

Abstract
Nickel compounds are environmental toxicants, prevalent in the atmosphere due to their widespread use in several industrial processes, extensive consumption of nickel containing products, as well as burning of fossil fuels. Exposure to nickel is associated with a multitude of chronic inflammatory lung diseases including asthma, chronic obstructive pulmonary disease (COPD) and pulmonary fibrosis. In addition, nickel exposure is implicated in the development of nasal and lung cancers. Interestingly, a common pathogenic mechanism underlying the development of diseases associated with nickel exposure is epithelial-mesenchymal transition (EMT). EMT is a process by which the epithelial cells lose their junctions and polarity and acquire mesenchymal traits including increased ability to migrate and invade. EMT is a normal and essential physiological process involved in differentiation, development and wound healing. However, EMT also contributes to a number of pathological conditions including fibrosis, cancer and metastasis. Growing evidence suggest that EMT induction could be an important outcome of nickel exposure. In this review, we discuss the role of EMT in nickel-induced lung diseases and the mechanisms associated with EMT induction by nickel exposure.
Keywords: Nickel, epithelial-mesenchymal transition, epigenetics, TGF-β, HIF-1
1. Introduction
Nickel compounds are environmental and occupational toxicants. Nickel occurs naturally in the atmosphere through volcanic activity, forest fires and weathering of rocks. However, industrial processes such as food processing, mining and refining, as well as extensive use of nickel-containing products such as stainless steel, jewelry and electric equipment, and medical implants increases its atmospheric levels [1–4]. In addition, sewage sludge incineration and fossil fuel combustion significantly contribute to atmospheric nickel levels [5]. While the average nickel concentration in ambient air in the United States is 2.22 ng/m3, in occupational settings, the concentrations of airborne nickel range from 1–60,000 μg/m3 [6]. Humans are primarily exposed to nickel through inhalation, ingestion and dermal contact [1, 4]. Furthermore, smoking cigarettes is an important source of human nickel exposure.
Nickel exposure is associated with a number of health hazards in humans. Pulmonary absorption is a major route of nickel exposure. Therefore, the lung is a major target organ of nickel toxicity and lung and nasal cancers are among the most serious consequences of exposure to nickel compounds [4, 7, 8]. Increase in mortality due to lung and nasal cancers has been reported in nickel refinery workers [6]. Furthermore, nasal cancer risk in nickel refinery workers increased with the duration of employment [9]. Both water-soluble and -insoluble nickel compounds cause neoplastic transformation of cells [10–12]. In animals, nickel compounds induced tumors at the sites of exposure [12]. Therefore, certain nickel compounds have been classified as Group 1 Carcinogens by the International Agency for Research on Cancer (IARC) [3, 13].
In addition to cancers, non-malignant respiratory diseases of nickel exposure have also been well-documented. Nickel exposure is associated with the development of a number of respiratory ailments including asthma, chronic bronchitis, emphysema, pulmonary fibrosis, pulmonary edema and reduced lung function [14–16]. Occupational asthma has been reported in electroplating workers exposed to nickel fumes [17]. Furthermore, inhalation of nickel salts has been experimentally shown to cause bronchoconstriction [18–20]. At occupational exposure levels, nickel sulphate, nickel subsulphate and nickel oxide caused lung inflammation and fibrosis in rats and mice. A positive correlation has been shown between the solubility of nickel compounds and the severity of lung inflammation and fibrosis, with nickel sulphate being the most toxic followed by nickel subsulphate and nickel oxide [6]. Furthermore, studies on nickel refinery workers showed association of nickel exposure with the development of chronic obstructive pulmonary disease (COPD) [21].
Interestingly, a key process that drives the pathogenesis of lung diseases associated with nickel exposure is epithelial-mesenchymal transition (EMT). EMT is a process during which the epithelial cells lose cell-cell adhesion and acquire mesenchymal properties, including increased ability to migrate and invade [22]. EMT is a normal cellular process that plays a major role during development and repair of damaged tissues. However, aberrant EMT induction results in organ fibrosis. Growing evidence suggest EMT induction as an important outcome of nickel exposure [23–26]. Here, we discuss the role of EMT in the development of diseases associated with nickel exposure and the potential mechanisms underlying EMT induction by nickel exposure.
2. Epithelial-mesenchymal transition (EMT) in the pathogenesis of lung diseases
EMT is a process by which the epithelial cells lose their cell polarity and tight junctions, reorganize their cytoskeleton and acquire mesenchymal characteristics including increased invasive and migratory potentials [22]. Downregulation of E-cadherin, an important cell-cell adhesion protein, is a hallmark of EMT. In addition, repression of epithelial markers such as claudins, occludin and desmoplakin, causing the dissolution of adherens junctions and tight junctions, is commonly observed during EMT. Downregulation of epithelial markers is accompanied by upregulation of mesenchymal markers including fibronectin, vimentin, α-smooth muscle actin (α-SMA), desmin, and N-cadherin [22, 27]. Furthermore, several transcription factor families such as SNAIL, ZEB and TWIST, which are identified as EMT master regulators, are also upregulated during EMT. EMT is a reversible process, with the mesenchymal cells capable of undergoing mesenchymal-epithelial transition (MET). During MET, the apico-basal polarity of the cells is established, junctional complexes are assembled and tight junctions are formed. MET generates epithelial cells at different developmental stages and also during metastatic colonization [28, 29].
EMT is categorized into three subtypes based on their distinct biological settings and different functional consequences. Type 1 EMT is associated with developmental processes such as gastrulation, neural crest formation and heart value formation. During type 1 EMT, the first set of mesenchymal cells known as primary mesenchyme are generated to create new tissues with diverse functions [30]. Type 2 EMT, which generates fibroblasts, is associated with wound healing and tissue regeneration. This type of EMT occurs as part of repair process to reconstruct tissues following trauma or inflammation. Although type 2 EMT is a normal physiological process, chronic inflammation and aberrations in myofibroblast activation could lead to increased deposition of extracellular matrix proteins, resulting in fibrosis and organ destruction [30–32]. Type 3 EMT is associated with the dissemination of cells from the primary tumors, thus playing a major role in epithelial cancer cell metastasis [22].
Emerging evidence implicates EMT in the pathogenesis of a number of lung diseases:
Pulmonary fibrosis is characterized by accumulation of myofibroblasts and increased deposition of extracellular matrix proteins. Injury to lung epithelium is an important event in the development of this disease. Upon injury, the epithelial cells undergo EMT, resulting in morphological changes which give rise to fibroblast-like cells [33]. Transforming growth factor-β (TGF-β) levels increase in the injured lung epithelial cells and has been identified as a major driver of EMT in lung fibrosis [33, 34]. TGF-β-exposed alveolar epithelial cells undergo EMT and exhibit fibroblast-like morphology. Furthermore, lung biopsies of idiopathic pulmonary fibrosis patients suggest contribution of EMT to pulmonary fibrosis [34].
COPD is a progressive airway obstructive disease, which involves destruction of lung parenchyma (emphysema) and chronic inflammation of large airways (bronchitis) [35]. In addition, pulmonary fibrosis is reported in COPD patients [36]. Emerging evidence strongly suggest EMT as a driver of COPD pathogenesis. In COPD associated with smoking, the epithelium is in an activated state and the structural changes to the underlying reticular basement membrane (Rbm) suggest EMT. Furthermore, increased vascularization of Rbm in large airways indicates type 3 EMT [37–40].
Asthma is a chronic disease characterized by progressive airway remodeling. In addition, chronic airway inflammation and airway hyperresponsiveness are major features of asthma [41, 42]. Recent studies have demonstrated the importance of EMT in airway remodeling in asthma, which include airway wall thickening, subepithelial fibrosis, increased smooth muscle mass and angiogenesis [42, 43]. Downregulation of cell adhesion proteins occur in asthmatic epithelium [44]. Furthermore, increased EMT has been observed in asthmatic patients as compared to normal individuals [42, 45].
In epithelial cancers, loss of cell-cell adhesion results in dissociation of the primary tumor mass and metastatic dissemination of the cancer cells. EMT is a critical process through which cancers acquire invasiveness and progress to a metastatic state. Increased production of TGF-β is normally observed in cancer cells and several studies suggest TGF-β signaling pathway as a major driver of EMT in cancer progression [46, 47]. Furthermore, recent studies have also identified cell dissemination from premalignant lesions, which suggests that EMT could be involved cancer initiation, in addition to being associated with metastasis [48–51].
Nickel, an EMT inducer, is implicated in the development of a number of lung diseases described above, including pulmonary fibrosis [52, 53], COPD [54], asthma [19, 55] and cancer and metastasis [9, 56]. This suggests that EMT induction by nickel exposure is likely a key mechanism associated with the pathogenesis of nickel-induced lung diseases. The following sections discuss molecular mechanisms underlying EMT induction by nickel exposure.
3. Transforming growth factor-β (TGF-β) signaling
TGF-β is a multifunctional cytokine that belongs to a superfamily of secreted factors. TGF-β is ubiquitously expressed and regulates a variety of physiological processes including embryogenesis, differentiation, cell proliferation, cytoskeletal organization and immune response [57, 58]. Aberrant TGF-β signaling is associated with a number of pathological conditions including autoimmune diseases, fibrotic diseases and cancer [57]. Mammalian cells have three TGF-β isoforms, TGF-β1, TGF-β2 and TGF-β3. TGF-β is synthesized as a large precursor, which is cleaved to release the active mature ligand in a dimeric form [57, 59]. Active TGF-β binds the type I (TGF-βR1) and type II (TGF-βRII) serine/threonine kinase receptors forming a heterotetrameric complex [57, 58]. Activation of the receptor complex by TGF-β leads to phosphorylation of SMAD proteins, which then translocate to the nucleus and regulate the expression of specific genes (Figure 1) [58, 60]. TGF-β signaling is also mediated by non-Smad pathways such as MAP Kinase, PI3K kinase, PP2A phosphatase and Rho GTPases [58, 61].
Figure 1. TGF-β signaling in nickel-induced EMT.
