

Abstract
Introduction
Many pathologies, including cancer, have been associated with aberrant phosphorylation-mediated signaling networks that drive altered cell proliferation, migration, metabolic regulation, and can lead to systemic inflammation. Phosphoproteomics, the large-scale analysis of protein phosphorylation sites, has emerged as a powerful tool to define signaling network regulation and dysregulation in normal and pathological conditions.
Areas Covered
We provide an overview of methodology for global phosphoproteomics as well as enrichment of specific subsets of the phosphoproteome, including phosphotyrosine and phospho-motif enrichment of kinase substrates. We review quantitative methods, advantages and limitations of different mass spectrometry acquisition formats, and computational approaches to extract biological insight from phosphoproteomics data. Throughout, we discuss various applications and their challenges in implementation.
Expert opinion
Over the past 20 years the field of phosphoproteomics has advanced to enable deep biological and clinical insight through the quantitative analysis of signaling networks. Future areas of development include Clinical Laboratory Improvement Amendments (CLIA)-approved methods for analysis of clinical samples, continued improvements in sensitivity to enable analysis of small numbers of rare cells and tissue microarrays, and computational methods to integrate data resulting from multiple systems-level quantitative analytical methods.
Keywords: cancer, cellular networks, kinase activity, mass spectrometry, molecular signaling, phosphoproteomics, phosphorylation
1. Introduction: Phosphoproteomics, signaling and cancer
Phosphorylation is a reversible post-translational modification(PTM) that is critical for regulating inter- and intracellular signaling networks [1]. In mammalian cells, phosphorylation typically occurs on the side chains of three amino acids; serine, threonine and tyrosine, although phosphorylation of other residues has been reported [2,3]. The addition of a phosphate group can have a variety of effects, as phosphorylated proteins may have altered conformation, stability, activity, subcellular localization, or protein-protein interactions. Due to its ability to modify protein function like a modular switch mechanism, phosphorylation is crucial to the regulation of signal transduction pathways [4].
Most major cellular processes, including proliferation, migration, apoptosis, and others, are regulated by protein phosphorylation-mediated signaling cascades that are also critical for relaying information about the external cell micro-environment and internal cell state. Dysregulated protein phosphorylation signaling due to aberrant kinase or phosphatase activity has been associated with a host of human pathologies [5–7]. Indeed, many hallmarks of cancer, including sustained proliferation, resistance to cell death, angiogenesis, avoiding growth suppression and invasion and metastasis can be linked to dysregulated signaling pathways and inappropriate kinase activity [8,9].
The transformative potential of multiple constitutively activated kinases, as well as the role of protein phosphorylation in regulating other aspects of biology, has fueled a deep interest in protein phosphorylation, including studies at the single protein level, protein complexes, enzyme-substrate relationships, or at the level of the phosphoproteome, the compendium of protein phosphorylation sites in a given biological sample. Phosphoproteomics, the large-scale analysis of protein phosphorylation sites, was pioneered in 2002 by Ficarro et al., and has developed rapidly over the past few decades [10]. Although phosphoproteomics may be performed using a variety of instruments and can encompass both targeted and discovery analyses, phosphoproteomics-based mapping of phosphorylation events in a large-scale, relatively unbiased manner mainly relies on mass spectrometry (MS)-based approaches [11,12]. Alternative techniques to measure protein phosphorylation include immunofluorescence / immunohistochemistry, phospho-flow, reverse-phase protein microarrays, and multiple different forms of western blotting. Although these techniques are widely used, they are dependent on antibody availability and specificity, and can be limited in the number of phosphorylation sites monitored per analysis [13,14]. By comparison, MS-based methods require minimal a priori knowledge, can identify and quantify >10,000 phosphorylation sites in a given sample, and provide high specificity by directly sequencing the site of protein phosphorylation.
MS-based phosphoproteomics has the potential to uncover activated signaling networks and novel targets in cancer cells, yet there are some inherent challenges. For instance, phosphorylation is a reversible modification that can be highly dynamic on the seconds-to minutes time scale [15]. Additionally, the phosphoproteome comprises approximately 0.1% of the proteome, and low-level phosphorylation events such as phosphotyrosine comprise only 0.1–1% of the phosphoproteome [15]; in many cases these ultra-low-abundance phosphorylation events are critical to decipher cellular signaling networks mediating oncogenic initiation and progression. Thus, MS-based phosphoproteomics must be able to identify and quantify ultra-low level, dynamic phosphorylation events. At the same time, some highly abundant proteins are phosphorylated at high stoichiometry, thus MS-based phosphoproteomics must also be able to handle a large dynamic range of phosphorylation.
Despite these challenges, MS-based phosphoproteomics has already proven to be capable of generating valuable phosphorylation data leading to biological insight [16,17]. It has been used to detect and validate potential biomarkers and drug targets, for example in identifying kinases and their phosphorylation sites not previously known to be involved in cellular signaling in multiple disease states [18–20]. Recently, progress was made in the mechanistic understanding of the hedgehog pathway; Scheidt et al. showed that aberrant signaling in this pathway is correlated with various cancers [21]. Phosphoproteomics may lead to new biomarkers for drug development, and also can elucidate resistance mechanisms and other mechanisms of action in disease [22–24]. For example, as cancer therapy development becomes increasingly focused on personalized medicine, phosphoproteomics has been used to uncover cancer cell signaling networks in patient tissues and signaling signatures of response to tyrosine kinase inhibitors [25–28]. Here we will focus on MS-based phosphoproteomics, including technical aspects of the analysis, challenges in implementation, and applications where phosphoproteomics has been used to uncover novel information in cancer signaling networks.
2. Sample preparation and phosphopeptide enrichment
While MS is the method of choice for large-scale analysis of phosphorylated proteins, high-yield sample preparation with minimal losses of specific classes of proteins is important, especially for analysis of ultra-low level phosphorylation sites (Figure 1). The first steps in MS-based sample preparation are cell lysis and protein extraction. Many protocols now utilize a urea-based buffer to lyse cells, as it rapidly denatures proteins to preserve the physiological modification state of proteins, and is easily removed by desalting in later steps. If heated, urea-based buffers can lead to the carbamylation of proteins; higher temperatures for longer times can exacerbate this effect. As an alternative, guanidine hydrochloride (Gnd-HCl) can be used, as this buffer allows for heating to high temperature for better solubilization, recovery and denaturation. However, Gnd-HCl can negatively impact digestion efficiency, so additional dilution needs to be performed prior to adding proteolytic enzymes. Although sodium dodecyl sulfate (SDS) is a powerful chaotropic agent that is widely used in molecular biology, western blotting, and protein arrays, it is challenging to remove in sample processing steps for MS analysis and can suppress protease activity and MS signal. Recent clean-up strategies like SP3 and S-trap have been developed that can overcome this hurdle, although complete removal of SDS can still be challenging [29,30]. Acid-labile surfactant is an MS-friendly alternative to SDS, yet due to the relatively high cost of this reagent and the additional clean-up steps required, most groups favor urea-based cell lysis.
Figure 1.
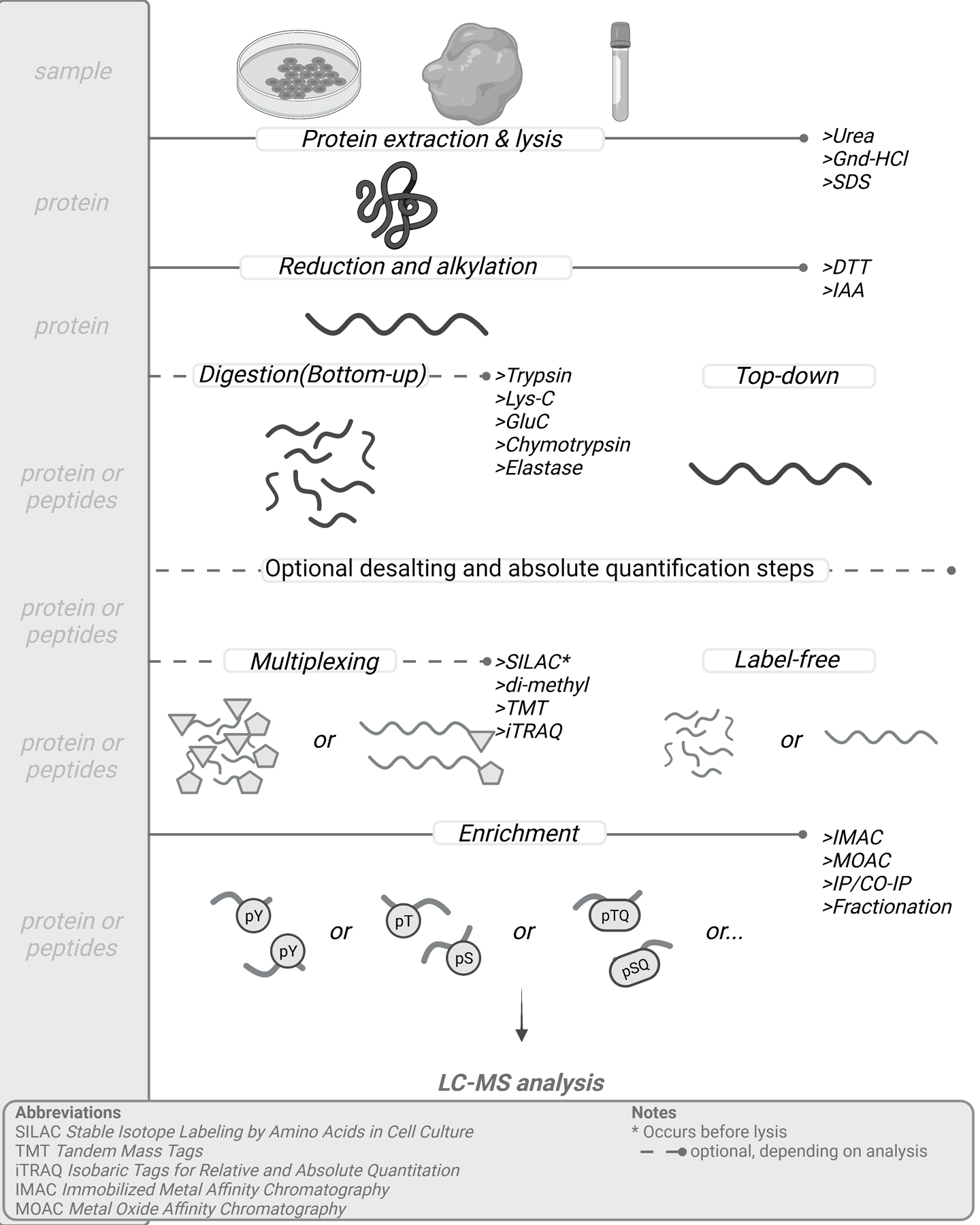
General sample preparation protocol for mass-spec based phosphoproteomic analysis. Protein is extracted from in vitro cell cultures or in vivo tissue or liquid samples, followed by chemical modification. In bottom-up proteomics, proteins are digested into peptides. Quantification can be performed by multiple methods, including non-isobaric and isobaric labeling, many of which are applied after digestion and before enrichment. Phosphopeptide enrichment purification can be performed at the global or subset-enrichment level depending on the biological question or experimental goals. Enrichment steps are critical in ensuring detection of these low-abundant peptides before analysis. Created with biorender.com
Following cell lysis, denatured proteins are typically reduced and alkylated, most commonly using dithiothreitol (DTT) and iodoacetamide (IAA), respectively.[18] After chemical modification, digestion of proteins into peptides is performed in bottom-up proteomics. Trypsin is the most widely used protease, as it cuts C-terminal to arginine and lysine residues; due to their frequency in the proteome, trypsin tends to produce peptide lengths that are compatible with MS analysis [31]. Additional benefits include the wide availability and high specificity of trypsin. Peptides generated by trypsin proteolysis tend to have improved ionization and fragmentation due to the basicity of the arginine and lysine residues at the C-terminus, thereby enhancing sequencing by MS. As an alternative choice, Lys-C cleaves C-terminal to lysine and is compatible with a range of different buffer conditions. Lys-C is commonly used in combination with trypsin in a double-digestion protocol, with the goal of reduced missed cleavages [32]. While trypsin is the most commonly preferred protease, it is worth noting that many phosphorylation sites fall in regions of the protein that are not amenable to MS-analysis following trypsin digestion. To gain deeper coverage of the phosphoproteome, some researchers combine data resulting from proteolysis with multiple enzymes (typically a single enzyme at a time), including GluC (V8 protease), Chymotrypsin and Elastase [33–35]. Regardless of the protease used, it must be ensured that the lysis buffer used is compatible with the protease or diluted sufficiently as not to inhibit protease activity [36].
