

Abstract
TGFβ signaling promotes progression of bone-metastatic (BMET) breast cancer (BCa) cells by driving tumor-associated osteolysis, a hallmark of BCa BMETs, thus allowing for tumor expansion within bone. Turmeric-derived bioactive curcumin, enriched in bone via local enzymatic deconjugation of inactive circulating curcumin-glucuronides, inhibits osteolysis and BMET progression in human xenograft BCa BMET models by blocking tumoral TGFβ signaling pathways mediating osteolysis. This is a unique anti-osteolytic mechanism in contrast to current osteoclast-targeting therapeutics. Therefore, experiments were undertaken to elucidate the mechanism for curcumin inhibition of BCa TGFβ signaling and the application of this finding across multiple BCa cell lines forming TGFβ-dependent BMETs, including a possible role for bioactive curcumin metabolites in mediating these effects. Immunoblot analysis of TGFβ signaling proteins in bone tropic human (MDA-SA, MDA-1833, MDA-2287) and murine (4T1) BCa cells revealed uniform curcumin blockade of TGFβ-induced Smad activation due to downregulation of plasma membrane associated TGFβR2 and cellular receptor Smad proteins that propagate Smad-mediated gene expression, resulting in downregulation of PTHrP expression, the osteolytic factor driving in vivo BMET progression. With the exception of early decreases in TGFβR2, inhibitory effects appeared to be mediated by oxidative metabolites of curcumin and involved inhibition of gene expression. Interestingly, while not contributing to changes in Smad-mediated TGFβ signaling, curcumin caused early activation of MAPK signaling in all cell lines, including JNK, an effect possibly involving interactions with TGFβR2 within lipid rafts. Treatment with curcumin or oxidizable analogs of curcumin may have clinical relevancy in the management of TGFβ-dependent BCa BMETs.
Keywords: curcumin, breast cancer, TGFβ, Smad, JNK, bone metastases
1. INTRODUCTION
Osteolysis is a hallmark of breast cancer (BCa) bone metastases (BMET), a tumor-driven process that promotes metastatic progression within the bone microenvironment [1,2]. Most women with advanced BCa develop osteolytic BMETs, which are essentially incurable, and may arise from asymptomatic disseminated tumor cells already present in bone at the time of initial diagnosis [3–5]. Tumoral TGFβ signaling in bone-disseminated BCa cells, fueled by TGFβ released from osteolytically-resorbed bone matrix, induces additional tumor-associated osteolysis by promoting the formation of more bone-resorbing osteoclasts, thus facilitating tumor expansion within bone in a feed-forward loop [1,2,6–21].
The importance of tumoral TGFβ signaling in promoting osteolytic BMET progression is supported by a large body of evidence from human BCa BMET xenograft models where neutralization of TGFβ, tumoral TGFβ signaling (including canonical Smad signaling, which is active in 75–100% of clinical BMET [18,22]), and/or TGFβ-inducible tumor-derived parathyroid hormone related protein (PTHrP), a osteolytic factor expressed in most clinical BMET [23–25], prevents tumor-associated osteoclast formation and in vivo metastatic progression. This inhibitory effect is independent of direct effects on tumor growth [1,2,6–21].
Of relevance for the current studies, curcumin, a naturally occurring turmeric-derived polyphenol, has also been reported by our laboratory and by others to block osteolysis in vivo and thus BCa BMET progression in these same pre-clinical models [26,27]. Notably, our laboratory has further demonstrated that curcumin inhibition of osteolysis and BMET progression, which contrasted with the lack of a direct effect on orthotopic tumor growth (i.e., proliferation), was associated with curcumin inhibition of tumoral TGFβ pathways driving osteolysis, most notably, TGFβ-inducible Smad signaling and PTHrP secretion [26,28,29].
Curcumin’s TGFβ-targeted anti-osteolytic effects are intriguing since drugs currently used to block osteolysis in BCa, which are beneficial but not curative, target osteoclasts but not the tumoral pathways driving their formation. Also of note, curcuminoids circulate in vivo as inactive glucuronides that do not inhibit tumoral TGFβ signaling [28,30,31]. However, adding to the potential promise of curcuminoids in targeting of TGFβ-driven tumor-associated osteolysis is recent evidence that bioactive curcumin can readily be formed in situ within the bone/tumor microenvironment via enzymatic deconjugation of curcumin-glucuronides [28,30,31], allowing for local targeting of TGFβ-signaling in bone while sparing systemic TGFβ activity. However, the mechanism by which curcumin suppresses BCa cell TGFβ signaling, critical for in vivo osteolysis and BMET progression [6,7,14,15,18,20,21], is not known.
Given the potential promise of curcuminoid polyphenols in blocking tumoral TGFβ signaling cascades mediating osteolysis and BCa BMET progression, current experiments were initiated to investigate the mechanism(s) responsible for the inhibitory effect of curcumin on Smad signaling in BCa cells forming TGFβ-dependent osteolytic BMET in vivo [6–9,14,15,18,20,21], and the uniformity of mechanism(s) across cell lines. In particular, these studies focus on MDA-SA cells, where pathologic roles of TGFβ-induced canonical (Smad) as well as non-canonical MAPK (p38, JNK, or ERK) signaling are well described, including the in vivo dependence of osteolysis and BMET progression on TGFβ-induced PTHrP [8,9,14,15,20], and in vivo protective effects of curcumin are established [26]. In addition, while deconjugation of circulating curcumin-glucuronide is required for curcumin bioactivity within bone [28,30,31], also explored here was the question of whether further oxidative metabolism of curcumin to form reactive intermediates is also required for inhibition of TGFβ signaling in bone metastatic BCa cells, as has been posited for other cellular curcumin targets [32–34].
2. MATERIALS AND METHODS
2.1. Materials
The chemical content of turmeric-derived curcumin (#218580100, Fisher Scientific) was verified using standard methods (see below), with assayed content 96.5% curcuminoids by weight, comprised of 80.6% curcumin with lesser amounts of demethoxycurcumin [13.5%] and bisdemethoxycurcumin [2.4%])) [35,36], and stock solutions prepared in DMSO. A non-oxidizable curcumin analog, diacetyl curcumin (DAC), was synthesized as previously described [34,37]. Cells were stimulated, as indicated, with recombinant human TGFβ1 (#240-B, R&D Systems). N-acetylcysteine (NAC, #A1540914) and nystatin (#J6048606) were purchased from Alfa Aesar. Cycloheximide (CHX, #C7698), (#SML1169), okadaic acid (OA, #459620), chloroquine (CQ, #C6628), and MG132 (#474791) were purchased from Millipore-Sigma. The MAPK inhibitors SP600125 (#S1077) and SB202190 (#S1460) were purchased from Selleckchem. Mini-PROTEAN TGX-PAGE gels (#4568046) were purchased from BioRad and PVDF membranes (#IPFL0010) from Millipore.
2.2. Cell Culture
Bone-metastatic (“bone tropic”) MDA-MD-231 BCa (MDA-SA) cells that secrete TGFβ-inducible PTHrP that is curcumin-inhibitable [8,14,15,20,26,28,29], and a MDA-SA-derived cell line stably transfected to overexpress TGFβR2 lacking the cytoplasmic domain required for TGFβ-mediated signal transduction (“TGFβR2Δcyt”) [20,38,39], were kindly provided by Dr. Theresa Guise, Indiana University [14,15]. The parent MDA-SA cells have been propagated from MDA-MB-231 cells initially obtained from Kent Osborne [40] (not ATCC) by Yoneda and Mundy at the University of Texas Health Sciences Center in San Antonio (SA) [41], who were the first to describe the cell’s bone tropic behavior and formation of osteolytic BMET following intracardiac injection. It should be noted that TGFβ induction of PTHrP in MDA-SA cells transfected with empty vector was indistinguishable from wild type cells used here [14]. Two bone-tropic cell lines derived from ATCC MDA-MB-231 cells by in vivo selection for an aggressive bone metastatic phenotype attributable to the overexpression of TGFβ-regulated genes (MDA-1833 and MDA-2287) were a kind gift from Dr. Joan Massagué, Sloan-Kettering [18,21]. Bone-tropic murine 4T1 cells, which form TGFβ-dependent BMET with evidence of active Smad signaling in immunocompetent mice [6,7,42–45] and secrete TGFβ-inducible PTHrP that is curcumin-inhibitable [28], were obtained directly from American Type Culture Collection (#CRL-2539, ATCC) and used within 10 passages. Human cell lines were authenticated using short tandem repeat profiling by the University of Arizona Genetics Core [46] and all cells were cultured in DMEM (#10–013-CV, Corning) supplemented with 10% FBS (#S11150, Atlanta Biologicals) and 1% penicillin/streptomycin (#15140, ThermoFisher). For analysis of curcumin effects on TGFβ signaling pathway proteins, unless otherwise stated, cells were pretreated with vehicle (DMSO) or curcumin followed by TGFβ1 (5 ng/ml) (or media alone) for specified times prior to isolation of whole cell lysates with RIPA buffer (#R0278, Millipore-Sigma) for Western analyses. Membrane-associated proteins were isolated from cells using the ProteoExtract Native Membrane Protein extraction kit (#444810, Millipore-Sigma), and plasma membrane proteins were isolated using the Pierce Cell Surface Isolation Kit (#89881, ThermoFisher), according to the manufacturers’ protocol. For analysis of effects on TGFβ-induced PTHrP, secreted PTHrP was assayed by commercial radioimmunoassay (RIA) as previously described [28]. Briefly conditioned media (treated with protease inhibitors prior to storage at −80°C) was obtained from cells pre-treated with curcumin, DAC, or vehicle for 4 hours prior to concurrent addition of TGFβ1 (5 ng/ml) for 24 hours. PTHrP was then quantified in the conditioned media using a PTHrP RIA (Beckman Coulter, #DSL-8100). Results are expressed either as absolute concentrations or relative to basal control cells or to TGFβ-stimulated cells in experiments where control cells were also included to verify TGFβ induction.
2.3. Protein Turnover Studies
To determine curcumin effects on protein degradation, cells were concurrently treated with cycloheximide (50 μg/mL) and curcumin (30 μM) (vs media alone) for up to 48 hours. Lysosomal or proteosomal protein degradation pathways were also blocked by pre-treatment with chloroquine (40 μM) or MG132 (10 μM), respectively, for 2–3 hours followed by treatment with curcumin (30 μM) vs media alone for 4h (for TGFβR2) or 16h (for Smad2). Curcumin effects on protein synthesis were determined by pre-treating cells with cycloheximide (50 μg/mL) for 24 hours to inhibit de novo protein synthesis and deplete endogenous protein levels, followed by treatment with curcumin (30 μM) vs media alone for up to 24 hours.
