

Abstract
BACKGROUND:
Autoimmune hemolytic anemias (AIHA) are characterized by the destruction of red cells following the production of autoantibodies directed against them. Although AIHA in children is usually self-limiting, many still succumb to the illness due to delay in the diagnosis and treatment. AIHA in children may be secondary to autoimmune diseases, drugs, or immune deficiencies. Early diagnosis and appropriate immunohematological evaluation can aid in the diagnosis and treatment.
OBJECTIVE:
To analyze the evaluation, treatment, and outcome of AIHA in children.
METHODS:
Prospective data of patients aged 0–18 years diagnosed with AIHA between June 2017 and May 2019 were collected.
INTERVENTION:
Prednisolone was the first-line agent in all; second-line agents included cyclosporine and rituximab. Red cell transfusion was given in those with severe anemia with cardiac decompensation.
RESULTS:
Eleven patients were diagnosed during the study period. Hemoglobin ranged from 1.2 to 9 g/dl. The initial presentation was severe anemia in 8 children and moderate anemia with thrombocytopenia in 3. The trigger was infection in 5. Polyspecific direct coomb's test (DCT) was positive in 10 patients. 2/10 polyspecific DCT-positive cases on further evaluation had immunoglobulin G (IgG) and C3d positivity, whereas rest 8 had only IgG. One infant was diagnosed with DCT-negative immunoglobulin A-mediated AIHA. 4/11 attained remission following the short course of prednisolone. Cyclosporine was used as the second-line agent in 2 and rituximab was used in 3. Seven children are in sustained remission and off medication. One died within 12 h of diagnosis.
CONCLUSION:
AIHA is not an uncommon problem in children and can vary in its clinical severity. Early and correct diagnosis helps in deciding appropriate treatment.
Keywords: Autoimmune, direct coomb's test, hemolytic anemia
Introduction
Autoimmune hemolytic anemias (AIHA) are characterized by the production of antibodies directed against one's own red blood cells (RBCs), resulting in their destruction by mononuclear phagocytic or complement system. AIHA observed in pediatrics is usually self-limiting and often precipitated by viral infections. The annual incidence in children is reported to be 1–3 cases/100,000 patients.[1,2] AIHA can be primary (37%; no underlying predisposition) or secondary (63%; due to autoimmune diseases, drugs, infections or underlying primary immunedeficiencies).[3] AIHA based on type and thermal properties (serological) of the auto-antibody attached to red cells can be warmreactive, paroxysmal cold hemoglobinuria, and cold agglutinin disease. In the present study, we describe the clinicopathological characteristics and treatment outcome of a series of children diagnosed with AIHA at our center.
Methods
Children aged 0–18 years, diagnosed with AIHA at a government pediatric institute in North India, over a period of 24 months (June 2017–May 2019) were included. Data regarding clinical presentation, evaluation, and response to treatment were collected prospectively. AIHA was diagnosed with anemia (<−2 standard deviation for age) with positive direct coomb's test (DCT) associated with corroborative evidence of hemolysis such as indirect hyperbilirubinemia (total bilirubin >2 mg/dl), raised lactate dehydrogenase (LDH) (>500 IU/ml), raised reticulocyte count, or red cell agglutination or spherocytosis on peripheral smear. Warm type AIHA was confirmed when the presence of immunoglobulin G (IgG) was established by monospecific DCT evaluation. Cold agglutinin was suspected in those with cold reactive IgM antibodies.
Extended immunohematology evaluation was done in all using Ethylene Diamine Tetra Acetic acid (EDTA) sample to establish the type of AIHA as well as the severity of hemolysis based on the type of immunoglobulin (Ig) attached to red cells. Positive or negative polyspecific DCT screening was followed by a monospecific DCT, identifying the type of IgG or immunoglobulin M (IgM) or immunoglobulin A (IgA) and complement (C3c or C3d) coating the red cells. In case of positive IgG, further assessment was done to determine the subclass of IgG (IgG1 or IgG3) [BioRad DCT IgG1/IgG3 cards] as well as the number of IgG subclass molecule attached DCT dilution cards].
