

Abstract
The bacterial communities of Caulerpa lentillifera were studied during an outbreak of an unknown disease in a sea grape farm from Vietnam. Clear differences between healthy and diseased cases were observed at the order, genus, and Operational Taxonomic Unit (OTU) level. A richer diversity was detected in the diseased thalli of C. lentillifera, as well as the dominance of the orders Flavobacteriales (phylum Bacteroidetes) and Phycisphaerales (Planctomycetes). Aquibacter, Winogradskyella, and other OTUs of the family Flavobacteriaceae were hypothesized as detrimental bacteria, this family comprises some well‐known seaweed pathogens. Phycisphaera together with other Planctomycetes and Woeseia were probably saprophytes of C. lentillifera. The Rhodobacteraceae and Rhodovulum dominated the bacterial community composition of healthy C. lentillifera. The likely beneficial role of Bradyrhizobium, Paracoccus, and Brevundimonas strains on nutrient cycling and phytohormone production was discussed. The bleaching of diseased C. lentillifera might not only be associated with pathogens but also with an oxidative response. This study offers pioneering insights on the co‐occurrence of C. lentillifera‐attached bacteria, potential detrimental or beneficial microbes, and a baseline for understanding the C. lentillifera holobiont. Further applied and basic research is urgently needed on C. lentillifera microbiome, shotgun metagenomic, metatranscriptomic, and metabolomic studies as well as bioactivity assays are recommended.
Keywords: 16S rRNA gene, aquaculture, green caviar, sea grape, seaweed, Vietnam
The co‐occurrence of bacteria in sea grape (green caviar) Caulerpa lentillifera under two health state conditions was studied. The role of potentially detrimental and beneficial bacteria was discussed. A baseline for understanding the C. lentillifera holobiont was presented.

1. INTRODUCTION
The sea grape or green caviar Caulerpa lentillifera J. Agardh 1837 (Ulvophyceae, Bryopsidales) is a green macroalga widely cultivated in tropical countries of the Indo‐Pacific region, and its market as a seafood delicacy has been expanded drastically in the last decades. Not only C. lentillifera unique texture, flavor, and appearance which let it resemble caviar (e.g., Ly et al., 2021), but also its elevated contents of polyunsaturated fatty acids, antioxidants, vitamins, and trace elements, comparatively higher than other edible macroalgae (de Gaillande et al., 2017; Paul et al., 2014; Saito et al., 2010; Yap et al., 2019), makes it a valuable functional food with exponentially increasing demand (Terada et al., 2021). Moreover, the combination of the high nutritional quality with the simple and low‐cost cultivation transform C. lentillifera into a promising candidate to enhance food security in tropical countries (de Gaillande et al., 2017; Stuthmann et al., 2021). However, one of the major factors contributing to the worldwide decline of macroalgal populations are microbial diseases (Egan et al., 2014) and C. lentillifera aquaculture is currently endangered by different outbreaks (Liang et al., 2019).
Macroalgae provide a nutrient‐rich niche for microbes and some bacteria are capable to invade algal tissue and causing disease by degrading complex polymers. Agarases, carrageenases, alginases, fucoidanases, fucanases, mannanases, cellulases, and pectinases are depolymerizing enzymes detected in several marine bacteria, responsible for the breakdown of algal cell walls (Goecke et al., 2010). Moreover, the low genetic diversity of cultivated algae and their tight distribution in aquaculture settings facilitate the rapid spread of detrimental microbes (Valero et al., 2017; Ward et al., 2020). Several bacterial diseases have been reported in commercial macroalgae such as the rote spot and hole‐rotten diseases caused by Pseudomonas spp. in Saccharina japonica, rotten thallus syndrome by Vibrio sp. in Gracilaria verrucosa, or Anaaki by Flavobacterium spp. in Pyropia yezoensis (reviewed by Egan et al., 2014; Goecke et al., 2010; Ward et al., 2020). Nevertheless, the role of microbes is not only detrimental but can also be beneficial.
Some bacteria are essential for algal health, growth, and development. Beneficial bacteria attached to macroalgae induce morphogenesis, fix nitrogen, produce phytohormones, release antifouling and antimicrobial compounds, transport metabolites and nutrients, detoxify pollutants, contribute to algal reproduction, and are key for their adaptation to new environments (Aires et al., 2013; Hollants et al., 2013; R. P. Singh & Reddy, 2014). The relationship between macroalgae and bacteria is generally mutualistic, and bacteria are mainly benefited from organic substrates for their metabolism and a stable microenvironment protected from predators and changing environmental conditions. The relationship between macroalgae and bacterial communities is so tight and reciprocal that the whole entity is considered a holobiont (Arnaud‐Haond et al., 2017; Califano et al., 2020; Egan et al., 2013). Furthermore, the bioactive compounds produced by microbes in the holobiont have an enormous potential for drug discovery and biotechnological applications (Friedrich, 2012; Luyen et al., 2019).
Despite the commercial importance of C. lentillifera and its relevance for human nutrition under future climate‐driven and overpopulation scenarios, studies about their microbiome are very limited. Liang et al. (2019) described a disease in C. lentillifera in Chinese aquaculture systems characterized by a dark‐green biofouling, pink‐colored and missing ramuli. In this particular case, Bacteroidetes and Cyanobacteria dominated the bacterial communities of diseased C. lentillifera. Although recent advances in the study of the macroalgal microbiome using next‐generation sequencing (NGS) techniques, the information is still limited and in several cases derived from culture methods with their intrinsic limitations (Friedrich, 2012). The 16S ribosomal RNA (rRNA) gene approach offers broader insights into the natural composition of bacterial communities attached to algae and plays an important role in understanding the holobiont during disease (Egan et al., 2014; Kopprio et al., 2021). The aims of this study were: (1) to characterize the bacterial communities of C. lentillifera in a Vietnamese sea grape farm and (2) to assess the bacterial community composition under two different health conditions. We hypothesize a higher diversity and a dominance of Bacteroidetes in the bacterial communities of diseased C. lentillifera.
2. METHODS
2.1. Culture conditions, sampling, and disease symptoms
C. lentillifera thalli were sampled in the facilities of the company VIJA at Van Phong Bay (N 12° 35′ 17.67″, E 109°13′ 39.76″) in the central‐eastern coastline of Vietnam. Water temperature in the ponds reached ∼31°C in May, with pH values of ∼8.1, conductivity ∼53 mS cm−1, and dissolved oxygen ∼5.6 mg L−1. The sea grapes were cultivated in ponds, which were used previously for shrimp farming and then placed for cleaning in tanks a few days before processing. The ponds and tanks were covered with shade cloths to avoid direct sunlight. Six healthy and seven diseased thalli of C. lentillifera from different tanks, originally harvested from the same pond, were collected during an outbreak of an unknown disease. In contrast to the bright green color of healthy individuals, the diseased C. lentillifera showed a white biofilm on the external circumference of the vesicular ramuli (Figure 1, Arrow A) followed by a marked tissue discoloration or bleaching (Figure 1, Arrow B). After the described symptoms, the diseased algae lost their turgid appearance and ramuli, and thereafter decayed (Figure 1, Arrow C).
Figure 1.

