

Abstract
Proteins are an impactful class of therapeutics but can exhibit suboptimal therapeutic performance arising from poor control over the timescale of clearance. Covalent PEGylation is one established strategy to extend circulation time, but often at the cost of reduced activity and increased immunogenicity. Supramolecular PEGylation may afford similar benefits without necessitating that the protein be permanently modified with a polymer. Here we show that insulin pharmacokinetics can be modulated by tuning the affinity-directed dynamics of a host–guest motif used to non-covalently endow insulin with a PEG chain. When administered subcutaneously, supramolecular PEGylation with higher binding affinities extended the time of total insulin exposure systemically. Pharmacokinetic modeling reveals the extension in the duration of exposure arises specifically from decreased absorption from the subcutaneous depot governed directly by the affinity and dynamics of host–guest exchange. The lifetime of the supramolecular interaction thus dictates the rate of absorption, with negligible impact attributed to association of the PEG upon rapid dilution of the supramolecular complex in circulation. This modular approach to supramolecular PEGylation offers a powerful tool to tune protein pharmacokinetics in response to the needs of different disease applications.
Keywords: Insulin, cucurbit[7]uril, host–guest chemistry, Drug delivery, Protein modification
Graphical Abstract
INTRODUCTION
Biopharmaceuticals, including peptides, proteins, and antibodies, offer impactful new therapies to treat a wide range of diseases.1–2 Unfortunately, these agents still face formulation and drug delivery challenges ranging from aggregation and short shelf-lives to poor circulation times.3 Covalent conjugation of poly(ethylene glycol) (PEG), an approach known as PEGylation, has been used to improve stability and increase the circulation time of protein and peptide therapeutics.1, 3–7 The increased size of the PEGylated agent reduces renal clearance, thereby extending circulation time, yet also introduces steric hindrance from the conjugated polymer that can reduce protein activity.1, 3, 7–8 The hydrodynamic radius of PEGylated therapeutics often exceeds the size limits for glomerular filtration and renal clearance (by design), shifting the route of elimination to hepatic clearance which can introduce unanticipated effects from targeted liver delivery.3–4, 7
By comparison, dynamic and non-covalent modification of proteins with PEG chains does not permanently modify the protein drug, thus mitigating many of the negative consequences of direct covalent PEGylation.9 Supramolecular PEGylation using macrocyclic host–guest interactions thus presents an attractive alternative.10–11 One particular macrocycle, cucurbit[7]uril (CB[7]), has proven especially useful for such applications due to its broad range of affinity in binding a variety of different guest chemistries as well as high water solubility relative to other macrocycles in the CB[n] class.12–16 Complexation of CB[7] and guest has been estimated to occur with dynamics near the diffusion limit, though guest binding is slowed slightly in the presence of cations (e.g., Na+ in serum) due to competition from portal interactions between the cation and CB[7].17–19 The association rate in physiologic conditions can therefore reasonably be approximated as kon ~ 107 M−1 s−1,17–19 with changes in guest binding affinity (Keq) thus enabling direct control over the exchange rate (koff) of the interaction. The N-terminal (B1) phenylalanine on insulin has been identified as a guest molecule for CB[7] binding (Keq = 1.5 × 106 M−1).20 Exploiting this interaction, the conjugation of PEG to CB[7] has recently demonstrated utility as a supramolecular route to endow insulin with PEG-specific function in formulation, improving insulin stability, altering insulin pharmacokinetics, and enabling co-formulation of insulin with an otherwise incompatible protein (amylin), all by leveraging dynamic non-covalent interactions.11, 21–22
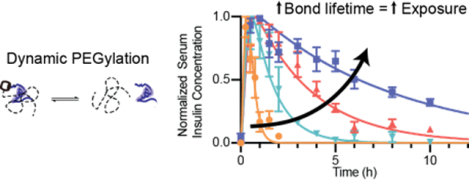
In this study, we demonstrate an approach for supramolecular PEGylation using tunable host–guest dynamics to alter the absorption and pharmacokinetic profile of insulin. As the first protein drug used in humans, insulin is vital in therapeutic management for over 20 million individuals with type-1 diabetes worldwide. Insulin variants with long-acting function are important to replace basal insulin and maintain glucose homeostasis between meals and overnight. Covalent PEGylation has been explored clinically for long-lasting insulin function with liver-biased activity (peglispro, Eli Lilly) by attaching a 20 kDa PEG chain to the B28 lysine on insulin lispro.8, 23–24 Regrettably, this PEGylated insulin candidate resulted in increased liver fat deposits in clinical trials, linked to its hepatopreferential action and hepatic clearance route, ultimately resulting in discontinuation of the program.23 Previous work demonstrated that supramolecular PEGylation of insulin can modulate its duration of action by varying PEG chain length.11 We therefore hypothesized that we could further modulate insulin pharmacokinetics, extending its duration of action, by altering the host–guest binding affinity used for appendage of the PEG chain to the therapeutic protein. Supramolecular PEGylation does not permanently increase the hydrodynamic size of the protein, and once the insulin dissociates from the PEG chain it is able to be absorbed into the blood and cleared from the body normally (Figure 1). This modular approach points to a powerful tool utilizing supramolecular affinity to tune pharmacokinetics, making therapeutics more responsive to the needs of different applications.
Figure 1. Schematic of insulin absorption.
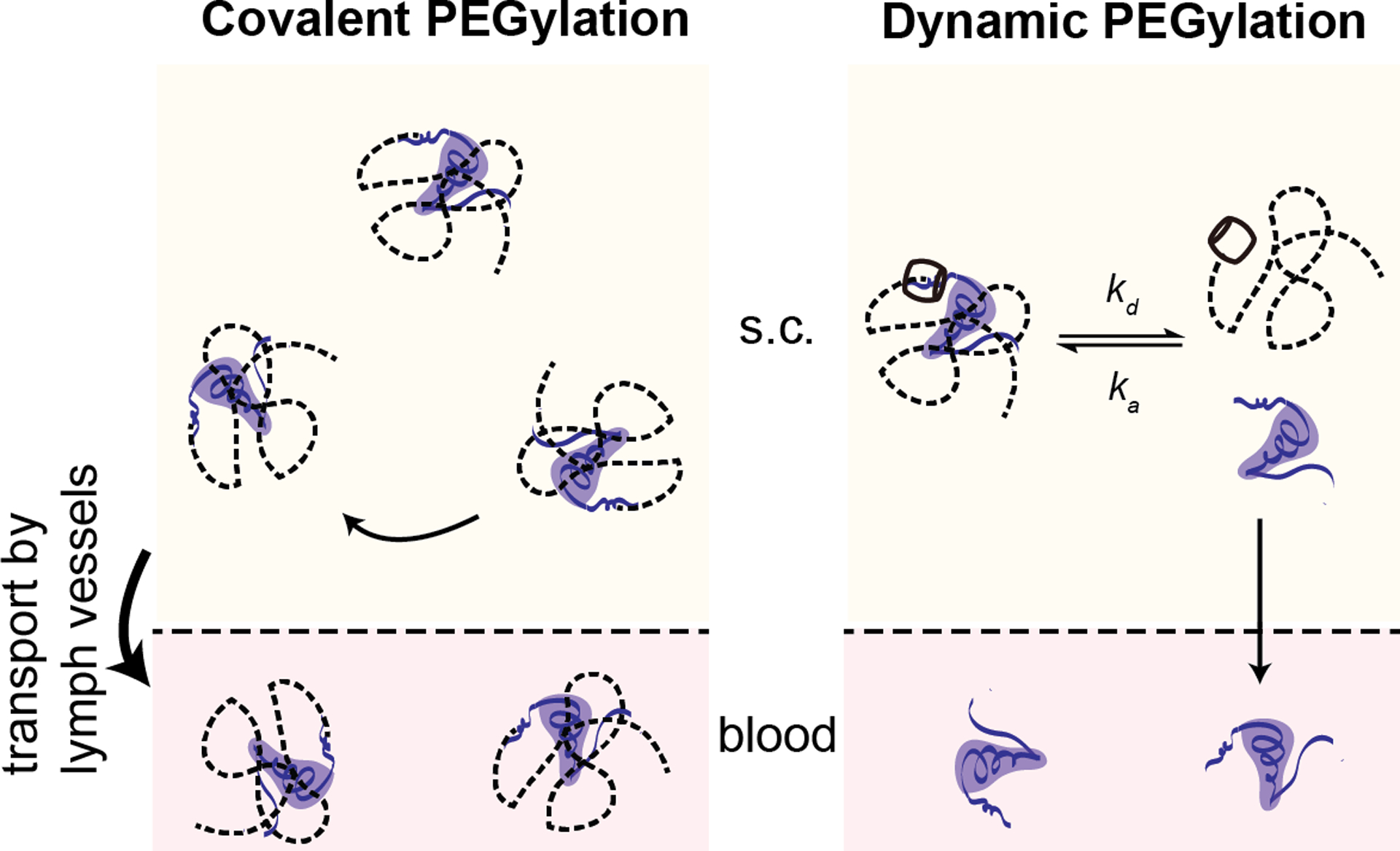
Covalent PEGylation permanently alters the hydrodynamic size of the insulin necessitating uptake from the subcutaneous space through lymphatic vessels and driving increased circulation time once in the blood. In contrast, supramolecular PEGylation, using host–guest binding with cucurbit[7]uril, results in an insulin/PEG complex with a larger hydrodynamic radius that is not readily absorbed from the subcutaneous space into the blood. However, as dynamic exchange of the host–guest binding occurs, some insulin will become available for absorption into the blood where it is unhindered by the bulky PEG chain.