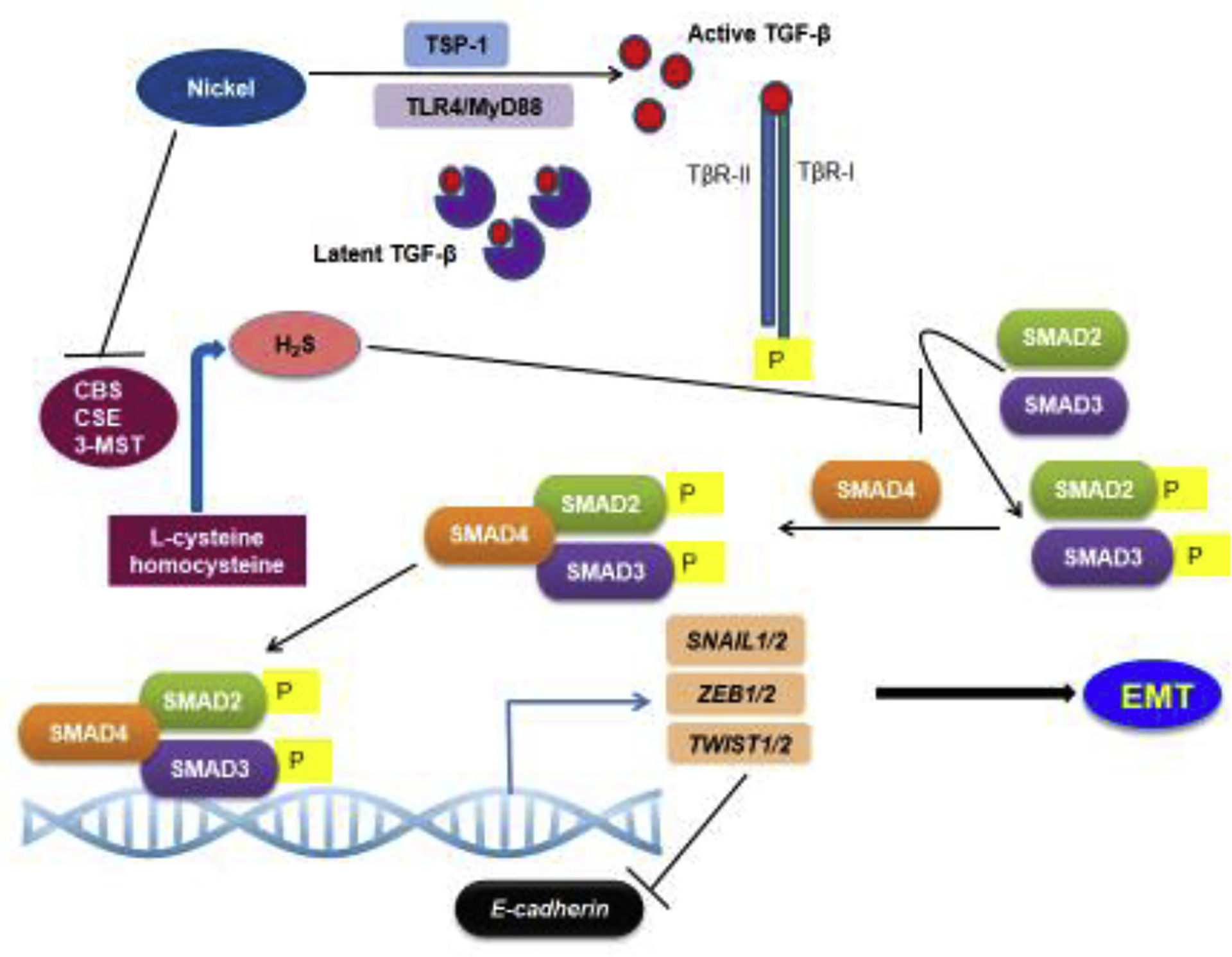
During TGF-β signaling, the active TGF-β binds the TGF-β receptors TGF-βR1 and TGF-βRII. The activation of the TGF-β receptor complex results in phosphorylation of the SMAD proteins, which then translocate to the nucleus and regulate target gene expression. Nickel exposure could activate TGF-β signaling via multiple mechanisms, including TLR4 signaling, TSP-1 upregulation and inhibition of H2S production. Nickel exposure induces TLR4 signaling. which has been shown to enhance TGF-β signaling. The extracellular matrix protein, TSP-1, an important activator of latent TGF-β, is one of the highest upregulated genes in the lungs of mice exposed to nickel, suggesting its role in nickel-induced TGF-β signaling. H2S is an important negative regulator of TGF-β signaling. Nickel exposure promoted TGF-β signaling by downregulating the enzymes involved in H2S production, CBS, CSE and 3MST, thereby increasing protein levels of TGF-β1 and phosphorylated SMAD2 and SMAD3.
TGF-β signaling plays an important role in the induction of EMT. TGF-β induces EMT during physiological processes such as embryogenesis and wound healing. However, under pathological conditions, TGF-β-induced EMT could be involved in tumor progression and metastasis, and the development of fibrotic diseases [60, 62, 63]. TGF-β signaling activates the EMT master regulators, SNAIL, ZEB and TWIST [62] and the mesenchymal markers, matrix metalloproteinases (MMPs), α-SMA, vimentin, fibronectin, and N-cadherin [64]. SNAIL, ZEB and TWIST repress E-cadherin expression by directly binding to the E-boxes at its promoter. [65–69]. SNAIL is also a repressor of claudins and occludin, which are involved in maintaining epithelial structure and function [70, 71]. In addition, SNAIL can also activate the expression of the mesenchymal proteins fibronectin, vimentin [72, 73] and N-cadherin [74].
Studies in mice exposed to nickel sulfate aerosol for 72 hours showed increase in TGF-β1 transcript and protein in the bronchoalveolar lavage fluid (BALF). Furthermore, expression of genes associated with TGF-β signaling and extracellular matrix function was significantly increased [46]. TGF-β1 repressed the mouse surfactant-associated protein B (Sftpb) via the TGF-β–responsive region at the Sftpb promoter [46]. Interestingly, Sftpb knockout has been shown to decrease the expression of E-cadherin and increase the expression of vimentin and fibronectin in the lung adenocarcinoma A549 cells. Furthermore, Sftpb knockout enhanced cell survival and migration, suggesting EMT [75]. This suggests that SFTPB downregulation by TGF-β induces EMT. Inhibition of TGF-β using the TGF-β type II receptor–IgG-Fc (TGFbRII-Fc) chimera decreased the nickel-induced total BALF protein levels in mice [46]. TGF-β inhibition also prevented NiCl2 induced E-cadherin downregulation in both immortalized normal human bronchial epithelial BEAS-2B cells and the lung adenocarcinoma A549 cells [26].
Nano nickel oxide (nano-NiO) induced EMT through activation of TGF-β signaling [76]. The lungs of rats exposed to nano-NiO showed an increase in TGF-β levels, which was associated with pulmonary fibrosis [76, 77]. In addition, several fibrosis associated factors including Smad2 and Smad4 were overexpressed [76]. Furthermore, higher protein levels of collagen I and III were observed in nano-NiO exposed rat lungs, suggesting higher collagen deposition. Collagen I promotes EMT in lung cancer cells via activation of TGF-β3 signaling [78]. Similarly, A549 cells, as well as the human fetal lung fibroblasts exposed to nano-NiO, showed increased TGF-β levels [79]. Furthermore, the nano-NiO-treated A549 cells showed mesenchymal characteristics, including increase in the levels of α-SMA, vimentin and fibronectin and decrease in the levels of E-cadherin, suggesting EMT [79].
Interestingly, although both nano-NiO (20 nm particles) and micro-NiO (1 μm particles) exposure increased the expression of TGF-β and other fibrosis-associated factors in rat lungs, the levels of upregulation were higher in nano-NiO exposed lungs compared to those of the micro-NiO exposed lungs [76]. Furthermore, no increase in collagen I and III was seen in the lungs of rats exposed to micro-NiO. This suggests increased propensity of nano-NiO to cause lung injury and induce EMT, and thus is potentially a more serious health hazard than micro-NiO.
One of the endogenous negative regulators of the EMT process is hydrogen sulfide (H2S) [80]. Endogenous H2S plays a number of physiological roles and has been recently identified as a gasotransmitter that is at least as important as the other well-studied gasotransmitters, nitric oxide (NO) and carbon monoxide (CO) [80–82]. In mammalian cells, endogenous H2S is produced through the metabolism of cysteine. The enzymes involved in this process include the two pyridoxal 5′-phospate- (PLP)-dependent enzymes: cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE); and the PLP-independent, 3-mercaptopyruvate sulfurtransferase (3MST)/cysteine aminotransferase (CAT) pathway [83–85].
H2S attenuates EMT by inhibiting TGF-β signaling. Inhibition of TGF-β signaling by H2S is involved in the anti-fibrotic mechanism in several tissues including the lung and kidney [86]. Several studies have shown that the exogenous H2S donor, sodium hydrosulfide (NaHS), could inhibit EMT and airway remodeling in lung epithelial cells [82, 86–89]. NaHS attenuated EMT induced by the herbicide, paraquat (PQ), in A549 cells by inhibiting the TGF-β1/Smad2/3 signaling pathway [87]. Furthermore, a recent study suggested loss of H2S-mediated inhibition of TGF-β-Smad signaling as a cause for NiCl2-induced EMT in A549 cells [88]. NiCl2-exposed A549 cells underwent EMT, as evidenced by the downregulation of E-cadherin and the upregulation of vimentin and increased migratory ability. Examination of the associated mechanisms showed that nickel exposure downregulated CBS, CSE and 3MST, the enzymes involved in H2S production. Concurrently, an increase was detected in the protein levels of TGF-β1 as well as in the levels of phosphorylated SMAD2 and SMAD3. However, pretreatment of A549 cells with exogenous NaHS prevented NiCl2-induced TGF-β1-Smad signaling [88]. These results suggest a novel mechanism of EMT induction by nickel exposure via impaired H2S signaling (Figure 1).
Nickel is an activator of human TLR4 signaling, which triggers an allergic inflammatory response [90, 91]. NiCl2 exposure induced TLR4 signaling in the lung cancer A549 and H1299 cells [8]. This study also uncovered the elevated expression of IL-8, TGF-β, MMP2 and MMP9 caused by nickel exposure and demonstrated that TLR4/MyD88 signaling was important in increasing the invasive potential of the cells [8]. TLR4 activation has been shown to enhance TGF-β signaling in hepatic fibrosis [92]. Based on these results, it is plausible that nickel-induced TLR4 activation could be a potential mechanism underlying TGF-β induction in nickel-exposed cells (Figure 1). However, although nickel is an activator of human TLR4, it does not activate mouse TLR4 due to the sequence variations between the human and mouse TLR4 [91]. This suggests existence of additional mechanisms of TGF-β activation by nickel.
TGF-β is produced as an inactive complex, which is activated by the degradation of the prosegments. The extracellular matrix protein, thrombospondin 1 (TSP-1), is an important activator of latent TGF-β [93, 94]. Interestingly, TSP-1 was one of the highest upregulated genes in the lungs of mice exposed to NiSO4 aerosol [46]. This suggests that TSP-1 could be associated with TGF-β activation upon nickel exposure (Figure 1).
4. DNA methylation
5-methylcytosine (5mC) is an important DNA modification that plays principal roles in a variety of cellular processes. [95]. Nickel-induced aberrant changes to DNA methylation have been well-characterized in a number of studies [25, 96–102]. The DNA methylation alterations caused by nickel exposure is likely a consequence of its ability to inhibit the iron- and 2-oxoglutarate-dependent (2-OG) dioxygenases. Notable among the 2-OG dioxygenases are the Ten-eleven translocation (TET) family of DNA hydroxylases. Removal of 5mC occurs through the TET family dioxygenases, which mediate oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) [103]. The TET enzymes contain Fe(II) at their catalytic center. Ni(II), being highly similar to Fe(II) in charge and radius, can replace Fe(II) at the catalytic center of the Tet dioxygenases, thus making them highly susceptible to nickel-mediated inhibition [104].
NiCl2 exposure downregulated E-cadherin in BEAS-2B cells. E-cadherin downregulation was associated with promoter DNA hypermethylation, and DNA methyltrasferase inhibitor 5-aza-2’-deoxycytidine restored its expression [25]. Furthermore, treatment of BEAS-2B cells with antioxidants or reactive oxygen species (ROS) inhibitors and scavengers reversed nickel-induced EMT, and pretreatment of the cells with the antioxidant N-acetylcysteine (NAC) inhibited nickel-induced E-cadherin downregulation. In addition, NAC treatment prevented DNA hypermethylation at the E-cadherin promoter. This suggests a role for ROS generated by nickel exposure in the hypermethylation of E-cadherin promoter [25]. ROS has also been shown to induce hypermethylation of E-cadherin promoter through the induction of SNAIL expression in Huh7 and Hep3B cells [105]. SNAIL directly binds the E-cadherin promoter and recruits the H3K9me2 methyltransferase, G9a, along with the DNA methyltransferase, DNMT1. SNAIL binding of the E-cadherin promoter is required for DNMT1 recruitment, and G9a functions as a bridge between SNAIL and DNMT1 [106].
Nickel sulfide exposure in mice repressed p16 gene expression through promoter hypermethylation [98]. The tumor suppressor p16 is an EMT repressor, which regulates EMT by transcriptionally activating miR-141 and miR-146b-5p, negative regulators of AUF1, an RNA binding protein. AUF1 stabilizes ZEB1 mRNA, thus increasing its protein levels and thereby functioning as an EMT inducer [107]. Ectopic expression of p16 in the highly invasive breast cancer cell line upregulated epithelial markers and downregulated mesenchymal markers [107] Therefore, it is plausible that nickel-exposure-induced p16 downregulation could contribute to EMT.
5. Histone modifications
Besides the TET family proteins, another category of major epigenetic regulators among the iron- and 2-oxoglutarate-dependent dioxygenases are the Jumonji C (JmjC)-domain containing family of histone demethylases [108]. In eukaryotes, DNA is wrapped around the four core histone proteins, H2A, H2B, H3 and H4, to form the fundamental structural unit of chromatin, the nucleosome. The N-terminal tails of histones are subject to a variety of post-translational modifications including acetylation, methylation, phosphorylation and ubiquitination [109–112]. Specific histone modifications are associated with the active or silent states of a particular genomic locus. For example, histone acetylation is associated with transcriptional activation, and histone methylation is associated with both gene activation and silencing. While histone H3 lysine 4 trimethylation (H3K4me3) is associated with gene activation, H3K9me3, H3K9me2 and H3K27me3 are associated with gene silencing [113, 114].
