

Abstract
Glucagon-like peptide-1 (GLP-1) is best known for its insulinotropic action following food intake. Its metabolite, GLP-1 (9–36), was assumed biologically inactive due to low GLP-1 receptor (GLP-1R) affinity and non-insulinotropic properties; however, recent studies contradict this assumption. Increased use of FDA approved GLP-1 analogues for treating metabolic disorders and neurodegenerative diseases raises interest in GLP-1 (9–36)’s biological role. We use human SH-SY5Y neuroblastoma cells and a GLP-1R overexpressing variety (#9), in both undifferentiated and differentiated states, to evaluate the neurotrophic/neuroprotective effects of GLP-1 (9–36) against toxic glutamate exposure and other oxidative stress models (via the MTS, LDH or ROS assays). In addition, we examine GLP-1 (9–36)’s signaling pathways, including cyclic-adenosine monophosphate (cAMP), protein kinase-A (PKA), and 5’ adenosine monophosphate activated protein kinase (AMPK) via use of ELISA, pharmacological inhibitors, or GLP-1R antagonist. Human HMC3 and mouse IMG microglial cell lines were used to study the anti-inflammatory effects of GLP-1 (9–36) against lipopolysaccharide (LPS) (via ELISA). Finally, we applied GLP-1 (9–36) to primary dissociation cultures challenged with α-synuclein or amyloid-β and assessed survival and morphology via immunochemistry. We demonstrate evidence of GLP-1R, cAMP, PKA, and AMPK mediated neurotrophic and neuroprotective effects of GLP-1 (9–36). The metabolite significantly reduced IL-6 and TNF-α levels in HMC3 and IMG microglial cells, respectively. Lastly, we show mild but significant effects of GLP-1 (9–36) in primary neuron cultures challenged with α-synuclein or amyloid-β. These studies enhance understanding of GLP-1 (9–36)’s effects on the nervous system and its potential as a primary or complementary treatment in pathological contexts.
Keywords: GLP-1, GLP-1 (9–36), Exendin-4, neuroinflammation, neuroprotection, neurodegeneration
Graphical Abstract
The current study evaluates the neurotrophic, neuroprotective, and anti-inflammatory properties of the glucagon-like peptide-1 (GLP-1) (7–36) metabolite, GLP-1 (9–36). Endogenous GLP-1 (9–36) is produced by the cleavage of GLP-1 (7–36) and has distinct properties from its parent peptide. Here, we characterize GLP-1 receptor (GLP-1R) mediated signaling properties of GLP-1 (9–36) against glutamate, H2O2, lipopolysaccharide and challenges associated with Alzheimer’s disease (amyloid-β) and Parkinson’s disease (α-synuclein). These studies enhance understanding of GLP-1 (9–36)’s effects on the nervous system and its potential as a primary or complementary treatment in pathological contexts.
We identified multiple mechanisms by which anti-microbial protein Regenerating Family Member 3 Alpha (REG3A) participates in the pathophysiologic response to large-vessel ischemic stroke. REG3A interacts with inflammatory cytokine Interleukin-6 (IL6) and immunomodulatory protein Interleukin-17C (IL17C) to induce other extracellular and intracellular effectors responsible for both driving and attenuating inflammation. These novel findings contribute to the ongoing investigation of inflammation and therapeutic strategies sequela of ischemic stroke.
Introduction
Regulation of blood sugar homeostasis and insulin release is largely driven by the postprandially produced incretin signaling hormones, including glucagon-like peptide-1 (GLP-1) (Nauck et al., 1986), and deficits in incretin signaling are hallmarks of type-2 diabetes mellitus (T2DM) (Holst and Ørskov, 2004). GLP-1 is produced and secreted in a glucose-dependent manner by the enteroendocrine L cells within the small intestine and acts upon its G-protein coupled receptors (GPCRs) on pancreatic β-cells to promote insulin secretion (Nauck et al., 1993). Its insulinotropic action is short-lived on the order of minutes. Membrane bound and soluble circulating dipeptidyl peptidase-4 (DPP-IV) efficiently cleaves the N-terminal dipeptide from polypeptides that express either an alanine or proline in their penultimate position; hence, it has a strong affinity for GLP-1 (Deacon, 2019). Non-insulinotropic metabolites are thereby generated, most predominantly GLP-1 (9–36) amide (Deacon, 2004) (henceforth referred to a GLP-1 (9–36) and shown in Fig. 1A).
Figure 1: GLP-1(9–36) induces intracellular cAMP levels in neuronal cells.
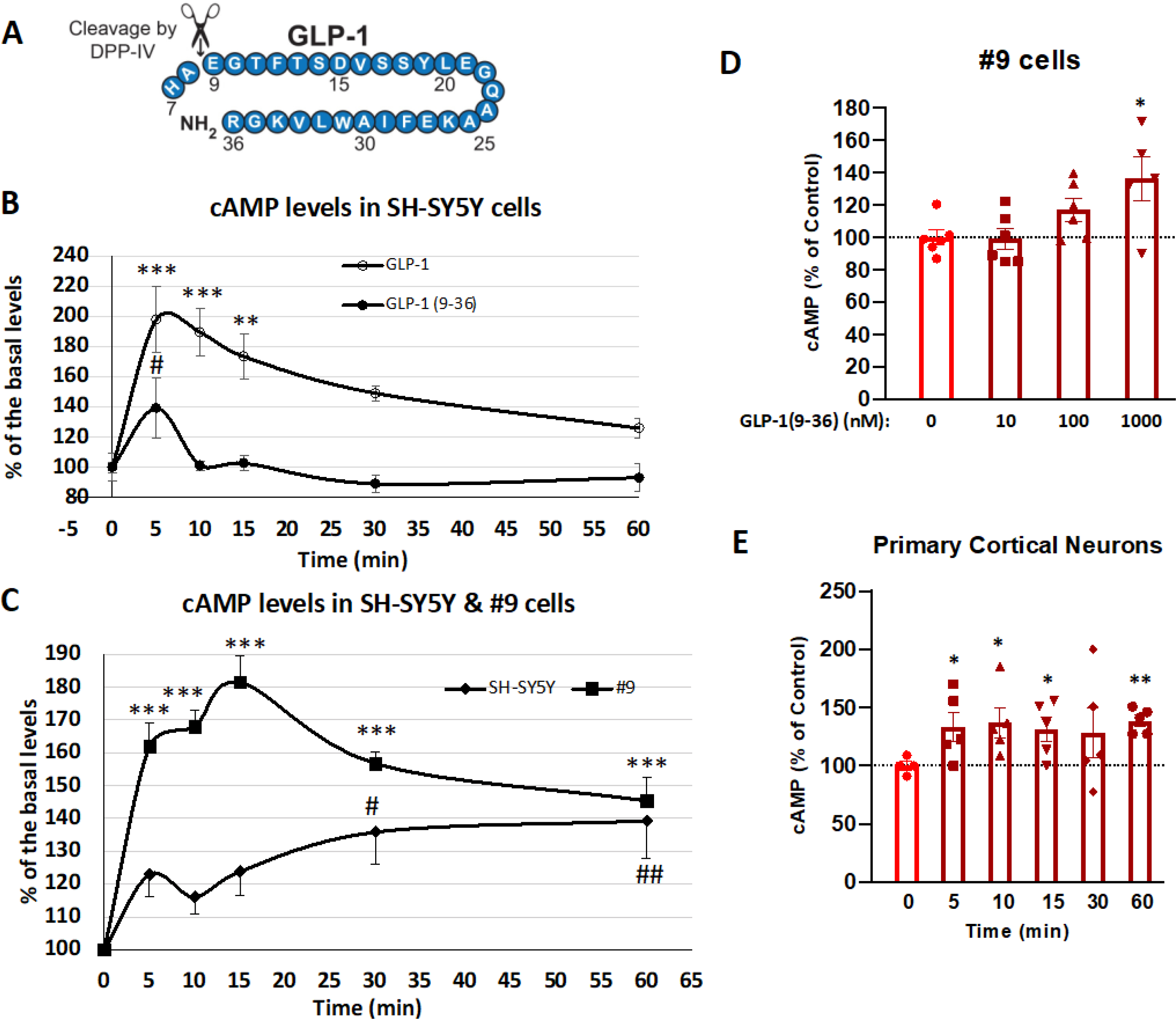
(A) Insulinotropic GLP-1 (7–36) is cleaved by DPP-IV at amino acid 2, forming the metabolite GLP-1 (9–36). (B) Comparison of cAMP response of GLP-1 and GLP-1(9–36) in SH-SY5Y cells. SH-SY5Y cells are treated with 100 nM of GLP-1 or GLP-1 (9–36) over a time course of 60 min (n = 6 for each time point). Intracellular cAMP levels were measured at 0, 5, 10, 15, 30 and 60 min; (C) Comparison of cAMP response of GLP-1(9–36) in SH-SY5Y and #9 (GLP-1R stable expression line) cells. SH-SY5Y or #9 cells are treated with 1000 nM of GLP-1 (9–36) over a time course of 60 min. Intracellular cAMP levels were measured at 0, 5, 10, 15, 30 and 60 min (n = 8 for each time point); (D) GLP-1(9–36) dose-dependently increases intracellular cAMP production in #9 cells. #9 cells were treated with GLP-1 (9–36) at concentrations of 10, 100 and 1000 nM for 15 min and intracellular cAMP production was quantified (n = 6 per condition); (E) GLP-1 (9–36) increases intracellular cAMP levels in primary cortical neurons. Primary cortical neuron cultures (at DIV 15) were treated with 1000 nM of GLP-1 (9–36) and intracellular cAMP levels were measured at 0, 5, 10, 15, 30 and 60 min (n = 5 for each time point). (One-way analysis of variance (ANOVA) tests were used for comparison of multiple samples, followed by post hoc Bonferroni’s tests. Statistical comparison vs. control (time zero or vehicle-treated), *P < 0.05; **P < 0.01; ***P < 0.001; # P < 0.05; ## P < 0.01).
The discovery of a naturally occurring DPP-IV resistant analogue of human GLP-1 (7–36) amide, Exendin-4 (Ex-4), was a breakthrough for the development of a new class of incretin-based diabetes medications (Eng et al., 1992). Synthetic Ex-4, known as Exenatide (Byetta™), became Food and Drug Administration (FDA) approved in 2005, and the following year, a DPP-IV inhibitor known as sitagliptin (Januvia™) was approved. Several others have since been FDA approved or are in clinical trials for treatment of metabolic disorders (for a comprehensive list, see Glotfelty et al., 2019). During the time that Exenatide was being developed clinically as a glucoregulatory agent for T2DM treatment, its neurotrophic and neuroprotective properties became evident (Gilman et al., 2003; Perry et al., 2003, 2002b, 2002a) and sparked interest in utilizing incretins as potential treatments for neurological disorders (Greig et al., 2004; Perry and Greig, 2003). Insulin dysregulation is common in a variety of neurodegenerative disorders (PD, AD, vascular dementia, Huntington disease, traumatic brain injury), and repurposing incretin therapies for treating these diseases may be ideal for future clinical trials (Cheong et al., 2020; Craft and Watson, 2004; Hölscher, 2020; Shaughness et al., 2020).
Whereas most focus has been given to the insulinotropic GLP-1 moiety [GLP-1 (7–36) amide], recent interest has been expressed in the peripheral effects of DPP-IV produced GLP-1 (9–36). DPP-IV expression is found in high concentrations in the brush border epithelium and in the nearby circulatory vessels proximal to the L-cells where GLP-1 is initially released (Hansen et al., 1999). GLP-1 (9–36) is rapidly produced as a cleavage product and exists in circulating levels 10-fold that of its precursor (Knudsen and Pridal, 1996). Although GLP-1 (9–36) is not insulinotropic (Elahi et al., 2008), it does have glucoregulatory properties; suppressing glucose production in hepatocytes through an insulin-independent mechanism (Tomas et al., 2010). This effect may be species specific as it was not observed in mice (Rolin et al., 2004) but was evident in pigs (Deacon et al., 2002) and humans (Meier et al., 2006). GLP-1 (9–36) binds to the GLP-1 receptor (GLP-1R) with an affinity less than 1% that of GLP-1 (7–36) (Knudsen and Pridal, 1996) further supporting a novel mechanism of action of this circulating metabolite.
In a mouse model of intracerebral hemorrhage, the GLP-1 (7–36) mimetic, liraglutide, blocked brain edema, neurological deficits and neuroinflammation; albeit, these positive benefits were found to be primarily mediated via liraglutide’s metabolite, GLP-1 (9–36). Co-administration of a GLP-1R antagonist and liraglutide did not block these positive outcomes; however, the effects were abolished when liraglutide was co-administered with a DPP-IV inhibitor. These effects were achieved via activation of 5’ adenosine monophosphate-activated protein kinase (AMPK) independent of the canonical GLP-1R agonism (Hou et al., 2012). This suggests a novel receptor or alternative activation of the GLP-1R by the metabolite. Recent research has provided evidence for GLP-1 (9–36) activating the glucagon receptor (GcgR) in the gut (Guida et al., 2020), insulin-like growth factor 1 receptor (IGF-1R) in astrocytes (Huang et al., 2020), and a GLP-1R independent cytoprotective mechanism in cardiomyocytes (Ban et al., 2010). However, Day et al. (2017) reported that synaptic plasticity effects observed in the brain are associated with GLP-1R activation. These studies demonstrate that GLP-1 (9–36) acts upon many cell types, although direct characterization in isolated neuronal cells has been limited.
Hallmarks of neurodegenerative disorders include high levels of oxidative stress, cell death, aberrant protein production [e.g., amyloid-β (Αβ) in Alzheimer’s disease (AD) and α-synuclein (α-syn) in Parkinson’s disease (PD)] and accompanying neuroinflammation. In the present study, we investigated mitigation of these disease paradigms across cell culture models (including differentiated and undifferentiated human SH-SY5Y neuroblastoma and a GLP-1R overexpressing variety of the same line), directly comparing the action of GLP-1 to its primary metabolite GLP-1 (9–36). Our studies evaluated cAMP activation of both GLP-1 and GLP-1 (9–36) over time and in several cell lines and primary neuronal cultures. We interrogated GLP-1 (9–36) mediated cell survival mechanisms, focusing on cAMP-dependent protein kinase (PKA) and AMPK signaling. The therapeutic potential of GLP-1 (9–36) in models of glutamate toxicity and oxidative stress was assessed, and our findings suggest a direct mechanism of action associated with GLP-1R. The anti-inflammatory capacity of GLP-1 (9–36) against an LPS challenge was examined in human and mouse microglial cell models. Finally, we evaluated the ability of Ex-4 and GLP-1 (9–36) to protect mouse primary brain cell cultures from toxic α-syn and Aβ challenges. Our studies advance understanding of the therapeutic potential of GLP-1 (9–36)-induced signaling and its physiologic roles in the nervous system.
Methods
The current study was not pre-registered. Note that n values represent single well replicates (biological replicates). For all assays (cAMP), culture media or supernatant from each biological replicate was measured in duplicates or triplicates, with the average of these measurements constituting each “n”.