EXPERIMENTAL METHODS
Synthesis of cucurbit[7]uril-PEG20k
Synthesis of PEG terminated with alkyne group (mPEG-alkyne).
In a dry round-bottom flask, sodium hydride (0.32g, 60% dispersion in mineral oil, Beantown Chemical) was slowly added to a solution of methoxypolyethylene glycol (mPEG, Alfa Aesar, Mn = 20,000, 1g) in 25 mL of dry THF. The mixture was stirred for 20 min at room temperature. Then, propargyl bromide (0.88 mL, 80 wt.% solution in toluene, Beantown Chemical) was added and the reaction mixture was stirred for two days at room temperature. After quenching with a small volume of water, the reaction mixture was transferred to a 50 mL centrifuge tube. The supernatant was collected by centrifuge and evaporated under reduced pressure. The residue was dissolved in 100 mL of DCM and washed with brine three times (100 mL each). The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure into a small volume which was precipitated into cold diethyl either. The product was obtained as a yellow powder in quantitative yield. 1H-NMR (400 MHz Bruker, 25°C, CDCl3): δ (ppm) = 4.20 (b, 2H), 3.83–3.45 (m, PEG chain), 3.38 (s, 3H), 2.44 (t, 1H). See Supplemental Information Figure S1 for synthesis scheme, and Figure S2 for 1H-NMR of mPEG20k-Alkyne.
Synthesis of PEG-CB[7].
CB[7]-N3 (20 mg) was synthesized according to previously published methods.25 The pure CB[7]-N3 product was combined along with mPEG-Alkyne (300 mg), copper(II) sulfate pentahydrate (CuSO4·5H2O, 1mg) and PMDETA (98%, 0.8 μL, Acros) and dissolved in 8 mL DMF/water (1/1, v/v) in a Schlenk flask. The flask was degassed with three freeze-pump-thaw cycles. On the last cycle, the flask was opened to quickly add sodium ascorbate (5 mg) into the flask before re-capping the flask. The flask was vacuumed and backfilled with N2 for 5 cycles before immersion in a 50 °C oil bath to thaw the solution and initiate the ‘click’ reaction. After 2 days, the reaction was quenched by exposure to air. The reaction mixture was then transferred into dialysis tubing (MWCO = 3500, Thermo Scientific) and dialyzed against water for two days. The pure product was obtained after lyophilization as yellow solid and was determined to be fully substituted with CB[7]. 1H-NMR (500 MHz Bruker, 25°C D2O, Figure S3): δ (ppm) = 8.04 (s, 1H), 7.49 (s, free probe), 6.58 (s, threaded probe), 5.76–5.62 (m, 14H), 5.55–5.40 (m, 12H), 4.67 (s, 2H), 4.46 (t, 2H), 4.34–4.12(m, 14H), 4.18 (s, free probe), 3.88 (s, threaded probe), 3.82–3.52 (m, PEG chain), 3.5 (s, 3H), 2.30 (s, 2H), 2.01 (s, 2H), 1.74 (s, 3H), 1.09 (s, 2H). See Supplemental Information Figure S1 for synthesis scheme and Figure S4 for MALDI-MS of mPEG20k-Alkyne and mPEG20k-CB[7].
Synthesis of azide-PEG20k
Synthesis of PEG terminated with mesylate (mPEG-Ms).
In a dry round-bottom flask, mPEG (Alfa Aesar, Mn = 20,000, 5g) was dissolved in 25 mL DCM with triethylamine (0.348 mL). The solution was cooled to 0°C in an ice bath and methanesulfonyl chloride (MsCl, 0.194 mL) was added slowly. The flask was then removed from the ice bath and the reaction mixture was stirred for two days at room temperature. After quenching with a small volume of water, the reaction mixture was diluted into 200 mL of DCM and washed with brine three times (200 mL each). The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure into a small volume which was precipitated into cold diethyl either. The product was obtained as a white powder (4.5g, 90% yield). 1H-NMR (500 MHz Bruker, 25°C, CDCl3): δ (ppm) = 4.28 (m, 2H), 3.79–3.49 (m, PEG chain), 3.38 (s, 3H), 3.09 (s, 3H). See Supplemental Information Figure S5 for synthesis scheme.
Synthesis of PEG terminated with azido group (mPEG-N3).
Sodium azide (32.5 mg), and PEG-Ms (2.0g) were dissolved in 20 mL of DMF and stirred at 60°C for 2 days. Then the reaction mixture was then diluted into 200 mL of DCM and washed with brine three times (200mL each). The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure into a small volume which was precipitated into cold diethyl either. The product was obtained as a white powder (1.8g, 90%). 1H-NMR (400 MHz Bruker, 25°C, CDCl3): δ (ppm) = 3.83–3.45 (m, PEG chain), 3.40–3.37 (t, 2H), 3.38 (s, 3H). See Supplemental Information Figure S5 for synthesis scheme and Figure S6 for 1H-NMR.
Synthesis of O-adamantane-PEG20k
An alkyne-modified O-linked adamantane (Alkyne-O-Ada) was synthesized as previously described.16 Then, mPEG20k-N3 (300 mg), Alkyne-O-Ada (3mg), copper(II) sulfate pentahydrate (CuSO4·5H2O, 1mg) and N,N,N’,N”,N”-pentamethyldiethylenetriamine (PMDETA, 98%, 0.8 μL, Acros) were dissolved in 10 mL of DMF in a Schlenk flask. The flask was degassed with three freeze-pump-thaw cycles. On the last cycle, the flask was opened to quickly add sodium ascorbate (5 mg) before re-capping. The flask was vacuumed and backfilled with N2 for 5 cycles before immersion in a 50 °C oil bath to thaw the solution and initiate the ‘click’ reaction. After 2 days, the reaction mixture was dialyzed against water for 2 days. Then the product was obtained by lyophilization as white powder. 1H-NMR (400 MHz Bruker, 25°C, CDCl3, Figure S7): δ (ppm) = 7.68 (s, 1H), 4.64 (bs, 2H), 4.51 (t, 2H), 3.88–3.45 (m, PEG chain), 3.38 (s, 3H), 2.21 (bs, 3H), 1.82 (bd, 6H), 1.68 (m, 6H). See Supplemental Information Figure S8 for synthesis scheme.
