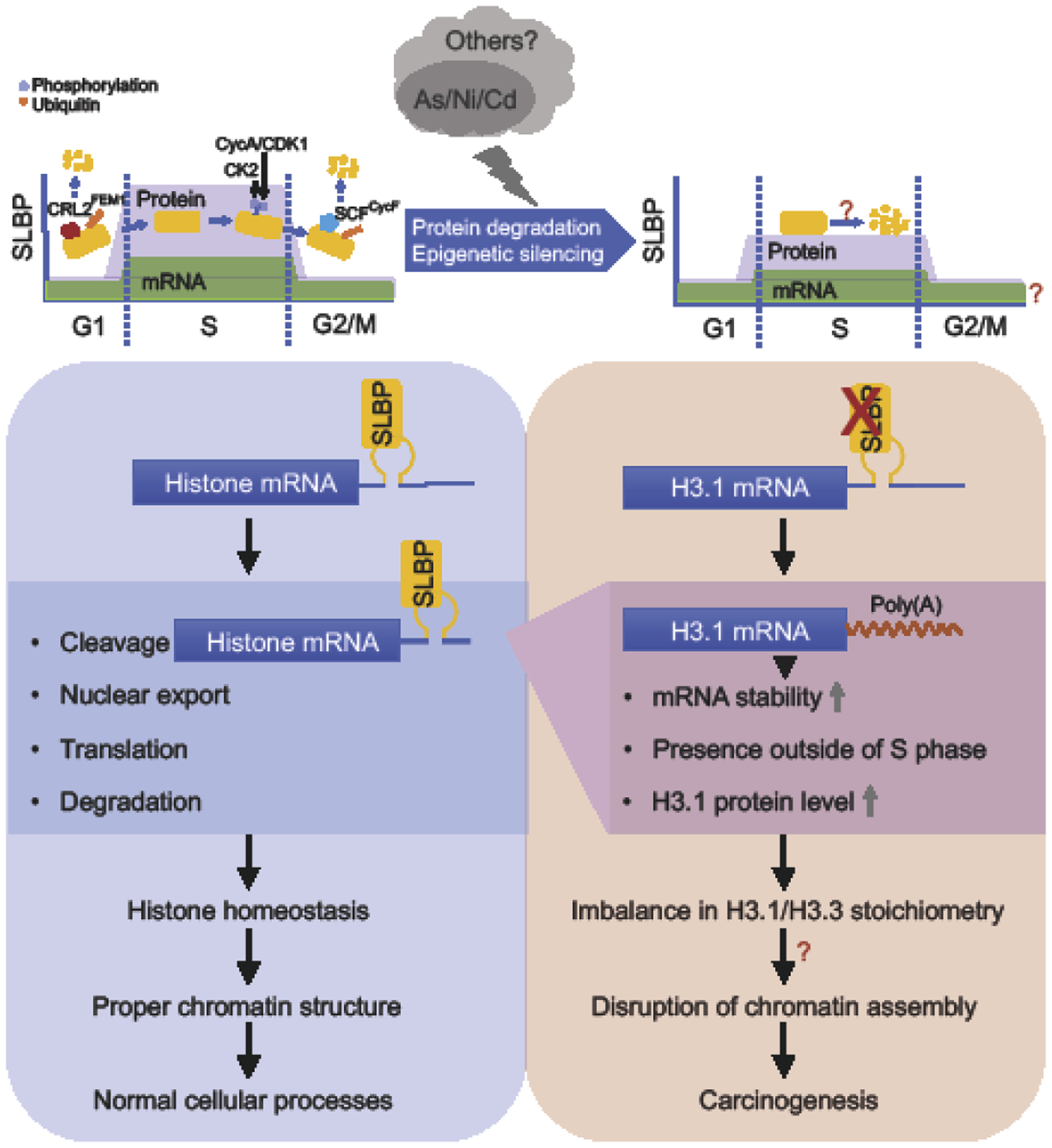
SLBP is a key regulator of canonical histone mRNAs. It binds to the stem-loop structure in the 3’ end of canonical histone pre-mRNAs, regulating their 3’ end processing, nuclear export, translation, and stability. SLBP is cell-cycle regulated with peak levels in S phase. SLBP protein is maintained at low levels outside of S phase, by proteasome-mediated degradation. SLBP protein is ubiquitinated and degraded by ubiquitin E3 ligase CRL2FEM1A/B/C in G1 and by SCFCyclin F in G2 phase, respectively. Phosphorylation at Thr62 by CDK1/Cyclin A primes SLBP for phosphorylation at Thr61 by CK2 at the end of S phase, initiating subsequent SCFCyclin F-mediated SLBP degradation in G2. SLBP is critical for the maintenance of histone homeostasis, proper chromatin structure and cellular processes in normal cells. Exposure to heavy metals, such as arsenic and nickel, induces depletion of SLBP by both proteasome-mediated protein degradation and transcriptional silencing by epigenetic mechanisms. Depletion of SLBP causes abnormal 3’ end processing by adding a poly(A) tail to canonical histone mRNAs. Polyadenylated canonical histone H3.1 mRNA is stable, leading to the increase in H3.1 protein level and subsequently an imbalance in H3.1 and H3.3 stoichiometry. This results in displacement of H3.3 at important gene regulatory regions, which causes abnormal transcription, cell cycle arrest, chromosome instability, and cell transformation. Further study is needed to be done in the future for a better understanding of SLBP regulation by environmental exposures and its roles in environmental diseases such as cancer.