Diseased Caulerpa lentillifera from the Vietnamese aquaculture. (A) White biofilm on the external circumference of the ramuli. (B) Bleaching of the ramuli. (C) Bleaching, shrinking, and loss of ramuli
2.2. DNA extraction and amplification
C. lentillifera thalli were collected in sterile vials and preserved at −20°C until further analysis. Within a few days, the samples were transported frozen and DNA was extracted according to Griffiths et al. (2000) under the laboratory conditions of the Leibniz Centre for Tropical Marine Research. For every sample, ∼10 ramuli together with the supporting rachis were immersed in 600 µl of hexadecyltrimethylammonium bromide with 60 µl of 10% sodium dodecyl sulfate and 60 µl of 10% N‐lauroylsarcosine. Glass beads were added and the tissue was homogenized in a FastPrep at 5 m s−1. Homogenates were rinsed with 600 µl of phenol–chloroform–isoamyl alcohol (25:24:1) and centrifuged at 16,000g and 4°C for 10 min. The upper aqueous layer was transferred to a clean reaction tube, mixed with two volumes of 30% polyethylene glycol 6000 and 1.6 M NaCl, and incubated at 4°C for 120 min. Subsequently, the sample was centrifuged at 17,000g and 4°C for 90 min, the supernatant was carefully removed and the pellet was washed with ice‐cold 70% ethanol. The pellet was air‐dry at 37°C for a few minutes, dissolved in 20 μl polymerase chain reaction grade water, and stored at −20°C. The hypervariable region V3‐V4 of the 16S rRNA gene was amplified with the set of primers according to Klindworth et al. (2013): Bact‐341F (5ʹ−3ʹ: CCT ACG GGN GGC WGC AG) and Bact‐785R (GAC TAC HVG GGT ATC TAA KCC)
2.3. Sequencing, bioinformatics, and statistical analysis
The amplicon V3‐V4 of the 16S rRNA gene was sequenced with a 2 × 300‐bp paired‐end run on an Illumina MiSeq platform. Sequencing, removal of primer sequences, and demultiplexing were conducted by the company LGC genomics. Sequences were trimmed and merged using Trimmomatic v0.36 (Bolger et al., 2014) and PEAR v0.9.8 (J. Zhang et al., 2014), respectively. A total of ∼373,000 reads with a mean of 28,700 reads per sample were detected after primer removal, and a total of ~272,000 reads with a mean of 20,900 reads remained after merging. Operational Taxonomic Units (OTUs) were clustered by minimum entropy decomposition (MED) MED v2.1 (Eren et al., 2015) and their representatives were submitted to SilvaNGS (v132; https://ngs.arb-silva.de/silvangs/) using a sequence similarity of one for clustering and the remaining variables as default. Singleton, doubleton, and sequences from mitochondria, chloroplasts, and archaea were removed from the analysis. Demultiplexed and primer‐clipped sequences were deposited at the European Nucleotide Archive using the data brokerage service of the German Federation for Biological Data (Diepenbroek et al., 2014) with the number PRJEB42826, in compliance with the minimal information about any (x) sequence standard (Yilmaz et al., 2011).
Pooling of taxa, removal of OTUs with poor alignment quality, and relative sequence abundance and diversity calculations were conducted in R v4.0.5 (R Core Team, 2021) and additional packages such as vegan (Oksanen et al., 2019) and iNEXT (Hsieh et al., 2019). The rarefaction curves were performed with iNEXT, reached a plateau and the mean number of reads per sample used for statistical analyses was ∼17,000. The diversity indexes: Chao, Shannon, and Inverse Simpson were calculated with iNEXT. Relative sequence abundances of the main orders, genera, and diversity indexes between the diseased and healthy cases were compared with a Mann–Whitney test. Data were transformed (x = log [(OTU number + OTU meansample)/OTU meansample]), a Bray Curtis similarity matrix was calculated, and differences between the states at order and OTU level were evaluated using permutational multivariate analysis of variance (PERMANOVA). In case of significant differences, similarity percentage analysis (SIMPER) was performed to detect the main taxa contributing to dissimilarities. Samples at the OTU level were ordinated by nonmetric multidimensional scaling (NMDS). A heat map based on the 60 most abundant OTUs, covering 64 ± 11% of the total relative sequence abundance, was performed with the software XLSTAT‐Ecology (Addinsoft, 2018) using the default parameters. The dendrogram at the top of the heat map indicated similarity between samples, while the dendrogram on the left side showed similarity between OTUs. Graphics and statistics were performed with R 4.0.5, Xact 7.21, PRIMER v6 + PERMANOVA, and XLSTAT‐Ecology.
3. RESULTS
After data curation, the means of the total OTU number per sample were 12,200 for the healthy cases and 21,300 for the diseased cases. A total of 810 unique OTUs were detected in C. lentillifera, from which 400 were shared between both cases and 380 were exclusively found in diseased C. lentillifera. The order Rhodobacterales (class Alphaproteobacteria) and its genus Rhodovulum presented the highest relative sequence abundance in C. lentillifera at each respective taxonomic level (Figure 2). The Rhodobacteraceae was the only family detected within the order Rhodobacterales. The mean relative abundance of Clostridiales (phylum Firmicutes) was approximately three times significantly higher in healthy C. lentillifera as confirmed by the Mann–Whitney test (Figure 2). The mean abundance of Flavobacteriales of the phylum Bacteroidetes was about two times higher in diseased C. lentillifera and significant differences were observed. The family Flavobacteriaceae dominated the order Flavobacteriales and comprised 99.7% of the OTUs for this order. The order Phycisphaerales (phylum Planctomycetes) and Steroidobacterales (class Gammaproteobacteria) characterized diseased C. lentillifera thalli. The general trend observed at order level was similar at genus level: The relative sequence abundance of Clostridium sensu stricto 7 (Clostridiales) was also three times higher in healthy than in diseased cases, while Aquibacter and Flavobacteriaceae unclassified (Flavobacteriales), Phycisphaera (Phycisphaerales) and Woeseia (Steroidobacterales) were typical of diseased cases. Without significant differences, other abundant genera were Rhodovulum, Cutibacterium, and Paracocccus for healthy C. lentillifera, while Thalassobius, Tropicibacter, and Blastopirellula for diseased C. lentillifera.
Figure 2.

Mean relative sequence abundance (±standard deviation) of the main bacterial orders and genera in healthy and diseased individuals of the sea grape Caulerpa lentillifera. *Significant differences at p < 0.05 according to the Mann–Whitney test. Caption superscript and color show correspondence between order and genus (e.g., Rho = Rhodobacterales). Aphaproteobact., Alphaproteobacteria; Cand. Kaiserbact., Candidatus Kaiserbacteria; Clostr., Clostridium; Flavobact., Flavobacteriaceae; mar. gr., marine group; Methyloligel., Methyloligellaceae; uncl., unclassified
PERMANOVA revealed significant differences between the healthy and diseased cases at the order level (Pseudo‐F = 3.89, p = 0.016). According to SIMPER analyses, the total dissimilarity between healthy and diseased cases was 41.3%. The orders which presented significant differences in the Mann–Whitney test were also important contributors to the dissimilarities between healthy and diseased thalli (Table 1). Other relevant orders with higher percentages of dissimilarities but without significant differences were Propionibacteriales and Betaproteobacteriales for the healthy cases, while Saccharimonadales and Verrucomicrobiales were relevant for the diseased cases. Furthermore, PERMANOVA detected significant differences at the OTU level between healthy and diseased C. lentillifera (Pseudo‐F = 3.12, p = 0.015). According to SIMPER analysis, the total dissimilarity at OTU level was 71.3% and the main OTUs contributing to this value are detailed in Table 1.
Table 1.