Synthesis of p-xylylenediamine-PEG20k
An alkyne-modified p-Xylylenediamine (Alkyne-diN-Xyl) was synthesized as previously described.16 mPEG20k-N3 (300 mg), Alkyne-diN-Xyl (4.67 mg), copper(II) sulfate pentahydrate (CuSO4·5H2O, 1mg) and PMDETA (98%, 0.8 μL, Acros) were dissolved in 10 mL of DMF in a Schlenk flask. The flask was degassed with three freeze-pump-thaw cycles. On the last cycle, the flask was opened to quickly add sodium ascorbate (5 mg) before re-capping. The flask was vacuumed and backfilled with N2 for 5 cycles before immersion in a 50 °C oil bath to thaw the solution and initiate the ‘click’ reaction. After 2 days, the reaction mixture was dialyzed against water for 2 days. Then the product was obtained by lyophilization. 1H-NMR (500 MHz Bruker, 25°C, D2O, Figure S9): δ (ppm) = 8.16–7.41 (m, 2H), 7.50 (bs, 1H), 7.41–7.11 (m, 2H), 5.28 (bs, 2H), 4.34 (m, 2H), 4.25 (m, 2H), 4.21 (m, 2H), 3.94 (m, 2H), 3.82–3.53 (m, PEG chain), 3.36 (s, 3H). See Supplemental Information Figure S10 for synthesis scheme. See Supplemental Figure S11 for MALDI-MS.
Insulin-cucurbit[7]uril conjugation
CB[7]-N3, prepared as described previously,25 was used for conjugation to insulin at the B1 position. First, an NHS-ester reaction was used to functionalize the insulin with a bicyclo[6.1.0]nonyne (BCN), enabling a subsequent azide-BCN copper-free click reaction with CB[7]-N3. An insulin stock solution of 10 mg/mL was prepared by adding 0.1 M aq HCl (conc) to 100 mg (17.2 μmol) of insulin until the dispersed powder was fully dissolved. Bis-tris was added to buffer the solution to pH~6.5, resulting in a temporarily cloudy precipitate that quickly dissipates. 1.2 eq (20.6 μmol) BCN-NHS ester was added to insulin from a freshly prepared stock solution. This solution was incubated at room temperature overnight. Buffer conditions (pH~6.5) were used to favor functionalization of the B1 phenylalanine with BCN over the B29 lysine. The B1 position was chosen for the conjugation so that the CB[7] would block the natural phenylalanine guest, preventing an insulin polymer from forming, and to keep the PEGylation site consistent between groups. CB[7]-BCN intermediate was confirmed using Thermo Exactive Orbitrap Liquid Chromatography Mass Spectrometry (Figure S12), H1-NMR (600 MHz, DMSO-d6, 298 K) (Figure S13–S16) and MALDI-MS (Figure 3b) and was purified using HPLC (Figure S17). HPLC purification was performed using an Agilent 1260 semi-preparative HPLC with a C18 column with dH2O and acetonitrile as the mobile phases. After HPLC purification, the insulin-BCN solution was concentrated to 5 mg/mL using an Amicon Ultra centrifugal filter (MWCO 3 kDa). For the click reaction 2 eq. CB[7]-N3 was added for every 1 eq of insulin-BCN (2 mL, 5 mg/mL) in a solution of approximately 40 wt.% acetonitrile 60 wt.% dH2O and incubated for 24 hours at room temperature. Following reaction, the insulin-CB[7] solution was diluted 10-fold with dH2O and concentrated using an Amicon Ultra centrifugal filter (MWCO 3 kDa). The wash step was performed three times to remove unreacted CB[7]- N3 and to remove acetonitrile content. MALDI was used to confirm the final insulin-CB[7] conjugate. Nanodrop measurements were used to estimate insulin concentrations for formulation. Insulin-CB[7] conjugate secondary structure was confirmed using circular dichroism (Figure S18).
Figure 3: Synthesis of insulin conjugates.
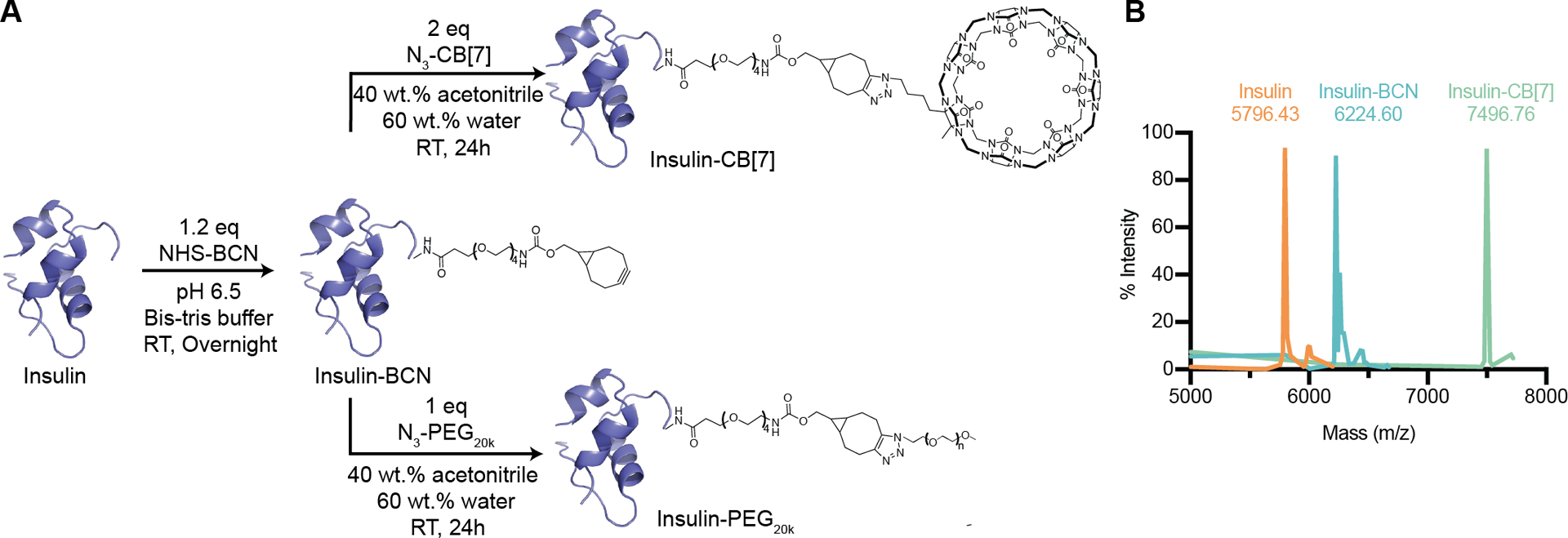
(A) Scheme showing insulin-CB[7] and insulin-PEG20k synthesis. Insulin was functionalized with bicyclo[6.1.0]nonyne (BCN) using a N-hydroxysuccinimide (NHS)-ester reaction followed by a copper-free click reaction between BCN and azide-modified CB[7] or azide-PEG20k, resulting in the final insulin conjugates. (B) MALDI-MS was used to verify formation of the intermediate (Insulin-BCN, expected m/z=6217.11) and final product (Insulin-CB[7], expected m/z=7491.29). MALDI-MS spectra were baseline corrected, with the minimum intensity value in the spectra subtracted from all other values.
Synthesis of Insulin-PEG20k
mPEG20k-N3 was added at a 1:1 molar ratio to the 5 mg/mL insulin-BCN intermediate prepared in the same way as the insulin-BCN for the CB[7]-N3 click reaction described above. The solution (40 wt.% acetonitrile 60 wt.% dH2O) was incubated at room temperature for 24 hours. Following incubation, insulin-PEG20k was diluted 10-fold with dH2O and concentrated using a 10 kDa Amicon Ultra centrifugal filter eight times to remove unconjugated insulin and acetonitrile. Conjugation was confirmed using MALDI-MS (Figure S19). Nanodrop measurements were used to estimate insulin concentrations for formulation.