Cold acid elution was done on DCT-positive red cells as per the standard technique.[4] The eluate was tested with cell panel using the gel cards to identify the antibody specificity. The specificity of the antibody was assessed for all patients by antibody screening using BioRad gel cards and panel cells. Evaluation for the secondary causes was done in those who were steroid dependent or refractory to treatment at the end of 3 months. ANA, dsDNA, C3, C4, and thyroid function were performed to rule out autoimmune diathesis. Infection screen was performed in cold reactive autoantibody for mycoplasma and Epstein–Barr virus (EBV) infections. Viral markers for HIV, hepatitis B and C were done in all. Basic primary immunodeficiency evaluation included IgG, IgA, IgM, lymphocyte subsets (T/B/natural killer [NK] cells), and double negative subsets when indicated. Prednisolone at 2 mg/kg/day in divided doses was given as the first line in all except in one case where methylprednisolone was used; second-line agents included rituximab, cyclosporine, mycophenolate mofetil, and mercaptopurine. Least incompatible red cell transfusion was given in those with severe anemia with cardiac decompensation. Folic acid supplementation was continued in all patients till sustained remission for more than 6 months.
Results
Eleven patients (5:6 male:female) with AIHA were consecutively diagnosed during the study period. Age at the onset of illness ranged from 10 months to 14 years. Four out of 10 patients had a history of blood transfusion before visit to our hospital. Hemoglobin (Hb) at presentation ranged from 1.2 to 9 g/dl. The initial presentation was severe anemia (Hb <7 g/dl) in 8 children and mild-moderate anemia with thrombocytopenia (Evan's syndrome [ES]) in 3. Dark-colored urine was noted in three patients only. Antecedent fever or evidence of infection (respiratory illness in 4 and urinary tract infection in 1) was present in 5 children. No significant correlation was noted with the presence of jaundice, serum bilirubin, or reticulocyte count. Splenomegaly was seen in all. Secondary AIHA evaluation was done in all steroid dependent and relapsing patients. Two were diagnosed with probable systemic lupus erythematosus (positive antinuclear antibody); however, on follow-up till date, no other criterion has been positive. Evaluation for primary immune deficiency was done in 4; combined immunodeficiency was identified in 1. A longer follow-up of this cohort will perhaps identify the evolution of more secondary diagnoses[1] [Table 1].
Table 1.
Presentation and outcome of AIHA patients
| Age at onset (years) | Sex | Presentation | Comorbidities | Hemoglobin (g/dl) | Platelet count | Polyspecific DCT | Monospecific DCT | Treatment | Outcome | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Anti IgG | Anti C3d | Anti IgA | |||||||||
| 1 | Female | Insidious | Infection (UTI) | 3 | Normal | 2+ | + | - | - | Prednisolone | Partial remission Prednisolone |
| 7 | Female | Acute | ITP | 7 | <10,000/µL | 2+ | + (IgG1) | - | - | Prednisolone Rituximab | Remission (6 months) |
| 13 | Female | Insidious | Probable lupus | 7.5 | Normal | 1+ | + | - | - | Prednisolone | Remission (18 months) |
| 6 | Male | Acute | CMV (PID ruled out) | 2.5 | Normal | 4+ | + (IgG1, IgG3) | - | - | Prednisolone Rituximab | Remission (12 months) |
| 14 | Male | Insidious | PID (combined immunodeficiency) | 5 | Normal | 3+ | + (IgG1, IgG3) | - | - | Prednisolone MMF 6 MP Cyclosporine | Partial remission on cyclosporine |
| 13 | Female | Insidious | ITP Probable lupus | 7.8 | 20,000/µL | 1+ | + | - | - | Prednisolone | Partial remission Prednisolone |
| 1.3 | Male | Acute | Infection | 3.5 | Normal | 4+ | + (IgG1, IgG3) | + | - | Prednisolone Rituximab | Remission (7 months) |
| 10 | Male | Insidious | ITP | 9 | 45,000/µL | 4+ | + | - | - | Prednisolone | Remission (14 months) |
| 2 | Female | Acute | Nil | 5 | Normal | 3+ | + | - | - | Prednisolone | Remission (9 months) |
| 0.8 | Male | Acute | Infection | 5.3 | Normal | - | Weak | - | 4+ | Prednisolone | Remission (8 months) |