2.4. Western Blot Analyses
Proteins, isolated from whole cell lysates and quantitated by Bradford assay for normalized loading (#5000002, Bio-Rad), were separated on Mini-PROTEAN TGX-PAGE gels (BioRad), and transferred to Immobilon-FL PVDF membranes (Millipore-Sigma), with even protein loading confirmed by stain-free imaging of UV-activated binding of gel trihalo compound binding to protein tryptophan residues (BioRad) and by detection of the housekeeping gene, β-actin (#4967, Cell Signaling Technology [CST]). Blots were probed with primary antibodies (obtained from CST, unless otherwise stated) directed against Smad2 (#5339), pSmad2 (S465/467, #3108), pSmad2-L (S245/250/255, #3104), Smad3 (#9523), pSmad3 (S423/425, #9520), Smad2/3 (#8685), SARA (#13285), Smad4 (#38454), Smad7 (#42–0400, ThermoFisher), JNK (#9252), p-JNK (T183/Y185, #4668), p38 (#8690), p-p38 (T180/Y182, #4511), ERK1/2 (#4695), p-ERK1/2 (T202/Y204, #4370), TGFβR2 (#79424 [CST] or #ab184948 [Abcam], as indicated), TGFβR1 (#ab31013, Abcam), or ETS-1 (#sc-350, Santa Cruz) followed by HRP-conjugated secondary antibody (#7074, CST). All anti-Smad and anti-MAPK antibodies recognize both human and murine proteins, as does the CST TGFβR2 antibody. While the Abcam antibody used to verify curcumin effects on TGFβR2 in human cells is only documented to recognize human. Effects of deglycosylation on the size of TGFβR2 proteins detected by Western were determined by treatment of lysates with PNGase F (#P0704, New England Biolabs) according to the manufacturer’s protocol. Blots were visualized with SuperSignal West Femto ECL substrate (#34095, ThermoFisher). Quantitative densitometry was determined using ImageJ (v2.0.0, NIH).
2.5. Real-time quantitative PCR
RNA was isolated from whole cell lysates using TRI-reagent (#T9424, Millipore-Sigma) with cDNA synthesized using BioRad iScript kit (#170–8890), as per manufacturers protocols. Real-time quantitative PCR (qPCR) was performed on a Roche LightCycler 96 system using BioRad iQ Supermix (#170–8860) and TaqMan primers against Smad2 (#Hs00183425_m1), TGFβR2 (#Hs00234253_m1), and 18S (#Hs9999901_s1, Thermo Fisher). Cycle thresholds (Ct) values were normalized to an endogenous housekeeping control (18S RNA), with mRNA levels in curcumin-treated cells expressed relative to vehicle controls using the 2−ΔΔCt method, as previously described [47]. Statistical analysis (t-test) was performed using ΔΔCt values and confidence intervals (CI) were calculated, as described elsewhere [48].
2.6. Statistical Analysis
Statistical analyses were performed using Prism v6.0h software (GraphPad, San Diego, CA), with data expressed as mean ± SEM, except where noted. Half-maximal inhibitory concentrations (IC50) were determined by using a four-parameter logistic equation. Significant differences were determined by t-test or by one-way or two-way ANOVA with post-hoc testing, as appropriate.
3. Results
3.1. Curcumin inhibition of TGFβ-induced Smad signaling was associated with decreased TGFβR2 and Smad proteins and activation of MAPKs
Curcumin reduced levels of C-terminal phosphorylated Smad2 and Smad3, the receptor Smads (R-Smads), in response to TGFβ stimulation in a concentration-dependent fashion (Fig. 1A). This required >4 hours of curcumin exposure prior to TGFβ activation (Fig. 1B) in MDA-SA cells. Curcumin inhibition of receptor-mediated Smad2/3 phosphorylation could not be attributed to decreases in SARA, which stabilizes phosphorylated Smad2/3 [39], nor increases in Smad7, which blocks Smad2/3 phosphorylation (Fig. 1A). Dephosphorylation of the R-Smads also did not appear to be enhanced by curcumin, as pre-treatment with okadaic acid (OA), an inhibitor of the protein phosphatase (PP1/2A) targeting phosphorylated Smad2/3 [49], did not alter curcumin’s inhibitory effect (Fig. S1). Smad4 and constitutively active ETS-1, two factors required for Smad-dependent TGFβ induction of PTHrP gene expression in MDA-SA cells [26,28,29,50], were also unchanged by curcumin (Fig. 1A).
Figure 1. Curcumin effects on Smad and MAPK signaling proteins in control and TGFβ-stimulated MDA-SA cells.
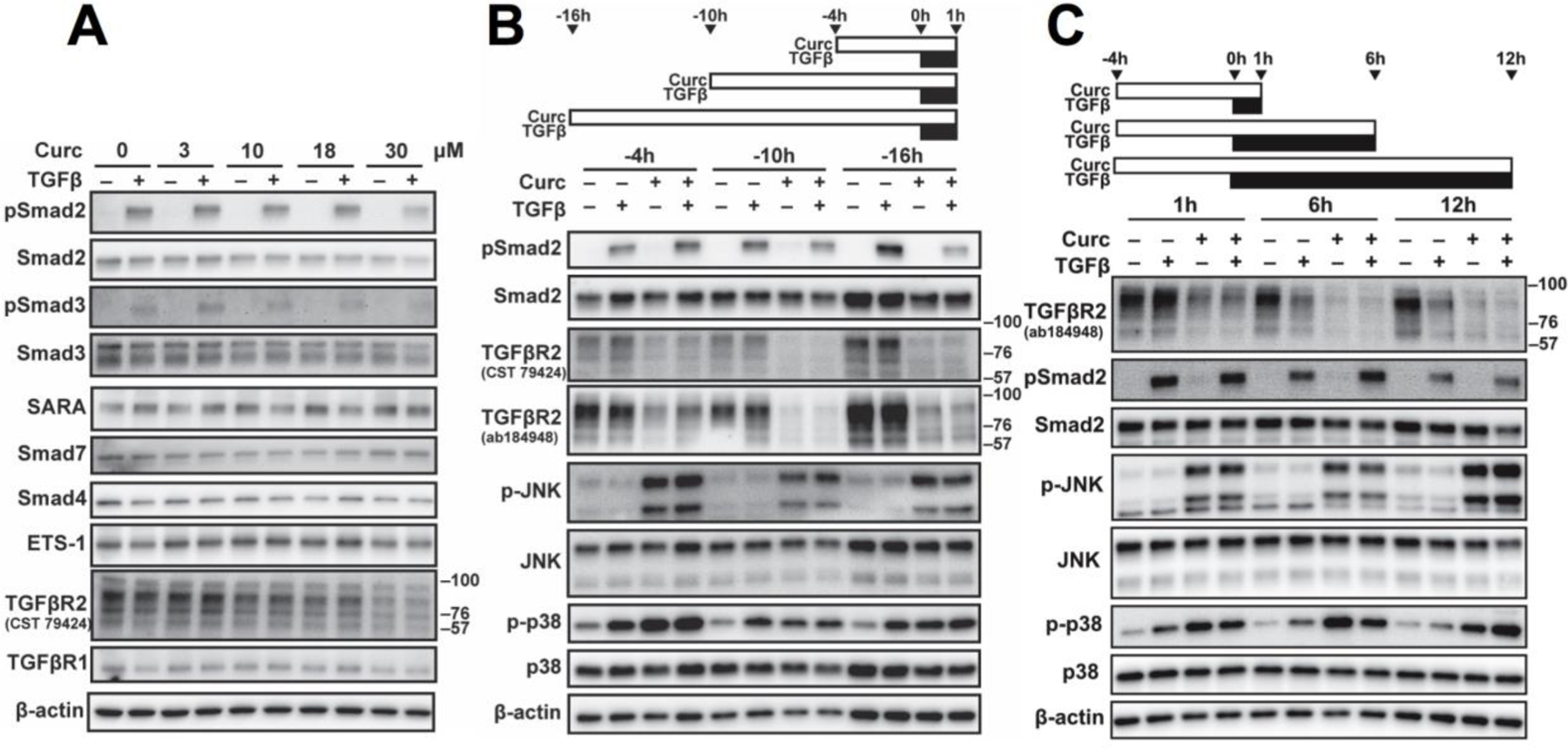
A) Concentration-dependent effects of curcumin (16h-pretreatment followed by 1h of concurrent TGFβ). B) Time-dependent effects of curcumin (30 μM) pre-treatment (indicated by the white bar), followed by 1h of concurrent TGFβ (black bar). C) Time dependent effects of TGFβ following 4h of curcumin (30 μM) pre-treatment. Results are representative of two or more experiments.
Notably, while changes in R-Smad phosphorylation/dephosphorylation did not appear to account for curcumin-induced decreases in TGFβ-stimulated R-Smad activation, constitutive Smad2/3 protein levels were reduced. This effect was concentration-dependent and required >4h of curcumin exposure (Fig. 1A,B). In addition, reduced pSmad2/Smad2 levels were preceded by curcumin-induced changes in TGFβ receptor 2 (TGFβR2) and MAPK signaling (Fig. 1A–C). Curcumin caused an early (4h), profound, and persistent (16h) decrease in TGFβR2 (but not TGFβR1), independent of TGFβ treatment (Fig. 1B,C). These findings were confirmed using antibodies directed against two different TGFβR2 epitopes with collapse of Western bands into an anticipated ~60 kDa protein [51] following deglycosylation (Fig. S2). The curcumin-mediated decrease in constitutive TGFβR2 protein levels, which continued for 12 hours after initiation of TGFβ Smad signaling (Fig. 1C), was associated with persistent activation of MAPK signaling (Fig. 1B,C). ERK signaling was constitutively active and unaltered by curcumin and/or TGFβ in MDA-SA cells (Fig. S3). However, p38 MAPK signaling (phosphorylation of p38), which is also induced by TGFβ in MDA-SA cells and contributes to TGFβ-regulated expression of PTHrP and other osteolytic factors [8,20,26], was independently activated by curcumin. JNK signaling, a pathway unaltered by TGFβ (Fig. 1B,C), was also independently activated by curcumin.
Essentially identical curcumin-induced changes in TGFβ signaling pathways were confirmed in all BCa cell lines forming TGFβ-dependent osteolytic BMET in vivo (Fig. 2). An associated decrease in constitutive Smad2/3 and TGFβ-induced pSmad2/3 levels was documented in human MDA-1833, human MDA-2287, and murine 4T1 cells (Fig. 2A), such that constitutive Smad2 levels in all four BCa cell lines (including MDA-SA) were reduced by ≥36.5% (p< 0.05, n=3–4) and pSmad2 levels by > 54.5% (p< 0.05, n=3–4) after 16 hours of curcumin (30 µM) treatment. Early and persistent curcumin-mediated decreases in TGFβR2 (Fig. 2B) confirmed using both antibodies, and activation of JNK and p38 MAPK signaling (Fig. 2B), were similarly documented in the entire panel of bone-tropic BCa cell lines forming TGFβ-dependent BMET in vivo. JNK signaling in human MDA-2287 cells was an exception, being constitutively active and unaltered by curcumin treatment.
Figure 2. Curcumin effects on TGFβ signaling proteins in other breast cancer cell lines forming TGFβ-dependent bone metastases.
