

Abstract
HIV-associated neurological dysfunction is observed in more than half of the HIV-infected population, even in the current antiretroviral era. The mechanisms by which HIV mediates CNS dysfunction are not well understood but have been associated with the presence of long-lasting HIV reservoirs. In the CNS, macrophage/microglia and a small population of astrocytes harbor the virus. However, the low number of HIV-infected cells does not correlate with the high degree of damage, suggesting that mechanisms of damage amplification may be involved.
Here, we demonstrate that the survival mechanism of HIV-infected cells and the apoptosis of surrounding uninfected cells is regulated by inter-organelle interactions among the mitochondria/Golgi/endoplasmic reticulum system and the associated signaling mediated by IP3 and calcium. We identified that latently HIV-infected astrocytes had elevated intracellular levels of IP3, a master regulator second messenger, which diffuses via gap junctions into neighboring uninfected astrocytes resulting in their apoptosis. In addition, using laser capture microdissection, we confirmed that bystander apoptosis of uninfected astrocytes and the survival of HIV-infected astrocytes were dependent on mitochondrial function, intracellular calcium, and IP3 signaling. Blocking gap junction channels did not prevent an increase in IP3 or inter-organelle dysfunction in HIV-infected cells but reduced the amplification of apoptosis into uninfected neighboring cells. Our data provide a mechanistic explanation for bystander damage induced by surviving infected cells that serve as viral reservoirs and provide potential targets for interventions to reduce the devastating consequences of HIV within the brain.
Keywords: NeuroHIV, dementia, reservoirs, HIV, apoptosis
Graphical Abstract
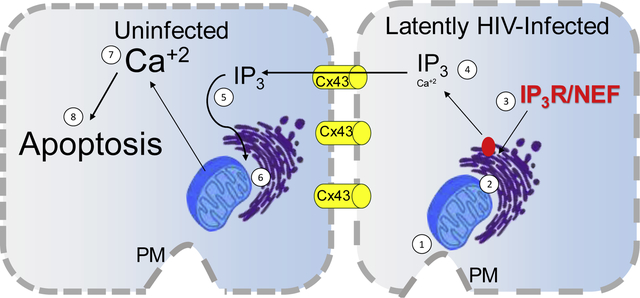
HIV cognitive impairment affects at least half of the HIV-infected population despite effective antiretroviral therapy. The brain disease mechanism in the HIV-infected population has been associated with viral reservoirs and unknown damage mechanisms. We have identified that HIV uses gap junctional communication to spread CNS toxicity, even in the absence of viral replication. We propose that HIV-infected astrocytes survive infection by altering the interactions between the plasma membrane (PM), mitochondria, and the ER (1, 2), binding of Nef to the IP3R1 (3), and increased intracellular IP3 levels (4) that do not result in calcium and apoptosis. The maintenance of Cx43 containing GJ enables the diffusion of IP3 into neighboring uninfected cells (5), resulting in proper signaling in the ER (6), increasing calcium (7), and activating the apoptotic process (8).
One Sentence Summary:
Gap junction channels amplify apoptosis from only a few HIV-infected astrocytes into surrounding uninfected astrocytes by an inter-organelle and IP3 mediated mechanism.
Introduction
Human Immunodeficiency virus-1 (HIV) enters the central nervous system (CNS) early after infection, and the virus persists in viral reservoirs even under effective antiretroviral treatment (ART), resulting in neurological abnormalities in at least half of the HIV-infected population (Heaton et al., 2011; Letendre, 2011). It is well-established that microglia/macrophages and a small population of astrocytes are the cell populations infected by the virus. However, viral production is too low to explain the extensive CNS pathology and cognitive disease often observed in the current ART era (Chan et al., 2016; Kaur et al., 2019; Tedaldi et al., 2015). Thus, the residual virus, secreted viral proteins, or the presence of viral reservoirs must be generating CNS damage in a viral replication-independent manner. We propose that one of these mechanisms is mediated by connexin (Cx) containing channels hijacked by low or residual viral protein, including HIV-Tat and HIV-Nef, in the absence of HIV-replication (Berman et al., 2016; Valdebenito et al., 2018).
Astrocytes are the most abundant cells within the CNS and actively participate in brain physiology, including maintaining blood-brain barrier (BBB) integrity, providing neuronal support, and facilitating neuroimmune interactions (Becher et al., 2000). These functions are mediated by soluble factors, cell-to-cell interactions, and intercellular signaling coordination through gap junction channels (Saez et al., 2003). Gap junctions (GJs) are conglomerates of channels that allow for the exchange of small molecules between the cytoplasm of adjacent cells, including current-carrying ions, second-messengers, metabolites, and small peptides/mRNAs (Saez et al., 2003). Each channel is formed by the docking of two hemichannels (HC), or connexons, located in opposing cell membranes, and each connexon is an assembly of six connexins (Cxs). GJs coordinate metabolic and electrical signals at long distances from the original stimulus site (Saez et al., 2003). In pathological conditions, astrocyte GJ inhibition has been shown either to reduce (Cotrina et al., 1998; Garcia-Dorado et al., 1997; Rawanduzy et al., 1997) or enhance damage (Blanc et al., 1998; Frantseva et al., 2002; Siushansian et al., 2001) between connected cells. GJs may mediate these effects by reducing the ability of the astrocytes to remove the extracellular toxic soluble factors and/or by maintaining the intercellular propagation of pro-apoptotic or survival signals between dying and healthy cells. However, the mechanism underlying HIV-associated CNS damage remains unknown.
