

Abstract
Protective immunity against COVID-19 likely depends on the production of SARS-CoV-2 specific plasma cells and memory B cells after infection or vaccination. Previous work has found that germinal center reactions are disrupted in severe COVID-19. This may adversely affect long-term immunity against re-infection. Consistent with an extrafollicular B cell response, patients with severe COVID-19 have elevated frequencies of clonally expanded, class switched, unmutated plasmablasts. However, it is unclear whether B cell populations in individuals with mild COVID-19 are similarly skewed. Here, we use single cell RNA sequencing of B cells to show that, in contrast to patients with severe COVID-19, subjects with mildly symptomatic COVID-19 have B cell repertoires enriched for clonally diverse, somatically-hypermutated memory B cells approximately 30 days after the onset of symptoms. This provides evidence that B cell responses are less disrupted in mild COVID-19, and result in the production of memory B cells.
Introduction
COVID-19 is a pandemic, life-threatening disease caused by the virus SARS-CoV-2. As of February 2021, there have been over 100 million confirmed cases of COVID-19 which have led to over 2 million deaths globally (1). The clinical course of SARS-CoV-2 infection is highly variable, ranging from often asymptomatic infection in healthy young adults to severe pneumonia and multi-system failure that is more prevalent in the elderly and those with comorbidities. The immunological response has been implicated both in overcoming infection and in contributing to severe disease (2). The quality and persistence of immunological memory after natural infection is a critical factor in suppressing re-infection and ending the pandemic. Immunological memory is mediated in part by B cells, which mature in germinal centers (GC) to produce neutralizing antibodies during initial infection (3). These B cells can then differentiate into long lived plasma cells which release antibodies into the blood, and memory B cells which can be quickly activated upon re-infection (4). Many efforts to develop successful vaccines depend on stimulating GCs to produce memory B cells, as well as plasma cells that release neutralizing antibodies (5).
Recent work has shown that the GC response, which is important for long term immunity, is disrupted in severe COVID-19 (6, 7). Supporting this, others have shown evidence of broad changes in B cell populations such as an expansion of B cell clones in COVID-19 that lack somatic hypermutation (8–10) (ref. 9 posted on preprint server). While much of this work has focused on severely ill COVID-19 patients, most COVID-19 cases are mild or asymptomatic. If previously documented disruptions in the B cell response are primarily found in severe COVID-19, it is possible that mild cases can produce effective long-term immunity through affinity maturation within GCs.
Here, we use single-cell RNA sequencing (scRNA-seq) of B cells from subjects with mild COVID-19 to investigate whether a successful B cell response is detected approximately one month after the onset of symptoms. We find that subjects with mild COVID-19 have high frequencies of clonally diverse, somatically hypermutated memory B cells compared to patients with severe COVID-19 and healthy controls. By contrast, 2/3 patients with severe COVID-19 at a similar timepoint showed a high frequency of unmutated plasmablasts and clonal expansions, consistent with an early and/or extrafollicular B cell response. Overall, our results provide evidence that previously documented disruptions in the B cell response during severe COVID-19 are less detectable in mild COVID-19, and that mild or asymptomatically infected individuals produce large populations of mutated memory B cells approximately one month after the onset of symptoms. These cells potentially form the basis of longer-term protective immunity to SARS-CoV-2.
Materials and Methods
Patient cohorts
Cryopreserved PBMCs were obtained from Precision for Medicine (Frederick, MD) which collected and processed samples from symptomatic SARS-CoV-2 patients at least 21 days post symptom onset (DPSO) after informed consent. All subjects were confirmed to have COVID-19 by RT-PCR testing of nasopharyngeal samples (Hologic Panther Fusion SARS-CoV-2 assay). All seven subjects included in this study expressed varying degrees of SARS-CoV-2 symptoms, but none required hospitalization. Normal human PBMCs from four healthy control subjects that were collected prior to the pandemic were obtained from the same vendor. Additionally, previously generated data from three hospitalized patients from the Yale COVID-19 IMPACT (Implementing Medical and Public Health Action Against Coronavirus CT) Biorepository were included in the analysis (9). We classified these patients as “severe” COVID-19 on the basis of hospitalization. The term “severe” includes both “progressive” patients that were hospitalized in the ICU and eventually succumbed to their disease, and “stable” patients hospitalized in Internal Medicine wards whose condition improved, as defined in (9) (posted on preprint server). Baseline characteristics of all patients are provided in Supplemental Table I.