Depletion of SLBP has been shown to result from exposure of cells to heavy metals including arsenic, nickel, and cadmium [13, 42]. It is suggested that depletion of SLBP which results from metal exposure, could be due to proteasome degradation of SLBP and altered transcription of SLBP mRNA [13, 42]. It is important to understand how arsenic and other metals induce SLBP degradation and if phosphorylation is required for the SLBP degradation. Identifying the phosphorylation site(s) and the modifying enzyme(s) as well as ubiquitination site(s) and the ubiquitin ligase(s) involved in metal-induced SLBP degradation are needed for better understanding of metal-induced SLBP depletion (Fig. 1). It has been known that arsenic selectively binds to C3H- or C4-type zinc fingers, displacing zinc within the proteins, thereby changing the functions of zinc finger-containing proteins [48]. Thus, it would be interesting to explore if any of identified kinase(s)/phosphatase(s) related to SLBP degradation by arsenic are enzymes with C3H- or C4-type zinc finger motif and are targeted by arsenic.
Studies also showed that heavy metal depletion of SLBP is correlated with an increase in H3.1poly(A) transcripts and H3 protein levels [13, 42]. However, it has not yet been determined if observed gain of polyadenylated canonical histone mRNAs and proteins is solely due to depletion of SLBP. While arsenic-induced polyadenylation of canonical histone mRNAs is likely due to reduction of SLBP, the mechanisms that underlie arsenic-induced increase in canonical histone protein levels need to be further investigated, given that other factors are also involved in pre-mRNA processing of canonical histones. As described above, a number of studies demonstrated that loss of SLBP function generally leads to reduction of histone protein levels. Although some studies showed accumulation of canonical histone mRNAs or no changes in histone protein levels due to the loss of SLBP function, one cannot rule out the possibilities that arsenic and other metals may also target factors other than SLBP to control histone homeostasis. Moreover, it is also necessary to examine if arsenic involves SLBP-regulated histone mRNA nuclear export, degradation, and translation, in addition to their 3’ processing (Fig. 1).
Arsenic exposure increased the level of polyadenylated H3.1 mRNA by about 3 folds as compared to that in unexposed cells in M phase [13]. This may lead to an increase in the protein level for H3.1 in M phase where no canonical H3 exists in normal condition, which might in turn affect the assembly of variant histones. In fact, it was demonstrated that the H3.3 variant was depleted from critical regulatory elements by arsenic exposure or by induction of polyadenylated H3.1 mRNA [11]. In the future, it would be important to dissect how arsenic exposure or polyadenylation of canonical histone H3.1 mRNA compromises H3.3 assembly at these genomic loci (Fig. 1). As polyadenylated H3.1 mRNA induced abnormal transcription, cell cycle arrest, and chromosome instability, the role for inhibition of H3.3 assembly in these processes by H3.1 polyadenylation needs further investigation. Furthermore, heavy metals, such as arsenic, causes carcinogenesis by multiple mechanism, including oxidative stress and epigenetic regulation, just name a few. Therefore, it is critical to clarify to what extent the SLBP depletion and subsequent canonical histone polyadenylation and histone variant displacement contributes to arsenic and other metal-induced carcinogenesis.