Materials
Ex-4 (cat#AS-24464), GLP-1(7–36) amide (cat#AS-22463), GLP-1(9–36) amide (cat#AS-65070), and the GLP-1R antagonist Exendin (9–39) (cat#AS-24468) were purchased from AnaSpec Inc. (Fremont, CA, USA). L-Glutamic acid monosodium salt hydrate (glutamate) (cat#G1626), Triton-X 100 (cat#T9284), and hydrogen peroxide solution 30% (w/w) in H2O (H2O2) (cat#H1009) were obtained from Sigma-Aldrich Corporation (St. Louis, MO, USA). Protein kinase A inhibitor H89 dihydrochloride (cat#2910) and AMP-activated protein kinase (AMPK) inhibitor Dorsomorphin dihydrochloride (also known as Compound C) (cat#3093) were from Tocris Bioscience (Minneapolis, MN, USA).
Cell Culture
None of the cell lines used in this study are listed as commonly misidentified cell lines by the International Cell Line Authentication Committee (ICLAC; http://iclac.org/databases/cross-contaminations/). All cell cultures were maintained in a 37°C incubator comprising 5% CO2 and 95% air, with media replacement every other day or as indicated.
SH-SY5Y Cells:
Immortal SH-SY5Y human neuroblastoma cells (ATCC® CRL-22669™) and associated growth media were purchased from American Type Culture Collection (ATCC) (Manassas, VA, USA). Only adherent cells were used throughout the current studies at a maximum of 15 passages. A human GLP-1R over expressing line of SH-SY5Y cells (#9 cells), previously described by our group (Li et al., 2010b), also was used in the current studies. Both lines were grown in a mixture of half Eagle’s Minimum Essential Medium (EMEM) (ATCC® 30–2003™) and half Ham’s F12-K (Kaighn’s) Medium (ATCC® 30–2004™) supplemented with 10% heat-inactivated fetal bovine serum (Gibco™ cat#10082147) and 100 U/mL penicillin/streptomycin (Gibco™ cat#15140148). The cells were split at a 1:3 ratio every 5 days using 0.05% trypsin and 0.53 mM ethylenediaminetetraacetic acid (EDTA) (Invitrogen, Carlsbad, CA, cat#AM9912).
Differentiated SH-SY5Y Cells:
To further establish the neuroblastoma cell cultures as a reliable neuronal model, we differentiated SH-SY5Y and #9 neuroblastoma cells by following a published 18-day protocol (Shipley et al., 2016). This protocol includes carefully timed supplementation with three separate differentiation medias, which allows for gradual serum-starvation and the addition of retinoic acid, extracellular matrix proteins and neurotrophic factors. Over time, the cells develop neuron-like morphologies, resulting in a representative expansive and networked neuronal culture (Fig. 6G). Cultures were plated on extracellular matrix coated plates or PDL coated coverslips (Neuvitro, Cat# GG121.5PDL) for the final step of differentiation and subsequent cellular assays and immunohistochemistry. Brightfield, live cell images were obtained with 20x magnification from a Zeiss Primovert inverted phase contrast microscope.
Figure 6: Neurotrophic and neuroprotective effects of GLP-1(9–36) in differentiated SH-SY5Y/#9 cells.

Representative live (inset) and anti-Map2 stained undifferentiated and differentiated #9 cells. The differentiated cells display a networked neuronal phenotype typical of primary cultures (A). GLP-1(9–36) (1 μM) treatment for 48 h significantly increased cell viabilities in differentiated #9 cells (C) but not differentiated SH-SY5Y cells (B); Similarly, 24 h pretreatment with GLP-1(9–36) (1 μM) significantly protected differentiated #9 cells from H2O2-induced reduction in cell viabilities (E) and increase in LDH levels in culture media (G), while no effect was observed in differentiated SH-SY5Y cells (D, F). (n = 10 per condition). (Student t-test or one-way analysis of variance (ANOVA) tests were used for comparison of multiple samples, followed by post hoc Tukey’s multiple tests. Statistical comparison vs. vehicle-treated control: *P < 0.05; **P < 0.01; ****P < 0.0001; Statistical comparison vs. H2O2 alone group: #P < 0.05; ##P < 0.01) (scale bar= 50 μm).
HMC3 Microglia:
The transformed human microglial cell line HMC3 was also from ATCC (cat# CRL-3304), which retain many of the properties of primary microglial cells. HMC3 cells were cultured with EMEM (ATCC) supplemented with 10% fetal bovine serum (Gibco™ cat#10082147) and 100 U/mL penicillin/streptomycin (Gibco™ cat#15140148) and maintained similarly as SH-SY5Y cells except 0.25% trypsin and 0.53 mM ethylenediaminetetraacetic acid (EDTA) (Invitrogen, cat#AM9912) was used for subculture. A maximum of 10 passages were used for these cells.
Immortalized mouse microglia (IMG):
The immortalized mouse microglia (IMG) (McCarthy et al., 2016) (Sigma-Aldrich, cat# SCC-134) cell line was used as an additional neuroinflammation model. IMG cells were cultured in High Glucose DMEM (Sigma Cat. No. D6546) with 10% fetal bovine serum (Gibco™ cat#10082147) and 100 U/mL penicillin/streptomycin (Gibco™ cat#15140148). For subculture, Accutase® (Sigma, cat#A6964) detachment media was used for subculture for a maximum of 10 passages.
Primary Cortical Neurons (used in cAMP assay)
All experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and followed current European Union regulations (Directive 2010 / 63 / EU) agreement number A1301310. Experimental animal protocols were approved by the Animal Care and Use Committee of the Intramural Research Program, National Institute on Aging (438-TGB-2022) and were in compliance with the guidelines for animal experimentation of the National Research Council (Committee for the Update of the Guide for the Care and Use of Laboratory Animals, 2011) and the National Institutes of Health (DHEW publication 85–23, revised, 1995). A timed-pregnant Sprague-Dawley rat (fetus age day 15) (Charles River Laboratories, Wilmington, MA, USA) was euthanized via 5% isoflurane and cervical dislocation to obtain primary cortical neurons (PCNs). Cortices from eight pups were carefully dissected, pooled, and dissociated by mild trypsinization and added at a density of approximately 6 × 104 cells/well on 96-well plates in the plating media (DMEM F-12 media (Gibco™ cat#11320033) containing 2% B27 supplement (Gibco™ cat#17504044), 10% Heat-inactivated-FBS, 0.5 mM L-glutamine, and 25 μM L-glutamate). From day 4 in vitro (DIV4), cultures were maintained in the feeding media [Neurobasal™ medium (Gibco™ cat#21103049) containing 2% B27 supplement (Fisher Scientific, cat# 11530536) and 0.5 mM L-glutamine (Gibco, cat#25030149)].
Primary dissociation cell cultures challenged with α-synuclein or amyloid-β:
Rat dopaminergic or cortical neuron mixed dissociation cultures were maintained as previously described by Callizot et al. (2019, 2013) and Zhang et al. (2005) with minor modifications. A timed-pregnant rat (Wistar) (Janvier Labs, France) was euthanized (via isoflurane induced deep anesthesia and cervical dislocation) upon fetus maturation of 14–15 days gestation. Fetuses (10 pups) were removed aseptically and immediately placed in ice-cold L15 Leibovitz medium (L15) (Dutscher, cat#P04–27055) containing 2% penicillin (10,000 U/mL)/streptomycin (10 mg/mL) (Dutscher cat#P06–07100)), and 1% bovine serum albumin (BSA) (Dutscher, cat#P06–1391100). The ventral portion of the mesencephalic flexure, a midbrain region rich in dopaminergic neurons or a piece of cortex tissue, was dissected and pooled in a trypsin/EDTA solution (0.05 % trypsin and 0.02 % EDTA) for 20 min at 37 °C. Following this chemical dissociation procedure, Dulbecco’s modified Eagle’s medium (DMEM) (Dutscher, cat#P04–03600) containing DNAase I grade II (0.5 mg/mL) (Dutscher, cat#P60–37780100) and 10 % of fetal calf serum (FCS) (Dutscher, cat#S181B-500) was added, and cells were dissociated mechanically by 3 passages through a 10 mL pipette. Thereafter, cells were centrifuged at 180 x g for 10 min at 4°C on a layer of BSA (3.5%) (Dutscher, cat#P06–1391100) in L15 medium. The cell pellet was resuspended in culture medium (Neurobasal medium, 2 % solution of B27 supplement, 2 mmol/liter of L-glutamine, 2% Penicillin/Streptomycin solution, 10 ng/ml of brain-derived neurotrophic factor (BDNF) (Dutscher, cat#CB-1115002), 1 ng/ml of glial cell line-derived neurotrophic factor (GDNF) (Dutscher, cat#Z02927–1), 4% heat-inactivated FCS (Dutscher, cat#S181B-500), 1 g/L of glucose (Sigma-Aldrich, cat#G8644), 1 mM of sodium pyruvate (Sigma-Aldrich, cat#S8636–100mL), and 100 μM of non-essential amino acids (Dutscher cat#P08–32100)). Cells were then seeded to a density of 80,000 cells/well for dopaminergic neurons or 45,000 cells/well for cortical neurons in poly-L-lysine pre-coated 96 well-plates and maintained in a humidified incubator. Half medium changes were made every 2 days.
In dopaminergic neuron-enriched mixed cell cultures (DIV7), cells were pre-incubated for 24 h with culture medium with known GLP-1 (9–36) or Ex-4 concentrations (10, 100, 300, or 1000 nM) or vehicle. Cells were then exposed to α-syn for 48 h (250 nM α-syn, n=6 per group: human recombinant α-syn 1–140 aa from rPeptide, Watkinsville, GA cat#S-1001–2). The α-syn used contains <1.3 U endotoxin/mg of peptide which does not produce significant effects in terms of neurotoxicity or induction of ROS production, as previously reported (Zhang et al., 2005). Prior to addition, the α-syn was prepared as a 4 μM solution in culture medium (vehicle) and slowly shaken at 37o C for 72 h in the dark to induce oligomerization. In cortical neuron mixed cell cultures (DIV 11), cells were pre-incubated for 1 h (time was optimized for treatment) with culture medium containing known GLP-1 (9–36) concentrations (10, 100, 300, or 1000 nM) or vehicle. Cells were then exposed to Aβ (1–42) (5 μM, 72 h, n=6 per group: Bachem, Torrance, CA cat#4147158). This Aβ was previously prepared as a 40 μM solution that was slowly shaken at 37° C for 72 h in the dark in defined culture media (vehicle) described above to induce its oligomerization (Callizot et al., 2013).
Following experimental incubation times, cell culture supernatants were removed and frozen for later assays.
cAMP assay
Intracellular cAMP content was determined using the Direct cAMP (acetylated) ELISA kit (Enzo Life Sciences, Inc., Farmingdale, NY cat# ADI-900–067) as per the manufacturer’s protocol. SH-SY5Y and #9 cell cultures were first grown to 80–90% confluency in 24-well plates and subsequently serum-deprived overnight (0.5% serum media). To measure cAMP induction over time, the morning following serum deprivation 100 nM of GLP-1(7–36) or GLP-1(9–36) dissolved in 0.5% serum media were added to cultures. At 0, 5, 10, 15, 30, and 60 min timepoints, cells were lysed with 0.1 M HCl containing 0.5% Triton X-100 (Sigma, cat# T8787) (150 μl/well) for 10 min at room temperature (RT). Cell lysates were collected and centrifuged at 600 g at RT to remove cell debris, and supernatants were directly used for cAMP measurement. To assess the #9 cells’ cAMP response to various doses of GLP-1(9–36) (0, 10, 100, or 1000 nM), cell lysates and supernatants were collected after 15 min incubations and analyzed as described above. For time course measurements of cAMP production induced by 1000 nM GLP-1(9–36) in DIV15 PCNs, cells were collected at 0, 5, 10, 15, 30, 45 and 60 min after treatment in feeding media, and processed as described above for the preparation of supernatants.
Cell viability assays (MTS)
Cell viability was assessed using the CellTiter 96® Aqueous One Solution Cell Proliferation Assay kit (MTS) (Promega, Madison, WI, cat#G3580), which measures the formazan product that is produced in proportion to viable cell populations (measured using absorbance at 490 nm). For all viability studies, SH-SY5Y and #9 cells were grown in 96-well plates and serum-starved (0.5%) overnight before experimentation. Neurotrophic properties following 24 h incubation of varying concentrations of GLP-1 (9–36) (0, 1, 10, 100, or 1000 nM) were first evaluated in SH-SY5Y cells. For neuroprotective studies, SH-SY5Y or #9 cells were incubated for 2 h with the varied concentrations of GLP-1 (9–36) (0, 1, 10, 100, or 1000 nM) before a 24 h exposure to varied concentrations of glutamate (100 or 150 mM) or H2O2 (800 μM). Glutamate and H2O2 concentrations were selected from previous concentration-dependent pilot studies focused to provide a statistically significant but submaximal loss of viability. To study the GLP-1R mediated effects observed in these studies, GLP-1R antagonist [GLP-1 (9–39)] was added in 10-fold excess (10 μM) to GLP-1(9–36) (1 μM) 2 h prior to 100 mM glutamate challenge in both SH-SY5Y and #9 cell lines. Viability in these cells was examined after 24 h. To study the signaling pathways initiated by GLP-1 (9–36), cells were pre-treated for 2 h with GLP-1(9–36) (1 μM) in the absence or presence of PKA inhibitor H89 (10, 20 or 50 μM) or AMPK inhibitor Compound C (5, 10 or 50 μM) against 150 mM glutamate challenges. Neuroprotective and neurotrophic effects were evaluated 24 h later via the cell viability MTS assay.
Cell membrane integrity assays (LDH)
We used the fluorometric CytoTox-ONE™ Homogeneous Membrane Integrity Assay (Promega, Cat.# G7892) to measure lactate dehydrogenase (LDH) levels in the culture media. LDH is released from cells that have damaged membranes, allowing for relative comparisons of toxicity in individual wells and treatments. Higher levels of LDH correlates with higher levels of non-viable cells. In brief, we performed the assay on 96-well plates by following manufacturer’s protocol. After completing the manufacturer’s protocol, plates wells were individually read with an excitation wavelength of 560 nm and an emission wavelength of 590 nm.
Cellular Reactive Oxygen Species (ROS) Assay
The Fluorescent DCFDA Cellular ROS Detection Assay (Abcam, cat#ab113851) was used, according to the manufacturer’s recommended protocol, to detect hydroxyl, peroxyl and other ROS activity within the cell. This assay utilizes a fluorogenic dye, 2’,7’ –dichlorofluorescin diacetate (DCFDA), which permeates the cell membrane and is proportionately oxidized by ROS into a highly fluorescent 2’, 7’ –dichlorofluorescin (DCF). Cell cultures (#9 cells) were plated in 96-well plates and serum starved (0.5% serum) overnight before treatment. A 1 h pre-treatment of #9 cells with 100 nM GLP-1 (9–36) was followed by a Tert-Butyl Hydrogen Peroxide (TBHP) challenge at concentrations of either 25 μM or 50 μM. These selected TBHP concentrations were optimized from pilot studies indicating significant upregulation of ROS production in the #9 cells. Under similar dosing procedures, 100 nM GLP-1 (9–36) was compared to 100 nM Ex-4 versus a 50 μM TBHP challenge. DCF levels were detected by fluorescence spectroscopy with maximum excitation and emission spectra of 495 nm and 529 nm respectively.