Following digestion, phosphoproteomic workflows can take multiple directions. Some groups choose to desalt peptides prior to chemical modification (e.g., non-isobaric or isobaric labeling, see below), while other applications may skip these steps and move straight to phosphopeptide enrichment. It is worth noting that the optimal workflow for a given application may not follow either of these approaches. We recommend developing a simple protocol with minimal unnecessary steps that could lead to increased peptide loss, especially for enrichment and analysis of low-level phosphopeptides from small amounts of starting material.
Due to the low stoichiometry of phosphoproteins compared to non-phosphorylated proteins, enrichment is a key step towards successful detection of phosphorylated peptides [37]. For large-scale phosphoproteomic analysis, phosphopeptide enrichment is typically performed by either immobilized metal affinity chromatography (IMAC) or by metal-oxide affinity chromatography (MOAC). Both of these techniques depend on the affinity and coordination of negatively charged phosphate groups towards positively charged metal ions such as Fe3+ (IMAC) and Ti4+ (MOAC). [38] Since binding to the metal occurs through the phosphate moiety, phosphorylated serine, threonine, and tyrosine appear to be enriched equivalently, although phosphorylated serine (pSer) and phosphorylated threonine (pThr) constitute approximately 90% and ~9–10% of the phosphoproteome, respectively, with phosphorylated tyrosine comprising the remaining 0.1–1%. Since the selectivity of most metal ions for phosphorylated peptides is often imperfect, nonspecific binding of non-phosphorylated peptides can confound the analysis, especially for low-level samples [10,39]. Non-specific binding can be mitigated through a variety of techniques, including addition of organic acids to MOAC-based enrichment, chemical modification of carboxylate groups, or by using a nitrilotriacetic acid (NTA)-based resin for Fe3+-IMAC. While several studies have pointed to the complementary nature of IMAC and MOAC in phosphopeptide enrichment, [40] using an excess of either material enables similar degree of enrichment, and thus selecting either IMAC or MOAC should suffice for most large-scale phosphoproteomic analyses [41]. For most applications, sufficient depth of coverage can be achieved with a simple one-step enrichment protocol (e.g., NTA-Fe3+ IMAC spin columns), and additional depth can be afforded by fractionation of the sample prior to phosphopeptide enrichment of each fraction [42].
Using any of the above strategies to enrich phosphopeptides from mammalian cell lysate for analysis on a high-resolution mass spectrometer, it is fairly straightforward to identify thousands of high confidence phosphopeptides from a single sample. While large-scale data generation has become relatively easy, unfortunately, gaining biological understanding from this data remains challenging, largely due to the complexity of the phosphoproteome. Moreover, the overwhelming abundance of pSer sites are generally static (e.g., unaltered by a given biological perturbation), and their role in regulating the biology of the system is poorly understood. For instance, in one of the early large scale phosphoproteomics analysis, Olsen et al. identified and quantified over 6,000 phosphosites in HeLa cells stimulated with EGF at several time points [43]. Despite this massive data set, ~85% of the phosphosites were unaffected by the stimulation conditions. As might be expected given their overall low abundance in the phosphoproteome only ~103 pTyr sites out of 6,600 total phosphorylation sites (~1.6%) were identified in this analysis, despite stimulation of the epidermal growth factor receptor (EGFR), a receptor tyrosine kinase (RTK) that is highly expressed in these cells. Similar results have been seen for a range of other systems, including recent large-scale phosphoproteomic studies of human tumors, where ~34,000 total phosphopeptides were identified and quantified, yet only ~2% of these were pTyr sites [44,45]. Since aberrantly activated tyrosine kinases are known to be oncogenic and tyrosine phosphorylation sites on kinase activation loops are known to be critical regulators of kinase activity, the paucity of tyrosine phosphorylation sites in these data sets would suggest that some of the potentially most informative signals might be missed in these large-scale studies [46–48].
3. Enrichment of subsets of the phosphoproteome
In order to access low-abundance phosphorylation sites within the complex, high dynamic range phosphoproteome, it is often necessary to perform an additional enrichment step. For instance, if the goal is to identify and quantify RTK-driven signaling networks in a given cell line or tissue specimen, specific enrichment for pTyr-containing proteins or peptides will significantly improve the analysis [49–51]. Perhaps the most common method for pTyr-enrichment involves using pan-specific anti-pTyr antibodies to immunoprecipitate pTyr-containing peptides following tryptic digestion [49,50]. There are multiple pan-specific anti-pTyr antibodies available commercially; while each one has affinity for a range of pTyr sites, each tends to have some degree of bias. We have found that mixing several antibodies together can give greater overall coverage of the pTyr sites in a given biological sample [52,53]. As with most enrichment methods, the amount of non-specific binding in pTyr-immunoprecipitations tends to increase with decreasing sample amount. To address this issue, a second stage of enrichment (e.g., with an Fe3+-NTA IMAC spin column) can be used to reduce the level of non-phosphorylated peptides present in the pTyr IP. Alternative strategies to enrich pTyr peptides or proteins have been explored. In cell signaling networks, Src-homology 2 (SH2) domains bind to pTyr sites within a protein or on a different protein to regulate activity and protein-protein interactions (i.e., in the context of recruitment of adaptor proteins to an activated RTK) [54]. Taking advantage of their binding affinity, several groups, including by Bian et al., have utilized SH2 domains for enrichment of pTyr containing peptides, and have also engineered SH2 domains for increased binding affinity [55,56]. These “SH2 superbinders” have enabled enrichment of thousands of pTyr sites, yet different SH2 domains appear to have different specificity, thus sequential analysis or mixing SH2 domains for a given analysis might provide increased depth of coverage [57].
The strategy of enriching phosphorylation site subsets for deeper analysis can be applied to a range of biological applications requiring cellular signaling network analyses. For instance, cells respond to many environmental contaminants and cytotoxic chemotherapies by initiating a DNA damage response; in both contexts this response is critical for halting the cell cycle, fixing DNA lesions / adducts, and enabling the cell to survive the damage. To identify critical regulators of the DNA damage response, cells were treated with ionizing radiation and phosphopeptides were enriched using a combination of phosphorylation site specific antibodies [58]. In this study, the non-specificity of the antibodies resulted in enrichment and identification of hundreds of phosphorylation sites potentially involved in DNA damage response signaling networks. An alternative approach is to use phospho-motif specific antibodies that recognize phosphorylation sites within a particular amino acid sequence. For instance, ATM and ATR kinases tend to phosphorylate serines or threonines followed by a glutamine; ATM/ATR substrate phospho-motif antibodies therefore enrich pSer or pThr followed by glutamine (e.g., pSQ/pTQ) [59]. Using these antibodies for peptide IP from proteolyzed cell lysate allows for the selective enrichment of hundreds of pSQ/pTQ containing peptides in a single analysis. Similarly, the ERK 1/2 mitogen activated kinases can regulate cell proliferation and tend to phosphorylate S or T residues in the context of a proline in the −2 position and in the +1 position. ERK 1/2 substrate motif antibodies can therefore be used to IP hundreds of peptides containing a PXXpSP or PXXpTP motif, where X represents any amino acid. In both of these cases, as with multiple other available phospho-motif specific antibodies, it is critical to note that all peptides that match these phosphorylation motifs are not necessarily substrates of the given kinase [60]. Indeed, many peptides that happen to have the motif of interest are quantitatively unaffected by activation or inhibition of the kinase of interest. As with pTyr enrichment, the complexity of the phosphoproteome tends to obscure many of these phosphomotif-containing peptides; recent large-scale phosphoproteomics data sets contain tens of pSQ/pTQ peptides, while specific enrichment using phospho-motif antibodies would likely provide over five hundred pSQ/pTQ-conatining peptides from the same sample [44,59]. It is worth noting that phosphorylation subset enrichments can be performed serially on the same sample to gain increased depth of coverage on multiple biological pathways [61]. For instance, cancer cells are often driven by aberrantly activated RTKs that signal through the ERK 1/2 MAP kinases to drive proliferation while also activating protein kinase B (AKT) to promote cell survival. Proliferating cells incur DNA damage during replication and therefore activate a DNA damage response. Each of these networks could be interrogated in a given biological sample, or across multiple biological samples, by serial IP using pan-specific and phospho-motif specific antibodies, followed by global phosphoproteomics on the resulting supernatant.