While the physical and chemical properties of SLBP have been discovered along with some of its mechanisms, there is still a lot unknown. Further investigation of the details of all the interactions of SLBP and its roles of such in cellular processes will help in understanding the role of this protein. Additionally, further research into the degradation mechanism which regulates SLBP mRNA and protein levels and which leads to depletion of these levels are necessary for gaining a better understanding of SLBP. Then stemming from this further investigation into what exposures could lead to altered levels of SLBP is also needed. This is important as altering SLBP levels has been shown to affect histone mRNA and protein levels within cells. Affected histone levels have been shown to lead to genomic instability and transformation of cells, thus highlighting how maintaining SLBP function and levels are important not only for the understanding of normal cellular processes but also for understanding carcinogenesis induced by metals and possibly by other environmental factors.
Acknowledgements
The results shown here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. Beatrix Bradford is supported by the NIEHS training grant T32 ES007324. This work was supported by grants from the US National Institutes of Health: R01ES029359 and R01ES030583 (C.J.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare that there are no conflicts of interest.
References
- [1].Marzluff WF, Wagner EJ, Duronio RJ, Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail, Nat Rev Genet 9(11) (2008) 843–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Marzluff WF, Koreski KP, Birth and Death of Histone mRNAs, Trends Genet 33(10) (2017) 745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hanson RJ, Sun J, Willis DG, Marzluff WF, Efficient extraction and partial purification of the polyribosome-associated stem-loop binding protein bound to the 3’ end of histone mRNA, Biochemistry 35(7) (1996) 2146–56. [DOI] [PubMed] [Google Scholar]
- [4].Martin F, Schaller A, Eglite S, Schumperli D, Muller B, The gene for histone RNA hairpin binding protein is located on human chromosome 4 and encodes a novel type of RNA binding protein, EMBO J 16(4) (1997) 769–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wang ZF, Whitfield ML, Ingledue TC 3rd, Dominski Z, Marzluff WF, The protein that binds the 3’ end of histone mRNA: a novel RNA-binding protein required for histone pre-mRNA processing, Genes Dev 10(23) (1996) 3028–40. [DOI] [PubMed] [Google Scholar]
- [6].Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y, et al. , The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses, Current Protocols in Bioinformatics 2016, pp. 1.30.1–1.30.33. [DOI] [PubMed] [Google Scholar]
- [7].Whitfield ML, Zheng LX, Baldwin A, Ohta T, Hurt MM, Marzluff WF, Stem-loop binding protein, the protein that binds the 3’ end of histone mRNA, is cell cycle regulated by both translational and posttranslational mechanisms, Mol Cell Biol 20(12) (2000) 4188–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kodama Y, Rothman JH, Sugimoto A, Yamamoto M, The stem-loop binding protein CDL-1 is required for chromosome condensation, progression of cell death and morphogenesis in Caenorhabditis elegans, Development 129(1) (2002) 187–96. [DOI] [PubMed] [Google Scholar]
- [9].Pettitt J, Crombie C, Schumperli D, Muller B, The Caenorhabditis elegans histone hairpin-binding protein is required for core histone gene expression and is essential for embryonic and postembryonic cell division, J Cell Sci 115(Pt 4) (2002) 857–66. [DOI] [PubMed] [Google Scholar]