| 1 | Female | Acute | Infection | 1.2 | Normal | 4+ | + | + | - | Methyl prednisolone IVIG | Died |
IgG subclass (IgG1/IgG3) screening can only be done in cases where initial titer of IgG (total) is more than 1:300 which was only in 4 cases. DCT=Direct coomb’s test, UTP=Urinary tract infection, ITP=Immunethrombocytopenia, CMV=Cytomegalovirus, PID=Pelvic inflammatory disease, IVIG=Intravenous immunoglobulin, MP=Methylprednisolone, MMF=Mycophenolate mofetil
Polyspecific DCT was positive in 10 out of 11 patients. One infant had DCT-negative IgA-mediated AIHA. Two out of 10 polyspecific DCT positive cases had IgG and C3d positivity on monospecific DCT testing, whereas rest 8 had only IgG coating the red cells. DCT titration was more than 1:300 in four patients, where only 1 of these four patients had both IgG1 and IgG3 coating and rest 3 had only IgG1. DCT was strongly positive in all patients with severe anemia (8/11). Alloantibody screen was negative in all except one. Specificity of autoantibody was found only in one case, which was against Rh blood group antigen (anti E).
Ten children received prednisolone at 2 mg/kg/day as the primary treatment. One child who was ventilated in view of cardiac decompensation (respiratory distress, hepatomegaly, and tachycardia) received a single dose of methylprednisolone. Intravenous immunoglobulin (IVIG) was also tried in this child at 1 g/kg dose. However, this child died within 12 h of hospital admission. Good treatment response to prednisolone was noted in all the other children. Three children attained remission (stable Hb >11 g/dl with no need for transfusion support) following which, prednisolone was tapered and stopped over the next 4–6 weeks. A drop in Hb was noted in four patients while tapering steroids. In those who were steroid dependent, the lowest steroid dose was identified and continued. Cyclosporine at 5–6 mg/kg/day in two divided doses orally was used as the second-line agent in 2 and Rituximab@375 mg/m2/dose weekly for 4 doses was used in 3. Rituximab was tolerated well with no infective or other adverse events. Screening for infections such as hepatitis B, C and tuberculosis was done before starting rituximab. Seven children are in sustained remission off any medication, whereas the rest are on low dose steroids with/without cyclosporine. DCT titers correlate with treatment response in majority. DCT continues to be positive in one child postrituximab despite being in clinical remission for over 12 months.
Discussion
Autoimmune hemolytic anemia is a relatively uncommon immune disorder resulting from the destruction of red cells following autoantibodies being produced against the red cell antigens. Most children present to emergency or intensive care, and the need for correct diagnosis and early initiation of treatment cannot be overemphasized. Based on the characteristics of the antibody, AIHA may be divided into warm reactive, cold hemagglutinin, mixed, and paroxysmal cold hemoglobinuria. In children, they present with sudden onset pallor associated with the features of cardiac decompensation. As observed in our series, evidence of “hemolysis” such as dark urine and jaundice need not be present in all. AIHA is primary when there is no underlying condition and secondary to an infectious, neoplastic or immune dysregulation in 63% of cases.[5] In our series, all patients had warm AIHA with no evidence of cold reactive AIHA (IgM antibody) or paroxysmal cold hemoglobinuria (DCT neg/C3d+, IgG).[5]
One patient in our series had DCT negative AIHA. This can occur in case of low density of IgG, low avidity of IgG, NK cell-mediated hemolysis and another antibody such as IgA, IgM or a different allotype of IgG which is not detected by polyclonal DCT.[8] Unless tested further with a high index of clinical suspicion, they can be missed and mis-labeled. In our patient, IgA autoantibody was attached to the red cells in a high number with a strong (4+) reaction on monospecific DCT gel cards, and the patient was managed with a short course of prednisolone.