4. Quantitative phosphoproteomics methods
As mentioned above, with either subset enrichment strategies or with global phosphoproteomics analyses, it is now possible to identify thousands to well over 10,000 phosphorylation sites from a given biological sample, respectively. Despite this plethora of data, gaining biological knowledge from these analyses is still quite challenging, as identification of a phosphorylation site on a protein in a given sample does not necessarily mean that the site is regulatory of the biological processes in that sample. To infer biological knowledge from phosphoproteomics, quantitative data comparing phosphorylation levels across different conditions (i.e., cell stimulation with a mitogen, treatment with a given kinase inhibitor, or treatment with a chemotherapy agent, among others) is often required. Multiple label-free or label-based quantification strategies can be employed to determine the relative abundance of phosphorylation sites between various conditions of a biological system [62,63]. The simplest of these approaches is label-free quantification, in which the amount of a given phosphorylation site, typically estimated by the area under the curve (AUC) of the chromatographic elution profile of the precursor ion, is compared across phosphoproteomic analyses of different biological conditions. This approach is straightforward, can be highly quantitative, does not require additional sample handling, and can be used to compare across hundreds of samples without requiring a normalization channel. Moreover, since quantification typically occurs from the precursor signal intensity, dynamic range signal compression, e.g., from MS/MS-based quantification techniques, see below, may be less of an issue. On the other hand, the vast number of phosphopeptides present in global phosphoproteomics experiments can lead to highly complex full scan mass spectra (MS1 spectra), and may result in overlap of isotope envelopes and inaccurate quantification, especially on mass spectrometers with lower resolving power. Additionally, sample handing and chromatographic reproducibility are critically important for accurate label-free quantification. More complex workflows, including phosphopeptide IP’s, can adversely affect the accuracy of label-free quantification. Early versions of label-free quantification relied on spectral counting, in which the number of MS/MS spectra for a given precursor were compared across different conditions. This approach is most accurate when applied to highly abundant peptides and can be confounded by low-level peptides (typically less than 5 spectral counts), where stochasticity between runs can confound quantitative accuracy.
As an alternative to label-free quantification, multiplexed, label-based strategies have been developed. These approaches fall into two categories: non-isobaric labels (MS1 quantification) and isobaric labels (MS/MS quantification). Non-isobaric labels, as the name implies, utilize different numbers of heavy isotopes on each tag to generate labels for each sample that differ in mass. Non-isobaric labels can be applied at multiple steps in sample generation or sample processing. Stable Isotope Labeling with Amino Acid in Cell Culture (SILAC), as described by Mann’s group, is one of the most commonly used non-isobaric multiplexing methods [64]. In SILAC, cells from one condition are cultured in media that contains one or more heavy-isotope labeled amino acids, while cells from another condition are cultured in media with corresponding light-isotope labeled amino acids. Lysine or arginine are most commonly used for labeling to ensure that tryptic peptides will be quantifiable. Although SILAC labeling can be expensive, especially for in vivo applications [65,66], the protocol is relatively easy to implement, and since labeling occurs during cell culture (or organism growth), samples can be mixed early in sample processing and quantification is relatively unaffected by variation in sample handling. However, SILAC is typically limited to multiplexing of 2 or 3 conditions and results in increased complexity of full scan mass spectra that can adversely affect quantification dynamic range [67]. SILAC has frequently been used in combination with phosphoproteomics to quantify cancer cell signaling networks [68–71]. For instance, Zhang et al. combined pTyr IP with SILAC to quantify the adaptive response of lung adenocarcinoma cells to EGFR inhibition [72], and Cunningham et al. combined SILAC with global phosphoproteomics to define fibroblast growth factor receptor (FGFR) signaling networks in triple negative breast cancer [73].
Beyond SILAC, non-isobaric chemical labels have also frequently been used for quantitative analysis of phosphorylation-mediated signaling networks. In this approach, peptides from each sample are chemically modified after proteolytic digestion, with each sample being tagged with the same label with different number of heavy isotopes. For instance, di-methyl labels, with one sample labeled with 12C-H2 formaldehyde and other samples labeled with 12C-D2 or 13C-D2 formaldehyde offer an inexpensive alternative to SILAC and have been used to elucidate phosphorylation changes following cell stimulation in a variety of contexts [74,75]. This approach is similarly limited to multiplexing of 2 or 3 samples and can be affected by complexity of MS1 spectra due to the combination of multiple non-isobarically tagged samples.
Isobaric tags label all samples with a tag of the same mass; quantification occurs in MS/MS through production and detection of reporter ions generated during fragmentation [76,77]. Isobaric tags enable multiplexing of up to 18 samples [78] in a single analysis with relatively minimal increase in the complexity of MS1 spectra. This high degree of multiplexing reduces MS analysis time while also reducing inter-analysis variability, including chromatographic differences and precursor selection for MS2. While inter-analysis irreproducibility in discovery-mode analyses can still be a problem for larger sample sets [27], 16- or 18-plex isobaric tags significantly reduce this potential problem. Since the m/z ratio for each tagged peptide is identical from all samples, isobaric tags have been used for ‘boost’ experiments for phosphoproteomics, among other applications, wherein one of the samples is present in much higher amount, improving the signal-to-noise ratio in full scan mass spectra and driving selection of peaks for MS/MS. This approach can improve sensitivity, enabling analysis of smaller amounts of samples in the ‘non-boost’ channel, but may lead to decreased quantitative accuracy [79]. Dynamic range compression, one of the major potential problems with isobaric labels, is thought to be due to co-isolation of multiple peptides for MS/MS. This problem is exacerbated in complex mixtures, including global phosphoproteomics, due to the massive number of peptides in the sample. Although narrowing the isolation window can reduce dynamic range compression, MS3 may provide more accurate quantification as demonstrated by McAllister et al. [80], but may not be applicable to low-abundance peptides including pTyr and phospho-motif enriched peptides [62,79]. Enriched subsets of the phosphoproteome tend to be much less complex, and thus dynamic range compression is less of an issue, although it can still adversely affect quantitative accuracy. In addition to dynamic range compression, isobaric labels tend to be similar in cost to SILAC, and introduce at least one additional step in the workflow, thereby risking sample loss. Even with these issues, isobaric labeling has enabled a wide range of quantitative phosphoproteomic analyses, including quantification of pTyr dynamics following growth factor stimulation [81,82], quantification of pTyr signaling networks from formalin fixed parrafin embedded (FFPE) sections of human tumor specimens by Kohale et al. [26], and quantification of adaptive response to therapy in cancer models and tumor tissues [63,83,84], among many others.
5. MS-based analysis of the phosphoproteome: applications and challenges
Proteomics experiments can be generally divided into top-down / middle down and bottom-up proteomics. In top-down proteomics, intact proteins are ionized and analyzed by mass spectrometry (MS) in the absence of a digestion step [85]. In middle-down proteomics, typically a single digestion step, e.g., Cyanogen bromide (CNBr)-digestion, is used to produce a few large protein fragments which are then ionized for MS analysis [86]. Both of these approaches enable identification of proteoforms: the post-translation modification ‘code’ present on a given protein, although middle-down approaches can suffer from data integration requirements to reconstitute the intact protein [87,88]. Although top-down and middle-down proteomics have been extensively used to analyze the post-translational modification code of histones, to date there are only a few applications of these approaches to the phosphoproteome [89–92], with no in-depth analysis of signaling network alterations between conditions. The dearth of top-down phosphoproteomics experiments might be due to the inherent challenges of top-down proteomics, as characterization of intact proteins, and especially PTMs on intact proteins, can require significant time, effort, and expertise. Additionally, dynamic PTMs such as phosphorylation are highly challenging, as each new phosphorylation site results in a new proteoform, and thus a dozen or more sites on a given protein may lead to a combinatorial explosion of protein states. With that said, top-down and middle-down phosphoproteomics are needed to define the associations between phosphorylation sites across a given protein and to answer fundamental questions, including whether EGFR or other RTKs are phosphorylated on multiple sites on the C-terminal tail in a given isoform, or whether each of the multiple sites are mostly exclusive to a given isoform. Almost all phosphoproteomics experiments have been performed by bottom-up proteomics, where proteins are proteolyzed to peptides, typically using enzymes such as trypsin (see above). In general, bottom-up proteomics experiments are much easier compared to top- or middle-down experiments, but this ease of analysis comes at the cost of information that may be critical for understanding protein function and signaling network mechanisms. For instance, can phosphorylation of an inhibitory site, e.g., the C-terminal phosphorylation site on Src-family kinases, co-occur with phosphorylation of the activation loop? Since these sites are separated by ~100 residues, this information is only available by top-down or middle-down analyses.
Bottom-up proteomics can be sub-divided into three subcategories based on the data acquisition method: data-dependent acquisition (DDA), data-independent acquisition (DIA) and targeted phosphoproteomics [93](Figure 2). These methods mainly differentiate from each other by the manner in which specific ions are chosen for fragmentation. DDA, also known as discovery mode, generally selects the most abundant peptides for isolation and fragmentation. When coupled with fractionation, DDA experiments can provide deep characterization of the phosphoproteome [61,94,95], and are still the most common approach to signaling network analyses. Although the total number of detected peptides is generally higher with DDA compared to DIA or targeted approaches, DDA tends to suffer from run-to-run irreproducibility of identified and quantified peptides, due to the stochastic nature of peptide sampling. DDA-based quantification of cancer signaling networks in cell lines and tumors can suffer from the ‘sparse-matrix’ problem, where only a subset of phosphorylation sites are quantified across all samples, even when coupled with multiplexed isobaric reagents [27,96,97]. In DIA, pre-set windows of m/z ratios are sequentially isolated, fragmented, and analyzed, with the goal of covering the full m/z ratio rapidly enough to fragment and analyze all peptides present in a given sample. Since peak selection is absent in DIA, run-to-run reproducibility tends to be much greater compared to DDA.[98] However, depending on the width of the isolation window and the sample complexity being analyzed, DIA can be adversely affected by dynamic range, e.g., loss of the lowest abundance fragments in the presence of highly abundant fragments. Due to the complex MS/MS spectra, DIA is also best applied with a library of spectra, typically obtained from multiple DDA experiments, and is suboptimal for discovery. However, more recent DIA experiments have utilized smaller isolation windows that are on-par with DDA experiments; these new settings suggest a powerful combination of high reproducibility with the potential for discovery of novel signaling components. Quantification with DIA tends to occur through label-free, as the duty cycle for full scan MS is suboptimal for non-isobaric labels, and the mixed MS/MS spectra are suboptimal for isobaric labels. As mentioned above, label-free quantification for DIA can be problematic when working with more complex workflows involving IP of pTyr or phospho-motifs, both of which are highly susceptible to slight variations between sample processing steps. Nevertheless, DIA methods have been applied to analysis of signaling networks for multiple applications [99–101], and new tools are emerging to facilitate these approaches [102].