Streptozotocin induced model of diabetes in rats
Male Sprague Dawley rats (Charles River) were used for experiments. Animal studies were performed in accordance with the guidelines for the care and use of laboratory animals; all protocols were approved by the Stanford Institutional Animal Care and Use Committee (Protocol #32873). The protocol used for diabetes induction using streptozotocin (STZ) was adapted from the protocol by Kenneth K. Wu and Youming Huan and was performed as previously reported.21–22, 26–27 Briefly, male Sprague Dawley rats 160–230 g (8–10 weeks) were weighed and fasted in the morning 6–8 hours prior to treatment with STZ (MedChemExpress). STZ was protected from light and diluted to 10 mg/mL in the sodium citrate buffer immediately before injection. STZ solution was injected intraperitoneally at 65mg/kg into each rat. Rats were provided with water containing 10% sucrose for 24 hours after injection with STZ and were given subcutaneous saline injections daily to prevent dehydration. Rat blood glucose levels were tested for hyperglycemia daily after the STZ treatment via tail vein blood collection using a handheld Bayer Contour Next glucose monitor (Bayer). Diabetes was defined as having three consecutive blood glucose measurements >400 mg/dL in non-fasted rats.
In vivo pharmacokinetics and pharmacodynamics in diabetic rats
Diabetic rats were fasted for 4–6 hours before injection. For subcutaneous pharmacokinetic and pharmacodynamic experiments 20 rats were randomly assigned to treatment groups and were injected with one of the following insulin formulations: (i) insulin (2 U/kg), (ii) insulin & CB[7]-PEG20k (10 U/kg) (iii) insulin-CB[7] & Xyl-PEG20k (10 U/kg) (iv) insulin-CB[7] & Ada-PEG20k (10 U/kg) (v) insulin-PEG20k (10 U/kg). All formulations were prepared in 10 mM phosphate buffer (pH ~ 7.4) with 2.6 wt.% glycerol as a tonicity agent and had concentrations of 586 μM insulin (similar to standard 100 U/mL formulation). For non-covalently PEGylated formulations a 1:1 molar ratio of the PEG conjugate:insulin was added. After injection, rats were provided food ad libitum to prevent hypoglycemia that could result from high insulin doses. After formulation administration, blood glucose measurements were taken using a handheld glucose monitor (Bayer Contour Next) and additional blood was collected (Sarstedt serum tubes) for analysis with ELISA. Timepoints were taken at 0, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 18, 24 hours following administration. Serum insulin concentrations were quantified using an ultra-sensitive human insulin ELISA (Mercodia) or iso-insulin ELISA (Mercodia). A second pharmacokinetics experiment was performed with intravenous injections to understand the elimination half-life of insulin and PEGylated insulins from the blood. In this experiment, 24 rats were randomly assigned to a formulation group. The same formulations as in the subcutaneous experiment were tested with the addition of a sixth formulation (vi) insulin-CB[7], which was added as a control. All formulations were administered at ~2 U/kg dose. Rats kinetic curves were excluded from analysis when there was a clear absorption phase present indicative of part of the injection entering subcutaneously rather than intravenously. Insulin-CB[7] was not tested subcutaneously because it is insoluble at formulation concentrations and when injected in the subcutaneous space (where dilution is minimal) will remain insoluble.
In vivo pharmacokinetics modeling
For this analysis a three-compartment model with first order rate equations was used to model the absorption and clearance of insulin into the blood. Insulin (bound to PEG through host–guest interactions) after subcutaneous injection (Iinj), insulin available for absorption (dissociated from PEG) (Ieq), and the plasma (Ip) were numerically solved using a system of differential equations, outlined below, as a function of time using the SciPy (version 1.2.1) odeint function in Python (version 3.6.8).28
| (1) |
| (2) |
| (3) |
Concentrations were initialized such that at t = 0 all insulin was present in Iinj. Kinetic rate constants were fit for the normalized rat pharmacokinetic curves by minimizing the sum of squared errors (SSE) between the generated, normalized insulin plasma concentrations derived from the model at the experimental time points from 0 to 48 hours and the normalized plasma insulin concentrations for each insulin formulation. We assume that based on the intravenous pharmacokinetic data that confirm that there is no difference in plasma elimination half-life between non-covalently PEGylated and unmodified insulin formulations. Thus, k3 can be calculated from the average of the four formulations observed elimination half-life. Since insulin and insulin-CB[7] are similar in size, we assume that once free from the PEG chain insulins will be absorbed from the subcutaneous space similarly, so k2 was constrained to be constant for all groups. That leaves k1 as the only formulation dependent parameter. The SSE was minimized by first employing a grid search using SciPy’s optimize brute function and subsequently refining the rate constants by employing SciPy’s optimize minimize function using the L-BFGS-B method.
Statistics
All data is reported as mean ± standard error mean (SEM) unless specified. GraphPad Prism 8 was used to fit a one-phase exponential decay non-linear regression to the intravenous pharmacokinetic data and to perform a one-way ANOVA with Tukey-Kramer post-hoc test for multiple comparisons to compare the elimination half-lives between groups.
RESULTS & DISCUSSION
Design of supramolecular PEGylation system
Previous work demonstrated supramolecular PEGylation via interaction between a PEG-linked CB[7] macrocycle and the N-terminal B1 phenylalanine of insulin, with increasing PEG length serving to extend the duration of insulin action.11 In this study, we hypothesized that modulating the binding affinity of PEG-appending host–guest interactions could be used to further control insulin pharmacokinetics. CB[7] is an ideal host molecule for these studies as it has a broad range of binding affinities for different guest molecules,12–16 including the N-terminal phenylalanine on insulin.20 In this work, PEG chain length (20 kDa) was kept constant to mirror the PEG length used in peglispro and limit absorption of PEGylated insulin directly into blood following subcutaneous administration. Then, by altering the CB[7]–guest affinity motif used to append the PEG chain, we could assess how changes in binding affinity, and by extension binding dynamics (e.g., the binding lifetime, τ, which is inversely related to koff), affect insulin pharmacokinetics. Three different guests for CB[7] were chosen with Keq values ranging from 106-1010 M−1. These guests were (i) N-terminal phenylalanine (Keq ~ 106 M−1) on the insulin B chain interacting with CB[7]–PEG20k; (ii) p-xylylenediamine (Xyl) (Keq ~ 109 M−1) and (iii) O-adamantane (O-Ada) (Keq ~ 1010 M−1),16 bound to PEG20k and interacting with CB[7]-modified insulin. As controls, unmodified recombinant human insulin and insulin covalently modified with PEG20k were used.
Two strategies were used for supramolecular PEGylation with CB[7]–guest motifs of three different affinities (Figure 2). The first took advantage of the native B1 phenylalanine on insulin as the guest moiety, combined with a CB[7]-PEG20k conjugate as the host (Figure 2a, Figure S1–S4). This system uses the same topology for supramolecular PEGylation described in previous work.11 To access the guests with higher affinity, p-xylylenediamine (Xyl) and O-linked adamantane (O-Ada), and limit the number of additional conjugation steps, the topology of the host–guest system was reversed. In the reversed system, CB[7] was conjugated to insulin while the two guests were coupled to PEG20k (Figure 2b, Figure S5–S11). CB[7] was conjugated to insulin at the B1 terminal amine using an N-hydroxysuccinimide (NHS)-ester reaction to first functionalize the insulin with a bicyclo[6.1.0]nonyne (BCN), followed by azide-BCN copper- free click reaction (Figure 3a), using a previously reported azide-modified CB[7].25 Buffer conditions (pH 6.5) were used to promote selective NHS-ester modification of the B1 phenylalanine over the B29 lysine.29–31 The B1 position was chosen for the conjugation to block the native phenylalanine guest of CB[7] and prevent the formation of insulin polymer species, as well as to keep the site of PEGylation consistent for the two supramolecular topologies. The ‘clicked’ CB[7]-BCN conjugate was confirmed using liquid chromatography mass spectrometry (LC-MS; Figure S12), matrix assisted laser desorption ionization–mass spectrometry (MALDI-MS; Figure 3B) and1H NMR (Figure S13–S16). Insulin-BCN was purified using semi-preparative high performance liquid chromatography (HPLC; Figure S17). Formation of insulin-CB[7] was confirmed using MALDI-MS (Figure 3b) and secondary structure of the conjugate was confirmed by circular dichroism (Figure S18). For the covalent insulin-PEG20k control, azide-PEG20k was conjugated to the same insulin-BCN intermediate via copper-free click reaction (Figure 3a), which was confirmed using MALDI-MS (Figure S19).