The role of astrocytes in NeuroHIV has received little attention to date because astrocytes support extremely limited HIV entry, low to undetectable HIV replication, and few become infected. However, our laboratory showed that during acute and chronic HIV infection, Cx43 expression, functional GJ communication, and HC opening between HIV-infected astrocytes and neighboring uninfected cells are maintained or activated. We observed bystander damage from these sparse HIV-infected astrocytes into uninfected astrocytes, neurons, and endothelial cells, demonstrating that damage can be widespread and extensive (Berman et al., 2016; Eugenin and Berman, 2013; Malik and Eugenin, 2019; Malik et al., 2017; Okafo et al., 2017). This mechanism is unique to HIV because infection with other viruses (Zika, Dengue, and HCV) or cell activation (cytokines, ROS, and chemokines) results in the shutdown of gap junctional communication. The maintenance of GJ communication enables the few HIV-infected astrocytes, even in the absence of HIV replication, to amplify inflammation and apoptosis into uninfected neighboring cells, as we demonstrated in vivo and in vitro (Berman et al., 2016; Eugenin and Berman, 2007, 2013; Eugenin et al., 2011; Lutgen et al., 2020; Malik et al., 2017; Orellana et al., 2014). The compromised HIV astrocyte network could reach distances up to 200±49.5 μm, denoting how efficient the HIV-GJ amplification system is for toxicity and associated inflammation (Malik et al., 2017; Valdebenito et al., 2021). GJ and HC communication maintenance in HIV-infected astrocytes and surrounding uninfected cells are HIV-Tat-dependent and require direct binding of HIV-Tat to the Cx43 promoter (Berman et al., 2016; Malik et al., 2017; Prevedel et al., 2017). Further, using a Cx43-GFAP knockdown animal model to knock out Cx43 only in astrocytes and microinjection of human HIV infected cells into the mouse cortex to examine GJ communication and associated bystander damage, we demonstrated that few human cells could effectively spread bystander damage into mice cells by an HIV and Cx43 dependent manner (Malik et al., 2017).
Here, we report that bystander apoptosis induced by the few HIV-infected astrocytes is GJ and IP3 receptor/calcium-dependent and associated with inter-organelle interaction compromise in an HIV replication-independent manner. In addition, HIV infection promotes the survival of the small population of HIV-infected astrocytes, allowing them to serve as viral reservoirs. Bystander damage of uninfected astrocytes and the survival of HIV-infected astrocytes were associated with compromised mitochondria/Golgi/ER interactions, IP3 and calcium-related metabolism, and apoptosis in HIV-infected and surrounding uninfected cells (up to 300 μm). We further determined that Nef protein binds to IP3 receptor 1 (IP3R1), preventing calcium release and subsequent apoptosis in HIV-infected astrocytes, but not in uninfected surrounding astrocytes, providing a unique mechanism for the survival of HIV reservoirs. Overall, our data identify a unique mechanism of HIV-associated damage orchestrated by HIV-infected astrocytes and amplified by Cx43 containing GJ channels, even in the absence of viral replication and large numbers of HIV-infected cells.
Materials and Methods.
Materials.
DMEM, fetal bovine serum (FBS), penicillin/streptomycin (P/S), and trypsin-EDTA were from Thermo-Fisher (Grand Island, NY). Purified mouse IgG2B and IgG1 myeloma proteins were from Cappel, ICN Pharmaceuticals (Aurora, OH). Monoclonal antibodies to GFAP and FITC or Cy3- conjugated anti-rabbit IgG, Cy3 or FITC, anti-mouse IgG antibodies, were from Sigma (St. Louis, MO). Purified mouse IgG2B and IgG1 myeloma proteins were from Cappel, ICN Pharmaceuticals. Secondary antibodies conjugated to Cy5 were obtained from Jackson Immuno-research laboratories, Inc. (West Grove, PA). DAPI, anti-rabbit, and anti-mouse conjugated to Alexa were purchased from Thermo-Fisher (Eugene, OR). The in situ cell death detection kit (TUNEL) was purchased from Roche (Mannheim, Germany).
Astrocyte cultures.
Cortical human fetal tissue was obtained as part of a research protocol approved by the Albert Einstein College of Medicine and Rutgers University. The preparation of cultures of astrocytes was performed as previously described (Eugenin et al., 2003; Eugenin et al., 2007). Human astrocytes were cultured in DMEM medium supplemented with 5–10 % FBS and pen/strep.
U87 cells.
Cell lines (U87MG) and U87 cells transfected with CD4 and CCR5 were obtained from the NIH repository (Bethesda, MD). U87 cells were grown in DMEM medium supplemented with 5–10% FBS and Pen/strep and maintained at 37°C in a humidified incubator supplied with 5% CO2. Mycoplasma tests were performed every four months.
HIV-infection of primary astrocyte cultures.
Confluent cultures of human astrocytes were infected by incubation with virus stocks (50–80 ng p24/ml/1×106 cells, MOI, 0.1 to 1), HIVADA, HIVJR-CSF, or HIV92UG021, using a previously described protocol (Ohagen et al., 1999). Briefly, astrocyte cultures were exposed to the virus for 24 h. Then, the medium was removed, and astrocytes were washed extensively to eliminate the unbound virus before the addition of fresh medium. Immunofluorescence analyses for GFAP, CD68, and HIV-p24 indicated that the cells infected with HIV were astrocytes and that no contamination with microglia, CD68 positive cells, was detected. Also, no HIV-p24 staining was detected 2 days post-infection, and new p24 was only detected at 5–7 days post-infection.
qRT-PCR.
Total RNA was extracted from GBM cells using TRIzol (Life Technologies Waltham, MA) and the phase-lock system (Eppendorf, Hauppauge, NY), following the manufacturer’s instructions. cDNA synthesis was performed using 2 μg total RNA using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions. The amplified cDNA was used to amplify and quantify IP3 receptors and GAPDH mRNA expression by qPCR using Absolute Blue qPCR SYBR low ROX mix in a StepOnePlus thermocycler (Applied Biosystems, Life Technologies, Waltham, MA). The primers used correspond to IP3 receptor 1 forward, 5’-GAA GAG CAC ATC AAG GAA GAA CAC-3’, IP3 receptor 1 reverse, 5’-TGC TGA CCA ATG ACA TGG CT-3’; IP3 receptor 2 forward, 5’-TAG TCC TGG TGA AAG TTA AAG ACC C-3’, IP3 receptor 2 reverse, 5’-CAG ACT CAT GGT CGA TTC CAA CT-3’; IP3 receptor 3 forward, 5’-TGT ACT TCA TTG TGC TGG TCC G-3’, IP3 receptor 3 reverse, 5’-CGA ATC TCA TTC TGC TCC CC-3’ and GAPDH forward: 5′-GAGAAGTATGACAACAGCCTCAA-3′, GAPDH reverse: 5′-AGTCCTTCCACGATACCAAAG-3′. The program used was denaturation for 15 min at 95°C and 40 cycles of denaturation, 15 s at 95°C; anneal, 30 s at 60°C; and amplification, 30 s at 72°C. According to the CT values, expression was determined using the ΔΔCT method (Applied Biosystems, Life Technologies).