Conclusion
The clinical presentation was found to predict treatment response. As discussed in immune thrombocytopenia (ITP), two distinct subgroups of patients are seen in AIHA also. Younger patients presented acutely (median age of 1.6 years in acute presentation vs. 13 years in insidious presentation). An acute transient presentation is usually seen in children <4 years where almost all entered remission and are off treatment. Among those who presented acutely (n = 6), all are in sustained remission at a median follow-up of 8 months except for one child who was brought with Hb of 1.2 g/dl and died of cardiac failure despite aggressive treatment. Among insidious presenters (n = 5), 3 are in partial remission on low dose prednisolone or cyclosporine. Immunohematological evaluation has been known to help both in treatment and prognostication. The presence of Ig+/C3d has a poor prognosis, whereas isolated C3d+ DCT has a better prognosis. We did not have any patient with isolated C3d+ status on immunohematological evaluation.
AIHA in association with auto-ITP or neutropenia is defined as ES. This distinct subtype named AIHA/ES has been described in 37% of the largest published childhood AIHA cohort. AIHA/ES is known to have high level of steroid resistance, longer treatment duration, and adverse immunological profile.
Underlying hereditary predisposition for immune cytopenias has been suggested in recent literature.[16] Primary immune deficiency evaluation was done in four of our patients. One patient was diagnosed to have a novel combined immunodeficiency involving the Janus Kinase 3 (JAK3) pathway.
Glucocorticoids were used as the first-line agent in all children. IVIG was used only in one patient at 1 g/kg dose (patient 11) in view of the critical presentation. It has been noted that the cardiac adaptation in children to severe anemia is remarkably better than adults.[1] This was observed in our series as well and only this child succumbed to severe anemia, mostly as a result of delayed presentation to the hospital rather than poor response to treatment. Mortality in pediatric AIHA has been reported in 11%–32%.[1] In all the rest, prednisolone alone in association with emergent monitoring, red cell transfusions, and other supportive care was able to result in stabilization of the condition. Least incompatible packed red cells were transfused in those with cardiac decompensation under the cover of steroids with monitoring for intravascular haemolysis and renal dysfunction. Prednisolone tapering was done slowly, and in those who achieved clinical remission, it was stopped after 6 weeks. It has been observed that 70%–80% of patients with warm AIHA respond to the first-line agent alone.[6] In our series, almost all patients responded to prednisolone; however, sustained remission was achieved with prednisolone only in 40%. In those who had a fall in Hb on attempted taper, the lowest dose of prednisolone was identified, and second-line agents were considered. Partial response was defined as improvement of Hb >2 g/dl from baseline but failure to achieve normal range. In previous studies, 20%–45% of patients with AIHA have required a second-line agent. In our series, 4/10 patients needed a second-line agent to attain remission. These patients also had high titer (>1:300) of IgG immunoglobulins attached to the RBCs. Rituximab was used in 3 of our patients after having had 2, 3, and 1 relapses [patient 2,4 and 7; Table 1] while on steroids alone. No significant adverse events were observed with rituximab, and sustained remission off all medications has been noted for 6, 12, and 7 months, respectively. Cyclosporine was used as a maintenance agent in one patient with unacceptable side effects of steroids. Mycophenolate and 6-mercaptopurine were used in the patient with combined immunodeficiency due to the difficulty in using rituximab which causes B-lymphocyte depletion; however, both agents were discontinued due to the lack of efficacy.[6,7] Bone marrow transplantation was advised in this child but has been deferred till date due to the lack of suitable donor.
In our experience, rituximab may be considered as a second-line agent in pediatric AIHA, after a thorough evaluation for secondary causes and infections considering its potential for cure.[13,14] This recommendation can also be for the cases that have higher titers of DCT positivity with IgG molecule. The association with the number and type of IgG subclass attached on RBCs to severity of presentation and need of initiate upfront second-line therapy cannot be commented upon due to the lack of any randomized controlled trial in this regard. Splenectomy was not considered in any patient of ours considering the long-term morbidity associated and the availability of better second-line agents.
Unlike established guidelines for the evaluation of AIHA, extensive investigations were not done in most of our patients due to resource constraints. The basic evaluation protocol followed at our center is as follows [Figure 1]:[5,6,7,8,9,10,11,12,13,14,15]
Figure 1.