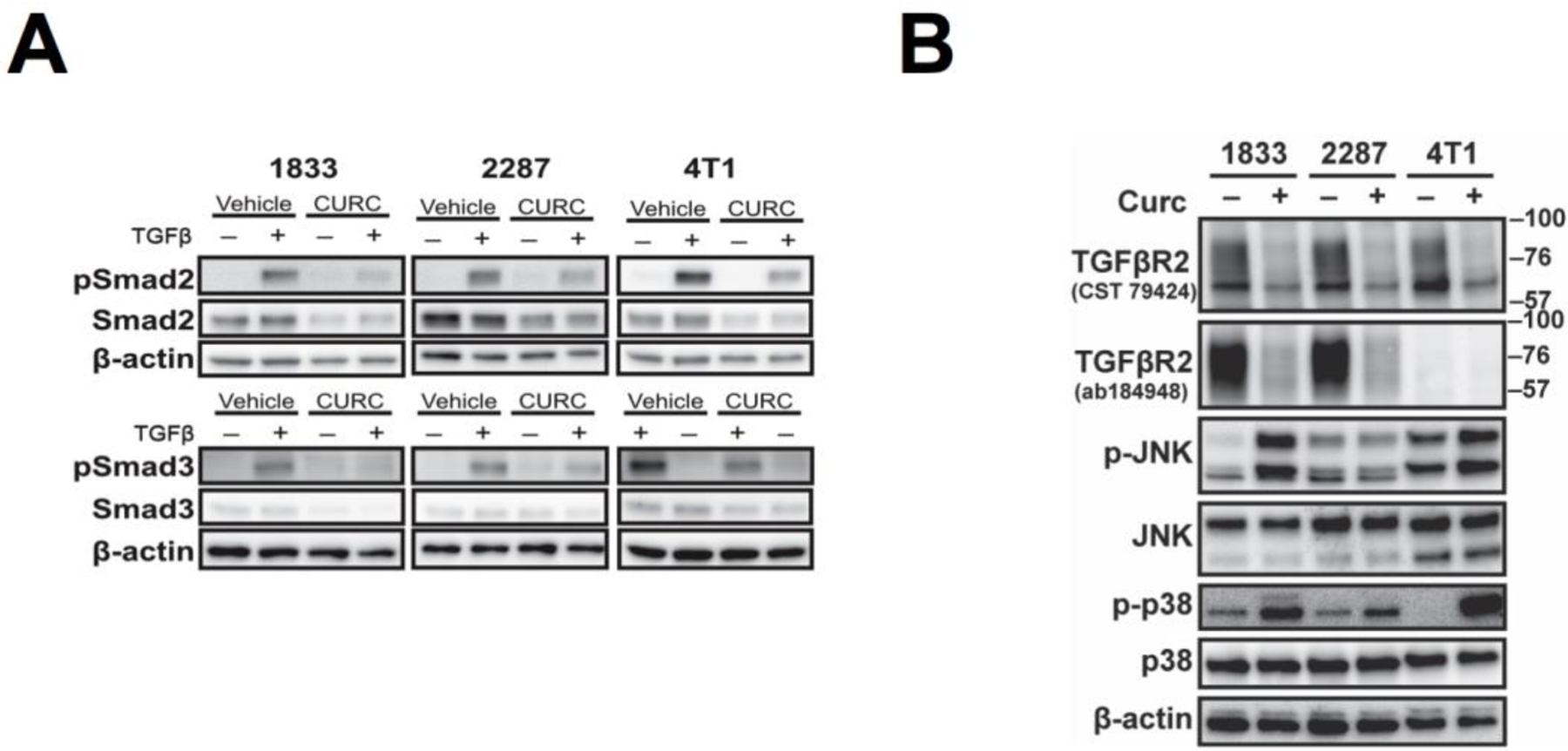
A) Smad signaling in human MDA-1833, MDA-2287, and murine 4T1 cells pre-treated with curcumin (30 μM) for 16h followed by 1h of concurrent TGFβ. B) TGFβ receptor and MAPK signaling following curcumin (30 μM) treatment for 16h. Note: As in MDA-SA cells, ERK signaling was constitutively active and unaltered by curcumin and/or TGFβ in these cell lines (data not shown).
3.2. Possible cross-talk between curcumin-activated MAPKs (p38 and JNK) and Smad-dependent TGFβ signaling
Experiments were next conducted to determine whether curcumin activation of MAPKs contributed to changes in TGFβR2 and/or R-Smads. A known MAPK-mediated means of altering the fate of pSmad2/3 [52–54], phosphorylation of serine residues in the linker region of Smad2/3 [Smad2/3-L]), was first examined as one possible mechanism. Curcumin stimulated Smad2-L phosphorylation in TGFβ-naïve cells, an effect evident within 4 hours of treatment (Fig. S4) that persisted for 16 hours (Fig. 3A). At both 4 and 16 hours, this effect was additive with TGFβ’s previously reported stimulatory effects on Smad linker phosphorylation [53] (Fig. 3A and S4). Curcumin-induced Smad2-L phosphorylation was mediated by JNK activation, as inhibition of JNK, but not p38 activity, reversed this effect (Fig. 3B). However, neither induction of p38 or JNK signaling by curcumin, including JNK-stimulated Smad linker phosphorylation, mediated curcumin reductions in TGFβR2, nor decreases in Smad2 or TGFβ-simulated pSmad2, as pretreatment with JNK or p38 inhibitors, whose efficacy was otherwise confirmed (data not shown), did not prevent these curcumin inhibitory effects (Fig. 3B). Additionally, while Smad linker region phosphorylation can alter nuclear trafficking/effects of activated R-Smads [54], JNK-mediated phosphorylation of Smad2-L did not appear responsible for curcumin inhibition of TGFβ-stimulated PTHrP expression. Inhibition of JNK suppressed constitutive (but not TGFβ-inducible) PTHrP secretion in MDA-SA cells, but did not prevent curcumin inhibition of TGFβ-inducible PTHrP (Fig. 3C), a driver of in vivo metastatic progression in the MDA-SA BMET model [15]. Additionally, p38 inhibition did not alter curcumin blockade of TGFβ-inducible PTHrP in MDA-SA cells (Fig 3C).
Figure 3. Effect of MAPK inhibitors on Smad2 phosphorylation (linker or C-terminus), TGFβR2, and PTHrP secretion in MDA-SA cells treated with curcumin and/or TGFβ.
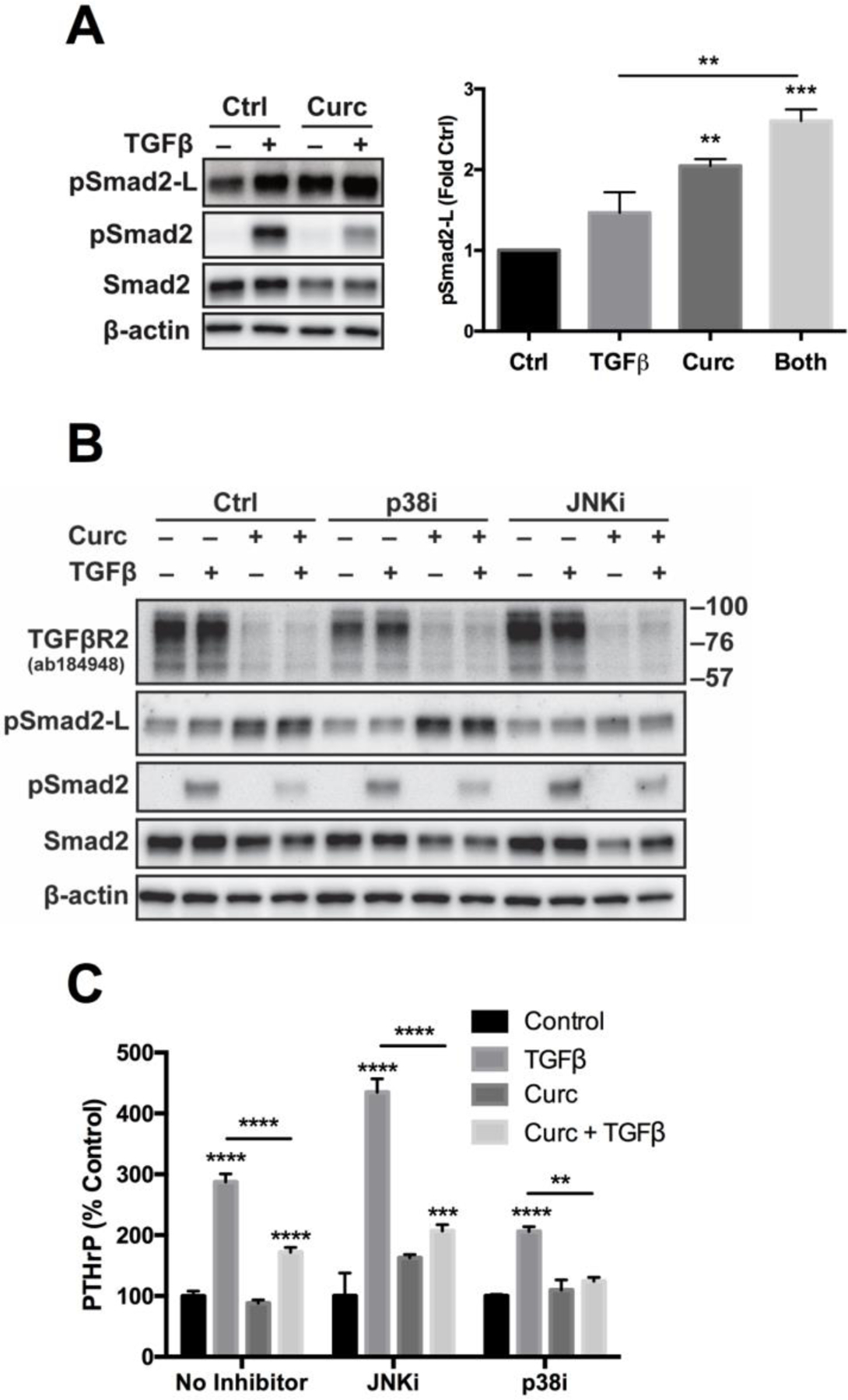
A) Effect of curcumin (30 uM, 16h pre-treatment) and/or TGFβ (1h) on both Smad2 linker-region phosphorylation (pSmad2-L) and receptor-mediated C-terminal phosphorylation (pSmad2) (densitometric quantitation [n=3] of pSmad2-L, right panel). B) Effect of MAPK inhibitors (3 h pre-treatment prior to curcumin) of p38 (SB202190, 10 μM) or JNK (SP600125, 25 μM) on Smad2 linker or C-terminal phosphorylation or TGFβR2 protein in curcumin and/or TGFβ-treated cells (16h curcumin followed by 1h TGFβ). Representative of two experiments. C) Effect of JNK or p38 inhibitor (1h pre-treatment, 25 μM SP600125 or 10 μM SB202190, respectively) on PTHrP secretion in cells treated with curcumin and/or TGFβ (30 μM curcumin, 4 hr prior to addition TGFβ; n=3–8/group). Because inhibitors decreased constitutive PTHrP levels, data are expressed as % change relative to constitutive (control) levels. ** p < 0.01, *** p < 0.001, **** p < 0.0001 vs control or as specified.
