

Abstract
Drugs that block N-methyl-D-aspartate receptors (NMDARs) suppress hippocampus-dependent memory formation; they also block long-term potentiation (LTP), a cellular model of learning and memory. However, the fractional block that is required to achieve these effects is unknown. Here, we measured the dose-dependent suppression of contextual memory in vivo by systemic administration of the competitive antagonist (R,S)-3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP); in parallel, we measured the concentration-dependent block by CPP of NMDAR-mediated synapses and LTP of excitatory synapses in hippocampal brain slices in vitro. We found that the dose of CPP that suppresses contextual memory in vivo (EC50 = 2.3 mg/kg) corresponds to a free concentration of 53 nM. Surprisingly, applying this concentration of CPP to hippocampal brain slices had no effect on the NMDAR component of evoked field excitatory postsynaptic potentials (fEPSPNMDA), or on LTP. Rather, the IC50 for blocking the fEPSPNMDA was 434 nM, and for blocking LTP was 361 nM – both nearly an order of magnitude higher. We conclude that memory impairment produced by systemically administered CPP is not due primarily to its blockade of NMDARs on hippocampal pyramidal neurons. Rather, systemic CPP suppresses memory formation by actions elsewhere in the memory-encoding circuitry.
Keywords: NMDA receptors; Contextual Fear Conditioning; (R,S)-3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP); Long Term Potentiation (LTP)
Graphical Abstract
1. Introduction
N-methyl-D-aspartate receptors (NMDARs) are glutamate-activated, cation-permeable, ionotropic receptors that contribute to excitatory synaptic transmission throughout the nervous system (Hansen et al., 2017; Traynelis et al., 2010). They play essential roles in normal nervous system development and higher cognitive functions – most notably in controlling the synaptic strength modifications that occur during short-term and long-term memory formation (Bliss and Collingridge, 2013; Cole et al., 1993; Collingridge et al., 1983b; Morris et al., 1986; Nicoll, 2017). They have also been implicated in a wide variety of disorders, including Alzheimer’s disease, Parkinson’s disease, autism spectrum disorders, epilepsy, chronic pain, and depression (Adell, 2020; Bliss et al., 2014; Hallett and Standaert, 2004; Hanada, 2020; Morsy and Trippier, 2019; Pagano et al., 2021; Petrenko et al., 2003).
Many of the studies that established the functional contributions of NMDARs employed drugs that block these receptors relatively selectively. One such compound, first introduced in 1986 (Davies et al., 1986) and still used extensively (Abraham and Mason, 1988; Bergeron and Rompré, 2013; Eger et al., 2006; Fung et al., 2016; Ge et al., 2010; Gemperline et al., 2014; Kentros et al., 1998; Kouvaros et al., 2015; McDonald et al., 2005; Medvedev et al., 2010; Neyama et al., 2020; Villarreal et al., 2007), is the competitive antagonist (R,S)-3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP). CPP was derived from the widely used compound, 2-amino-5-phosphonopentanoic acid (AP5, also known as APV), but it is approximately five times as potent (Davies et al., 1986; Lehmann et al., 1987). CPP has proven to be exceptionally useful in experimental studies, because – unlike AP5 – it crosses the blood-brain barrier. Therefore, it can be administered systemically (typically IP or IV) (Lehmann et al., 1987). In addition, it is water soluble, so it is readily used for in vitro studies of expressed receptors or brain slices (Harris et al., 1986).
Because of its advantageous characteristics, we sought to use CPP as a receptor-specific standard to assess the relevance of NMDAR block to memory suppression by general anesthetics. Many anesthetic agents block NMDAR-mediated synaptic transmission (Petrenko et al., 2014). Some directly act on postsynaptic receptors, including the dissociative anesthetics phencyclidine and ketamine (MacDonald et al., 1991), the anesthetic gasses nitrous oxide and xenon (de Sousa et al., 2000; Jevtovic-Todorovic et al., 1998), and the volatile hydrocarbon cyclopropane (Ogata et al., 2006). Others reduce neurotransmitter release, such as the halogenated hydrocarbons halothane and isoflurane (Hemmings et al., 2005; Perouansky et al., 1995; Zimin et al., 2018), or act in both ways (Kirson et al., 1998; MacIver and Roth, 1988). Importantly, however, clinically relevant anesthetic concentrations block NMDAR synapses only partially, and many general anesthetics also modulate other CNS targets, such as GABAA receptors. These other effects may also contribute to anesthetic-induced amnesia.
In this study we sought to establish the quantitative relationship between partial block of NMDARs and its effects on memory and on long-term potentiation (LTP), an in vitro model of learning and memory. We measured the dose-dependent effects of intraperitoneal administration of CPP on contextual fear conditioning (CFC), a hippocampus-dependent learning paradigm, and the effects of corresponding CPP concentrations on NMDAR synaptic function and LTP in hippocampal brain slices. Surprisingly, at the concentration corresponding to its EC50-CFC in vivo (53 nM), CPP did not measurably suppress NMDARs or LTP. Instead, the EC50 concentrations that blocked the fEPSPNMDA and LTP in hippocampal brain slices were 434 nM and 361 nM respectively – nearly an order of magnitude higher. These results indicate that CPP does not suppress hippocampus-dependent memory formation by blocking NMDARs on CA1 pyramidal neurons, but by acting at other sites (most likely NR2A subunit-expressing NMDARs) elsewhere in the memory-encoding circuitry.
2. Materials and Methods
All studies were conducted following institutional IACUC approval (behavioral studies at University of California San Francisco, and brain slices studies at the University of Wisconsin-Madison) in accordance with the National Institutes for Health (NIH) guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978).
2.1. Contextual Fear Conditioning (CFC)
The effect of CPP on learning and memory was assessed using contextual fear conditioning, a widely employed hippocampus-dependent task (Kim and Fanselow, 1992; Phillips and LeDoux, 1992). In this task, mice are trained to associate an aversive event (e.g., a shock) with the context in which it is given. The “context” is the chamber in which the mouse receives the shock; it can include visual, auditory, olfactory and tactile cues. After presentation of the shock in the training context, the mouse learns that this context predicts shock. It will exhibit its innate fear response – immobility, or “freezing” – when returned to the training context (in the absence of shock). Quantifying freezing behavior during this context test assesses the memory of the association between the context and shock.
Experiments were conducted on a total of 55 three- to four-week-old male hybrid C57BL/6J x 129SF2/J mice (Jackson Laboratories, Bar Harbor, ME). The mice were housed five per cage (26 cm long x 13 cm wide x 16 cm high), and food and water were available ad libitum. The housing area was kept on a 12:12-hr light-dark cycle, with lights coming on at 6:00 am, and all experimental procedures took place during the light cycle. Seven to 8 mice were assessed at 0, 1, 2, 2.5, 3, and 9 mg/kg intraperitoneal CPP (Tocris Bioscience, Bio-Techne Corp., Minneapolis, MN, USA). The CPP was dissolved in sterile saline and administered at a volume of 10 ml/kg body weight. Sixty minutes after CPP injection, groups of four mice at a time were transferred to fear conditioning chambers (32 cm L x 25 cm W x 25 cm H) constructed of clear acrylic. The grid floor used to deliver shock was composed of 19 stainless steel bars, each 4 mm in diameter, spaced 16 mm center to center. These floors were connected to a shock delivery system (Med Associates; St. Albans, VT). The chambers were wiped down with a pine-scented cleaner (5% Pine Scented Disinfectant, Midland, Inc.; Sweetwater, TN) before and after each session. The overhead fluorescent bulbs were left on and a ventilation fan provided background noise (65 db) in the training room. After a 3-min baseline period, mice received a series of three shocks (1 mA, 2 s) separated by 64 seconds (with the exception of one no-shock control group). After the last shock, mice remained in the chambers for an additional 64 seconds. The following day, mice were placed back in the training chambers and observed for freezing behavior. Each animal’s behavior was scored every 8 seconds during the 8-min test period, and a percentage of freezing was calculated by dividing the number of freezing observations a mouse had by the total number possible during the observation period and multiplying by 100.