Sample preparation and single cell RNA + BCR sequencing
All sample processing steps were done in a biosafety level 4 facility of the Galveston National Laboratory. Cryopreserved PBMCs were thawed before being slowly transferred to a 50 ml conical tube and diluted in PBS containing 0.5 mM EDTA buffer (PEB). The cells were then centrifuged at 300 x g for 5 minutes at 4°C, washed twice and resuspended in 0.5 ml PEB. B cells were isolated from PBMCs by Human Pan B cell immunomagnetic negative selection kit (StemCell Technologies, MA) per manufacturer’s instructions. Cell concentration was determined using Trypan blue staining with a TC20 automated cell counter (Bio-Rad, CA) and adjusted if necessary to achieve the desired concentration of 700–1200 cells/μl.
10x barcoding, library preparation and sequencing
Approximately 7,000 purified B-cells from each sample were combined with Chromium Next GEM Single Cell 5ʹ Gel Beads v1.1 and Reverse Transcription master mix (10x Genomics, CA) to generate barcoded Gel Beads in Emulsion (GEM) according to manufacturer’s recommendations. The GEMs were recovered and purified using silane magnetic beads (Thermo Fisher Scientific, MA). The full-length cDNA along with cell-barcode identifiers were PCR amplified for 11 cycles and then purified using SPRISelect magnetic beads (Beckman Coulter, CA). The purified cDNA was inactivated in 80% ethanol before being removed from BSL-4. The quality of ethanol precipitated DNA was assessed by Bioanalyzer, and both B-cell receptor (BCR) and 5’ gene expression libraries (GEX) were prepared as per manufacturer’s instructions. Libraries were quantified, both GEX and BCR libraries were pooled according to equivalent molar concentrations and sequenced on Illumina NovaSeq6000 sequencing platform. BCR libraries were sequenced by Illumina Novaseq and the output binary base call (BCL) files were demultiplexed and converted into FASTQ files with bcl2fastq. We then used Cell Ranger pipeline v4.0 (11) commands count and vdj respectively for the single cell gene expression libraries and B-cell receptor libraries to obtain a data matrix of expression for all genes and all cells.
B cell subtype identification and B cell receptor analysis
Detailed descriptions of B cell subtype identification and B cell receptor (BCR) repertoire analysis are provided in Supplemental Fig. 1. Briefly, scRNA-seq gene expression information was preprocessed and clustered using Seurat v3.2.2 (12). Only barcodes corresponding to B cell subtypes with associated BCRs were included. B cell subtypes were assigned using the immunoStates database (13) and known marker genes for plasmablasts (PRDM1, XBP1), memory B cells (CD24, TNFRSF13B) and naive B cells (IGHD, IL4R, TCL1A) (Fig. 1). We identified one plasmablast cluster, as well as four naive and three memory B cell clusters which were merged. We also identified two small doublet clusters which were removed. Cell type annotations from patients with severe COVID-19 were largely concordant (1132/1186 cells) with their previous annotations in (9) (Supplemental Fig. 3E). BCR repertoire analysis was performed using the Immcantation suite. To obtain V, D, and J gene assignments, BCR sequences were aligned to the IMGT v3.1.24 germline reference database using IgBLAST v1.13.0 (14, 15). Single-linkage hierarchical clustering among cells with common V genes, J genes, and junction lengths was used to group clonal clusters (16, 17). SHM level was calculated as the frequency of non-ambiguous mismatches from each cell to the V gene of its inferred germline sequence. Clonal diversity was performed using uniform downsampling with Alakazam v1.0.2.999 (18).
Figure 1:
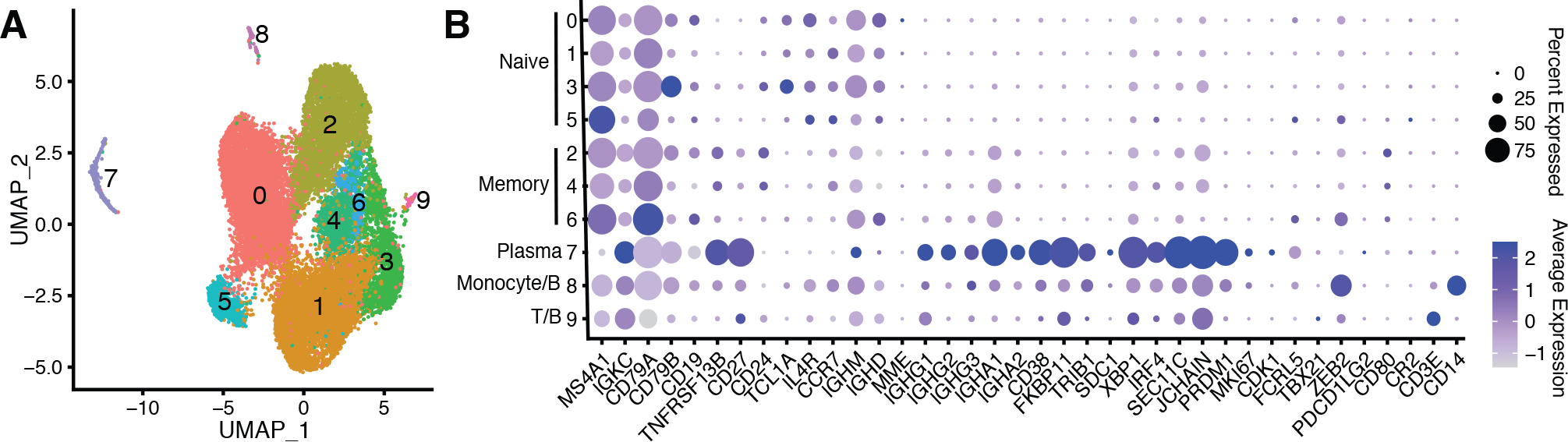
A) UMAP of cells obtained from patients with mild and severe COVID-19, as well as healthy control subjects. Each point represents a single cell. B) Dot plots showing expression of B cell marker genes for naive, memory, and plasmablast B cell subsets for the clusters identified in panel A.