3.3. Effect of curcumin on TGFβR2 trafficking
TGFβR2 protein was primarily membrane-associated (e.g. plasma membrane or endocytic vesicles), in agreement with prior reports [55], and was reduced by curcumin in MDA-SA cells (Fig. 4A) independent of TGFβ stimulation in both the membrane and soluble fractions. Soluble localization of Smad2/3 and MAPK proteins confirming successful fractionation (Fig. 4A). TGFβR2 within the plasma membrane, critical for initiation of TGFβ signaling, was also specifically reduced by curcumin (Fig. 4B). Because TGFβR2/R1 complexes in lipid rafts vs. clathrin-coated pits are reported to mediate MAPK vs. Smad signaling, respectively [39,55–58], MDA-SA cells were treated with inhibitors of each endocytic process to assess effects on curcumin-induced changes. Nystatin treatment to disrupt lipid rafts [39] did not prevent early (Fig. 4C and S5) or late (Fig. 4D) curcumin reductions in TGFβR2. However, nystatin did block curcumin-induced JNK activation (Fig. 4C and S5) and subsequent Smad2-L phosphorylation (Fig. S5), while having no effect on curcumin-induced p38 activation (Fig. 4C and S5). Treatment with an inhibitor of clathrin-coated pit endocytosis (Pitstop) did not alter curcumin effects on TGFβR2, TGFβ-mediated Smad2 phosphorylation, or MAPK signaling (Fig. 4C,D).
Figure 4. Effects of curcumin on TGFβR2 localization and signaling in cells expressing wildtype TGFβR2 (MDA-SA) vs. a truncated TGFβR2 lacking the cytoplasmic domain (MDA-SA-TGFβR2Δcyt).
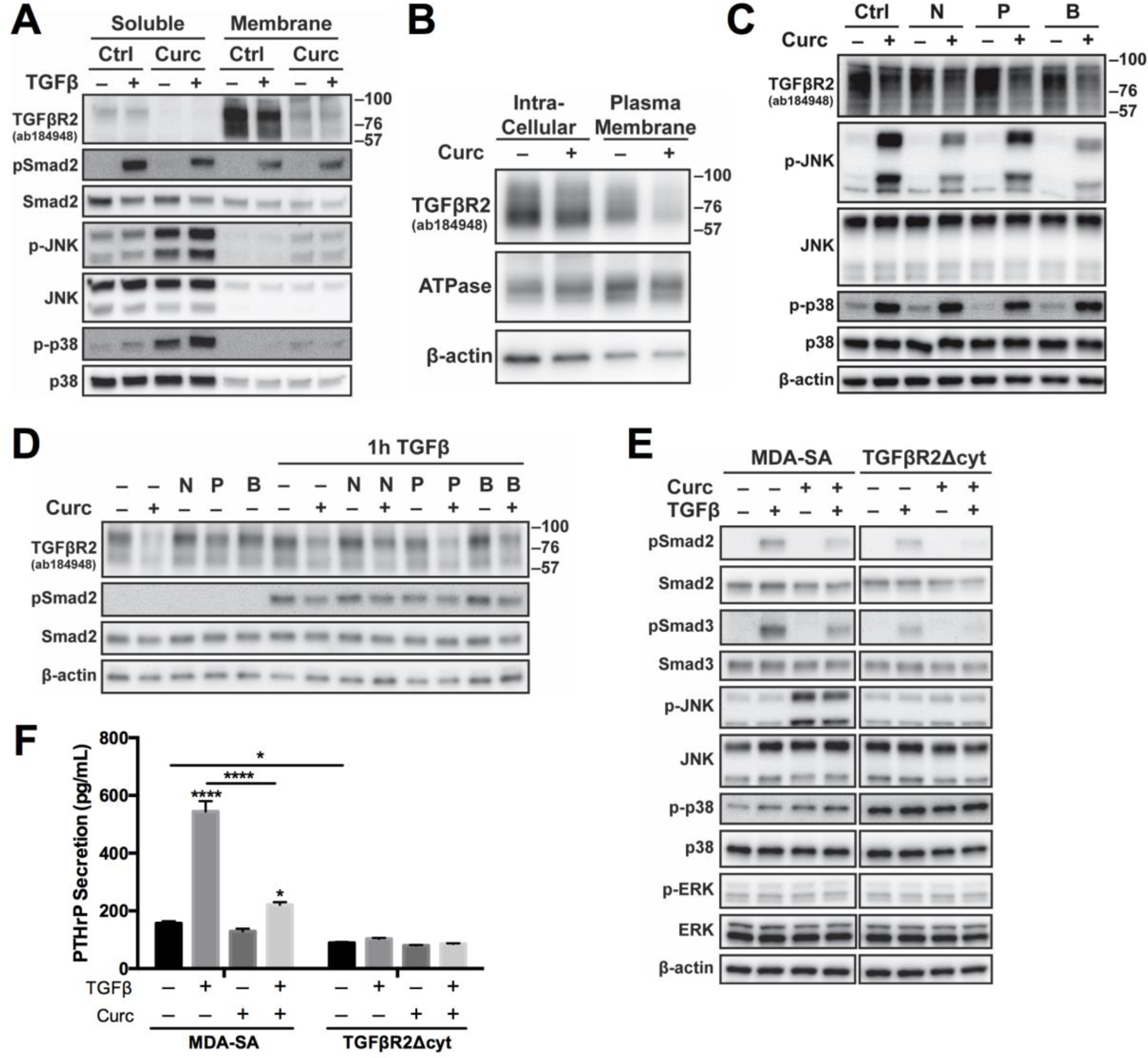
A) Membrane-associated (vs soluble) TGFβR2, Smad2, and MAPK proteins in MDA-SA cells treated with curcumin (30 μM, 4h pre-treatment) and/or TGFβ (1h). B) TGFβR2 in sodium/potassium ATPase-enriched plasma membrane (vs. intracellular) actin-enriched fractions from curcumin-treated (30 μM, 4h) MDA-SA cells. C) Early (4h) or D) late (16h) effects of curcumin (30 uM) on TGFβR2, MAPK and/or Smad signaling in cells pre-treated for 1h with a lipid raft disrupter (nystatin, 50 μg/mL [39]) and/or inhibitor of clathrin-dependent endocytosis (Pitstop 2, 20 μM [60]). Note: A single blot is shown in D with intervening unrelated data omitted. E) Effect of absent TGFβR2 cytoplasmic domain on Smad and MAPK signaling proteins or F) PTHrP secretion (n=4/group) in MDA-SA vs. MDA-TGFβR2Δcyt cells pre-treated with curcumin (30 μM) prior to TGFβ stimulation. * p < 0.05, **** p < 0.0001 vs control or as specified. N = nystatin, P = Pitstop 2, B = nystatin and Pitstop 2
TGFβ-stimulated Smad2/3 phosphorylation was considerably reduced in MDA-SA cells overexpressing TGFβR2 with a truncated cytoplasmic region (“TGFβR2Δcyt”) vs wild type cells) but remained curcumin inhibitable (Fig. 4E). This truncation prevents TGFβR2-mediated signal transduction [20,38] but likely does not alter the predominant lipid raft location of plasma membrane TGFβR2 receptors [39,59]. Notably, JNK activation by curcumin was also prevented by overexpression of TGFβR2Δcyt, suggesting a possible role for ligand-independent TGFβR2 signaling in mediating curcumin induction of JNK (Fig. 4E). In contrast, activation of p38, which was increased at baseline relative to wild type cells, was no longer induced by curcumin or TGFβ in TGFβR2Δcyt-transfected cells (Fig. 4E). TGFβ-stimulated secretion of PTHrP in TGFβR2Δcyt-transfected cells was also completely abrogated and unaffected by addition of curcumin (Fig. 4F).
3.4. Effect of curcumin on TGFβR2 and Smad2 protein stability and synthesis
TGFβR2 protein had a short 2.2h half-life in cycloheximide chase experiments, in agreement with previous estimates [61], and receptor stability was unchanged by curcumin treatment (Fig. 5A, top). Similarly, Smad2 protein levels (14.5h half-life), while trending slightly lower in response to curcumin treatment, were not statistically different from controls (Fig. 5A, bottom). Consistent with a lack of effect on protein degradation, blockade of proteasomal or lysosomal degradation pathways with MG-132 or chloroquine, respectively, did not prevent curcumin-mediated reductions in TGFβR2 or Smad2/3 proteins (Fig S6). In experiments examining curcumin effects on protein synthesis following 24h of cycloheximide pre-treatment to deplete proteins, increases in TGFβR2 significantly lagged in curcumin-treated (vs. control) cells and remained 2-fold lower at 24h (Fig. 5B, top). Similarly, Smad2 protein levels were significantly lower in curcumin-treated cells (Fig. 5B, bottom), remaining statistically unchanged, while levels in control cells increased significantly within 24h (p < 0.05). In parallel with decreased protein synthesis, TGFβR2 and Smad2 mRNA levels were significantly lower in curcumin-treated (vs. control) MDA-SA cells (Fig. 5C).
Figure 5. Effect of curcumin on TGFβR2 and Smad2 stability, synthesis and expression.
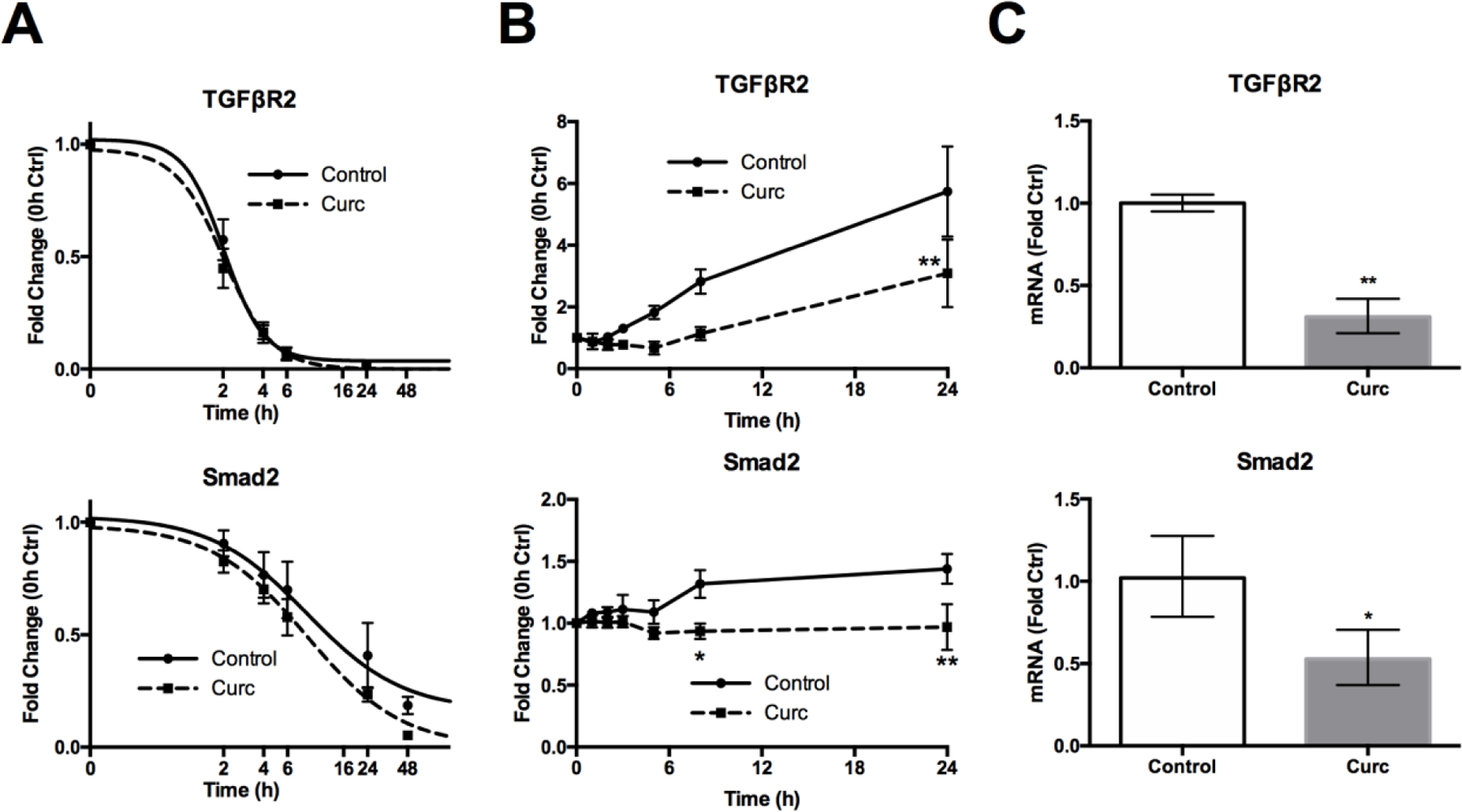
A) TGFβR2 and Smad protein stability (Western blot densitometry, n = 3/time) in control vs curcumin-treated (30 μM) MDA-SA cells concurrently treated with cycloheximide (CHX) to block protein synthesis. B) TGFβR2 and Smad protein synthesis (Western blot densitometry, n = 4/time) in control vs curcumin-treated (30 μM) MDA-SA cells pre-treated with cycloheximide for 24 h. C) TGFβR2 and Smad2 mRNA levels in curcumin-treated (30 μM, 16h) vs. control MDA-SA cells, reported as mean ± CI * p < 0.05, ** p < 0.01 vs. control.