Human Interleukin-6 (IL-6) levels in HMC3 culture media by ELISA
To study the anti-neuroinflammatory effect of GLP-1 (9–36) in human microglia HMC3 cells, we first pre-treated HMC3 cells with 1 μM of GLP-1(9–36) for 24 h in 96-well plates and then challenged cells with LPS (10 ng/ml) (Sigma, cat# L2880) for another 24 h. Four groups were included in this experiment: Vehicle-treated control group, LPS-alone group, GLP-1 (9–36) group and LPS + GLP-1 (9–36) group (n=6 each group, experiment repeated 3 times). At the end of experiment, cell culture media were saved for the measurement of cytokine IL-6 and the MTS assay were also performed to access cell viabilities. Human IL-6 levels were measured by using BioLegend’s ELISA MAX™ Deluxe Set (cat#430504) with samples diluted in a 1:2 ratio, the results of IL-6 concentration in the media were normalized (IL-6 results divided by relative cell viability, assessed via the MTS assay).
Mouse TNF-α levels in IMG culture media by ELISA
To test the capacity for GLP-1 (9–36) to affect other inflammatory mediators, including TNF-α (which HMC3 lack the capability to produce) (Dello Russo et al., 2018; Janabi et al., 1995), mouse IMG microglia cells were used as an additional neuroinflammation model (McCarthy et al., 2016). Cells were grown in 24-well culture plates and treated with GLP-1 (9–36) (100 nM and 1μM) as described in the HMC3 experiments. Vehicle-treated control group (culture media was used as vehicle), LPS-alone group, and LPS + GLP-1 (9–36) groups were used in these experiments. LPS (1 ng/mL) was added to cells after 24 h incubation in the two doses of GLP-1(9–36) similarly in these experiments (n=5–6, experiment repeated 3 times). Mouse TNF-α levels were measured using BioLegend’s ELISA MAX™ Deluxe Set (cat#430904) according to the manufacturer’s recommendations. Vehicle control samples were diluted 1:2 with all other samples diluted 1:10. The results were normalized by cell viability, assessed via the MTS assay.
Immunochemistry
Primary cell cultures:
Primary cell cultures were washed in PBS and fixed in 4% paraformaldehyde (PFA) in PBS (pH 7.3, 21° C, 20 min), washed twice in PBS, and then permeabilized. PBS containing 0.1 % of saponin and 1% of FCS (“staining buffer”) was added (21° C, 15 min) to block non-specific binding, and cells were subsequently incubated in staining buffer (2 h at RT) with either (i) a chicken polyclonal antibody, anti-microtubule- associated-protein 2 (MAP-2) (Abcam cat#ab5392,RRID: AB_2138153) (1/1000 dilution) to quantify cortical neurons; (ii) rabbit polyclonal anti-TH (Millipore cat#AB152, RRID: AB_390204) (1:2000 dilution) to quantify dopaminergic neurons and neurites; or (iii) mouse monoclonal anti-OX-41(Novus cat#NB100–65530, RRID: AB_960866) (1:500 dilution) to visualize microglia. After washing the sections, they were incubated in staining buffer with corresponding secondary antibodies (Sigma-Aldrich) at a 1/400 dilution: anti-rabbit IgG CF™ 568A (cat#SAB4600084), anti-mouse IgG CF™ 488A (cat#SAB4600042, RRID: AB_2532075), anti-chicken IgY highly cross adsorbed CF™ 568a (cat#SAB4600079) in staining buffer for 1 h at RT. Photomicrographs were automatically acquired by ImageXpress(R) Micro-Confocal High Content Imaging System (Molecular Devices, San Jose, CA) and were analyzed via a custom designed algorithm in the automated software (Custom Module Editor®, MetaXpress, by Molecular Devices). All images were acquired under the same capture parameters. For α-syn treatment dopaminergic neuron survival, the number of TH positive neurons was quantified (20 images/well at 10x magnification). These same images were used to determine dopaminergic neuron “neurite network” via quantification of total TH positive neurite length in each image, as described in Callizot et al. (2019). Microglial presence was quantified via area of microglial cells (80% of the well of a 96-well plate was imaged, or 0.272 cm2/well and quantified for OX-41 staining in μm2) and compared to control levels. For Aβ treatment, total neuron number/survival was quantified from photomicrographs (30 images/well at 20x magnification) via the number of MAP-2 positive neurons, as in Callizot et al. (2013). For images, average number of Map-2 positive cells per image (~900 cells/well) were used for comparison.
HMC3 and IMG Microglia:
HMC3 and IMG cells were plated on 12 mm sterile glass coverslips (Fisher Scientific, cat#12–545-81) in a 24 well plate, at a density of 40,000 cells per coverslip. The cells grew for two days in their respective growth medias described above. Cells were washed twice with PBS before a 10- minute fixation using 4% paraformaldehyde (PFA) (Fisher Scientific, cat#AAJ19943K2). PFA was removed and cells were washed an additional two times with PBS. A one-hour block/permeabilization step was performed using PBS with 3% bovine serum albumin (Sigma, cat# A7030) and 0.1% saponin (Sigma, cat#47036) (microglia staining buffer). Primary antibodies anti-P2Ry12 (clone S16007D) (Biolegend, cat#848002, RRID: AB_2650633, 1:200 dilution, raised in rat), anti-Iba1 (Synaptic Systems, cat#234004, RRID: AB_2493179, 1:500 dilution, raised in guinea pig), and anti-GLP-1R (Invitrogen, cat#PA5–97789, RRID: AB_2812404, 1:100 dilution, raised in rabbit) were incubated on coverslips in microglia staining buffer for 1 h at RT. Coverslips were subsequently washed three times with 0.1% saponin in PBS (wash buffer) for 5 min/wash. Corresponding highly cross adsorbed Alexa Fluor® (Thermo Scientific) secondary antibodies were diluted (1:500) in microglia staining buffer and applied to cells for 1 h at RT: donkey anti-rat IgG 488 (cat#A-21208, RRID: AB_141709), goat anti-guinea pig IgG 555 (cat#A-21435, RRID: AB_2535856), and goat anti-rabbit 647 IgG (cat# A-21244, RRID: AB_2535812). Secondary only controls were used for to ensure antibody specificity (available upon request). Cells were washed three times in wash buffer for 5 mins/wash with a fourth wash with PBS alone. Coverslips were mounted on slides with Prolong Diamond with DAPI (Invitrogen, cat#P36962), with DAPI used to visualize cell nuclei. Cells were imaged at 20x using an LSM 880 confocal microscope (Zeiss). A Z-stack was collected through the thickness of each cell culture and collapsed into a maximum intensity projection for the final image (pinhole set to 1 for each channel) Imaging parameters were calibrated using the secondary only controls as a baseline for laser power and gain.
Differentiated and undifferentiated SH-SY5Y cell cultures:
The same fixing and staining procedure, as outlined for the IMG cells, was followed for the differentiated and undifferentiated cell cultures. The primary antibody for the neuronal marker anti-Map2 (Synaptic Systems, cat#188003, RRID: AB_2281442, raised in rabbit) was used for visualization of cell structure, along with DAPI to visualize the nuclei. Goat anti-rabbit 647 IgG (cat# A-21244, RRID: AB_2535812) secondary antibody was used to conjugate the primary anti-Map2 antibody. Imaging was performed similarly to the IMG cells with the Zeiss LSM880 Confocal microscope.
Statistics
Data are presented as mean ± standard error of the mean (SEM) and were analyzed by Prism software v. 8. Normality was assumed for statistical analysis and one-way analysis of variance (ANOVA) tests were used for comparison of multiple samples, followed by post hoc tests (as indicated in each figure) with appropriate corrections for serial measurements if required. A value of p <0.05 or less is considered statistically significant. Sample size was selected based on our prior studies (Li et al., 2020, 2017, 2009) and a power analysis (Charan and Kantharia, 2013). Blinding was performed in relation to α-syn and Aβ challenge studies, where a different experimenter analyzed results. Other experiments were not blinded. Grubbs’ test was used to determine outliers in experiments. One replicate in the TNF-α quantification was removed via Grubbs’ parameters. Wells that provided data out of range of detection were not included in statistical analysis for figures 6 and 7. No exclusion criteria was predetermined.
Figure 7: GLP-1 (9–36) is anti-inflammatory in human and mouse microglial cells.

(A) Immortalized human (HMC3) and adult murine (IMG) microglia express canonical microglial protein markers Ibal and P2Ry12 in addition to GLP-1R. Both cell lines respond robustly to LPS challenge, inducing significant release of IL-6 (B) or TNF-α (C). These elevations of IL-6 (in HMC3 cells, n=6 per condition, B) and TNF-α (in IMG cells, n=5–6 per condition, C) are significantly reduced by 24 h preincubation in 1 μM GLP-1 (9–36). (One-way analysis of variance (ANOVA) tests were used for comparison of multiple samples, followed by post hoc Tukey’s multiple tests. Statistical comparison vs. vehicle-treated control: **P < 0.01; ****P < 0.0001; comparison vs. LPS alone: #P < 0.05; ##P < 0.01) (scale bar= 100 μm)
Results
GLP-1 metabolite GLP-1 (9–36) is a weak GLP-1R agonist in neuronal cells
GLP-1 can stimulate intracellular cAMP production in neuronal cells through GLP-1R. In primary cortical neurons, GLP-1 at concentrations as low as 10 nM rapidly increased intracellular cAMP levels within minutes (Li et al., 2009). To evaluate whether the GLP-1 metabolite GLP-1 (9–36) (Fig. 1A) preserves this cAMP stimulating function, we first compared treatment between GLP-1 and GLP-1 (9–36) in neuroblastoma SH-SY5Y cells at concentrations of 100 nM in a time-dependent study (as our preliminary data indicated that lower concentrations of GLP-1 (9–36) did not alter cAMP levels in SH-SY5Y cells) (Fig. 1B). Compared to the dramatic rise in intracellular cAMP levels achieved by 100 nM GLP-1 (198% of basal levels at 5 min, p<0.001), the same concentration of GLP-1 (9–36) transiently and modestly increased the cAMP level in SH-SY5Y (Fig. 1B) (139% of basal levels at 5 min, p<0.05). To enhance this cAMP effect, we next used a human GLP-1 receptor overexpression #9 (SH-SY5Y) cell line and compared 1 μM GLP-1 (9–36) treatment in both the original SH-SY5Y and #9 cells. In contrast to the original SH-SY5Y cells, #9 cells express 2-fold human GLP-1R protein, as previously reported (Li et al., 2010b). As expected, the same concentration of GLP-1 (9–36) induced a much more robust cAMP response in #9 cells than in SH-SY5Y cells (maximal cAMP elevation original SH-SY5Y: 139% (p<0.05 to 0.01) and #9: 182% (p<0.001) of basal levels), indicating that this GLP-1 (9–36) action is mediated by GLP-1R signaling (Fig. 1C). To confirm the cAMP effect of GLP-1 (9–36), we treated #9 cells with different concentrations of GLP-1 (9–36) for 15 min (concentrations: 10, 100, 1000 nM). We observed that only the highest concentration of GLP-1 (9–36) (1000 nM) induced a significant rise of intracellular cAMP, indicating a dose-dependent response (Fig. 1D). Finally, we evaluated rat primary cortical neuron cultures (PCNs) to confirm our findings in neuroblastoma cells. PCNs at DIV 15 were treated with 1000 nM of GLP-1 (9–36) and cAMP levels were measured time-dependently (0, 5, 10, 15, 30 and 60 min). GLP-1 (9–36) induced a rise in intracellular cAMP levels in PCNs as early as 5 min (133% of basal levels), and cAMP levels remained elevated over the course of 1 h (Fig. 1E).
Neurotrophic and neuroprotective actions of GLP-1 (9–36)
We previously demonstrated that GLP-1 receptor agonists (including dual and triagonist mimetics) are neurotrophic and neuroprotective across numerous physiological and pathological insults (Bader et al., 2020, 2018; Chen et al., 2018; Li et al., 2020, 2017, 2009). To characterize the neurotrophic and neuroprotective effects of GLP-1 (9–36), we first treated SH-SY5Y cells in a concentration-dependent (1, 10, 100, 1000 nM) manner for 24 h and assessed cell survival by MTS assay. No differences in cell viabilities were evident after GLP-1 (9–36) treatment, as shown in Figure 2A. Furthermore, separate experiments of prolonged treatment with GLP-1 (9–36) to 48 h, likewise, did not alter cell viability (data not shown), suggesting a lack of GLP-1 (9–36) mediated neurotrophic action in these cells. To evaluate potential neuroprotective GLP-1 (9–36) effects, we first pre-treated SH-SY5Y cells in a concentration-dependent (10, 100, 1000 nM) manner for 2 h, and then challenged them with glutamate (final concentrations: 100 mM or 150 mM) for 24 h. Cell viability was then assessed. Glutamate challenge induced dose-dependent cell death of approximately 24% and 45% (100 mM and 150 mM glutamate, Fig. 2B (i) top and 2B (ii) bottom, respectively), compared to controls. Pretreatment with GLP-1 (9–36) modestly protected the cells, with improved cell viabilities reaching statistical significance only at higher GLP-1 (9–36) doses (Fig. 2B) (GLP-1 (9–36) at 1000 nM provided 33% mitigation of 100 mM glutamate toxicity, and GLP-1 (9–36) at 100 and 1000 nM provide 18% mitigation of 150 mM glutamate toxicity). Alternatively, #9 cells were pre-treated with GLP-1 (9–36) dose-dependently (1, 10, 100, 1000 nM) for 2 h, and challenged with H2O2 at a final concentration of 800 μM for 24 h. Whereas 800 μM H2O2 decreased cell viability by approximately 16%, versus controls, all evaluated concentrations of GLP-1 (9–36) mitigated H2O2-induced cell death (Fig. 2C) (providing from 38% to 69% mitigation for GLP-1 (9–36) 1 nM to 1000 nM).
Figure 2: Neurotrophic and neuroprotective effects of GLP-1(9–36) in SH-SY5Y/#9 cells.