Figure 2. Schematic of PEGylated insulin formulations.
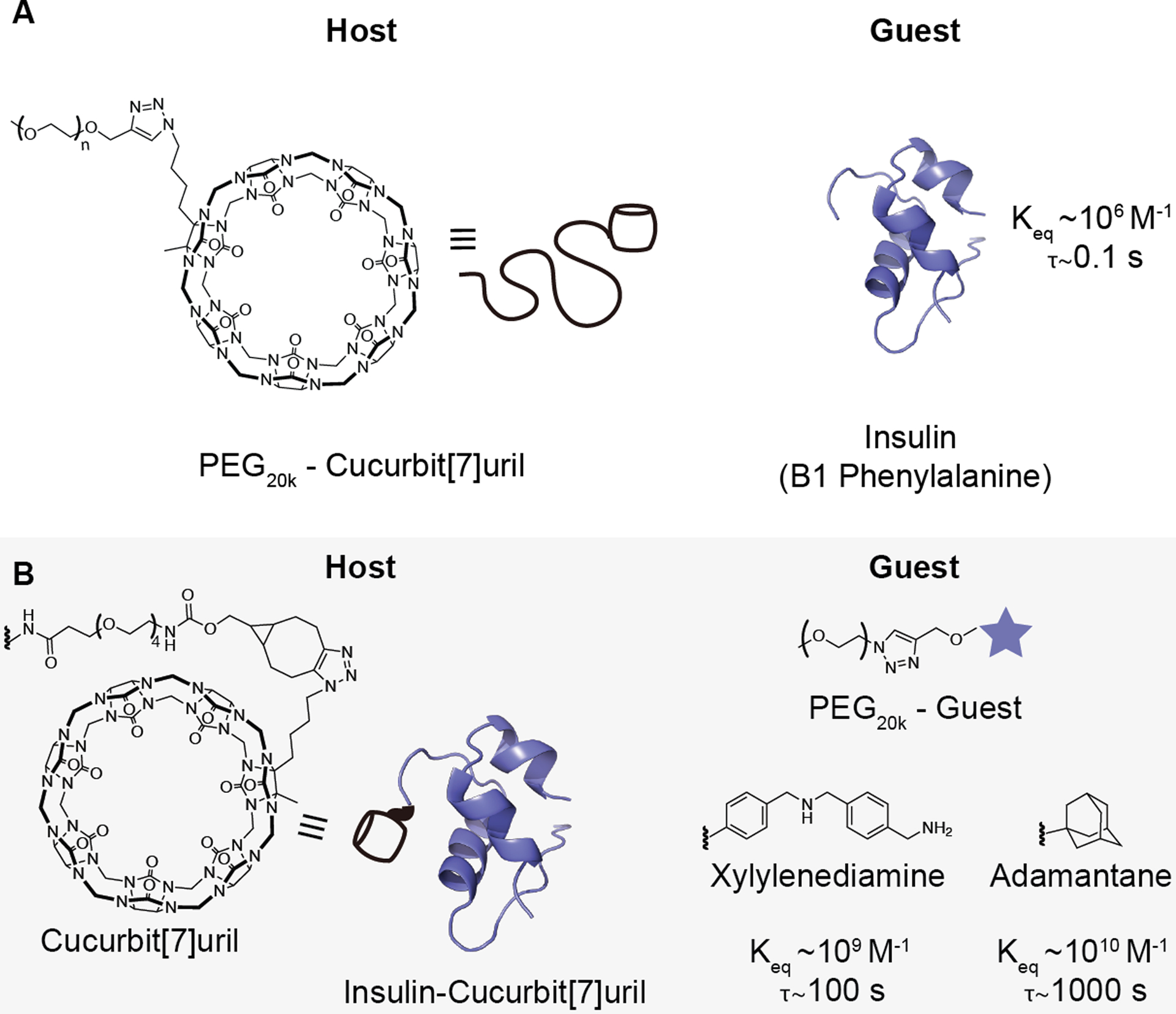
To assess the effect of host–guest binding affinity on the pharmacokinetics of dynamically PEGylated insulin, two non-covalently PEGylated insulin systems were tested. (A) The N-terminal B1 phenylalanine of insulin can bind to cucurbit[7]uril (CB[7]) with a Keq~106 M−1. A PEG20k-CB[7] can thus be used to non-covalently PEGylate the phenylalanine on insulin. (B) To test host–guest motifs with higher binding affinity, a covalent insulin-CB[7] conjugate was combined with a guest-linked PEG20k. To depict the conjugation between CB[7] and insulin, the chemical structures of CB[7], bicyclo[6.1.0]nonyne (BCN), and the PEG linker used are shown (left). PEG20k-xylylenediamine (Keq~109 M−1) or PEG20k-O-adamantane (Keq~1010 M−1) were selected as guests. Bond lifetime (τ), is inversely related to the dissociation rate koff and was calculated based on the following relationships and assumptions: Keq = kon/koff; kon~107; τ = 1/koff. See Table S1 for a complete list of values.
Pharmacokinetics in diabetic rats
Having confirmed conjugation of insulin-CB[7], formulations were prepared for in vivo testing in diabetic rats. Unmodified insulin and covalently PEGylated insulin-PEG20k were used as short-acting and long-acting controls, respectively. These were compared to supramolecular PEGylation of insulin using formulations of (i) insulin & CB[7]-PEG20k, (ii) insulin-CB[7] & Xyl-PEG20k, and (iii) insulin-CB[7] & O-Ada-PEG20k. The insulin-CB[7] conjugate alone was not used as a control for subcutaneous administration because it was not sufficiently soluble at formulation concentrations (all insulin-CB[7] & guest-PEG formulations were soluble). All formulations had concentrations of 586 μM insulin (comparable to a standard 100 U/mL insulin formulation), and for host–guest PEGylated formulations a 1:1 molar ratio of the PEG conjugate:insulin was used. We hypothesized that a greater binding affinity, and thus longer bond lifetime, between the PEG and insulin would result in increased residence time of insulin in the subcutaneous depot and translate to an extended duration of insulin exposure. Here, duration of exposure is defined as peak width at 25% peak height ((Time to 25% peak down) – (Time to 25% peak up) of the individually normalized curves). After subcutaneous administration of insulin formulations in diabetic rats, we observed that while all host–guest PEGylated formulations had a similar time to peak (~1 h), the duration of exposure increased with increasing binding affinity of the host–guest interaction (Duration: Phe = 2.2 ± 0.3 h; Xyl = 5.5 ± 0.7 h; O-Ada = 12.4 ± 0.9 h). Covalent insulin-PEG20k exhibited a longer time to peak (7 ± 1 h) and the longest duration of exposure (28 ± 8 h), consistent with the behavior expected for covalently PEGylated insulin. The larger hydrodynamic radius introduced by the covalent PEG chain limits direct absorption into the blood, instead requiring more prolonged absorption through the lymphatic system and extending the time to peak.8 Similarly, the prolonged duration of exposure observed for insulin-PEG20k is characteristic of the extension in circulation time afforded by covalent PEGylation.
The affinity regime explored for these guests is below what would be expected to readily form complexes under dilution in the body.32 As such, the elevated concentration when confined within the subcutaneous depot is likely key to the observed affinity-dependent differences in duration of insulin exposure, rather than supramolecular PEGylation serving to directly increase circulation time in the blood. To test this hypothesis of a subcutaneous depot effect, we administered each formulation intravenously to determine insulin elimination half-life. Insulin-CB[7] alone (with no PEG-guest) was also tested to verify that functionalization with CB[7] did not alter circulation time. As expected, there was no difference between the half-lives of host–guest PEGylated insulin formulations, insulin-CB[7], and insulin alone, suggesting that dissociation of the host–guest interaction happens rapidly under dilution and does not measurably affect circulation time. In contrast, covalent insulin-PEG20k demonstrated an extended half-life of 37 minutes compared to the ~4–8 minutes range observed for the other formulations (p<0.001) (Figure S20).