3.5. Possible role for oxidative metabolites of curcumin in blocking TGFβ signaling
After 4 hours of 10 μM curcumin treatment, bicyclopentadione (BCP, 0.9 μM), a stable end product of multistep curcumin oxidation [32], was detected in MDA-SA cell conditioned media as curcumin levels decreased (Fig. S7). Pre-treatment of MDA-SA cells with N-acetylcysteine (NAC) – a precursor of glutathione that scavenges and neutralizes unstable curcumin-derived oxidative intermediates [32,62] – reversed all changes induced by curcumin at 16 hours, normalizing TGFβR2 and Smad2 levels, restoring TGFβ-induced pSmad levels, and preventing curcumin activation of MAPK signaling (Fig. 6A). All early curcumin effects were also reversed by NAC, with the notable exception of early decreases in TGFβR2 (Fig. 6B). To further assess a possible role of oxidative metabolites, curcumin effects were compared to those of diacetyl curcumin (DAC) [34], a curcumin analog (Fig. 6C) that did not undergo oxidative metabolism during 24 hours of incubation with MDA-SA cells (data not shown). In contrast to curcumin-induced JNK and p38 phosphorylation and decreasing TGFβR2 levels, non-oxidizable DAC was without effect (Fig. 6B). Similarly, while curcumin inhibited TGFβ-stimulated PTHrP secretion, DAC was without effect (Fig. 6D).
Figure 6. Possible role of oxidative metabolites in mediating curcumin effects.
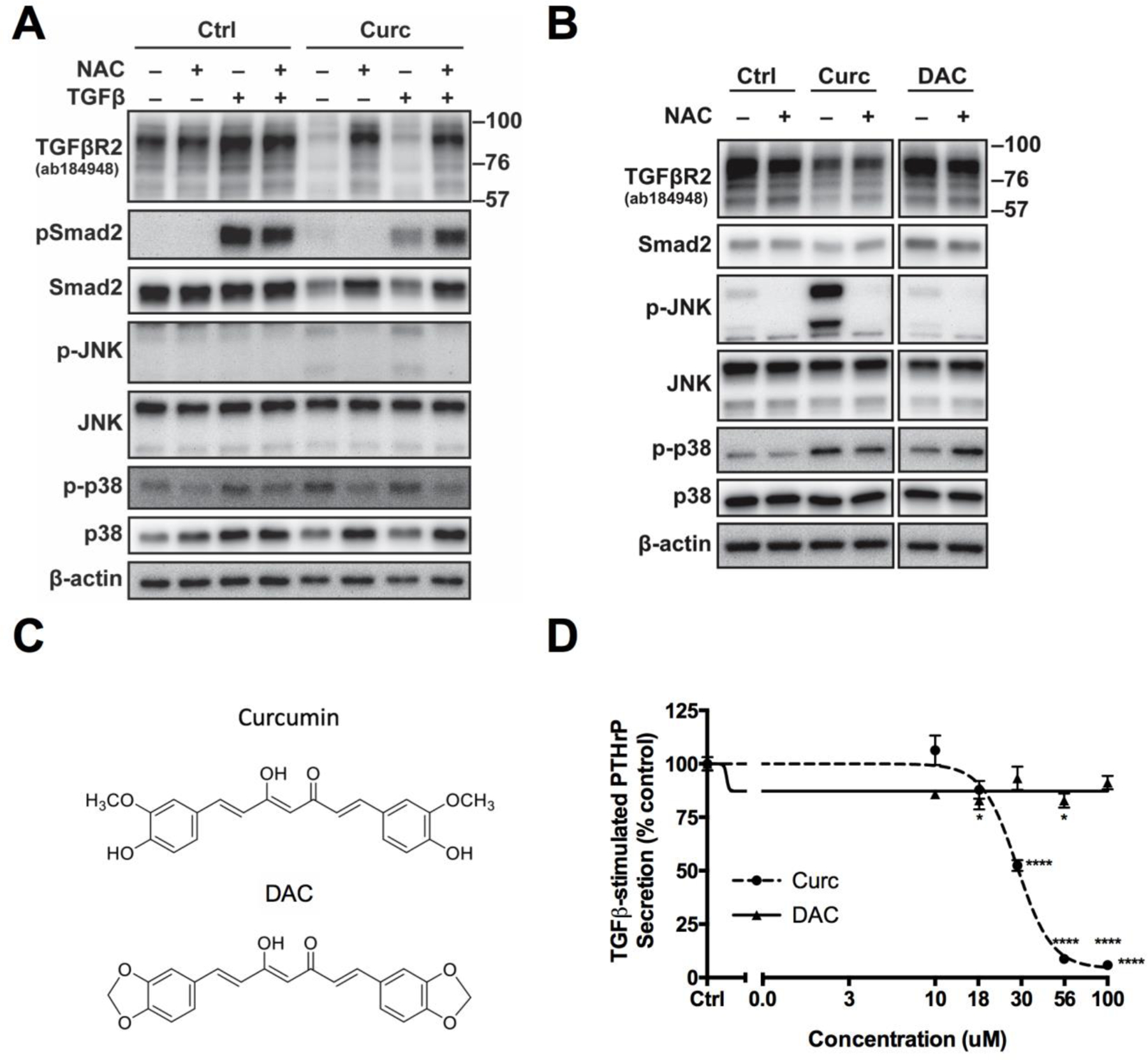
A) Effect of N-acetylcysteine (10 mM NAC, 1 h pretreatment) on late (16 h curcumin [30 μM] prior to 1h TGFβ stimulation) curcumin-induced changes in TGFβR2, Smad and MAPK signaling in control vs. TGFβ-stimulated cells B) Early (4h) effects of curcumin vs. diacetyl curcumin (DAC) (30 μM) on TGFβR2, Smad2, or MAPK signaling proteins in control vs NAC-pretreated MDA-SA cells. Note: This is a single blot with unrelated intervening data omitted. C) Structure of curcumin vs. DAC, a non-oxidizable analog of curcumin. D) Effect of curcumin vs. DAC (30 µM, 4h pre-treatment) on TGFβ-stimulated PTHrP secretion in MDA-SA cells. * p < 0.05, **** p < 0.0001 vs. control.
4. Discussion
Effects of TGFβ in cancer are complex, with possible beneficial effects as a tumor suppressor during early oncogenesis vs. detrimental effects at later stages, including a well-defined role in promoting tumor-associated osteolysis in BCa metastatic progression within bone [1,63]. In metastatic breast cancer, where osteolytic bone metastases occur in most cases, TGFβ signaling in bone-disseminated cells is a well-documented driver of osteolysis and metastatic progression, independent of effects on tumor cell proliferation [6–8,14,15,18–21]. Because of recent evidence of in situ metabolism of inactive circulating curcumin-glucuronides to form the active aglycone within bone [30,31], as well as inhibitory effects of curcumin on breast cancer cell osteolytic TGFβ signaling pathways and TGFβ-dependent osteolysis and BMET progression in vivo [26,28,29], the current experiments were undertaken to examine the mechanism(s) by which curcumin inhibits TGFs signaling in breast cancer cells known to form TGFβ dependent BMET in vivo, all of which represent the triple negative (TNBC) BCa subtype, for which targeted therapeutics (e.g. antiestrogens) are not available. Curcumin uniformly decreased TGFβ-stimulated Smad signaling in TNBC bone-tropic cell lines dependent on this signaling pathway for osteolytic in vivo BMET progression [6,7,14,15,18,20,21]. Decreased TGFβ-stimulated, receptor-mediated (C-terminal) Smad phosphorylation in curcumin-treated BCa cells were not attributable to altered accessory proteins (e.g., SARA or Smad7) or phosphorylation stability (e.g., phosphatases). Instead, curcumin specifically targeted TGFβR2 and R-Smads (Fig. 7), two key signaling proteins required for continuous propagation of TGFβ-induced Smad signaling [39,55,64,65], which persisted for up to 12 hours in bone metastatic cells. Early and persistent curcumin-mediated decreases in total and plasma-membrane associated TGFβR2 protein, the latter being the initiation site for TGFβ binding and signal transduction [55,65], were documented in all tested BCa cell lines. In contrast, TGFβR1 levels forming an active complex with TGFβ-bound TGFβR2, were unchanged. Decreased TGFβR2 did not appear mediated by curcumin-induced changes in receptor trafficking or degradation, but rather occurred secondary to decreased synthesis of this short lived (t1/2 = 2h) receptor (Fig. 7). A targeted decrease in R-Smad protein levels (Smad2 and Smad3) was also documented in all curcumin-treated bone-tropic BCa cell lines, which was most apparent at later times following curcumin treatment likely due to their longer half-life [65], and was also attributable to decreased synthesis (Fig. 7).
Figure 7. Overview of curcumin effects on TGFβ signaling pathways in bone-disseminated human breast cancer cells that drive tumor-associated osteolysis and promote BMET expansion.