Principal orders and OTUs in the sea grape Caulerpa lentillifera contributing to the highest dissimilarity values (41% and 71% of the total dissimilarity, respectively) according to SIMPER analyses
| Healthy | Diseased | |||
|---|---|---|---|---|
| Taxa | % | Taxa | % | |
| Orders | ClostridialesCl | 5.3 | PhycisphaeralesPh | 4.4 |
| PropionibacterialesPr | 4.0 | Saccharimonadales | 3.3 | |
| BetaproteobacterialesBe | 3.1 | FlavobacterialesFl | 3.2 | |
| MicrococcalesMc | 2.6 | VerrucomicrobialesVe | 2.8 | |
| EnterobacterialesEn | 2.5 | PirellulalesPi | 2.6 | |
| MicrotrichalesMt | 2.4 | PlanctomycetalesPl | 2.6 | |
| BacillalesBa | 2.2 | CaulobacteralesCa | 2.3 | |
| ChitinophagalesCh | 1.7 | SteroidobacteralesSt | 2.3 | |
| OTUs | Paracoccus 2980 | 0.6 | Phycisphaera 3117Ph | 0.6 |
| Spongiimonas 0219Fl | 0.5 | Methyloligellaceae uncl. 0867 | 0.5 | |
| Clostridium sensu stricto 7 4975Cl | 0.5 | Thalassobius 2935 | 0.5 | |
| Cutibacterium 2507Pr | 0.5 | Pir4 lineage 0189Pi | 0.5 | |
| Aliihoeflea 1742 | 0.5 | Flavobacteriaceae uncl. 2304Fl | 0.5 | |
| Pelomonas 1964Be | 0.5 | Aquibacter 3662Fl | 0.5 | |
| Brevundimonas 1698Ca | 0.5 | JGI 069‐P22 0837 uncl. | 0.5 | |
| Bradyrhizobium 1899 | 0.4 | Thalassobius 1635 | 0.5 | |
| Proteus 2600En | 0.4 | Winogradskyella 2272Fl | 0.5 | |
| Paracoccus 4667 | 0.4 | Rubritalea 2117Ve | 0.5 | |
| Clostridium sensu stricto 7 1787Cl | 0.4 | Aureicoccus 3615Fl | 0.5 | |
| Micrococcus 3447Mc | 0.4 | Woeseia 0634St | 0.4 | |
| Rubritalea 3432Ve | 0.4 | Blastopirellula 1161Pi | 0.4 | |
| Woeseia 3512St | 0.4 | Coraliomargarita 3905 | 0.4 | |
| Suttonella 1166 | 0.4 | Psychrobacter 1144 | 0.4 | |
| Afipia 1915 | 0.4 | Cyclobacteriaceae uncl. 1240 | 0.4 | |
| Ralstonia 0332Be | 0.3 | Ulvibacter 0198Fl | 0.4 | |
| Escherichia‐Shigella 2098En | 0.3 | Alphaproteobacteria uncl.1689 | 0.4 | |
| Staphylococcus 0913Ba | 0.3 | Sva0996 1388Mt | 0.4 | |
| Kocuria 3438Mc | 0.3 | Aquibacter 2359Fl | 0.4 | |
| Enhydrobacter 0637 | 0.3 | Blastopirellula 3839Pi | 0.4 | |
| Methyloligellaceae uncl. 3194 | 0.3 | Pir4 lineage 0659Pi | 0.4 | |
| Vibrionimonas 3429Ch | 0.3 | Thalassobius 4280 | 0.4 | |
| Blastopirellula 3811Pi | 0.3 | Phycisphaera 3114Ph | 0.4 | |
Note: Taxa are grouped based on their dominance in healthy or diseased cases. Caption superscript: indicates correspondence between order and genus (e.g., Cl = Clostridiales). The number after each genus refers to the sequence number or MED node.
Abbreviations: OTU, Operational Taxonomic Unit; uncl., unclassified.
The OTUs of order Clostridiales, typical of healthy C. lentillifera, were Clostridium sensu stricto 7 sq (sequence number or MED node) 4975 and sq 1787 (Table 1). Other OTUs dominating with elevated percentages of dissimilarity in the healthy cases were those of the genera: Paracoccus, Spongiimonas, Cutibacterium, Aliihoeflea, Pelomonas, Brevundimonas, and Bradyrhizobium. In the case of diseased cases, the major OTUs were those of Flavobacteriaceae unidentified, Aquibacter, Winogradskyella, Aureicoccus, and Ulvibacter for the order Flavobacteriales (all belonging to the family Flavobacteriaceae), Phycisphaera for the order Phycisphaerales and Woeseia for the order Steroidobacterales. Other OTUs with elevated percentages of dissimilarity in the diseased cases were Methyloligellaceae unclassified sq 0867, Thalassobius sq 2935, Pir4 lineage sq 0189, and JGI 069‐P22 unclassified sq 0837. A list with other relevant OTUs according to their dissimilarity is presented in Table 1. Moreover, the NMDS ordination of C. lentillifera samples at the OTU level (Figure 3) suggested contrasting bacterial communities between healthy and diseased cases.
Figure 3.

Nonmetric multidimensional scaling ordination of healthy and diseased individuals of the sea grape Caulerpa lentillifera at Operational Taxonomic Unit level
The heat map (Figure 4) showed a similar cluster of four diseased C. lentillifera, while a variable ordination of the other cases. OTUs associated exclusively with diseased status were Aquibacter sq 3662, Flavobacteriaceae unclassified sq 2304, Woeseia sq 0634, Phycisphaera sq 3117, and Muricauda sq 2354; while those exclusively detected in the healthy status were Bradyrhizobium sq 1899, Paracoccus sq 4667, and Paracoccus sq 2980. OTUs of the genera JGI 069‐P22 unclassified, Thalassobius, Saccharimonadales unclassified, Flavobacteriaceae unclassified, Winogradskyella, Aureicoccus, Pir4 lineage, Tropicibacter, Algisphaera, Cellvibrionaceae unclassified, and Aquibacter dominated mainly in the diseased cases. Other OTUs which dominated but not exclusively in the healthy cases are displayed in the heat map. The box plots of the diversity indexes (Figure 5) evidenced higher values in diseased C. lentillifera and significant differences according to Mann–Whitney comparisons.
Figure 4.

Heatmap of the main Operational Taxonomic Units on healthy (H) and diseased (D) individuals of the sea grape Caulerpa lentillifera. (*) Unclassified (**) sensu stricto 7. The number after each genus refers to the sequence number or minimum entropy decomposition node
Figure 5.
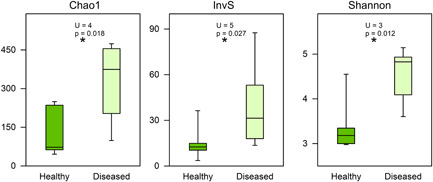
Diversity comparisons between healthy and diseased states of Caulerpa lentillifera according to the Chao1, Inverse Simpson (InvS), and Shannon indexes. *Significant differences at p < 0.05 according to the Mann–Whitney test
4. DISCUSSION
4.1. Main composition
The bacterial communities of C. lentillifera presented a similar composition as other green algae at higher taxonomic levels. The dominance of Alphaproteobacteria and Bacteroidetes has been reported in green macroalgae and some Caulerpa species (Aires et al., 2013; Friedrich, 2012; Singh & Reddy, 2014). The order Rhodobacterales (Alphaproteobacteria) and family Rhodobacteraceae dominated the bacterial communities of C. lentillifera in this particular case from the Vietnamese aquaculture. The bacterial communities at lower taxonomic levels are generally highly variable in seaweeds and their composition may be associated with a particular function within the holobiont. The microbiome composition depends generally on the geographic location, environmental factors, seaweed tissues, developmental stage, and even among local individuals (reviewed by Friedrich, 2012). Burke et al. (2011) explain the pattern of bacterial colonization on green macroalgae with the competitive lottery model, according to this model different species with similar functional traits can occupy the same niche depending on a stochastic probability. The highest relative abundance of Rhodobacterales coincided with the results obtained in C. lentillifera from a study case in China (Liang et al., 2019); nevertheless, one of the differences was in the dominant genus within Rhodobacterales: Leisingera in contrast to Rhodovulum in our study.
4.2. Potential detrimental bacteria
The pattern of higher diversity in diseased C. lentillifera was also observed in a case study from China (Liang et al., 2019), where the spherical ramuli turned pink‐red, detached, and decayed, and the orders Flavobacteriales (Bacteroidetes), Phycisphaerales, and Cellvibrionales dominated the thalli of diseased C. lentillifera (Liang et al., 2019). With a different coloration, a similar pattern was observed in our study: Flavobacteriales, Phycisphaerales, and an unidentified genus of the family Cellvibrionaceae were typical of diseased C. lentillifera. The family Flavobacteriaceae has the potential to break down algal walls, invade tissues, and cause disease under certain conditions. Bacteroidetes and particularly Flavobacteriaceae are degraders of complex biopolymers like sulfated polysaccharides from algal tissues (Jain et al., 2019; Yilmaz et al., 2016).