- [10].Sullivan E, Santiago C, Parker ED, Dominski Z, Yang X, Lanzotti DJ, Ingledue TC, Marzluff WF, Duronio RJ, Drosophila stem loop binding protein coordinates accumulation of mature histone mRNA with cell cycle progression, Genes Dev 15(2) (2001) 173–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen D, Chen QY, Wang Z, Zhu Y, Kluz T, Tan W, Li J, Wu F, Fang L, Zhang X, He R, Shen S, Sun H, Zang C, Jin C, Costa M, Polyadenylation of Histone H3.1 mRNA Promotes Cell Transformation by Displacing H3.3 from Gene Regulatory Elements, iScience 23(9) (2020) 101518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Brocato J, Chen D, Liu J, Fang L, Jin C, Costa M, A Potential New Mechanism of Arsenic Carcinogenesis: Depletion of Stem-Loop Binding Protein and Increase in Polyadenylated Canonical Histone H3.1 mRNA, Biol Trace Elem Res 166(1) (2015) 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Brocato J, Fang L, Chervona Y, Chen D, Kiok K, Sun H, Tseng HC, Xu D, Shamy M, Jin C, Costa M, Arsenic induces polyadenylation of canonical histone mRNA by down-regulating stem-loop-binding protein gene expression, J Biol Chem 289(46) (2014) 31751–31764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tan D, Marzluff WF, Dominski Z, Tong L, Structure of histone mRNA stem-loop, human stem-loop binding protein, and 3’hExo ternary complex, Science 339(6117) (2013) 318–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Thapar R, Structural basis for regulation of RNA-binding proteins by phosphorylation, ACS Chem Biol 10(3) (2015) 652–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Thapar R, Structure-specific nucleic acid recognition by L-motifs and their diverse roles in expression and regulation of the genome, Biochim Biophys Acta 1849(6) (2015) 677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang J, Tan D, DeRose EF, Perera L, Dominski Z, Marzluff WF, Tong L, Hall TM, Molecular mechanisms for the regulation of histone mRNA stem-loop-binding protein by phosphorylation, Proc Natl Acad Sci U S A 111(29) (2014) E2937–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Thapar R, Roles of Prolyl Isomerases in RNA-Mediated Gene Expression, Biomolecules 5(2) (2015) 974–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Thapar R, Contribution of protein phosphorylation to binding-induced folding of the SLBP-histone mRNA complex probed by phosphorus-31 NMR, FEBS Open Bio 4 (2014) 853–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Koseoglu MM, Graves LM, Marzluff WF, Phosphorylation of threonine 61 by cyclin a/Cdk1 triggers degradation of stem-loop binding protein at the end of S phase, Mol Cell Biol 28(14) (2008) 4469–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Consortium TU, UniProt: the universal protein knowledgebase in 2021, Nucleix Acids Research, 2021, pp. D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dankert JF, Rona G, Clijsters L, Geter P, Skaar JR, Bermudez-Hernandez K, Sassani E, Fenyo D, Ueberheide B, Schneider R, Pagano M, Cyclin F-Mediated Degradation of SLBP Limits H2A.X Accumulation and Apoptosis upon Genotoxic Stress in G2, Mol Cell 64(3) (2016) 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang J, Liu J, Wei W, “FEM1”nism controls SLBP stability during cell cycle, Cell Cycle 16(7) (2017) 597–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Djakbarova U, Marzluff WF, Koseoglu MM, Translation regulation and proteasome mediated degradation cooperate to keep stem-loop binding protein low in G1-phase, J Cell Biochem 115(3) (2014) 523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dankert JF, Pagan JK, Starostina NG, Kipreos ET, Pagano M, FEM1 proteins are ancient regulators of SLBP degradation, Cell Cycle 16(6) (2017) 556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang Y, Huang H, Li Y, Knocking down miR-384 promotes growth and metastasis of osteosarcoma MG63 cells by targeting SLBP, Artif Cells Nanomed Biotechnol 47(1) (2019) 1458–1465. [DOI] [PubMed] [Google Scholar]
- [27].Townley-Tilson WH, Pendergrass SA, Marzluff WF, Whitfield ML Genome-wide analysis of mRNAs bound to the histone stem-loop binding protein RNA, 12 (10) (2006), pp. 1853–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lanzotti DJ, Kaygun H, Yang X, Duronio RJ, Marzluff WF, Developmental control of histone mRNA and dSLBP synthesis during Drosophila embryogenesis and the role of dSLBP in histone mRNA 3’ end processing in vivo, Mol Cell Biol 22(7) (2002) 2267–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sullivan KD, Mullen TE, Marzluff WF, Wagner EJ, Knockdown of SLBP results in nuclear retention of histone mRNA, RNA 15(3) (2009) 