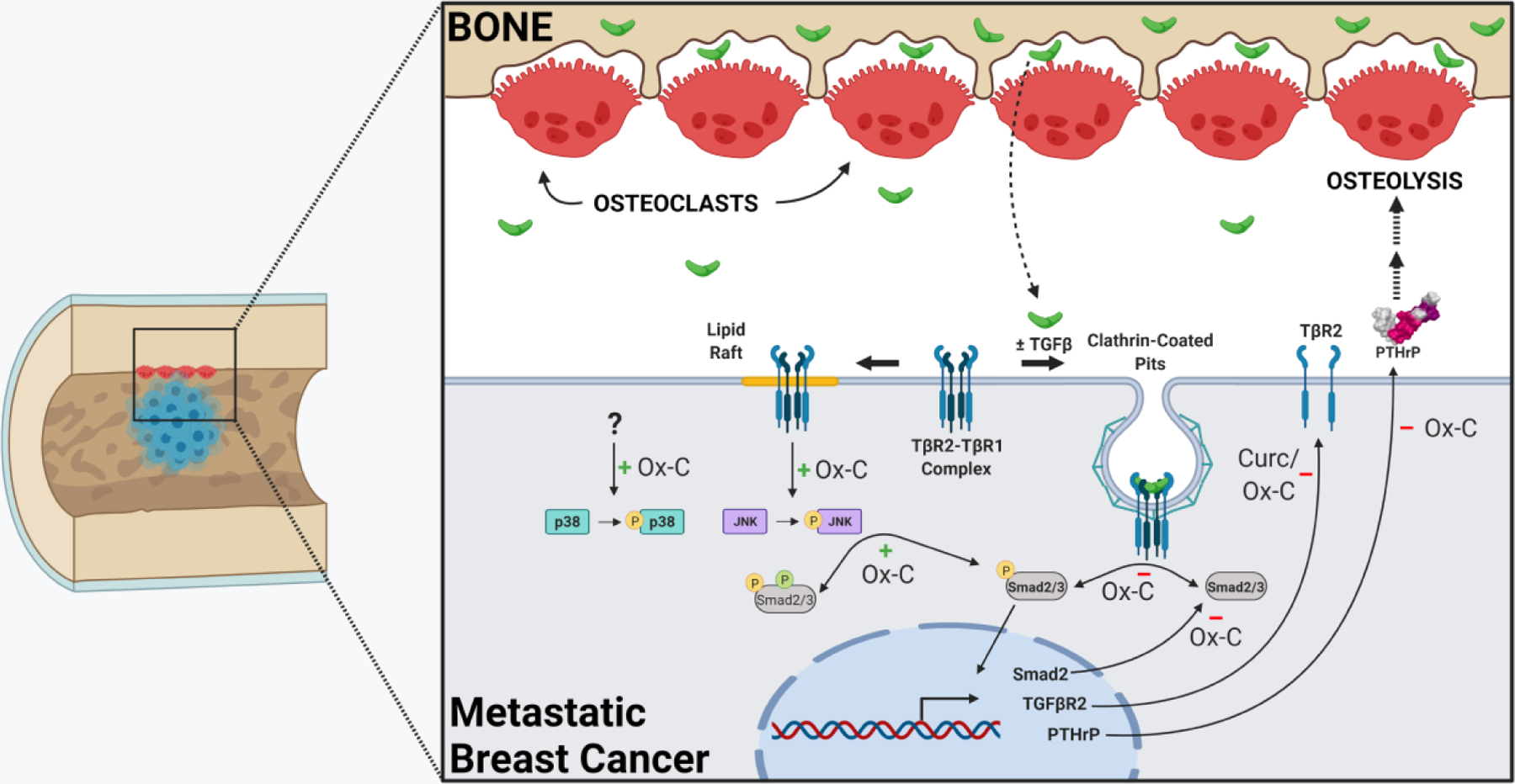
Curcumin inhibited TGFβ-stimulated receptor-mediated Smad activation (e.g. phosphorylation, denoted by yellow P), which requires internalization of TGFβ-bound TGFβR2/R1 complexes via clathrin-coated pits, directly (via decreased-constitutive Smad proteins) and indirectly (via decreased TGFβR2 proteins). Curcumin-mediated reductions in TGFβR2 and Smad2 proteins were likely attributable to decreased gene expression and were dependent on curcumin oxidation, with the exception of early reductions in TGFβR2. Curcumin inhibition of TGFβ-inducible Smad signaling had the ultimate effect of blocking TGFβ-stimulated secretion of osteolytic PTHrP, without which osteolysis and BMET progression do not occur in vivo. Independent of curcumin effects on Smad-mediated TGFβ-inducible PTHrP, oxidative intermediates of curcumin induced (green positive sign) JNK activation (see yellow P), and subsequent Smad linker region phosphorylation (green P), in a manner possibly dependent on lipid raft localization of TGFβR2 with functional cytoplasmic domains, albeit independent of ligand binding. Curcumin also induced phosphorylation of p38 through an unknown mechanism also dependent on curcumin oxidation. Created with BioRender.com.
While regulated changes in TGFβR2 and R-Smad attributable to protein trafficking and degradation are well described following TGFβ stimulation [39,55], much less is known about regulation of their constitutive expression [66,67]. While future studies are needed to elucidate the specific cellular target(s) of curcumin responsible for reducing constitutive levels of TGFβR2 and R-Smad, results here clearly demonstrated that inhibitory effects of curcumin on the synthesis of these proteins were: 1) specific, targeting only TGFβR2 and R-Smads; 2) uniform across all BCa cell lines forming Smad-mediated TGFβ-dependent BMETs; and 3) due, at least in part, to curcumin inhibition of TGFβR2 and Smad2 gene expression.
A role for reactive curcumin oxidative metabolites as mediators of curcumin effects on TGFs signaling pathways in bone tropic BCa cells was elucidated using MDA-SA cells, whose TGFβ-dependent signaling pathways driving in vivo BMET progression are particularly well described and emblematic of the other BCa BMET models [6,7,9,14,15,18,20,21]. NAC has previously been used to scavenge and block the effects of reactive curcumin oxidative intermediates on protein function in other curcumin-treated cell types [32,62]. Thus, this evidence that NAC prevents reductions in TGFβR2 and pSmad/Smad levels in response to curcumin treatment in MDA-SA cells (with the exception of early decreases in TGFβR2) suggests that reactive intermediates of curcumin oxidative degradation may play an important role in blocking the Smad signaling driving MDA-SA BMET progression in vivo. Alternatively, it is possible that NAC is simply achieving this effect by decreasing oxidative stress. In the absence of a specific target(s) of reactive curcumin metabolites mediating effects on TGFβ signaling, it is difficult to elucidate which NAC mechanism is prevailing. However, the detection of BCP, a final stable end-product of curcumin oxidation [32] and biomarker for reactive “upstream” intermediates (e.g., quinone methide and spiroepoxide) [34], in curcumin-treated BCa cell conditioned media and the lack of activity of a non-oxidizable curcumin analog, provide additional support for a possible role of reactive curcumin metabolites in blocking Smad-signaling that drives BMET progression in these, and potentially other, TGFβ-dependent bone metastatic BCa cells. Since reactive curcumin intermediates are capable of forming stable adducts with proteins that alter protein function, as has been reported for DNA topoisomerase and IKKβ [33,34], future studies will be required to query the possible role of specific protein/curcumin adduct(s) in mediating curcumin reductions in TGFβR2 and Smad protein gene expression and synthesis (Fig. 7).
One additional and unanticipated effect of curcumin was its early and persistent induction of MAPK signaling in all BCa cell lines studied (Fig. 7), also attributable to oxidative intermediates. While crosstalk between Smad and MAPK signaling pathways is well documented [52,54,58], in MDA-SA cells neither JNK nor p38 activation by curcumin was responsible for curcumin’s inhibitory effects on TGFβR2 or Smad signaling, or for curcumin inhibition of TGFβ-inducible PTHrP, the primary TGFβ-regulated gene responsibility for BMET progression in the MDA-SA in vivo model [15]. This, of course, does not rule out a possible role for curcumin activation of MAPK signaling in altering the expression of other TGFβ-regulated genes in this or other bone tropic cell lines. In this regard, it is of interest that curcumin induction of JNK signaling led to phosphorylation of the linker region of the R-Smads, a modification that can alter the nuclear trafficking of receptor-activated Smad complexes and thus, Smad-mediated gene expression [52,54]. Evidence that curcumin activation of JNK signaling was dependent on intact lipid rafts and abrogated by overexpression of TGFβR2 lacking the cytoplasmic domain was also intriguing. Prior studies suggest that loss of the TGFβR2 cytoplasmic domain does not alter the receptor’s preferential location in lipid rafts [39,59], a compartment mediating TGFβR2/R1 MAPK signaling (contrasting with clathrin-coated pit TGFβR2/R1 complexes mediating Smad signaling) [57,58], but does abrogate TGFβR2/R1 signal transduction (ligand-dependent or -independent) [20,38,68]. Therefore, the lipid raft/TGFβR2 cytoplasmic-domain dependence of JNK activation by curcumin in the absence of TGFβ suggests the possibility that ligand-independent activation of TGFβR2/TGFβR1 complexes by curcumin, or more specifically oxidized intermediates of curcumin degradation, within lipid rafts could be mediating JNK activation in bone-tropic BCa cells (Fig. 7). Consistent with this postulate, structural changes to plasma membrane lipid rafts or proteins within these domains in response to curcumin treatment have been reported in other cell types [69–71].
While in vivo bioactivity of curcumin, including bone protective effects, has been reported in humans and in animal models [26,35,72,73], the relationship between in vivo curcumin metabolism and bioactivity appears complex. Following curcumin ingestion, the major circulating metabolite, curcumin-glucuronide is inactive, but can be enzymatically deconjugated within bone to form bioactive aglycone curcumin [30,31]. We have posited that this enzymatic enrichment of the active aglycone within bone could promote bone bioactivity, including TGFβ inhibitory effects of curcumin in BCa cells disseminated to this site, which also is notably enriched for the ligand driving tumor-induced osteolysis (TGFβ) due to its release from resorbed bone matrix [30]. These current studies suggest that curcumin formed in situ in bone must also undergo subsequent, irreversible metabolism to form short-lived reactive oxidative intermediates to block Smad-mediated TGFβ signaling in tumor cells metastatic to this site, further increasing the bone-specificity of curcumin’s anti-TGFβ effects. To date, in vivo evidence of curcumin oxidation is lacking, potentially due to the insensitivity of current methods, which rely on detection of an end product (BCP) that is only formed if upstream intermediates do not covalently attach to biomolecules (e.g., protein) [32–34]. However, given the potential clinical utility of a bone-specific TGFβ inhibitor in BCa [9,10] and curcumin’s uniform inhibitory effects on TGFβ-induced Smad signaling in all TGFβ-dependent bone-metastatic BCa cell lines studied, the results reported here suggest potential promise for curcumin or oxidizable curcumin analogs in specifically blocking BMET progression in BCa. The necessity of oxidative metabolism in the bioactivity of curcumin has been previously reported for other curcumin effects [34], including inhibition of NF-κB signaling [74] or poisoning of topoisomerase [33]. However, to our knowledge this is the first report of oxidative metabolites of curcumin being required for the inhibition of TGFβ signaling.
Given the ready availability of curcumin for over the counter use [75], it is important to note that osteolytic effects of tumoral TGFβ signaling have primarily been documented in pre-clinical “triple-negative” BCa (TNBC) BMET models lacking expression of estrogen receptor (ER-), progesterone receptor (PR-), and HER2, including the cell lines studied here [1,2,6–10,14–21,26,28,76]. However, most breast cancer bone metastases are associated with tumors expressing ER, PR, or HER2 [77], which provides a clinical caveat for the applicability of the current findings. As research examining TGFβ signaling in non-TNBC osteolytic BMET progression progresses [78,79], additional research will be warranted to determine the utility of TGFβ inhibition and, thus, use of curcumin or similar oxidizable analogs, for the management of BMET risk across a range of clinical BCa subtypes.
