

Abstract
Background:
Drug induced liver injury (DILI) remains a prominent global issue and acetaminophen (APAP) overdose represents a common cause of hepatic injury and DILI. Transient alanine aminotransferase (ALT) elevations have been documented while adhering to recommended daily dosing. However, no metabolites have been identified in pre-treatment samples predicting which patients will develop these transient increases.
Methods:
This was a secondary analysis of samples collected from a parent study describing the course of ALT levels in subjects receiving therapeutic APAP dosing. Two hundred and four subjects recruited from Denver, Colorado received 4 g APAP/daily for at least 16 days. Subjects were grouped by ALT at any monitored time point above 60 units/L (n = 25) vs. no increase (n = 179). Serum samples from days 0, 7, 16, and 31 were run on ultra-high performance liquid chromatography mass spectrometry. We report the metabolomic results of samples analyzed prior to APAP administration and over time. Significant changes in metabolite and demographic variable expressions were explored using t-tests with false discovery rate correction, chi square, and partial least squares discriminant analyses.
Results:
Within pre-treatment day 0 samples, allantoate and ornithine were significantly elevated in subjects of the ALT elevation group (p = .032). Baseline ALT (p = .011) and alkaline phosphatase (p = .006) were also significant. These metabolites were significant independent of race, ethnicity, gender, or BMI.
Conclusions:
Allantoate and ornithine are directly involved in pathways related to nitrogen release and urea production. Further investigation into alterations in the glutathione metabolism and urea cycle pathways may lead to a greater understanding of the mechanisms associated with hepatic adaptation for a variety of pharmaceuticals.
Keywords: Urea cycle, glutathione metabolism, ornithine, DILI, alanine transaminase
Introduction
Globally, an estimated 24 per 100,000 people will develop drug induced liver injury (DILI) annually, which is the most common cause of acute liver failure (ALF) in the United States and Europe [1–4]. While some cases of DILI are caused by dose related hepatotoxins, many cases are due to allergic hypersensitivity or idiosyncratic events that are well tolerated in most patients. Development of DILI is often a result of failed hepatic adaptation in these patients [5]. There are multiple physiological events that could contribute to diminished or failed hepatic adaptation including those related to allergic, idiosyncratic, or toxic etiologies [6].
Acetaminophen (APAP) has served as a model hepatotoxin for many years. Direct hepatocellular injury is the most common mechanism suspected to be the cause associated with APAP induced toxicity [7,8]. In addition to its dose-related hepatotoxicity, prolonged (>3 days) therapeutic dosing of APAP (4 g/day for more than three days) will cause transient elevations in alanine transaminase (ALT) levels in half the population [9,10]. Over time, the ALT level almost always returns to baseline with no clinical adverse effects while still consuming APAP [10], which highlights the importance of hepatic adaptation. Given the extensive understanding of APAP metabolism and toxicity, APAP represents an optimal model to describe and further explore hepatic adaptation.
The phenomenon of transient ALT elevation during treatment is also noted with multiple other pharmaceuticals that are viewed as safe when taken at therapeutic doses [11]. Understanding the metabolic processes that occur during adaptation to APAP may provide insight into how failure to adapt occurs with other pharmaceuticals. Hepatic adaptation explains and is requisite for people to continue taking these medications without sustained hepatic injury. Exploration of the mechanisms and pathways associated with hepatic adaptation provides an understanding of which patients are likely to asymptomatically experience a transient elevation in ALT. Identification of experimental drugs inducing ALT elevations related to hepatic adaptation rather than DILI would allow more drugs under development to continue through to clinical trials [2]. Additionally, the ability to predict these markers could aid clinicians in closer monitoring of their patients taking medications known to induce hepatic adaptation processes. This may ultimately help clinicians monitor their patients at greater risk of developing DILI and/or ALF. Unfortunately, no pre-treatment biomarkers have been identified to predict adverse outcomes associated with therapeutic administration [12–14], but metabolomics offers a possible solution [11]. Metabolomics is a rapidly growing field that aims to measure end stage metabolites of upstream biologic processes to predict individual responses to therapeutics and direct clinical interventions.
Identification of metabolic pathways associated with transient ALT elevation during therapeutic administration of APAP could be useful to identify other drugs likely to elicit the same elevations. This study uses metabolomics in conjunction with subject clinical data collected in the parent study to identify metabolites and their associated biologic pathways in pre-APAP administration serum samples and provocative samples during administration to identify the presence and mechanisms of hepatic adaptation.
Methods
Study design
We performed secondary metabolomic analyses from serum samples collected from a parent study describing the course of alanine aminotransferase (ALT) levels in subjects receiving therapeutic dosing of APAP for at least 16 days (NCT00743093) [10]. Subjects were instructed to take 2 × 500mg tablets every 4 h for four doses each day; start time varied from day to day, but four-hour intervals were consistent, and subjects documented this in their study diary. They also recorded all other medications taken and any alcohol consumption throughout the study period. Compliance was verified by study diary and pill counts at each study visit. In addition to the diaries, clinical parameters collected included: race, ethnicity, age, sex, body mass index (BMI), significant medical history, and adverse drug events (as outlined in the U.S. Food and Drug Administration, FDA 21 312.32 Code of Federal regulations) [15]. Samples were stored at −80 °C after blood chemistry analysis. The parent study also included a placebo arm whose subjects were not investigated as part of this study.
Subject enrollment
The parent study and this metabolomic analysis were approved by the Colorado Multiple Institutional Review Board and followed the ethical Declaration of Helsinki. The study population for this secondary analysis consisted of the same inclusion and exclusion criteria as the parent study. Briefly, subjects were recruited from the Denver, Colorado community and were ≥18 years of age. Subjects were excluded if there was APAP use within four days preceding study initiation, new medications started within the month prior, clinical lab work suggestive of impaired liver function, increased alcohol consumption or inability to give informed consent [10]. Blood was drawn on study days 0 (baseline, pre-APAP administration), 4, 7, 10, 16, and 31. Blood chemistry values collected at each visit include: ALT, alkaline phosphatase (ALKP), international normalized ratio (INR), total protein, albumin, total and direct bilirubin, aspartate transaminase (AST), and triglycerides.