2.2. Motor Activity
To assess the effect of CPP on motor activity, we monitored the number of crossovers during the 3-min period before tone-shock pairing on the conditioning day. Crossovers were scored from videotape by blinded observers and defined as full crossing of both the fore and hind paws across the midpoint of each cage (designated by a black vertical stripe placed on the video monitors).
2.3. Brain CPP Concentrations by UPLC –MS/MS
To determine the CPP concentration present in brain slices after incubation in CPP solution, we incubated 400 μm-thick coronal brain slices in beakers containing artificial cerebrospinal fluid (aCSF) plus CPP (50 nM, 150 nM, 500 nM, or 10 μM). Beakers were set upon a platform that maintained a gentle rocking motion (“Belly Dancer”) so that slices would remain suspended in solution, for durations ranging from 1-120 minutes. Immediately upon removal of brain slices from the incubation beakers they were placed onto filter paper and wicked dry with a Kimwipe, then weighed. Groups of 10 brain slices incubated under identical conditions were combined and homogenized in glass test tubes with 2 ml 0.01N HCl using a rotor-stator homogenizer. The brain homogenate was transferred to 15 ml centrifuge tubes and centrifuged at 15,000 g for 25 minutes at 4°C. The supernatant was transferred to a fresh 15 ml conical polypropylene tube. The pelleted tissue was resuspended in 2 mL 0.01N HCl and centrifuged for a second time at 15,000 g for 25 minutes at 4°C. The supernatant from the second spin was combined with the supernatant from the first spin. 3.5 mL 5% NH4OH (v/v) was then added to basify the solution prior to solid-phase extraction (SPE). SPE was performed using Oasis MAX SPE cartridges from Waters (Milford, MA). The SPE cartridge was conditioned with methanol, equilibrated with water, and then the basified sample was loaded. The cartridge was washed with 5% NH4OH and then washed with methanol. Analyte was eluted with 8% formic acid/77% methanol (v/v). Samples were resuspended in 50 μL of 20 mM HFBA in 99:1 water:ACN . Samples were then vortexed, spun down in a centrifuge for 10 s and transferred to HPLC vials. Quantitation of CPP from brain tissue slices was carried out as previously described. (Gemperline et al., 2014) Briefly, 7 μL of the sample was injected onto a Waters Acquity UPLC BEH C18 column (100 x 2.1 mm ID, 1.7 μm) using 20 nM HFBA in water and 20 mM HFBA in acetonitrile as mobile phase A and B respectively. The following gradient was used to separate the analyte and internal standard (IS) (time/minute, % mobile phase B): (0, 1), (4, 6), (4.1, 95), (5.5, 95), (5.6, 1), (8, 1). The flow rate was set to 0.35 mL/min and the column and sample temperatures were maintained at 35 °C and 10 °C respectively throughout the experiment. A Waters Acquity binary pump UPLC system (Milford, MA) interfaced to an AB Sciex QTrap 5500 (Framingham, MA) mass spectrometer with a Turbo V ™ source was used for LC-MS/MS acquisition. The mass spectrometer was operated in positive ion ESI mode and multiple reaction monitoring (MRM) mode was utilized to detect the precursor to product ions used for quantitation of CPP and IS: m/z 252.958→207.100 and 334.955→136.033, respectively. Isotopically-labeled cyclic adenosine monophosphate (13C5 cAMP) was used as the internal standard. MultiQuant 2.1 software from AB Sciex was used to generate calibration curves based on the peak area ratios of CPP and IS from blank tissue samples spiked with known amounts of CPP; these curves were used to back-calculate CPP concentration in the mouse brain slices.
2.4. Electrophysiology
Coronal 400 μm thick hippocampal slices were prepared from three- to four-week-old male and female F1 hybrid (C57BL/6J x 129SF2/J) mice (Jackson Laboratories, Bar Harbor, ME). Mice were anesthetized with 2.5% isoflurane (Novaplus, Hospira, Inc., Lake Forest, IL), then the brain was removed, blocked by removing the cerebellum and olfactory cortex, glued to a microtome slice tray with cyanoacrylate glue (Krazy Glue Instant, Westerville, OH), and placed for slicing in ice-cold cutting artificial cerebrospinal fluid containing (in mM) 124 NaCl, 3 KCl, 3.6 MgSO4, 0.6 CaCl2, 25 NaHCO3, 1.25 NaH2PO4, 1 ascorbic acid, 10 glucose, bubbled with 95% O2/5% CO2. 400 μm thick slices were cut using a vibratome (Leica VT 100S, Leica Microsystems Nussloch GmbH, Nussloch, Germany). Brain slices recovered in a holding chamber filled with carbogenated recording aCSF containing (in mM) 124 NaCl, 3 KCl, 25 NaHCO3, 1.25 NaH2PO4, 1.3 MgSO4, 2.5 CaCl2, 15 glucose, 1 ascorbic acid for 30 minutes at 32°C and remained in the recovery chamber at room temperature (20–22 °C) for at least 45 minutes before being transferred to the recording chamber.
Extracellular recordings were performed in a submerged chamber perfused by aCSF at a flow rate of 3.0 ml/min at 30±1°C. Recording electrodes were constructed from borosilicate glass pipettes with a filament (1.5 mm outer diameter × 0.86 mm inner diameter; Sutter Instruments), pulled using a horizontal puller (P-97; Sutter Instruments) to achieve a tip diameter of ~ 1 μm and filled with 1M NaCl, resulting in pipette resistances between 0.4-0.8 MΩ. Recording electrodes were placed in the stratum radiatum of CA1 hippocampus, approximately 50-100μm beneath the slice surface. Recordings were amplified using a differential amplifier, AC Microelectrode Amplifier Model 1800 (A-M Systems, Carlsborg, WA), digitized at 20 kHz using a NI USB-6341, X-series AD board (National Instruments, Austin, TX), and acquired using WinLTP software (Version 2.10; William W. Anderson, Bristol, England). Field EPSPs (fEPSPs) were evoked electrically by a bipolar tungsten stimulating electrode (0.5 MΩ; World Precision Instruments) placed in the stratum radiatum to activate Schaffer collateral fibers. Stimuli 0.1 ms in duration were delivered using a constant-current stimulus isolator (model A365D; World Precision Instruments) at 0.05 Hz using a stimulus intensity (“baseline”) adjusted to evoke responses below half-maximal fEPSP slope. Baseline stimulus amplitude ranged from 30–125 μA and was typically between 30 and 90 μA.
To measure CPP suppression of LTP, once a stable baseline response amplitude was achieved (defined as <10% change in fEPSP amplitude over 30 min), LTP was evoked using a theta burst stimulus (TBS) protocol consisting of three stimulus trains separated by 20 seconds. Each train consisted of 10 bursts of 4 pulses at 100 Hz, delivered with a 200ms train period, at baseline stimulus intensity. LTP was expressed as a percentage of the pre-tetanus baseline, calculated as the average fEPSP slope over the last 10 min of recording divided by the average fEPSP slope measured during the 10 min before TBS, times 100.
To measure CPP block of NMDARs, once a stable baseline response amplitude was achieved, NMDAR-mediated fEPSPs were isolated by switching to an aCSF perfusate containing 10 μM CNQX, 10 μM bicuculline, 3μM glycine, and 0.1 mM MgSO4. CPP was then added to the aCSF following stabilization of the fEPSPNMDA response. To demonstrate that the evoked fEPSPs were triggered by NMDARs, in a separate set of experiments we added the selective NMDAR antagonist (2R)-amino-5-phosphonovaleric acid, (AP5, 40 μM) to the low-Mg++ aCSF.