Pharmacokinetic modeling
To further understand the impact of the host–guest affinity linking insulin and PEG on duration of exposure, we used a first order three-compartment model to fit the pharmacokinetic data for the three host–guest PEGylated insulin formulations (Figure 5a). The model assumes complete complex formation between insulin and PEG at the time of injection. Though reassociation of the host–guest interaction will occur due to the relatively high concentrations of both the host and guest species in the depot, combined with the steric hindrance to absorption provided by the PEG chain, ultimately some insulin will become free and available for absorption into the blood stream. While this equilibrium between PEG-bound and unbound insulin is complex, for the purposes of modeling the system we have approximated the transition of insulin from PEGylated to available for absorption with a first order rate constant (k1). The absorption of insulin into the blood and its elimination from the blood are also modeled by first order rate constants. The rate constant for elimination (k3) can be calculated using the elimination half-life from intravenous administration of the formulations. Since the elimination half-life was consistent for all host–guest PEGylated formulations as well as unmodified insulin, k3 can be estimated from the average of these four elimination half-lives (k3 = 7.44 h−1). Further, if we assume that only the free insulin is absorbed, then the absorption rate of insulin from the subcutaneous depot into the blood is similar for all formulations; thus, we can constrain the elimination rate from the subcutaneous space (k2) to be constant for all formulations. This leaves k1, the rate constant representing the transition from the bound PEG-insulin complex to free insulin (or free insulin-CB[7]), as the only parameter that will differ between formulations (Table S2). The resulting predicted curves show excellent alignment with the experimental data (Figure 5b), corroborating our hypothesis that the pharmacokinetic differences observed between formulations are dependent on a subcutaneous depot effect modulated by the host–guest binding affinity. We likewise observed a clear relationship between k1 and the predicted dissociation rate (koff) of the specific host–guest interaction (Figure 5c). The binding affinity used for supramolecular PEGylation, which dictates the complex lifetime (τ), therefore directly controls the rate of absorption and subsequent activity of insulin following subcutaneous administration.
Figure 5: Modeling of formulation pharmacokinetics.
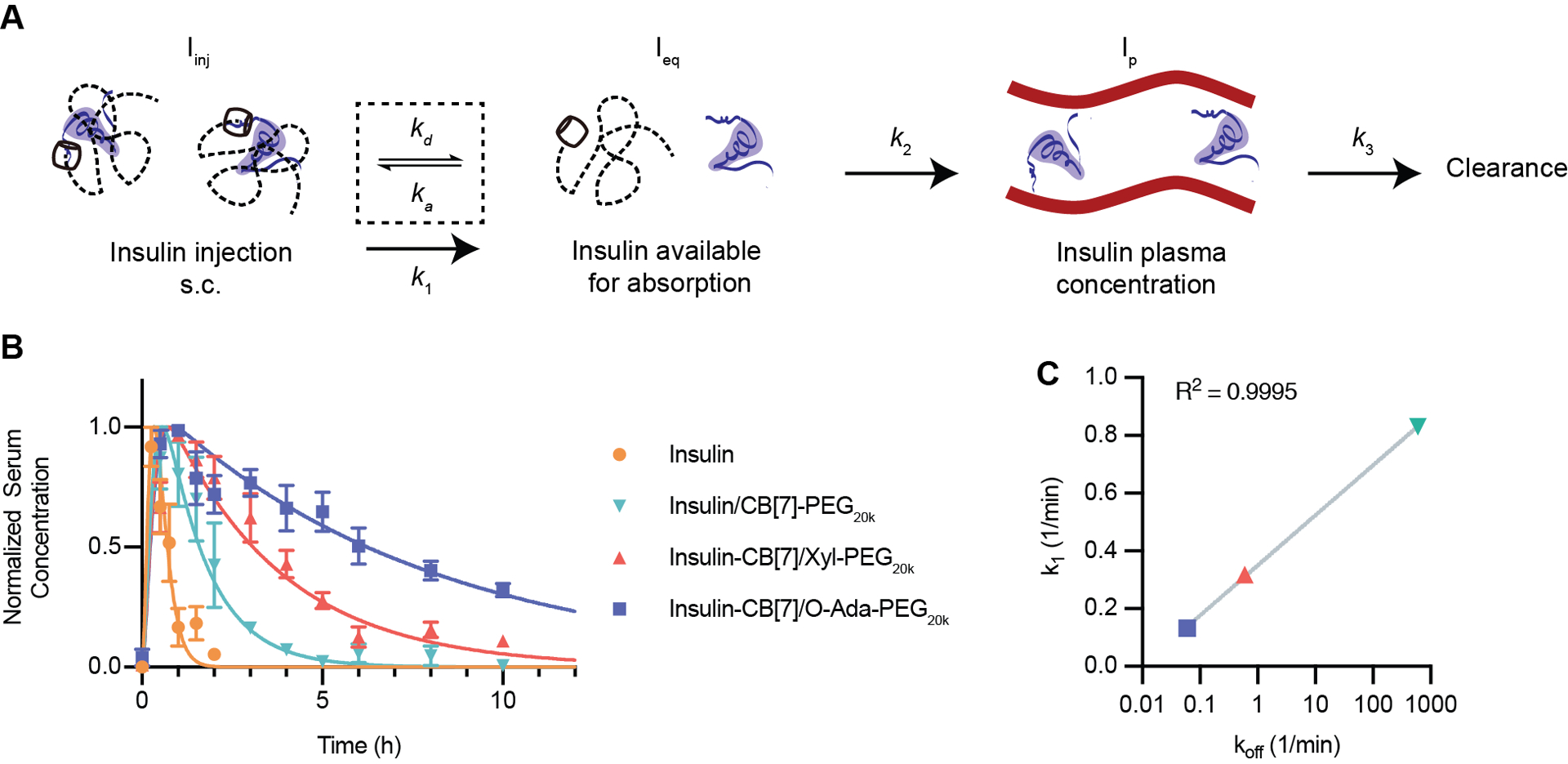
(A) Pharmacokinetic scheme showing a three-compartment model. After injection, insulin will transition dynamically between a PEGylated state (not readily absorbed) and a free state where it is available for absorption into the blood. Available insulin will be absorbed into the blood from the subcutaneous space and then cleared from the blood. (B) Normalized pharmacokinetics in diabetic rats modeled using a least-squares fit to determine k1, and k2, with k3 based on the elimination half-life calculated from intravenous injection experiments. Data is shown as mean ± SEM. (C) k1 (fitted parameter from experimental data) plotted versus koff (calculated based on host–guest binding affinity). A semi-log line was fit to the data using GraphPad Prism 9 best fit: Y=0.1735*log(X)+0.1761, Adjusted R2=0.9995.
CONCLUSIONS
Impactful therapeutics for a number of diseases come from protein and peptide drugs, though formulation and drug delivery challenges remain. One such challenge arises in ensuring tunable and application-specific duration of action. While covalent PEGylation is an established approach to increase circulation time of protein therapeutics, these benefits often come at the cost of reduced activity and immunogenicity. Here, we demonstrate a route for host–guest PEGylation of insulin wherein varying the binding affinity, and concomitant dynamics, of the interaction enables direct modulation of pharmacokinetics. When administered subcutaneously, higher binding affinities corresponded to slower absorption from the subcutaneous depot, thereby extending the duration of insulin exposure in the blood. Intravenous administration of these host–guest PEGylated formulations showed no change in the circulation time of insulin, suggesting that the insulin rapidly dissociates from the PEG chain upon dilution in the blood. These results indicate that the increased duration of insulin exposure observed after subcutaneous administration is a result of a depot effect, wherein high local concentrations allow for dynamic reassociation of insulin and PEG through host–guest interactions. Pharmacokinetic modeling further supports affinity-dependent differences, manifest in modulation of the dissociation rate (koff) for each complex, as the key to realizing these differences in insulin absorption. Combined with previously demonstrated control of function through the selection of PEG chain length, modulating host–guest binding affinity offers another useful tool to tune protein pharmacokinetics through supramolecular PEGylation. As the host–guest interactions investigated in this work still have relatively short bond lifetimes (~100 s for O-Ada), future work to investigate ultra-high affinity host–guest interactions may be interesting to uncover the limits of affinity required to approach covalent-like PEGylation over timescales relevant in the body. Supramolecular PEGylation to extend duration of action by exploiting host–guest binding affinity, and the associated differences in dynamics, thus offers an exciting avenue to increase the exposure time to biopharmaceutics without the risks entailed in covalent modification to alter function or clearance.