Data availability
RNA sequencing data have been deposited in NCBI’s Gene Expression Omnibus and are available at the GEO Series accession number GSE164381 (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164381). Scripts to reproduce these analyses are available at: https://bitbucket.org/kleinstein/projects.
Results
B cell repertoires of subjects with mild COVID-19 have elevated levels of memory B cells compared to patients with severe COVID-19
B cells are divided into distinct functional subtypes. Naive B cells are antigen-inexperienced precursors to other B cell subtypes, plasmablasts are antibody producing B cells released into the blood, and memory B cells are antigen-experienced cells produced during an immune response that can be activated upon re-infection (4). To characterize these B cell subtypes in subjects with mild COVID-19, we obtained scRNA-seq and paired BCR sequence data from the peripheral blood of four healthy subjects and seven subjects with mild COVID-19. These subjects with mild COVID-19 were sampled between 24 and 37 (mean = 29.3) days after reported onset of symptoms. These subjects reported few symptoms and none were hospitalized. To compare the B cell response in mild and severe COVID-19 at similar timepoints, we also obtained scRNA-seq data from a prior study of three hospitalized COVID-19 patients sampled between 20 and 23 (mean = 21) days after reported onset of symptoms (9) (posted on preprint server). We categorized these hospitalized patients as having “severe” COVID-19. These patients included one “progressive” patient, who eventually succumbed to his disease, and two “stable” patients whose condition improved, as defined in (9). We identified naive, memory, and plasmablast B cell subsets in each of these cohorts by clustering cells with similar gene expression patterns (Methods). We identified a single plasmablast cluster, multiple naive and memory B cell clusters which were later merged, and two B cell doublet clusters which were discarded (Methods, Fig. 1, Supplemental Figs. 2 and 3).
Prior work has shown that severe COVID-19 can cause large changes in B cell populations 1 – 3 weeks after the onset of symptoms, such as increased plasmablast frequency (9, 19, 20) (ref. 9 posted on preprint server). It is not known if subjects with mild COVID-19 have similar changes. To test this, we quantified the frequency of naive, memory, and plasmablast B cells in each cohort. Consistent with a resting immune system, healthy controls had a significantly higher frequency of naive B cells compared to subjects with mild COVID-19 (Fig. 2A, p = 0.0061, Wilcoxon test). By contrast, patients with severe COVID-19 had a higher frequency of plasmablasts compared to mild COVID-19, though this difference did not reach significance (Fig. 2A, p = 0.067, Wilcoxon test). These plasmablast expansions are consistent with a severe infection that is ongoing or recently resolved. Subjects with mild COVID-19 had a significantly higher frequency of memory B cells than either healthy subjects or patients with severe COVID-19 (Fig. 2A, p = 0.012 and 0.033 respectively, Wilcoxon test), consistent with recently resolved infection. These results show that, ~30 days after symptom onset, mild COVID-19 induces large scale changes in B cell populations characterized by increased frequency of memory B cells, in contrast to severe COVID-19, which is characterized by an increased plasmablast frequency at a similar time-point.