2.5. Statistical analysis
A one-way analysis of variance (ANOVA) was used to analyze freezing during the context test. Fisher protected least significance test (PLSD) was used for all planned group comparisons. An alpha level of less than 0.05 was accepted as significant. Statview statistical software (Berkeley, CA) was used to perform these analyses. The freezing response as a function of CPP dose was fit to a logistic equation (Hill equation) using the nonlinear curve fit tool in Graphpad Prism 9 for MACOS (GraphPad Software, Inc., San Diego, CA). Motor activity as a function of CPP dose was analyzed using the linear regression tool in Graphpad Prism 9.
For electrophysiological experiments, Graphpad Prism 9 was used to test for statistical significance between groups and for fitting concentration-response curve of the CPP block of the fEPSPNMDA. The independence of the CPP partition coefficient vs. concentration in aCSF was evaluated by calculating the Pearson correlation coefficient, using a two-tailed P-value to test for significance. Differences between LTP blockade at different CPP concentrations were tested using one-way ANOVA with Dunnett’s post-test and multiplicity-adjusted p-values. All values are presented as the mean ± SEM.
3. Results
3.1. Contextual fear conditioning
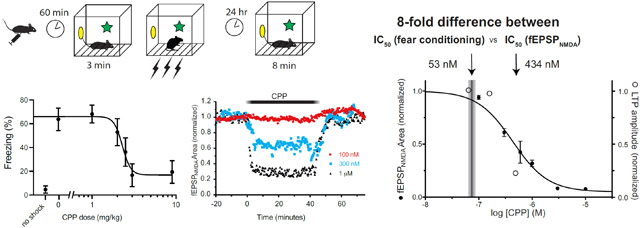
To assess the effect of CPP on contextual fear conditioning, we injected doses of 0 (saline control), 1, 2, 3, and 9 mg/kg IP to groups of 8 mice and placed them in the conditioning chamber one hour later. After three minutes we administered a series of three shocks and then returned them to their home cages. The next day we placed the mice back in the conditioning chamber measured their freezing behavior over 8 minutes (Fig. 1A). We found that CPP induced a dose-dependent suppression of freezing (Fig. 1C), with complete suppression (no difference compared to no-shock control) at 3 and 9 mg/kg (ANOVA: F(6,49) = 7.1546, p<0.0001), and no effect (compared to saline control) at 1 and 2 mg/kg. Upon seeing this steep dose-dependence, we added a group of 7 mice at a dose of 2.5 mg/kg. We found that this dose reduced freezing by approximately 50% compared to saline control (65% in the saline control group versus 36% in the 2.5 mg/kg group). Fitting the full dataset to a Hill equation yielded a IC50 dose of 2.3 mg/kg (95% CI: 1.5-2.9 mg/kg).
Fig. 1.

Effects of CPP on activity and contextual fear conditioning. A) Behavioral paradigm used to study contextual conditioning, in groups of 7-8 mice. B) Freezing scores during the context test on day 2. Increased freezing indicates better memory. CPP suppressed learning with an EC50 dose of 2.3 mg/kg IP. C) Exploratory activity during the first three minutes of the context exposure on day 1, as indicated by number of crossovers of a light beam.
3.2. Activity
To assess the effect of CPP on overall motor activity, we measured the number of crossovers in the activity grid after mice had been injected with 0 (saline control), 1, 2, 2.5, 3, and 9 mg/kg IP of CPP (Fig. 1B). All groups showed a similar level of activity during the observation period (approximately 15 to 20 crossovers). Linear regression analysis showed no significant difference between the slope of the line of best-fit and “0” (F(1,45) = 2.08, p=0.16), indicating that CPP did not dose-dependently alter motor activity.
3.3. Tissue concentrations of CPP in vivo and in vitro
To establish in vitro concentrations of CPP in aCSF that correspond to administration of amnestic doses in vivo, we performed a series of brain slice equilibration studies using a mass spectrometry-based method. (Gemperline et al., 2014) We conducted two sets of studies. For the first set, we incubated brain slices in aCSF containing 10 μM CPP, for durations up to two hours (Fig. 2A). A monoexponential fit of concentration vs. time yielded a time constant of 8.3 min, which is close to the time constant of equilibration for etomidate (9.5 min) that we reported previously, corresponding to a diffusion coefficient for CPP in brain slice tissue of ~0.2 x 10−6 cm2s−1 (Benkwitz et al., 2007). The final concentration of CPP in brain tissue was 1092 ng CPP/g tissue, or 3.9 μM CPP (using a brain slice density of 1.04), yielding a partition coefficient λtissue:aCSF = 0.39.
Figure 2.
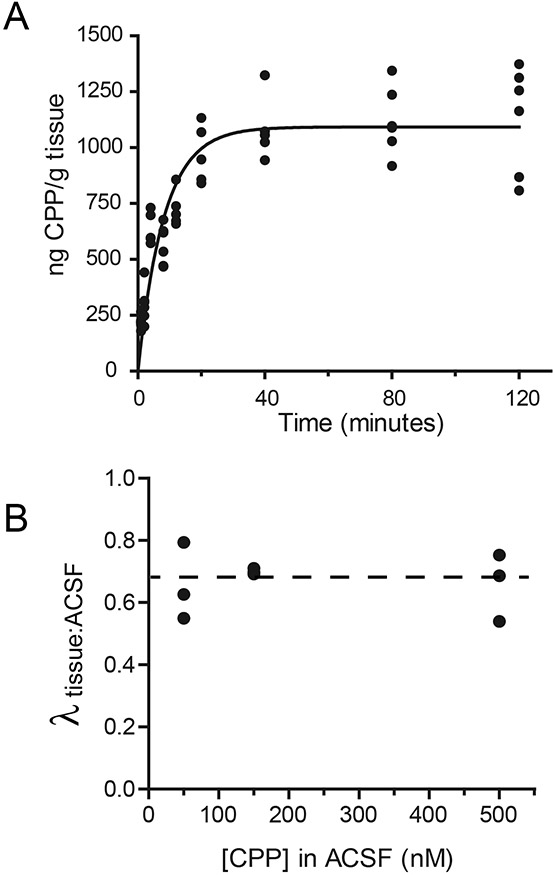
Determination of diffusion of CPP into brain slices, and partition coefficient between artificial cerebrospinal fluid (aCSF) and brain tissue. A) 400 μm-thick brain slices were incubated with gentle agitation for times ranging from 1-120 minutes. Following incubation, slices were dried, weighed, and total CPP content was determined using the same UPLC-MS/MS method used to measure CPP in brain tissue following in vivo administration. The solid line shows an exponential fit to the data, yielding τ=8.3 min. B) Relative concentrations of CPP in brain tissue (measured using incubation times of 80 minutes) versus aCSF was used to derive partition coefficients at different aCSF concentrations. The partition coefficient was independent of aCSF concentration.
Since the concentration of CPP in brain tissue produced by incubation in aCSF containing 10 μM CPP (1092 ng CPP / g tissue) far exceeded the brain concentration one hour after injection of 9 mg/kg IP (65 ng CPP / g tissue), (Gemperline et al., 2014) we conducted a second series of studies using lower concentrations of CPP in aCSF that would match more closely the brain tissue concentrations that correspond to amnestic doses. Thus, we incubated brain slices in aCSF containing 50, 150, and 500 nM CPP for 80 minutes (Fig. 2B). We found that λtissue:aCSF was independent of concentration over this range (Pearson’s correlation coefficient r = −0.2435; p = 0.8434), and averaged 0.67 ± 0.03 (Fig. 2B, dashed line).
Using this value (λtissue:aCSF = 0.67) together with the brain concentration 60 minutes after an IP injection of 3 mg/kg (16.8±3.8 ng CPP/g brain tissue; 95% CI 13.6-20) , and the IC50 (CFC) dose of 2.3 mg/kg (Fig. 1), we calculate the aCSF concentration in vitro that corresponds to a 50% effective dose of CPP in vivo as 53 nM (95%CI 18-67 nM).