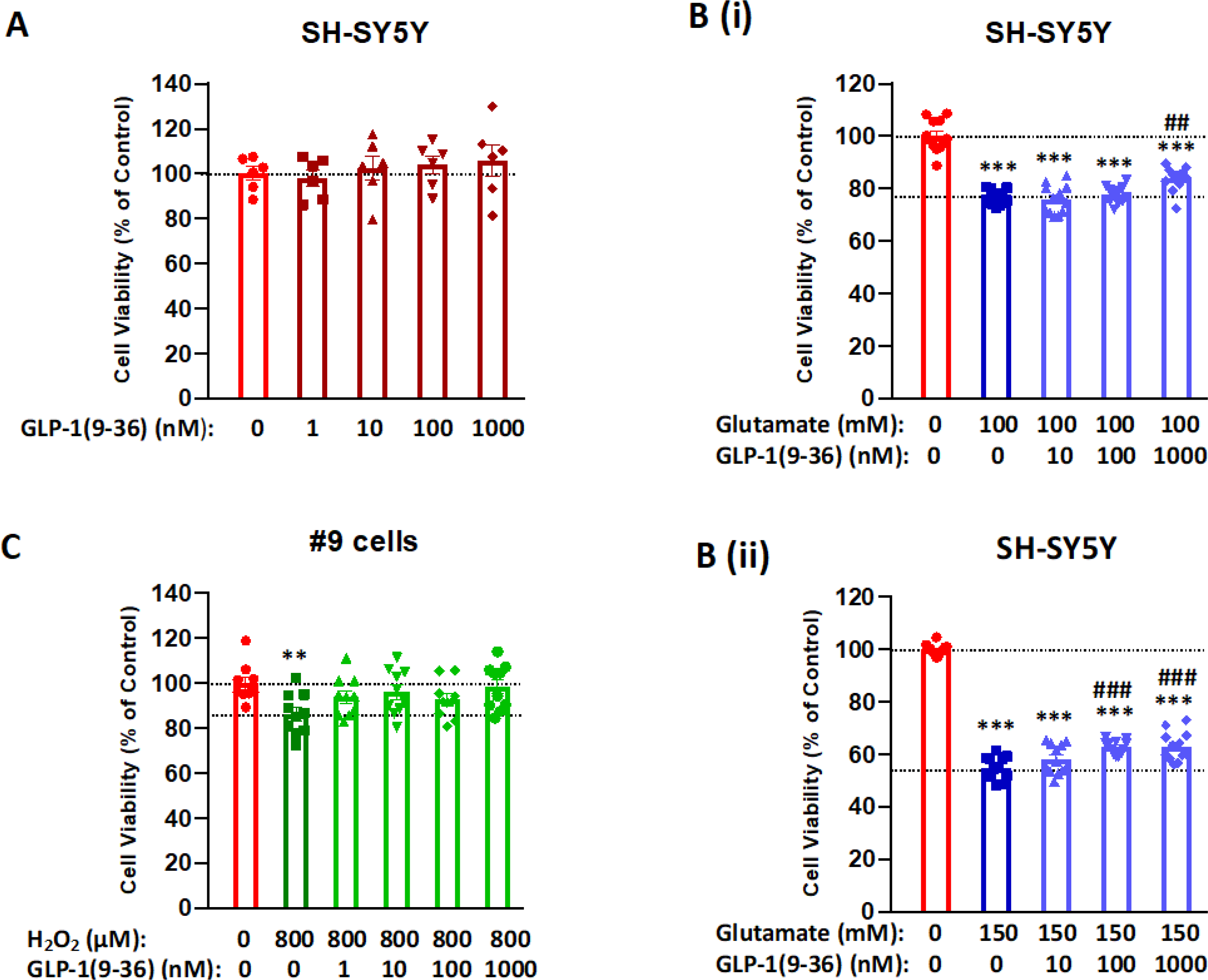
(A) GLP-1(9–36) lacked neurotrophic action in SH-SY5Y cells. Cells were treated with GLP-1(9–36) at concentrations of 1, 10, 100 and 1000 nM for 24 h, cell viabilities were not significantly different from the vehicle-treated control (n = 6 per condition); (B) GLP-1(9–36) dose-dependently protected SH-SY5Y cells from glutamate toxicity. Cells were pre-treated with various concentrations (1, 10, 100 and 1000 nM) of GLP-1(9–36) for 2 h and then challenged with two concentrations of glutamate (upper panel: 100 mM; lower panel: 150 mM) for 24 h. Higher concentrations of GLP-1(9–36) (1000 nM in upper panel and 100 and 1000 nM in lower panel) significantly protected the cell viability when compared with glutamate treatment alone (n = 10 per condition); (C) GLP-1(9–36) treatment protected #9 cells from oxidative stress challenge of H2O2. Cells were pre-treated with various concentrations (1, 10, 100 and 1000 nM) of GLP-1(9–36) for 2 h and then challenged with 800 μM H2O2 for 24 h, all concentrations of GLP-1(9–36) treatment prevented H2O2-induced cell death (n = 6 per condition). (One-way analysis of variance (ANOVA) tests were used for comparison of multiple samples, followed by post hoc Tukey’s multiple tests. Statistical comparison vs. vehicle-treated control: **P < 0.01; ***P < 0.001; comparison vs. glutamate alone: ##P < 0.01; ###P < 0.001).
The neuroprotective effect of GLP-1 (9–36) in neuronal cells is mediated through the GLP-1R
To investigate whether the observed neuroprotective effect of GLP-1 (9–36) in neuronal cells is mediated through the GLP-1R, we used GLP-1R antagonist Exendin 9–39 to block GLP-1R signaling. In both SH-SY5Y and #9 cell lines, 100 mM glutamate challenge for 24 h induced a small but significant reduction in cell viability (14% in SH-SY5Y cells and 10% in #9 cells, respectively), which was prevented by 1 μM GLP-1(9–36) pre-treatment for 2h in both cell lines (providing 44.5% and 70% protection from the glutamate cell loss in SH-SY5Y and #9 cells, respectively). However, this GLP-1 (9–36) protective effect was abolished by the presence of 10 μM Ex-9–39, which was added simultaneously with GLP-1 (9–36) (Fig. 3). Interestingly, we also observed that a neurotrophic effect of GLP-1(9–36) was significant in #9 cells (Fig. 3B) but not in SH-SY5Y cells (117% vs 104% of their respective controls in viabilities) (Fig. 3A) when comparing the GLP-1 (9–36) only group with the control group; thereby cross-validating our earlier SH-SY5Y study (Fig. 2).
Figure 3: Neuroprotective effects of GLP-1(9–36) in SH-SY5Y/#9 cells depend on GLP-1 receptor.
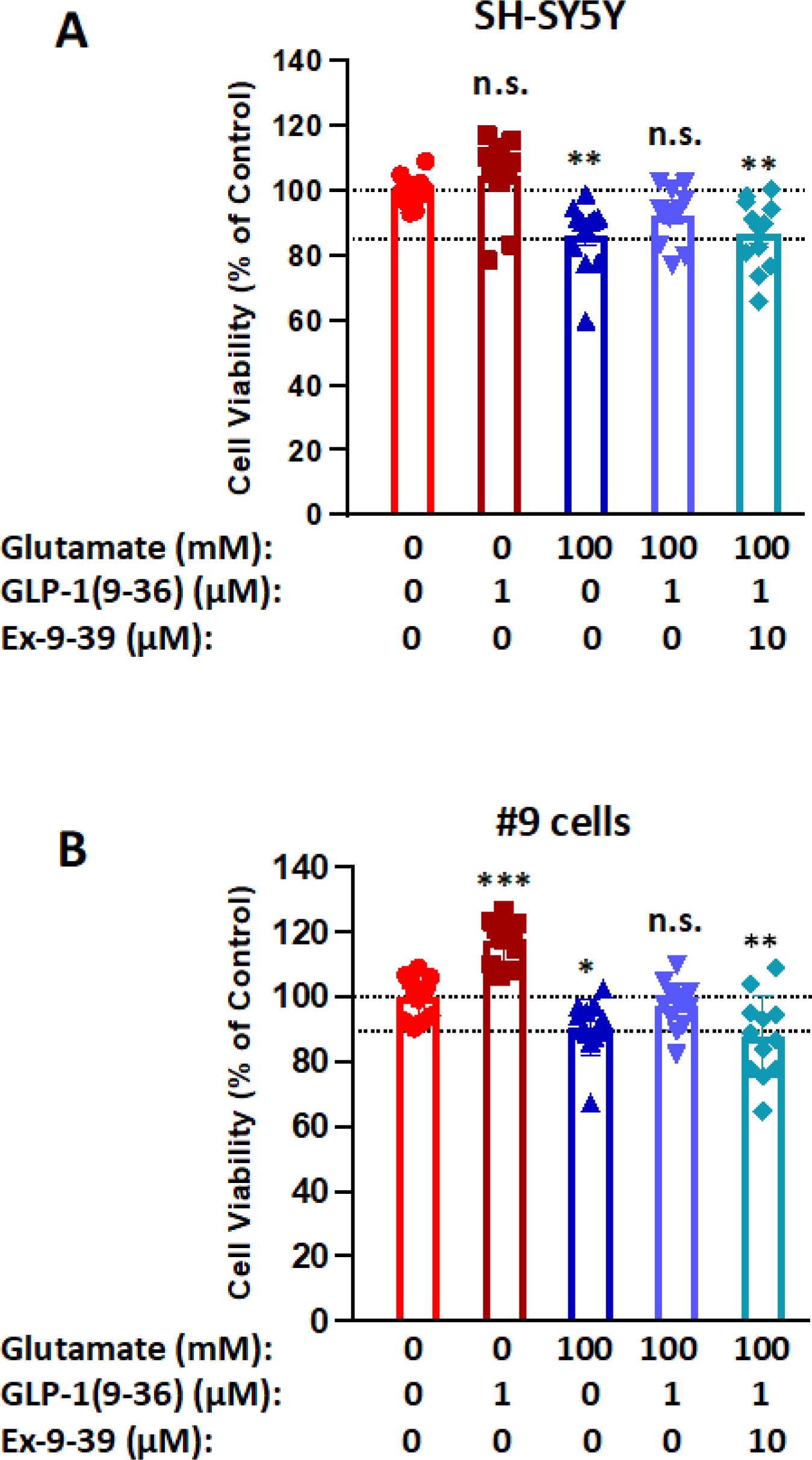
Glutamate (100 mM)-induced cell death (24 h) in both SH-SY5Y (A) and #9 (B) cells was prevented by 1 μM GLP-1(9–36) pre-treatment for 2 h. However, this effect was abolished by the presence of 10 μM Ex-9–39 (a GLP-1R antagonist). The neurotrophic effect of GLP-1(9–36) was significant in #9 cells (B) but not SH-SY5Y cells (A) (n = 10 per condition). (One-way analysis of variance (ANOVA) tests were used for comparison of multiple samples, followed by post hoc Tukey’s multiple tests. Statistical comparison vs. vehicle-treated control: *P < 0.05; **P < 0.01; ***P < 0.001)
GLP-1 (9–36) reduces cellular ROS production in neuronal cells
Elevated ROS production is associated with many neurological disorders. To evaluate whether GLP-1 (9–36) can reduce ROS production in neuronal cells, we pre-treated #9 cells with 100 nM GLP-1 (9–36) or Ex-4 and then challenged the cells with Tert-Butyl Hydrogen Peroxide (TBHP) at a final concentration of either 25 μM or 50 μM. A fluorescent DCFDA Cellular ROS Detection Assay was used to measure intracellular ROS levels at the end of the experiment. Whereas TBHP dose-dependently elevated intracellular ROS levels to 152% (25 μM TBHP) or 187% (50 μM TBHP) of the control level, GLP-1 (9–36) pretreatment significantly prevented 23% or 36.8% of these elevated ROS levels (Fig 4A) respectively. During a 50 μM TBHP challenge, 100 nM GLP-1 (9–36) and 100 nM Ex-4 (Fig 4B) significantly reduced ROS (by 37% and 36% respectively) levels induced by TBHP, suggesting similar protective effects against elevated intracellular ROS levels.
Figure 4: GLP-1 (9–36) reduces cellular ROS production in SH-SY5Y cells.
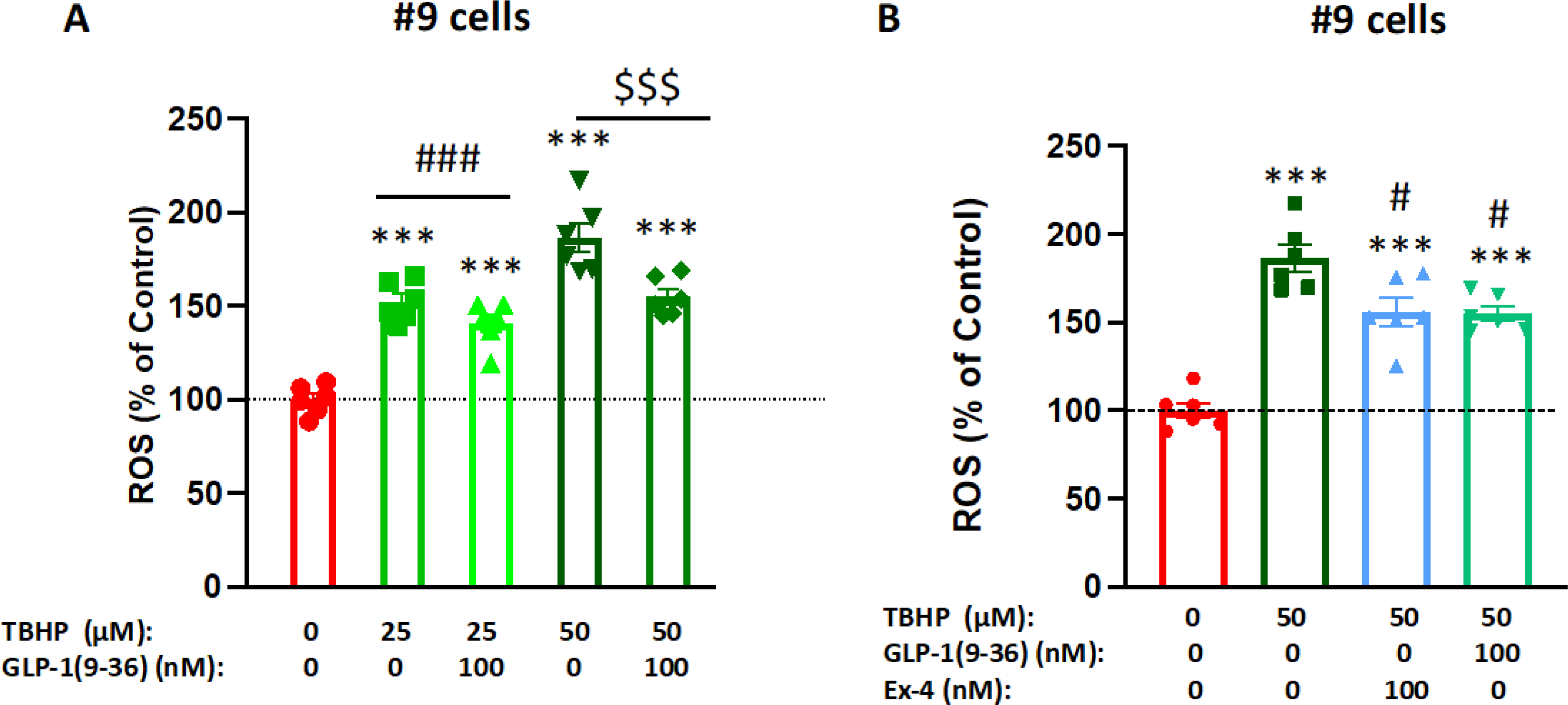
SH-SY5Y cells treated with 25 μM or 50 μM tertbutyl hydrogen peroxide (TBHP) for 2 h significantly increased intracellular ROS production in a dose-dependent manner. Pre-treatment (1 h) of the cells with 100 nM GLP-1 (9–36) significantly reduced cellular ROS level in the presence of 25 or 50 μM TBHP (A), Pre-treatment (1h) of the cells with 100 nM Ex-4 or GLP-1 (9–36) similarly reduced cellular ROS levels in the presence of 50 μM TBHP (B). (n = 6 per condition). (One-way analysis of variance (ANOVA) tests are used for comparison of multiple samples, followed by post hoc Tukey’s multiple tests. Statistical comparison vs. vehicle-treated control: ***P < 0.001; comparison vs. TBHP alone: ###P < 0.001; $ $ $P < 0.001).
GLP-1 (9–36) mediates neurotrophic/neuroprotective properties via PKA and AMPK in neuronal cells
To interrogate which signaling pathways are involved in the observed neurotrophic/neuroprotective effects of GLP-1 (9–36) in neuronal cells, selective pathway inhibitors were added to GLP-1R overexpressing cells (#9 cells) together with the metabolite and neurotrophic/neuroprotective effects were then quantified. In #9 cells, treatment with 1 μM GLP-1 (9–36) induced a significant increase in cell viabilities, approximately 115% of control (Fig. 5A and B). However, presence of PKA inhibitor H89 (Fig. 5A) or AMPK inhibitor Compound C (Fig. 5C) abolished this effect, indicating both pathways are involved in the neurotrophic activity of GLP-1 (9–36). As expected, pre-incubation of GLP-1 (9–36) (1 μM, 2 h) significantly rescued glutamate (150 mM)-induced cell death (increasing viability by approximately 50%). This effect was also abolished in the presence of H89 or Compound C (Fig. 5A and B), suggesting that GLP-1 (9–36) may mediate its neuroprotective activity via both PKA and AMPK pathways. Lower doses of H89 (10 μM) (Fig. 5C) and Compound C (5 μM) (Fig 5D), which do not cause any toxicity, similarly abolished the protective effect of the metabolite from glutamate toxicity.