The JmjC domain histone demethylases (KDM2 to KDM7), target a number of both activating histone methylation marks, including H3K4me2/3 and H3K36me1/2/3, and silencing histone methylation marks, including H3K9me3 and H3K27me3 [115]. Similar to TET enzymes, the JmjC domain histone demethylases are sensitive to nickel exposure due to the ability of nickel to inhibit their activities by replacing iron at the iron-binding sites of these enzymes [116, 117]. Nickel inhibited the activity of the H3K9 demethylase, JMJD1A/KDM3A in a dose-dependent manner, with approximately one molecule of Ni(II) inhibiting one molecule of JMJD1A. The inhibitory effect was significantly amplified in the absence of iron. However, the addition of iron ions could not rescue JMJD1A activity, suggesting that Ni(II) binds the enzyme with a higher affinity as compared to Fe(II) [117].
SPRY2, a tumor suppressor gene, is an important suppressor of ERK1/2 activation, and its silencing is often observed in a number of human cancers especially in the metastatic stages [118, 119]. Nickel exposure inhibited SPRY2 expression in BEAS-2B cells, which was associated with increased H3K9me2 levels at its promoter [116]. SPRY2 repression in nickel-exposed BEAS-2B cells resulted in ERK activation and neoplastic transformation. JMJD1A overexpression and knockdown studies showed that JMJD1A, which directly binds the SPRY2 promoter was essential for its expression. SPRY2 repression is often observed in a number of human cancers. This suggests downregulation of SPRY2 through JMJD1A inactivation as one of the mechanisms underlying nickel carcinogenesis [116]. SPRY2 has also been demonstrated to negatively regulate TGF-β signaling and TGF-β-induced EMT by inhibiting ERK activation [118]. These studies suggest that nickel could induce EMT by inhibiting JMJD1A demethylase activity. Furthermore, nickel exposure caused spreading of the repressive H3K9me2 to adjacent active chromatin regions via inhibition of the DNA binding activity of the insulator binding protein CTCF, resulting in the downregulation of a number of genes [113]. It is likely that nickel-induced inhibition of H3K9me2 demethylase activity contributes to H3K9me2 spreading and aberrant gene silencing.
KDM6B/JMJD3 is a histone demethylase that mediates the removal of repressive histone modification H3K27me3. TGF-β exposure upregulated KDM6B expression in the mammary epithelial NMuMG cells, resulting in H3K27me3 demethylation at SNAIL1 promoter. This resulted in the transcriptional activation of SNAIL and induction of EMT [120]. Interestingly, NiCl2 exposure increased KDM6B expression in the human embryonic kidney cell line HEK293T and renal carcinoma cell line 786-0. There was a concomitant decrease in the levels of H3K27me3 in both these cell lines [121]. Therefore, it is plausible that KDM6B upregulation by nickel could be associated with EMT induction.
Lysine-specific demethylase (LSD1/KDM1A) is a transcriptional repressor that demethylates H3K4me1 and H3K4me2 [122]. LSD1 is associated with the silencing of epithelial genes. LSD1 overexpression occurs in a number of solid tumors and levels of LSD1 expression correlates with tumor aggressiveness and EMT induction [122–127]. Nickel exposure has been shown to upregulate SNAIL in BEAS-2B cells [24]. SNAIL represses epithelial genes by recruiting LSD1, which recognizes H3K4 methylation through its interaction with the protein SFMBT1, resulting in its demethylation [128]. Interestingly, the protein levels of LSD1 remained unaltered due to nickel exposure. However, the levels of LSD1 acetylation significantly decreased [127]. In A549 cells, LSD1 protein is acetylated by the histone acetyl transferase MOF, which prevents its binding to the epithelial gene regulatory elements. Upon nickel exposure, MOF was significantly downregulated, resulting in LSD1 deacetylation. This caused increased binding of LSD1 at the epithelial genes, E-cadherin and KRT8, and caused H3K4me2 demethylation. Overexpression of MOF could counteract the nickel-induced EMT. Although MOF depletion facilitated nickel-induced EMT induction, in the absence of nickel exposure, depletion of MOF could not induce EMT in A549 cells. This suggests that in addition to MOF downregulation, other signals provided by nickel exposure are important for EMT induction [127].
NiCl2-induced EMT in the lung epithelial BEAS-2B cells is associated with increased ZEB1 expression [24]. In the epithelial cells, the ZEB1 promoter is marked by both the activating histone modification, H3K4me3, and the repressive histone modification, H3K27me3, thus existing in a bivalent chromatin environment [24]. Bivalent genes are normally transcriptionally silent, but are considered to be in a poised state [129]. When appropriate signals are received, the promoter bivalency resolves to monovalency through loss of the activating or the repressing marks, leading to the corresponding gene expression changes [129]. Nickel exposure resolved the bivalent chromatin at ZEB1 promoter to a monovalent status by inducing loss of H3K27me3, resulting in active gene transcription [24].
ZEB1 is a negative regulator of E-cadherin [24]. The nickel-exposed BEAS-2B cells with high ZEB1 expression exhibit EMT characteristics, including downregulation of epithelial markers, upregulation of mesenchymal markers and metastatic phenotype [24]. Interestingly, the loss of H3K27me3 was stable, and chromatin environment at the ZEB1 gene did not revert to epithelial state even after the termination of nickel exposure, resulting in continued ZEB1 overexpression post exposure [24]. This resulted in persistent mesenchymal phenotype even after the termination of exposure. Nickel exposure caused persistent changes to the active histone modification H3K4me3 as well [130]. These results suggest that epigenetic alterations that persist after the cessation of exposure could be a common phenomenon associated with nickel exposure.
ZEB1 is a negative regulator of the microRNAs (miRNAs) associated with epithelial phenotype, miR-200 family and miR-205. On the other hand, miR-200/205 are negative regulators of ZEB1. Thus, ZEB1 and miR-200/205 exist in a double negative feedback loop [131, 132]. In BEAS-2B cells, ZEB1 overexpression due to nickel exposure caused downregulation of miR-200/205, which likely caused further increase in ZEB1 expression. Continued downregulation of miR-200/205 even after the termination of nickel exposure contributed to the continuous overexpression of ZEB1 resulting in the persistent EMT phenotype. However, ZEB1 knockdown after the termination of nickel exposure abrogated miR-200/205 downregulation and re-established the epithelial phenotype in BEAS-2B cells. These results suggest ZEB1 as an important mediator of nickel-induced persistent EMT [24].
Bivalent chromatin at ZEB1 gene has also been observed in the non-CSC population of the human mammary basal epithelial cells (HMECs). EMT signals such as TGF-β quickly converted the bivalent chromatin to an active chromatin configuration, and the cells enter CSC state [133]. These findings suggest that the bivalent chromatin configuration at the ZEB1 promoter enables its rapid upregulation upon receiving appropriate environmental cues. Nickel exposure could be one such environmental cue, which activates ZEB1 gene in order to trigger the wound healing process via EMT induction. However, persistent ZEB1 overexpression could result in pathogenesis. The molecular basis of irreversible alteration to the chromatin at ZEB1 promoter, which enables its persistent overexpression, remains to be investigated.
6. MicroRNAs (miRNAs)
MicroRNAs (miRNAs) are non-coding RNAs, which are 18–25 nucleotides in length. miRNAs negatively regulate gene expression either by targeting the 3′ UTR of target mRNAs and causing their degradation or translation inhibition. Regulation of EMT associated transcription factors by miRNAs are well documented. miR-203, miR-30a and miR-34 family downregulate SNAIL expression [134–136]. ZEB family genes are well-studied targets of miR-200 family and miR-205. TWIST1 is also a direct target of several miRNAs, including miR-15, -16, -186, -381 and -495 in various cell types [137–141]. Furthermore, miR-9 is a repressor of E-cadherin. In addition to the miRNAs that directly target the EMT transcription factors, several miRNAs that target other transcription factors such as MYC and SP1 are also described as pro- or anti-metastatic [134]. These studies suggest that deregulation of miRNAs could have significant consequences in the development of EMT-associated diseases and cancer metastasis.
Prominent among the EMT-regulating miRNAs are the miR-200 family and miR-205, which are negative regulators of EMT master regulators ZEB1 and ZEB2. miR-200/205 are downregulated by nickel exposure, which contributed to nickel-induced EMT [24]. In the nickel-exposed BEAS-2B cells, miR-200/205 downregulation is likely caused by ZEB1 upregulation, since ZEB1 knockdown restored the levels of miR-200/205 [24] (see section ‘Histone modifications’ for more information on ZEB1-miR-200/205 interaction). TGF-β treatment has been shown to upregulate several EMT associated transcription factors including ZEB1 and ZEB2 [142, 143]. Therefore, TGF-β signaling activated by nickel exposure could also contribute to miR-200/205 downregulation and EMT.
Upregulation of miR-21 is seen in multiple cancers, including cancers of the lung, and elevated expression of miR-21 promoted EMT [144, 145]. In A549 cells, miR-21 inhibition increased E-cadherin expression while miR-21 mimics decreased it [144]. Exposure to nickel nanoparticles (nano-Ni) caused upregulation of miR-21 in mouse lungs [53]. Nano-Ni exposure also caused increased expression of fibrosis-associated factors TGF-β1, p-Smad2, COL1A1, and COL3A1. Furthermore, severe lung inflammation and fibrosis were detected in the lungs [53]. In the miR-21 knockout mice, nano-Ni-induced upregulation of pSmad2, COL1A1 and COL3A1 was significantly diminished, although TGF-β1 levels remained unaltered. Furthermore, pulmonary inflammation and fibrosis decreased in miR-21-knockout mice suggesting a role for miR-21 in nano-Ni-induced EMT [53]. Though the mechanism of miR-21 overexpression by nano-Ni exposure remains unclear, studies have shown that it can be upregulated by TGF-β [146–149]. Therefore, TGF-β induced by nano-Ni exposure could be associated with miR-21 upregulation and fibrogenesis.
miR-152 is a negative regulator of DNA methyltransferase 1 (DNMT1) [150]. Downregulation of miR-152 is associated with EMT induction [151]. Nickel sulfide (NiS) exposure downregulated miR-152 in human bronchial epithelial 16HBE cells through promoter DNA hypermethylation. While ectopic expression of miR-152 inhibited cell proliferation in the NiS-transformed cells 16HBE, anti-miR-152 promoted it [152]. This suggests that NiS-induced miR-152 downregulation could potentially induce EMT.
7. Long non-coding RNAs (LncRNAs)
LncRNAs are non-coding RNAs, which are over 200 nucleotides in length. Recent studies have begun to uncover the role of lncRNAs in regulating EMT and metastasis. A number of lncRNAs such as HOTAIR, H19 and MALAT1 are identified as EMT promoters [153–157]. Some lncRNAs function as competing endogenous RNAs (ceRNAs), which impede binding of miRNAs to their targets. LncRNA-ATB upregulated ZEB1/2 and promoted hepatocellular carcinoma metastasis by functioning as a ceRNA for the miR-200 family members. Similarly, HOTTIP upregulated SNAIL and promoted EMT and metastasis in esophageal carcinoma cells by functioning as a ceRNA for miR-30b, a SNAIL repressor [158]. Furthermore, MEG8 suppressed the expression of MIR34a and MIR203 by facilitating EZH2 mediated H3K27me3 enrichment at their promoters, resulting in the upregulation of SNAIL [159].
Although a few lncRNAs, such as NRG1 [160] and MEG3 [161], are implicated in nickel-exposure-induced tumorigenesis, their role in nickel-induced EMT has not been thoroughly investigated. Nickel exposure downregulated MEG3 through promoter DNA hypermethylation, resulting in malignant transformation of BEAS-2B cells [161]. MEG3 overexpression reversed the nickel-induced cell transformation. Lung tissues from lung squamous cell carcinoma (SCC) also showed a significant downregulation of MEG3, suggesting a role for MEG3 inhibition in lung tumorigenesis [161]. A study on gastric cancer tissues also suggested the anti-tumorigenic effects of MEG3. Interestingly, this study showed that MEG3 upregulated E-cadherin while downregulating the mesenchymal markers N-cadherin, Snail and β-catenin [162], thereby decreasing the cell’s ability to invade and migrate. Moreover, this study showed that the anti-EMT effect of MEG3 was mediated by its ability to negatively regulate miR-21, a promoter of EMT [162].