3.4. Brain Slice Electrophysiology
To determine how strongly NMDARs on hippocampal pyramidal neurons are blocked at the concentration corresponding to IC50 (CFC), we measured the concentration-dependent effects of CPP on NMDAR-mediated field EPSPs (fEPSPNMDA) in hippocampal brain slices. fEPSPNMDA responses were isolated pharmacologically by including 10 μM CNQX, 10 μM bicuculline, 3μM glycine, and 0.1 mM MgSO4 in the recording aCSF. Examples of individual responses are shown in Fig. 3A. Extracellular potentials with a rapid onset, approximately −1 mV in amplitude and 50 ms in duration, were observed following single electrical stimuli of the Schaffer collateral pathway in stratum radiatum. Responses often had multiple small spikes superimposed on the negative deflection, likely reflecting the prolonged time course of NMDAR-mediated depolarization of pyramidal neurons and the lack of feedforward inhibition with AMPA and GABAAR antagonists in the recording aCSF. In separate experiments, responses were blocked nearly completely (95 ± 3%, mean ± SD, n=5) following addition of 40 μM AP5 to the aCSF, demonstrating that they were triggered by NMDARs (Fig. 4). Applying CPP at concentrations ranging from 100 nM to 10 μM reduced the fEPSPNMDA amplitude in a concentration-dependent manner, and the effect reversed rapidly upon washout of CPP (Fig. 3B). Higher concentrations of CPP also eliminated the superimposed spiking. A concentration-response curve of the CPP block of the fEPSPNMDA is shown in Fig. 3C. Non-linear curve fit of the data to the sigmoidal equation Y=100/(1+10^( Log10IC50-X)*HillSlope) yielded a mean log10IC50 = −6.36 ± 0.05, or IC50 = 434 nM, and a Hill slope of −1.3.
Figure 3.

Concentration-dependent block by CPP of the EPSPNMDA in vitro. A) Evoked field potentials were measured in the CA1 area of hippocampal brain slices, in the presence pharmacological blockers of AMPA (10 μM CNQX) and GABAA receptors (10 μM bicuculline), the NMDAR co-agonist glycine (3μM), and a low concentration of Mg++ (0.1 mM). Calibration bars 0.3 mV (300 nM) or 0.5 mV (100 nM, 1 μM), 50 mS. B) Time series plot of evoked responses during wash-in and washout of CPP. C) Summary of CPP block of at EPSPNMDA (filled circles, mean ± sem) at concentrations from 100 nM – 10 μM. n=2-5 per concentration. The shaded vertical bar shows the estimated CPP concentration corresponding to IC50 for fear conditioning to context in vivo. The open circles show relative block of long-term potentiation, for comparison (see Fig. 5 for details).
Figure 4.

Block of the EPSPNMDA by the NMDAR antagonist 2-amino-5-phosphonopentanoic acid (AP5) in vitro. A) Example of evoked field potentials measured in the CA1 area of hippocampal brain slices, in the presence pharmacological blockers of AMPA (10 μM CNQX) and GABAA receptors (10 μM bicuculline), the NMDAR co-agonist glycine (3μM), and a low concentration of Mg++ (0.1 mM), before and after addition of AP5. B) Time series plot of evoked responses during wash-in of AP5 (40 μM), normalized to the average response in the five minutes prior to addition of AP5 to the aCSF. Filled symbols and error bars represent mean ± SEM, n=5.
Surprisingly, this IC50 concentration for CPP block of the fEPSPNMDA (434 nM) is nearly an order of magnitude greater than the concentration that that blocked memory in vivo (60 nM). To test whether the conditions we used to isolate the fEPSPNMDA altered its sensitivity to CPP in a more functionally integrated environment, we tested the effects of CPP on LTP of the Schaffer collateral pathway – an NMDAR-dependent process (Collingridge et al., 1983a; Morris et al., 1986) – in brain slices maintained in standard aCSF (Fig. 5). Overall, there was a significant effect of CPP on LTP, as assessed 80-90 minutes after a theta-burst stimulus (aCSF control 127±5.1%, n=16; CPP 67 nM 132±6.1%, n=11; CPP 150 nM 122±3.9%, n=15; CPP 500 nM 106±2.2%, n=10; ordinary one-way ANOVA, F(3, 48) = 4.6, p = 0.0062). However, this suppression was significant only at the highest concentration tested (Dunnett’s multiple comparison test, DF=48, p>0.05 for CPP 67nM vs aCSF, p>0.05 for CPP 150nM vs aCSF, p<0.05 for CPP 500 nM vs aCSF). Fitting these data to a sigmoid equation, with Hill Coefficient fixed to 1.3 (not enough concentrations were tested to provide an accurate slope measure), yielded a mean log10IC50 = −6.44 ± 0.28, or IC50 = 361 nM. Thus, the degree of suppression of LTP approximately matches the fractional block of the isolated fEPSPNMDA (open circles on Fig 3C), indicating that the conditions used to isolate the fEPSPNMDA did not strongly influence its sensitivity to receptor block.
Figure 5.
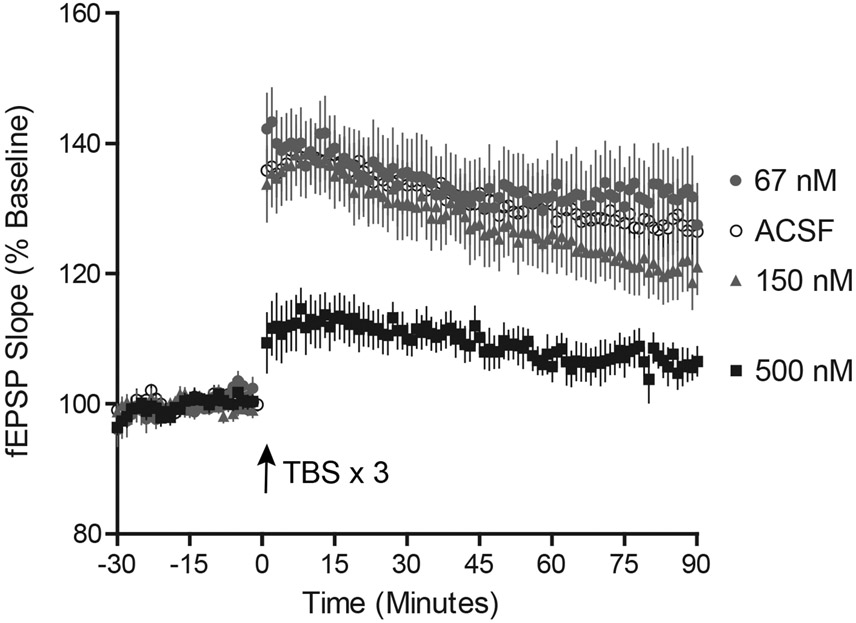
Suppression of long-term potentiation by CPP in vitro. Hippocampal brain slices (400 μm thick) were maintained in standard aCSF (n=16) or in the continuous presence of CPP (67 nM, n=11; 150 nM, n=15; 500 nM n=10). The slope of the evoked fEPSP (mean ± sem) was plotted as a function of time. Each point represents the mean ± sem. Three one-second trains of theta-burst stimuli were applied every 20 s at t=0. Only the concentration of 500 nM CPP suppressed the average amplitude over the last 10 min of the recording.
4. Discussion
We sought to establish the degree to which NMDA receptors must be blocked to impair memory formation. We obtained the surprising result that the concentration of CPP in vitro that corresponds to the IC50 dose that suppresses contextual fear conditioning in vivo does not measurably reduce the amplitude of the isolated evoked NMDAR-fEPSP, and it does not suppress LTP of the Schaffer collateral synapse. Below, we consider possible explanations for the mismatch between in vivo and in vitro effects of CPP.
4.1. Measurement errors or technical artefacts
Before considering the possible physiological explanations for the resistance of the NMDAR-fEPSP and LTP of the Schaffer collateral pathway to CPP, we first address whether some potential sources of measurement error or technical artefacts might have led to an underestimate of the effect of CPP in brain slice recordings.