Figure 2:
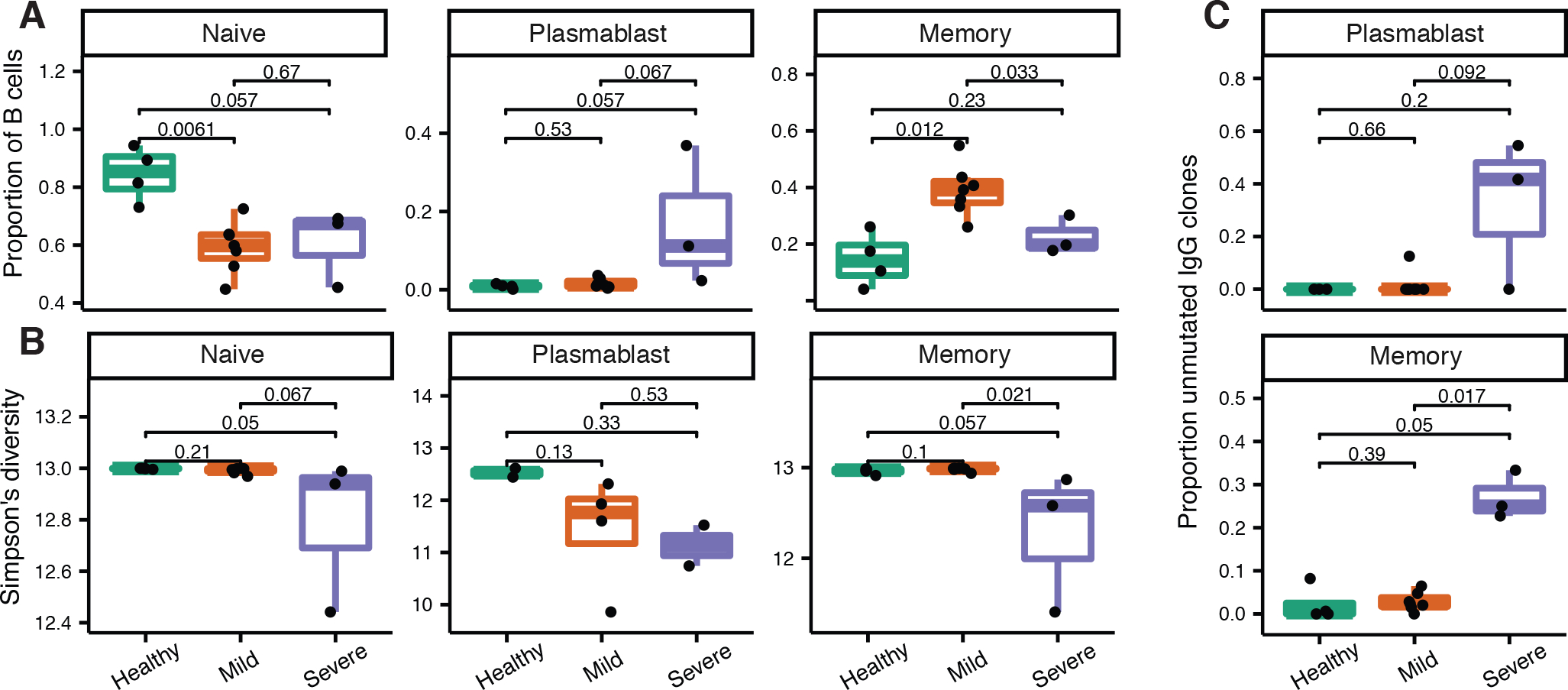
Frequency of B cell subtypes and unmutated B cell clones in all patient cohorts. A) Naive (clusters 0, 1, 3, 5), plasmablast (cluster 7), and memory (clusters 2, 4, 6) B cell frequencies among total B cells in each cohort. B) Clonal diversity of each B cell subset in each cohort. To account for differences in sequence depth among samples, Simpson’s diversity was calculated as the mean over 1000 resampling realizations to a uniform sequence depth among samples and cell type. C) Proportion of IgG B cell clones that are unmutated (median SHM < 1%) in each cohort. The proportion of unmutated IgG clones was not considered for cells within naive B cell clusters, because these likely represent a small number of mis-clustered cells (Supplemental Fig. 3A). P values in all panels were calculated using a Wilcoxon test.
Subjects with mild COVID-19 have more clonally diverse B cell repertoires than patients with severe COVID-19
During a GC reaction, B cells rapidly proliferate and undergo somatic hypermutation (SHM), which alters the DNA sequence of their BCRs. A group of B cells that descend from the same naive B cell but potentially differ by SHM variants is referred to as a “clone.” B cell clones can be identified bioinformatically by clustering B cells based on BCR nucleotide sequence similarity (Methods). While B cell repertoires in the blood of healthy subjects typically have high clonal diversity (i.e. many clones with few cells each), the repertoires of patients with severe COVID-19 often have lower clonal diversity, with a small number of large clones (9, 20) (ref. 9 posted on preprint server). These large B cell clones are consistent with a substantial and ongoing B cell response. To test if subjects with mild COVID-19 also have low clonal diversity, we calculated Simpson’s diversity for each subject and each cell type after down-sampling to the same sequence depth for each subject and cell type. This analysis showed that the memory B cell repertoires of subjects with mild COVID-19 were significantly more clonally diverse than those of patients with severe COVID-19 (Fig. 2B, p=0.021, Wilcoxon test), and similarly diverse as those of healthy controls (Fig. 2B, p=0.1). Interestingly, we observed a similar trend towards lower diversity in naive B cells in patients with severe COVID-19 compared to healthy and mild COVID-19 subjects (Fig. 2B). This is unlikely to be driven by mis-clustered naive cells, as all B cells in non-singleton naive clones in severe patients were unmutated and IgM or IgD. Overall, these results indicate that the clonal diversity of mild COVID-19 is more similar to that of healthy, resting repertoires than of severe COVID-19.