Image analysis.
Raw data for confocal analysis, 3D deconvolutions were obtained using NIS elements (Nikon, Japan). Quantification of colocalization, intensities, lengths, and stability were performed in NIS elements and Image J (NIH).
Laser capture microdissection and associated experiments.
Cultures or cocultures of the astrocytoma cells U87MG (not susceptible to HIV infection) and U87 cells transfected with CD4 and CCR5 (susceptible to HIV infection) were performed. Both cell types were separated by a silicon ring or barrier with a width of 10–20 μm to enable gap junction communication after 12 h post-removal of the silicon ring between two different cell populations. U87 cells transfected with CD4 and CCR5 were seeded in the center of the plate and then infected with HIV for five days to enable low viral replication levels. Around the ring, only U87 MG was cultured. Thus, the neighbor cells lacked the receptors to become infected with HIV. After 3 days in co-culture, different concentric fractions were isolated by laser capture. To perform the isolations, we used LMD6000 equipment with the respective software (Leica, Germany). The isolated fractions were subjected to western blots and co-immunoprecipitation. In parallel plates, confocal for cytochrome C, mitotracker, and nucleus were performed to examine colocalization of cytochrome C and mitochondria as well as TUNEL staining to determine apoptosis. Other parallel plates were subjected to calcium imaging using Fura-2 to correlate the laser captured material with functional data. For western blots and co-immunoprecipitation, samples from both sides of the co-culture were isolated and run for IP3R1, interacting IP3R1 with Nef and cytochrome C and APAF-1 as a sign of apoptosis. To perform these experiments, material from at least 18 plates was needed.
Western blot analysis.
Cells or laser captured material were lysed with RIPA buffer (Cell Signaling, Beverly, MA) containing protease and phosphate inhibitors (Cell Signaling, Danvers, MA), and 100 μg (or total collected material in the case of laser capture microdissection) of protein were electrophoresed on a 4–20% gradient polyacrylamide gel (Bio-Rad, Hercules, CA), and transferred to nitrocellulose membranes. Membranes were probed with antibodies against INPP5A, IPMK, IPTKA, IPTKC, and IP6K1, as well as GAPDH as a loading control (Thermo-Fisher, Carlsbad, CA). Each membrane was probed sequentially to identify several IP enzymes from the same sample. This information is indicated in the figure legends. Densitometric analysis was performed using ImageJ (NIH).
Immunofluorescence.
Human primary astrocytes and the astrocytoma cell lines U87MG and U87CD4CCR5 were grown on glass coverslips or plates, fixed, and permeabilized in 70% ethanol for 20 min at −20°C. Cells were incubated in blocking solution for 30 min at room temperature and then in primary antibody or dye (anti-Nef, anti-cytochrome C, anti-APAF-1, anti-IP3R1, anti-IP3R2, anti-IP3R3, anti-INPP5A, anti-IPMK, anti-IPTKA, anti-IPTKC, anti-IP6K1, mitotracker dye, or isotype/serum controls: both 1:50) overnight at 4°C. Cells were washed several times with PBS at room temperature and incubated with phalloidin conjugated to Texas Red to identify actin filaments and/or the appropriate secondary antibody (Sigma, St. Louis, MO) for 1 h at room temperature, followed by another wash in PBS for 1 h. Cells were then mounted using an antifade reagent with DAPI and examined by confocal microscopy using an A1 Nikon confocal microscope equipped with a spectral detection system (Tokyo, Japan).
Electron microscopy.
Cells were fixed for 30 min at RT using 4% paraformaldehyde, 2% glutaraldehyde, buffered with 0.1 M sodium cacodylate. Cells were dried with hexamethyldisilazane until fully dry under a fume hood. The cells were analyzed using a JEOL1200EX TEM accelerating voltage 20 kV to 120 kV, microprocessor control, equipped with side-entry goniometer stage, side-mounted Gatan 2k × 2k Orius CCD camera. To perform the EM analyses and quantifications for the inter-organelle interactions, we followed the seminal approach described in detail in neuronal and non-neuronal cells using TEM and cryoEM, including the complexes among plasma membrane, ER, Golgi, and the mitochondria (Frohlich et al., 2014; Sun et al., 2019).
Metabolic Measures.
The extracellular acidification rate (ECAR) and cellular oxygen consumption rate (OCR) were determined using the Seahorse XF Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, MA) following the manufacturer’s protocols. ECAR and OCR were examined using the Seahorse XF Cell Mito Stress Test Kit. The Seahorse XF-96 Wave software was used to analyze the data. OCR is shown in pmol/minute and ECAR in mpH/minute. All values were normalized to the protein concentration to standardize the determinations.
IP3 and DAG ELISA.
Cells were cultured at 3 × 106 cells per well in 6-well plates. Cell confluence was ~90% to enable the formation of gap junctions. Cell lysate and laser captured fractions were collected, and the levels of IP3 and DAG were determined by ELISA (Biosource, Camarillo, CA). Detection limits were 9.375 and 141 pg/ml, respectively.
Statistical analysis.
Information on the statistical tests used and the exact values of n (number of experiments) can be found in Figure Legends. All data were expressed as mean ± standard deviation (mean ± SD). All statistical analyses were performed using Prism 6.0 (GraphPad Software Inc.). The statistical tests were chosen according to the following: two-tailed paired or unpaired t-tests were applied on datasets with a normal distribution (Kolmogorov-Smirnov test), whereas two-tailed Mann-Whitney (unpaired test) or Wilcoxon matched-paired signed-rank tests were used otherwise. p< 0.05 was considered as the level of statistical significance.
Results
HIV infection of a few susceptible astrocytes triggers bystander apoptosis of neighboring uninfected astrocytes by an IP3, calcium, and GJ-dependent mechanism, even in the absence of active HIV replication.