MEG3 is also downregulated by nano-NiO exposure [163]. Nano-NiO exposure induced EMT, causing collagen deposition in A549 cells and pulmonary fibrosis in rats in a TGF-β dependent manner. MEG3 overexpression decreased TGF-β1 expression and suppressed EMT. Therefore, MEG3 downregulation has been implicated in the EMT induction and fibrosis by nano-NiO exposure [163]. Contrarily, MEG3 has also been shown to be essential for TGF-β-induced EMT in the lung cancer cell lines, A549 and LC-2/ad, where it recruits repressive epigenetic marks to the regulatory regions of E-cadherin and MIR200 [164].
8. Hypoxia-inducible factor-1 (HIF-1) signaling
Cellular response to low oxygen is mainly mediated by HIF-1 signaling [165]. HIF-1 is a heterodimeric transcription factor consisting of a hypoxically inducible α subunit and a constitutively expressed β subunit. Under normoxia, the proline residues in the oxygen-dependent degradation domain (ODDD) of HIF-1α are hydroxylated by prolyl-hydroxylases (PHDs). The hydroxylated proline residues are recognized by von Hippel-Lindau protein (pVHL), which mediates HIF-1α ubiquitination, leading to its proteosomal degradation [166]. Hypoxia prevents HIF-1α hydroxylation due to inhibition of PHD activity, resulting in lack of interaction with pVHL and HIF-1α stabilization. The stabilized HIF-1α translocates to the nucleus, dimerizes with HIF-1β and activates target genes by binding the hypoxia-response elements (HREs) at their promoters [167–169] (Figure 2). HIF-1α is stabilized in solid tumors due to low oxygen availability and is associated with a number of cancers [170–173].
Figure 2. Activation of HIF-1 signaling in nickel-exposed cells contributes to EMT.
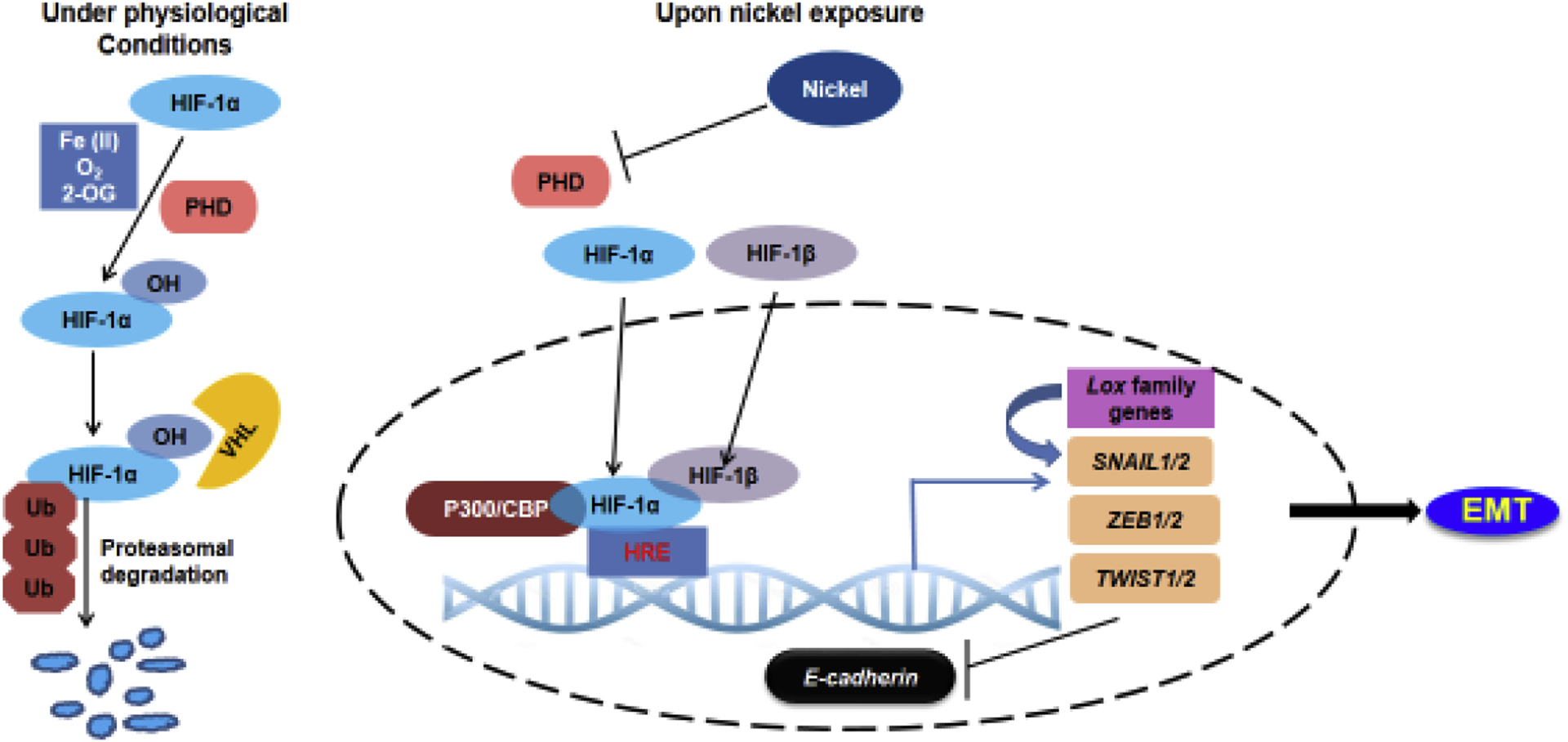
Under physiological conditions, HIF-1α is rapidly degraded via hydroxylation of its oxygen-dependent degradation domain by prolyl-hydroxylases (PHDs). Upon nickel exposure, Ni(II) replaces Fe(II) at the iron-binding site of PHDs causing their inactivation. This leads to the stabilization of HIF-1α, which translocates to the nucleus, dimerizes with HIF-1β and binds the HREs at the promoters of target genes, resulting in their activation. HIF-1 could directly activate EMT master regulators, SNAIL, ZEB and TWIST, by directly binding their promoters. HIF-1 could also upregulate lysyl oxidase genes Lox and LoxL2, which interact with SNAIL and cause E-cadherin repression.
PHDs are 2-oxoglutarate (2-OG)-dependent dioxygenases, which require Fe(II) and ascorbate as cofactors [174]. Ni(II) can compete with Fe(II) and replace it at the iron-binding site of PHDs causing their inactivation. This results in HIF-1α stabilization [117, 175, 176]. Therefore, nickel exposure stabilizes HIF-1α even under normoxia [170]. HIF-1 has been well-documented to regulate a large number of cellular processes including cell survival, proliferation, motility, metabolism, extracellular matrix function and angiogenesis [177–180]. Emerging evidence suggest a major role for HIF signaling in the induction of metastasis. A number of studies have demonstrated the role of HIF-1 in inducing EMT in multiple cell-types [181–184]. EMT can be induced by increased HIF-1α expression either due to hypoxia or due to the constitutive expression of HIF-1α mutant containing a deletion in the ODD domain [185, 186]. Moreover, HIF-1α knockdown in hypoxic cells reversed both the expression of EMT markers and the metastatic ability [185, 187, 188].
The effect of HIF-1 signaling on EMT is mediated by a number of downstream targets of HIF-1 such as the EMT master regulators, SNAIL, ZEB and TWIST. HIF-1 directly targets these EMT factors by binding the HREs at their promoters and activating their expression (Figure 2) [184–186, 189–191]. Suggesting major roles for the EMT master regulators in mediating HIF-1 signaling, ZEB1 inhibition abolished HIF-1α-induced EMT in metastatic colorectal cancer cells [189]. Similarly, TWIST knockdown in lung and hypopharyngeal cancer cells abolished the EMT phenotype and inhibited EMT induced by hypoxia or HIF-1α overexpression [185, 186]. Inhibition of SNAIL by shRNAs reduced HIF-1α-induced EMT in gastric CSCs [192]. These studies also suggest that the downstream effectors of HIF-1-induced EMT could be cell-type specific. In addition, HIF-1 can also regulate EMT markers in an indirect fashion. HIF-1α promoted EMT and fibrogenesis in mice renal epithelial cells by upregulating lysyl oxidase genes Lox and LoxL2 [193]. The lysyl oxidases have been shown to functionally interact with SNAIL, causing E-cadherin repression in MDCK cells [194].
Stabilization of HIF-1α by nickel compounds is well documented [25, 170, 195–197]. NiCl2 exposure significantly increased the protein levels of HIF-1α, which was associated with E-cadherin repression in BEAS-2B cells [25]. ShRNA-mediated HIF1α knockdown in BEAS-2B cells diminished NiCl2-induced E-cadherin downregulation and fibronectin upregulation, suggesting a role for HIF-1 signaling in NiCl2-induced EMT in the lung [25]. Nano-NiO exposure increased the levels of HIF-1α and TGF-β1 in human fetal lung fibroblasts, which contributed to the development of pulmonary fibrosis [77]. Exposure of human fetal lung fibroblast cells to HIF1-α inhibitor 2-deoxy-d-glucose (2-DG) reduced the nano-NiO-induced overexpression of TGF-β1. On the other hand, TGF-β-Smad inhibitor treatment diminished the nano-NiO-induced increase in HIF-1α protein levels [77]. This suggests a synergistic interaction between the HIF-1 and TGF-β signaling pathways in the development of nickel-induced pulmonary fibrosis [77]. Although the role of HIF-1 signaling in nickel-induced EMT has not been extensively investigated, given the well-established role of HIF-1 signaling in EMT induction, it is reasonable to speculate that HIF-1α stabilization by nickel exposure could play a major role in EMT induction.
9. Conclusions and future perspectives
Evidence from recent studies clearly indicate the importance of EMT induction in the etiology of lung diseases associated with nickel exposure. TGF-β is an important regulator of a number of pathogenic processes [198]. It is clear that the activation of TGF-β signaling by nickel exposure is an important event in nickel pathogenesis. Although some mechanisms such as impaired H2S production [88] and activation of TLR4 signaling [8] could begin to explain the underlying mechanisms, further investigation is necessary to fully understand this critically important process. Similarly, HIF-1 signaling, which could directly regulate the expression of EMT associated factors including ZEB, TWIST and SNAIL, is an important EMT inducer. The mechanisms underlying nickel-mediated HIF-1α stabilization is well understood. Future studies will reveal the importance of HIF-1 signaling in nickel-induced lung diseases.
In addition to TGF-β and HIF-1 signaling, epigenetic deregulation constitutes an important phenomenon that is associated with nickel-induced EMT. The mutagenic potential of nickel compounds is low and accumulating evidence suggest aberrant epigenetic changes as major drivers of nickel toxicity. Consequently, nickel-induced epigenetic changes have been extensively characterized by a number of studies [113, 130]. However, epigenetic changes related to EMT induction is only beginning to be understood. Although inactivation of 2-OG dioxygenases potentially plays a major role in the aberrant epigenetic changes caused by nickel exposure, the mechanisms associated with nickel-induced dysregulation of the epigenome are not fully understood. Bivalency resolution via loss of H3K27me3 at the ZEB1 promoter plays an important role in EMT induction by nickel. However, the underlying mechanisms remain unclear. Bivalent chromatin, which poises genes for activation upon receiving appropriate stimuli, is present in all cell types. However, it is more prevalent in the embryonic stem (ES) cells, where the developmental gene promoters are marked by both active and repressive epigenetic marks [111, 129, 199]. EMT is involved in a number of early developmental processes such as gastrulation and neural crest formation. Therefore, it is reasonable to speculate that nickel exposure could have significant deleterious consequences during early development, and this warrants in-depth investigation in the future. Thus, while the current studies have shed important light on the contribution of EMT to nickel-induced lung diseases, there are a number of open questions and exciting avenues for future research.