Phenotype stratification
Subjects were defined as “ALT elevation” when ALT increased >60 IU/L per the chemistry analysis completed in the parent study as an estimation of 1.5× the clinically relevant upper limit of the ALT reference range (40 IU/L). For this study, the “ALT elevation” group are the subjects who had a transient elevation in ALT and therefore were assumed to undergo hepatic adaptation. As the ALT elevation with therapeutic APAP administration is transient, 1.5× the upper limit of the clinically relevant reference range was used to identify subjects with an APAP induced ALT elevation rather than normal physiologic fluctuations of ALT over time. All other subjects were defined as “No ALT elevation.” Samples chosen for analysis came from subjects in the treatment arm of the parent study and included the following: baseline samples from all subjects, samples from study day that subject reached their peak ALT level (day 7 or 10 for each subject), and a subset of samples from study days 16 and 31. This subset population was chosen as a representative sampling of demographically matched subjects who developed an ALT elevation and those that did not to prevent population bias in our analyses based on age, sex, race, or ethnicity.
Metabolite extraction and identification
Samples were prepared for ultra-high performance liquid chromatography mass spectrometry (UHPLC–MS) metabolomics as previously described [16]. Briefly, metabolites were extracted from serum (20 μL) in ice-cold lysis/extraction buffer (methanol:acetonitrile:water 5:3:2) at a 1:25 dilution. Isotopically labeled standards (Supplemental Table 1) were added at expected biological concentrations to the lysis buffer for absolute quantitation. Samples were agitated (30 min 4°C) and centrifuged (18,213×g, 10min, 4°C). Protein pellets were discarded. Supernatants were diluted 1:1 using 10mM ammonium and stored at −80 °C prior to metabolomic analyses.
Plasma extracts were injected into a Thermo Vanquish UHPLC system (San Jose, CA) coupled to a Thermo Q Exactive mass spectrometer with electrospray ionization (Bremen, Germany). Metabolites were separated on a Kinetex C18 column (150 × 2.1mm, 1.7 μ – Phenomenex, Torrance, CA) at 45°C using a five-minute gradient method [17] at 450 μL/min and mobile phases (A: water/0.1% formic acid; B: acetonitrile/0.1% formic acid) for positive ion mode. Negative ion mode used the same five-minute gradient method at 450 μL, with the following changes: 1 mM ammonium acetate (NH4OAc) substituted for 0.1% formic acid (A: 95/5 water/acetonitrile 1 mM NH4OAc; B: 5/95 water/acetonitrile 1 mM NH4OAc). Solvent gradient: 0–0.5 min 0% B; 0.5–1.1 min 0–100% B, 1.1–2.75 min hold at 100% B, 2.75–3 min 100–0% B, 3–5 min hold at 0% B. Quality controls were generated from pooled aliquots of extracts and ran every 10 analytical runs to control for technical variability, as judged by coefficients of variation. To correct for technical variability, quantitative measurements across samples were normalized against quality controls through the software MetaboDrift [18]. There were 164 metabolites identified and validated as previously described [19,20].
Metabolomic statistical analyses
Supervised targeted analyses were completed using MetaboAnalyst (Version 4.0) [21] to identify significant differences in baseline metabolomes between subjects who developed an ALT elevation and those who did not as well as between those groups over the course of therapy for each (days 0, 7/10, 16, and 31). ANOVA simultaneous comparison analyses (ASCAs) were completed for multivariate analysis of our subset population to observe changes simultaneously between time and ALT class - significance of results generated were determined by leverage and squared prediction error (SPE) values. Independent t tests with false discovery rate (FDR) multiple comparisons corrections, fold changes, and partial least squares discriminant analyses (PLS-DA) plots were created after data transformation using auto scaling (mean-centered and divided by the standard deviation of each variable). Fold changes of at least twofold difference between groups or p<.05 after multiple comparisons correction were considered significant. PLS-DA plots represented the differences between metabolomes of the different groups. Cross validation of the PLS-DA model was completed with permutation testing. Area under the receiver operating characteristic (AUROC) curves were generated for significant variables to show the diagnostic ability of those variables.
Statistical analyses for clinical and demographic variables
Independent t tests with FDR for multiple comparisons correction were used to identify significant clinical variables and chi-square tests were used to compare categorical clinical variables. Significant metabolites and clinical variables groups were investigated together using logistic regression tests to predict which subjects would require and undergo hepatic adaptation with APAP administration. All statistical analyses were completed in R (version 4.0.2, Vienna, Austria).
Results
Subject demographic and clinical profiles
Of the entire study population (n = 204) taking therapeutic APAP dosing, 179 (87.7%) never had an ALT ≥60 IU/L and were grouped as “No ALT elevation.” All other study participants had an ALT of at least 60 IU/L and were grouped as “ALT elevation.” There were no significant differences in standard demographics between response groups (Table 1).
Table 1.
Subject demographic and clinical profiles: there were no significant differences in demographics between ALT elevation and no ALT elevation groups.
| Demographic/clinical parameter | No ALT elevation | ALT elevation | p Value |
|---|---|---|---|
| N (%) | 179 (87.7) | 25 (12.3) | |
| Female, n (%) | 130 (72.6) | 17 (68) | .629 |
| Race, Caucasian, n (%) | 125 (69.8) | 16 (64) | .554 |
| Race, other, n (%) | 54 (30.2) | 9 (36) | .554 |
| Ethnicity, non-Hispanic, n (%) | 152 (84.9) | 19 (76) | .257 |
| Age, median (IQR) | 35 (18,74) | 30 (20,58) | .938 |
| BMI (kg/m2), mean (st.dev.) | 26.5 (5.7) | 28.9 (7.6) | .061 |
| Never drink, no, n (%) | 146 (81.6) | 20 (80) | .851 |
| Baseline ALT, mean (st.dev.) | 19.7 (7.1) | 25.8 (8.4) | <.001 |
| Baseline ALKP, mean (st.dev.) | 58.3 (15.2) | 72.2 (18.2) | <.001 |
| Peak ALT, median (IQR) | 26 (16) | 75 (28) | <.0001 |
IQR: interquartile range; st.dev.: standard deviation.
ALT and ALKP were significantly elevated at baseline for the ALT elevation group.
The serum samples used for the peak ALT elevation study time point were chosen for each subject from either their day 7 or day 10 sample, based on whichever was higher, because this was the timepoint most likely to demonstrate discordant biology from subject baseline. There was no significant difference between treatment group and sample day (chi-square = 0.304, p = .581).
Pertinent baseline clinical parameters are reported in Table 1. Subjects in the ALT elevation group had significantly higher baseline ALT levels (difference in means of 6.1 IU/L, p = <.001) than the no ALT elevation cohort. This suggests that there is a subset of baseline (pre-APAP administration) ALT levels that are considered not clinically actionable (within reference range), but could still be predictive of an increase in ALT with APAP administration. Subjects in the ALT elevation group also had significantly higher baseline ALKP levels (difference in means of 13.9 IU/L) than the no ALT elevation cohort. While ALKP is not specific to liver damage, this difference between groups suggests that baseline serum ALKP levels that are not considered clinically actionable can still identify underlying hepatic physiology that makes a patient more susceptible to developing increased ALT with APAP administration. There were no significant differences between baseline INR, albumin, AST, or bilirubin levels nor related to subject medical history that predicted subsequent ALT elevation. No significant differences were observed when comparing alcohol consumption or concurrent medication use during the study period.