Many members of the Flavobacteriaceae have been confirmed or suspected as causative agents of seaweed diseases (Goecke et al., 2010; Kumar et al., 2016; Ward et al., 2020). In our particular case, we hypothesize as potential detrimental bacteria, strains of Aquibacter, Winogradskyella, Aureicoccus, Ulvibacter, and Muricauda. For example, Winogradskyella species have been suggested as detrimental bacteria on P. yezoensis with yellow spot disease (Liu et al., 2020) and on Delisea pulchra with a bleaching disease (Kumar et al., 2016). The bleaching and white coloration symptom in C. lentillifera may not only be linked to a pathogen invasion but also an excessive oxidative burst response. According to Box et al. (2008), some Caulerpa species produce reactive oxygen species (ROS) against invasive organisms and if the ROS production is excessive, a situation of oxidative stress appears in the macroalga.
We hypothesize that Phycisphaera and Algisphaera spp. of the order Phycisphaerales were opportunistic pathogens or saprophytes of C. lentillifera. As mentioned before, the order Phycisphaerales was typical of the disease cases from the Chinese aquaculture, and strains of these genera were isolated from marine macroalgae presenting agarolitic activity (Fukunaga et al., 2009; Yoon et al., 2014). Nevertheless, Planctomycetes are widespread in the epiphytic microbial community of macroalgae, possess potential beneficial effects for their host like the production of bioactive compounds, and are rarely identified as seaweed pathogens (Lage & Bondoso, 2014). Some strains of Phycisphaera, Algisphaera, Pir4 lineage, and Blastopirellula may be using their higher number of sulfatases for the degradation of algal polysaccharides (Bondoso et al., 2017) from the dead or senescent tissue of C. lentillifera, which could be injured by other pathogen or degraded because of excessive production of ROS.
Woeseia species of the order Steroidobacterales can utilize a broad range of energy‐yielding metabolisms and substrates (Mußmann et al., 2017) and some strains may be saprophytes in C. lentillifera. Other bacteria may be specialized in small molecules derived from the breakdown of algal polymers and benefited from the degradation of algal tissue. Members of the family Methyloligellaceae oxidize generally one carbon atom molecules (Walker et al., 2021) and Patescibacteria (JGI 069‐P22 and some Saccharimonadales in this study) may be using monosaccharides because they have reduced genes for polysaccharide catabolism in their simple genomes (Tian et al., 2020).
The classification of potential detrimental bacteria at higher taxonomic levels in C. lentillifera is not clear. The order Verrucomicrobiales is well‐known as active polysaccharide degraders (Martinez‐Garcia et al., 2012) but only a few genera were linked to algal disease. For example, several OTUs of Rubritalea within this order were typical of S. japonica with symptoms of rotten hole disease (R. Zhang et al., 2020). Furthermore, the ability to metabolize algal compounds of Rhodobacteraceae in a mutualistic relationship with seaweeds may change to detrimental with some members of this family such as Thalassobius and Tropicibacter. Thalassobius spp. characterized diseased Delisea pulchra (Kumar et al., 2016) and were responsible for the direct lysis of red‐tide microalgae (Wang et al., 2010). Tropicibacter multivorans has been detected in the microbiome of Caulerpa cylindraceae but little information is available about its function (Rizzo et al., 2016). In this study case, Tropicibacter spp. might contribute to the degradation of algal polymers.
4.3. Potential beneficial bacteria
A beneficial role was suspected on bacteria of the order Rhodobacterales and the genus Rhodovulum, with the highest relative sequence abundance at each respective taxonomic level in C. lentillifera. Rhodobacterales are generally surface colonizers and facilitate the settlement of microbial communities with the production of extracellular polymeric substances (Dang et al., 2008). According to Simon et al. (2017), marine Rhodobacteraceae are characterized by genes for the degradation of sulfated polysaccharides from algae, production of phytohormones, metabolism of osmolytes, transport of metals, and detoxification. Rhodovulum species have a high metabolic versatility with the capability to degrade organic pollutants, produce polymers, and contribute with photosynthetic functions (Baker et al., 2021; Foong et al., 2019; Khandavalli et al., 2018).
Paracoccus is a common Rhodobacteraceae among seaweed‐associated bacteria with a key role in the cycling of nitrogen and the production of siderophores (Mei et al., 2019). Paracoccus strains promote the growth and the morphogenesis in Ulva species (Ghaderiardakani et al., 2017) and were described as auxin producers (Kurepin et al., 2014). Plant hormone production seems to be widespread in various genera of marine bacteria (Goecke et al., 2010). Furthermore, Paracoccus sp. presented algicidal activity against microalgae (Zhang et al., 2018) and hypothetically, some Paracoccus strains may prevent microalgae fouling on C. lentillifera. The macroalgal tissues constitute a highly competitive niche for nutrients and space.
The presumptive production of the antimicrobial compound of C. lentillifera may explain partially the low diversity in healthy C. lentillifera compared to diseased cases. The trend of higher diversity in diseased C. lentillifera was reported comparing C. lentillifera of contrasting health states in a case from the Chinese aquaculture (Liang et al., 2019). Some Caulerpa species contain antibacterial compounds such as alkaloids, terpenoids, phenols, and flavonoids (Goecke et al., 2010; Yap et al., 2019; Zainuddin et al., 2019). Moreover, strains of Clostridium sensu stricto 7 and Cutibacterium might reduce diversity with the production of antimicrobial metabolites in healthy C. lentillifera. Some green algae provide habitat for Clostridium species and even for potential pathogens (Chun et al., 2013).
Clostridium spp. can catabolize several algal polymers (Song et al., 2011), have a likely function of copper detoxification in Codium tormentosum (Le Pennec & Gall, 2019), and are sources of novel antimicrobial compounds (Pahalagedara et al., 2020; Schieferdecker et al., 2019). A new thiopeptide antibiotic as a microbiota modulator was detected on Cutibacterium sp. (Claesen et al., 2020) and some species of this genus might produce antibiotics on Kappaphycus striatus (Kopprio et al., 2021). A similar role on algal defense is suspected in the other Actinobacteria Micrococcus spp. (e.g., Hollants et al., 2013). In addition, this genus together with Bradyrhizobium were reported as beneficial endophytic bacteria in many terrestrial plants with agronomic value (Afzal et al., 2019).
Bradyrhizobium and Allihoeflea of the order Rhizobiales may participate in the cycling of nitrogen and the production of phytohormones. “Rhizobacteria” were extensively studied in terrestrial plants because of their ability to fix nitrogen and to modify genetically their host. Some organisms of the family Rhizobiaceae presented growth‐enhancing and probiotic properties on green microalgae (Rivas et al., 2010). Furthermore, an endosymbiotic bacterium of this taxa was responsible for the nitrogen supply in Caulerpa taxifolia (Chisholm et al., 1996). Bradyrhizobium japonicum increases the biomass and starch content of the green microalgae Chlamydomonas reinhardtii (Xu et al., 2016) and some strains of B. japonicum are producers of several phytohormones such as indole‐3‐acetic acid, gibberellic acid, zeatin, abscisic acid, and ethylene (Boiero et al., 2007).