Acknowledgements
This work was supported by National Institutes of Environmental Health Sciences (NIEHS), National Institutes of Health (NIH) grants R01ES024727, R01ES031402 to S.C. and the core Center Grant P30ES000260 from NIEHS, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors declare that there are no conflicts of interest
References
- [1].Cameron KS, Buchner V, Tchounwou PB, Exploring the molecular mechanisms of nickel-induced genotoxicity and carcinogenicity: a literature review, Rev Environ Health 26(2) (2011) 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Eliades T, Athanasiou AE, In vivo aging of orthodontic alloys: implications for corrosion potential, nickel release, and biocompatibility, Angle Orthod 72(3) (2002) 222–37. [DOI] [PubMed] [Google Scholar]
- [3].Salnikow K, Zhitkovich A, Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium, Chem Res Toxicol 21(1) (2008) 28–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Aleksandra D, Urszula B, The impact of nickel on human health, J Elem 13(4) (2008) 685–696. [Google Scholar]
- [5].Khodadoust AP, Reddy KR, Maturi K, Removal of nickel and phenanthrene from kaolin soil using different extractants, Environmental Engineering Science 21(6) (2004) 691–704. [Google Scholar]
- [6].Agency for Toxic Substances and Disease Registry (ASTDR), Toxicological Profile for Nickel., Department of Health and Human Services, Public Health Service: Atlanta, GA, USA., 2005, pp. 1–397. [Google Scholar]
- [7].Oller AR, Costa M, Oberdorster G, Carcinogenicity assessment of selected nickel compounds, Toxicol Appl Pharmacol 143(1) (1997) 152–66. [DOI] [PubMed] [Google Scholar]
- [8].Xu Z, Ren T, Xiao C, Li H, Wu T, Nickel promotes the invasive potential of human lung cancer cells via TLR4/MyD88 signaling, Toxicology 285(1–2) (2011) 25–30. [DOI] [PubMed] [Google Scholar]
- [9].Anttila A, Pukkala E, Aitio A, Rantanen T, Karjalainen S, Update of cancer incidence among workers at a copper/nickel smelter and nickel refinery, Int Arch Occup Environ Health 71(4) (1998) 245–50. [DOI] [PubMed] [Google Scholar]
- [10].Coogan TP, Latta DM, Snow ET, Costa M, Toxicity and carcinogenicity of nickel compounds, Crit Rev Toxicol 19(4) (1989) 341–84. [DOI] [PubMed] [Google Scholar]
- [11].Grimsrud TK, Berge SR, Haldorsen T, Andersen A, Exposure to different forms of nickel and risk of lung cancer, Am J Epidemiol 156(12) (2002) 1123–32. [DOI] [PubMed] [Google Scholar]
- [12].Ke Q, Davidson T, Chen H, Kluz T, Costa M, Alterations of histone modifications and transgene silencing by nickel chloride, Carcinogenesis 27(7) (2006) 1481–8. [DOI] [PubMed] [Google Scholar]
- [13].Beyersmann D, Hartwig A, Carcinogenic metal compounds: recent insight into molecular and cellular mechanisms, Arch Toxicol 82(8) (2008) 493–512. [DOI] [PubMed] [Google Scholar]
- [14].Mittal R, Gupta P, Dash DJ, Prasad R, Chhabra SK, Occupational emphysema following long-term exposure to metal fumes during electroplating in a non-smoker, Indian J Chest Dis Allied Sci 58(2) (2016) 123–5. [PubMed] [Google Scholar]
- [15].Gul U, Cakmak SK, Olcay I, Kilic A, Gonul M, Nickel sensitivity in asthma patients, J Asthma 44(5) (2007) 383–4. [DOI] [PubMed] [Google Scholar]
- [16].Berge SR, Skyberg K, Radiographic evidence of pulmonary fibrosis and possible etiologic factors at a nickel refinery in Norway, J Environ Monit 5(4) (2003) 681–8. [DOI] [PubMed] [Google Scholar]
- [17].Bright P, Burge PS, O’Hickey SP, Gannon PF, Robertson AS, Boran A, Occupational asthma due to chrome and nickel electroplating, Thorax 52(1) (1997) 28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fernandez-Nieto M, Quirce S, Carnes J, Sastre J, Occupational asthma due to chromium and nickel salts, Int Arch Occup Environ Health 79(6) (2006) 483–6. [DOI] [PubMed] [Google Scholar]
- [19].Block GT, Yeung M, Asthma induced by nickel, JAMA 247(11) (1982) 1600–2. [PubMed] [Google Scholar]
- [20].Hong CS, Oh SH, Lee HC, Huh KB, Lee SY, Occupational asthma caused by nickel and zinc, Korean J Intern Med 1(2) (1986) 259–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vinnikov D, Semizhon S, Rybina T, Zaitsev V, Pleshkova A, Rybina A, Occupational exposure to metals and other elements in the tractor production, PLoS One 13(12) (2018) e0208932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kalluri R, Weinberg RA, The basics of epithelial-mesenchymal transition, J Clin Invest 119(6) (2009) 1420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhu Y, Chen QY, Li AH, Costa M, The Role of Non-Coding RNAs Involved in Nickel-Induced Lung Carcinogenic Mechanisms, Inorganics 7(7) (2019) 81. [Google Scholar]
- [24].Jose CC, Jagannathan L, Tanwar VS, Zhang X, Zang C, Cuddapah S, Nickel exposure induces persistent mesenchymal phenotype in human lung epithelial cells through epigenetic activation of ZEB1, Mol Carcinog 57(6) (2018) 794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wu CH, Tang SC, Wang PH, Lee H, Ko JL, Nickel-induced epithelial-mesenchymal transition by reactive oxygen species generation and E-cadherin promoter hypermethylation, J Biol Chem 287(30) (2012) 25292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wu CH, Hsiao YM, Yeh KT, Tsou TC, Chen CY, Wu MF, Ko JL, Upregulation of microRNA-4417 and Its Target Genes Contribute to Nickel Chloride-promoted Lung Epithelial Cell Fibrogenesis and Tumorigenesis, Sci Rep 7(1) (2017) 15320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu Y, New insights into epithelial-mesenchymal transition in kidney fibrosis, J Am Soc Nephrol 21(2) (2010) 212–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gunasinghe NP, Wells A, Thompson EW, Hugo HJ, Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer, Cancer Metastasis Rev 31(3–4) (2012) 469–78. [DOI] [PubMed] [Google Scholar]
- [29].Pei D, Shu X, Gassama-Diagne A, Thiery JP, Mesenchymal-epithelial transition in development and reprogramming, Nat Cell Biol 21(1) (2019) 44–53. [DOI] [PubMed] [Google Scholar]
- [30].Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA, Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease, J Clin Invest 119(6) (2009) 1438–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tennakoon AH, Izawa T, Kuwamura M, Yamate J, Pathogenesis of Type 2 Epithelial to Mesenchymal Transition (EMT) in Renal and Hepatic Fibrosis, J Clin Med 5(1) (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hung CF, Origin of Myofibroblasts in Lung Fibrosis, Current Tissue Microenvironment Reports 1(4) (2020) 155–162. [Google Scholar]
- [33].Pozharskaya V, Torres-Gonzalez E, Rojas M, Gal A, Amin M, Dollard S, Roman J, Stecenko AA, Mora AL, Twist: a regulator of epithelial-mesenchymal transition in lung fibrosis, PLoS One 4(10) (2009) e7559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Willis BC, Borok Z, TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease, Am J Physiol Lung Cell Mol Physiol 293(3) (2007) L525–34. [DOI] [PubMed] [Google Scholar]
- [35].Nowrin K, Sohal SS, Peterson G, Patel R, Walters EH, Epithelial-mesenchymal transition as a fundamental underlying pathogenic process in COPD airways: fibrosis, remodeling and cancer, Expert Rev Respir Med 8(5) (2014) 547–59. [DOI] [PubMed] [Google Scholar]
- [36].Wang H, Liao Z, Wan C, Wen F, Chen L, Epithelial-mesenchymal transition: a key mechanism for cigarette smoke-associated pulmonary fibrosis in COPD?, Eur J Intern Med 26(2) (2015) 143. [DOI] [PubMed] [Google Scholar]
- [37].Sohal SS, Mahmood MQ, Walters EH, Clinical significance of epithelial mesenchymal transition (EMT) in chronic obstructive pulmonary disease (COPD): potential target for prevention of airway fibrosis and lung cancer, Clin Transl Med 3(1) (2014) 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Soltani A, Muller HK, Sohal SS, Reid DW, Weston S, Wood-Baker R, Walters EH, Distinctive characteristics of bronchial reticular basement membrane and vessel remodelling in chronic obstructive pulmonary disease (COPD) and in asthma: they are not the same disease, Histopathology 60(6) (2012) 964–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Soltani A, Reid DW, Sohal SS, Wood-Baker R, Weston S, Muller HK, Walters EH, Basement membrane and vascular remodelling in smokers and chronic obstructive pulmonary disease: a cross-sectional study, Respir Res 11 (2010) 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Soltani A, Sohal SS, Reid D, Weston S, Wood-Baker R, Walters EH, Vessel-associated transforming growth factor-beta1 (TGF-beta1) is increased in the bronchial reticular basement membrane in COPD and normal smokers, PLoS One 7(6) (2012) e39736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Park JA, Fredberg JJ, Cell Jamming in the Airway Epithelium, Ann Am Thorac Soc 13 Suppl 1 (2016) S64–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sun Z, Ji N, Ma Q, Zhu R, Chen Z, Wang Z, Qian Y, Wu C, Hu F, Huang M, Zhang M, Epithelial-Mesenchymal Transition in Asthma Airway Remodeling Is Regulated by the IL-33/CD146 Axis, Front Immunol 11 (2020) 1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yang ZC, Qu ZH, Yi MJ, Shan YC, Ran N, Xu L, Liu XJ, MiR-448-5p inhibits TGF-beta1-induced epithelial-mesenchymal transition and pulmonary fibrosis by targeting Six1 in asthma, J Cell Physiol 234(6) (2019) 8804–8814. [DOI] [PubMed] [Google Scholar]
- [44].Hackett TL, Epithelial-mesenchymal transition in the pathophysiology of airway remodelling in asthma, Curr Opin Allergy Clin Immunol 12(1) (2012) 53–9. [DOI] [PubMed] [Google Scholar]
- [45].Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky DV, Murray LA, Argentieri R, Kicic A, Stick SM, Bai TR, Knight DA, Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1, Am J Respir Crit Care Med 180(2) (2009) 122–33. [DOI] [PubMed] [Google Scholar]
- [46].Wesselkamper SC, Case LM, Henning LN, Borchers MT, Tichelaar JW, Mason JM, Dragin N, Medvedovic M, Sartor MA, Tomlinson CR, Leikauf GD, Gene expression changes during the development of acute lung injury: role of transforming growth factor beta, Am J Respir Crit Care Med 172(11) (2005) 1399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Inamura S, Sakurai Y, Kawada S, Shohtsu A, Kuribayashi S, [Improvement of heart failure after intracardiac operation for tetralogy of Fallot by transcatheter embolization of major aortopulmonary collateral arteries], Kyobu Geka 43(3) (1990) 219–21. [PubMed] [Google Scholar]
- [48].Tellez CS, Juri DE, Do K, Bernauer AM, Thomas CL, Damiani LA, Tessema M, Leng S, Belinsky SA, EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells, Cancer Res 71(8) (2011) 3087–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Klein CA, Parallel progression of primary tumours and metastases, Nat Rev Cancer 9(4) (2009) 302–12. [DOI] [PubMed] [Google Scholar]
- [50].Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation, Cell 144(5) (2011) 646–74. [DOI] [PubMed] [Google Scholar]