Figure 5: GLP-1(9–36) is neurotrophic and neuroprotective via PKA and AMPK activation.
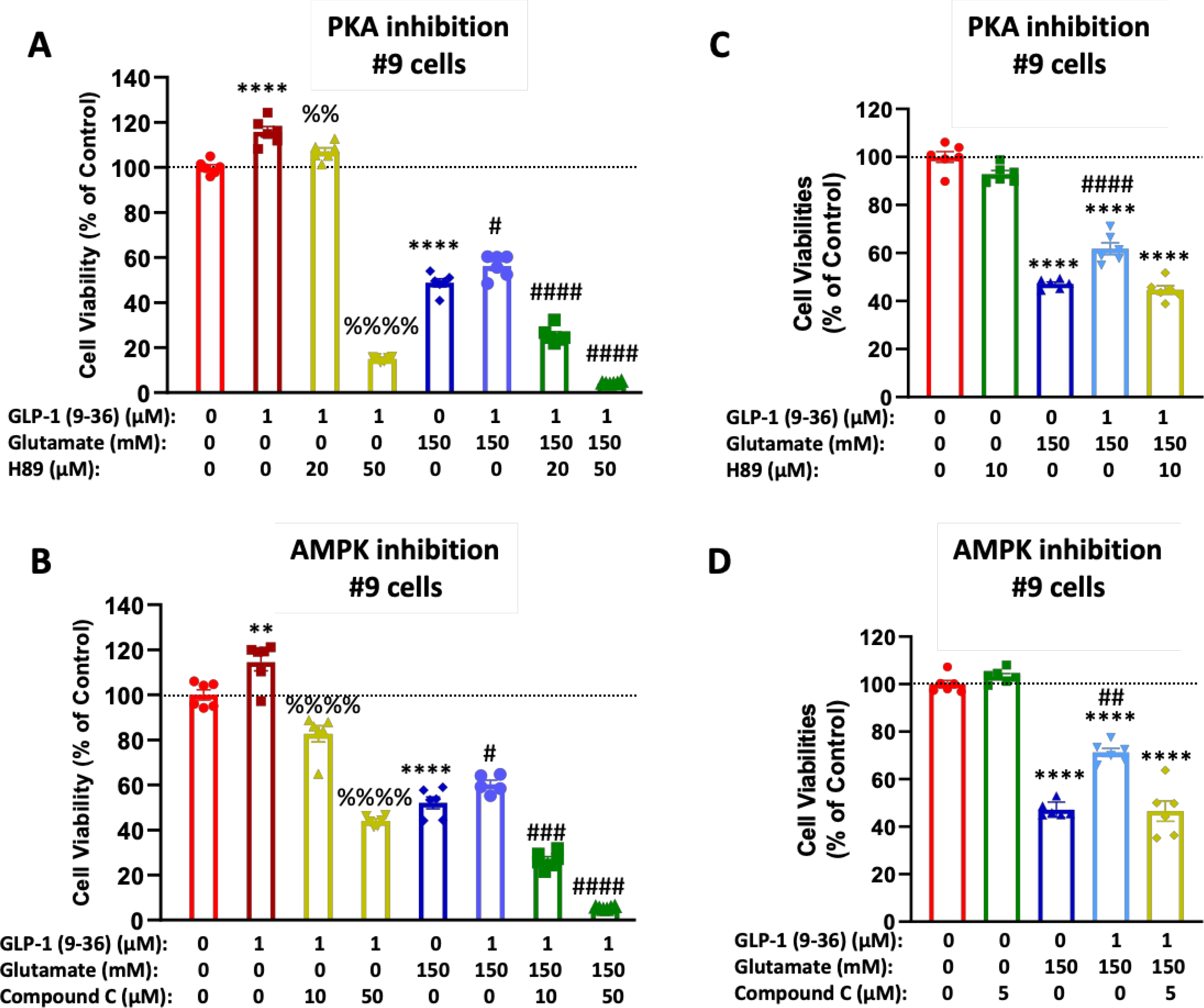
#9 cells treated with 1 μM of GLP-1 (9–36) showed both a neurotrophic effect and neuroprotective effect against glutamate-induced cell death (A, B). These effects were abolished by the presence of PKA inhibitor H89 (at concentrations of 20 or 50 μM) (A) or AMPK inhibitor Compound C (at concentrations of 10 or 50 μM) (B) Similarly, the neuroprotective effects of GLP-1(9–36) were abolished at lower concentrations of H89 (10 μM) or Compound C (5 μM) when inhibitors have no self-toxicity (C, D).(n = 6 per condition). (One-way analysis of variance (ANOVA) tests were used for comparison of multiple samples, followed by post hoc Tukey’s multiple tests. Statistical comparison vs. vehicle-treated control: **P < 0.01; ****P < 0.0001; comparison vs. GLP-1 (9–36) alone: %%P < 0.01; %%%%P < 0.0001; comparison vs. glutamate alone: #P < 0.05; ###P < 0.001; ####P < 0.0001)
Neurotrophic and neuroprotective effects of GLP-1 (9–36) in differentiated SH-SY5Y/#9 cells
We demonstrated neurotrophic and neuroprotective effects of GLP-1 (9–36) in undifferentiated neuroblastoma cells, which are easy to obtain and grow but have limitations. To validate our model, we differentiated SH-SY5Y and #9 cells into a homogeneous, networked, neuronal population. Following a well-established method, we were able to differentiate SH-SY5Y and #9 cells from a neuroblast-like state into mature human neurons (Shipley, 2016). Differentiated SH-SY5Y/#9 cells were treated with 1 μM GLP-1 (9–36) for 48 h. Cell viabilities were assessed by MTS assay as previously described. Treatment with GLP-1(9–36) induced a significant increase (~17%) in cell viabilities compared to controls in differentiated #9 cells (Fig. 6B), but this effect was not observed in differentiated SH-SY5Y cells (Fig. 6A), in line with our results obtained from undifferentiated cells. This implies that the neurotrophic effect detected was at least partially mediated through the GLP-1R. Consistently, the oxidative stress insult of 75 μM H2O2 killed more cells in differentiated SH-SY5Y cells (62% of cell loss) (Fig. 6C) than in differentiated #9 cells (39% of cell loss) (Fig. 6D). A 24 h pretreatment with 1 μM GLP-1 (9–36) significantly rescued cell viabilities in differentiated #9 cells only (by 21%) (Fig. 6D). In concurrence with the MTS assay results following the 75 μM H2O2 insult, LDH levels rose to 180% of controls in differentiated SH-SY5Y cells compared to 145% in differentiated #9 cells (Fig. 6E, 6F). Pretreatment of 1 μM GLP-1 (9–36) for 24 h decreased LDH levels in the media to 120% of controls only in differentiated #9 cells (Fig. 6F), suggesting the importance of GLP-1R in neuroprotective effects observed.
GLP-1 (9–36) is anti-inflammatory in human and mouse microglia challenged with LPS
Immortalized human embryonic (HMC3) and adult murine (IMG) microglial cells treated with LPS (10 or 1 ng/mL respectively) were used to model neuroinflammation. Importantly, both the HMC3 and IMG cells express canonical microglial markers Iba1 and P2Ry12, in addition to GLP-1R protein (Fig 7A). HMC3 cells are a well characterized microglia cell model (Dello Russo et al., 2018; Janabi et al., 1995), while the IMG cells are a relatively new immortalized microglia model (McCarthy et al., 2016). The IMG cells have the capacity to produce a wider range of cytokines than the HMC3 cells, including TNF-α, and thus were used in addition to the HMC3 cells to confirm the anti-inflammatory capacity of GLP-1 (9–36). Microglial cells preincubated in 1 μM GLP-1 (9–36) for 24 h significantly reduced levels of IL-6 (in HMC3 cells, Fig. 7B) and TNF- α (IMG cells, Fig. 7C) released into cell culture media following a 24 h 10 ng/mL and 1 ng/mL LPS challenges, respectively.
GLP-1 (9–36) ameliorates multiple deterioration in mixed primary cell culture models of neurodegeneration (neuronal loss)
Mixed brain cell primary dissociation cultures were pre-treated with various concentrations (10, 100, 300 and 1000 nM) of GLP-1 (9–36) or Ex-4 for 24 h and then challenged with oligomeric α-syn (250 nM for 48 h (Zhang et al., 2005)) to induce an unsurmountable neuronal loss of approximately 36.4% (Fig. 8A). Thereafter, neurite length (a measure of the total length of surviving TH+ cell processes) and OX-41 staining (a measure of the area of microglial immunostaining) were evaluated. As illustrated in Figure 8B and 8C, oligomeric α-syn induced a reduction in neurite length in surviving TH-positive neurons and an increase of microglia, respectively, both of which were ameliorated by GLP-1 (9–36) and Ex-4 in a concentration-dependent manner. Specifically, α-syn induced a 47.9% loss of neurites in surviving neurons and increased OX-41 staining (a marker of microglia/macrophages) (Taylor et al., 2011) to 137%, versus unchallenged mixed primary cultures. Pre-incubation with 300 nM of GLP-1 (9–36) and Ex-4 significantly ameliorated this α-syn induced neurite loss by 23.2% and 20.9%, respectively. Moreover, both 300 nM and 1000 nM of GLP-1 (9–36) and all concentrations of Ex-4 used significantly mitigated microglial presence by up to 63.6% (with 1000 nM of GLP-1 (9–36)) and 84.6% (with 1000 nM of Ex-4), respectively (p<0.05). In addition, dissociated mixed primary cortical cell cultures were pre-treated with various concentrations (10, 100, 300 and 1000 nM) of GLP-1 (9–36) for 1 h and then challenged with 5 μM Aβ for 72 h (Callizot et al., 2013) to induce a neuronal loss of approximately 35.9% (Fig. 8D). Pre-incubation with GLP-1 (9–36) dose-dependently ameliorated this effect, specifically 300 nM of GLP-1 (9–36) significantly reduced Aβ- induced neuron loss by 26.7% (p<0.05).
Figure 8: GLP-1(9–36) in chronic neurodegeneration models on mixed-cell primary cultures: preservation of neurite network and reduced microglia presence (α-syn challenge) and increased neuron survival (amyloid-β challenge).

In primary culture of mesencephalic neurons (dopaminergic neurons) co-cultured with astrocytes and microglial cells, treatment with 250 nM of α-synuclein induced neuronal loss as measured by TH+ neuron numbers compared to control treatment (A) and, importantly, neurite length (B) in the surviving dopaminergic neurons (accompanied by representative TH staining of neurons). Whereas pretreatment with GLP-1 (9–36) or Ex-4 (doses:10 to 1000 nM for 24 h) did not alter the TH number of surviving neurons (A) (n = 5–6 per condition), it notably dose-dependently mitigated the neurite network loss (B) (n = 4–6 per condition). α-Synuclein treatment also increased microglial cell presence (μm2 per image), shown by increased microglial marker OX41 (representative staining shown) (C) (n = 4–6 per condition), which pretreatment with GLP-1 (9–36) or Ex-4 dose-dependently decreased. (D) GLP-1(9–36) increases surviving primary neurons following amyloid-β challenge. In primary culture of cortical mixed dissociation cells, treatment with 5 μM of Aβ induced neuronal loss. Pretreatment with GLP-1 (9–36) (doses: 10 to 300 nM for 1 h) dose-dependently increased the number of Map2 positive neurons (n=5–6 per condition). (One-way analysis of variance (ANOVA) tests were used for comparison of multiple samples, followed by post hoc Dunnett’s tests Statistical comparison vs. α-synuclein or Aβ alone: *P < 0.05, scale bars= 50 μm)
Discussion
Following food intake, endogenous GLP-1 has a short active window for gluco-regulation due to DPP-IV cleavage of its insulinotropic moiety, GLP-1 (7–36), to its non-insulinotropic form, GLP-1 (9–36). The role of GLP-1 (9–36) has yet to be fully characterized, and it was once considered to be an inert waste product, although it may be a primary agonist for GLP-1Rs that exist within the peripheral (PNS) and central nervous system (CNS) (Richards et al., 2014). Whereas, the GLP-1 receptor (GLP-1R) resides ubiquitously in the pancreas, it is also expressed in other tissues (Pyke et al., 2014), including the brain where its activation produces cell type specific pleiotropic effects (Rowlands et al., 2018). Our current studies seek to further understand the roles GLP-1 (9–36) may play in the brain, specifically in neuronal and microglial cells, and thereby provide us understanding of what a deficiency in this ubiquitous peptide may have on the nervous system, especially in pathological situations. This would also shed a light on the currently available GLP-1 mimetics in clinical use that may or may not affect physiological levels of GLP-1 (9–36).
With increased age, prevalence of metabolic disorders such as T2DM is also increased. T2DM is characterized by a blunted incretin response and insulin resistance, both of which are linked to development of neurodegenerative disorders, including AD and PD (Cheong et al., 2020; Hogg et al., 2018; Hölscher, 2020). The connection between disrupted metabolic homeostasis and neurodegeneration has increasingly made the class of drugs known as “incretin mimetics” of interest in treating diseases of the CNS. Incretin mimetics have been used to treat metabolic disorders since the early 2000s - most notably the GLP-1 analogue Ex-4. A myriad of preclinical in vivo and in vitro studies demonstrate that GLP-1- based therapies have efficacy in treating a variety of neurological disorders, including traumatic brain injury (Glotfelty et al., 2019; Greig et al., 2014), PD (Athauda and Foltynie, 2016; Glotfelty et al., 2020; Kim et al., 2017), AD (Hölscher, 2020), stroke (Chen et al., 2018; Li et al., 2009; Zhang et al., 2015) and amyotrophic lateral sclerosis (Salcedo et al., 2012). Incretin mimetics are currently being tested in a wide variety of animal models of neurodegenerative disorders, with several in human clinical trials (Glotfelty et al., 2020, 2019).
GLP-1R activation is known to activate a variety of downstream neuroprotective secondary messengers, most notably cAMP (Rowlands et al., 2018). Thus, we first sought to compare cAMP signaling of GLP-1 (9–36) to its parent peptide, GLP-1 (7–36). In SH-SY5Y cells, GLP-1 (9–36) increases cAMP to a significantly lower degree than GLP-1 (7–36) at a 100 nM dose (Fig. 1B). Using a GLP-1R overexpressing (#9) SH-SY5Y cell line (Li et al., 2010b), we demonstrated that GLP-1 (9–36) significantly augments cAMP levels (Fig.1C) and establish the GLP-1R as a target of the metabolite. This cAMP effect is dose-dependent in #9 cells (Fig. 1D) and is time-dependently maintained in PCNs for up to 60-minutes following application (Fig. 1E), further confirming that GLP-1 (9–36) can signal through GLP-1R. Previous studies show that GLP-1 (9–36) is capable of increasing cAMP production (Ban et al., 2010) and this cAMP response is dependent on levels of GLP-1R expression (Knudsen et al., 2012), which our work confirms. This is particularly relevant in the context of PD, as Glp-1r mRNA in the substantia nigra of PD patients is found at 10-fold the levels of control humans, with increased GLP-1R protein expression in the same brain region of PD model mice (Yun et al., 2018). Interestingly, allosteric modulation of the GLP-1R enhances the cAMP response to the metabolite (Li et al., 2012), and allosteric modulation of the metabolite itself can increase cAMP production to significantly higher levels than that of its parent peptide [GLP-1 (7–36)] (Wootten et al., 2012). All together, these findings point to potential development of highly active GLP-1 (9–36) analogues that utilize different signaling paradigms compared to existing incretin mimetics (Wootten et al., 2012; Wootten and Miller, 2020).