A beneficial role for C. lentillifera growth and health is hypothesized on Brevundimonas strains. Members of this genus establish a symbiotic relationship with green microalgae promoting their growth, producing indole‐3‐acetic acid, and enhancing nutrient uptake (Sforza et al., 2018; Tate et al., 2013; Zhang et al., 2021). Some Brevundimonas strains have potential antifouling activity against cyanobacteria (Lin et al., 2014) and alleviate toxicity, fix nitrogen, and promote growth in some terrestrial plants (Naqqash et al., 2020; Singh et al., 2016). Pelomonas sq 1964 may contribute to a healthy status on C. lentillifera. This genus is common in the rhizosphere of some plants with a nitrogen fixation function (Terakado‐Tonooka et al., 2008) and some of their aquatic strains produce antibacterial compounds such as pelopuradazole (He et al., 2014). Surprisingly, potential human pathogens like Escherichia‐Shigella and Staphylococcus were related to the healthy state of C. lentillifera according to SIMPER analysis. Hollants et al. (2013) reported these bacteria with the beneficial functions of defense and morphogenesis in seaweeds, respectively.
5. CONCLUSIONS
A common composition of bacterial communities was observed in diseased C. lentillifera from Vietnamese and Chinese study cases. A disease in C. lentillifera may be caused by a community of detrimental bacteria together with changes in algal response, and not only by a particular pathogen (e.g., Kumar et al., 2016). Moreover, in our study was not possible to differentiate pathogens from saprophytes. A mutualistic relationship with particular bacteria may change and become detrimental under certain conditions; nevertheless, clear differences between healthy and diseased states were observed at several taxonomic levels. This study explores changes in the bacterial community composition of C. lentillifera under two conditions, detects co‐occurrence but not causality, we cannot discard other etiological agents not covered by the selected amplicon. Common patterns and roles were inferred in this initial study from the Vietnamese aquaculture, a valuable insight for the understanding of potential key players in the sea grape holobiont. The microbiome research on C. lentillifera is still in its infancy and the results of this study are valuable but limited, we recommend further basic and applied research using NGS techniques on culturable and nonculturable microorganisms. Bioactivity studies may confirm the antimicrobial properties of C. lentillifera and attached microorganisms, and shotgun metagenomics, metatranscriptomics, and metabolomics may provide valuable insights into the functions of the C. lentillifera microbiome and potential biotechnological applications.
CONFLICT OF INTERESTS
None declared.
ETHICS STATEMENT
None required.
AUTHOR CONTRIBUTIONS
Germán Kopprio: Conceptualization (equal), data curation (equal), formal analysis (equal), investigation (equal), methodology (equal), project administration (equal), writing‐original draft (equal). Nguyen Dinh Luyen: Investigation (equal), methodology (equal). Le Huu Cuong: Conceptualization (equal), Investigation (equal), methodology (equal). Tran Mai Duc: Investigation (equal), methodology (equal). Anna Fricke: Investigation (equal), methodology (equal), visualization (equal), writing‐original draft (equal). Andreas Kunzmann: Conceptualization (equal), funding acquisition (equal), investigation (equal). Le Mai Huong: Funding acquisition (equal), investigation (equal), supervision (equal), visualization (equal). Astrid Gärdes: Conceptualization (equal), formal analysis (equal), funding acquisition (equal), investigation (equal), methodology (equal), project administration (equal), supervision (equal), visualization (equal).
ACKNOWLEDGMENT
This study was financed by the BMBF project Aquaweed (031B0121). Open access funding enabled and organized by Projekt DEAL.
Kopprio, G. A. , Luyen, N. D. , Cuong, L. H. , Duc, T. M. , Fricke, A. , Kunzmann, A. , Huong, L. M. , & Gärdes, A. (2021). Insights into the bacterial community composition of farmed Caulerpa lentillifera: A comparison between contrasting health states. MicrobiologyOpen, 10, e1253. 10.1002/mbo3.1253
DATA AVAILABILITY STATEMENT
Sequencing data are available at European Nucleotide Archive (https://www.ebi.ac.uk/ena/data/view/PRJEB42826).
REFERENCES
- Addinsoft . (2018). XLSTAT statistical and data analysis solution. https://www.xlstat.com
- Afzal, I. , Shinwari, Z. K. , Sikandar, S. , & Shahzad, S. (2019). Plant beneficial endophytic bacteria: Mechanisms, diversity, host range and genetic determinants. Microbiological Research, 221, 36–49. 10.1016/j.micres.2019.02.001 [DOI] [PubMed] [Google Scholar]
- Aires, T. , Serrão, E. A. , Kendrick, G. , Duarte, C. M. , & Arnaud‐Haond, S. (2013). Invasion is a community affair: Clandestine followers in the bacterial community associated to green algae, Caulerpa racemosa, track the invasion source. PLoS One, 8, 68429. 10.1371/journal.pone.0068429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud‐Haond, S. , Aires, T. , Candeias, R. , Teixeira, S. J. L. , Duarte, C. M. , Valero, M. , & Serrão, E. A. (2017). Entangled fates of holobiont genomes during invasion: Nested bacterial and host diversities in Caulerpa taxifolia . Molecular Ecology, 26, 2379–2391. 10.1111/mec.14030 [DOI] [PubMed] [Google Scholar]
- Baker, A. , Ahmad, B. , Alarjani, K. M. , Aldosri, N. S. , & Khan, M. S. (2021). Biostimulation of Rhodovulum sp., for enhanced degradation of di‐n‐butyl phthalate under optimum conditions. Chemosphere, 266, 128998. 10.1016/j.chemosphere.2020.128998 [DOI] [PubMed] [Google Scholar]
- Boiero, L. , Perrig, D. , Masciarelli, O. , Penna, C. , Cassán, F. , & Luna, V. (2007). Phytohormone production by three strains of Bradyrhizobium japonicum and possible physiological and technological implications. Applied Microbiology and Biotechnology, 74, 874–880. 10.1007/s00253-006-0731-9 [DOI] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondoso, J. , Godoy‐Vitorino, F. , Balagué, V. , Gasol, J. M. , Harder, J. , & Lage, O. M. (2017). Epiphytic Planctomycetes communities associated with three main groups of macroalgae. FEMS Microbiology Ecology, 93, fiw255. 10.1093/femsec/fiw255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Box, A. , Sureda, A. , Terrados, J. , Pons, A. , & Deudero, S. (2008). Antioxidant response and caulerpenyne production of the alien Caulerpa taxifolia (Vahl) epiphytized by the invasive algae Lophocladia lallemandii (Montagne). Journal of Experimental Marine Biology and Ecology, 364, 24–28. 10.1016/j.jembe.2008.06.029 [DOI] [Google Scholar]
- Burke, C. , Thomas, T. , Lewis, M. , Steinberg, P. , & Kjelleberg, S. (2011). Composition, uniqueness and variability of the epiphytic bacterial community of the green alga Ulva australis . ISME Journal, 5, 590–600. 10.1038/ismej.2010.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Califano, G. , Kwantes, M. , Abreu, M. H. , Costa, R. , & Wichard, T. (2020). Cultivating the macroalgal holobiont: Effects of integrated multi‐trophic aquaculture on the microbiome of Ulva rigida (Chlorophyta). Frontiers in Marine Science, 7, 52. 10.3389/fmars.2020.00052 [DOI] [Google Scholar]
- Chisholm, J. R. M. , Dauga, C. , Ageron, E. , Grimont, P. A. D. , & Jaubert, J. M. (1996). “Roots” in mixotrophic algae. Nature, 381, 382. 10.1038/381382a0 [DOI] [Google Scholar]
- Chun, C. L. , Ochsner, U. , Byappanahalli, M. N. , Whitman, R. L. , Tepp, W. H. , Lin, G. , Johnson, E. A. , Peller, J. , & Sadowsky, M. J. (2013). Association of toxin‐producing Clostridium botulinum with the macroalga Cladophora in the Great Lakes. Environmental Science and Technology, 47, 2587–2594. 10.1021/es304743m [DOI] [PubMed] [Google Scholar]
- Claesen, J. , Spagnolo, J. B. , Ramos, S. F. , Kurita, K. L. , Byrd, A. L. , Aksenov, A. A. , Melnik, A. V. , Wong, W. R. , Wang, S. , Hernandez, R. D. , Donia, M. S. , Dorrestein, P. C. , Kong, H. H. , Segre, J. A. , Linington, R. G. , Fischbach, M. A. , & Lemon, K. P. (2020). A Cutibacterium acnes antibiotic modulates human skin microbiota composition in hair follicles. Science Translational Medicine, 12, aay5445. 10.1126/scitranslmed.aay5445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang, H. , Li, T. , Chen, M. , & Huang, G. (2008). Cross‐ocean distribution of Rhodobacterales bacteria as primary surface colonizers in temperate coastal marine waters. Applied and Environmental Microbiology, 74, 52–60. 10.1128/aem.01400-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Gaillande, C. , Payri, C. , Remoissenet, G. , & Zubia, M. (2017). Caulerpa consumption, nutritional value and farming in the Indo‐Pacific region. Journal of Applied Phycology, 29, 2249–2266. 10.1007/s10811-016-0912-6 [DOI] [Google Scholar]
- Diepenbroek, M. , Glöckner, F. O. , Grobe, P. , Güntsch, A. , Huber, R. , König‐Ries, B. , Kostadinov, I. , Nieschulze, J. , Seeger, B. , Tolksdorf, R. , & Triebel, D. (2014). Towards an integrated biodiversity and ecological research data management and archiving platform: The German federation for the curation of biological data (GFBio). GI‐Jahrestagung.