Patients with severe COVID-19 have increased unmutated IgG memory B cell and plasmablast clones compared to those with mild COVID-19
During acute infection, naive B cells can class switch and differentiate outside of GCs (21). Previous work has shown that GCs can be lost in severe COVID-19 (6), suggesting the B cell response is primarily extrafollicular. This is supported by other studies showing a high frequency of unmutated IgG clones in patients with severe COVID-19 (7, 9, 10). To determine whether this high frequency of unmutated IgG B cells is also present in mild COVID-19 and healthy controls, we calculated the proportion of B cell clones that were unmutated (defined as having a median SHM frequency < 1%) separated by cell types and isotypes in each cohort. Patients with severe COVID-19 had a significantly higher proportion of unmutated IgG memory B cell clones compared to mild COVID-19 (Fig. 2C, p = 0.017, Wilcoxon test), as well as a non-significantly higher proportion of unmutated IgG plasmablast clones compared to mild COVID-19 (Fig. 2C, p = 0.092, Wilcoxon test). The proportion of either unmutated IgG plasmablast or memory B cell clones was not detectably different in subjects with mild COVID-19 compared to healthy controls (p = 0.66 and 0.39, respectively, Wilcoxon test). We did not find any significant differences among cohorts when repeating these analyses with IgA B cells (Supplemental Fig. 2E). These results suggest subjects with mild COVID-19 undergo affinity maturation which produces class-switched, somatically hypermutated memory B cell clones by 3 – 4 weeks after the onset of symptoms, a process which is potentially impaired in patients with severe COVID-19.
Discussion
An appropriate B cell response is critical to the development of long-term protective immunity. Previous studies have shown evidence that GC B cell responses are disrupted in severe COVID-19 (6). Patients with severe COVID-19 show signs of extrafollicular B cell response, with clonal expansions, an elevated frequency of plasmablasts, as well as an elevated frequency of unmutated memory B cell and plasmablast clones approximately three weeks after symptom onset. By contrast, we show here that individuals with mild COVID-19 have elevated frequencies of clonally diverse, somatically hypermutated memory B cells at a similar timepoint.
B cells play a critical role in the development of long term protective immunity, and previous work has shown that GCs are frequently lost during severe COVID-19 (6). Disruption of GCs has been shown to correlate with COVID-19 morbidity (7), reinforcing our conclusions that extrafollicular B cell responses are a hallmark of severe rather than mild COVID-19. Woodruff et al. (7) also showed that extrafollicular responses correlate with the production of neutralizing antibodies, showing GC reactions may not be necessary for short term neutralizing antibody production against SARS-CoV-2. It is unclear whether disruption of GCs in patients with severe COVID-19 reduces the development of SARS-CoV-2 specific memory B cells. Indeed, the extent to which extrafollicular responses produce memory B cells is not clear in general (22). While we find that B cell populations in patients with severe COVID-19 are skewed towards plasmablasts, it is possible these patients develop more memory B cells at a later time than sampled in this study. In general, it is important to note that all results in this study pertain to a cohort of COVID-19 patients sampled 3 – 4 weeks after the onset of symptoms. The blood compartment in most patients is likely to return to a baseline state closely resembling healthy controls at later timepoints.