In aged brains and across neurodegenerative disorders, oxidative stress and glutamate toxicity are common. Co-morbid metabolic disorders such as T2DM may disrupt the known neuroprotective benefits conferred by metabolic hormones, including the incretins and insulin, exacerbating these diseases. In the present study, we establish a neuroprotective effect for GLP-1 (9–36), which is the most prevalent circulating GLP-1 moiety in the body. Albeit GLP-1 (9–36) does not significantly increase cell viability in SH-SY5Y cells (Fig. 2A), a high dose (1000 nM) significantly increased cell viability against a glutamate challenge (100 mM) (Fig. 2Bi). Interestingly, cell viabilities are significantly improved against an increased glutamate challenge (150 mM) with concentrations of GLP-1 (9–36) as low as 100 nM (Fig. 2Bii). This effect is abolished in both the SH-SY5Y cells and #9 cells when excess (10 μM) GLP-1R antagonist is co-applied with the metabolite (Fig. 3A and 3B), further establishing the GLP-1R as the primary target of the metabolite in these cells.
Several studies argue that GLP-1 (9–36) relies on a receptor independent from GLP-1R (Ban et al., 2010; Guida et al., 2020; Hou et al., 2012; Huang et al., 2020), although receptor engagement may be tissue dependent (Rowlands et al., 2018). Radiolabeled GLP-1 (9–36) binds to the hippocampus at similar levels as radiolabeled GLP-1 (7–36) in the mouse brain; however, it did not localize other areas of the brain where radiolabeled GLP-1 (7–36) was ubiquitous (Kuc et al., 2014). Furthermore, IGF-1R in astrocytes appears to mediate GLP-1 (9–36)’s anti-stroke properties in a mouse model (Huang et al., 2020). Clearly, much is not understood about the mechanisms by which the GLP-1 (9–36) fragment engages the nervous system.
Nevertheless, in the human neuroblastoma cells used in the current study, we show that increased GLP-1R expression drives an enhanced cAMP response to GLP-1 (9–36), in addition to the neuroprotective and neurotrophic properties of the peptide. The utility of GLP-1 (9–36) as a neuro-modulatory agent is supported by its rescue of synaptic plasticity and cognitive deficits in mouse models of AD (Ma et al., 2012) and Down’s syndrome (Day et al., 2019). Improved neuronal function was also demonstrated, with enhancement (Day et al., 2017) and rescue (Day et al., 2019) of hippocampal long-term potentiation (LTP). The Day et al. (2017) study contradicts previous work which shows that GLP-1 (9–36) does not enhance LTP (Gault and Hölscher, 2008) and shows synaptic plasticity effects were mediated via the GLP-1R (via antagonist studies), similar to the GLP-1R mediated effects we observe. This implies that, at least in part, the canonical receptor is of interest in neuronal cells. Additional research is needed to delineate whether other multiple receptors are involved in the action of GLP-1 (9–36) in the brain.
Our results suggest GLP-1 (9–36) restores the ability of cellular programs to respond to oxidative stress, which is particularly important for the brain as it is highly susceptible to damage from ROS due to its high oxygen consumption (Salim, 2017). Our glutamate toxicity studies in the SH-SY5Y cells do not directly recapitulate an excitotoxic primary neuron response to glutamate due to their lack of key glutamate receptors (Sun et al., 2010). Instead, these studies should be considered complementary to the oxidative stress experiments using H2O2 and TBHP, as the levels of glutamate are high enough to reverse the cysteine-glutamate transporter and thus deprive the cells of glutathione, an important antioxidant (Maher and Schubert, 2000). It is likely that the presence and abundance of ionotropic glutamate receptors various across cell types and their differentiation state (Kritis et al., 2015), which prompted the use of both undifferentiated and differentiated SH-SY5Y cells to add to our understanding of the neurotrophic/protective actions of GLP-1 (9–36) (Nicolini et al., 1998).
In #9 cells, an oxidative stress challenge using H2O2 resulted in significant cell death that was ameliorated with doses of metabolite as low as 1 nM (Fig. 2C). We then challenged SH-SY5Y cells to TBHP (25 and 50 μM), an organic peroxide widely used to induce oxidation, and showed a significant increase in intracellular ROS. In response to the more severe challenge (50 μM TBHP), a 100 nM GLP-1 (9–36) or Ex-4 pretreatment significantly decreased ROS (Fig. 4). Previous work describes the ability of GLP-1 (9–36) to mitigate elevated ROS production in response to transient hyperglycemia in cultured endothelial cells and in mice (Giacco et al., 2015). Furthermore, GLP-1 (9–36) pretreatment of transgenic AD mice normalized mitochondrial superoxide levels in hippocampus, and, thereby mitigated Aβ associated impairments in synaptic plasticity (Ma et al., 2012). Similarly, in a mouse model of Down’s syndrome, GLP-1 (9–36) reversed ROS to near wildtype levels in the hippocampus (Day et al., 2019). In the light of these findings, our study builds on the body of evidence that this metabolite of GLP-1 plays important physiologic roles in maintaining homeostasis potentially throughout the body.
GLP-1 (9–36) can suppress cell death pathways, notably restoring protein kinase B (Akt) inhibition of glycogen synthase kinase-3b (GSK3b) in a mouse model of AD (Ma et al., 2012). Another study has demonstrated cytoprotective effects of GLP-1R agonism in SH-SY5Y cells through the relief of chronic endoplasmic reticulum stress via an Akt and signal transducer and activator of transcription 3 (STAT3) driven mechanism (Panagaki et al., 2017). In vivo administration of GLP-1 (9–36) decreases eukaryotic elongation factor-2 (eEF2) phosphorylation in the mouse hippocampus, increasing cellular potential for protein synthesis, purportedly a mechanism by which the metabolite enhances hippocampal LTP (Day et al., 2017). Day et. al (2017) additionally demonstrated in acutely treated hippocampal slices a significant GLP-1 (9–36)-induced increase in phosphorylated (activated) AMPK, an upstream regulator for eEF2. Comparably, GLP-1R independent activation of AMPK by GLP-1 (9–36) was observed in a mouse model of intracerebral hemorrhage (Hou et al., 2012). This can occur independently (Han et al., 2019) or in conjunction (Wei et al., 2016) with the commonly described GLP-1R mediated activation of cAMP. In our GLP-1R overexpressing #9 cells, we confirm PKA (Fig. 5A) and AMPK (Fig. 5B) activation as primary downstream signaling molecules of GLP-1 (9–36), contributing to restored cell viability in response to a glutamate challenge. Additionally, the cytotoxic effects of glutamate were exacerbated when PKA and AMPK are inhibited in a dose-dependent manner. Our demonstration of GLP-1R mediated rescue of cell death from the glutamate challenge and its inhibition by Ex (9–39) (Fig. 3) points to the PKA and AMPK signaling as GLP-1R mediated.
We addressed concerns regarding use of a neuroblastoma cell line in our studies by repeating some basic viability assays and oxidative stress challenges in a differentiated version of the SH-SY5Y and #9 cells (pictured in Fig. 6A). Concurring with our initial studies (Fig. 3A and 3B), 1 μM GLP-1 (9–36) similarly induced a significant increase in viability in the differentiated #9 cells but not the SH-SY5Y cells (Fig. 6B and 6C). Differentiated #9 cells experience less cell death from a 75 μM H2O2 challenge than differentiated SH-SY5Y cells and show significantly increased viability with 1 μM GLP-1 (9–36) against the challenge, an even more robust effect than shown previously in Fig. 2C with undifferentiated cells. The differentiated #9 cells, but not the differentiated SH-SY5Y cells, are rescued from oxidative stress challenge with GLP-1 (9–36) (Fig 6D and E). Consistent with MTS data from the 75 μM H2O2 challenge, LDH levels assayed in culture media, which correlates to the level of non-viable cells, were significantly reduced from the GLP-1 (9–36) treatment in differentiated #9 cells but not the SH-SY5Y cells (Fig. 6F and 6G). Our data indicates that the undifferentiated cell cultures behave similarly to the more neuronal like differentiated cultures, adding confidence to the results of our model system and studies.
GLP-1 (9–36) has previously not been evaluated independently for anti-inflammatory capacity. One in vivo mouse model of intracerebral hemorrhage notes considerable contributions of the GLP-1 (9–36) metabolite (derived from the GLP-1 analogue, Liraglutide) to anti-inflammatory action observed (Hou et al., 2012); however, direct action of the metabolite on microglia has not been studied. In our current studies, we have employed human embryonic (HMC3) and adult murine microglial cell lines to investigate the anti-inflammatory capacity of GLP-1 (9–36). Both cell lines express canonical microglial markers Iba1 and P2Ry12 along with the GLP-1R, which has previously not been reported (Fig 7A). Both have been used to model resident microglia of the brain (Lepiarz and Olajide, 2019; McCarthy et al., 2016; Rai et al., 2020). Among inflammatory mediators produced by these cell lines, embryonic derived HMC3 cells most prominently have the capacity to produce IL-6, while IMG cells produce more diverse sets of cytokines (McCarthy et al., 2016), including TNF-α. Therefore, we measured the levels of these proteins released into cell culture media in response to an LPS challenge. IL-6 (in HMC3 cells) and TNF-α (in IMG cells) were significantly reduced with a 24 h pretreatment of 1 μM GLP-1 (9–36) (Fig. 7B and 7C). Our studies hence demonstrate that GLP-1 (9–36) has the capacity to blunt neuroinflammation from a mild challenge.
GLP-1 analogues have previously demonstrated efficacy in reversing dopaminergic cell death in animal models of PD (Chen et al., 2018; Glotfelty et al., 2020; Li et al., 2009; Salcedo et al., 2012) and recently have shown benefits in a human PD clinical trial (Athauda et al., 2017), with further ones planned or ongoing (Glotfelty et al., 2020). Preliminary in vivo studies show that incretin mimetics may provide respite to levodopa-induced dyskinesia (LID), a condition caused by prolonged exposure to Levodopa, the primary treatment for PD symptoms (Abuirmeileh et al., 2012; Yu et al., 2020). Similar beneficial effects of incretin therapies were previously demonstrated in animal models of AD (Hölscher, 2020; Li et al., 2018, 2010a; Tai et al., 2018) and, to a limited extent, in human clinical trials (Cairns, 2020; Gejl et al., 2016; Mullins et al., 2019). An evaluation of cognitive effects in T2DM patients treated with incretin mimetics showed a reduced incidence of cognitive impairment (Cukierman-Yaffe et al., 2020). Furthermore, meta-analyses of patients using incretin-based therapies as metabolic disorder treatments show a possible prophylactic effect from the drugs for the development of PD (Brauer et al., 2020; Svenningsson et al., 2016) and stroke (Barkas et al., 2019; Malhotra et al., 2020).
GLP-1 (9–36) may hence be an important part of the incretin axis against neurodegeneration as it does not readily degrade and remains in circulation, allowing for direct access to the brain. Thus, we sought to understand how this metabolite interacts with disease challenges associated with PD (α-syn) and AD (Aβ). Dopaminergic neurons co-cultured with astrocytes and microglia confronted with an unsurmountable α-syn challenge (250 nM: inducing 36.4% dopaminergic neuron loss (Fig. 8A)) demonstrated a loss of neurites in surviving cells, which was significantly mitigated at 300 nM doses of both GLP-1 (9–36) and Ex-4 (Fig. 8B). In addition, significant reductions in microglial activation were observed at higher doses of the metabolite (300 and 1000 nm) (Fig. 8C). Ex-4 was used as a positive control, and significantly reduced microglial presence as we (Bader et al., 2018; Chen et al., 2018) and others (Yun et al., 2018) have previously shown. Recent evidence purports that a major anti-inflammatory effect of GLP-1 (9–36) is astrocyte-driven, resulting in significant decreases in TNF-α, IL-6, and IL-1b protein release (Huang et al., 2020). The crosstalk between astrocytes and microglia in the neuroinflammatory process (Liddelow et al., 2017) was not captured in our current studies, but should be considered in future experiments.
The focus of our studies was to obtain knowledge of the endogenous metabolite of GLP-1 that is lacking in the field. We consider that GLP-1 (9–36) represents a component of an endogenous trophic system to optimize cellular survival in response to physiological challenges and to, thereby, maintain homeostasis. In this regard, it may provide a therapeutic approach itself, or represent an important consideration when using other incretin mimetics (for example, do the other mimetics break down to generate the 9–36 moiety or work in conjunction with the metabolite or affect the endogenous metabolite’s level?). Often, incretin signaling is impaired in individuals at risk for chronic neurological disorders, and it is possible that the metabolite provides some form of protection or activates cellular signaling pathways that are not engaged with full length incretin peptides.
Conclusion
Our findings support previous studies that indicate an important role for GLP-1 (9–36) in brain health. As this peptide was previously considered to be inert consequent to its weak GLP-1R binding, our studies strengthen the importance of this predominantly circulating incretin protein as a key component of neuronal cell health. We show that GLP-1 (9–36) is capable of activating disease modifying neuroprotective and neurotrophic components of cell signaling associated with the GLP-1R. Notably, further work is needed to understand whether multiple receptors are involved in the activation of these pathways, particularly in the light of prior studies of other GLP-1 metabolites, epitomized by GLP-1 (28–36) amide, targeting mitochondria to modulate oxidative phosphorylation, inhibit oxidative stress, and suppress apoptosis in pancreatic β-cells independently of the GLP-1R (Liu et al., 2012). As GLP-1 (9–36) cannot induce hypoglycemia and is a well-tolerated endogenous peptide (Guglielmi and Sbraccia, 2017), it may represent an ideal novel or complementary treatment option for neurodegeneration.
Acknowledgements:
The authors thank Drs. Noëlle Callizot and Alexandre Henriques (Department of Pharmacology, Neuro-Sys SAS, Gardanne, France) in relation to mixed culture studies involving Aβ and α-syn.
Additional thanks are to Lauren Brick, Visual Media Core, National Institute on Drug Abuse, National Institutes of Health, USA, in relation to illustrations.
Funding and Additional Information:
The research of the authors was supported in part by (i) the Intramural Research Programs of the National Institute on Aging (NIA) and National Institute on Drug Abuse (NIDA), National Institutes of Health, USA, (ii) the Swedish Research Council K2012-62X-03185-42-4, and (iii) the Swedish Brain Foundation. E Glotfelty is supported by the National Institutes of Health—Karolinska Institute Graduate Partnership Program.