- Egan, S. , Fernandes, N. D. , Kumar, V. , Gardiner, M. , & Thomas, T. (2014). Bacterial pathogens, virulence mechanism and host defence in marine macroalgae. Environmental Microbiology, 16, 925–938. 10.1111/1462-2920.12288 [DOI] [PubMed] [Google Scholar]
- Egan, S. , Harder, T. , Burke, C. , Steinberg, P. , Kjelleberg, S. , & Thomas, T. (2013). The seaweed holobiont: Understanding seaweed–bacteria interactions. FEMS Microbiology Reviews, 37, 462–476. 10.1111/1574-6976.12011 [DOI] [PubMed] [Google Scholar]
- Eren, A. M. , Morrison, H. G. , Lescault, P. J. , Reveillaud, J. , Vineis, J. H. , & Sogin, M. L. (2015). Minimum entropy decomposition: Unsupervised oligotyping for sensitive partitioning of high‐throughput marker gene sequences. ISME Journal, 9, 968–979. 10.1038/ismej.2014.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foong, C. P. , Higuchi‐Takeuchi, M. , & Numata, K. (2019). Optimal iron concentrations for growth‐associated polyhydroxyalkanoate biosynthesis in the marine photosynthetic purple bacterium Rhodovulum sulfidophilum under photoheterotrophic condition. PLoS One, 14, 212654. 10.1371/journal.pone.0212654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich, M. W. (2012). Bacterial communities on macroalgae. In Wiencke C., & Bischof K. (Eds.), Seaweed biology: Novel insights into ecophysiology, ecology and utilization, ecological studies (pp. 189–201). Springer. 10.1007/978-3-642-28451-9_10 [DOI] [Google Scholar]
- Fukunaga, Y. , Kurahashi, M. , Sakiyama, Y. , Ohuchi, M. , Yokota, A. , & Harayama, S. (2009). Phycisphaera mikurensis gen. nov., sp. nov., isolated from a marine alga, and proposal of Phycisphaeraceae fam. nov., Phycisphaerales ord. nov and Phycisphaerae classis nov. in the phylum Planctomycetes. Journal of General and Applied Microbiology, 55, 267–275. 10.2323/jgam.55.267 [DOI] [PubMed] [Google Scholar]
- Ghaderiardakani, F. , Coates, J. C. , & Wichard, T. (2017). Bacteria‐induced morphogenesis of Ulva intestinalis and Ulva mutabilis (Chlorophyta): A contribution to the lottery theory. FEMS Microbiology Ecology, 93, fix094. 10.1093/femsec/fix094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecke, F. , Labes, A. , Wiese, J. , & Imhoff, J. F. (2010). Chemical interactions between marine macroalgae and bacteria. Marine Ecology Progress Series, 409, 267–299. 10.3354/meps08607 [DOI] [Google Scholar]
- Griffiths, R. I. , Whiteley, A. S. , O'Donnell, A. G. , & Bailey, M. J. (2000). Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA‐ and rRNA‐based microbial community composition. Applied and Environmental Microbiology, 66, 5488–5491. 10.1128/AEM.66.12.5488-5491.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, X.‐X. , Chen, X.‐J. , Peng, G.‐T. , Guan, S.‐Y. , Lei, L.‐F. , Yao, J.‐H. , Liu, B.‐X. , & Zhang, C.‐X. (2014). Pelopuradazole, a new imidazole derivative alkaloid from the marine bacteria Pelomonas puraquae sp. nov. Natural Product Research, 28, 680–682. 10.1080/14786419.2014.891591 [DOI] [PubMed] [Google Scholar]
- Hollants, J. , Leliaert, F. , Clerck, O. D. , & Willems, A. (2013). What we can learn from sushi: A review on seaweed–bacterial associations. FEMS Microbiology Ecology, 83, 1–16. 10.1111/j.1574-6941.2012.01446.x [DOI] [PubMed] [Google Scholar]
- Hsieh, T. C. , Ma, K. H. , & Chao, A. (2019) iNEXT: iNterpolation and EXTrapolation for species diversity. R package version 2.0.19. http://chao.stat.nthu.edu.tw/blog/software-download/
- Jain, A. , Krishnan, K. P. , Singh, A. , Thomas, F. A. , Begum, N. , Tiwari, M. , Bhaskar, V. P. , & Gopinath, A. (2019). Biochemical composition of particles shape particle‐attached bacterial community structure in a high Arctic fjord. Ecological Indicators, 102, 581–592. 10.1016/j.ecolind.2019.03.015 [DOI] [Google Scholar]
- Khandavalli, L. V. N. S. , Lodha, T. , Abdullah, M. , Guruprasad, L. , Chintalapati, S. , & Chintalapati, V. R. (2018). Insights into the carbonic anhydrases and autotrophic carbon dioxide fixation pathways of high CO2 tolerant Rhodovulum viride JA756. Microbiological Research, 215, 130–140. 10.1016/j.micres.2018.07.006 [DOI] [PubMed] [Google Scholar]
- Klindworth, A. , Pruesse, E. , Schweer, T. , Peplies, J. , Quast, C. , Horn, M. , & Glöckner, F. O. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next‐generation sequencing‐based diversity studies. Nucleic Acids Research, 41, 1–e1. 10.1093/nar/gks808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopprio, G. A. , Cuong, L. H. , Luyen, N. D. , Duc, T. M. , Ha, T. H. , Huong, L. M. , & Gärdes, A. (2021). Carrageenophyte‐attached and planktonic bacterial communities in two distinct bays of Vietnam: Eutrophication indicators and insights on ice‐ice disease. Ecological Indicators, 121, 107067. 10.1016/j.ecolind.2020.107067 [DOI] [Google Scholar]
- Kumar, V. , Zozaya‐Valdes, E. , Kjelleberg, S. , Thomas, T. , & Egan, S. (2016). Multiple opportunistic pathogens can cause a bleaching disease in the red seaweed Delisea pulchra . Environmental Microbiology, 18, 3962–3975. 10.1111/1462-2920.13403 [DOI] [PubMed] [Google Scholar]
- Kurepin, L. V. , Zaman, M. , & Pharis, R. P. (2014). Phytohormonal basis for the plant growth promoting action of naturally occurring biostimulators. Journal of the Science of Food and Agriculture, 94, 1715–1722. 10.1002/jsfa.6545 [DOI] [PubMed] [Google Scholar]
- Lage, O. M. , & Bondoso, J. (2014). Planctomycetes and macroalgae, a striking association. Frontiers in Microbiology, 5, 267. 10.3389/fmicb.2014.00267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, Z. , Liu, F. , Wang, W. , Zhang, P. , Sun, X. , Wang, F. , & Kell, H. (2019). High‐throughput sequencing revealed differences of microbial community structure and diversity between healthy and diseased Caulerpa lentillifera . BMC Microbiology, 19, 225. 10.1186/s12866-019-1605-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S. , Pan, J. , Li, Z. , Liu, X. , Tan, J. , & Yang, H. (2014). Characterization of an algicidal bacterium Brevundimonas J4 and chemical defense of Synechococcus sp. BN60 against bacterium J4. Harmful Algae, 37, 1–7. 10.1016/j.hal.2014.05.002 [DOI] [Google Scholar]