Figure 2.
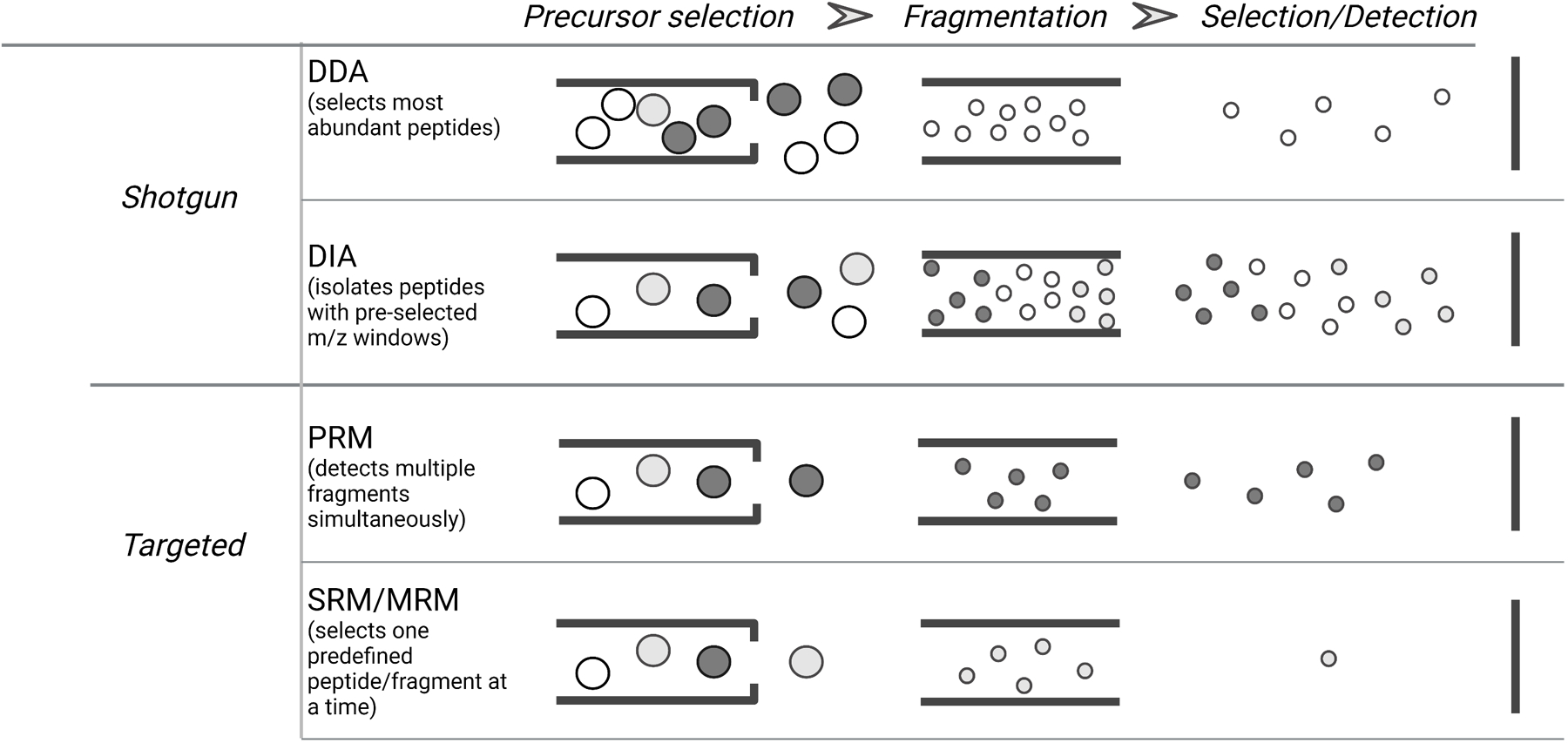
Types of MS data acquisition modes in bottom-up proteomics. data dependent acquisition (DDA) selects the most abundant peptides which are then isolated and fragmented sequentially. Data Independent Acquisition (DIA) isolates and fragments peptides within defined m/z windows. In targeted acquisition mode, the instrument is set up to detect and fragment selected peptides that are defined a priori. In parallel reaction monitoring (PRM), multiple fragments per precursor can be detected at the same time. In SRM or MRM, fragment ions from a given precursor are typically detected sequentially. Created with biorender.com
Targeted phosphoproteomics provides consistent reproducibility with high accuracy quantification and can provide high sensitivity, especially when coupled with internal standard trigger peptides, but it comes at the cost of discovery and coverage as shown in our research group [27]. As the name implies, targeted methods require prior knowledge about the signals (e.g., phosphopeptides) of interest, as the instrument method is typically constructed to select the precursor m/z ratios of interest for fragmentation and quantification. Targeted methods typically utilize multiple reaction monitoring (MRM) or parallel reaction monitoring (PRM) based methods to isolate and fragment the precursor of interest. In MRM, specific fragment ions are detected sequentially, typically on a triple quadrupole MS, while in PRM, all fragment ions are detected in the same analysis, on an ion trap or TOF MS. Although targeted methods have historically been used to quantify a few targets per analysis, MRM-based targeted phosphoproteomics have been used to monitor signaling networks. In one example application by Wolf-Yadlin et al., 226 pTyr-containing peptides were quantified by MRM to assess the dynamic signaling network response to EGF stimulation at multiple time points [103]. MRM methods have also been used to quantify the DNA damage response to chemotherapy; in this example, a combination of phospho- and non-phosphopeptides were targeted for quantification [104,105]. In targeted analyses with a large number of targets, elution time windows have allowed for longer detection times while maintaining high duty cycles. More recently, heavy-isotope labeled internal standard (IS) trigger peptides have bypassed the need for elution time windows; in this approach, fragmentation of the heavy labeled IS trigger peptide is coupled with on-the-fly pseudo-spectral matching to a known fragment ion fingerprint [106]. Matching of multiple fragment ions initiates high resolution MS/MS of the endogenous peptide, and quantitative data can be generated by comparing the light:heavy ratio of multiple fragment ion peaks. The SureQuant framework available on selected ThermoFisher MS instruments allows for monitoring of hundreds of IS-peptides. We have recently developed an approach (SureQuant pTyr) to quantify pTyr signaling networks comprised of ~400 pTyr peptides in EGF-stimulated cell lines and across dozens of human colorectal tumor specimens [27]. Although SureQuant pTyr provides quantitative data for each detected peptide in each sample, quantification relies on one-point calibration and assumes a linear dynamic range. To generate absolute quantification data with internal standard curves for each peptide, we developed Multiplex Absolute Regressed Quantification with Internal Standards (MARQUIS), a multiplexed MRM- or PRM-based targeted approach in which different amounts of heavy-isotope labeled IS phosphopeptides were added to each biological sample. Following the addition of these reference peptides, multiple samples were isobarically labeled and combined for analysis [107]. Reporter ions from MS/MS of the TMT-labeled endogenous peptides could then be regressed against the internal standard curve provided by MS/MS of the TMT-labeled IS peptides. Application of this approach to ~20 pTyr phosphorylation in the EGFR signaling network provided absolute quantification, e.g., copies/cell, for the temporal dynamic profiles for each phosphorylation site and relative stoichiometry information for multiple sites on the EGFR C-terminal tail. In the future, combining a MARQUIS-style approach with SureQuant pTyr may provide absolute quantification of hundreds of nodes in the network, enabling more mechanistic computational modeling of cancer cell signaling networks.
6. Computational analysis of phosphoproteomic data
While technological and methodological advancements have greatly expanded the capabilities of phosphoproteomics, the ultimate impact depends on the biological knowledge that can be gleaned from the data. In this section, steps in data analysis as well as potential tools to gain biological insight will be discussed (Figure 3).
Figure 3.
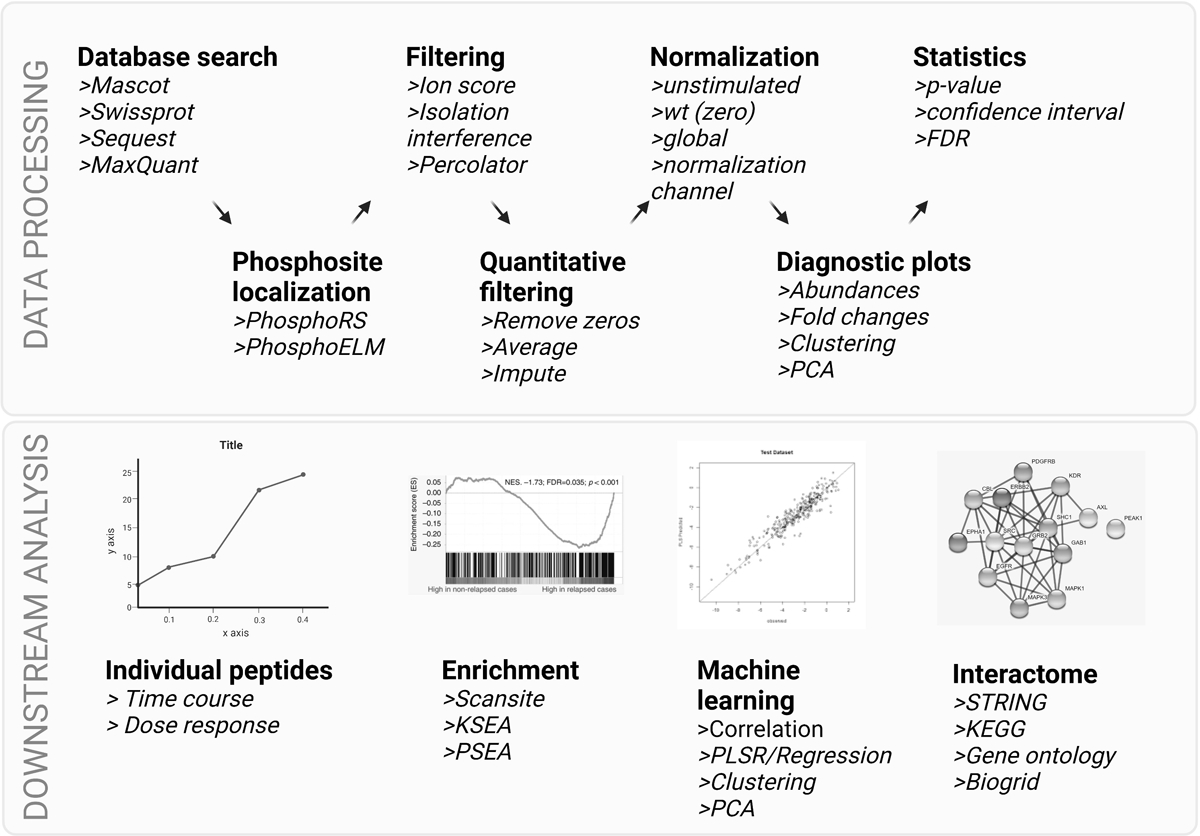
Steps in data processing and downstream analysis tools for phosphoproteomic data. Top: multiple data processing steps are needed in order to identify and quantify phosphorylation sites from MS/MS data, including database searching, site localization and quality filtering for identifications. Depending on the quantitative method, data may go through additional filtering, normalization, clustering, and statistical analysis as a first pass at identifying differentially phosphorylated peptides. Bottom: more nuanced biological information can be gained through additional computational analysis, including temporal analysis, kinase / substrate or pathway enrichment of phosphorylation subsets, and machine learning approaches to identify modules and pathways of phosphorylation-mediated signaling networks. Each of these tools can lead to predicted functions for phosphorylation sites in the data. Validation experiments should be performed to confirm analysis results. Created with BioRender.com
High quality / high accuracy phosphopeptide identification and quantification can be critical for defining cancer cell signaling networks from phosphoproteomic data [108]. Since each phosphopeptide is typically a ‘one-hit wonder’, high stringency in data filtering is recommended to remove as many false positive assignments as possible and to localize the phosphorylation site to the correct residue, if possible. While restrictive filters may compromise the ultimate data set size, high quality data facilitates computational data analysis and ultimate biological insight.