The subset analysis of 14 subjects who had serum samples for all post-ingestion time points, included seven subjects from each group (ALT elevation vs. no ALT elevation). There were no significant differences in demographics for this population.
Metabolites in the glutathione metabolism and urea cycle pathways were prominently identified using multivariate analyses
Investigating the subset subject cohort (n = 14), ASCA was used to identify major metabolome changes, specifically with four metabolites identified as being significant in this model (Figure 1(A)). This analysis allowed for a combined analysis based both on ALT class and by time series (days 0 vs. peak (day 7 or 10) vs. 16 vs. 31). Significant metabolites identified using ASCA were included using a combination of SPE and leverage (Figure 1(B)). As metabolites identified by a low SPE are associated with decreased prediction error within the developed model, a low SPE is desired to confirm their relevance in the model. Increased leverage for each metabolite highlights the importance and impact it has on the final model. Running a 1000 permutation test confirmed the significance of the model based on ALT class (p = <.05). Two metabolites involved within the glutathione metabolism pathway were significant: glycine and 5-oxoproline (Figure 1(C)). As well, there were two metabolites that were significant within the urea cycle: 5-l-glutamyl-l-glutamine and l-citrulline (Figure 1(D)). All of these metabolites displayed lower expression levels in the subjects that did not develop ALT elevations. This suggests that there is less glutathione and urea cycling within the subjects that did not experience an ALT elevation. Leverage and SPE values for each metabolite within this model can be found in Supplemental Table 2.
Figure 1.
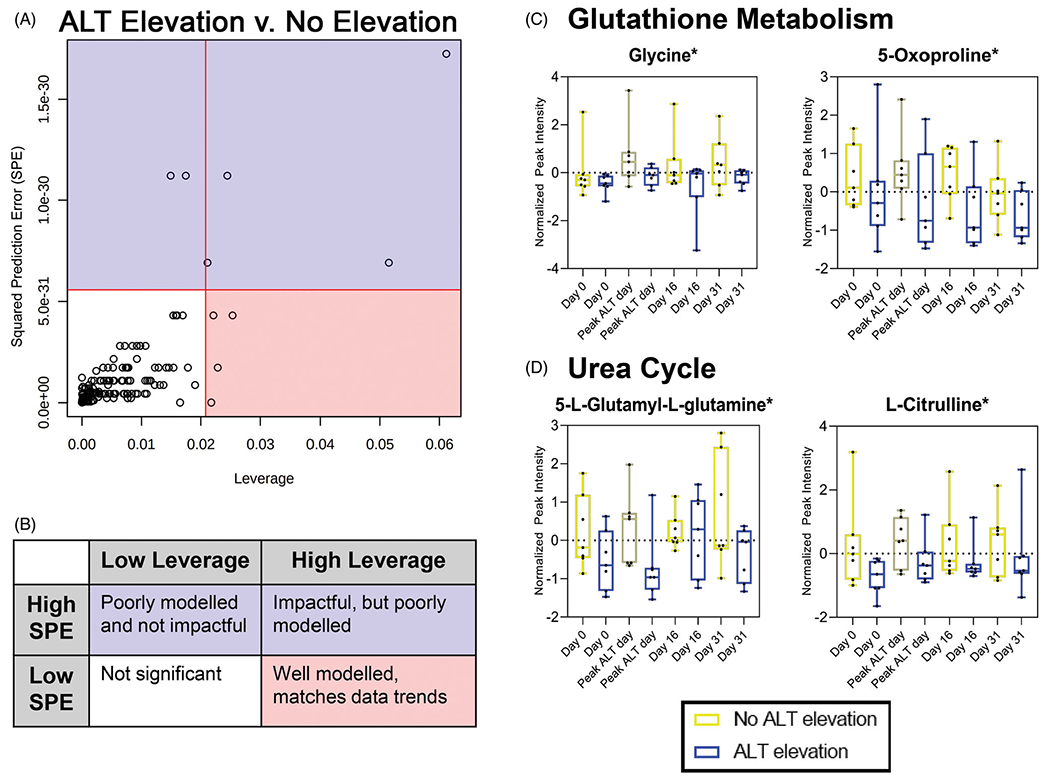
Metabolites involved with glutathione metabolism and urea cycle associated with ALT class: (A) four metabolites identified as being significant and well modeled using ASCA to look at ALT class and time in a multivariate analysis using SPE and leverage as criteria to determine significance. (B) Key to interpret ASCA matched spatially and by color to plot in A. (C) Metabolites identified as significant in ASCA related to glutathione metabolism. (D) Metabolites identified as significant in ASCA related to urea cycle. *Significance determined by high leverage and low SPE (see Supplemental Table 2 for details). ALT elevation, n = 7; no ALT elevation, n = 7.
Metabolome changes over time associated with glutathione, starch and sucrose metabolism and fatty acid oxidation were identified through ASCA
Looking at the subset subject cohort (n = 14), ASCA was used to identify major metabolome changes based on the interaction between ALT class and time where five metabolites had significantly different expression levels (Figure 2(A)). These metabolites were identified as significant using a combination of criteria: higher leverage, a marker of increasing significance within the model; and lower SPE, denoting decreased prediction error and increased model fit. Performing a 1000 permutation test did not reveal the interaction model to be significant on its own (p = .45), but did identify significant metabolites as determined by leverage and SPE. Glutathione, a metabolite relevant to APAP toxicity, was significant in this model with lower expression in the no ALT elevation group at the first two time points then noticeably higher at days 16 and 31 compared with the group that had ALT elevations (Figure 2(B)). Two metabolites were identified as significant within this model that are both involved in the starch and sucrose metabolism pathways: maltose and d-glucose 6-phosphate (Figure 2(C)). In the early time points, maltose was expressed more significantly in the no ALT elevation group compared with ALT elevation, but this switched at day 31 while both groups had d-glucose 6-phosphate levels that fluctuated over time. Subjects with no ALT elevation showed that expression levels of a medium chain fatty acid (10-hydroxydecanoic acid) exhibited relatively stable levels, while those with ALT elevations experienced changes over time suggesting changes to fatty acid metabolism over time in this population (Figure 2(D)). Supporting this change in energy utilization, subjects with no ALT elevation experienced a decrease in an acylcarnitine (tetradecenoyl carnitine) over time while subjects that did have an elevation experienced less change in expression level of this metabolite (Figure 2(D)). Leverage and SPE values for each metabolite within this model can be found in Supplemental Table 3.