4.1.1 The most obvious candidate is the method used to translate amnestic brain concentrations in vivo into equivalent aCSF concentrations in vitro. We established this relationship by measuring drug content in whole brain tissue when CPP was applied either by IP injection or by soaking brain slices in a known concentration. Since we used exactly the same extraction and mass spectrometry-based measurements for both, (Gemperline et al., 2014) it seems unlikely that a systematic error between preparations could have been introduced. We did use a high aCSF concentration initially to establish the partition coefficient in vitro, but we also adjusted the aCSF concentration to approximately match in vivo measurements, providing further confidence in this value.
4.1.2 A second source of technical artefact to consider is the timing of measurements. When extracting the brain following in vivo administration, we matched the time course used for fear conditioning experiments (one hour after IP administration), and our prior study showed that brain concentrations are approximately stable between 45, 60, and 75 minutes (Gemperline et al., 2014). Moreover, we first measured the equilibration rate in vitro and then used this information to assure full equilibration when we measured the partition coefficient. Therefore, errors due to unmatched or rapidly changing conditions in vivo or in vitro are unlikely.
4.1.3 Intravascular blood was present in the brain we harvested in vivo but it was not in vitro. Although the blood volume is only one-tenth that of neuronal tissue, plasma concentrations 60 minutes after IP injection are approximately ten times whole brain concentrations (Gemperline et al., 2014). We took care to minimize intravascular CPP by blocking the brain to exclude major blood vessels, but any ‘extra CPP’ that was included would mean that the tissue concentration is even lower than the value that we measured, further accentuating the mismatch. In addition, we measured whole-brain concentrations of CPP, but it is possible that distribution is not entirely uniform across the brain, whether due to differences in vascularization or blood brain barrier permeability, so that the concentration within the hippocampus might have been higher than elsewhere in the brain. By the same token, its rapid equilibration and the relatively stability of brain concentrations between 45-75 minutes following IP injection argues against this possibility.
4.1.4 The age and sexes of the mice used for the in vivo vs. in vivo studies were not perfectly matched. We used 3- to 4-week-old male mice for the in vivo studies, but 3- to 5-week-old mice of both sexes for in vitro studies. Although mice are sexually immature at these ages, pubertal changes are observed as early as postnatal day 25 (Mayer et al., 2010), and changes in inhibitory signaling (though not intrinsic excitability) occur during the estrus cycle (Piekarski et al., 2017). Thus, although it is unlikely that substantial differences in CPP sensitivity of LTP or fEPSPs vs. fear conditioning were imparted by this mismatch, the possibility cannot be excluded.
4.1.5 The experimental procedures used to conduct behavioral studies of memory versus brain slice measurements of fEPSPs and LTP might have influenced our results. For example, administration of isoflurane anesthesia prior to decapitation (Pekny et al., 2014), or changes in brain circuitry induced by brain slice preparation itself (Ting et al., 2014), might have influenced susceptibility to block by CPP. While the many possible changes are impossible to discount, we would note that brain slice physiology has been used widely for decades in investigations of cellular and network properties that underlie brain function (Collingridge, 1995), and it has been found to be generally reliable.
4.1.6 A final type of technical artefact to consider is whether the concentration of CPP required to block NMDARs in vitro could have been influenced by the method we used to measure NMDAR-fEPSPs. In particular, there is a complex interaction between the glycine site and the agonist site, wherein an agonist-induced decrease in glycine affinity leads to receptor desensitization, with a time course that matches that of NMDAR deactivation following a burst of afferent inputs (Mayer et al., 1989; Parsons et al., 1998; Parsons et al., 1993). If the affinity for the antagonist CPP is similarly influenced, then the concentration of glycine in the aCSF that we used could have displaced CPP compared to its steady-state receptor occupancy in vivo. However, it seems unlikely that this effect could be great enough to account for the observed mismatch, because additional glycine was not included in the aCSF when we measured suppression of LTP by CPP, and the concentrations required to block NMDAR-fEPSPs and LTP showed good correspondence (Figs 3,4). Similarly, the correspondence between NMDAR-fEPSP block and LTP suppression argues against other similar technical factors related to method for measuring NMDAR function, since NMDARs were operating in an approximately physiologically relevant context for the LTP experiments.
Based upon these considerations, we conclude that it is unlikely that measurement errors or technical artefacts caused the NMDAR-fEPSP or LTP to resist CPP.
4.2. Physiological factors
An alternative explanation is that CPP acts preferentially on NMDARs that incorporate GluN2A subunits (GluN2A-NMDARs), but that these receptors represent only a minority of receptors in hippocampal pyramidal neurons. NMDARs are made up of four subunits: two obligatory GluN1 subunits, which bind the co-agonist glycine, and two GluN2 subunits, which form the binding sites for glutamate. There are four types of GluN2 subunits, and they differ substantially in their physiological properties and pharmacological sensitivities. In the forebrain, most receptors incorporate GluN2A and/or GluN2B. It has been known for many years that a developmental switch from GluN2B to GluN2A occurs in the early postnatal period, coincident with use-dependent changes that occur during development. However, both subunits remain present in many or most brain regions, and in adult rodents, more than half of all hippocampal and cortical NDMARs exist as triheteromeric GluN1/Glun2A/GluN2B NMDARs (Rauner and Kohr, 2011). The expression levels of GluN2A and GluN2B subunits are regulated by distinct but highly coordinated biochemical mechanisms (Kellermayer et al., 2018), and rapid changes in their postsynaptic localization can occur, modifying susceptibility to synaptic plasticity and memory (Bellone and Nicoll, 2007; Dupuis et al., 2014).
Although CPP is not as highly selective as the GluN2B antagonist ifenprodil and some of its derivatives (Neyton and Paoletti, 2006), it does exhibit some degree of GluN2 subunit-dependent block (Feng et al., 2005; Lozovaya et al., 2004), with the rank order of potency GluN2A (EC50 41 nM) > GluN2B (270 nM) > GluN2C (630 nM) > GluN2D (1.9 μM). Our finding that CPP blocked CFC with an EC50 concentration of 53 nM thus suggest that CPP acts in vivo by targeting GluN2A-NMDARs, whereas in vitro it blocked fEPSPs and LTP through actions on GluN2B-NMDARs or triheteromeric GluN1/GluN2A/GluN2B receptors. The concentration-dependence of CPP block of triheteromeric receptors has not been established; however, based on the finding that the sensitivity of N1/N2A/N2B triheteromers to block by NVP-AAM077 (Auberson et al., 2002), another GluN2A-preferring competitive antagonist, falls between the diheteromeric 2A vs. 2B receptors (Tovar et al., 2013), suggest that they would similarly display intermediate sensitivity.
If CPP does indeed block contextual fear conditioning primarily through GluN2A-NMDARs, where might the instrumental receptors be located?
4.2.1. Extrahippocampal sites?
Although the contextual learning paradigm that we used is known to engage the hippocampus (Kim and Fanselow, 1992; Moita et al., 2004; Phillips and LeDoux, 1992), it does also engage the amygdala to encode associations between the context and foot shock (Kim and Cho, 2020; Phillips and LeDoux, 1992; Wahlstrom et al., 2018). Some circuits in the amygdala continue to express GluN2A-NMDARs even into adulthood (Aroniadou-Anderjaska et al., 2018). Therefore, since both hippocampus and amygdala would have been subject to CPP modulation during the acquisition phase of CFC, it is possible that disrupted GluN2A-dependent signaling in amygdala was responsible for the reduced freezing response. Along the same vein, other brain regions, including the striatum (Ikegami et al., 2014) and medial prefrontal cortex (Giustino and Maren, 2015), are engaged during contextual fear conditioning, so CPP disruption of NMDAR signaling at any of these sites might plausibly have contributed to its behavioral effects.