Supplementary Material
Figure 4: Pharmacokinetics and pharmacodynamics in diabetic rats.
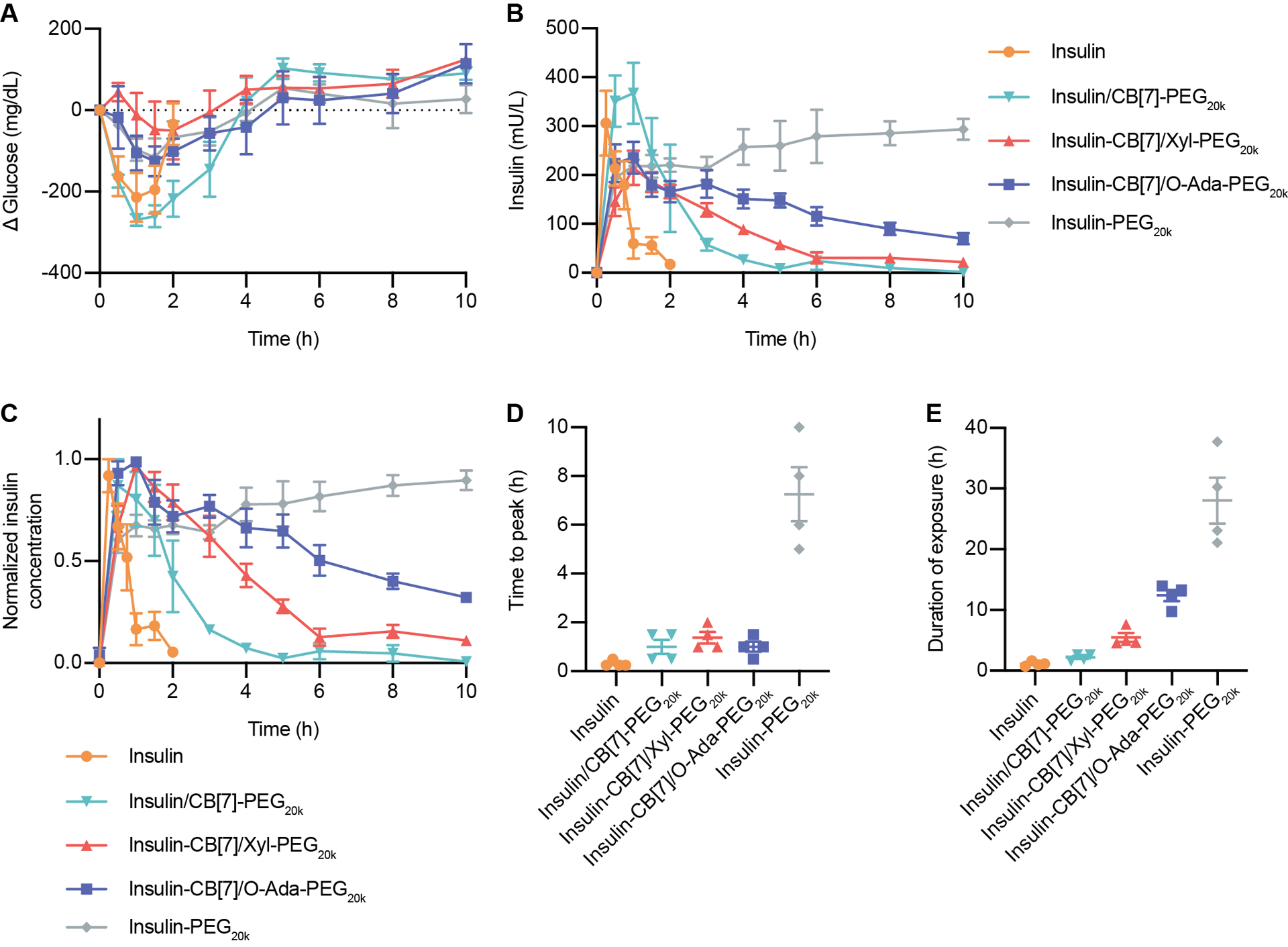
Fasted diabetic rats were administered one of five insulin formulations subcutaneously: (i) insulin (2 U/kg), (ii) insulin & CB[7]-PEG20k (10 U/kg), (iii) insulin-CB[7] & Xyl-PEG20k (10 U/kg), (iv) insulin-CB[7] & O-Ada-PEG20k (10 U/kg), (v) insulin-PEG20k (10 U/kg). Following administration, rats were given access to food. (A) Change in blood glucose levels following insulin administration. (B) Pharmacokinetics in terms of serum insulin concentrations following insulin administration. (C) Pharmacokinetics for each rat was normalized individually, and normalized values were averaged for insulin concentration for each treatment group. (D) Time to reach peak insulin concentrations. (E) Duration of exposure, defined as time to depletion of 25% of insulin peak concentration. Error bars indicate means ± SEM with n = 4 for all groups.
ACKNOWLEDGMENT
This work was funded in part by NIDDK R01 (NIH grant #R01DK119254), and a Pilot and Feasibility funding from the Stanford Diabetes Research Center (NIH grant #P30DK116074), as well as the American Diabetes Association Grant (1-18-JDF-011) and a Research Starter Grant from the PhRMA. Support is also provided by the Stanford Maternal and Child Health Research Institute through the SPARK Translational Research Program. M.J.W. acknowledges funding support from the American Diabetes Association Pathway Award (1-19-ACE-31), The Helmsley Charitable Trust (2019PG-T1D016), and the Juvenile Diabetes Research Foundation (5-CDA-2020-947-A-N). C.L.M. was supported by the NSERC Postgraduate Scholarship and the Stanford BioX Bowes Graduate Student Fellowship. A.I.D. was supported by the Schmidt Science Fellows Award. J.L.M was supported Department of Defense NDSEG Fellowship and by a Stanford Graduate Fellowship. A.A.A.A. was funded by grant NNF18OC0030896 from the Novo Nordisk Foundation and the Stanford Bio-X Program. The authors thank the Veterinary Service Centre staff for their technical assistance.
ABBREVIATIONS
- CB[7]
cucurbit[7]uril
- PEG
poly(ethylene glycol)
- Xyl
p-Xylylenediamine
- O-Ada
O-linked adamantane
- Phe
Phenylalanine
Footnotes
Supporting Information.
The following files are available free of charge.
Conflicts of interest
There are no conflicts to declare.