A vast number of computational methods have been developed to extract cellular signaling network information from phosphoproteomic data [109]; here we will highlight a few general computational data analysis strategies as well as some specific instances where tools have been applied to provide novel biological insight. As a first pass approach, clustering methods, including hierarchical clustering, k-means-clustering, self-organizing maps, and others, have been applied to reveal associations between phosphorylation sites and to thereby define ‘modules’ of co-regulated sites across different biological conditions. In some cases, clustering has helped to predict the function of unknown phosphorylation sites, for instance when they are co-regulated with multiple sites of known function [43,49,110]. Although different clustering methods can be applied to extract additional information from a given data set, combining the results from multiple clustering methods can reveal consensus modules and provide novel insight. As one example of this strategy, the multiple cluster analysis methodology (MCAM) algorithm was developed by Naegle et al. Application of MCAM to dynamic phosphotyrosine data uncovered phospho-site specific interactions, including a novel interaction between EGFR phosphorylation and the PDLIM1 cytoskeletal protein [111,112]. Co-correlation can also be used to identify phosphorylation sites that are co-regulated across multiple biological conditions. This approach was applied to studies of pTyr signaling networks in glioblastoma and non-small cell lung cancer tumor tissues to highlight patient-specific activated signaling networks [26,113,114]. To gain additional insight into signaling networks from large-scale phosphoproteomic datasets, multiple computational approaches have been developed to predict kinase-phosphorylation site associations. These algorithms, e.g., ptmRS and Thesaurus, typically rely on kinase motifs, known substrates, or a combination of motifs, substrates, and protein-protein interactions to predict kinases that may be responsible for phosphorylating particular sites in the data [115–122]. As with all predictive algorithms, the results from these analyses should be confirmed through additional experiments, e.g., by chemical or genetic perturbation of the kinase.
Integrative ‘omics, combining information from several different types of data, can provide additional insight into the structure and function of phosphorylation mediated signaling networks. As one example, proteogenomics, combining proteomic and phosphoproteomic data with genomic and transcriptomic data has been used to classify subtypes of different human tumors and identify putative activated signaling networks and central nodes [44,123–125]. Other integrative ‘omics tools have combined proteomics, phosphoproteomics, transcriptional profiling, and metabolomics to attempt to characterize the role of phosphorylation sites in regulating the state of a given system and to identify targeted nodes [44,126–128]. Another computational tool that allows integration of multiple datatypes is partial least squares regression (PLSR), which utilizes input matrices X (e.g., phosphoproteomic dataset) and Y (e.g., phenotypic outcome such as proliferation) to build a predictive model correlating associations between phosphorylation sites and phenotypic outcome. We have previously used this approach to identify pTyr sites associated with migration and proliferation in HER2 overexpressing mammary epithelial cells and to highlight a counter-intuitive role for the ERK 1/2 MAP kinases in EGFRvIII expressing glioma cells [96,97,129]. Frejno et al. used a PLSR-based analysis on multi-omics data to determine the landscape of proteome activity in a large set of cancer cell lines, with the goal of predicting drug response and novel functional associations from these networks [130]. Other approaches have been developed over the years to integrate different types of omics as well as non-omics data, such as immunohistochemical data [83,131].Computational analysis can be challenging with discovery-mode proteomic or phosphoproteomic datasets, due to the sparse matrix problem associated with missing observations between biological replicates or different biological conditions. From a computational perspective, one option is to disregard all peptides that are not quantified across all conditions; unfortunately this approach radically reduces the dataset size [96]. [An alternative approach is to impute quantitative values for the missing data, typically zeros, or an average of the remaining channels. Although this allows conservation of more peptide information, it risks false hypothesis generation in downstream analysis, as it assumes high similarity between biological conditions [132]. Targeted data acquisition approaches (see above) help to reduce the sparse matrix problem and should enable improved computational analysis.
7. Future of phosphoproteomics
MS-based phosphoproteomics has made great strides over the past decade in terms of detection limits, speed, accuracy and resolution. Phosphoproteomics has emerged as a powerful tool for analysis of signaling networks in diseased tissues and model systems, both in vivo and in vitro. Nonetheless, one of the main challenges in MS-based phosphoproteomics is the application to limited sample amounts / low abundance model systems. Progress in this area has enabled successful phosphoproteomic analysis on extracellular vesicles secreted from cancer cells to identify potential biomarkers in glioblastoma-EGFRVIII variant [133]. Additionally, methods have emerged that address the challenge of limited starting material when working with patient tissue specimens [134], and phosphoproteomic analyses have now been successfully performed on FFPE tissue samples [26,135]. Simultaneously, given the value of patient samples and the importance of multi-omics analysis, methods are being developed that allow for simultaneous extraction of DNA, RNA and protein from samples [136].
Another challenge for phosphoproteomics is spatial analysis, due to the highly dynamic nature of this PTM. With the use of proximity labeling strategies, Liu et al. demonstrated the ability to monitor altered phosphorylation patterns due to ER stress in in vitro and in vivo systems [137]. We previously used phosphoproteomics to characterize the immediate-early signaling dynamics in the EGFR network and proximity ligation assays (PLA) to characterize dynamic recruitment of adaptor proteins to the membrane [82], or total internal reflection fluorescence (TIRF) microscopy to monitor in vivo SH2 binding dynamics and binding site kinetics [138]. Being able to directly quantify spatially resolved signaling networks by MS-based phosphoproteomic analysis has yet to be accomplished.
Another fascinating development in the field is single cell proteomics (scProteomics) [139]. This type of analysis has the potential to provide information about co-occurrence of phosphorylation sites and states of individual cells and would therefore enable significantly improved definition of cellular signaling networks. Although progress has been made at the proteome level using nanodroplet sample preparation platforms, phosphopeptide analysis remains challenging, as most signaling nodes are well below detection limits thus far [140].
Continued improvements in MS instrumentation, phosphoproteomic methodology, and computational modeling algorithms will facilitate the application of MS-based phosphoproteomics to additional systems, including tissue microarrays, needle biopsies, and rare liquid tumors, and the interpretation of this data to define activated signaling networks in these tissues.
8. Expert opinion
Quantitative MS-based phosphoproteomics has begun to emerge as a powerful technique enabling the identification of activated oncogenic signaling networks from a variety of biological systems, including in vitro and in vivo model systems as well as patient tumor tissue specimens. While global phosphoproteomics can provide an impressive overview of the phosphorylation state of thousands of proteins within a given sample, this analysis method may fail to identify and quantify low-abundance sites, e.g., pTyr and many oncogenic kinase substrates, that may be critical regulators of biological function, especially in cancer cell signaling. Accessing these low abundance sites requires enrichment of selected subsets of the phosphoproteome, often through immunoprecipitation. These experiments can be more technically challenging compared to global phosphoproteomics, as the low amount of phosphorylated peptides are strongly impacted by sample losses during processing steps. Nonetheless, pTyr phosphoproteomics can provide insight into activated tyrosine kinases regulating a range of oncogenic phenotypes, including cellular migration, proliferation, invasion, therapeutic resistance, among others. Advances in pTyr proteomics, including higher affinity reagents for pTyr enrichment, targeted approaches for monitoring selected pTyr phosphorylation sites, and MS-instrumentation improvements to provide enhanced sensitivity, have begun to make this approach more turn-key and accessible to a wider range of proteomics research labs and core facilities.
The next step in the evolution of phosphoproteomics and especially for pTyr proteomics may be development of a CLIA (Clinical Laboratory Improvement Amendments) approved analysis workflow to provide clinical insight for patient stratification and therapeutic selection based on signaling network activation in clinical samples. Moving to CLIA approval will require standardization of standard operating procedure (SOPs), dedicated instrumentation, and highly reproducible quantification. Although difficult to implement, CLIA approval may be facilitated by targeted approaches such as SureQuant pTyr, where heavy-isotope encoded standard peptides are included in every analysis, yielding accurate quantification even in the context of sub-optimal pTyr enrichment. Recently we implemented SureQuant pTyr to identify activated signaling networks in dozens of human colorectal tumor specimens; importantly, this method was performed with commercially available columns, reagents, and instrumentation, and thus should be accessible to most, if not all, proteomics facilities.