4.2.2. DG or CA3?
A second possibility is that CPP targets GluN2A-NMDARs that are located elsewhere in the in hippocampus, such as the dentate gyrus (DG) or CA3 region. Arguing against this possibility are the many studies showing that that the CFC paradigm we used is sensitive to manipulation of CA1 circuitry (Daumas et al., 2005; Jimenez et al., 2020; Lee and Kesner, 2004), whereas the DG becomes most strongly engaged in that tasks that entail pattern separation (McHugh et al., 2007). Moreover, targeted deletion of NMDARs in DG granule cells does not impair spatial memory formation (Bannerman et al., 2012). Taken together, these findings indicate that selective suppression of the DG by CPP is unlikely. Similarly, the CA3 region does become engaged in contextual learning (Hunsaker et al., 2009), but its modulation most strongly impacts single-trial learning tasks (Lee and Kesner, 2004). Nevertheless, selective action of CPP within the CA3 circuit does remain a possibility.
4.2.3. Hippocampal interneurons?
A third possibility, and the one that appears to us to be most likely, is that the critical GluN2A-NMDRs are embedded within the interneuron circuits that support and control memory. NMDARs play multiple and critical roles in interneurons, both during development and throughout adulthood (Pelkey et al., 2017). They serve varied roles, such as control of spike timing during network oscillations (Klausberger and Somogyi, 2008; Maccaferri and Dingledine, 2002) and supporting plasticity in interneurons themselves (Pelletier and Lacaille, 2008). Some interneurons, such as neurogliaform cells, rely heavily on NMDARs for their glutamate-mediated excitation (Chittajallu et al., 2017). Interestingly, interneurons originating from the medial ganglionic eminence (MGE) versus caudal ganglionic eminence (CGE) express different complements of excitatory glutamatergic receptors: the MGE-derived parvalbumin (PV)-expressing basket cells, somatostatin (Sst)-expressing cells, and a subset of nitric oxide synthase (NOS)-expressing interneurons, undergo a use-dependent switch from GluN2B to GluN2A during the second and third postnatal weeks (juvenile period); by contrast, CGE-derived interneurons, including cells that express cholecystokinin, vasoactive intestinal peptide, reelin, and the remainder of NOS-expressing cells, retain GluN2B into adulthood (Matta et al., 2013). Thus, CPP antagonism of GluN2A-NMDARs on dendrite-targeting interneurons, including neurogliaform cells, ivy cells, and O-LM cells, via GluN2A-NMDRs, might account for its ability to block contextual memory.
4.3. Summary and conclusions
Regarding the original motivation of this study – to use CPP as a standard to assess the relevance of NMDAR block to memory suppression by general anesthetics – what conclusions can we draw? The primary lesson is that LTP of the prototypical Schaffer collateral/commissural (SCC) synapse from CA3 onto CA1 pyramidal cells, which has revealed a wealth of information about the molecular underpinnings of synaptic plasticity (Nicoll, 2017), does not serve as a good model of CPP-induced amnesia. Based upon previous reports that CPP blocks both LTP and the NMDAR component of SCC excitatory synapses (Abraham and Mason, 1988; Fung et al., 2016), and that CFC depends upon intact NMDAR signaling in the CA1 region (McHugh et al., 1996; Tsien et al., 1996), we had expected a simple causal relationship between CPP block of NMDARs, LTP, and memory; clearly, that is not the case. A second is that even the modest degree of selectivity of CPP for NR2A-NMDARs may be behaviorally meaningful, and perhaps useful. Although memory impairment is generally considered a liability for many drugs that are developed as therapeutics, in the context of anesthesia and surgery – during which the complete elimination of memory is desirable – the ability of a subunit-selective agent to suppress memory with high potency might indeed represent a therapeutic opportunity. Finally, the possibility that CPP interferes with memory formation by modulating NMDARs on interneurons presents an interesting confluence with memory suppression by etomidate, which acts through α5-GABAARs on non-pyramidal cells (most likely interneurons) to block LTP (Rodgers et al., 2015). Thus, modulation of interneurons might serve as a common theme for physiological and pharmacological control of learning and memory.
Highlights.
CPP blocks contextual fear conditioning (CFC) with an ED50 dose of 2.3 mg/kg
The ED50 (CFC) dose of CPP corresponds to a free aCSF concentration of 53 nM CPP
- CPP blocks NMDAR-EPSPs and LTP with ED50 concentrations of 434 nM and 361 nM
- Implication: CPP acts on non-CA1-pyramidal-neuron NMDARs to block learning
ACKNOWLEDGEMENTS
We would like to acknowledge the support of the late Edmond I. Eger, 2nd, in whose laboratory at UCSF the behavioral experiments were conducted, and excellent technical support from Mark Perkins and Marissa Dreger.
Funding:
This work was supported by the National Institutes of Health R01 GM101497 (RAP), R01 GM118801 (RAP), R01 DK071801 (LL), F32 GM106670 (KTL), and RO1 MH62122 (MSF); and the Ralph M. Waters, M.D., Distinguished Chair at the University of Wisconsin-Madison (RAP)
Abbreviations
- (CPP)
(R,S)-3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid
- (NMDARs)
N-methyl-D-aspartate receptors
- (fEPSPNMDA)
NMDAR component of evoked field excitatory postsynaptic potentials
- (LTP)
Long-term potentiation
- (aCSF)
Artificial cerebrospinal fluid
- (CFC)
Contextual fear conditioning
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: The authors declare no competing interests.
REFERENCES
- Abraham WC, Mason SE, 1988. Effects of the NMDA receptor/channel antagonists CPP and MK801 on hippocampal field potentials and long-term potentiation in anesthetized rats. Brain Research 462, 40–46. [DOI] [PubMed] [Google Scholar]
- Adell A, 2020. Brain NMDA Receptors in Schizophrenia and Depression. Biomolecules 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V, Pidoplichko VI, Figueiredo TH, Braga MFM, 2018. Oscillatory Synchronous Inhibition in the Basolateral Amygdala and its Primary Dependence on NR2A-containing NMDA Receptors. Neuroscience 373, 145–158. [DOI] [PubMed] [Google Scholar]
- Auberson YP, Allgeier H, Bischoff S, Lingenhoehl K, Moretti R, Schmutz M, 2002. 5-Phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorganic and Medicinal Chemistry Letters 12, 1099–1102. [DOI] [PubMed] [Google Scholar]
- Bannerman DM, Bus T, Taylor A, Sanderson DJ, Schwarz I, Jensen V, Hvalby O, Rawlins JN, Seeburg PH, Sprengel R, 2012. Dissecting spatial knowledge from spatial choice by hippocampal NMDA receptor deletion. Nature Neuroscience 15, 1153–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C, Nicoll RA, 2007. Rapid bidirectional switching of synaptic NMDA receptors. Neuron 55, 779–785. [DOI] [PubMed] [Google Scholar]
- Benkwitz C, Liao M, Laster MJ, Sonner JM, Eger EI, Pearce RA, 2007. Determination of the EC50 amnesic concentration of etomidate and its diffusion profile in brain tissue: implications for in vitro studies. Anesthesiology 106, 114–123. [DOI] [PubMed] [Google Scholar]