Data Availability
All data supporting the results in this study are available within the Article and its Supplementary Information. Raw data files are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Mitragotri S; Burke PA; Langer R, Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat. Rev.s Drug Discov 2014, 13 (9), 655–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee AC-L; Harris JL; Khanna KK; Hong J-H, A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci 2019, 20 (10), 2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang JS; DeLuca PP; Lee KC, Emerging PEGylated drugs. Expert Opin. Emerg. Drugs 2009, 14 (2), 363–380. [DOI] [PubMed] [Google Scholar]
- 4.Hamidi M; Azadi A; Rafiei P, Pharmacokinetic Consequences of Pegylation. Drug Deliv 2006, 13 (6), 399–409. [DOI] [PubMed] [Google Scholar]
- 5.Veronese FM; Mero A, The Impact of PEGylation on Biological Therapies. BioDrugs 2008, 22 (5), 315–329. [DOI] [PubMed] [Google Scholar]
- 6.Jevševar S; Kunstelj M; Porekar VG, PEGylation of therapeutic proteins. Biotechnol. J 2010, 5 (1), 113–128. [DOI] [PubMed] [Google Scholar]
- 7.Paola M; Franco D; Luigi C, PEGylation of Proteins and Liposomes: a Powerful and Flexible Strategy to Improve the Drug Delivery. Curr. Drug Metab 2012, 13 (1), 105–119. [DOI] [PubMed] [Google Scholar]
- 8.Knadler MP; Nguyen T-H; Campanale K; De Veer MJ; Beals JM; Li S; Hansen R; Siesky A; Michael MD; Porter CJH, Addition of 20-kDa PEG to Insulin Lispro Alters Absorption and Decreases Clearance in Animals. Pharm. Res 2016, 33 (12), 2920–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reichert C; Borchard G, Noncovalent PEGylation, An Innovative Subchapter in the Field of Protein Modification. J. Pharm. Sci 2016, 105 (2), 386–390. [DOI] [PubMed] [Google Scholar]
- 10.Biedermann F; Rauwald U; Zayed JM; Scherman OA, A supramolecular route for reversible protein-polymer conjugation. Chem. Sci 2011, 2 (2), 279–286. [Google Scholar]
- 11.Webber MJ; Appel EA; Vinciguerra B; Cortinas AB; Thapa LS; Jhunjhunwala S; Isaacs L; Langer R; Anderson DG, Supramolecular PEGylation of biopharmaceuticals. Proc. Natl. Acad. Sci. U. S. A 2016, 113 (50), 14189–14194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biedermann F; Uzunova VD; Scherman OA; Nau WM; De Simone A, Release of High-Energy Water as an Essential Driving Force for the High-Affinity Binding of Cucurbit[n]urils. J. Am. Chem. Soc 2012, 134 (37), 15318–15323. [DOI] [PubMed] [Google Scholar]
- 13.Cao L; Šekutor M; Zavalij PY; Mlinarić-Majerski K; Glaser R; Isaacs L, Cucurbit[7]uril⋅Guest Pair with an Attomolar Dissociation Constant. Angew. Chem. Int. Ed 2014, 53 (4), 988–993. [DOI] [PubMed] [Google Scholar]
- 14.Assaf KI; Nau WM, Cucurbiturils: from synthesis to high-affinity binding and catalysis. Chem. Soc. Rev 2015, 44 (2), 394–418. [DOI] [PubMed] [Google Scholar]
- 15.Barrow SJ; Kasera S; Rowland MJ; del Barrio J; Scherman OA, Cucurbituril-Based Molecular Recognition. Chem. Rev 2015, 115 (22), 12320–12406. [DOI] [PubMed] [Google Scholar]
- 16.Zou L; Braegelman AS; Webber MJ, Dynamic Supramolecular Hydrogels Spanning an Unprecedented Range of Host–Guest Affinity. ACS Appl. Mater. Interfaces 2019, 11 (6), 5695–5700. [DOI] [PubMed] [Google Scholar]
- 17.Tang H; Fuentealba D; Ko YH; Selvapalam N; Kim K; Bohne C, Guest binding dynamics with cucurbit[7]uril in the presence of cations. J. Am. Chem. Soc 2011, 133 (50), 20623–33. [DOI] [PubMed] [Google Scholar]
- 18.Miskolczy Z; Biczók L, Kinetics and Thermodynamics of Berberine Inclusion in Cucurbit[7]uril. J. Phys. Chem. B 2014, 118 (9), 2499–2505. [DOI] [PubMed] [Google Scholar]
- 19.Hu C; Grimm L; Prabodh A; Baksi A; Siennicka A; Levkin PA; Kappes MM; Biedermann F, Covalent cucurbit[7]uril–dye conjugates for sensing in aqueous saline media and biofluids. Chem. Sci 2020, 11 (41), 11142–11153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chinai JM; Taylor AB; Ryno LM; Hargreaves ND; Morris CA; Hart PJ; Urbach AR, Molecular recognition of insulin by a synthetic receptor. J. Am. Chem. Soc 2011, 133 (23), 8810–8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maikawa CL; Smith AAA; Zou L; Meis CM; Mann JL; Webber MJ; Appel EA, Stable Monomeric Insulin Formulations Enabled by Supramolecular PEGylation of Insulin Analogues. Adv. Ther 2019, 75, 1900094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maikawa CL; Smith AAA; Zou L; Roth GA; Gale EC; Stapleton LM; Baker SW; Mann JL; Yu AC; Correa S; Grosskopf AK; Liong CS; Meis CM; Chan D; Troxell MD; Maahs DM; Buckingham BA; Webber MJ; Appel EA, A co-formulation of supramolecularly stabilized insulin and pramlintide enhances mealtime glucagon suppression in diabetic pigs. Nat. Biomed. Eng 2020, 4, 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacober SJ; Prince MJ; Beals JM; Hartman ML; Qu Y; Linnebjerg H; Garhyan P; Haupt A, Basal insulin peglispro: Overview of a novel long-acting insulin with reduced peripheral effect resulting in a hepato-preferential action. Diabetes Obes. Metab 2016, 18 (1463–1326 (Electronic)), 3–16. [DOI] [PubMed] [Google Scholar]
- 24.Owens RA; Hansen RJ; Kahl SD; Zhang C; Ruan X; Koester A; Li S; Qian H-R; Farmen MW; Michael MD; Moyers JS; Cutler GB Jr.; Vick A; Beals JM, In Vivo and In Vitro Characterization of Basal Insulin Peglispro: A Novel Insulin Analog. J. Pharmacol. Exp. Ther 2016, 357 (3), 459–465. [DOI] [PubMed] [Google Scholar]
- 25.Vinciguerra B; Cao L; Cannon JR; Zavalij PY; Fenselau C; Isaacs L, Synthesis and self-assembly processes of monofunctionalized cucurbit[7]uril. J. Am. Chem. Soc 2012, 134 (31), 13133–13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu KK; Huan Y, Streptozotocin-induced diabetic models in mice and rats. Curr. Protoc. Pharmacol 2008, 5.47.41–45.47.14. [DOI] [PubMed] [Google Scholar]
- 27.Maikawa CL; Mann JL; Kannan A; Meis CM; Grosskopf AK; Ou BS; Autzen AAA; Fuller GG; Maahs DM; Appel EA, Engineering Insulin Cold Chain Resilience to Improve Global Access. Biomacromolecules 2021, In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mann JL; Maikawa CL; Smith AAA; Grosskopf AK; Baker SW; Roth GA; Meis CM; Gale EC; Liong CS; Correa S; Chan D; Stapleton LM; Yu AC; Muir B; Howard S; Postma A; Appel EA, An Ultra-fast Insulin Formulation Enabled by High Throughput Screening of Polymeric Excipients. Sci. Transl. Med 2020, 12, eaba6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen D; Disotuar MM; Xiong X; Wang Y; Chou DH-C, Selective N-terminal functionalization of native peptides and proteins. Chem. Sci 2017, 8 (4), 2717–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosen CB; Francis MB, Targeting the N terminus for site-selective protein modification. Nat. Chem. Bio 2017, 13 (7), 697–705. [DOI] [PubMed] [Google Scholar]
- 31.Smith AAA; Maikawa CL; Roth GA; Appel EA, Site-selective modification of proteins using cucurbit[7]uril as supramolecular protection for N-terminal aromatic amino acids. Org. Biomol. Chem 2020, 18 (23), 4371–4375. [DOI] [PubMed] [Google Scholar]
- 32.Zou L; Braegelman AS; Webber MJ, Spatially Defined Drug Targeting by in Situ Host-Guest Chemistry in a Living Animal. ACS Cent. Sci 2019, 5 (6), 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the results in this study are available within the Article and its Supplementary Information. Raw data files are available from the corresponding author upon reasonable request.