Phosphoproteomics continues to be limited by sample requirements, with many studies requiring relatively large amounts of frozen tissue specimens for in vivo studies, or multiple plates of cells per condition for in vitro studies. It is worth noting that multiple labs have been developing improved methods to enable analysis of signaling networks from smaller amounts of starting material, and continued developments over the next several years should enable the analysis of pTyr signaling networks from tissue microarray (TMA) samples, or from small numbers of rare cell types. While starting material requirements may not rival genomic or transcriptional profiling experiments, both of these technologies have amplification steps that are currently not possible with proteomic approaches. Importantly, without amplification it is challenging to perform phosphoproteomics at the single cell level. While biologically relevant phosphorylation sites span a large range of copy numbers in different cell types, we can take, as an example, a phosphorylation site that may be present at a reasonably high expression level of 10,000 copies/cell. Detection of this site in a single cell would require low-zeptomole sensitivity (10,000 copies/cell equates to ~1.7×10−20 moles/cell (17 zeptomoles/cell), assuming no losses in sample processing and high efficiency ionization. At this point, it seems that single cell phosphoproteomics might be limited to very high expression phosphorylation sites, which may be less informative regarding activated signaling networks. Continued technological development is clearly required to advance this aspect of the field forward. Non-MS-based approaches, including immunohistochemistry, immunofluorescence, and phosphoflow can accomplish single-cell signaling network analysis, albeit with limited and targeted detection of selected phosphorylation sites.
One other challenge for phosphoproteomics is data integration with other systems-level measurements that may be performed on additional aliquots of the same samples. For instance, integration of phosphorylation data with transcript expression (bulk or single cell), metabolomics, methylation, genome sequencing, and other ‘omics measurements, will provide significant insight into the co-regulation of these often dynamic regulatory networks. While some methods (see above) have been developed to attempt to integrate these data, our understanding of how transcript expression is related to signaling network regulation, and vice-versa, is still elementary. Development of improved computational algorithms, including artificial intelligence / machine learning approaches, are needed to define the linkages between these often highly complex data sets.
Over the past two decades the field of phosphoproteomics has experienced explosive growth in new tools, technologies, algorithms, and applications that have advanced the field to enable deep biological and clinical insight. Continued development of the field will lead to more stable technologies that can be applied to identify therapeutic targets, track therapeutic efficacy in clinical trials, and identify non-genomic adaptive response / resistance mechanisms that adversely impact patient survival.
Article Highlights:
Phosphopeptide enrichment methods
Tyrosine phosphoproteomics
Phospho-motif enrichment for kinase substrates
Quantitative phosphoproteomics
Data acquisition methods for phosphoproteomics
Computational approaches to identify activated signaling networks
Acknowledgments
Funding
This manuscript was funded in part by NIH grants U54 CA210180, U01 CA238720, U01 CA215709, and R01 GM139998.
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. Funders of this work had no influence on its content.
References Cited
- [1].Humphrey SJ, James DE, Mann M. Protein Phosphorylation: A Major Switch Mechanism for Metabolic Regulation. Trends Endocrinol Metab 2015. p. 676–687. [DOI] [PubMed] [Google Scholar]
- [2].Fuhs SR, Hunter T. pHisphorylation: the emergence of histidine phosphorylation as a reversible regulatory modification. Curr. Opin. Cell Biol 2017;45:8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fuhs SR, Meisenhelder J, Aslanian A, et al. Monoclonal 1- and 3-Phosphohistidine Antibodies: New Tools to Study Histidine Phosphorylation. Cell 2015;162:198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schmelzle K, White FM. Phosphoproteomic approaches to elucidate cellular signaling networks. Curr Opin Biotechnol 2006. p. 406–414. [DOI] [PubMed] [Google Scholar]
- [5].Tsuji S, Ohno Y, Nakamura S, et al. Temozolomide has anti-tumor effects through the phosphorylation of cPLA2 on glioblastoma cells. Brain Res 2019;1723. [DOI] [PubMed] [Google Scholar]
- [6].Mao L, Zhan Y, Wu B, et al. ULK1 phosphorylates Exo70 to suppress breast cancer metastasis. Nat. Commun 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Xiao M, Xie J, Wu Y, et al. The eEF2 kinase-induced STAT3 inactivation inhibits lung cancer cell proliferation by phosphorylation of PKM2. Cell Commun. Signal 2020;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell; 2011. p. 646–674. [DOI] [PubMed] [Google Scholar]
- [9].Ardito F, Giuliani M, Perrone D, et al. The crucial role of protein phosphorylation in cell signalingand its use as targeted therapy (Review). Int. J. Mol 2017. 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10]. Ficarro SB, McCleland M, Stukenberg PT, et al. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol 2002;20:301–305. ● This manuscript describes a method for radically reducing non-specific binding during phosphopeptide enrichment, thereby enabling large-scale phosphoproteomics analyses.
- [11].Carter AM, Tan C, Pozo K, et al. Phosphoprotein-based biomarkers as predictors for cancer therapy. Proc. Natl. Acad. Sci. U.S.A 2020;117:18401–18411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bekker-Jensen DB, Bernhardt OM, Hogrebe A, et al. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]. Sevecka M, MacBeath G. State-based discovery: a multidimensional screen for small-molecule modulators of EGF signaling. Nat. Methods 2006;3:825–831. ● In addition to highlighting the potential for analysis of signaling networks with reverse-phase protein arrays (RPPA), this manuscript also showed that most phospho-specific antibodies do not provide high accuracy quantitative data in the RPPA format.
- [14]. Krutzik PO, Crane JM, Clutter MR, et al. High-content single-cell drug screening with phosphospecific flow cytometry. Nat. Chem. Biol 2008;4:132–142. ● Single-cell phosphoproteomics using phospho-specific antibodies and mass cytometry.
- [15]. Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc. Natl. Acad. Sci. U.S.A 1980;77:1311–1315. ●● This manuscript was the first report of tyrosine phosphorylation and also estimated the frequency of tyrosine phosphorylation to be ~0.1–1% of the phosphoproteome.
- [16].Raffel S, Klimmeck D, Falcone M, et al. Quantitative proteomics reveals specific metabolic features of acute myeloid leukemia stem cells. Blood 2020;136:1507–1519. [DOI] [PubMed] [Google Scholar]
- [17].Nelson ME, Parker BL, Burchfield JG, et al. Phosphoproteomics reveals conserved exercise‐stimulated signaling and AMPK regulation of store‐operated calcium entry. EMBO J 2019;38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Morshed N, Ralvenius WT, Nott A, et al. Phosphoproteomics identifies microglial Siglec‐F inflammatory response during neurodegeneration. Mol Syst Biol 2020;16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kubiniok P, Finicle BT, Piffaretti F, et al. Dynamic Phosphoproteomics Uncovers Signaling Pathways Modulated by Anti-oncogenic Sphingolipid Analogs. Mol Cell Proteom 2019;18:408–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hu Y, Sun L, Zhang Y, et al. Phosphoproteomics reveals key regulatory kinases and modulated pathways associated with ovarian cancer tumors. Onco Targets Ther 2020;13:3595–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Scheidt T, Alka O, Gonczarowska-Jorge H, et al. Phosphoproteomics of short-term hedgehog signaling in human medulloblastoma cells. Cell Commun Signal 2020;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Malik N, Nirujogi RS, Peltier J, et al. Phosphoproteomics reveals that the hVPS34 regulated SGK3 kinase specifically phosphorylates endosomal proteins including Syntaxin-7, Syntaxin-12, RFIP4 and WDR44. Biochem J 2019;476:3081–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Batth TS, Papetti M, Pfeiffer A, et al. Large-Scale Phosphoproteomics Reveals Shp-2 Phosphatase-Dependent Regulators of Pdgf Receptor Signaling. Cell Rep 2018;22:2784–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hijazi M, Smith R, Rajeeve V, et al. Reconstructing kinase network topologies from phosphoproteomics data reveals cancer-associated rewiring. Nat Biotechnol 2020;38:493–502. [DOI] [PubMed] [Google Scholar]
- [25].van Alphen C, Cloos J, Beekhof R, et al. Phosphotyrosine-based Phosphoproteomics for Target Identification and Drug Response Prediction in AML Cell Lines. Mol Cell Proteom 2020;19:884–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kohale IN, Burgenske DM, Mladek AC, et al. Quantitative Analysis of Tyrosine Phosphorylation from FFPE Tissues Reveals Patient-Specific Signaling Networks. Cancer Res 2021;81:3930–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27]. Stopfer LE, Flower CT, Gajadhar AS, et al. High-density, targeted monitoring of tyrosine phosphorylation reveals activated signaling networks in human tumors. Cancer Res 2021;81:2495–2509. ● This manuscript describes the targeted analysis of hundreds of tyrosine phosphorylation sites through SureQuant pTyr
- [28].Tuncbag N, Milani P, Pokorny JL, et al. Network Modeling Identifies Patient-specific Pathways in Glioblastoma. Sci Rep 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ludwig KR, Schroll MM, Hummon AB. Comparison of In-Solution, FASP, and S-Trap Based Digestion Methods for Bottom-Up Proteomic Studies. J Proteome Res 2018;17:2480–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hailemariam M, Eguez RV, Singh H, et al. S-Trap, an Ultrafast Sample-Preparation Approach for Shotgun Proteomics. J Proteome Res 2018;17:2917–2924. [DOI] [PubMed] [Google Scholar]
- [31].Bubis JA, Gorshkov V, Gorshkov MV., et al. PhosphoShield: Improving Trypsin Digestion of Phosphoproteins by Shielding the Negatively Charged Phosphate Moiety. J Am Soc Mass Spectrom 2020;31:2053–2060. [DOI] [PubMed] [Google Scholar]
- [32].Lundby A, Franciosa G, Emdal KB, et al. Oncogenic Mutations Rewire Signaling Pathways by Switching Protein Recruitment to Phosphotyrosine Sites. Cell 2019;179:543–560.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Giansanti P, Tsiatsiani L, Low TY, et al. Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat Protoc 2016;11:993–1006. [DOI] [PubMed] [Google Scholar]
- [34].van der Laarse SAM, van Gelder CAGH, Bern M, et al. Targeting proline in (phospho)proteomics. FEBS J 2020;287:2979–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pankow S, Bamberger C, Yates JR. A posttranslational modification code for CFTR maturation is altered in cystic fibrosis. Sci Signal 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gao X, Li Q, Liu Y, et al. Multi-in-One: Multiple-Proteases, One-Hour-Shot Strategy for Fast and High-Coverage Phosphoproteomic Investigation. Anal Chem 2020;92:8943–8951. [DOI] [PubMed] [Google Scholar]
- [37]. Humphrey SJ, Karayel O, James DE, et al. High-throughput and high-sensitivity phosphoproteomics with the EasyPhos platform. Nat Protoc 2018;13:1897–1916. ● Demonstrates a rapid and facile method for deep characterization of the phosphoproteome
- [38].Gao Y, Ha YS, Kwon TG, et al. Characterization of Kinase Expression Related to Increased Migration of PC-3M Cells Using Global Comparative Phosphoproteome Analysis. Cancer Genom Proteom 2020;17:543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].CM P, MH L, AJR H, et al. Defeating Major Contaminants in Fe 3+- Immobilized Metal Ion Affinity Chromatography (IMAC) Phosphopeptide Enrichment. Mol Cell Proteom 2018;17:1028–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bodenmiller B, Mueller LN, Mueller M, et al. Reproducible isolation of distinct, overlapping segments of the phosphoproteome. Nat Methods 2007;4:231–237. [DOI] [PubMed] [Google Scholar]
- [41].Ruprecht B, Koch H, Medard G, et al. Comprehensive and reproducible phosphopeptide enrichment using iron immobilized metal ion affinity chromatography (Fe-IMAC) columns. Mol Cell Proteom 2015;14:205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yeh T, Ho M, Chen W, et al. Comparison of different fractionation strategies for in-depth phosphoproteomics by liquid chromatography tandem mass spectrometry. Anal Bioanal. Chem 2019;411:3417–3424. [DOI] [PubMed] [Google Scholar]
- [43]. Olsen JV, Blagoy Blagoev, Gnad F, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006;127:635–648. ● Among the first very large scale phosphoproteomics papers, reporting quantitative data for over 6,000 phosphorylation sites in cells stimulated with epidermal growth factor.