Figure 2.
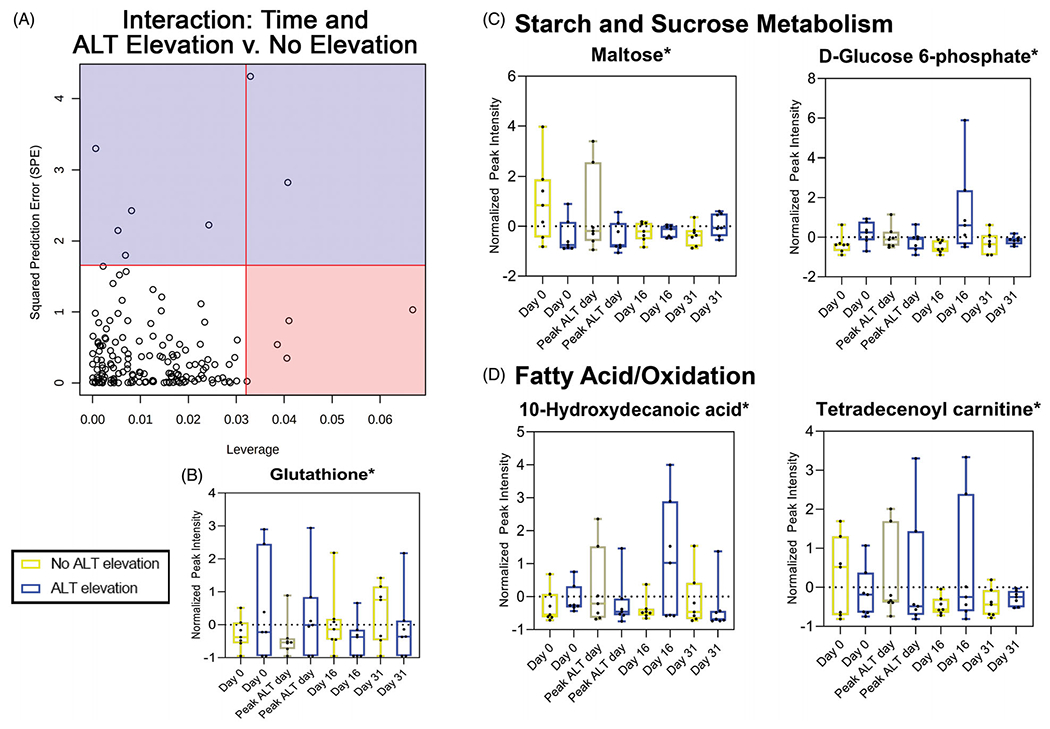
Glutathione and metabolites that are part of starch and sucrose metabolism or fatty acids/oxidation pathways are associated with the interaction between ALT class and time: (A) five metabolites identified as significant and well modeled using ASCA to investigate metabolome changes of the interaction between ALT class and time. (B) Glutathione identified as having significantly different expression levels between time points and ALT class. (C) Two metabolites associated with starch and sucrose metabolism were identified as being significant with the interaction of ALT class and time. (D) One medium chain fatty acid and one acylcarnitine were identified as being significantly different based on the interaction between ALT class and time. *Significance determined by high leverage and low SPE (see Supplemental Table 3 for details). ALT elevation, n = 7; no ALT elevation, n = 7.
Significant metabolome differences identified in pre-treatment samples of subjects who developed ALT elevation with APAP administration compared with those who did not
Investigating the overall cohort (n = 204), there were 164 measured metabolites present in the serum between both groups. Hierarchal clustering of the 20 most significant metabolites shows a distinct metabolome for subjects who developed ALT elevation from those who did not (Figure 3(A)). Using partial least squares regression analyses, component 1 explains 21.4% of the variance within the metabolomics dataset between those that developed ALT elevations and those that did not (Figure 3(B)). Component 2 explains 5.3% of the variance within this dataset across the same groups (Figure 3(B)). Notably, three metabolites, as measured by relative quantitation, were identified as significantly elevated in subjects who developed an ALT elevation compared with those who did not: ornithine, allantoate, and nicotinamide. Ornithine (p = .032) is found downstream of allantoate (p = .032) in the urea cycle where they are both involved in the removal of excess nitrogen (Figure 3(C)). The nucleotide, nicotinamide (p = .032) as a component of nicotinamide adenine dinucleotide (NAD) is involved in many physiologic pathways including the excretion of ammonia through the urea cycle (Figure 3(D)).
Figure 3.