Astrocytes are the most abundant cell type within the CNS. Yet, the low prevalence of HIV-infected astrocytes and lack of significant HIV-1 replication within them led to the common impression that these cells did not play a significant role in NeuroHIV. Remarkably, however, our laboratory demonstrated that viral replication and large numbers of HIV-infected astrocytes are not required for the bystander damage mediated by GJ and HC (Eugenin and Berman, 2007; Eugenin et al., 2011). Normally, astrocyte networks have one of the most effective communication and signaling systems within the CNS, reaching several millimeters from the site of action even in vivo (Kielian, 2008; Vejar et al., 2019; Xing et al., 2019). Our laboratory identified that HIV uses GJs and HCs not only to amplify inflammation/apoptosis into neighboring uninfected cells, as we described (Berman et al., 2016; Eugenin and Berman, 2007, 2013; Eugenin et al., 2011). However, the mechanisms of survival and bystander apoptosis were not fully addressed.
Upon HIV infection, we detected three different stages of astrocyte-related pathogenesis: first, an apoptotic stage with bystander apoptosis of neighboring uninfected astrocytes (up to 7 days); second, a resolution stage characterized by lower bystander apoptosis and repopulation of the areas damaged by glial proliferation (14 to 21 days), and third, a stable cell-to-cell interaction without apoptosis but with significant inflammation and dysfunction of surrounding cells (Berman et al., 2016; Eugenin and Berman, 2007, 2013; Eugenin et al., 2011; Lutgen et al., 2020; Malik et al., 2017). Further, analysis of bystander damage mediated by astrocytes using in vivo models (human tissues, mice microinjected with human cells, and monkeys) indicates that HIV-infected astrocytes aggregate in small cell clusters (3–7 cells) (Berman et al., 2016; Eugenin and Berman, 2007, 2013; Eugenin et al., 2011; Lutgen et al., 2020; Malik et al., 2017). Bystander apoptosis and signaling dysfunction could spread up to 300 μm by a GJ- and HC-dependent mechanism. Astrocytes at further distances showed minimal to no involvement (Berman et al., 2016; Eugenin and Berman, 2007, 2013; Eugenin et al., 2011; Lutgen et al., 2020; Malik et al., 2017). In this manuscript, we focus on the early stages of HIV infection and bystander damage.
To determine if different HIV isolates induced similar bystander apoptosis, human primary astrocyte cultures were infected with HIVADA, HIVJR-CSF, HIVBal, or HIV92UG021 using 50–80 ng p24/ml/1×106 cells equivalent to an MOI, 0.1 to 1, as described in the methods section. Apoptosis was determined 7 days post-infection using TUNEL staining. HIV infection of human primary astrocytes with HIVADA (R5), HIVJRCSF (R5), HIVBAL (R5), or HIV92UG021 (X4) for 7 days resulted in bystander apoptosis of uninfected neighboring astrocytes in contact with HIV-p24 positive astrocytes (Fig. 1A, *p≤0.004, n=9–12 independent experiments). In agreement with our published data and based on the long-term expression of HIV-p24 protein, we detected that HIV infects only 3.25±2.8 % of the total number of astrocytes (Eugenin and Berman, 2007; Eugenin et al., 2011). Uninfected astrocyte cultures treated with the inactivated virus or the medium used to amplify the viruses showed low baseline apoptosis of 5.1±2.3 % (Fig. 1A). HIV infection of astrocytes resulted in 19.65±3.68 (HIVADA), 18.57±5.45 (HIVJR-CSF), 16.89±2.98 (HIVBal), and 15.98±2.98% (HIV92UG021) of apoptosis, especially among uninfected astrocytes in contact with HIV-infected astrocyte clusters (Fig. 1A).
Figure 1. HIV infection of astrocytes induces bystander apoptosis of uninfected cells by a mechanism involving gap junctional communication, IP3 receptors, and intracellular calcium.
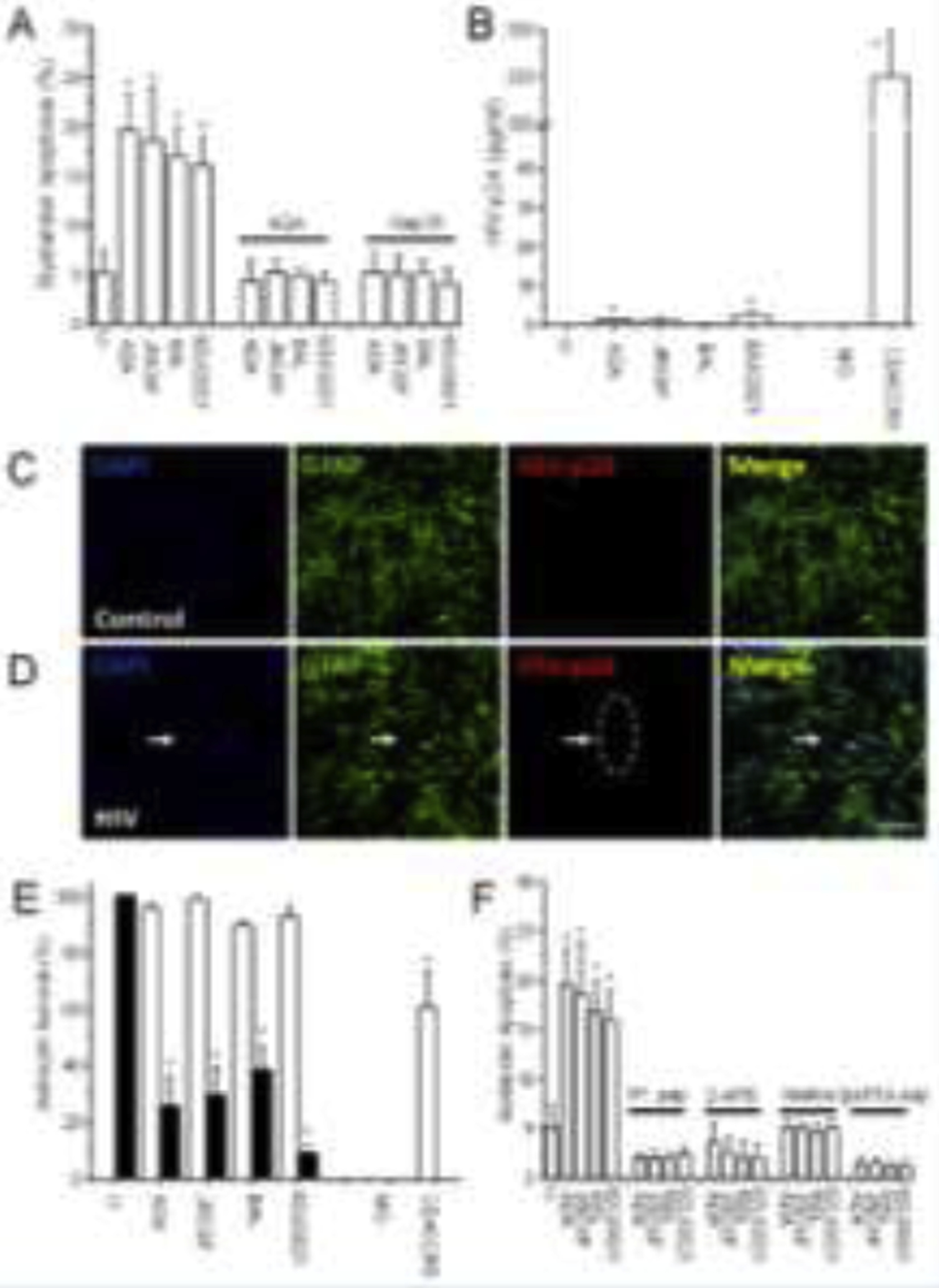
HIV infection of human primary astrocytes was performed for 24 h. The viral excess was washed out, and bystander apoptosis, viral replication, and astrocyte survival were evaluated 7 days post-infection. Cells were fixed, then stained for DAPI, TUNEL, and HIV-p24 to quantify apoptosis and infection. (A) Quantification of apoptosis using different viral isolates (ADA, JRCSF, Bal, or 92UG021) seven days post-infection (*p≤0.004 as compared to control conditions, n=9–12). In these experiments, we detected 3.25±2.8 % of the astrocytes in the culture produced HIV-p24. Application of 18-α-glycyrrhetinic acid (AGA, 35 μM) or the Cx43 blocking peptide (gap26, 300 μM) every 2 days after HIV infection prevented bystander apoptosis induced by the few HIV-infected astrocytes into the surrounding cells. (B) Quantification of HIV replication by HIV-p24 ELISA. Supernatants from uninfected, after washout of the virus (24 h post washout), and at 7 days post-infection were quantified by ultrasensitive