- [44].Wang LB, Karpova A, Gritsenko MA, et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021;39:509–528.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Clark DJ, Dhanasekaran SM, Petralia F, et al. Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 2019;179:964–983.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46]. Pawson T. Protein modules and signalling networks. Nature 1995;373:573–580. ●● An excellent review of the fundamentals of cellular signaling networks
- [47].Pawson T, Hunter T. Signal transduction and growth control in normal and cancer cells. Current Opin Genet Dev 1994;4:1–4. [DOI] [PubMed] [Google Scholar]
- [48].Rozakis-Adcock M, Fernley R, Wade J, et al. The SH2 and SH3 domains of mammalian Grb2 couple the EGF receptor to the Ras activator mSos1. Nature 1993;363:83–85. [DOI] [PubMed] [Google Scholar]
- [49].Zhang Y, Wolf-Yadlin A, Ross PL, et al. Time-resolved mass spectrometry of tyrosine phosphorylation sites in the epidermal growth factor receptor signaling network reveals dynamic modules. Mol Cell Proteom 2005;4:1240–1250. [DOI] [PubMed] [Google Scholar]
- [50]. Rush J, Moritz A, Lee KA, et al. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol 2005;23:94–101. ● The first report of immunoprecipitation of phosphotyrosine-containing peptides for subsequent proteomic analysis
- [51].Blagoy Blagoev, Ong SE, Kratchmarova I, et al. Temporal analysis of phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat Biotechnol 2004;22:1139–1145. [DOI] [PubMed] [Google Scholar]
- [52].Reddy R, Curran TG, Zhang Y, et al. Measurement of Phosphorylated Peptides with Absolute Quantification. Methods Mol Biol 2016;1410:281–292. [DOI] [PubMed] [Google Scholar]
- [53].Zhang Y, Wolf-Yadlin A, White FM. Quantitative proteomic analysis of phosphotyrosine-mediated cellular signaling networks. Methods Mol Biol 2007;359:203–212. [DOI] [PubMed] [Google Scholar]
- [54].Pawson T SH2 and SH3 domains in signal transduction. Adv Cancer Res 1994;64:87–110. [DOI] [PubMed] [Google Scholar]
- [55].Bian Y, Li L, Dong M, et al. Ultra-deep tyrosine phosphoproteomics enabled by a phosphotyrosine superbinder. Nat Chem Biol 2016;12:959–966. [DOI] [PubMed] [Google Scholar]
- [56].Tong J, Cao B, Martyn G, et al. Protein-phosphotyrosine proteome profiling by superbinder-SH2 domain affinity purification mass spectrometry, sSH2-AP-MS. Proteomics 2017;17. [DOI] [PubMed] [Google Scholar]
- [57].Li S, Zou Y, Zhao D, et al. Revisiting the phosphotyrosine binding pocket of Fyn SH2 domain led to the identification of novel SH2 superbinders. Protein Sci 2021;30:558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007;316:1160–1166. [DOI] [PubMed] [Google Scholar]
- [59].Stokes MP, Rush J, MacNeill J, et al. Profiling of UV-induced ATM/ATR signaling pathways. Proc. Natl. Acad. Sci. U.S.A 2007;104:19855–19860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Carlson SM, Chouinard CR, Labadorf A, et al. Large-scale discovery of ERK2 substrates identifies ERK-mediated transcriptional regulation by ETV3. Sci Signal 2011;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mertins P, Qiao J, Patel J, et al. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods 2013;10:634–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hogrebe A, von Stechow L, Bekker-Jensen DB, et al. Benchmarking common quantification strategies for large-scale phosphoproteomics. Nat Commun 2018;9:1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Stepath M, Zulch B, Maghnouj A,, et al. Systematic Comparison of Label-Free, SILAC, and TMT Techniques to Study Early Adaption toward Inhibition of EGFR Signaling in the Colorectal Cancer Cell Line DiFi. J Proteome Res 2020;19:926–937. [DOI] [PubMed] [Google Scholar]
- [64]. Ong SE, Blagoev B, Kratchmarova I, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteom 2002;1:376–386. ●● This manuscript describes the use of stable isotope encoded amino acids for labeling of proteins in cell culture, now one of the most common methods for non-isobaric multiplexing of biological samples.
- [65].Westman-Bringmalm A, Abramsson A, Pannee J, et al. SILAC zebrafish for quantitative analysis of protein turnover and tissue regeneration. J Proteomics 2011;75:425–434. [DOI] [PubMed] [Google Scholar]
- [66].Kruger M, Moser M, Ussar S, et al. SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell 2008;134:353–364. [DOI] [PubMed] [Google Scholar]
- [67].Altelaar A, Frese C, Preisinger C, et al. Benchmarking stable isotope labeling based quantitative proteomics. J Proteomics 2013;88:14–26. [DOI] [PubMed] [Google Scholar]
- [68].Osinalde N, Moss H, Arrizabalaga O, et al. Interleukin-2 signaling pathway analysis by quantitative phosphoproteomics. J Proteomics 2011;75:177–191. [DOI] [PubMed] [Google Scholar]
- [69].Hammond DE, Hyde R, Kratchmarova I, et al. Quantitative analysis of HGF and EGF-dependent phosphotyrosine signaling networks. J Proteome Res 2010;9:2734–2742. [DOI] [PubMed] [Google Scholar]
- [70].Zhang G, Neubert TA. Use of stable isotope labeling by amino acids in cell culture (SILAC) for phosphotyrosine protein identification and quantitation. Methods Mol Biol 2009;527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Dengjel J, Aimov V, Olsen JV, et al. Quantitative proteomic assessment of very early cellular signaling events. Nat Biotechnol 2007;25:566–568. [DOI] [PubMed] [Google Scholar]
- [72].Zhang X, Maity T, Kashyap MK, et al. Quantitative Tyrosine Phosphoproteomics of Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitor-treated Lung Adenocarcinoma Cells Reveals Potential Novel Biomarkers of Therapeutic Response. Mol Cell Proteom 2017;16:891–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Cunningham DL, Sarhan AR, Creese AJ, et al. Differential responses to kinase inhibition in FGFR2-addicted triple negative breast cancer cells: a quantitative phosphoproteomics study. Sci Rep 2020;10:7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Boersema PJ, Foong LY, Ding VMY, et al. In-depth qualitative and quantitative profiling of tyrosine phosphorylation using a combination of phosphopeptide immunoaffinity purification and stable isotope dimethyl labeling. Mol Cell Proteom 2010;9:84–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Polat AN, Kraiczek K, Heck AJR, et al. Fully automated isotopic dimethyl labeling and phosphopeptide enrichment using a microfluidic HPLC phosphochip. Anal Bioanal Chem 2012;404:2507–2512. [DOI] [PubMed] [Google Scholar]
- [76]. Ross PL, Huang YN, Marchese JN, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteom 2004;3:1154–1169. ●● Describes the multiplexed analysis of samples using isobaric tags; this manuscript generated a great deal of interest in multiplexed analyses.
- [77].Thompson A, Schafer J, Kuhn K, et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 2003;75:1895–1904. [DOI] [PubMed] [Google Scholar]
- [78].Li J, Cai Z, Bomgarden RD, et al. TMTpro-18plex: The Expanded and Complete Set of TMTpro Reagents for Sample Multiplexing. J Proteome Res 2021;20:2964–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Stopfer LE, Conage-Pough JE, White FM. Quantitative Consequences of Protein Carriers in Immunopeptidomics and Tyrosine Phosphorylation MS 2 Analyses. Mol Cell Proteom 2021;20:100104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80]. McAlister GC, Nusinow DP, Jedrychowski MP, et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 2014;86:7150–7158. ● Dynamic range compression can adversely affect the quantitative accuracy of multiplexed analyses using isobaric tags. This manuscript describes the development and application of multinotch MS3 to improve sensitivity and quantitative accuracy for these samples.
- [81].Schmelzle K, Kane S, Gridley S, et al. Temporal dynamics of tyrosine phosphorylation in insulin signaling. Diabetes 2006;55:2171–2179. [DOI] [PubMed] [Google Scholar]
- [82]. Reddy RJ, Gajadhar AS, Swenson EJ, et al. Early signaling dynamics of the epidermal growth factor receptor. Proc. Natl. Acad. Sci. U.S.A 2016;113:3114–3119. ●● Describes the use of quantitative pTyr phosphoproteomics to quantify signaling network dynamics with second-scale temporal resolution, highlighting the rapid activation of EGFR signaling.