- Liu, Q. , Zhi, Y. , He, Y. , Ren, Z. , Chen, H. , & Yang, R. (2020). Changes in phycospheric and environmental microbes associated with an outbreak of yellow spot disease on Pyropia yezoensis . Aquaculture, 529, 735651. 10.1016/j.aquaculture.2020.735651 [DOI] [Google Scholar]
- Luyen, N. D. , Huong, L. M. , Thi Hong Ha, T. , Cuong, L. H. , Thi Hai Yen, D. , Nhiem, N. X. , Tai, B. H. , Gardes, A. , Kopprio, G. , & Van Kiem, P. (2019). Aspermicrones A‐C, novel dibenzospiroketals from the seaweed‐derived endophytic fungus Aspergillus micronesiensis . Journal of Antibiotics, 72, 843–847. 10.1038/s41429-019-0214-8 [DOI] [PubMed] [Google Scholar]
- Ly, K. V. , Murungu, D. K. , Nguyen, D. P. , & Nguyen, N. A. T. (2021). Effects of different densities of sea grape Caulerpa lentillifera on water quality, growth and survival of the whiteleg shrimp Litopenaeus vannamei in polyculture system. Fishes, 6, 19. 10.3390/fishes6020019 [DOI] [Google Scholar]
- Martinez‐Garcia, M. , Brazel, D. M. , Swan, B. K. , Arnosti, C. , Chain, P. S. , Reitenga, K. G. , Xie, G. , Poulton, N. J. , Lluesma Gomez, M. , Masland, D. E. , Thompson, B. , Bellows, W. K. , Ziervogel, K. , Lo, C. C. , Ahmed, S. , Gleasner, C. D. , Detter, C. J. , & Stepanauskas, R. (2012). Capturing single cell genomes of active polysaccharide degraders: An unexpected contribution of Verrucomicrobia. PLoS One, 7, 35314. 10.1371/journal.pone.0035314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei, X. , Wu, C. , Zhao, J. , Yan, T. , & Jiang, P. (2019). Community structure of bacteria associated with drifting Sargassum horneri, the causative species of golden tide in the Yellow Sea. Frontiers in Microbiology, 10, 1192. 10.3389/fmicb.2019.01192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mußmann, M. , Pjevac, P. , Krüger, K. , & Dyksma, S. (2017). Genomic repertoire of the Woeseiaceae/JTB255, cosmopolitan and abundant core members of microbial communities in marine sediments. ISME Journal, 11, 1276–1281. 10.1038/ismej.2016.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqqash, T. , Imran, A. , Hameed, S. , Shahid, M. , Majeed, A. , Iqbal, J. , Hanif, M. K. , Ejaz, S. , & Malik, K. A. (2020). First report of diazotrophic Brevundimonas spp. as growth enhancer and root colonizer of potato. Scientific Reports, 10, 12893. 10.1038/s41598-020-69782-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen, J. , Blanchet, F. G. , Friendly, M. , Kindt, R. , Legendre, P. , McGlinn, D. , Minchin, P. R. , O'Hara, R. B. , Simpson, G. L. , Solymos, P. , Stevens, M. H. H. , Szoecs, E. & Wagner, H. (2019). vegan: Community Ecology Package. R package version 2.5‐4. https://CRAN.R-project.org/package=vegan
- Pahalagedara, A. S. N. W. , Flint, S. , Palmer, J. , Brightwell, G. , & Gupta, T. B. (2020). Antimicrobial production by strictly anaerobic Clostridium spp. International Journal of Antimicrobial Agents, 55, 105910. 10.1016/j.ijantimicag.2020.105910 [DOI] [PubMed] [Google Scholar]
- Paul, N. A. , Neveux, N. , Magnusson, M. , & de Nys, R. (2014). Comparative production and nutritional value of “sea grapes”—the tropical green seaweeds Caulerpa lentillifera and C. racemosa . Journal of Applied Phycology, 26, 1833–1844. 10.1007/s10811-013-0227-9 [DOI] [Google Scholar]
- Le Pennec, G. , & Gall, E. A. (2019). The microbiome of Codium tomentosum: Original state and in the presence of copper. World Journal of Microbiology and Biotechnology, 35, 167. 10.1007/s11274-019-2740-8 [DOI] [PubMed] [Google Scholar]
- Rivas, M. O. , Vargas, P. , & Riquelme, C. E. (2010). Interactions of Botryococcus braunii cultures with bacterial biofilms. Microbial Ecology, 60, 628–635. 10.1007/s00248-010-9686-6 [DOI] [PubMed] [Google Scholar]
- Rizzo, L. , Fraschetti, S. , Alifano, P. , Pizzolante, G. , & Stabili, L. (2016). The alien species Caulerpa cylindracea and its associated bacteria in the Mediterranean Sea. Marine Biology, 163, 4. 10.1007/s00227-015-2775-9 [DOI] [PubMed] [Google Scholar]
- Saito, H. , Xue, C. , Yamashiro, R. , Moromizato, S. , & Itabashi, Y. (2010). High polyunsaturated fatty acid levels in two subtropical macroalgae, Cladosiphon okamuranus and Caulerpa lentillifera . Journal of Phycology, 46, 665–673. 10.1111/j.1529-8817.2010.00848.x [DOI] [Google Scholar]
- Schieferdecker, S. , Shabuer, G. , Knuepfer, U. , & Hertweck, C. (2019). Clostrindolin is an antimycobacterial pyrone alkaloid from Clostridium beijerinckii . Organic and Biomolecular Chemistry, 17, 6119–6121. 10.1039/c9ob00968j [DOI] [PubMed] [Google Scholar]
- Sforza, E. , Pastore, M. , Santeufemia Sanchez, S. , & Bertucco, A. (2018). Bioaugmentation as a strategy to enhance nutrient removal: Symbiosis between Chlorella protothecoides and Brevundimonas diminuta . Bioresource Technology Reports, 4, 153–158. 10.1016/j.biteb.2018.10.007 [DOI] [Google Scholar]
- Simon, M. , Scheuner, C. , Meier‐Kolthoff, J. P. , Brinkhoff, T. , Wagner‐Döbler, I. , Ulbrich, M. , Klenk, H.‐P. , Schomburg, D. , Petersen, J. , & Göker, M. (2017). Phylogenomics of Rhodobacteraceae reveals evolutionary adaptation to marine and non‐marine habitats. ISME Journal, 11, 1483–1499. 10.1038/ismej.2016.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, N. , Marwa, N. , Mishra, S. k , Mishra, J. , Verma, P. C. , Rathaur, S. , & Singh, N. (2016). Brevundimonas diminuta mediated alleviation of arsenic toxicity and plant growth promotion in Oryza sativa L. Ecotoxicology and Environmental Safety, 125, 25–34. 10.1016/j.ecoenv.2015.11.020 [DOI] [PubMed] [Google Scholar]
- Singh, R. P. , & Reddy, C. R. K. (2014). Seaweed–microbial interactions: Key functions of seaweed‐associated bacteria. FEMS Microbiology Ecology, 88, 213–230. 10.1111/1574-6941.12297 [DOI] [PubMed] [Google Scholar]
- Song, J.‐H. , Ventura, J.‐R. S. , Lee, C.‐H. , & Jahng, D. (2011). Butyric acid production from brown algae using Clostridium tyrobutyricum ATCC 25755. Biotechnology and Bioprocess Engineering, 16, 42–49. 10.1007/s12257-010-0177-x [DOI] [Google Scholar]