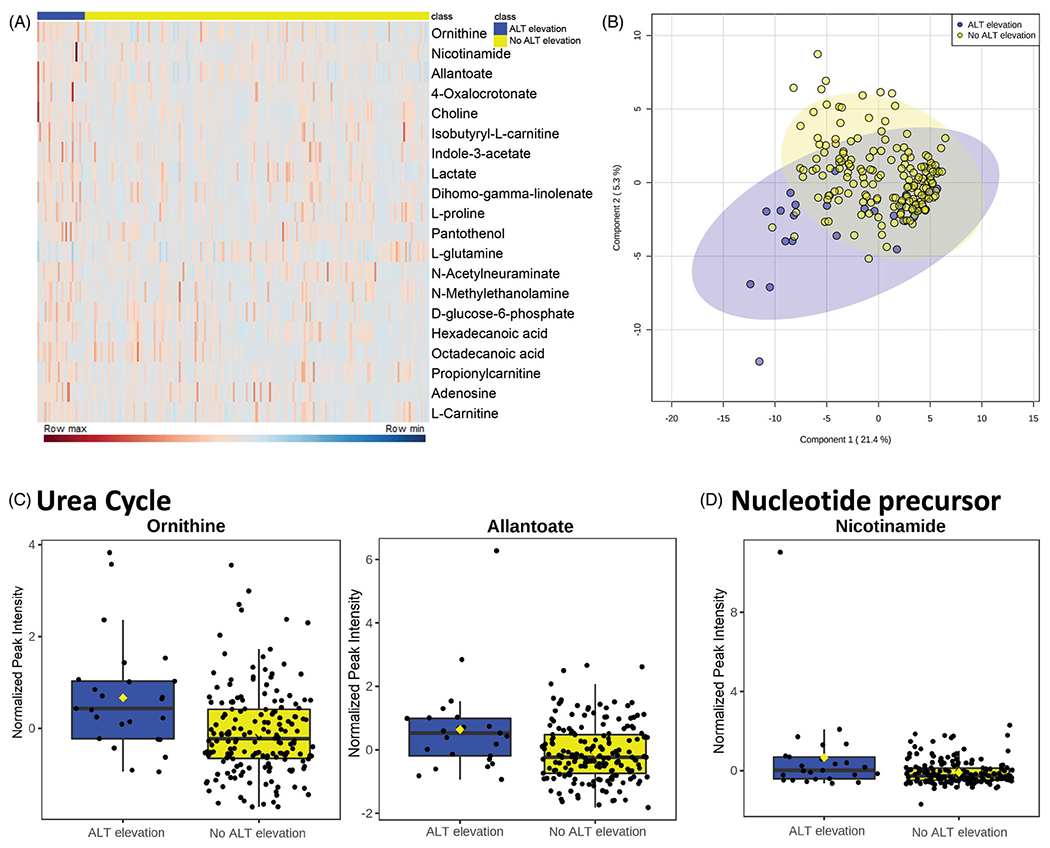
Changes in metabolome of pre-APAP treatment serum samples between subjects who had an ALT elevation during course of therapy to those who did not: (A) top 20 metabolites as determined by p value displayed in heatmap identifying the difference in relative metabolite levels between subjects who developed an ALT elevation during treatment and those who did not. Rows indicate individual metabolites with levels of expression from highest (row max) to lowest (row min) across each row. Each column describes the metabolome of an individual subject. (B) PLS-DA 2D image - principal component analysis between subjects who developed an ALT elevation and those that did not identifying pattern of metabolite expression in pre-APAP treated samples. (C, D) Significant (p = .032) metabolites identified by independent t tests with FDR multiple comparisons correction. ALT elevation, n = 25; no ALT elevation, n = 179.
Multiple metabolites with altered expression levels between treatment groups are found within glutathione metabolism and urea pathways
Many of the metabolites identified as having significantly different expression levels between treatment groups through ASCA (subset cohort, n = 14) and PLS-DA (entire cohort, n = 204) are linked to show the relevance of both pathways in this study (Figure 4). There are five metabolites identified within the glutathione metabolism pathway and three associated with the urea cycle that achieved significance between our different cohorts. While there was not a detectable difference in expression levels between our treatment groups in the analyses performed, the amino acid glutamine links and plays a critical role in each of these pathways. Glutathione is known to be involved with APAP metabolism, as an antioxidant acting on N-acetyl-p-benzoquinone imine (NAPQI) [22]. Urea and metabolites of the urea cycle have not previously been used to guide therapy of APAP toxicity or predict adverse response to APAP administration. Abnormalities in this pathway may be indicative of mitochondrial dysfunction, which has been associated with APAP induced hepatotoxicity [23,24]. The link between these pathways could lead to the identification of a novel biomarker or pathway associated with hepatic adaptation.
Figure 4.
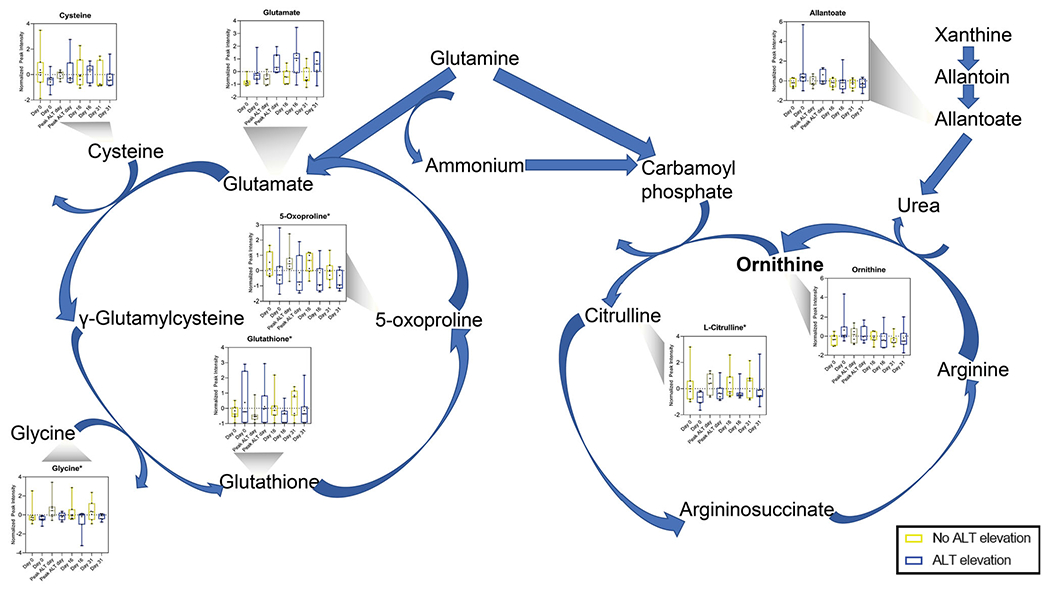
Many metabolites in glutathione metabolism and urea cycle pathways have significantly different expressions between ALT class: urea cycle and glutathione metabolism pathways linked by glutamine. Graphs of metabolite expression over time and between ALT class used data from subset cohort matched to associated pathway. *Significant metabolite determined by high leverage and low SPE in ASCA. ALT elevation, n = 7; No ALT elevation, n = 7.
The combination of ALT, ALKP and ornithine levels is predictive of subjects that will develop increased ALT levels with APAP administration
We used logistic regression in the overall cohort (n = 204) to investigate the impact of significant clinical and metabolomic variables to identify a predictive model for hepatic adaptation in response to APAP administration. Predictors that remain significant in our model include: ALT (p = .011), ALKP (p = .006), and ornithine (p = .022) (Figure 5(A,B)). This model demonstrates fair predictive ability for identifying patients that will have an increase in ALT during the course of APAP treatment with an AUROC curve of 0.799 (confidence interval: 0.715, 0.883; p = <.0001) (Figure 5(C)).
Figure 5.