- Bergeron S, Rompré PP, 2013. Blockade of ventral midbrain NMDA receptors enhances brain stimulation reward: a preferential role for GluN2A subunits. European Neuropsychopharmacology 23, 1623–1635. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL, 2013. Expression of NMDA receptor-dependent LTP in the hippocampus: bridging the divide. Mol Brain 6, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL, Morris RG, 2014. Synaptic plasticity in health and disease: introduction and overview. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 369, 20130129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittajallu R, Wester JC, Craig MT, Barksdale E, Yuan XQ, Akgul G, Fang C, Collins D, Hunt S, Pelkey KA, McBain CJ, 2017. Afferent specific role of NMDA receptors for the circuit integration of hippocampal neurogliaform cells. Nat Commun 8, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole BJ, Klewer M, Jones GH, Stephens DN, 1993. Contrasting effects of the competitive NMDA antagonist CPP and the non-competitive NMDA antagonist MK 801 on performance of an operant delayed matching to position task in rats. Psychopharmacology 111, 465–471. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, 1995. The brain slice preparation: a tribute to the pioneer Henry McIlwain. Journal of Neuroscience Methods 59, 5–9. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H, 1983a. The antagonism of amino acid-induced excitations of rat hippocampal CA1 neurones in vitro. Jounal of Physiology (London) 334, 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H, 1983b. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. Jounal of Physiology (London) 334, 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daumas S, Halley H, Frances B, Lassalle JM, 2005. Encoding, consolidation, and retrieval of contextual memory: differential involvement of dorsal CA3 and CA1 hippocampal subregions. Learning and Memory 12, 375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J, Evans RH, Herrling PL, Jones AW, Olverman HJ, Pook P, Watkins JC, 1986. CPP, a new potent and selective NMDA antagonist. Depression of central neuron responses, affinity for [3H]D-AP5 binding sites on brain membranes and anticonvulsant activity. Brain Research 382, 169–173. [DOI] [PubMed] [Google Scholar]
- de Sousa SL, Dickinson R, Lieb WR, Franks NP, 2000. Contrasting synaptic actions of the inhalational general anesthetics isoflurane and xenon.[comment]. Anesthesiology 92, 1055–1066. [DOI] [PubMed] [Google Scholar]
- Dupuis JP, Ladepeche L, Seth H, Bard L, Varela J, Mikasova L, Bouchet D, Rogemond V, Honnorat J, Hanse E, Groc L, 2014. Surface dynamics of GluN2B-NMDA receptors controls plasticity of maturing glutamate synapses. EMBO Journal 33, 842–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eger EI, Liao M, Laster MJ, Won A, Popovich J, Raines DE, Solt K, Dutton RC, Cobos FV, Sonner JM, 2006. Contrasting roles of the N-methyl-d-aspartate receptor in the production of immobilization by conventional and aromatic anesthetics. Anesthesia and Analgesia 102, 1397–1406. [DOI] [PubMed] [Google Scholar]
- Feng B, Morley RM, Jane DE, Monaghan DT, 2005. The effect of competitive antagonist chain length on NMDA receptor subunit selectivity. Neuropharmacology 48, 354–359. [DOI] [PubMed] [Google Scholar]
- Fung TK, Law CS, Leung LS, 2016. Associative spike timing-dependent potentiation of the basal dendritic excitatory synapses in the hippocampus in vivo. Journal of Neurophysiology 115, 3264–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Dong Z, Bagot RC, Howland JG, Phillips AG, Wong TP, Wang YT, 2010. Hippocampal long-term depression is required for the consolidation of spatial memory. Proceedings of the National Academy of Sciences of the United States of America 107, 16697–16702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemperline E, Laha K, Scarlett CO, Pearce RA, Li L, 2014. Measurement of NMDA Receptor Antagonist, CPP, in Mouse Plasma and Brain Tissue Following Systematic Administration Using Ion-Pair LCMS/MS. Anal Methods 6, 6389–6396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustino TF, Maren S, 2015. The Role of the Medial Prefrontal Cortex in the Conditioning and Extinction of Fear. Front Behav Neurosci 9, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett PJ, Standaert DG, 2004. Rationale for and use of NMDA receptor antagonists in Parkinson's disease. Pharmacology and Therapeutics 102, 155–174. [DOI] [PubMed] [Google Scholar]
- Hanada T, 2020. Ionotropic Glutamate Receptors in Epilepsy: A Review Focusing on AMPA and NMDA Receptors. Biomolecules 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Yi F, Perszyk RE, Menniti FS, Traynelis SF, 2017. NMDA Receptors in the Central Nervous System. Methods in Molecular Biology 1677, 1–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris EW, Ganong AH, Monaghan DT, Watkins JC, Cotman CW, 1986. Action of 3-((+/−)-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP): a new and highly potent antagonist of N-methyl-D-aspartate receptors in the hippocampus. Brain Research 382, 174–177. [DOI] [PubMed] [Google Scholar]
- Hemmings HC Jr., Yan W, Westphalen RI, Ryan TA, 2005. The general anesthetic isoflurane depresses synaptic vesicle exocytosis. Molecular Pharmacology 67, 1591–1599. [DOI] [PubMed] [Google Scholar]
- Hunsaker MR, Tran GT, Kesner RP, 2009. A behavioral analysis of the role of CA3 and CA1 subcortical efferents during classical fear conditioning. Behavioral Neuroscience 123, 624–630. [DOI] [PubMed] [Google Scholar]
- Ikegami M, Uemura T, Kishioka A, Sakimura K, Mishina M, 2014. Striatal dopamine D1 receptor is essential for contextual fear conditioning. Sci Rep 4, 3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevtovic-Todorovic V, Todorovic SM, Mennerick S, Powell S, Dikranian K, Benshoff N, Zorumski CF, Olney JW, 1998. Nitrous oxide (laughing gas) is an NMDA antagonist, neuroprotectant and neurotoxin. Nature Medicine, 460–463. [DOI] [PubMed] [Google Scholar]
- Jimenez JC, Berry JE, Lim SC, Ong SK, Kheirbek MA, Hen R, 2020. Contextual fear memory retrieval by correlated ensembles of ventral CA1 neurons. Nat Commun 11, 3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellermayer B, Ferreira JS, Dupuis J, Levet F, Grillo-Bosch D, Bard L, Linares-Loyez J, Bouchet D, Choquet D, Rusakov DA, Bon P, Sibarita JB, Cognet L, Sainlos M, Carvalho AL, Groc L, 2018. Differential Nanoscale Topography and Functional Role of GluN2-NMDA Receptor Subtypes at Glutamatergic Synapses. Neuron 100, 106–119 e107. [DOI] [PubMed] [Google Scholar]
- Kentros C, Hargreaves E, Hawkins RD, Kandel ER, Shapiro M, Muller RV, 1998. Abolition of long-term stability of new hippocampal place cell maps by NMDA receptor blockade. Science 280, 2121–2126. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS, 1992. Modality-specific retrograde amnesia of fear. Science 256, 675–677. [DOI] [PubMed] [Google Scholar]
- Kim WB, Cho JH, 2020. Encoding of contextual fear memory in hippocampal-amygdala circuit. Nat Commun 11, 1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirson ED, Yaari Y, Perouansky M, 1998. Presynaptic and postsynaptic actions of halothane at glutamatergic synapses in the mouse hippocampus. British Journal of Pharmacology. 124, 1607–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P, 2008. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouvaros S, Kotzadimitriou D, Papatheodoropoulos C, 2015. Hippocampal sharp waves and ripples: Effects of aging and modulation by NMDA receptors and L-type Ca2+ channels. Neuroscience 298, 26–41. [DOI] [PubMed] [Google Scholar]
- Lee I, Kesner RP, 2004. Encoding versus retrieval of spatial memory: double dissociation between the dentate gyrus and the perforant path inputs into CA3 in the dorsal hippocampus. Hippocampus 14, 66–76. [DOI] [PubMed] [Google Scholar]
- Lehmann J, Schneider J, McPherson S, Murphy DE, Bernard P, Tsai C, Bennett DA, Pastor G, Steel DJ, Boehm C, et al. , 1987. CPP, a selective N-methyl-D-aspartate (NMDA)-type receptor antagonist: characterization in vitro and in vivo. Journal of Pharmacology and Experimental Therapeutics 240, 737–746. [PubMed] [Google Scholar]
- Lozovaya NA, Grebenyuk SE, Tsintsadze T, Feng B, Monaghan DT, Krishtal OA, 2004. Extrasynaptic NR2B and NR2D subunits of NMDA receptors shape 'superslow' afterburst EPSC in rat hippocampus. J Physiol 558, 451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri G, Dingledine R, 2002. Control of feedforward dendritic inhibition by NMDA receptor-dependent spike timing in hippocampal interneurons. Journal of Neuroscience. 22, 5462–5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JF, Bartlett MC, Mody I, Pahapill P, Reynolds JN, Salter MW, Schneiderman JH, Pennefather PS, 1991. Actions of ketamine, phencyclidine and MK-801 on NMDA receptor currents in cultured mouse hippocampal neurones. Journal of Physiology 432, 483–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver MB, Roth SH, 1988. Inhalation anaesthetics exhibit pathway-specific and differential actions on hippocampal synaptic responses in vitro. British Journal of Anaesthesia 60, 680–691. [DOI] [PubMed] [Google Scholar]