- Stuthmann, L. E. , Springer, K. , & Kunzmann, A. (2021). Cultured and packed sea grapes (Caulerpa lentillifera): Effect of different irradiances on photosynthesis. Journal of Applied Phycology, 33, 1125–1136. 10.1007/s10811-020-02322-x [DOI] [Google Scholar]
- Tate, J. J. , Gutierrez‐Wing, M. T. , Rusch, K. A. , & Benton, M. G. (2013). The effects of plant growth substances and mixed cultures on growth and metabolite production of green algae Chlorella sp.: A review. Journal of Plant Growth Regulation, 32, 417–428. 10.1007/s00344-012-9302-8 [DOI] [Google Scholar]
- Terada, R. , Takaesu, M. , Borlongan, I. A. , & Nishihara, G. N. (2021). The photosynthetic performance of a cultivated Japanese green alga Caulerpa lentillifera in response to three different stressors, temperature, irradiance, and desiccation. Journal of Applied Phycology, 33, 2547–2559. 10.1007/s10811-021-02439-7 [DOI] [Google Scholar]
- Terakado‐Tonooka, J. , Ohwaki, Y. , Yamakawa, H. , Tanaka, F. , Yoneyama, T. , & Fujihara, S. (2008). Expressed nifH genes of endophytic bacteria detected in field‐grown sweet potatoes (Ipomoea batatas L.). Microbes and Environments, 23, 89–93. 10.1264/jsme2.23.89 [DOI] [PubMed] [Google Scholar]
- Tian, R. , Ning, D. , He, Z. , Zhang, P. , Spencer, S. J. , Gao, S. , Shi, W. , Wu, L. , Zhang, Y. , Yang, Y. , Adams, B. G. , Rocha, A. M. , Detienne, B. L. , Lowe, K. A. , Joyner, D. C. , Klingeman, D. M. , Arkin, A. P. , Fields, M. W. , Hazen, T. C. , … Zhou, J. (2020). Small and mighty: Adaptation of superphylum Patescibacteria to groundwater environment drives their genome simplicity. Microbiome, 8, 51. 10.1186/s40168-020-00825-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valero, M. , Guillemin, M.‐L. , Destombe, C. , Jacquemin, B. , Gachon, C. M. M. , Badis, Y. , Buschmann, A. H. , Carolina, C. , & Sylvain, F. (2017). Perspectives on domestication research for sustainable seaweed aquaculture. Perspectives in Phycology, 4, 33–46. 10.1127/pip/2017/0066 [DOI] [Google Scholar]
- Walker, A. M. , Leigh, M. B. , & Mincks, S. L. (2021). Patterns in benthic microbial community structure across environmental gradients in the Beaufort Sea shelf and slope. Frontiers in Microbiology, 12, 581124. 10.3389/fmicb.2021.581124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Li, Z. , Su, J. , Tian, Y. , Ning, X. , Hong, H. , & Zheng, T. (2010). Lysis of a red‐tide causing alga, Alexandrium tamarense, caused by bacteria from its phycosphere. Biological Control, 52, 123–130. 10.1016/j.biocontrol.2009.10.004 [DOI] [Google Scholar]
- Ward, G. M. , Faisan, J. P. , Cottier‐Cook, E. J. , Gachon, C. , Hurtado, A. Q. , Lim, P. E. , Matoju, I. , Msuya, F. E. , Bass, D. , & Brodie, J. (2020). A review of reported seaweed diseases and pests in aquaculture in Asia. Journal of the World Aquaculture Society, 51, 815–828. 10.1111/jwas.12649 [DOI] [Google Scholar]
- Xu, L. , Li, D. , Wang, Q. , & Wu, S. (2016). Improved hydrogen production and biomass through the co‐cultivation of Chlamydomonas reinhardtii and Bradyrhizobium japonicum . International Journal of Hydrogen Energy, 41, 9276–9283. 10.1016/j.ijhydene.2016.04.009 [DOI] [Google Scholar]
- Yap, W.‐F. , Tay, V. , Tan, S.‐H. , Yow, Y.‐Y. , & Chew, J. (2019). Decoding antioxidant and antibacterial potentials of Malaysian green seaweeds: Caulerpa racemosa and Caulerpa lentillifera . Antibiotics (USSR), 8, 152. 10.3390/antibiotics8030152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz, P. , Kottmann, R. , Field, D. , Knight, R. , Cole, J. R. , Amaral‐Zettler, L. , Gilbert, J. A. , Karsch‐Mizrachi, I. , Johnston, A. , Cochrane, G. , Vaughan, R. , Hunter, C. , Park, J. , Morrison, N. , Rocca‐Serra, P. , Sterk, P. , Arumugam, M. , Bailey, M. , Baumgartner, L. , … Glöckner, F. O. (2011). Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nature Biotechnology, 29, 415–420. 10.1038/nbt.1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz, P. , Yarza, P. , Rapp, J. Z. , & Glöckner, F. O. (2016). Expanding the world of marine bacterial and archaeal clades. Frontiers in Microbiology, 6, 1524. 10.3389/fmicb.2015.01524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, J. , Jang, J.‐H. , & Kasai, H. (2014). Algisphaera agarilytica gen. nov., sp. nov., a novel representative of the class Phycisphaerae within the phylum Planctomycetes isolated from a marine alga. Antonie Van Leeuwenhoek, 105, 317–324. 10.1007/s10482-013-0076-1 [DOI] [PubMed] [Google Scholar]
- Zainuddin, E. N. , Anshary, H. , Huyyirnah, H. , Hiola, R. , & Baxa, D. V. (2019). Antibacterial activity of Caulerpa racemosa against pathogenic bacteria promoting “ice‐ice” disease in the red alga Gracilaria verrucosa . Journal of Applied Phycology, 31, 3201–3212. 10.1007/s10811-019-01805-w [DOI] [Google Scholar]
- Zhang, B. , Chen, J. , Su, Y. , Sun, W. , & Zhang, A. (2021). Utilization of Indole‐3‐acetic acid–secreting bacteria in algal environment to increase biomass accumulation of Ochromonas and Chlorella . BioEnergy Research, 27, 435–444. 10.1007/s12155-021-10246-8 [DOI] [Google Scholar]
- Zhang, F. , Ye, Q. , Chen, Q. , Yang, K. , Zhang, D. , Chen, Z. , Lu, S. , Shao, X. , Fan, Y. , Yao, L. , Ke, L. , Zheng, T. , & Xu, H. (2018). Algicidal activity of novel marine bacterium Paracoccus sp. Strain Y42 against a harmful algal‐bloom‐causing dinoflagellate, Prorocentrum donghaiense . Applied and Environmental Microbiology, 84, 1015–1018. 10.1128/AEM.01015-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Kobert, K. , Flouri, T. , & Stamatakis, A. (2014). PEAR: A fast and accurate Illumina Paired‐End reAd mergeR. Bioinformatics, 30, 614–620. 10.1093/bioinformatics/btt593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, R. , Chang, L. , Xiao, L. , Zhang, X. , Han, Q. , Li, N. , Egan, S. , & Wang, G. (2020). Diversity of the epiphytic bacterial communities associated with commercially cultivated healthy and diseased Saccharina japonica during the harvest season. Journal of Applied Phycology, 32, 2071–2080. 10.1007/s10811-019-02025-y [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Sequencing data are available at European Nucleotide Archive (https://www.ebi.ac.uk/ena/data/view/PRJEB42826).