Abbreviations:
- AD
Alzheimer’s Disease
- Akt
protein kinase B
- AMPK
5’ adenosine monophosphate-activated protein kinase
- Aβ
Amyloid β
- BDNF
brain-derived neurotrophic factor
- BSA
bovine serum albumin
- cAMP
cyclic-adenosine monophosphate
- DCF
2’, 7’ –dichlorofluorescin
- DCFDA
2’,7’ –dichlorofluorescin diacetate
- DPP-IV
dipeptidyl peptidase-4
- EDTA
ethylenediaminetetraacetic acid
- EMEM
Eagle’s Minimum Essential Medium
- Ex-4
Exendin-4
- FCS
fetal calf serum
- FDA
Food and Drug Administration
- GDNF
glial cell line-derived neurotrophic factor
- GLP-1(R)
glucagon-like peptide-1 (receptor)
- GPCR
G-protein coupled receptor
- GSK3b
glycogen synthase kinase-3b
- HMC3
human microglial cell clone 3
- IGF-1R
insulin-like growth factor 1 receptor
- IL-6
interleukin-6
- IMG
immortalized microglial
- LDH
lactate dehydrogenase
- LID
levodopa-induced dyskinesia
- LPS
lipopolysaccharide
- LTP
long term potentiation
- PCNs
primary cortical neuron cultures
- PD
Parkinson’s Disease
- PFA
paraformaldehyde
- PKA
protein kinase A
- ROS
reactive oxygen species
- RRID
Research Resource Identifier
- RT
room temperature
- STAT3
signal transducer and activator of transcription 3
- T2DM
type-2 diabetes mellitus
- TBHP
Tert-Butyl Hydrogen Peroxide
- TH
tyrosine hydroxylase
- TNF-α
tumor necrosis factor- alpha
- α-syn
α-synuclein
- #9 cells
human GLP-1R over-expressing SH-SY5Y cell clone 9
Footnotes
Conflict of Interest:
NHG is a co-inventor on patents related to the use of GLP-1R agonists for the treatment of neurodegenerative disorders and has assigned all rights to the National Institutes of Health, USA.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- Abuirmeileh A, Harkavyi A, Rampersaud N, Lever R, Tadross JA, Bloom SR, Whitton PS, 2012. Exendin-4 treatment enhances L-DOPA evoked release of striatal dopamine and decreases dyskinetic movements in the 6-hydoxydopamine lesioned rat. J. Pharm. Pharmacol 64, 637–643. 10.1111/j.2042-7158.2011.01394.x [DOI] [PubMed] [Google Scholar]
- Athauda D, Foltynie T, 2016. The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson’s disease: mechanisms of action. Drug Discov. Today 21, 802–818. 10.1016/j.drudis.2016.01.013 [DOI] [PubMed] [Google Scholar]
- Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, Hibbert S, Budnik N, Zampedri L, Dickson J, Li Y, Aviles-Olmos I, Warner TT, Limousin P, Lees AJ, Greig NH, Tebbs S, Foltynie T, 2017. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet 390, 1664–1675. 10.1016/S0140-6736(17)31585-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader M, Choi H-I, Rubovitch V, Li Y, Kim HK, Glotfelty E, Pick CG, Lecca D, Hoffer BJ, Kim DS, Tweedie D, Greig NH, Rachmany L, 2018. Pharmacokinetics and efficacy of PT302, a sustained-release Exenatide formulation, in a murine model of mild traumatic brain injury. Neurobiol. Dis 124, 439–453. 10.1016/j.nbd.2018.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader M, Li Y, Tweedie D, Shlobin NA, Bernstein A, Rubovitch V, Tovar-y-Romo LB, DiMarchi RD, Hoffer BJ, Greig NH, Pick CG, 2020. Neuroprotective Effects and Treatment Potential of Incretin Mimetics in a Murine Model of Mild Traumatic Brain Injury. Front. Cell Dev. Biol 7, 356. 10.3389/fcell.2019.00356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban K, Kim KH, Cho CK, Sauvé M, Diamandis EP, Backx PH, Drucker DJ, Husain M, 2010. Glucagon-Like Peptide (GLP)-1(9–36)amide-mediated cytoprotection is blocked by exendin(9–39) yet does not require the known GLP-1 receptor. Endocrinology 151, 1520–1531. htt 10.1210/en.2009-1197 [DOI] [PubMed] [Google Scholar]
- Barkas F, Elisaf M, Milionis H, 2019. Protection against stroke with glucagon-like peptide 1 receptor agonists: a systematic review and meta-analysis. Eur. J. Neurol 10.1111/ene.13905 [DOI] [PubMed] [Google Scholar]
- Brauer R, Wei L, Ma T, Athauda D, Girges C, Vijiaratnam N, Auld G, Whittlesea C, Wong I, Foltynie T, 2020. Diabetes medications and risk of Parkinson’s disease: a cohort study of patients with diabetes. Brain 143, 3067–3076. 10.1093/brain/awaa262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns E, 2020. CTAD 2020 – Elad fails, but GLP-1s could still have a future in Alzheimer’s | Evaluate [WWW Document]. Eval. Vantage. URL https://www.evaluate.com/vantage/articles/news/snippets/ctad-2020-elad-fails-glp-1s-could-still-have-future-alzheimers (accessed 11.16.20). [Google Scholar]
- Callizot N, Combes M, Henriques A, Poindron P, 2019. Necrosis, apoptosis, necroptosis, three modes of action of dopaminergic neuron neurotoxins. PLoS One 14, e0215277. 10.1371/journal.pone.0215277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callizot N, Combes M, Steinschneider R, Poindron P, 2013. Operational dissection of β-amyloid cytopathic effects on cultured neurons. J. Neurosci. Res 91, 706–716. 10.1002/jnr.23193 [DOI] [PubMed] [Google Scholar]
- Charan J, Kantharia N, 2013. How to calculate sample size in animal studies? J. Pharmacol. Pharmacother 10.4103/0976-500X.119726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Yu S-J, Li Y, Lecca D, Glotfelty E, Kim HK, Choi H-I, Hoffer BJ, Greig NH, Kim D-S, Wang Y, 2018. Post-treatment with PT302, a long-acting Exendin-4 sustained release formulation, reduces dopaminergic neurodegeneration in a 6-Hydroxydopamine rat model of Parkinson’s disease. Sci. Rep 8, 10722. 10.1038/s41598-018-28449-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong JLY, de Pablo-Fernandez E, Foltynie T, Noyce AJ, 2020. The Association Between Type 2 Diabetes Mellitus and Parkinson’s Disease. J. Parkinsons. Dis 10, 775–789. 10.3233/jpd-191900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft S, Watson GS, 2004. Insulin and neurodegenerative disease: Shared and specific mechanisms. Lancet Neurol. 10.1016/S1474-4422(04)00681-7 [DOI] [PubMed] [Google Scholar]
- Cukierman-Yaffe T, Gerstein HC, Colhoun HM, Diaz R, García-Pérez LE, Lakshmanan M, Bethel A, Xavier D, Probstfield J, Riddle MC, Rydén L, Atisso CM, Hall S, Rao-Melacini P, Basile J, Cushman WC, Franek E, Keltai M, Lanas F, Leiter LA, Lopez-Jaramillo P, Pirags V, Pogosova N, Raubenheimer PJ, Shaw JE, Sheu WHH, Temelkova-Kurktschiev T, 2020. Effect of dulaglutide on cognitive impairment in type 2 diabetes: an exploratory analysis of the REWIND trial. Lancet Neurol. 19, 582–590. 10.1016/S1474-4422(20)30173-3 [DOI] [PubMed] [Google Scholar]
- Day SM, Yang W, Ewin S, Zhou X, Ma T, 2017. Glucagon-like peptide-1 cleavage product GLP-1 (9–36) amide enhances hippocampal long-term synaptic plasticity in correlation with suppression of Kv4.2 expression and eEF2 phosphorylation. Hippocampus 27, 1264–1274. 10.1002/hipo.22795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day SM, Yang W, Wang X, Stern JE, Zhou X, Macauley SL, Ma T, 2019. Glucagon-Like Peptide-1 Cleavage Product Improves Cognitive Function in a Mouse Model of Down Syndrome. eNeuro 6. 10.1523/ENEURO.0031-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon CF, 2019. Physiology and Pharmacology of DPP-4 in Glucose Homeostasis and the Treatment of Type 2 Diabetes. Front. Endocrinol. (Lausanne) 10. 10.3389/fendo.2019.00080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon CF, 2004. Circulation and degradation of GIP and GLP-1. Horm. Metab. Res 10.1055/s-2004-826160 [DOI] [PubMed] [Google Scholar]
- Deacon CF, Plamboeck A, Møller S, Holst JJ, 2002. GLP-1-(9–36) amide reduces blood glucose in anesthetized pigs by a mechanism that does not involve insulin secretion. Am. J. Physiol. - Endocrinol. Metab 282, E873–E879. 10.1152/ajpendo.00452.2001 [DOI] [PubMed] [Google Scholar]
- Dello Russo C, Cappoli N, Coletta I, Mezzogori D, Paciello F, Pozzoli G, Navarra P, Battaglia A, 2018. The human microglial HMC3 cell line: Where do we stand? A systematic literature review. J. Neuroinflammation 10.1186/s12974-018-1288-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elahi D, Egan JM, Shannon RP, Meneilly GS, Khatri A, Habener JF, Andersen DK, 2008. GLP-1 (9–36) amide, cleavage product of GLP-1 (7–36) amide, is a glucoregulatory peptide. Obesity 16, 1501–1509. 10.1038/oby.2008.229 [DOI] [PubMed] [Google Scholar]
- Eng J, Kleinman WA, Singh L, Singh G, Raufman JP, 1992. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom: Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J. Biol. Chem 267, 7402–7405. 10.1007/s10529-011-0745-y [DOI] [PubMed] [Google Scholar]
- Gault VA, Hölscher C, 2008. GLP-1 agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta-amyloid. Eur. J. Pharmacol 587, 112–117. 10.1016/j.ejphar.2008.03.025 [DOI] [PubMed] [Google Scholar]
- Gejl M, Gjedde A, Egefjord L, Møller A, Hansen SB, Vang K, Rodell A, Brændgaard H, Gottrup H, Schacht A, Møller N, Brock B, Rungby J, 2016. In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: Randomized, placebo-controlled, double-blind clinical trial. Front. Aging Neurosci 8. 10.3389/fnagi.2016.00108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacco F, Du X, Carratu A, Gerfen GJ, D’Apolito M, Giardino I, Rasola A, Marin O, Divakaruni AS, Murphy AN, Shah MS, Brownlee M, 2015. GLP-1 cleavage product reverses persistent ROS generation after transient hyperglycemia by disrupting an ROS-generating feedback loop. Diabetes 64, 3273–3284. 10.2337/db15-0084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman CP, Perry TA, Furukawa K, Grieg NH, Egan JM, Mattson MP, 2003. Glucagon-like peptide 1 modulates calcium responses to glutamate and membrane depolarization in hippocampal neurons. J. Neurochem 87, 1137–1144. 10.1046/j.1471-4159.2003.02073.x [DOI] [PubMed] [Google Scholar]
- Glotfelty EJ, Delgado TE, Tovar-y-Romo LB, Luo Y, Hoffer BJ, Olson L, Karlsson TE, Mattson MP, Harvey BK, Tweedie D, Li Y, Greig NH, 2019. Incretin Mimetics as Rational Candidates for the Treatment of Traumatic Brain Injury. ACS Pharmacol. Transl. Sci 2, 66–91. 10.1021/acsptsci.9b00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glotfelty EJ, Olson L, Karlsson TE, Li Y, Greig NH, 2020. Glucagon-like peptide-1 (GLP-1)-based receptor agonists as a treatment for Parkinson’s disease. Expert Opin. Investig. Drugs 1–8. 10.1080/13543784.2020.1764534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig NH, Mattson MP, Perry T, Chan SL, Giordano T, Sambamurti K, Rogers JT, Ovadia H, Lahiri DK, 2004. New therapeutic strategies and drug candidates for neurodegenerative diseases: p53 and TNF-α inhibitors, and GLP-1 receptor agonists. Ann. N. Y. Acad. Sci 1035, 290–315. 10.1196/annals.1332.018 [DOI] [PubMed] [Google Scholar]
- Greig NH, Tweedie D, Rachmany L, Li Y, Rubovitch V, Schreiber S, Chiang Y-H, Hoffer BJ, Miller J, Lahiri DK, Sambamurti K, Becker RE, Pick CG, 2014. Incretin mimetics as pharmacologic tools to elucidate and as a new drug strategy to treat traumatic brain injury. Alzheimer’s Dement. 10, S62–75. 10.1016/j.jalz.2013.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielmi V, Sbraccia P, 2017. GLP-1 receptor independent pathways: emerging beneficial effects of GLP-1 breakdown products. Eat. Weight Disord 10.1007/s40519-016-0352-y [DOI] [PubMed] [Google Scholar]
- Guida C, Miranda C, Asterholm IW, Basco D, Benrick A, Chanclon B, Chibalina M, Harris M, Kellard J, McCulloch L, Real J, Rorsman N, Yeung HY, Reimann F, Shigeto M, Clark A, Thorens B, Rorsman P, Ladds G, Ramracheya R, 2020. GLP-1(9–36) mediates the glucagonostatic effect of GLP-1 by promiscuous activation of the glucagon receptor. bioRxiv 785667. 10.1101/785667 [DOI] [Google Scholar]
- Han F, Hou N, Liu Y, Huang N, Pan R, Zhang X, Mao E, Sun X, 2019. Liraglutide improves vascular dysfunction by regulating a cAMP-independent PKA-AMPK pathway in perivascular adipose tissue in obese mice. Biomed. Pharmacother. 120, 109537. 10.1016/j.biopha.2019.109537 [DOI] [PubMed] [Google Scholar]
- Hansen L, Deacon CF, Ørskov C, Holst JJ, 1999. Glucagon-like peptide-1-(7–36)amide is transformed to glucagon-like peptide-1-(9–36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology 140, 5356–5363. 10.1210/endo.140.11.7143 [DOI] [PubMed] [Google Scholar]
- Hogg E, Athreya K, Basile C, Tan EE, Kaminski J, Tagliati M, 2018. High prevalence of undiagnosed insulin resistance in non-diabetic subjects with Parkinson’s disease. J. Parkinsons. Dis 8, 259–265. 10.3233/JPD-181305 [DOI] [PubMed] [Google Scholar]
- Hölscher C, 2020. Brain insulin resistance: role in neurodegenerative disease and potential for targeting. Expert Opin. Investig. Drugs 10.1080/13543784.2020.1738383 [DOI] [PubMed] [Google Scholar]
- Holst JJ, Ørskov C, 2004. The incretin approach for diabetes treatment: Modulation of islet hormone release by GLP-1 agonism, in: Diabetes. American Diabetes Association, pp. S197–S204. 10.2337/diabetes.53.suppl_3.S197 [DOI] [PubMed] [Google Scholar]
- Hou J, Manaenko A, Hakon J, Hansen-Schwartz J, Tang J, Zhang JH, 2012. Liraglutide, a long-acting GLP-1 mimetic, and its metabolite attenuate inflammation after intracerebral hemorrhage. J. Cereb. Blood Flow Metab 32, 2201–2210. 10.1038/jcbfm.2012.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liu Y, Cheng L, Li J, Zhang T, Zhao G, Zhang H, 2020. Glucagon-like peptide-1 cleavage product GLP-1(9–36) reduces neuroinflammation from stroke via the activation of insulin-like growth factor 1 receptor in astrocytes. Eur. J. Pharmacol 887, 173581. 10.1016/j.ejphar.2020.173581 [DOI] [PubMed] [Google Scholar]