- Matta JA, Pelkey KA, Craig MT, Chittajallu R, Jeffries BW, McBain CJ, 2013. Developmental origin dictates interneuron AMPA and NMDA receptor subunit composition and plasticity. Nature Neuroscience 16, 1032–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C, Acosta-Martinez M, Dubois SL, Wolfe A, Radovick S, Boehm U, Levine JE, 2010. Timing and completion of puberty in female mice depend on estrogen receptor alpha-signaling in kisspeptin neurons. Proceedings of the National Academy of Sciences of the United States of America 107, 22693–22698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Vyklicky L Jr., Clements J, 1989. Regulation of NMDA receptor desensitization in mouse hippocampal neurons by glycine. Nature 338, 425–427. [DOI] [PubMed] [Google Scholar]
- McDonald RJ, Hong NS, Craig LA, Holahan MR, Louis M, Muller RU, 2005. NMDA-receptor blockade by CPP impairs post-training consolidation of a rapidly acquired spatial representation in rat hippocampus. European Journal of Neuroscience 22, 1201–1213. [DOI] [PubMed] [Google Scholar]
- McHugh TJ, Blum KI, Tsien JZ, Tonegawa S, Wilson MA, 1996. Impaired hippocampal representation of space in CA1-specific NMDAR1 knockout mice. Cell 87, 1339–1349. [DOI] [PubMed] [Google Scholar]
- McHugh TJ, Jones MW, Quinn JJ, Balthasar N, Coppari R, Elmquist JK, Lowell BB, Fanselow MS, Wilson MA, Tonegawa S, 2007. Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science 317, 94–99. [DOI] [PubMed] [Google Scholar]
- Medvedev NI, Popov VI, Rodriguez Arellano JJ, Dallérac G, Davies HA, Gabbott PL, Laroche S, Kraev IV, Doyère V, Stewart MG, 2010. The N-methyl-D-aspartate receptor antagonist CPP alters synapse and spine structure and impairs long-term potentiation and long-term depression induced morphological plasticity in dentate gyrus of the awake rat. Neuroscience 165, 1170–1181. [DOI] [PubMed] [Google Scholar]
- Moita MA, Rosis S, Zhou Y, LeDoux JE, Blair HT, 2004. Putting fear in its place: remapping of hippocampal place cells during fear conditioning. Journal of Neuroscience 24, 7015–7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M, 1986. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature 319, 774–776. [DOI] [PubMed] [Google Scholar]
- Morsy A, Trippier PC, 2019. Current and Emerging Pharmacological Targets for the Treatment of Alzheimer's Disease. J Alzheimers Dis 72, S145–s176. [DOI] [PubMed] [Google Scholar]
- Neyama H, Dozono N, Ueda H, 2020. NR2A-NMDA Receptor Blockade Reverses the Lack of Morphine Analgesia Without Affecting Chronic Pain Status in a Fibromyalgia-Like Mouse Model. Journal of Pharmacology and Experimental Therapeutics 373, 103–112. [DOI] [PubMed] [Google Scholar]
- Neyton J, Paoletti P, 2006. Relating NMDA Receptor Function to Receptor Subunit Composition: Limitations of the Pharmacological Approach. Journal of Neuroscience 26, 1331–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, 2017. A Brief History of Long-Term Potentiation. Neuron 93, 281–290. [DOI] [PubMed] [Google Scholar]
- Ogata J, Shiraishi M, Namba T, Smothers CT, Woodward JJ, Harris RA, 2006. Effects of anesthetics on mutant N-methyl-D-aspartate receptors expressed in Xenopus oocytes. J Pharmacol.Exp.Ther 318, 434–443. [DOI] [PubMed] [Google Scholar]
- Pagano J, Giona F, Beretta S, Verpelli C, Sala C, 2021. N-methyl-d-aspartate receptor function in neuronal and synaptic development and signaling. Curr Opin Pharmacol 56, 93–101. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Danysz W, Hesselink M, Hartmann S, Lorenz B, Wollenburg C, Quack G, 1998. Modulation of NMDA receptors by glycine--introduction to some basic aspects and recent developments. Amino Acids 14, 207–216. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Zong X, Lux HD, 1993. Whole cell and single channel analysis of the kinetics of glycine-sensitive N-methyl-D-aspartate receptor desensitization. British Journal of Pharmacology 109, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny T, Andersson D, Wilhelmsson U, Pekna M, Pekny M, 2014. Short general anaesthesia induces prolonged changes in gene expression in the mouse hippocampus. Acta Anaesthesiologica Scandinavica 58, 1127–1133. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, McBain CJ, 2017. Hippocampal GABAergic Inhibitory Interneurons. Physiological Reviews 97, 1619–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier JG, Lacaille JC, 2008. Long-term synaptic plasticity in hippocampal feedback inhibitory networks. Progress in Brain Research 169, 241–250. [DOI] [PubMed] [Google Scholar]
- Perouansky M, Baranov D, Salman M, Yaari Y, 1995. Effects of halothane on glutamate receptor-mediated excitatory postsynaptic currents. a patch-clamp study in adult mouse hippocampal slices. Anesthesiology 83, 109–119. [DOI] [PubMed] [Google Scholar]
- Petrenko AB, Yamakura T, Baba H, Shimoji K, 2003. The role of N-methyl-D-aspartate (NMDA) receptors in pain: a review. Anesthesia and Analgesia 97, 1108–1116. [DOI] [PubMed] [Google Scholar]
- Petrenko AB, Yamakura T, Sakimura K, Baba H, 2014. Defining the role of NMDA receptors in anesthesia: are we there yet? European Journal of Pharmacology 723, 29–37. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE, 1992. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behavioral Neuroscience 106, 274–285. [DOI] [PubMed] [Google Scholar]
- Piekarski DJ, Boivin JR, Wilbrecht L, 2017. Ovarian Hormones Organize the Maturation of Inhibitory Neurotransmission in the Frontal Cortex at Puberty Onset in Female Mice. Current Biology 27, 1735–1745 e1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauner C, Kohr G, 2011. Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-methyl-D-aspartate receptor population in adult hippocampal synapses. Journal of Biological Chemistry 286, 7558–7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers FC, Zarnowska ED, Laha KT, Engin E, Zeller A, Keist R, Rudolph U, Pearce RA, 2015. Etomidate Impairs Long-Term Potentiation In Vitro by Targeting alpha5-Subunit Containing GABAA Receptors on Nonpyramidal Cells. Journal of Neuroscience 35, 9707–9716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, Feng G, 2014. Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods in Molecular Biology 1183, 221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, McGinley MJ, Westbrook GL, 2013. Triheteromeric NMDA Receptors at Hippocampal Synapses. Journal of Neuroscience 33, 9150–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R, 2010. Glutamate receptor ion channels: structure, regulation, and function. Pharmacological Reviews 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S, 1996. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87, 1327–1338. [DOI] [PubMed] [Google Scholar]
- Villarreal DM, Gross AL, Derrick BE, 2007. Modulation of CA3 afferent inputs by novelty and theta rhythm. Journal of Neuroscience 27, 13457–13467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlstrom KL, Huff ML, Emmons EB, Freeman JH, Narayanan NS, McIntyre CK, LaLumiere RT, 2018. Basolateral Amygdala Inputs to the Medial Entorhinal Cortex Selectively Modulate the Consolidation of Spatial and Contextual Learning. Journal of Neuroscience 38, 2698–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin PI, Woods CB, Kayser EB, Ramirez JM, Morgan PG, Sedensky MM, 2018. Isoflurane disrupts excitatory neurotransmitter dynamics via inhibition of mitochondrial complex I. British Journal of Anaesthesia 120, 1019–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]