Our results are consistent with other recent studies describing changes in B cell repertoires in subjects with COVID-19. Several studies have documented plasmablast expansions in severe COVID-19 (9, 19, 20) (ref. 9 posted on preprint server). Our results show that mildly symptomatic subjects, by contrast, have signs of affinity maturation such as mutated memory B cells 3 – 4 weeks after the onset of symptoms. This is consistent with recent studies showing memory B cell production following mild COVID-19. Rodda et al. (23) documented mutated, SARS-CoV-2 binding memory B cells 1 – 3 months following onset of symptoms. Gaebler et al. (24) also documented SARS-CoV-2 binding memory B cells and ongoing affinity maturation in a large cohort of primarily mild COVID-19 subjects. Consistent with our results, Kuri-Cervantes et al. (20) showed that recovered subjects with mild COVID-19 had plasmablast frequencies similar to healthy controls. Kuri-Cervantes et al. (20) did not explicitly compare total memory B cell proportions among patient cohorts, so it is not clear how those results compare to ours. Together with these studies, our results provide further evidence that mildly infected SARS-CoV-2 subjects develop immunological memory after infection. Our results show that these changes are detectable even without filtering to SARS-CoV-2 binding B cells. One advantage of the scRNA-seq + BCR approach taken in this study is that BCR sequences can be paired with B cell subtypes for individual cells. This shows, for instance, memory B cells in subjects with mild COVID-19 are both somatically hypermutated and clonally diverse.
There are a few caveats to consider in interpreting these results. First, only three patients with severe COVID-19 were included from (9) (posted to preprint server) due to the earlier timepoint of most patients in that study. However, that study’s other patients show similar patterns of unmutated plasmablast expansions, so the trends we observed are unlikely to be unique to the patients we selected. Second, the patients with severe COVID-19 included were sampled on average 7 days earlier after the onset of symptoms than subjects with mild COVID-19. It is possible the observed differences in these cohorts are due to differences in sampling time. However, if this were true, we would expect subjects with mild COVID-19 sampled earlier to have more plasmablasts and fewer memory B cells. By contrast, the two subjects with mild COVID-19 sampled the earliest (24 days post symptoms) showed similarly high frequencies of memory B cells (33% and 36%) as the full mild COVID-19 cohort (mean = 39%, range = 26% - 55%). Patients with severe COVID-19 were also older than the mildly infected individuals (mean = 72 vs 38 years old, respectively), and were treated with the IL-6 inhibitor tocilizumab. It is possible the observed differences in B cell repertoires are due to those factors. However, we do not believe this is due to tocilizumab treatment because a similarly high frequency of plasmablasts and unmutated IgG clones was also observed in patients not treated with tocilizumab in (9). Further, previous work did not find a relationship between age and plasmablast expansion in severe COVID-19 (20). While prior work has shown a decline in repertoire diversity with age (25), we did not observe a significant decrease in diversity with age within cohorts (Supplemental Fig. 2D). Finally, while our clustering is driven largely by heathy and mild cohorts (Supplemental Fig. 2C), we do not expect this to affect major B cell subtype classification, particularly because annotations in the severe cohort are concordant with those in their original study (Supplemental Fig. 3E).
There are several limitations beyond the scope of this study. It is not known whether sample preparation and sequencing protocols used were affected by COVID-19 pathogenesis, which may affect cell type frequencies. Further, the data we used are from peripheral blood samples, which may poorly represent the total B cell repertoire during COVID-19. Finally, because we do not have antigen specificity information for the cells we observe, we cannot conclude that the memory B cell populations in the mild COVID-19 cohort (or any cohort or cell type) are SARS-CoV-2 specific. Future studies with larger and better-controlled cohorts, validation through other methods such as flow cytometry, and antigen specificity information are warranted.
Overall, the results of this study show that B cell repertoires in patients with mild COVID-19 are not as disrupted as in severe COVID-19 approximately one month after the onset of symptoms. This study further demonstrates that subjects with mildly symptomatic COVID-19 produce somatically hypermutated memory B cells after infection, which potentially form a key component of immunological memory to SARS-CoV-2.
Supplementary Material
Key points:
Mild COVID-19 patients have increased memory B cells 1 month after symptom onset.
BCR repertoires of mild COVID-19 patients are clonally diverse and mutated.
Acknowledgements
We would like to thank Dr. Yongchao Ge for assistance with data processing and submission.
Funded in part by the National Institutes of Health, National Institute of Allergy and Infectious Diseases grant R01 AI104739. S.C.S., A.B., and S.H.K. were supported by Defense Advanced Research Projects Agency contract number N6600119C4022. NK is funded by NIH NHLBI grants R01HL127349, R01HL141852, U01HL145567, UH2HL123886.
Footnotes
Competing interests
K.B.H. receives consulting fees from Prellis Biologics. S.H.K. receives consulting fees from Northrop Grumman. N.K. served as a consultant to Boehringer Ingelheim, Third Rock, Pliant, Samumed, NuMedii, Theravance, LifeMax, Three Lake Partners, Optikira, Astra Zeneca, Augmanity over the last 3 years, reports Equity in Pliant and a grant from Veracyte and Boehringer Ingelheim and non-financial support from MiRagen and Astra Zeneca. N.K. has IP on novel biomarkers and therapeutics in IPF licensed to Biotech, all of which are outside the scope of this paper.