- [51].Larsen JE, Nathan V, Osborne JK, Farrow RK, Deb D, Sullivan JP, Dospoy PD, Augustyn A, Hight SK, Sato M, Girard L, Behrens C, Wistuba II, Gazdar AF, Hayward NK, Minna JD, ZEB1 drives epithelial-to-mesenchymal transition in lung cancer, J Clin Invest 126(9) (2016) 3219–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Mo Y, Jiang M, Zhang Y, Wan R, Li J, Zhong CJ, Li H, Tang S, Zhang Q, Comparative mouse lung injury by nickel nanoparticles with differential surface modification, J Nanobiotechnology 17(1) (2019) 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mo Y, Zhang Y, Wan R, Jiang M, Xu Y, Zhang Q, miR-21 mediates nickel nanoparticle-induced pulmonary injury and fibrosis, Nanotoxicology 14(9) (2020) 1175–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Syurin SA, Nikanov AN, Frolova NM, [The importance of behavioral risk factors for bronchopulmonary pathology in copper-nickel industry workers], Med Tr Prom Ekol (8) (2013) 22–6. [PubMed] [Google Scholar]
- [55].Malo JL, Cartier A, Doepner M, Nieboer E, Evans S, Dolovich J, Occupational asthma caused by nickel sulfate, J Allergy Clin Immunol 69(1 Pt 1) (1982) 55–9. [DOI] [PubMed] [Google Scholar]
- [56].Andersen A, Berge SR, Engeland A, Norseth T, Exposure to nickel compounds and smoking in relation to incidence of lung and nasal cancer among nickel refinery workers, Occup Environ Med 53(10) (1996) 708–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Weiss A, Attisano L, The TGFbeta superfamily signaling pathway, Wiley Interdiscip Rev Dev Biol 2(1) (2013) 47–63. [DOI] [PubMed] [Google Scholar]
- [58].Li MO, Flavell RA, TGF-beta: a master of all T cell trades, Cell 134(3) (2008) 392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Xu J, Lamouille S, Derynck R, TGF-beta-induced epithelial to mesenchymal transition, Cell Res 19(2) (2009) 156–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Heldin CH, Moustakas A, Signaling Receptors for TGF-beta Family Members, Cold Spring Harb Perspect Biol 8(8) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Moustakas A, Heldin CH, Non-Smad TGF-beta signals, J Cell Sci 118(Pt 16) (2005) 3573–84. [DOI] [PubMed] [Google Scholar]
- [62].Hao Y, Baker D, Ten Dijke P, TGF-beta-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis, Int J Mol Sci 20(11) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Nieto MA, Huang RY, Jackson RA, Thiery JP, Emt: 2016, Cell 166(1) (2016) 21–45. [DOI] [PubMed] [Google Scholar]
- [64].Wendt MK, Allington TM, Schiemann WP, Mechanisms of the epithelial-mesenchymal transition by TGF-beta, Future Oncol 5(8) (2009) 1145–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Choi J, Park SY, Joo CK, Transforming growth factor-beta1 represses E-cadherin production via slug expression in lens epithelial cells, Invest Ophthalmol Vis Sci 48(6) (2007) 2708–18. [DOI] [PubMed] [Google Scholar]
- [66].Shirakihara T, Saitoh M, Miyazono K, Differential regulation of epithelial and mesenchymal markers by deltaEF1 proteins in epithelial mesenchymal transition induced by TGF-beta, Mol Biol Cell 18(9) (2007) 3533–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A, The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells, Nat Cell Biol 2(2) (2000) 84–9. [DOI] [PubMed] [Google Scholar]
- [68].Wong TS, Gao W, Chan JY, Transcription regulation of E-cadherin by zinc finger E-box binding homeobox proteins in solid tumors, Biomed Res Int 2014 (2014) 921564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA, Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis, Cell 117(7) (2004) 927–39. [DOI] [PubMed] [Google Scholar]
- [70].De Craene B, Gilbert B, Stove C, Bruyneel E, van Roy F, Berx G, The transcription factor snail induces tumor cell invasion through modulation of the epithelial cell differentiation program, Cancer Res 65(14) (2005) 6237–44. [DOI] [PubMed] [Google Scholar]
- [71].Ikenouchi J, Matsuda M, Furuse M, Tsukita S, Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail, J Cell Sci 116(Pt 10) (2003) 1959–67. [DOI] [PubMed] [Google Scholar]
- [72].Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA, The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression, Nat Cell Biol 2(2) (2000) 76–83. [DOI] [PubMed] [Google Scholar]
- [73].Olmeda D, Jorda M, Peinado H, Fabra A, Cano A, Snail silencing effectively suppresses tumour growth and invasiveness, Oncogene 26(13) (2007) 1862–74. [DOI] [PubMed] [Google Scholar]
- [74].Moreno-Bueno G, Cubillo E, Sarrio D, Peinado H, Rodriguez-Pinilla SM, Villa S, Bolos V, Jorda M, Fabra A, Portillo F, Palacios J, Cano A, Genetic profiling of epithelial cells expressing E-cadherin repressors reveals a distinct role for Snail, Slug, and E47 factors in epithelial-mesenchymal transition, Cancer Res 66(19) (2006) 9543–56. [DOI] [PubMed] [Google Scholar]
- [75].Lee S, Kim D, Kang J, Kim E, Kim W, Youn H, Youn B, Surfactant Protein B Suppresses Lung Cancer Progression by Inhibiting Secretory Phospholipase A2 Activity and Arachidonic Acid Production, Cell Physiol Biochem 42(4) (2017) 1684–1700. [DOI] [PubMed] [Google Scholar]
- [76].Chang XH, Zhu A, Liu FF, Zou LY, Su L, Liu SK, Zhou HH, Sun YY, Han AJ, Sun YF, Li S, Li J, Sun YB, Nickel oxide nanoparticles induced pulmonary fibrosis via TGF-beta1 activation in rats, Hum Exp Toxicol 36(8) (2017) 802–812. [DOI] [PubMed] [Google Scholar]
- [77].Qian F, He M, Duan W, Mao L, Li Q, Yu Z, Zhou Z, Zhang Y, Cross regulation between hypoxia-inducible transcription factor-1alpha (HIF-1alpha) and transforming growth factor (TGF)-ss1 mediates nickel oxide nanoparticles (NiONPs)-induced pulmonary fibrosis, Am J Transl Res 7(11) (2015) 2364–78. [PMC free article] [PubMed] [Google Scholar]
- [78].Shintani Y, Maeda M, Chaika N, Johnson KR, Wheelock MJ, Collagen I promotes epithelial-to-mesenchymal transition in lung cancer cells via transforming growth factor-beta signaling, Am J Respir Cell Mol Biol 38(1) (2008) 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Chang X, Tian M, Zhang Q, Gao J, Li S, Sun Y, Nano nickel oxide promotes epithelial-mesenchymal transition through transforming growth factor beta1/smads signaling pathway in A549 cells, Environ Toxicol 35(12) (2020) 1308–1317. [DOI] [PubMed] [Google Scholar]
- [80].Fang LP, Lin Q, Tang CS, Liu XM, Hydrogen sulfide attenuates epithelial-mesenchymal transition of human alveolar epithelial cells, Pharmacol Res 61(4) (2010) 298–305. [DOI] [PubMed] [Google Scholar]
- [81].Gadalla MM, Snyder SH, Hydrogen sulfide as a gasotransmitter, J Neurochem 113(1) (2010) 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Guan R, Wang J, Cai Z, Li Z, Wang L, Li Y, Xu J, Li D, Yao H, Liu W, Deng B, Lu W, Hydrogen sulfide attenuates cigarette smoke-induced airway remodeling by upregulating SIRT1 signaling pathway, Redox Biol 28 (2020) 101356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kimura H, Hydrogen sulfide: its production, release and functions, Amino Acids 41(1) (2011) 113–21. [DOI] [PubMed] [Google Scholar]
- [84].Shibuya N, Koike S, Tanaka M, Ishigami-Yuasa M, Kimura Y, Ogasawara Y, Fukui K, Nagahara N, Kimura H, A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells, Nat Commun 4 (2013) 1366. [DOI] [PubMed] [Google Scholar]
- [85].Singh S, Padovani D, Leslie RA, Chiku T, Banerjee R, Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions, J Biol Chem 284(33) (2009) 22457–22466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Guo L, Peng W, Tao J, Lan Z, Hei H, Tian L, Pan W, Wang L, Zhang X, Hydrogen Sulfide Inhibits Transforming Growth Factor-beta1-Induced EMT via Wnt/Catenin Pathway, PLoS One 11(1) (2016) e0147018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Bai YW, Ye MJ, Yang DL, Yu MP, Zhou CF, Shen T, Hydrogen sulfide attenuates paraquat-induced epithelial-mesenchymal transition of human alveolar epithelial cells through regulating transforming growth factor-beta1/Smad2/3 signaling pathway, J Appl Toxicol 39(3) (2019) 432–440. [DOI] [PubMed] [Google Scholar]
- [88].Ye M, Yu M, Yang D, Li J, Wang H, Chen F, Yu H, Shen T, Zhu Q, Zhou C, Exogenous hydrogen sulfide donor NaHS alleviates nickel-induced epithelial-mesenchymal transition and the migration of A549 cells by regulating TGF-beta1/Smad2/Smad3 signaling, Ecotoxicol Environ Saf 195 (2020) 110464. [DOI] [PubMed] [Google Scholar]
- [89].Lv M, Li Y, Ji MH, Zhuang M, Tang JH, Inhibition of invasion and epithelial-mesenchymal transition of human breast cancer cells by hydrogen sulfide through decreased phospho-p38 expression, Mol Med Rep 10(1) (2014) 341–6. [DOI] [PubMed] [Google Scholar]
- [90].Rothenberg ME, Innate sensing of nickel, Nat Immunol 11(9) (2010) 781–2. [DOI] [PubMed] [Google Scholar]
- [91].Schmidt M, Raghavan B, Muller V, Vogl T, Fejer G, Tchaptchet S, Keck S, Kalis C, Nielsen PJ, Galanos C, Roth J, Skerra A, Martin SF, Freudenberg MA, Goebeler M, Crucial role for human Toll-like receptor 4 in the development of contact allergy to nickel, Nat Immunol 11(9) (2010) 814–9. [DOI] [PubMed] [Google Scholar]
- [92].Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF, TLR4 enhances TGF-beta signaling and hepatic fibrosis, Nat Med 13(11) (2007) 1324–32. [DOI] [PubMed] [Google Scholar]
- [93].Murphy-Ullrich JE, Suto MJ, Thrombospondin-1 regulation of latent TGF-beta activation: A therapeutic target for fibrotic disease, Matrix Biol 68–69 (2018) 28–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Mir FA, Contreras-Ruiz L, Masli S, Thrombospondin-1-dependent immune regulation by transforming growth factor-beta2-exposed antigen-presenting cells, Immunology 146(4) (2015) 547–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Hashimoto H, Zhang X, Vertino PM, Cheng X, The Mechanisms of Generation, Recognition, and Erasure of DNA 5-Methylcytosine and Thymine Oxidations, J Biol Chem 290(34) (2015) 20723–20733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Lee YW, Klein CB, Kargacin B, Salnikow K, Kitahara J, Dowjat K, Zhitkovich A, Christie NT, Costa M, Carcinogenic nickel silences gene expression by chromatin condensation and DNA methylation: a new model for epigenetic carcinogens, Mol Cell Biol 15(5) (1995) 2547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Lee YW, Broday L, Costa M, Effects of nickel on DNA methyltransferase activity and genomic DNA methylation levels, Mutat Res 415(3) (1998) 213–8. [DOI] [PubMed] [Google Scholar]
- [98].Govindarajan B, Klafter R, Miller MS, Mansur C, Mizesko M, Bai X, LaMontagne K Jr., Arbiser JL, Reactive oxygen-induced carcinogenesis causes hypermethylation of p16(Ink4a) and activation of MAP kinase, Mol Med 8(1) (2002) 1–8. [PMC free article] [PubMed] [Google Scholar]
- [99].Yan Y, Kluz T, Zhang P, Chen HB, Costa M, Analysis of specific lysine histone H3 and H4 acetylation and methylation status in clones of cells with a gene silenced by nickel exposure, Toxicol Appl Pharmacol 190(3) (2003) 272–7. [DOI] [PubMed] [Google Scholar]
- [100].Ji W, Yang L, Yu L, Yuan J, Hu D, Zhang W, Yang J, Pang Y, Li W, Lu J, Fu J, Chen J, Lin Z, Chen W, Zhuang Z, Epigenetic silencing of O6-methylguanine DNA methyltransferase gene in NiS-transformed cells, Carcinogenesis 29(6) (2008) 1267–75. [DOI] [PubMed] [Google Scholar]