- Janabi N, Peudenier S, Héron B, Ng KH, Tardieu M, 1995. Establishment of human microglial cell lines after transfection of primary cultures of embryonic microglial cells with the SV40 large T antigen. Neurosci. Lett 195, 105–108. 10.1016/0304-3940(94)11792-H [DOI] [PubMed] [Google Scholar]
- Kim DS, Choi H-I, Wang Y, Luo Y, Hoffer BJ, Greig NH, 2017. A New Treatment Strategy for Parkinson’s Disease through the Gut-Brain Axis: The Glucagon-Like Peptide-1 Receptor Pathway. Cell Transplant. 26, 1560–1571. 10.1177/0963689717721234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen LB, Hastrup S, Underwood CR, Wulff BS, Fleckner J, 2012. Functional importance of GLP-1 receptor species and expression levels in cell lines. Regul. Pept 175, 21–29. 10.1016/j.regpep.2011.12.006 [DOI] [PubMed] [Google Scholar]
- Knudsen LB, Pridal L, 1996. Glucagon-like peptide-1-(9–36) amide is a major metabolite of glucagon-like peptide-1-(7–36) amide after in vivo administration to dogs, and it acts as an antagonist on the pancreatic receptor. Eur. J. Pharmacol 318, 429–435. 10.1016/S0014-2999(96)00795-9 [DOI] [PubMed] [Google Scholar]
- Kritis AA, Stamoula EG, Paniskaki KA, Vavilis TD, 2015. Researching glutamate - induced cytotoxicity in different cell lines: a comparative/collective analysis/study. Front Cell Neurosci. 9:91. 10.3389/fncel.2015.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuc RE, Maguire JJ, Siew K, Patel S, Derksen DR, Margaret Jackson V, O’Shaughnessey KM, Davenport AP, 2014. Characterization of [125I]GLP-1(9–36), a novel radiolabeled analog of the major metabolite of glucagon-like peptide 1 to a receptor distinct from GLP1-R and function of the peptide in murine aorta. Life Sci. 102, 134–138. 10.1016/j.lfs.2014.03.011 [DOI] [PubMed] [Google Scholar]
- Lepiarz I, Olajide O, 2019. The human microglia (HMC-3) as a cellular model of neuroinflammation. IBRO Reports 6, S92. 10.1016/j.ibror.2019.07.299 [DOI] [Google Scholar]
- Li N, Lu J, Willars GB, 2012. Allosteric Modulation of the Activity of the Glucagon-like Peptide-1 (GLP-1) Metabolite GLP-1 9–36 Amide at the GLP-1 Receptor. PLoS One 7, e47936. 10.1371/journal.pone.0047936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Jiao JJ, Hölscher C, Wu MN, Zhang J, Tong JQ, Dong XF, Qu XS, Cao Y, Cai HY, Su Q, Qi JS, 2018. A novel GLP-1/GIP/Gcg triagonist reduces cognitive deficits and pathology in the 3xTg mouse model of Alzheimer’s disease. Hippocampus 28, 358–372. 10.1002/hipo.22837 [DOI] [PubMed] [Google Scholar]
- Li Y, Duffy KB, Ottinger MA, Ray B, Bailey JA, Holloway HW, Tweedie D, Perry T, Mattson MP, Kapogiannis D, Sambamurti K, Lahiri DK, Greig NH, 2010a. GLP-1 receptor stimulation reduces amyloid-β peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J. Alzheimer’s Dis 19, 1205–1219. 10.3233/JAD-2010-1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Glotfelty EJ, Namdar I, Tweedie D, Olson L, Hoffer BJ, DiMarchi RD, Pick CG, Greig NH, 2020. Neurotrophic and neuroprotective effects of a monomeric GLP-1/GIP/Gcg receptor triagonist in cellular and rodent models of mild traumatic brain injury. Exp. Neurol 324, 113113. 10.1016/j.expneurol.2019.113113 [DOI] [PubMed] [Google Scholar]
- Li Y, Perry T, Kindy MS, Harvey BK, Tweedie D, Holloway HW, Powers K, Shen H, Egan JM, Sambamurti K, Brossi A, Lahiri DK, Mattson MP, Hoffer BJ, Wang Y, Greig NH, 2009. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc. Natl. Acad. Sci 106, 1285–90. 10.1073/pnas.0806720106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tweedie D, Mattson MP, Holloway HW, Greig NH, 2010b. Enhancing the GLP-1 receptor signaling pathway leads to proliferation and neuroprotection in human neuroblastoma cells. J. Neurochem 113, 1621–31. 10.1111/j.1471-4159.2010.06731.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wu KJ, Yu SJ, Tamargo IA, Wang Y, Greig NH, 2017. Neurotrophic and neuroprotective effects of oxyntomodulin in neuronal cells and a rat model of stroke. Exp. Neurol 288, 104–113. 10.1016/j.expneurol.2016.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA, 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Stanojevic V, Brindamour LJ, Habener JF, 2012. GLP1-derived nonapeptide GLP1(28–36)amide protects pancreatic beta;-cells from glucolipotoxicity. J. Endocrinol 213, 143–154. 10.1530/JOE-11-0328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Du X, Pick JE, Sui G, Brownlee M, Klann E, 2012. Glucagon-like peptide-1 cleavage product GLP-1(9–36) amide rescues synaptic plasticity and memory deficits in Alzheimer’s disease model mice. J. Neurosci 32, 13701–13708. 10.1523/JNEUROSCI.2107-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher P, Schubert D, 2000. Signaling by reactive oxygen species in the nervous system. Cell. Mol. Life Sci. 10.1007/PL00000766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra K, Katsanos AH, Lambadiari V, Goyal N, Palaiodimou L, Kosmidou M, Krogias C, Alexandrov AV, Tsivgoulis G, 2020. GLP-1 receptor agonists in diabetes for stroke prevention: a systematic review and meta-analysis. J. Neurol 1, 3. 10.1007/s00415-020-09813-4 [DOI] [PubMed] [Google Scholar]
- McCarthy RC, Lu DY, Alkhateeb A, Gardeck AM, Lee CH, Wessling-Resnick M, 2016. Characterization of a novel adult murine immortalized microglial cell line and its activation by amyloid-beta. J. Neuroinflammation 13, 21. 10.1186/s12974-016-0484-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JJ, Gethmann A, Nauck MA, Götze O, Schmitz F, Deacon CF, Gallwitz B, Schmidt WE, Holst JJ, 2006. The glucagon-like peptide-1 metabolite GLP-1-(9–36) amide reduces postprandial glycemia independently of gastric emptying and insulin secretion in humans. Am. J. Physiol. - Endocrinol. Metab 290, E1118–E1123. 10.1152/ajpendo.00576.2005 [DOI] [PubMed] [Google Scholar]
- Mullins RJ, Mustapic M, Chia CW, Carlson O, Gulyani S, Tran J, Li Y, Mattson MP, Resnick S, Egan JM, Greig NH, Kapogiannis D, 2019. A Pilot Study of Exenatide Actions in Alzheimer’s Disease. Curr. Alzheimer Res 16, 741–752. 10.2174/1567205016666190913155950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W, 1993. Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type- 2 diabetes mellitus. J. Clin. Invest 91, 301–307. 10.1172/JCI116186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauck MA, Homberger E, Siegel EG, Allen RC, Eaton RP, Ebert R, Creutzfeldt W, 1986. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J. Clin. Endocrinol. Metab 63, 492–498. 10.1210/jcem-63-2-492 [DOI] [PubMed] [Google Scholar]
- Nicolini G, Miloso M, Zoia C, Di Silvestro A, Cavaletti G, Tredici G, 1998. Retinoic acid differentiated SH-SY5Y human neuroblastoma cells: An in vitro model to assess drug neurotoxicity. Anticancer Res. 18, 2477–2481. [PubMed] [Google Scholar]
- Panagaki T, Michael M, Hölscher C, 2017. Liraglutide restores chronic ER stress, autophagy impairments and apoptotic signalling in SH-SY5Y cells. Sci. Rep 7, 1–16. 10.1038/s41598-017-16488-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry T, Haughey NJ, Mattson MP, Egan JM, Greig NH, 2002a. Protection and Reversal of Excitotoxic Neuronal Damage by Glucagon-Like Peptide-1 and Exendin-4. J. Pharmacol. Exp. Ther 302, 881–888. 10.1124/jpet.102.037481 [DOI] [PubMed] [Google Scholar]
- Perry T, Lahiri DK, Chen D, Zhou J, Shaw KTY, Egan JM, Greig NH, 2002b. A novel neurotrophic property of glucagon-like peptide 1: a promoter of nerve growth factor-mediated differentiation in PC12 cells. J. Pharmacol. Exp. Ther 300, 958–66. 10.1124/jpet.300.3.958 [DOI] [PubMed] [Google Scholar]
- Perry T, Lahiri DK, Sambamurti K, Chen D, Mattson MP, Egan JM, Greig NH, 2003. Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (Abeta) levels and protects hippocampal neurons from death induced by Abeta and iron. J. Neurosci. Res 72, 603–612. 10.1002/jnr.10611 [DOI] [PubMed] [Google Scholar]
- Perry TA, Greig NH, 2003. The glucagon-like peptides: A double-edged therapeutic sword? Trends Pharmacol. Sci 10.1016/S0165-6147(03)00160-3 [DOI] [PubMed] [Google Scholar]
- Pyke C, Heller RS, Kirk RK, Ørskov C, Reedtz-Runge S, Kaastrup P, Hvelplund A, Bardram L, Calatayud D, Knudsen LB, 2014. GLP-1 receptor localization in monkey and human tissue: Novel distribution revealed with extensively validated monoclonal antibody. Endocrinology 155, 1280–1290. 10.1210/en.2013-1934 [DOI] [PubMed] [Google Scholar]
- Rai MA, Hammonds J, Pujato M, Mayhew C, Roskin K, Spearman P, 2020. Comparative analysis of human microglial models for studies of HIV replication and pathogenesis. Retrovirology 17, 35. 10.1186/s12977-020-00544-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards P, Parker HE, Adriaenssens AE, Hodgson JM, Cork SC, Trapp S, Gribble FM, Reimann F, 2014. Identification and characterization of GLP-1 receptor-expressing cells using a new transgenic mouse model. Diabetes 63, 1224–33. 10.2337/db13-1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolin B, Deacon CF, Carr RD, Ahrén B, 2004. The major glucagon-like peptide-1 metabolite, GLP-1-(9–36)-amide, does not affect glucose or insulin levels in mice. Eur. J. Pharmacol 494, 283–288. 10.1016/j.ejphar.2004.05.013 [DOI] [PubMed] [Google Scholar]
- Rowlands J, Heng J, Newsholme P, Carlessi R, 2018. Pleiotropic Effects of GLP-1 and Analogs on Cell Signaling, Metabolism, and Function. Front. Endocrinol. (Lausanne) 9, 672. 10.3389/fendo.2018.00672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo I, Tweedie D, Li Y, Greig NH, 2012. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: An emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br. J. Pharmacol 10.1111/j.1476-5381.2012.01971.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim S, 2017. Oxidative stress and the central nervous system. J. Pharmacol. Exp. Ther 10.1124/jpet.116.237503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaughness M, Acs D, Brabazon F, Hockenbury N, Byrnes KR, 2020. Role of Insulin in Neurotrauma and Neurodegeneration: A Review. Front. Neurosci 10.3389/fnins.2020.547175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipley MM, Mangold CA, Szpara ML, 2016. Differentiation of the SH-SY5Y human neuroblastoma cell line. J. Vis. Exp 2016. 10.3791/53193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z-W, Zhang L, Zhu S-J, Chen W-C, Mei B, 2010. Excitotoxicity effects of glutamate on human neuroblastoma SH-SY5Y cells via oxidative damage. Neurosci. Bull 26, 8–16. 10.1007/s12264-010-0813-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Wirdefeldt K, Yin L, Fang F, Markaki I, Efendic S, Ludvigsson JF, 2016. Reduced incidence of Parkinson’s disease after dipeptidyl peptidase-4 inhibitors—A nationwide case-control study. Mov. Disord 10.1002/mds.26734 [DOI] [PubMed] [Google Scholar]
- Tai J, Liu W, Li Y, Li L, Hölscher C, 2018. Neuroprotective effects of a triple GLP-1/GIP/glucagon receptor agonist in the APP/PS1 transgenic mouse model of Alzheimer’s disease. Brain Res. 1678, 64–74. 10.1016/j.brainres.2017.10.012 [DOI] [PubMed] [Google Scholar]
- Taylor S, Calder CJ, Albon J, Erichsen JT, Boulton ME, Morgan JE, 2011. Involvement of the CD200 receptor complex in microglia activation in experimental glaucoma. Exp. Eye Res 92, 338–343. 10.1016/j.exer.2011.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas E, Stanojevic V, Habener JF, 2010. GLP-1 (936) amide metabolite suppression of glucose production in isolated mouse hepatocytes. Horm. Metab. Res 42, 657–662. 10.1055/s-0030-1253421 [DOI] [PubMed] [Google Scholar]
- Wei R, Ma S, Wang C, Ke J, Yang J, Li W, Liu Y, Hou W, Feng X, Wang G, Hong T, 2016. Exenatide exerts direct protective effects on endothelial cells through the AMPK/Akt/eNOS pathway in a GLP-1 receptor-dependent manner. Am. J. Physiol. - Endocrinol. Metab 310, E947–E957. 10.1152/ajpendo.00400.2015 [DOI] [PubMed] [Google Scholar]
- Wootten D, Miller LJ, 2020. Structural basis for allosteric modulation of class b g protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol 10.1146/annurev-pharmtox-010919-023301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootten D, Savage EE, Valant C, May LT, Sloop KW, Ficorilli J, Showalter AD, Willard FS, Christopoulos A, Sexton PM, 2012. Allosteric modulation of endogenous metabolites as an avenue for drug discovery. Mol. Pharmacol 82, 281–290. 10.1124/mol.112.079319 [DOI] [PubMed] [Google Scholar]
- Yu S-J, Chen S, Yang Y-Y, Glotfelty EJ, Jung J, Kim HK, Choi H-I, Choi D-S, Hoffer BJ, Greig NH, Wang Y, 2020. PT320, sustained release Exendin-4, mitigates L-DOPA-induced dyskinesia in a rat 6-hydroxydopamine model of Parkinson’s disease. Front. Neurosci 14. 10.3389/fnins.2020.00785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun SP, Kam TI, Panicker N, Kim Sangmin, Oh Y, Park JS, Kwon SH, Park YJ, Karuppagounder SS, Park H, Kim Sangjune, Oh N, Kim NA, Lee Saebom, Brahmachari S, Mao X, Lee JH, Kumar M, An D, Kang SU, Lee Y, Lee KC, Na DH, Kim D, Lee SH, Roschke VV, Liddelow SA, Mari Z, Barres BA, Dawson VL, Lee Seulki, Dawson TM, Ko HS, 2018. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med 24, 931–938. 10.1038/s41591-018-0051-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Wei, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang Wanqin, Zhou Y, Hong J-S, Zhang J, 2005. Aggregated α-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 19, 533–542. 10.1096/fj.04-2751com [DOI] [PubMed] [Google Scholar]
- Zhang YF, Chen YM, Li L, Hölscher C, 2015. Neuroprotective effects of (Val8)GLP-1-Glu-PAL in the MPTP Parkinson’s disease mouse model. Behav. Brain Res 293, 107–113. 10.1016/j.bbr.2015.07.021 [DOI] [PubMed] [Google Scholar]