Supplementary Material
Highlights.
Curcumin blocks breast cancer TGFβ-mediated Smad signaling driving osteolysis in bone
Curcumin mediates this effect by downregulating TGFβR2 and Smad2/3 gene expression
Oxidative curcumin metabolites appear responsible for the majority of these effects.
Curcumin also activated JNK signaling in a lipid-raft dependent manner
Acknowledgements
We would like to acknowledge Timothy M. Panknin, medical student at the University of Arizona, for his assistance with experimental data acquisition, which was supported by the National Heart Lung and Blood Institute at the NIH (T35HL007479).
Funding:
This work was supported by the National Cancer Institute (NCI), the National Center for Complementary and Integrative Health (NCCIH), and the Office of Dietary Supplements (ODS), at the National Institutes of Health (NIH) (R01CA174926 and R34AT007837 to JLF, R01AT006896 to CS, and F31AT009938 to AK); the United States Department of Agriculture (2014-38420-21799 National Needs Fellowship to AK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Guise TA, Kozlow WM, Heras-Herzig A, Padalecki SS, Yin JJ, Chirgwin JM. Molecular Mechanisms of Breast Cancer Metastases to Bone. Clin Breast Cancer 2008;5:S46–53. doi: 10.3816/cbc.2005.s.004. [DOI] [PubMed] [Google Scholar]
- [2].Esposito M, Guise T, Kang Y. The biology of bone metastasis. Cold Spring Harb Perspect Med 2018;8:a031252. doi: 10.1101/cshperspect.a031252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Braun S, Auer D, Marth C. The prognostic impact of bone marrow micrometastases in women with breast cancer. Cancer Invest 2009;27:598–603. doi: 10.1080/07357900802574496. [DOI] [PubMed] [Google Scholar]
- [4].Wong MH, Stockler MR, Pavlakis N. Bisphosphonates and other bone agents for breast cancer. Cochrane Database Syst Rev 2012;2. doi: 10.1002/14651858.CD003474.pub3. [DOI] [PubMed] [Google Scholar]
- [5].Drieling RL, LaCroix AZ, Beresford SAA, Boudreau DM, Kooperberg C, Chlebowski RT, et al. Long-term oral bisphosphonate use in relation to fracture risk in postmenopausal women with breast cancer. Menopause 2016;23:1. doi: 10.1097/GME.0000000000000696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhang Z, Hu Z, Gupta J, Krimmel JD, Gerseny HM, Berg AF, et al. Intravenous administration of adenoviruses targeting transforming growth factor beta signaling inhibits established bone metastases in 4T1 mouse mammary tumor model in an immunocompetent syngeneic host. Cancer Gene Ther 2012;19:630–6. doi: 10.1038/cgt.2012.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Futakuchi M, Nannuru KC, Varney ML, Sadanandam A, Nakao K, Asai K, et al. Transforming growth factor-β signaling at the tumor-bone interface promotes mammary tumor growth and osteoclast activation. Cancer Sci 2009;100:71–81. doi: 10.1111/j.1349-7006.2008.01012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gupta J, Robbins J, Jilling T, Seth P. TGFβ-dependent induction of interleukin-11 and interleukin-8 involves SMAD and p38 MAPK pathways in breast tumor models with varied bone metastases potential. Cancer Biol Ther 2011;11:311–6. doi: 10.4161/cbt.11.3.14096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chiechi A, Waning DL, Stayrook KR, Buijs J, Guise TA, Mohammad KS. Role of TGF-β in breast cancer bone metastases. Adv Biosci Biotechnol 2014;4:15–30. doi: 10.4236/abb.2013.410A4003.Role. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Colak S, ten Dijke P. Targeting TGF-β Signaling in Cancer. Trends in Cancer 2017;3:56–71. doi: 10.1016/j.trecan.2016.11.008. [DOI] [PubMed] [Google Scholar]
- [11].Deckers M, Van Dinther M, Buijs J, Que I, Löwik C, Van Der Pluijm G, et al. The tumor suppressor Smad4 is required for transforming growth factor β-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res 2006;66:2202–9. doi: 10.1158/0008-5472.CAN-05-3560. [DOI] [PubMed] [Google Scholar]
- [12].Ganapathy V, Ge R, Grazioli A, Xie W, Banach-Petrosky W, Kang Y, et al. Targeting the Transforming Growth Factor-β pathway inhibits human basal-like breast cancer metastasis. Mol Cancer 2010;9:1–16. doi: 10.1186/1476-4598-9-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Biswas S, Nyman JS, Alvarez JA, Chakrabarti A, Ayres A, Sterling J, et al. Anti-transforming growth factor ß antibody treatment rescues bone loss and prevents breast cancer metastasis to bone. PLoS One 2011;6:1–12. doi: 10.1371/journal.pone.0027090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, et al. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Guise TA, Yin JJ, Taylor SD, Kumagai Y, Dallas M, Boyce BF, et al. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Invest 1996;98:1544–9. doi: 10.1172/JCI118947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Juárez P, Guise TA. TGF-β in cancer and bone: Implications for treatment of bone metastases. Bone 2011;48:23–9. doi: 10.1016/j.bone.2010.08.004. [DOI] [PubMed] [Google Scholar]
- [17].Sterling JA, Edwards JR, Martin TJ, Mundy GR. Advances in the biology of bone metastasis: How the skeleton affects tumor behavior. Bone 2011;48:6–15. doi: 10.1016/j.bone.2010.07.015. [DOI] [PubMed] [Google Scholar]
- [18].Kang Y, He W, Tulley S, Gupta GP, Serganova I, Chen C-R, et al. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci U S A 2005;102:13909–14. doi: 10.1073/pnas.0506517102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Korpal M, Yan J, Lu X, Xu S, Lerit DA, Kang Y. Imaging transforming growth factor-Β signaling dynamics and therapeutic response in breast cancer bone metastasis. Nat Med 2009;15:960–6. doi: 10.1038/nm.1943. [DOI] [PubMed] [Google Scholar]
- [20].Käkönen S-M, Selander KS, Chirgwin JM, Yin JJ, Burns S, Rankin WA, et al. Transforming Growth Factor-β Stimulates Parathyroid Hormone-related Protein and Osteolytic Metastases via Smad and Mitogen-activated Protein Kinase Signaling Pathways. J Biol Chem 2002;277:24571–8. doi: 10.1074/jbc.M202561200. [DOI] [PubMed] [Google Scholar]
- [21].Kang Y, Siegel PM, Shu A, Drobnjak M, Kakonen SM, Cordón C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003;3:537–49. [DOI] [PubMed] [Google Scholar]
- [22].Meng X, Vander Ark A, Lee P, Hostetter G, Bhowmick NA, Matrisian LM, et al. Myeloid-specific TGF-β signaling in bone promotes basic-FGF and breast cancer bone metastasis. Oncogene 2016;35:2370–8. doi: 10.1038/onc.2015.297. [DOI] [PubMed] [Google Scholar]
- [23].Powell GJ, Southby J, Danks JA, Stillwell RG, Hayman J a, Henderson MA, et al. Localization of Parathyroid Hormone-related Protein in Breast Cancer Metastases: Increased Incidence in Bone Compared with Other Sites. Cancer Res 1991;51:3059–61. [PubMed] [Google Scholar]
- [24].Xu C, Wang Z, Cui R, He H, Lin X, Sheng Y, et al. Co-expression of parathyroid hormone related protein and TGF-beta in breast cancer predicts poor survival outcome. BMC Cancer 2015;15:1–10. doi: 10.1186/s12885-015-1873-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Southby J, Kissin MW, Danks JA, John AI, Moseley JM, Henderson MA, et al. Immunohistochemical Localization of Parathyroid Hormone-related Protein in Human Breast Cancer. Cancer Res 1990;50:7710–6. [PubMed] [Google Scholar]
- [26].Wright LE, Frye JB, Lukefahr AL, Timmermann BN, Mohammad KS, Guise TA, et al. Curcuminoids Block TGF-β Signaling In Human Breast Cancer Cells And Limit Osteolysis In A Murine Model Of Breast Cancer Bone Metastasis. J Nat Prod 2013;76:316–21. doi: 10.1021/np300663v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang K, Guo C, Dong X, Yu Y, Wang B, Liu W, et al. In Vivo Evaluation of Reduction-Responsive Alendronate-Hyaluronan-Curcumin Polymer-Drug Conjugates for Targeted Therapy of Bone Metastatic Breast Cancer. Mol Pharm 2018;15:2764–9. [DOI] [PubMed] [Google Scholar]
- [28].Kunihiro AG, Brickey JA, Frye JB, Luis PB, Schneider C, Funk JL. Curcumin, but not curcumin-glucuronide, inhibits Smad-signaling in TGFβ-dependent bone metastatic breast cancer cells and is enriched in bone compared to other tissues. J Nutr Biochem 2019;63:150–6. doi: 10.1016/j.jnutbio.2018.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wright LE, Frye JB, Gorti B, Timmermann BN, Funk JL. Bioactivity of Turmeric-Derived Curcuminoids and Related Metabolites in Breast Cancer. Curr Pharm Des 2013;19:6218–25. doi: 10.2174/1381612811319340013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kunihiro AG, Luis PB, Brickey JA, Frye JB, Chow HHS, Schneider C, et al. Beta-Glucuronidase Catalyzes Deconjugation and Activation of Curcumin-Glucuronide in Bone. J Nat Prod 2019;82:500–9. doi: 10.1021/acs.jnatprod.8b00873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kunihiro AG, Luis PB, Frye JB, Chew W, Chow H-HS, Schneider C, et al. Bone-specific metabolism of dietary polyphenols in resorptive bone diseases. Mol Nutr Food Res 2020;64. doi: 10.1002/mnfr.202000072.R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gordon ON, Luis PB, Sintim HO, Schneider C. Unraveling curcumin degradation: Autoxidation proceeds through spiroepoxide and vinylether intermediates en route to the main bicyclopentadione. J Biol Chem 2015;290:4817–28. doi: 10.1074/jbc.M114.618785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ketron AC, Gordon ON, Schneider C, Osheroff N. Oxidative metabolites of curcumin poison human type II topoisomerases. Biochemistry 2013;52:221–7. doi: 10.1021/bi3014455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Edwards RL, Luis PB, Varuzza PV., Joseph AI, Presley SH, Chaturvedi R, et al. The anti-inflammatory activity of curcumin is mediated by its oxidative metabolites. J Biol Chem 2017;292:21243–52. doi: 10.1074/jbc.RA117.000123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Funk JL, Oyarzo JN, Frye JB, Chen G, Clark R, Jolad SD, et al. Turmeric Extracts Containing Curcuminoids Prevent Experimental Rheumatoid Arthritis. J Nat Prod 2006;69:351–5. doi: 10.1021/np050327j.Turmeric. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Luis PB, Gordon ON, Nakashima F, Joseph AI, Shibata T, Uchida K, et al. Oxidative metabolism of curcumin-glucuronide by peroxidases and isolated human leukocytes. Biochem Pharmacol 2017;132:143–9. doi: 10.1016/j.bcp.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Griesser M, Pistis V, Suzuki T, Tejera N, Pratt DA, Schneider C. Autoxidative and cyclooxygenase-2 catalyzed transformation of the dietary chemopreventive agent curcumin. J Biol Chem 2011;286:1114–24. doi: 10.1074/jbc.M110.178806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wieser R, Attisano L, Wrana JL, Massagué J. Signaling activity of transforming growth factor beta type II receptors lacking specific domains in the cytoplasmic region. Mol Cell Biol 1993;13:7239–47. doi: 10.1128/mcb.13.12.7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat Cell Biol 2003;5:410–21. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- [40].Osborne C, Hamilton B, Titus G, Livingston R. Epidermal growth factor stimulation of human breast cancer cells in culture. Cancer Res 1980;40:2361–6. [PubMed] [Google Scholar]