- [101].Yasaei H, Gilham E, Pickles JC, Roberts TP, O’Donovan M, Newbold RF, Carcinogen-specific mutational and epigenetic alterations in INK4A, INK4B and p53 tumour-suppressor genes drive induced senescence bypass in normal diploid mammalian cells, Oncogene 32(2) (2013) 171–9. [DOI] [PubMed] [Google Scholar]
- [102].Zhang J, Zhang J, Li M, Wu Y, Fan Y, Zhou Y, Tan L, Shao Z, Shi H, Methylation of RAR-beta2, RASSF1A, and CDKN2A genes induced by nickel subsulfide and nickel-carcinogenesis in rats, Biomed Environ Sci 24(2) (2011) 163–71. [DOI] [PubMed] [Google Scholar]
- [103].Wu X, Zhang Y, TET-mediated active DNA demethylation: mechanism, function and beyond, Nat Rev Genet 18(9) (2017) 517–534. [DOI] [PubMed] [Google Scholar]
- [104].Yin R, Mo J, Dai J, Wang H, Nickel(II) Inhibits Tet-Mediated 5-Methylcytosine Oxidation by High Affinity Displacement of the Cofactor Iron(II), ACS Chem Biol 12(6) (2017) 1494–1498. [DOI] [PubMed] [Google Scholar]
- [105].Lim SO, Gu JM, Kim MS, Kim HS, Park YN, Park CK, Cho JW, Park YM, Jung G, Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: methylation of the E-cadherin promoter, Gastroenterology 135(6) (2008) 2128–40, 2140 e1–8. [DOI] [PubMed] [Google Scholar]
- [106].Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, Evers BM, Zhou BP, G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer, J Clin Invest 122(4) (2012) 1469–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Al-Khalaf HH, Aboussekhra A, p16(INK4A) induces senescence and inhibits EMT through microRNA-141/microRNA-146b-5p-dependent repression of AUF1, Mol Carcinog 56(3) (2017) 985–999. [DOI] [PubMed] [Google Scholar]
- [108].Johansson C, Tumber A, Che K, Cain P, Nowak R, Gileadi C, Oppermann U, The roles of Jumonji-type oxygenases in human disease, Epigenomics 6(1) (2014) 89–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Kouzarides T, Chromatin modifications and their function, Cell 128(4) (2007) 693–705. [DOI] [PubMed] [Google Scholar]
- [110].Jenuwein T, Allis CD, Translating the histone code, Science 293(5532) (2001) 1074–80. [DOI] [PubMed] [Google Scholar]
- [111].Roh TY, Cuddapah S, Cui K, Zhao K, The genomic landscape of histone modifications in human T cells, Proc Natl Acad Sci U S A 103(43) (2006) 15782–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Felsenfeld G, Groudine M, Controlling the double helix, Nature 421(6921) (2003) 448–53. [DOI] [PubMed] [Google Scholar]
- [113].Jose CC, Xu B, Jagannathan L, Trac C, Mallela RK, Hattori T, Lai D, Koide S, Schones DE, Cuddapah S, Epigenetic dysregulation by nickel through repressive chromatin domain disruption, Proc Natl Acad Sci U S A 111(40) (2014) 14631–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K, High-resolution profiling of histone methylations in the human genome, Cell 129(4) (2007) 823–37. [DOI] [PubMed] [Google Scholar]
- [115].Accari SL, Fisher PR, Emerging Roles of JmjC Domain-Containing Proteins, Int Rev Cell Mol Biol 319 (2015) 165–220. [DOI] [PubMed] [Google Scholar]
- [116].Chen H, Kluz T, Zhang R, Costa M, Hypoxia and nickel inhibit histone demethylase JMJD1A and repress Spry2 expression in human bronchial epithelial BEAS-2B cells, Carcinogenesis 31(12) (2010) 2136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Chen H, Giri NC, Zhang R, Yamane K, Zhang Y, Maroney M, Costa M, Nickel ions inhibit histone demethylase JMJD1A and DNA repair enzyme ABH2 by replacing the ferrous iron in the catalytic centers, J Biol Chem 285(10) (2010) 7374–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Tan X, Zhu Y, Chen C, Chen X, Qin Y, Qu B, Luo L, Lin H, Wu M, Chen W, Liu Y, Sprouty2 Suppresses Epithelial-Mesenchymal Transition of Human Lens Epithelial Cells through Blockade of Smad2 and ERK1/2 Pathways, PLoS One 11(7) (2016) e0159275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Masoumi-Moghaddam S, Amini A, Morris DL, The developing story of Sprouty and cancer, Cancer Metastasis Rev 33(2–3) (2014) 695–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Ramadoss S, Chen X, Wang CY, Histone demethylase KDM6B promotes epithelial-mesenchymal transition, J Biol Chem 287(53) (2012) 44508–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Guo X, Zhang Y, Zhang Q, Fa P, Gui Y, Gao G, Cai Z, The regulatory role of nickel on H3K27 demethylase JMJD3 in kidney cancer cells, Toxicol Ind Health 32(7) (2016) 1286–92. [DOI] [PubMed] [Google Scholar]
- [122].Shi Y, Histone lysine demethylases: emerging roles in development, physiology and disease, Nat Rev Genet 8(11) (2007) 829–33. [DOI] [PubMed] [Google Scholar]
- [123].Hojfeldt JW, Agger K, Helin K, Histone lysine demethylases as targets for anticancer therapy, Nat Rev Drug Discov 12(12) (2013) 917–30. [DOI] [PubMed] [Google Scholar]
- [124].Lim S, Metzger E, Schule R, Kirfel J, Buettner R, Epigenetic regulation of cancer growth by histone demethylases, Int J Cancer 127(9) (2010) 1991–8. [DOI] [PubMed] [Google Scholar]
- [125].Lin T, Ponn A, Hu X, Law BK, Lu J, Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition, Oncogene 29(35) (2010) 4896–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Lin Y, Wu Y, Li J, Dong C, Ye X, Chi YI, Evers BM, Zhou BP, The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1, EMBO J 29(11) (2010) 1803–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Luo H, Shenoy AK, Li X, Jin Y, Jin L, Cai Q, Tang M, Liu Y, Chen H, Reisman D, Wu L, Seto E, Qiu Y, Dou Y, Casero RA Jr., Lu J, MOF Acetylates the Histone Demethylase LSD1 to Suppress Epithelial-to-Mesenchymal Transition, Cell Rep 15(12) (2016) 2665–78. [DOI] [PubMed] [Google Scholar]
- [128].Tang M, Shen H, Jin Y, Lin T, Cai Q, Pinard MA, Biswas S, Tran Q, Li G, Shenoy AK, Tongdee E, Lin S, Gu Y, Law BK, Zhou L, McKenna R, Wu L, Lu J, The malignant brain tumor (MBT) domain protein SFMBT1 is an integral histone reader subunit of the LSD1 demethylase complex for chromatin association and epithelial-to-mesenchymal transition, J Biol Chem 288(38) (2013) 27680–27691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Voigt P, Tee WW, Reinberg D, A double take on bivalent promoters, Genes Dev 27(12) (2013) 1318–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Jose CC, Wang Z, Tanwar VS, Zhang X, Zang C, Cuddapah S, Nickel-induced transcriptional changes persist post exposure through epigenetic reprogramming, Epigenetics Chromatin 12(1) (2019) 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ, A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition, Cancer Res 68(19) (2008) 7846–54. [DOI] [PubMed] [Google Scholar]
- [132].Diaz-Lopez A, Moreno-Bueno G, Cano A, Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives, Cancer Manag Res 6 (2014) 205–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, D’Alessio AC, Young RA, Weinberg RA, Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity, Cell 154(1) (2013) 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Zhang J, Ma L, MicroRNA control of epithelial-mesenchymal transition and metastasis, Cancer Metastasis Rev 31(3–4) (2012) 653–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Siemens H, Jackstadt R, Hunten S, Kaller M, Menssen A, Gotz U, Hermeking H, miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions, Cell Cycle 10(24) (2011) 4256–71. [DOI] [PubMed] [Google Scholar]
- [136].Moes M, Le Bechec A, Crespo I, Laurini C, Halavatyi A, Vetter G, Del Sol A, Friederich E, A novel network integrating a miRNA-203/SNAI1 feedback loop which regulates epithelial to mesenchymal transition, PLoS One 7(4) (2012) e35440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Landeros N, Santoro PM, Carrasco-Avino G, Corvalan AH, Competing Endogenous RNA Networks in the Epithelial to Mesenchymal Transition in Diffuse-Type of Gastric Cancer, Cancers (Basel) 12(10) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Wang T, Hou J, Li Z, Zheng Z, Wei J, Song D, Hu T, Wu Q, Yang JY, Cai JC, miR-15a-3p and miR-16-1-3p Negatively Regulate Twist1 to Repress Gastric Cancer Cell Invasion and Metastasis, Int J Biol Sci 13(1) (2017) 122–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Cao C, Sun D, Zhang L, Song L, miR-186 affects the proliferation, invasion and migration of human gastric cancer by inhibition of Twist1, Oncotarget 7(48) (2016) 79956–79963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Liu C, Jian M, Qi H, Mao WZ, MicroRNA 495 Inhibits Proliferation and Metastasis and Promotes Apoptosis by Targeting Twist1 in Gastric Cancer Cells, Oncol Res 27(3) (2019) 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [141].Yin Y, Li X, Guo Z, Zhou F, MicroRNA381 regulates the growth of gastric cancer cell by targeting TWIST1, Mol Med Rep 20(5) (2019) 4376–4382. [DOI] [PubMed] [Google Scholar]
- [142].Gregory PA, Bracken CP, Smith E, Bert AG, Wright JA, Roslan S, Morris M, Wyatt L, Farshid G, Lim YY, Lindeman GJ, Shannon MF, Drew PA, Khew-Goodall Y, Goodall GJ, An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition, Mol Biol Cell 22(10) (2011) 1686–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Peinado H, Olmeda D, Cano A, Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype?, Nat Rev Cancer 7(6) (2007) 415–28. [DOI] [PubMed] [Google Scholar]
- [144].Dai L, Chen F, Zheng Y, Zhang D, Qian B, Ji H, Long F, Cretoiu D, miR-21 regulates growth and EMT in lung cancer cells via PTEN/Akt/GSK3beta signaling, Front Biosci (Landmark Ed) 24 (2019) 1426–1439. [DOI] [PubMed] [Google Scholar]
- [145].Wu ZH, Tao ZH, Zhang J, Li T, Ni C, Xie J, Zhang JF, Hu XC, MiRNA-21 induces epithelial to mesenchymal transition and gemcitabine resistance via the PTEN/AKT pathway in breast cancer, Tumour Biol 37(6) (2016) 7245–54. [DOI] [PubMed] [Google Scholar]
- [146].Wang T, Zhang L, Shi C, Sun H, Wang J, Li R, Zou Z, Ran X, Su Y, TGF-beta-induced miR-21 negatively regulates the antiproliferative activity but has no effect on EMT of TGF-beta in HaCaT cells, Int J Biochem Cell Biol 44(2) (2012) 366–76. [DOI] [PubMed] [Google Scholar]
- [147].Davis BN, Hilyard AC, Lagna G, Hata A, SMAD proteins control DROSHA-mediated microRNA maturation, Nature 454(7200) (2008) 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A, Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha, Mol Cell 39(3) (2010) 373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Qian B, Katsaros D, Lu L, Preti M, Durando A, Arisio R, Mu L, Yu H, High miR-21 expression in breast cancer associated with poor disease-free survival in early stage disease and high TGF-beta1, Breast Cancer Res Treat 117(1) (2009) 131–40. [DOI] [PubMed] [Google Scholar]
- [150].Zhang H, Qi D, Li J, Peng T, Yang L, Yuan J, Zhang Y, Hu Y, Su J, Que B, Li M, Zhou G, Chen Y, Zhang W, Ji W, A novel regulatory circuit of miR152 and DNMT1 in human bladder cancer, Oncol Rep 40(3) (2018) 1803–1812. [DOI] [PubMed] [Google Scholar]
- [151].Ning YX, Wang XY, Wang JQ, Zeng R, Wang GQ, miR152 regulates TGFbeta1induced epithelialmesenchymal transition by targeting HPIP in tubular epithelial cells, Mol Med Rep 17(6) (2018) 7973–7979. [DOI] [PubMed] [Google Scholar]