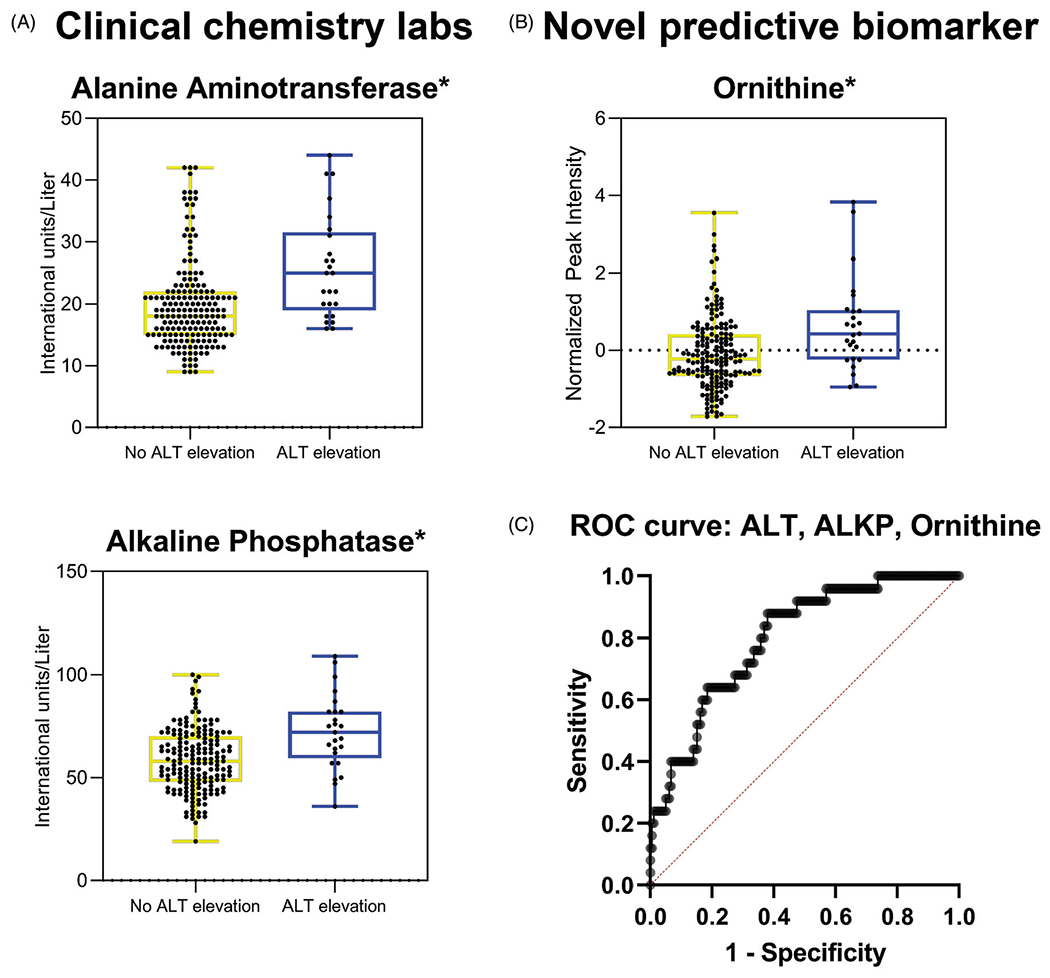
Logistic regression identified a model predictive of APAP response that includes pre-treatment ALT, ALKP, and ornithine: (A) increased ALT and ALKP IU/L levels in pre-treatment samples for subjects who developed an ALT elevation compared to those who did not remained significant in predictive model developed with logistic regression (p=.011 and p=.006, respectively). (B) Increased ornithine peak intensity in pre-treatment samples for subjects who developed an ALT elevation compared to those who did not remained significant in modeling (p=.022). (C) ROC curve that includes pre-treatment serum ALT, ALKP, and ornithine as predictors of ALT elevation with APAP administration, AUROC = 0.799 (confidence interval: 0.715, 0.883; p= <.0001). *p<.05; ALT elevation, n = 25; no ALT elevation, n = 179.
Discussion
Using pre-treatment samples, this study has identified a combination of clinical chemistry and metabolomic biomarkers that are predictive of subjects who will develop transient ALT elevation and subsequent hepatic adaptation. In this study, the adaptation cohort was defined as having a transiently elevated ALT when APAP therapy was administered at maximum recommended therapeutic dosing for 16 consecutive days. Biomarkers and biological pathways were identified for subjects who required hepatic adaptation. The metabolome observed in pre-treatment samples suggests alterations in the glutathione metabolism and the urea cycle associated with hepatic adaptation. These alterations may be indicative of underlying mitochondrial dysfunction, leading to ALT elevation and subsequent hepatic adaptation.
Subjects who required and successfully developed hepatic adaptation in this study had higher ALT and ALKP levels prior to APAP administration than those who never developed an ALT elevation. While more specific to cholestasis than to hepatic injury, as compared with ALT, increased ALKP levels can also suggest hepatic injury. Higher ALKP in the hepatic adaptation group suggests that these patients may already have a subclinical hepatic pathology that leaves these subjects at higher risk of hepatic insult with exposure to drugs that elicit transient ALT elevations. These patients subsequently adapt, allowing ALT to return to baseline. Thus, it is unclear if APAP leads to death of at risk senescent hepatocytes, due to unrecognized pathology, or if those that never elevate ALT are adapted to the drug insult at baseline.
As ALT levels returned to baseline in all but one of the subjects in this study, the transient ALT elevations observed are not clinically significant (Supplemental Figure 1). Hepatotoxicity from therapeutic APAP is exceptionally rare, in fact, it has never been reported in prospective trials [25]. This study further supports therapeutic APAP safety since both the subjects who required hepatic adaptation as well as those who did not experienced no symptoms and labs returned to baseline. Thus, the metabolome observed in the subjects who developed hepatic adaptation may be used as a guide to identify patterns for other drugs that elicit a transient elevation in ALT. Ultimately, this knowledge may provide insight into the mechanisms associated with hepatic adaptation.
Additionally, the knowledge of which subjects will experience asymptomatic increases in ALT with pharmacologic therapy but undergo successful hepatic adaptation could allow more drugs under development to continue through safety trials [2]. Liver injury in trial subjects has been responsible for the cessation of clinical trials for approximately 15% of drug compounds between 1969 and 2002 [26]. Therefore, knowledge of which subjects will undergo hepatic adaptation could guide monitoring for these subjects during the trial and for patients taking the medication once it is publicly available. Cost savings during drug development and recovery of drugs that previously failed clinical trials could result from metabolomic analyses that demonstrate these mechanisms of hepatic adaptation.
There are two main pathways of APAP metabolism – one related to toxic exposure and one related to therapeutic exposure [22]. In both pathways, APAP is metabolized through glucuronidation, sulfation, and oxidation processes in descending proportional order. With toxic exposures due to the excess of substrate (APAP) present and diminished sulfate availability [27], the sulfation pathway becomes saturated and the rate of glucuronidation increases. Additionally, NAPQI production is increased due to oxidation of APAP via cytochrome P450 2E1 (CYP2E1). Glutathione is the primary antioxidant responsible for mitigating the oxidative effects of NAPQI.