459–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lyons SM, Cunningham CH, Welch JD, Groh B, Guo AY, Wei B, Whitfield ML, Xiong Y, Marzluff WF, A subset of replication-dependent histone mRNAs are expressed as polyadenylated RNAs in terminally differentiated tissues, Nucleic Acids Res 44(19) (2016) 9190–9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Saldi T, Fong N, Bentley DL, Transcription elongation rate affects nascent histone pre-mRNA folding and 3’ end processing, Genes Dev 32(3–4) (2018) 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jimeno-Gonzalez S, Payan-Bravo L, Munoz-Cabello AM, Guijo M, Gutierrez G, Prado F, Reyes JC, Defective histone supply causes changes in RNA polymerase II elongation rate and cotranscriptional pre-mRNA splicing, Proc Natl Acad Sci U S A 112(48) (2015) 14840–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cakmakci NG, Lerner RS, Wagner EJ, Zheng L, Marzluff WF, SLIP1, a factor required for activation of histone mRNA translation by the stem-loop binding protein, Mol Cell Biol 28(3) (2008) 1182–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Salzler HR, Davidson JM, Montgomery ND, Duronio RJ, Loss of the histone pre-mRNA processing factor stem-loop binding protein in Drosophila causes genomic instability and impaired cellular proliferation, PloS one 4(12) (2009) e8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wagner EJ, Berkow A, Marzluff WF, Expression of an RNAi-resistant SLBP restores proper S-phase progression, Biochem Soc Trans 33(Pt 3) (2005) 471–3. [DOI] [PubMed] [Google Scholar]
- [36].Zhao X, McKillop-Smith S, Muller B, The human histone gene expression regulator HBP/SLBP is required for histone and DNA synthesis, cell cycle progression and cell proliferation in mitotic cells, J Cell Sci 117(Pt 25) (2004) 6043–51. [DOI] [PubMed] [Google Scholar]
- [37].Z.C. Uhlen M., Lee S, Sjostedt E, Fagerberg L, Bidkhori G, Benfeitas R, et al. , A pathology atlas of the human cancer transcriptome Science, 2017. [DOI] [PubMed]
- [38].Henikoff S, Nucleosome destabilization in the epigenetic regulation of gene expression, Nat Rev Genet 9(1) (2008) 15–26. [DOI] [PubMed] [Google Scholar]
- [39].Talbert PB, Henikoff S, Histone variants--ancient wrap artists of the epigenome, Nat Rev Mol Cell Biol 11(4) (2010) 264–75. [DOI] [PubMed] [Google Scholar]
- [40].Soria G, Polo SE, Almouzni G, Prime, repair, restore: the active role of chromatin in the DNA damage response, Mol Cell 46(6) (2012) 722–34. [DOI] [PubMed] [Google Scholar]
- [41].Bush KM, Yuen BT, Barrilleaux BL, Riggs JW, O’Geen H, Cotterman RF, Knoepfler PS, Endogenous mammalian histone H3.3 exhibits chromatin-related functions during development, Epigenetics & chromatin 6(1) (2013) 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Jordan A, Zhang X, Li J, Laulicht-Glick F, Sun H, Costa M, Nickel and cadmium-induced SLBP depletion: A potential pathway to metal mediated cellular transformation, PloS one 12(3) (2017) e0173624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].U.D.o.H.a.H. Services, Toxicological Profile for Nickel, Toxicological Profile ATSDR, 2005, pp. 1–10. [Google Scholar]
- [44].U.D.o.H.a.H. Services, Toxicological Profile for Cadmium, Toxicological Profile ATSDR, 2012, pp. 1–11. [Google Scholar]
- [45].Costa M, Klein CB, Toxicity and carcinogenicity of chromium compounds in humans, Crit Rev Toxicol 36(2) (2006) 155–63. [DOI] [PubMed] [Google Scholar]
- [46].NTP Toxicology and Carcinogenesis Studies of Sodium Dichromate Dihydrate (CAS No. 7789-12-0) in F344/N Rats and B6C3F1 Mice (Drinking Water Studies), Natl Toxicol Program Tech Rep Ser (546) (2008) 1–192. [PubMed] [Google Scholar]
- [47].Argos M, Ahsan H, Graziano JH, Arsenic and human health: epidemiologic progress and public health implications, Rev Environ Health 27(4) (2012) 191–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhou X, Sun X, Mobarak C, Gandolfi AJ, Burchiel SW, Hudson LG, Liu KJ, Differential binding of monomethylarsonous acid compared to arsenite and arsenic trioxide with zinc finger peptides and proteins, Chem Res Toxicol 27(4) (2014) 690–8. [DOI] [PMC free article] [PubMed] [Google Scholar]