HIV-p24 ELISA. In addition, U87 cells transfected with CD4 and CCR5 were used as a positive control (CD4CCR5). U87 MG cells (MG) were used as a negative control because these cells are not infectable with HIV (*p≤0.002, n=7). No virus was detected at 48 h post-infection, indicating that HIV detection after seven days corresponds to new HIV-p24 production (data not shown). Even when we concentrated the medium, it did not change the data or modify infection detection. (C) A representative confocal image of uninfected human primary cultures of astrocytes (control) stained for DAPI, GFAP, and HIV-p24. (D) Corresponds to a representative image of HIV-infected cultures at 7 days post-exposure. The arrow indicates a cluster of HIV-p24 positive cells. Bar: 50 μm. (E) Quantification of apoptosis in clusters of HIV-infected and neighboring astrocytes. HIV-p24 positive cells did not undergo apoptosis (white bars), indicating that HIV-infected cells are protected from cell death. However, uninfected astrocytes around HIV-infected astrocytes undergo apoptosis (black bars, quantification of cells in direct contact with HIV-p24 positive cells, n=13–15 independent experiments). (F) HIV-mediated bystander apoptosis was dependent on IP3 receptors and the release of intracellular calcium. HIV-infected cultures with different viral isolates (ADA, JRCSF, Bal, and 92UG021) were treated two days post-infection with a Cytochrome C-induced activation and IP3 and Calcium release-blocking peptide (IP3 pep, 200 μM), 2-APB (80 μM, inhibitor of IP3 dependent calcium release), Xestospongin (B and C, data combined, 5 μM, an inhibitor of IP3 dependent calcium release), or BAPTA-AM (5 μM, a membrane-permeable calcium chelator) and bystander apoptosis was evaluated seven days post-infection (* p≤0.004 as compared to control conditions, n=9–12). All data points are represented as mean ± SD.
The addition of 18-α-glycyrrhetinic acid (AGA, 35 μM, a GJ, and HC blocker) or the Cx43 blocking peptide, gap26 (300 μM, a GJ, and HC blocker), every two days after HIV infection to block GJ and HC communication prevented bystander apoptosis of the surrounding uninfected astrocytes (Fig. 1A). AGA and gap26 peptides did not change the total percentage of HIV-infected cells but efficiently prevented bystander apoptosis for all HIV strains used (4.06 to 5.2 % apoptosis levels). Neither the control scrambled peptide nor the AGA vehicle, DMSO, prevented bystander apoptosis (data not shown). These data indicate that HIV bystander apoptosis of uninfected astrocytes is GJ- and HC-dependent.
To determine if the HIV infection of astrocytes was productive, high-sensitivity HIV-p24 ELISA was used to measure virus replication. The medium of uninfected and HIV-infected cultures was collected after 2–7 days. Only HIVADA and HIV92UG021 infection of astrocyte cultures resulted in replication, which was low but significant at 1–2 pg/ml (Fig. 1B, *p≤0.002 compared to uninfected astrocyte cultures, n=7). Minimal or undetectable replication was obtained 7 days post-infection with the HIV isolates JRCSF and Bal (Fig. 1B). A representative example of HIV infection, HIV-p24 staining, denotes the typical cluster distribution of HIV-infected astrocytes (red staining corresponds to HIV-p24 in Fig. 1D, HIV). Apoptosis amplifies into surrounding uninfected cells from these few HIV-infected astrocyte clusters, as demonstrated in vivo and in vitro (Eugenin and Berman, 2007, 2013; Eugenin et al., 2011; Malik et al., 2017). Even though the medium obtained from U87MG cells (cells lacking CD4 and CCR5 and resistant to HIV infection) and HIVBal derived supernatant were concentrated four to eight times, using 10 kDa Amicon filters, no HIV-p24 antigen was detected. However, all cultures had newly produced HIV-p24 positive cells when staining was performed 7 days post-infection (Fig. 1C and D, control, and HIV infection). No HIV-p24 staining was detected in astrocyte cultures (Fig.1C). However, upon infection, small clusters of HIV-p24 positive cells were detected (Fig. 1D, arrow indicates a cluster). To clarify whether the HIV-p24 detected after 7 days in culture (microscopy and ELISA) was from the incoming virus or required synthesis of new viral proteins, we analyzed HIV-p24 expression at 48 h post-infection. Overall, ELISA and immunofluorescence could not detect HIV-p24 at 48 h post-infection, indicating that the HIV-p24 detected 7 days post-infection required new viral protein synthesis. Furthermore, astrocyte exposure to inactivated viruses did not show any staining or positive ELISA results. Thus, the HIV-p24 detected in the supernatant and inside the astrocyte clusters does not correspond to the input virus. This point will become more evident during the analysis of the electron microscopy data described below.