- [152].Ji W, Yang L, Yuan J, Yang L, Zhang M, Qi D, Duan X, Xuan A, Zhang W, Lu J, Zhuang Z, Zeng G, MicroRNA-152 targets DNA methyltransferase 1 in NiS-transformed cells via a feedback mechanism, Carcinogenesis 34(2) (2013) 446–53. [DOI] [PubMed] [Google Scholar]
- [153].Gugnoni M, Ciarrocchi A, Long Noncoding RNA and Epithelial Mesenchymal Transition in Cancer, Int J Mol Sci 20(8) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Niinuma T, Suzuki H, Nojima M, Nosho K, Yamamoto H, Takamaru H, Yamamoto E, Maruyama R, Nobuoka T, Miyazaki Y, Nishida T, Bamba T, Kanda T, Ajioka Y, Taguchi T, Okahara S, Takahashi H, Nishida Y, Hosokawa M, Hasegawa T, Tokino T, Hirata K, Imai K, Toyota M, Shinomura Y, Upregulation of miR-196a and HOTAIR drive malignant character in gastrointestinal stromal tumors, Cancer Res 72(5) (2012) 1126–36. [DOI] [PubMed] [Google Scholar]
- [155].Song Y, Wang R, Li LW, Liu X, Wang YF, Wang QX, Zhang Q, Long non-coding RNA HOTAIR mediates the switching of histone H3 lysine 27 acetylation to methylation to promote epithelial-to-mesenchymal transition in gastric cancer, Int J Oncol 54(1) (2019) 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Gao H, Hao G, Sun Y, Li L, Wang Y, Long noncoding RNA H19 mediated the chemosensitivity of breast cancer cells via Wnt pathway and EMT process, Onco Targets Ther 11 (2018) 8001–8012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Hirata H, Hinoda Y, Shahryari V, Deng G, Nakajima K, Tabatabai ZL, Ishii N, Dahiya R, Long Noncoding RNA MALAT1 Promotes Aggressive Renal Cell Carcinoma through Ezh2 and Interacts with miR-205, Cancer Res 75(7) (2015) 1322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Lin C, Wang Y, Wang Y, Zhang S, Yu L, Guo C, Xu H, Transcriptional and posttranscriptional regulation of HOXA13 by lncRNA HOTTIP facilitates tumorigenesis and metastasis in esophageal squamous carcinoma cells, Oncogene 36(38) (2017) 5392–5406. [DOI] [PubMed] [Google Scholar]
- [159].Terashima M, Ishimura A, Wanna-Udom S, Suzuki T, MEG8 long noncoding RNA contributes to epigenetic progression of the epithelial-mesenchymal transition of lung and pancreatic cancer cells, J Biol Chem 293(47) (2018) 18016–18030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Zhang J, Zhou Y, Wu Y, Ma L, Fan Y, Kang X, Shi H, Zhang J, Isolation and characterization of a novel noncoding RNA from nickel-induced lung cancer, Biol Trace Elem Res 150(1–3) (2012) 258–63. [DOI] [PubMed] [Google Scholar]
- [161].Zhou C, Huang C, Wang J, Huang H, Li J, Xie Q, Liu Y, Zhu J, Li Y, Zhang D, Zhu Q, Huang C, LncRNA MEG3 downregulation mediated by DNMT3b contributes to nickel malignant transformation of human bronchial epithelial cells via modulating PHLPP1 transcription and HIF-1alpha translation, Oncogene 36(27) (2017) 3878–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Xu G, Meng L, Yuan D, Li K, Zhang Y, Dang C, Zhu K, MEG3/miR21 axis affects cell mobility by suppressing epithelialmesenchymal transition in gastric cancer, Oncol Rep 40(1) (2018) 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [163].Zhan H, Chang X, Wang X, Yang M, Gao Q, Liu H, Li C, Li S, Sun Y, LncRNA MEG3 mediates nickel oxide nanoparticles-induced pulmonary fibrosis via suppressing TGF-beta1 expression and epithelial-mesenchymal transition process, Environ Toxicol (2021). [DOI] [PubMed] [Google Scholar]
- [164].Terashima M, Tange S, Ishimura A, Suzuki T, MEG3 Long Noncoding RNA Contributes to the Epigenetic Regulation of Epithelial-Mesenchymal Transition in Lung Cancer Cell Lines, J Biol Chem 292(1) (2017) 82–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Majmundar AJ, Wong WJ, Simon MC, Hypoxia-inducible factors and the response to hypoxic stress, Mol Cell 40(2) (2010) 294–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Ke Q, Costa M, Hypoxia-inducible factor-1 (HIF-1), Mol Pharmacol 70(5) (2006) 1469–80. [DOI] [PubMed] [Google Scholar]
- [167].Loboda A, Jozkowicz A, Dulak J, HIF-1 and HIF-2 transcription factors--similar but not identical, Mol Cells 29(5) (2010) 435–42. [DOI] [PubMed] [Google Scholar]
- [168].Kaluz S, Kaluzova M, Stanbridge EJ, Regulation of gene expression by hypoxia: integration of the HIF-transduced hypoxic signal at the hypoxia-responsive element, Clin Chim Acta 395(1–2) (2008) 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Dengler VL, Galbraith M, Espinosa JM, Transcriptional regulation by hypoxia inducible factors, Crit Rev Biochem Mol Biol 49(1) (2014) 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Davidson TL, Chen H, Di Toro DM, D’Angelo G, Costa M, Soluble nickel inhibits HIF-prolyl-hydroxylases creating persistent hypoxic signaling in A549 cells, Mol Carcinog 45(7) (2006) 479–89. [DOI] [PubMed] [Google Scholar]
- [171].Paul SA, Simons JW, Mabjeesh NJ, HIF at the crossroads between ischemia and carcinogenesis, J Cell Physiol 200(1) (2004) 20–30. [DOI] [PubMed] [Google Scholar]
- [172].Lv X, Li J, Zhang C, Hu T, Li S, He S, Yan H, Tan Y, Lei M, Wen M, Zuo J, The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism, Genes Dis 4(1) (2017) 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Wigerup C, Pahlman S, Bexell D, Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer, Pharmacol Ther 164 (2016) 152–69. [DOI] [PubMed] [Google Scholar]
- [174].Schofield CJ, Zhang Z, Structural and mechanistic studies on 2-oxoglutarate-dependent oxygenases and related enzymes, Curr Opin Struct Biol 9(6) (1999) 722–31. [DOI] [PubMed] [Google Scholar]
- [175].Yao Y, Costa M, Toxicogenomic effect of nickel and beyond, Arch Toxicol 88(9) (2014) 1645–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Davidson T, Chen H, Garrick MD, D’Angelo G, Costa M, Soluble nickel interferes with cellular iron homeostasis, Mol Cell Biochem 279(1–2) (2005) 157–62. [DOI] [PubMed] [Google Scholar]
- [177].Semenza GL, Targeting HIF-1 for cancer therapy, Nat Rev Cancer 3(10) (2003) 721–32. [DOI] [PubMed] [Google Scholar]
- [178].Joseph JP, Harishankar MK, Pillai AA, Devi A, Hypoxia induced EMT: A review on the mechanism of tumor progression and metastasis in OSCC, Oral Oncol 80 (2018) 23–32. [DOI] [PubMed] [Google Scholar]
- [179].Gilkes DM, Semenza GL, Wirtz D, Hypoxia and the extracellular matrix: drivers of tumour metastasis, Nat Rev Cancer 14(6) (2014) 430–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Hubbi ME, Semenza GL, Regulation of cell proliferation by hypoxia-inducible factors, Am J Physiol Cell Physiol 309(12) (2015) C775–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Tam SY, Wu VWC, Law HKW, Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1alpha and Beyond, Front Oncol 10 (2020) 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Emami Nejad A, Najafgholian S, Rostami A, Sistani A, Shojaeifar S, Esparvarinha M, Nedaeinia R, Haghjooy Javanmard S, Taherian M, Ahmadlou M, Salehi R, Sadeghi B, Manian M, The role of hypoxia in the tumor microenvironment and development of cancer stem cell: a novel approach to developing treatment, Cancer Cell Int 21(1) (2021) 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Su Q, Fan M, Wang J, Ullah A, Ghauri MA, Dai B, Zhan Y, Zhang D, Zhang Y, Sanguinarine inhibits epithelial-mesenchymal transition via targeting HIF-1alpha/TGF-beta feed-forward loop in hepatocellular carcinoma, Cell Death Dis 10(12) (2019) 939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Zhang L, Huang G, Li X, Zhang Y, Jiang Y, Shen J, Liu J, Wang Q, Zhu J, Feng X, Dong J, Qian C, Hypoxia induces epithelial-mesenchymal transition via activation of SNAI1 by hypoxia-inducible factor -1alpha in hepatocellular carcinoma, BMC Cancer 13 (2013) 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Yang MH, Wu KJ, TWIST activation by hypoxia inducible factor-1 (HIF-1): implications in metastasis and development, Cell Cycle 7(14) (2008) 2090–6. [DOI] [PubMed] [Google Scholar]
- [186].Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC, Wu KJ, Direct regulation of TWIST by HIF-1alpha promotes metastasis, Nat Cell Biol 10(3) (2008) 295–305. [DOI] [PubMed] [Google Scholar]
- [187].Xiong Y, Liu Y, Xiong W, Zhang L, Liu H, Du Y, Li N, Hypoxia-inducible factor 1alpha-induced epithelial-mesenchymal transition of endometrial epithelial cells may contribute to the development of endometriosis, Hum Reprod 31(6) (2016) 1327–38. [DOI] [PubMed] [Google Scholar]
- [188].Liu Z, Tu K, Wang Y, Yao B, Li Q, Wang L, Dou C, Liu Q, Zheng X, Hypoxia Accelerates Aggressiveness of Hepatocellular Carcinoma Cells Involving Oxidative Stress, Epithelial-Mesenchymal Transition and Non-Canonical Hedgehog Signaling, Cell Physiol Biochem 44(5) (2017) 1856–1868. [DOI] [PubMed] [Google Scholar]
- [189].Zhang W, Shi X, Peng Y, Wu M, Zhang P, Xie R, Wu Y, Yan Q, Liu S, Wang J, HIF-1alpha Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer, PLoS One 10(6) (2015) e0129603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Storci G, Sansone P, Mari S, D’Uva G, Tavolari S, Guarnieri T, Taffurelli M, Ceccarelli C, Santini D, Chieco P, Marcu KB, Bonafe M, TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype, J Cell Physiol 225(3) (2010) 682–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Nakuluri K, Mukhi D, Nishad R, Saleem MA, Mungamuri SK, Menon RK, Pasupulati AK, Hypoxia induces ZEB2 in podocytes: Implications in the pathogenesis of proteinuria, J Cell Physiol 234(5) (2019) 6503–6518. [DOI] [PubMed] [Google Scholar]
- [192].Yang SW, Zhang ZG, Hao YX, Zhao YL, Qian F, Shi Y, Li PA, Liu CY, Yu PW, HIF-1alpha induces the epithelial-mesenchymal transition in gastric cancer stem cells through the Snail pathway, Oncotarget 8(6) (2017) 9535–9545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH, Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition, J Clin Invest 117(12) (2007) 3810–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Peinado H, Del Carmen Iglesias-de la Cruz M, Olmeda D, Csiszar K, Fong KS, Vega S, Nieto MA, Cano A, Portillo F, A molecular role for lysyl oxidase-like 2 enzyme in snail regulation and tumor progression, EMBO J 24(19) (2005) 3446–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Salnikow K, Donald SP, Bruick RK, Zhitkovich A, Phang JM, Kasprzak KS, Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress, J Biol Chem 279(39) (2004) 40337–44. [DOI] [PubMed] [Google Scholar]
- [196].Li Q, Chen H, Huang X, Costa M, Effects of 12 metal ions on iron regulatory protein 1 (IRP-1) and hypoxia-inducible factor-1 alpha (HIF-1alpha) and HIF-regulated genes, Toxicol Appl Pharmacol 213(3) (2006) 245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Salnikow K, Davidson T, Costa M, The role of hypoxia-inducible signaling pathway in nickel carcinogenesis, Environ Health Perspect 110 Suppl 5 (2002) 831–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Gordon KJ, Blobe GC, Role of transforming growth factor-beta superfamily signaling pathways in human disease, Biochim Biophys Acta 1782(4) (2008) 197–228. [DOI] [PubMed] [Google Scholar]
- [199].Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES, A bivalent chromatin structure marks key developmental genes in embryonic stem cells, Cell 125(2) (2006) 315–26. [DOI] [PubMed] [Google Scholar]