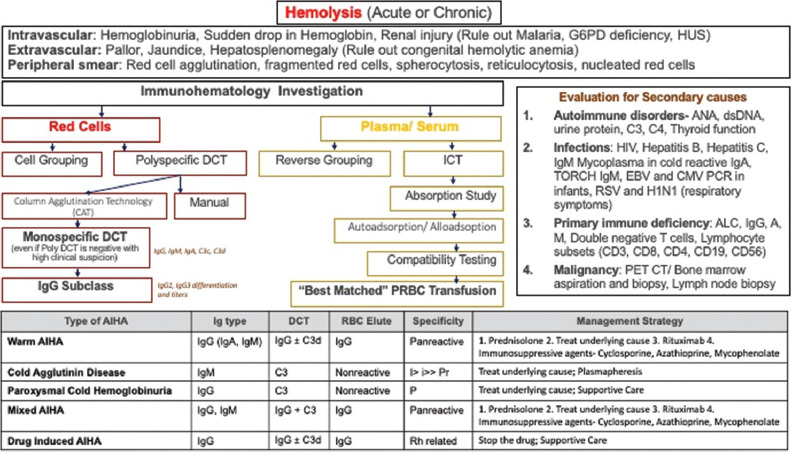
Flowchart of immunohematological workup for a suspected autoimmune hemolytic anemias in a child
Suspect hemolysis: Sudden drop in Hb often associated with cardiac decompensation, dark urine, raised bilirubin, raised reticulocyte count (may not be seen always), raised LDH, agglutination or spherocytosis on peripheral smear, raised mean corpuscular volume (due to agglutinated red cells or raised reticulocyte count), and hemoglobinuria (only in intravascular hemolysis)
Consider immune hemolysis: Positive polyspecific DCT
Type of immune hemolysis: Monospecific DCT in all cases of positive DCT and even in negative DCT with a high index of suspicion, Donath Landsteiner test in suspected paroxysmal cold hemoglobinuria
Look for secondary causes.
Autoimmune disorders: ANA, dsDNA, urine protein, C3, C4, and thyroid function.
Infections: HIV, hepatitis B, hepatitis C, IgM mycoplasma in cold reactive IgA, TORCH IgM, EBV and cytomegalovirus polymerase chain reaction in infants, respiratory syncytial virus, and H1N1 (respiratory symptoms).
Primary immunodeficiency, ALC, IgG, A, M, double negative T-cells, and lymphocyte subsets (CD3, CD8, CD4, CD19, and CD56).
We report this series in view of the rarity of this condition and the rarity of published literature in managing these patients. Although there are few case series reported globally and from India, a pediatric alone series with the use of advanced evaluation and advanced treatment strategies has not been reported so far.[9,10,11] Our series although small is well worked up and algorithmic treatment that is evidence based but adapted to resource-constrained settings has been used.
What is already known?
Autoimmune hemolytic anemia is a rare entity in children
Agents for treatment include prednisolone and other immunosuppressive agents.
What this study adds?
Glucocorticoids are efficacious for AIHA both for primary and secondary cases
Older age, associated thrombocytopenia, and secondary AIHA have refractory course
Unlike in ITP, identifying the characteristics of the autoantibody can aid in the treatment
Evaluation of the number of IgG molecule (titers) as well as type of IgG (IgG1/IgG3) molecule attached to the RBCs at the time of presentation can help in assessing the risk of hemolysis and possibility of need of a second-line of therapy with or without resistance to steroid alone.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Aladjidi N, Leverger G, Leblanc T, Picat MQ, Michel G, Bertrand Y, et al. New insights into childhood autoimmune hemolytic anemia: A French national observational study of 265 children. Haematologica. 2011;96:655–63. doi: 10.3324/haematol.2010.036053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ware RE, Rose WF. Autoimmune hemolytic anemia. In: Nathan DG, Orkin SH, editors. Nathan and Oski's Hematology of Infancy and Childhood. Philadelphia, PA: Saunders; 1998. pp. 499–522. [Google Scholar]
- 3.Vaglio S, Arista MC, Perrone MP, Tomei G, Testi AM, Coluzzi S, et al. Autoimmune hemolytic anemia in childhood: Serologic features in 100 cases. Transfusion. 2007;47:50–4. doi: 10.1111/j.1537-2995.2007.01062.x. [DOI] [PubMed] [Google Scholar]
- 4.Rekvig OP, Hannestad K. Acid elution of blood group antibodies from intact erythrocytes. Vox Sang. 1977;33:280–5. doi: 10.1111/j.1423-0410.1977.tb04476.x. [DOI] [PubMed] [Google Scholar]