The addition of raltegravir (5 μM, RAL, Supplemental Fig. 1 A and B, an integrase inhibitor) or dolutegravir (5 μM, DOL, Supplemental Fig. 1C and D, an integrase inhibitor) 24 h post-infection prevented the small increase in HIV-p24 induced by HIVADA and HIV92UG021 (see Fig. 1B), suggesting that viral integration is required to identify significant amounts of HIV-p24 within the cells or release to the medium after 7 days. However, HIV-mediated bystander apoptosis only required early integration (at least 24 h) to occur. The early use (24 h post-infection) of integrase inhibitors did not prevent bystander apoptosis (see supplemental Fig. 1A and C, respectively and compare Supplemental Fig. 1A and B or Fig. 1 E and F, n=5–9). These results support our data in murine and non-human primate models, indicating that the first infection cycles were essential to generate a long-lasting population of latently HIV-infected cells (Okafo et al., 2020).
Treatment with raltegravir or dolutegravir (10 μM, both protease inhibitors) of U87CD4CCR5 cultures exposed to HIV for 24 h prevented the production of HIV-p24 by 50.24±18.03% and 62.51±17.35%, respectively, as determined 7 days post-infection (Supplemental Fig. 1). Thus, both ART drugs were effective at preventing residual viral replication. However, bystander apoptosis was not affected by the decrease in replication achieved by the protease inhibitors (Supplemental Fig. 1A, bystander apoptosis as compared to viral replication, Supplemental Fig. 1B). In conclusion, early HIV integration and infection, but not replication, is required to induce HIV-associated bystander damage. Our data align with the in vivo data that significant CNS damage occurs in the HIV-infected population despite effective ART.
To examine the apoptosis levels of HIV-infected (HIV-p24 producing clusters, Fig. 1D) and uninfected astrocytes, we quantified apoptosis in both cell types based on HIV-p24 expression. Analysis of the uninfected astrocytes surrounding HIV-p24 positive astrocyte clusters indicates that 60–80% undergo apoptosis 7–14 days post-infection depending on the viral isolate used (Fig. 1E, black bars, *p≤0.001 as compared to 100 % survival, black bars, n=13–15, this phenomenon was already described in vivo and in vitro, but without a clear mechanism (Eugenin and Berman, 2007; Eugenin et al., 2011; Lutgen et al., 2020; Malik et al., 2017)). Quantification of apoptosis in HIV-infected cultures away from the sites with HIV-infected astrocyte clusters shows minimal apoptosis (3.67±2.069 %, distances more than 300–400 μm), suggesting that bystander apoptosis is cell-to-cell contact-dependent and not spread by soluble factors. Our data indicate that cell-to-cell contact is required to trigger bystander apoptosis of uninfected cells, as previously described in vivo and in vitro (Eugenin and Berman, 2007; Eugenin et al., 2011; Lutgen et al., 2020; Malik et al., 2017). In contrast, the low level of apoptosis in HIV-p24 positive astrocytes (with HIVADA, HIVJR-CSF, HIVBal, or HIV92UG021) shows that most infected astrocytes survive infection for all the viral isolates tested (Fig. 1E, white bars).
To examine whether HIV-infected astrocytes are protected from apoptosis, we treated uninfected and HIV-infected cultures with high concentrations of TNF-α (500 ng/ml), CD40L (1 μg/ml), or H2O2 (100 μM) to induce apoptosis (Aktas et al., 2007; O’Connor, 2013; Pender and Rist, 2001; Van der Meide and Schellekens, 1996). These treatments in uninfected astrocyte cultures resulted in 100 % toxicity after 24–48 h. However, the toxic treatments did not alter the survival of HIV-p24 positive astrocytes, which exhibit 96.34±2.45 % survival.
To further examine the mechanisms of bystander apoptosis, we used the astrocytoma cell line U87MG for several reasons: first, viral entry can be controlled; second, they form functional GJs; and third, transfection with CD4 and CCR5 made U87MG cells that establish GJ with infected cells susceptible to HIV infection. Indeed, while wild-type U87MG cells are HIV-resistant, even when a high concentration of virus was used (20–1000 ng/ml, MOI 0.1 to 100, 50 ng/ml is shown in Fig. 1E, MG), we did not detect any infection or bystander apoptosis in U87MG cells. In contrast, U87MG cells transfected with CD4 and CCR5 became susceptible to HIV infection and ~30% underwent apoptosis after 3–7 days of HIV exposure (Fig. 1E, *p≤0.001 compared to 100 % survival, CD4CCR5, n=13–15). These cell lines thus enable us to obtain more uniform and controlled data to examine the nature of bystander toxicity and apoptosis.
HIV-induced bystander apoptosis is mediated by an IP3 and calcium-dependent mechanism.