- [83].Randall EC, Emdal KB, Laramy JK, et al. Integrated mapping of pharmacokinetics and pharmacodynamics in a patient-derived xenograft model of glioblastoma. Nat Commun 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Shen L, Li Z, Shen L. Quantitative Tyrosine Phosphoproteomic Analysis of Resistance to Radiotherapy in Nasopharyngeal Carcinoma Cells. Cancer Manag Res 2020;12:12667–12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Donnelly DP, Rawlins CM, DeHart CJ, et al. Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat Methods 2019;16:587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Sidoli S, Garcia BA. Middle-down proteomics: a still unexploited resource for chromatin biology. Expert Rev Proteom 2017;14:617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Zhen Y, Huang X, Kelleher NL. Epiproteomics: quantitative analysis of histone marks and codes by mass spectrometry. Curr Opin Chem Biol 2016;33:142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Lu C, Coradin M, Porter EG, et al. Accelerating the Field of Epigenetic Histone Modification Through Mass Spectrometry-Based Approaches. Mol Cell Proteom 2020;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Roberts DS, Chen B, Tiambeng TN, et al. Reproducible Large-Scale Synthesis of Surface Silanized Nanoparticles as an Enabling Nanoproteomics Platform: Enrichment of the Human Heart Phosphoproteome. Nano Res 2019;12:1473–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Chen B, Hwang L, Ochowicz W, et al. Coupling functionalized cobalt ferrite nanoparticle enrichment with online LC/MS/MS for top-down phosphoproteomics. Chem Sci 2017;8:4306–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Riley NM, Hebert AS, Durnberger G, et al. Phosphoproteomics with Activated Ion Electron Transfer Dissociation. Anal Chem 2017;89:6367–6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Wu S, Yang F, Zhao R, et al. Integrated workflow for characterizing intact phosphoproteins from complex mixtures. Anal Chem 2009;81:4210–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].White FM, Wolf-Yadlin A. Methods for the Analysis of Protein Phosphorylation-Mediated Cellular Signaling Networks. Annu Rev Anal Chem (Palo Alto, Calif) 2016;9:295–315. [DOI] [PubMed] [Google Scholar]
- [94].Sharma K, D’Souza RCJ, Tyanova S, et al. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep 2014;8:1583–1594. [DOI] [PubMed] [Google Scholar]
- [95].Batth TS, Olsen JV. Offline High pH Reversed-Phase Peptide Fractionation for Deep Phosphoproteome Coverage. Methods Mol Biol 2016;1355:179–192. [DOI] [PubMed] [Google Scholar]
- [96].Kumar N, Wolf-Yadlin A, White FM, et al. Modeling HER2 effects on cell behavior from mass spectrometry phosphotyrosine data. PLoS Comput Biol 2007;3:0035–0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97]. Wolf-Yadlin A, Kumar N, Zhang Y, et al. Effects of HER2 overexpression on cell signaling networks governing proliferation and migration. Mol Syst Biol 2006;2. ●● Describes the use of partial least squares regression (PLSR) to integrate quantitative phosphoproteomics data with quantitative phenotypic data from cells under the same conditions to highlight network nodes associated with phenotype.
- [98].Kitata RB, Choong W-K, Tsai C-F, et al. A data-independent acquisition-based global phosphoproteomics system enables deep profiling. Nat Commun 2021. 12:1. 2021;12:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Gao E, Li W, Wu C, et al. Data-independent acquisition-based proteome and phosphoproteome profiling across six melanoma cell lines reveals determinants of proteotypes. Mol Omics 2021;17:413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Hu Y, Wang M, Ren S, et al. Quantitative proteomics and phosphoproteomic analyses of mouse livers after tick-borne Babesia microti infection. Int J Parasitol 2021;51:167–182. [DOI] [PubMed] [Google Scholar]
- [101].Parker BL, Yang G, Humphrey SJ, et al. Targeted phosphoproteomics of insulin signaling using data-independent acquisition mass spectrometry. Sci Signal 2015;8. [DOI] [PubMed] [Google Scholar]
- [102].Martinez-Val A, Bekker-Jensen DB, Hogrebe A, et al. Data Processing and Analysis for DIA-Based Phosphoproteomics Using Spectronaut. Methods Mol Biol 2021;2361:95–107. [DOI] [PubMed] [Google Scholar]
- [103]. Wolf-Yadlin A, Hautaniemi S, Lauffenburger DA, et al. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc. Natl. Acad. Sci. U.S.A 2007;104:5860–5865. ● The first report of MRM-based targeted pTyr phosphoproteomics on hundreds of phosphorylation sites in the EGFR signaling network.
- [104].Whiteaker JR, Zhao L, Saul R, et al. A Multiplexed Mass Spectrometry-Based Assay for Robust Quantification of Phosphosignaling in Response to DNA Damage. Radiat Res 2018;189:505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Whiteaker JR, Zhao L, Schoenherr RM, et al. Peptide Immunoaffinity Enrichment with Targeted Mass Spectrometry: Application to Quantification of ATM Kinase Phospho-Signaling. Methods Mol Biol 2017;1599:197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Gallien S, Kim SY, Domon B. Large-Scale Targeted Proteomics Using Internal Standard Triggered-Parallel Reaction Monitoring (IS-PRM). Mol Cell Proteom. 2015;14:1630–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Curran TG, Zhang Y, Ma DJ, et al. MARQUIS: A multiplex method for absolute quantification of peptides and posttranslational modifications. Nat Commun 2015;6:5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].White FM. The potential cost of high-throughput proteomics. Sci Signal 2011;4. [DOI] [PubMed] [Google Scholar]
- [109].Savage SR, Zhang B. Using phosphoproteomics data to understand cellular signaling: A comprehensive guide to bioinformatics resources. Clin Proteom 2020. p. 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Emdal KB, Dittmann A, Reddy RJ, et al. Characterization of in vivo resistance to osimertinib and JNJ-61186372, an EGFR/Met bispecific antibody, reveals unique and consensus mechanisms of resistance. Mol Cancer Ther 2017;16:2572–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Naegle KM, White FM, Lauffenburger DA, et al. Robust co-regulation of tyrosine phosphorylation sites on proteins reveals novel protein interactions. Mol Biosyst 2012;8:2771–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Naegle KM, Welsch RE, Yaffe MB, et al. MCAM: multiple clustering analysis methodology for deriving hypotheses and insights from high-throughput proteomic datasets. PLoS Comput. Biol 2011;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113]. Archer TC, Ehrenberger T, Mundt F, et al. Proteomics, Post-translational Modifications, and Integrative Analyses Reveal Molecular Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2018;34:396–410.e8. ● Combines multi-omic analysis with computational modeling to identify subtypes within medulloblastoma and highlights some of the markers important for each subtype.
- [114].Johnson H, White FM. Quantitative analysis of signaling networks across differentially embedded tumors highlights interpatient heterogeneity in human glioblastoma. J Proteome Res 2014;13:4581–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Linding R, Jensen LJ, Ostheimer GJ, et al. Systematic discovery of in vivo phosphorylation networks. Cell 2007;129:1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Kuleshov MV, Xie Z, London ABK, et al. KEA3: improved kinase enrichment analysis via data integration. Nucleic Acids Res 2021;49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Strasser SD, Ghazi PC, Starchenko A, et al. Substrate-based kinase activity inference identifies MK2 as driver of colitis. Integr Biol(Camb) 2019;11:301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Krug K, Mertins P, Zhang B, et al. A Curated Resource for Phosphosite-specific Signature Analysis. Mol Cell Proteom 2019;18:576–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Storey AJ, Naceanceno KS, Lan RS, et al. ProteoViz: A tool for the analysis and interactive visualization of phosphoproteomics data. Mol Omics 2020;16:316–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Pedersen AK, Pfeiffer A, Karemore G, et al. Proteomic investigation of Cbl and Cbl-b in neuroblastoma cell differentiation highlights roles for SHP-2 and CDK16. iScience 2021;24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Wirbel J, Cutillas P, Saez-Rodriguez J. Phosphoproteomics-based profiling of kinase activities in cancer cells. Methods Mol Biol 2018:103–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Searle BC, Lawrence RT, MacCoss MJ, et al. Thesaurus: quantifying phosphopeptide positional isomers. Nat Methods 2019;16:703–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Krug K, Jaehnig EJ, Satpathy S, et al. Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 2020;183:1436–1456.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Vasaikar S, Huang C, Wang X, et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019;177:1035–1049.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Stewart PA, Welsh EA, Slebos RJC, et al. Proteogenomic landscape of squamous cell lung cancer. Nat Commun 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Kedaigle A, Fraenkel E. Turning omics data into therapeutic insights. Curr Opin Pharmacol 2018;42:95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Soltis AR, Kennedy NJ, Xin X, et al. Hepatic Dysfunction Caused by Consumption of a High-Fat Diet. Cell Rep 2017;21:3317–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Pirhaji L, Milani P, Leidl M, et al. Revealing disease-associated pathways by network integration of untargeted metabolomics. Nat Methods 2016;13:770–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Huang PH, Miraldi ER, Xu AM, et al. Phosphotyrosine signaling analysis of site-specific mutations on EGFRvIII identifies determinants governing glioblastoma cell growth. Mol Biosyst 2010;6:1227–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Frejno M, Meng C, Ruprecht B, et al. Proteome activity landscapes of tumor cell lines determine drug responses. Nat Commun 2020 11:1. 2020;11:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Patel-Murray NL, Adam M, Huynh N, et al. A Multi-Omics Interpretable Machine Learning Model Reveals Modes of Action of Small Molecules. Sci Rep 2020;10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Wang S, Li W, Hu L, et al. NAguideR: performing and prioritizing missing value imputations for consistent bottom-up proteomic analyses. Nucleic Acids Res 2020;48:e83–e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Bijnsdorp IV, Schelfhorst T, Luinenburg M, et al. Feasibility of phosphoproteomics to uncover oncogenic signalling in secreted extracellular vesicles using glioblastoma-EGFRVIII cells as a model. J Proteomics 2021;232. [DOI] [PubMed] [Google Scholar]
- [134].Murilo JR, Kuras M, Rezeli M, et al. Automated phosphopeptide enrichment from minute quantities of frozen malignant melanoma tissue. PloS One 2018;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Coscia F, Doll S, Bech JM, et al. A streamlined mass spectrometry–based proteomics workflow for large-scale FFPE tissue analysis. J Pathol 2020;251:100–112. [DOI] [PubMed] [Google Scholar]
- [136].Sanchez-Quiles V, Shi MJ, Dingli F, et al. Triple extraction method enables high quality mass spectrometry-based proteomics and phospho-proteomics for eventual multi-omics integration studies. Proteomics 2021;2000303. [DOI] [PubMed] [Google Scholar]
- [137].Liu Y, Zeng R, Wang R, et al. Spatiotemporally resolved subcellular phosphoproteomics. Proc. Natl. Acad. Sci. U.S.A 2021;118:e2025299118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Jadwin JA, Oh D, Curran TG, et al. Time-resolved multimodal analysis of Src Homology 2 (SH2) domain binding in signaling by receptor tyrosine kinases. ELife 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139]. Budnik B, Levy E, Harmange G, et al. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018;19. ● Describes the use of single-cell proteomics to distinguish different cell states
- [140].Dou M, Clair G, Tsai CF, et al. High-Throughput Single Cell Proteomics Enabled by Multiplex Isobaric Labeling in a Nanodroplet Sample Preparation Platform. Anal Chem 2019;91:13119–13127. [DOI] [PMC free article] [PubMed] [Google Scholar]