References
- 1.Dong E, Du H, and Gardner L. 2020. An interactive web-based dashboard to track COVID-19 in real time. The Lancet Infectious Diseases 20: 533–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.García LF 2020. Immune Response, Inflammation, and the Clinical Spectrum of COVID-19. Front. Immunol. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Victora GD, and Nussenzweig MC. 2012. Germinal Centers. Annual Review of Immunology 30: 429–457. [DOI] [PubMed] [Google Scholar]
- 4.Murphy K, Travers P, Walport M, and Janeway C. 2012. Janeway’s immunobiology,. Garland Science, New York. [Google Scholar]
- 5.Haynes BF, Kelsoe G, Harrison SC, and Kepler TB. 2012. B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat Biotech 30: 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaneko N, Kuo H-H, Boucau J, Farmer JR, Allard-Chamard H, Mahajan VS, Piechocka-Trocha A, Lefteri K, Osborn M, Bals J, Bartsch YC, Bonheur N, Caradonna TM, Chevalier J, Chowdhury F, Diefenbach TJ, Einkauf K, Fallon J, Feldman J, Finn KK, Garcia-Broncano P, Hartana CA, Hauser BM, Jiang C, Kaplonek P, Karpell M, Koscher EC, Lian X, Liu H, Liu J, Ly NL, Michell AR, Rassadkina Y, Seiger K, Sessa L, Shin S, Singh N, Sun W, Sun X, Ticheli HJ, Waring MT, Zhu AL, Alter G, Li JZ, Lingwood D, Schmidt AG, Lichterfeld M, Walker BD, Yu XG, Padera RF, and Pillai S. 2020. Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell 183: 143–157.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woodruff MC, Ramonell RP, Nguyen DC, Cashman KS, Saini AS, Haddad NS, Ley AM, Kyu S, Howell JC, Ozturk T, Lee S, Suryadevara N, Case JB, Bugrovsky R, Chen W, Estrada J, Morrison-Porter A, Derrico A, Anam FA, Sharma M, Wu HM, Le SN, Jenks SA, Tipton CM, Staitieh B, Daiss JL, Ghosn E, Diamond MS, Carnahan RH, Crowe JE, Hu WT, Lee FE-H, and Sanz I. 2020. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nature Immunology 21: 1506–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kreer C, Zehner M, Weber T, Ercanoglu MS, Gieselmann L, Rohde C, Halwe S, Korenkov M, Schommers P, Vanshylla K, Cristanziano VD, Janicki H, Brinker R, Ashurov A, Krähling V, Kupke A, Cohen-Dvashi H, Koch M, Eckert JM, Lederer S, Pfeifer N, Wolf T, Vehreschild MJGT, Wendtner C, Diskin R, Gruell H, Becker S, and Klein F. 2020. Longitudinal Isolation of Potent Near-Germline SARS-CoV-2-Neutralizing Antibodies from COVID-19 Patients. Cell 182: 843–854.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Unterman A, Sumida TS, Nouri N, Yan X, Zhao AY, Gasque V, Schupp JC, Asashima H, Liu Y, Cosme C, Deng W, Chen M, Raredon MSB, Hoehn K, Wang G, Wang Z, Deiuliis G, Ravindra NG, Li N, Castaldi C, Wong P, Fournier J, Bermejo S, Sharma L, Casanovas-Massana A, Vogels CBF, Wyllie AL, Grubaugh ND, Melillo A, Meng H, Minasyan M, research Team TYI, Niklason LE, Ko AI, Montgomery RR, Farhadian SF, Iwasaki A, Shaw AC, van Dijk D, Zhao H, Kleinstein SH, Hafler DA, Kaminski N, and Cruz CSD. 2020. Single-Cell Omics Reveals Dyssynchrony of the Innate and Adaptive Immune System in Progressive COVID-19. medRxiv 2020.07.16.20153437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nielsen SCA, Yang F, Jackson KJL, Hoh RA, Röltgen K, Jean GH, Stevens BA, Lee J-Y, Rustagi A, Rogers AJ, Powell AE, Hunter M, Najeeb J, Otrelo-Cardoso AR, Yost KE, Daniel B, Nadeau KC, Chang HY, Satpathy AT, Jardetzky TS, Kim PS, Wang TT, Pinsky BA, Blish CA, and Boyd SD. 2020. Human B Cell Clonal Expansion and Convergent Antibody Responses to SARS-CoV-2. Cell Host & Microbe 28: 516–525.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall-Levin M, Wyatt PW, Hindson CM, Bharadwaj R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C, Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, and Bielas JH. 2017. Massively parallel digital transcriptional profiling of single cells. Nat Commun 8: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R. 2018. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology 36: 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vallania F, Tam A, Lofgren S, Schaffert S, Azad TD, Bongen E, Haynes W, Alsup M, Alonso M, Davis M, Engleman E, and Khatri P. 2018. Leveraging heterogeneity across multiple datasets increases cell-mixture deconvolution accuracy and reduces biological and technical biases. Nature Communications 9: 4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giudicelli V, Chaume D, and Lefranc M-P. 2005. IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucl. Acids Res. 33: D256–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye J, Ma N, Madden TL, and Ostell JM. 2013. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucl. Acids Res. 41: W34–W40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta NT, Adams KD, Briggs AW, Timberlake SC, Vigneault F, and Kleinstein SH. 2017. Hierarchical Clustering Can Identify B Cell Clones with High Confidence in Ig Repertoire Sequencing Data. The Journal of Immunology 198: 2489–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yaari G, Vander Heiden JA, Uduman M, Gadala-Maria D, Gupta N, Stern JNH, O’Connor KC, Hafler DA, Laserson U, Vigneault F, and Kleinstein SH. 2013. Models of Somatic Hypermutation Targeting and Substitution Based on Synonymous Mutations from High-Throughput Immunoglobulin Sequencing Data. Front Immunol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, and Kleinstein SH. 2015. Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31: 3356–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martínez-Colón GJ, McKechnie JL, Ivison GT, Ranganath T, Vergara R, Hollis T, Simpson LJ, Grant P, Subramanian A, Rogers AJ, and Blish CA. 2020. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nature Medicine 26: 1070–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuri-Cervantes L, Pampena MB, Meng W, Rosenfeld AM, Ittner CAG, Weisman AR, Agyekum RS, Mathew D, Baxter AE, Vella LA, Kuthuru O, Apostolidis SA, Bershaw L, Dougherty J, Greenplate AR, Pattekar A, Kim J, Han N, Gouma S, Weirick ME, Arevalo CP, Bolton MJ, Goodwin EC, Anderson EM, Hensley SE, Jones TK, Mangalmurti NS, Luning Prak ET, Wherry EJ, Meyer NJ, and Betts MR. 2020. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 5: eabd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete PF, Ellyard J, Shen Q, Robert PA, Cappello J, Vohra H, Zhang Y, Nowosad CR, Schiepers A, Corcoran LM, Toellner K-M, Polo JM, Meyer-Hermann M, Victora GD, and Vinuesa CG. 2019. Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity 51: 337–350.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elsner RA, and Shlomchik MJ. 2020. Germinal Center and Extrafollicular B Cell Responses in Vaccination, Immunity, and Autoimmunity. Immunity 53: 1136–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodda LB, Netland J, Shehata L, Pruner KB, Morawski PA, Thouvenel CD, Takehara KK, Eggenberger J, Hemann EA, Waterman HR, Fahning ML, Chen Y, Hale M, Rathe J, Stokes C, Wrenn S, Fiala B, Carter L, Hamerman JA, King NP, Gale M, Campbell DJ, Rawlings DJ, and Pepper M. 2021. Functional SARS-CoV-2-Specific Immune Memory Persists after Mild COVID-19. Cell 184: 169–183.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaebler C, Wang Z, Lorenzi JCC, Muecksch F, Finkin S, Tokuyama M, Cho A, Jankovic M, Schaefer-Babajew D, Oliveira TY, Cipolla M, Viant C, Barnes CO, Bram Y, Breton G, Hägglöf T, Mendoza P, Hurley A, Turroja M, Gordon K, Millard KG, Ramos V, Schmidt F, Weisblum Y, Jha D, Tankelevich M, Martinez-Delgado G, Yee J, Patel R, Dizon J, Unson-O’Brien C, Shimeliovich I, Robbiani DF, Zhao Z, Gazumyan A, Schwartz RE, Hatziioannou T, Bjorkman PJ, Mehandru S, Bieniasz PD, Caskey M, and Nussenzweig MC. 2021. Evolution of antibody immunity to SARS-CoV-2. Nature 591: 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tabibian-Keissar H, Hazanov L, Schiby G, Rosenthal N, Rakovsky A, Michaeli M, Shahaf GL, Pickman Y, Rosenblatt K, Melamed D, Dunn-Walters D, Mehr R, and Barshack I. 2016. Aging affects B-cell antigen receptor repertoire diversity in primary and secondary lymphoid tissues: Molecular Immunology. Eur. J. Immunol. 46: 480–492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data have been deposited in NCBI’s Gene Expression Omnibus and are available at the GEO Series accession number GSE164381 (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164381). Scripts to reproduce these analyses are available at: https://bitbucket.org/kleinstein/projects.