To examine the mechanism of HIV bystander apoptosis, we treated the HIV-infected astrocyte cultures with a Cytochrome C (CytC) enhanced IP3 activation blocking peptide (IP3 pep), DNKTVTFEEHIKEEHN-BIOPY- 577/618 maleimide (Boehning et al., 2005), two days post-infection to prevent further IP3 receptor activation (IP3 peptide, IP3 pep, 200 μM, Fig. 1F). This peptide prevented the opening of the IP3R in response to CytC even if the ligand, IP3, was present in the cytoplasm (Boehning et al., 2005). Treatment of HIV-infected astrocyte cultures with the IP3 peptide prevented bystander apoptosis for all four viral isolates examined (IP3 pep, Fig. 1F). Also, the addition of 2-APB (80 μM) or Xestospongin B or C (5 μM, the data for both inhibitors were combined due to no significant differences), both inhibitors of IP3-dependent calcium release, prevented bystander apoptosis (Fig. 1F). These inhibitors, especially 2-APB, could block GJ, ryanodine receptors, and voltage-gated Ca2+ channels (VGCC) depending on the concentration used (Bai et al., 2006; Chang et al., 2018; Doengi et al., 2009; Meini et al., 2008; Sticozzi et al., 2013; Wang et al., 2006). Thus, we evaluated the potentially toxic effects of these compounds. First, we determined that these compounds did not change the degree of GJ communication as assessed by scrape loading in uninfected and HIV-infected astrocyte cultures. Second, the concentration used was calibrated based on the expression levels of VGCC and ryanodine receptors and the ratio to IP3 receptors in our primary human astrocytes. Most astrocyte cultures had a significant expression of IP3 receptors (as described here); however, expression of VGCC and ryanodine receptors was low and highly variable depending on the donor analyzed. To address this point, we performed the analysis using 5 μM 2-APB or Xestospongin B or C, which yielded similar results (data not shown), suggesting that APB is targeting mostly IP3 receptors in primary cultures of astrocytes. These individual variations need to be explored, but we are open to the possibility that 2-APB treatment can alter VGCC and ryanodine receptors.
In addition, incubation with BAPTA-AM (5 μM, an intracellular calcium chelator) also prevented bystander apoptosis induced by HIV infection (Fig. 1F, BAPTA-AM). None of the treatments affected the survival of HIV-infected astrocytes. Our results indicate that HIV infection of primary astrocytes is infrequent with minimal to undetectable replication in infected cells; however, GJs and HCs can amplify apoptosis to neighboring uninfected astrocytes by an IP3 receptor- and calcium-dependent mechanism.
HIV-infection of astrocytes results in compromised mitochondria/endoplasmic reticulum/Golgi complex.
Several groups have described that the inter-organelle complexes among the ER-Golgi-mitochondria-plasma membrane are regulated by lipids and inositol phosphate metabolites (Baskin et al., 2016; Chung et al., 2015; Dong et al., 2016; Giordano et al., 2013; Kumar et al., 2018; Saheki and De Camilli, 2017). These inter-organelle/lipid/second messenger interactions are critical for regulating metabolism, lipid/protein trafficking, and associated signaling. We propose that HIV infection could alter these organelles and associated functions to promote survival of HIV-infected and apoptosis of uninfected astrocytes. Our studies in different cell types (or reservoirs) indicate that at least one of the following is altered during infection: mitochondria-mediated apoptosis, the apoptosome’s formation, and calcium dysregulation (Castellano et al., 2017; Castellano et al., 2019; Eugenin and Berman, 2013; Eugenin et al., 2011; Ganor et al., 2019). However, whether interorganelle dysregulation is associated with HIV infection is unknown.
We performed TEM and subsequent image analysis to examine the structure, distribution, interactions, and function of these organelle complexes in uninfected and HIV-infected astrocyte cultures. To perform these analyses, we followed the seminal work investigating multiple inter-organelle interactions in neurons, including between the complexes among plasma membrane, ER, Golgi, and the mitochondria that have been proposed to be essential for synaptic remodeling (Frohlich et al., 2014; Sun et al., 2019).
TEM analysis of uninfected and HIV-infected astrocyte cultures indicates that there are at least two specific areas affected by HIV infection: organelles close to the nucleus (Fig. 2A and C under control and HIV-infected conditions, see higher magnification in Supplemental Fig. 2) and those close to the plasma membrane (Fig. 2B in HIV conditions, see higher magnification in Supplemental Fig. 2). We found that compromised inter-organelle interactions were present in HIV-infected astrocytes (HIV-p24 positive cells) and surrounding uninfected cells (up to 300 μm). Inter-organelle interactions were similar to those in uninfected cultures after 400 μm from HIV-infected astrocytes, suggesting that inter-organelle dysfunction follows a similar pattern as the bystander apoptosis and dysfunction induced by HIV-astrocytes.