- [41].Sasaki A, Boyce BF, Story B, Wright KR, Chapman M, Boyce R, et al. Bisphosphonate risedronate reduces metastatic human breast cancer burden in bone in nude mice. Cancer Res 1995;55:3551–7. [PubMed] [Google Scholar]
- [42].McEarchern JA, Kobie JJ, Mack V, Wu RS, Meade-Tollin L, Arteaga CL, et al. Invasion and metastasis of a mammary tumor involves TGF-β signaling. Int J Cancer 2001;91:76–82. doi:. [DOI] [PubMed] [Google Scholar]
- [43].Liu J, Liao S, Diop-Frimpong B, Chen W, Goel S, Naxerova K, et al. TGF-β blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc Natl Acad Sci 2012;109:16618–23. doi: 10.1073/pnas.1117610109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hiraga T, Williams PJ, Ueda A, Tamura D, Yoneda T. Zoledronic Acid Inhibits Visceral Metastases in the 4T1/luc Mouse Breast Cancer Model. Clin Cancer Res 2004;10:4559–67. doi: 10.1158/1078-0432.CCR-03-0325. [DOI] [PubMed] [Google Scholar]
- [45].Lelekakis M, Moseley JM, Martin TJ, Hards D, Williams E, Lowen D, et al. A novel orthotopic model of breast cancer metastasis to bone. Clin Exp Metastasis 1999;17:163–70. [DOI] [PubMed] [Google Scholar]
- [46].Masters JR, Thomson JA, Daly-Burns B, Reid YA, Dirks WG, Packer P, et al. Short tandem repeat profiling provides an international reference standard for human cell lines. Proc Natl Acad Sci U S A 2001;98:8012–7. doi: 10.1073/pnas.121616198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ritter L, Davidson L, Henry M, Davis-Gorman G, Morrison H, Frye JB, et al. Exaggerated Neutrophil-Mediated Reperfusion Injury after Ischemic Stroke in a Rodent Model of Type 2 Diabetes. Microcirculation 2011;18:552–61. doi: 10.1111/j.1549-8719.2011.00115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- [49].Fernandez J, Candenas M, Souto M, Trujillo M, Norte M. Okadaic Acid, Useful Tool for Studying Cellular Processes. Curr Med Chem 2012;9:229–62. doi: 10.2174/0929867023371247. [DOI] [PubMed] [Google Scholar]
- [50].Lindemann RK, Ballschmieter P, Nordheim A, Dittmer J. Transforming Growth Factor β Regulates Parathyroid Hormone-related Protein Expression in MDA-MB-231 Breast Cancer Cells through a Novel Smad/Ets Synergism. J Biol Chem 2001;276:46661–70. doi: 10.1074/jbc.M105816200. [DOI] [PubMed] [Google Scholar]
- [51].Koli KM, Arteaga CL. Processing of the transforming growth factor β type I and II receptors. Biosynthesis and ligand-induced regulation. J Biol Chem 1997;272:6423–7. doi: 10.1074/jbc.272.10.6423. [DOI] [PubMed] [Google Scholar]
- [52].Massagué J Integration of Smad and MAPK pathways: A link and a linker revisited. Genes Dev 2003;17:2993–7. doi: 10.1101/gad.1167003. [DOI] [PubMed] [Google Scholar]
- [53].Matsuzaki K Smad phospho-isoforms direct context-dependent TGF-β signaling. Cytokine Growth Factor Rev 2013;24:385–99. doi: 10.1016/j.cytogfr.2013.06.002. [DOI] [PubMed] [Google Scholar]
- [54].Kamato D, Burch ML, Piva TJ, Rezaei HB, Rostam MA, Xu S, et al. Transforming growth factor-β signalling: Role and consequences of Smad linker region phosphorylation. Cell Signal 2013;25:2017–24. doi: 10.1016/j.cellsig.2013.06.001. [DOI] [PubMed] [Google Scholar]
- [55].Yakymovych I, Yakymovych M, Heldin CH. Intracellular trafficking of transforming growth factor β receptors. Acta Biochim Biophys Sin (Shanghai) 2018;50:3–11. doi: 10.1093/abbs/gmx119. [DOI] [PubMed] [Google Scholar]
- [56].Huang F, Chen Y-G. Regulation of TGF-β receptor activity. Cell Biosci 2012;2:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hata A, Chen Y-G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb Perspect Biol 2016;8:a022061. doi: 10.1101/cshperspect.a022061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhang YE. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb Perspect Biol 2017;9:ao22129. doi: 10.1101/cshperspect.a022129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Luga V, McLean S, Le Roy C, O’Connor-McCourt M, Wrana JL, Di Guglielmo GM. The extracellular domain of the TGFβ type II receptor regulates membrane raft partitioning. Biochem J 2009;421:119–31. doi: 10.1042/BJ20081131. [DOI] [PubMed] [Google Scholar]
- [60].Von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, et al. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 2011;146:471–84. doi: 10.1016/j.cell.2011.06.025. [DOI] [PubMed] [Google Scholar]
- [61].Juárez P, Fournier PGJ, Mohammad KS, McKenna RC, Davis HW, Peng XH, et al. Halofuginone inhibits TGF-β/BMP signaling and in combination with zoledronic acid enhances inhibition of breast cancer bone metastasis. Oncotarget 2017;8:86447–62. doi: 10.18632/oncotarget.21200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Luis PB, Boeglin WE, Schneider C. Thiol Reactivity of Curcumin and Its Oxidation Products. Chem Res Toxicol 2018;31:269–76. doi: 10.1021/acs.chemrestox.7b00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Liu S, Ren J, ten Dijke P. Targeting TGFβ signal transduction for cancer therapy. Signal Transduct Target Ther 2021;6:1–20. doi: 10.1038/s41392-020-00436-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lin X, Duan X, Liang YY, Su Y, Wrighton KH, Long J, et al. PPM1A Functions as a Smad Phosphatase to Terminate TGFβ Signaling. Cell 2006;125:915–28. doi: 10.1016/j.cell.2006.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zi Z, Klipp E. Constraint-based modeling and kinetic analysis of the Smad dependent TGF-beta signaling pathway. PLoS One 2007;2:e936. doi: 10.1371/journal.pone.0000936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bae HW, Geiser AG, Kim DH, Chung MT, Burmester JK, Sporn MB, et al. Characterization of the promoter region of the human transforming growth factor-β type II receptor gene. J Biol Chem 1995;270:29460–8. doi: 10.1074/jbc.270.49.29460. [DOI] [PubMed] [Google Scholar]
- [67].Chowdhury S, Ammanamanchi S, Howell GM. Epigenetic Targeting of Transforming Growth Factor β Receptor II and Implications for Cancer Therapy. Mol Cell Pharmacol 2009;1:57–70. doi: 10.4255/mcpharmacol.09.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Feng X-H, Derynck R. Ligand-independent activation of transforming growth factor (TGF) beta signaling pathways by heteromeric cytoplasmic domains of TGF-beta receptors. J Biol Chem 1996;271:13123–9. doi: 10.1074/jbc.271.22.13123. [DOI] [PubMed] [Google Scholar]
- [69].Coleman DT, Soung YH, Surh Y-J, Cardelli JA, Chung J. Curcumin Prevents Palmitoylation of Integrin β4 in Breast Cancer Cells. PLoS One 2015;10:e0125399. doi: 10.1371/journal.pone.0125399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Tsukamoto M, Kuroda K, Ramamoorthy A, Yasuhara K. Modulation of raft domains in a lipid bilayer by boundary-active curcumin. Chem Commun 2014;50:3427–30. doi: 10.1039/c3cc47738j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lin M-L, Lu Y-C, Chen H-Y, Lee C-C, Chung J-G, Chen S-S. Suppressing the formation of lipid raft-associated Rac1/PI3K/Akt signaling complexes by curcumin inhibits SDF-1α-induced invasion of human esophageal carcinoma cells. Mol Carcinog 2014;53:360–79. doi: 10.1002/mc.21984. [DOI] [PubMed] [Google Scholar]
- [72].Wright LE, Frye JB, Timmermann BN, Funk JL. Protection of trabecular bone in ovariectomized rats by turmeric (Curcuma longa L.) Is dependent on extract composition. J Agric Food Chem 2010;58:9498–504. doi: 10.1021/jf101873f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Khanizadeh F, Rahmani A, Asadollahi K, Ahmadi MRH. Combination therapy of curcumin and alendronate modulates bone turnover markers and enhances bone mineral density in postmenopausal women with osteoporosis. Arch Endocrinol Metab 2018;62:438–45. doi: 10.20945/2359-3997000000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Edwards RL, Luis PB, Nakashima F, Kunihiro AG, Presley SH, Funk JL, et al. Mechanistic Differences in the Inhibition of NF-κB by Turmeric and Its Curcuminoid Constituents. J Agric Food Chem 2020;68:6154–60. doi: 10.1021/acs.jafc.0c02607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Skiba MB, Luis PB, Alfafara C, Billheimer D, Schneider C, Funk JL. Curcuminoid Content and Safety-Related Markers of Quality of Turmeric Dietary Supplements Sold in an Urban Retail Marketplace in the United States. Mol Nutr Food Res 2018;62:1–10. doi: 10.1002/mnfr.201800143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wright LE, Ottewell PD, Rucci N, Peyruchaud O, Pagnotti GM, Chiechi A, et al. Murine models of breast cancer bone metastasis. Bonekey Rep 2016;5:1–11. doi: 10.1038/bonekey.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Kennecke H, Yerushalmi R, Woods R, Cheang MCU, Voduc D, Speers CH, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol 2010;28:3271–7. doi: 10.1200/JCO.2009.25.9820. [DOI] [PubMed] [Google Scholar]
- [78].Cheng JN, Frye JB, Whitman SA, Kunihiro AG, Pandey R, Funk JL. A role for TGFβ signaling in preclinical osteolytic estrogen receptor-positive breast cancer bone metastases progression. Int J Mol Sci 2021;22. doi: 10.3390/ijms22094463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Ganapathy V, Banach-Petrosky W, Xie W, Kareddula A, Neinhuis H, Miles G, et al. Luminal breast cancer metastasis is dependent on estrogen signaling. Clin Exp Metastasis 2012;29:493–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Frederic M, Hamroun D, Faivre L, Boileau C, Jondeau G, Claustres M, et al. A new locus-specific database (LSDB) for mutations in the TGFBR2 gene: UMD-TGFBR2. Hum Mutat 2008;29:3–8. doi: 10.1002/humu.20602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.