Ornithine concentrations, a metabolite involved with the urea cycle, in pre APAP administration samples was predictive of subjects that would need to undergo hepatic adaptation while taking the drug. Since it has been shown that APAP will increase ornithine levels [28], it makes sense that elevated ornithine prior to APAP administration would be associated with subsequent ALT elevation. Allantoate, also involved in urea production, was identified as being significantly elevated in pre APAP administration samples for subjects who would undergo hepatic adaptation. When adjusting for time series, citrulline concentration decreases were also significant, which further highlights the urea cycle relevance to the transient ALT elevation and subsequent adaptation. Increased levels of 5-l-glutamyl-l-glutamine have been observed to be associated with hyperammonemia and urea cycle dysfunction [29]. While there have been prior investigations into the predictive nature of urea as a marker of APAP-associated hepatic injury, they have not led to significant results [30,31]. Therefore, the significant involvement of the urea cycle is likely related to mitochondrial dysfunction, hepatocyte death, and the hepatic adaptation pathway. This may represent variable expression of ornithine transcarbamylase, therefore, we will follow up with analyses that examine genetic variants in this enzyme.
A study showing a link between the arginine metabolism pathway and the glutathione metabolism pathways supports the link between the urea cycle and glutathione pathways via ornithine and glutamate [32]. This suggests that multiple linked pathways are altered in subjects who develop hepatic adaptation with therapeutic APAP exposure. This underscores the multifactorial complex nature of liver injury and recovery highlighting the need for techniques that characterize multiple aspects of the associated biologic pathways to elucidate mechanisms.
The ASCA of time and ALT elevation suggests an alteration to mitochondrial function and energy utilization. Over the course of the study, there were significant changes in expression of 10-hydroxydecanoic acid and tetradecenoyl carnitine over time that differed between groups. Initially, levels of 10-hydroxydecanoic acid decreased in the ALT elevation group suggesting that it was used as an energy source during the initiation of APAP therapy up until peak ALT levels were achieved. By day 16, levels were noticeably increased indicating a shift in the primary energy source for the hepatocytes. The consistently low level of tetradecenoyl carnitine over time, except for a brief increase around day 16, in the ALT elevation group further corroborates the idea of a change in energy utilization due to mitochondrial dysfunction. Mitochondrial damage is a known contributor of APAP toxicity; after NAPQI production through hepatic cytochrome CYP2E1 biotransformation, NAPQI targets mitochondria leading to mitochondrial permeability and subsequent hepatocyte death [8,23].
The results of this study suggest that a combination of routinely collected clinical chemistry values and the biomarker ornithine are able to provide insight into the mechanisms of APAP-induced hepatic adaptation. Alterations in glutathione metabolism and urea cycles discriminate subjects who require hepatic adaptation while taking therapeutic doses of APAP.
There are important limitations to this study that must be considered. First, the number of subjects within this study that showed elevations in ALT throughout the study were few in number and therefore there is a risk that the associations we are observing are by chance. There is a risk that outliers could skew the data. There was only one subject that developed ALT elevation that did not return to baseline during the study timeline. The metabolites in this subject did not appear different from other subjects that had transient ALT elevation. However, the pathophysiology in this subject may be different in subjects with this pattern, though a statistical comparison in a single subject was impractical. This is a rare phenomenon and thousands of subjects would need to be enrolled to determine differences in biologic processes associated with this pattern. Second, this study was unable to control for diet or lifestyle choices which often significantly alter the results of metabolomics studies. However, these factors are present in the general population taking APAP and are thus representative of real-world drug dosing. Third, this study would benefit from having run isotopically labeled standards for metabolites found as significant within the urea cycle – radiolabeled ornithine and citrulline could further identify biologically and clinically relevant metabolite levels. A secondary benefit to using these standards is that they will provide a resource to determine differences in absolute concentrations of metabolites for potential clinical use. The conclusions in this study based on differences in relative quantitation do not provide immediately actionable results. Additionally, validation studies with larger cohorts will be required for absolute metabolite concentrations to have diagnostic utility. Fourth, it is necessary to confirm that metabolome changes identified here are associated broadly with hepatic adaptation rather than specifically related to APAP-associated hepatic adaptation. In addition, it will be important to validate that these changes in metabolite expression are specific to hepatic metabolism and associated drug-induced liver injury. Due to these samples being analyzed from serum, this step will be critical to rule out the possibility that metabolite expressions observed in this study are related to metabolism within other organ systems. These validation studies may include in vitro or in vivo animal studies to verify the mechanistic and physiologic associations observed. This will require a secondary study investigating other drugs that are also known to cause transient ALT elevations. Lastly, there were subjects in both cohorts that showed significant levels of APAP-glucuronide as well as APAP-sulfate at the pre APAP administration study day. Therefore, it might be necessary to require a larger washout period for subjects prior to study initiation and sample collection.
Conclusions
In summary, millions of Americans take over-the-counter APAP for pain and or fever relief. Based on data collected during clinical trials, it is expected that roughly half of the global population will experience an asymptomatic and transient elevation in ALT as defined by an increase in ALT of > 10 IU/L which is a lower threshold than the criteria of ALT >60 IU/L used in this study. There are biomarkers present in serum prior to the initiation of APAP therapy that predict which patients will develop a transiently increased ALT due to therapeutic APAP dosing and subsequent hepatic adaptation. These markers highlight the novel impact of alterations in the urea cycle in response to APAP administration in addition to the known role of the glutathione metabolism pathway. Further investigation into the impact of metabolites associated with the urea cycle, using isotopically labeled standards, is necessary to confirm and elucidate the impact on clinical practice. Further investigation into the mechanisms and pathways associated with hepatic adaptation could improve the understanding of DILI for a wide variety of pharmaceuticals.
Supplementary Material
Funding
Brandon Sonn is funded by CCTSI grant TL1TR002533. Brandon Sonn and Dr Monte are each partially funded by NIH CTSA R35GM124939-01 and NIH CTSI TR001082. The Rocky Mountain Poison & Drug Safety has contracts with McNeil Consumer Healthcare, the maker of Tylenol (acetaminophen). Otherwise, all authors have nothing to disclose.