- 5.Ladogana S, Maruzzi M, Samperi P, Perrotta S, Del Vecchio GC, Notarangelo LD, et al. Diagnosis and management of newly diagnosed childhood autoimmune hemolytic anemia. Recommendations from the red cell study groups of the pediatric haemato-oncology Italian association. Blood Transfus. 2017;15:259–67. doi: 10.2450/2016.0072-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ladogana S, Maruzzi M, Samperi P, Condorelli A, Casale M, Giordano P, et al. Second-line therapy in paediatric warm autoimmune haemolytic anaemia. Guidelines from the Associazione Italiana Onco-Ematologia Pediatrica (AIEOP) Blood Transfus. 2018;16:352–7. doi: 10.2450/2018.0024-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sobota A, Neufeld EJ, Lapsia S, Bennett CM. Response to mercaptopurine for refractory autoimmune cytopenias in children. Pediatr Blood Cancer. 2009;52:80–4. doi: 10.1002/pbc.21729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller J, Cai W, Andrews J, Narla A. A case series of pediatric patients with direct antiglobulin test negative autoimmune hemolytic anemia. Transfusion. 2019;59:2528–31. doi: 10.1111/trf.15350. [DOI] [PubMed] [Google Scholar]
- 9.Prabhu R, Bhaskaran R, Shenoy V, Rema G, Sidharthan N. Clinical characteristics and treatment outcomes of primary autoimmune hemolytic anemia: A single center study from South India. Blood Res. 2016;51:88–94. doi: 10.5045/br.2016.51.2.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naithani R, Agrawal N, Mahapatra M, Pati H, Kumar R, Choudhary VP. Autoimmune hemolytic anemia in India: Clinico-hematological spectrum of 79 cases. Hematology. 2006;11:73–6. doi: 10.1080/10245330500345587. [DOI] [PubMed] [Google Scholar]
- 11.Das SS, Nityanand S, Chaudhary R. Clinical and serological characterization of autoimmune hemolytic anemia in a tertiary care hospital in North India. Ann Hematol. 2009;88:727–32. doi: 10.1007/s00277-008-0674-6. [DOI] [PubMed] [Google Scholar]
- 12.Yaralı N, Bilir ÖA, Erdem AY, Çulha V, Kara A, Özbek N. Clinical features and treatment of primary autoimmune hemolytic anemia in childhood. Transfus Apher Sci. 2018;57:665–8. doi: 10.1016/j.transci.2018.07.014. [DOI] [PubMed] [Google Scholar]
- 13.Fan J, He H, Zhao W, Wang Y, Lu J, Li J, et al. Clinical features and treatment outcomes of childhood autoimmune haemolytic anemia: A retrospective analysis of 68 cases. J Pediatr Hematol Oncol. 2016;38:e50–5. doi: 10.1097/MPH.0000000000000476. [DOI] [PubMed] [Google Scholar]
- 14.Barcellini W, Zaja F, Zaninoni A, Imperiali FG, Battista ML, Di Bona E, et al. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: Clinical efficacy and biologic studies. Blood. 2012;119:3691–7. doi: 10.1182/blood-2011-06-363556. [DOI] [PubMed] [Google Scholar]
- 15.Hill QA, Stamps R, Massey E, Grainger JD, Provan D, Hill A, et al. The diagnosis and management of primary autoimmune haemolytic anaemia. Br J Haematol. 2017;176:395–411. doi: 10.1111/bjh.14478. [DOI] [PubMed] [Google Scholar]
- 16.Rotz SJ, Ware RE, Kumar A. Diagnosis and management of chronic and refractory immune cytopenias in children, adolescents, and young adults. Pediatr Blood Cancer. 2018;65:e27260. doi: 10.1002/pbc.27260. [DOI] [PubMed] [Google Scholar]