Footnotes
Supplemental data for this article can be accessed here.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Gayam V, Khalid M, Shrestha B, et al. Drug-induced liver injury: an institutional case series and review of literature. J Investig Med High Impact Case Rep. 2018;6:2324709618761754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kullak-Ublick GA, Andrade RJ, Merz M, et al. Drug-induced liver injury: recent advances in diagnosis and risk assessment. Gut. 2017;66(6):1154–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Suk KT, Kim DJ. Drug-induced liver injury: present and future. Clin Mol Hepatol. 2012;18(3):249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].McGill MR, Jaeschke H. Mechanistic biomarkers in acetaminophen-induced hepatotoxicity and acute liver failure: from preclinical models to patients. Expert Opin Drug Metab Toxicol. 2014;10(7):1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dara L, Liu Z-X, Kaplowitz N. Mechanisms of adaptation and progression in idiosyncratic drug induced liver injury, clinical implications. Liver Int. 2016;36(2):158–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Maeda M, Tanaka R, Aso M. Hepatic adaptation to therapeutic doses of acetaminophen: an exploratory study in healthy individuals. Clin Ther. 2020;42(7):1276–1291e1. [DOI] [PubMed] [Google Scholar]
- [7].Yoon E, Babar A, Choudhary M, et al. Acetaminophen-induced hepatotoxicity: a comprehensive update. J Clin Transl Hepatol. 2016;4(2):131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hinson JA, James LP. Mechanisms of acetaminophen-induced liver necrosis. Handb Exp Pharmacol. 2010;196:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Watkins PB, Slattery JT, Colonese CR, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily - a randomized controlled trial. J Am Med Assoc. 2006;296:7. [DOI] [PubMed] [Google Scholar]
- [10].Heard K, Green JL, Anderson V, et al. A randomized, placebo-controlled trial to determine the course of aminotransferase elevation during prolonged acetaminophen administration. BMC Pharmacol Toxicol. 2014;15:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].O’Connell TM, Watkins PB. The application of metabonomics to predict drug-induced liver injury. Clin Pharmacol Ther. 2010;88(3):394–399. [DOI] [PubMed] [Google Scholar]
- [12].Ganetsky M, Berg AH, Solano JJ, et al. Metabolomic analysis of acetaminophen induced subclinical liver injury. Clin Toxicol (Phila). 2019;9:1–9. [DOI] [PubMed] [Google Scholar]
- [13].Jetten MJ, Gaj S, Ruiz-Aracama A, et al. Omics analysis of low dose acetaminophen intake demonstrates novel response pathways in humans. Toxicol Appl Pharmacol. 2012;259(3):320–328. [DOI] [PubMed] [Google Scholar]
- [14].Wong A, Graudins A. Risk prediction of hepatotoxicity in paracetamol poisoning. Clin Toxicol (Phila). 2017;55(8):879–892. [DOI] [PubMed] [Google Scholar]
- [15].US Food Drug Administration. Guidance for industry and investigators - safety reporting requirements for INDs and BA/BE studies. Silver Spring (MD): US Food Drug Administration; 2012. p. 32. [Google Scholar]
- [16].Sonn BJ, Saben JL, McWilliams G, et al. Predicting response to lisinopril in treating hypertension: a pilot study. Metabolomics. 2019;15(10):133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nemkov T, Reisz JA, Gehrke S, et al. High-throughput metabolomics: isocratic and gradient mass spectrometry-based methods. In: D’Alessandro A, editor. High-throughput metabolomics: methods and protocols. New York (NY): Springer New York; 2019. p. 13–26. [DOI] [PubMed] [Google Scholar]
- [18].Thonusin C, IglayReger HB, Soni T, et al. Evaluation of intensity drift correction strategies using MetaboDrift, a normalization tool for multi-batch metabolomics data. J Chromatogr A. 2017;1523:265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nemkov T, D’Alessandro A, Hansen KC. Three-minute method for amino acid analysis by UHPLC and high-resolution quadrupole orbitrap mass spectrometry. Amino Acids. 2015;47(11):2345–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nemkov T, Hansen KC, D’Alessandro A. A three-minute method for high-throughput quantitative metabolomics and quantitative tracing experiments of central carbon and nitrogen pathways. Rapid Commun Mass Spectrom. 2017;31(8):663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chong J, Soufan O, Li C. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018;46(W1):W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mazaleuskaya LL, Sangkuhl K, Thorn CF, et al. PharmGKB summary: pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet Genomics. 2015;25(8):416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Anup Ramachandran LD, Akakpo JY, Jaeschke H. Mitochondrial dysfunction as a mechanism of drug-induced hepatotoxicity: current understanding and future perspectives. J Clin Transl Res. 2018;4(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ramachandran A, Jaeschke H. Acetaminophen toxicity: novel insights into mechanisms and future perspectives. Gene Expr. 2018;18(1):19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Richard C, Dart EB. Does therapeutic use of acetaminophen cause acute liver failure. Pharmacotherapy. 2007;27(9):12. [DOI] [PubMed] [Google Scholar]
- [26].Diane K, Wysowski LS. Adverse drug event surveillance and drug withdrawals in the United States, 1969–2002. JAMA Intern Med. 2005;165(12):7. [DOI] [PubMed] [Google Scholar]
- [27].Li J, Chiew AL, Isbister GK, et al. Sulfate conjugation may be the key to hepatotoxicity in paracetamol overdose. Br J Clin Pharmacol. 2020;87(5):2392–2396. [DOI] [PubMed] [Google Scholar]
- [28].Eda Ozcelik SU, Burukoglu D, Musmul A. Chitosan and blueberry treatment induces arginase activity and inhibits nitric oxide production during acetaminophen-induced hepatotoxicity. Phamacogn Mag. 2014;10(38):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hammond JW, Potter M, Truscott R, et al. Gamma-glutamylglutamine identified in plasma and cerebrospinal fluid from hyperammonaemic patients. Clin Chim Acta. 1990;194(2–3):173–183. [DOI] [PubMed] [Google Scholar]
- [30].Waring WS, Stephen AF, Robinson OD, et al. Serum urea concentration and the risk of hepatotoxicity after paracetamol overdose. QJM. 2008;101(5):359–363. [DOI] [PubMed] [Google Scholar]
- [31].Zyoud SH, Awang R, Sulaiman SA, et al. Impact of serum acetaminophen concentration on changes in serum potassium, creatinine and urea concentrations among patients with acetaminophen overdose. Pharmacoepidemiol Drug Saf. 2011;20(2):203–208. [DOI] [PubMed] [Google Scholar]
- [32].Liang M, Wang Z, Li H, et al. l-Arginine induces antioxidant response to prevent oxidative stress via stimulation of glutathione synthesis and activation of Nrf2 pathway. Food Chem Toxicol. 2018;115:315–328. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.