Figure 2. HIV infection of human astrocytes compromised the interactions and cooperation among mitochondria, endoplasmic reticulum (ER), Golgi apparatus (Golgi), and the plasma membrane (PM).
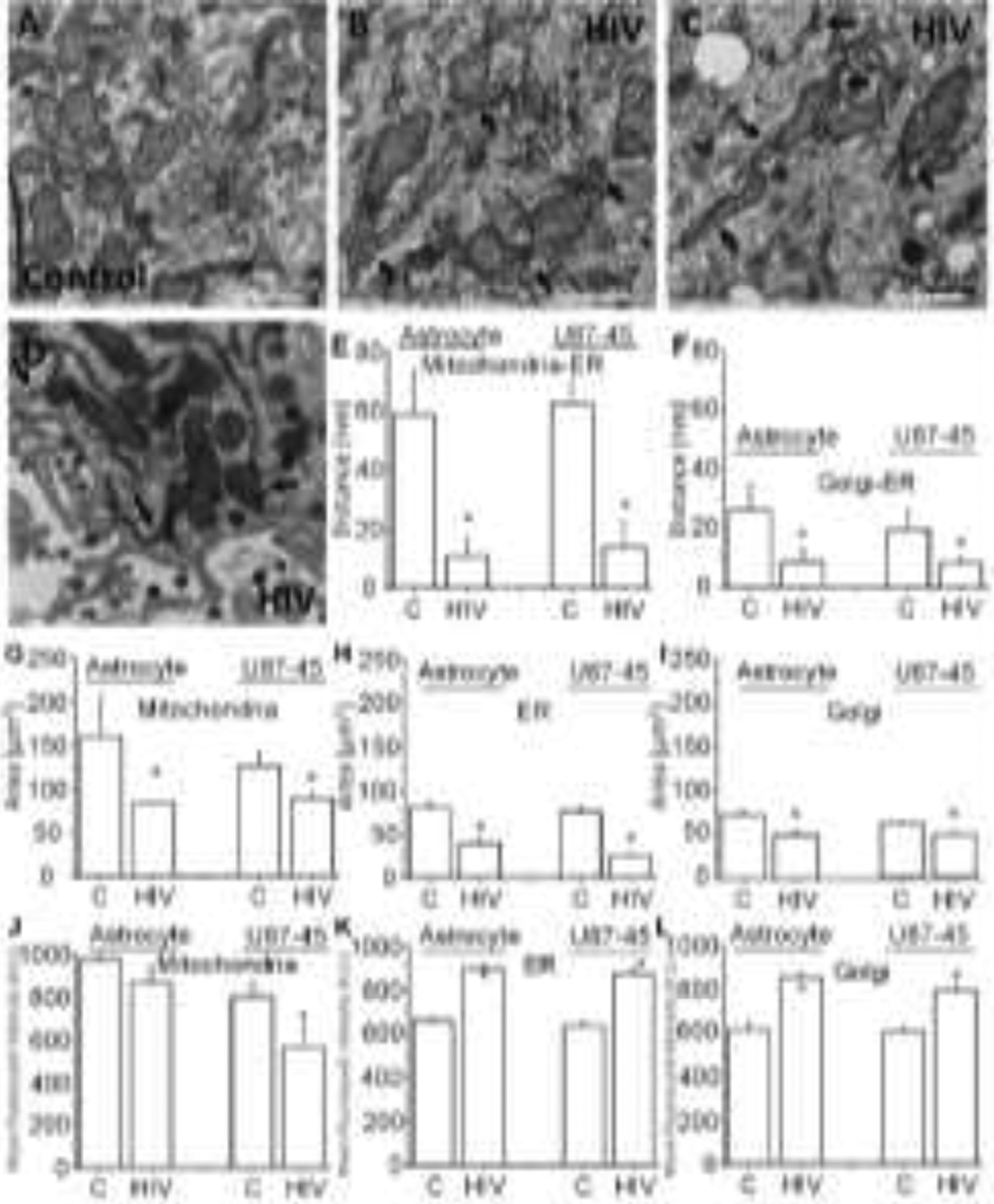
TEM micrographs showing two different areas of the cells. (A) an area close to the nucleus under uninfected control conditions. (B and C) Correspond to representative TEM from HIV-infected cultures. (D) Corresponds to a rare but consistent event found in HIV-infected astrocytes: the localized accumulation of mitochondria-ER-PM in areas of virus release. This event only happened in 1 % of the cells examined, and there was not enough replication to be detected by ultra-sensitive HIV-p24 ELISA, as described in Fig. 1. (E) Quantification of the distance of the mitochondrial membrane and the ER membrane in human primary astrocytes, uninfected controls (C), and HIV-infected cultures (HIV). (F) Quantification of the membrane distances between Golgi and the ER in both control and HIV-infected cell types. (G, H, and I) Quantification of the mitochondrial, ER, and Golgi area occupied by these organelles in control and HIV-infected conditions. (J, K, and L) Correspond to quantifying the mean fluorescent intensity of mitochondria, ER, and Golgi after cell staining and confocal microscopy for mitotracker, protein disulfide isomerase (PDI), and GOLPH3, respectively. (*p≤0.006 as compared to control conditions, n=23–26 independent experiments). All data points are represented as mean ± SD. bar=0.5 μm for the photomicrographs.
Our data indicate that uninfected astrocyte cultures have a conserved inter-organelle organization among the Golgi apparatus, ER, mitochondria, and the plasma membrane (Fig. 2A, under control conditions. See higher magnification in Supplemental Fig. 2). Quantification of the membrane distances between mitochondria-ER (59±15 and 62±8 nm, respectively for both cell types) and Golgi-ER (26±8.7 and 19.5±6.5 nm, respectively) in uninfected cultures of astrocytes or the astrocytoma cell line U87 transfected with CD4 and CCR5 (U87CD4CCR5) indicated a stable distance (Fig. 2E, control, C). HIV infection of human primary astrocyte cultures or U87CD4CCR5 resulted in long and thick actin filaments and enlarged lysosomes as identified by TEM (Fig. 2B and C). Elongated-constricted mitochondria and loss of ER/Golgi organization were also observed (Fig. 2B and C, EM close to the nucleus and the membrane, respectively; see higher magnification and details in Supplemental Fig. 2). Quantified inter-organelle distances between the membranes of the mitochondria-ER (10.9±5.7 and 13.5±8.5 nm, respectively for primary and the U87CD4CCR5 cell line) and Golgi-ER (8.6±4.3 and 7.9±2.1 nm, respectively for primary and the U87CD4CCR5 cell line) were reduced as compared to uninfected cultures (Fig. 2E and F, *p≤0.006, n=23–26). Thus, HIV infection resulted in a decreased distance between the different organelles examined in HIV-infected cells producing HIV-p24 and surrounding uninfected cells up to 300 μm, suggesting significant inter-organelle communication and associated signaling alterations.
We observed an increase in the number of inter-organelle contacts in HIV-infected conditions: 2.75±0.19 versus 1.27±0.38 contacts in uninfected conditions (data not shown). A rare but consistent observation (1 in 10 TEM grid preparations) was the highly localized plasma membrane production of virions in close association with mitochondria, ER, Golgi, and plasma membrane dysfunction, as indicated in Fig. 2D, suggesting that specific areas of the infected astrocytes are specialized for the release of low levels of HIV virions. This extremely localized viral production was only detected 5–7 days post-infection. However, we could not detect the soluble virus by highly-sensitivity HIV-p24 ELISA, as described in Fig. 1B at the same time points analyzed, suggesting that HIV infection and production were extremely low. In conclusion, the interactions among different organelle membranes were profoundly altered in response to HIV infection in a localized manner and were associated with bystander apoptosis.
Endoplasmic reticulum and Golgi interactions are compromised in HIV-infected astrocytes and surrounding uninfected cells.
We performed immunofluorescent confocal microscopy and image analysis to quantify the distribution, area, and protein expression/function/interaction among these organelles. To perform these analyses, mitotracker (for mitochondrial staining and membrane potential, Fig. 2G), Protein Disulfide Isomerase (PDI, ER, Fig. 2H), and GOLPH3 (Golgi marker, Fig. 2I) were used in addition to HIV-p24 staining to identify HIV-infected cells and define the areas with infection. Also, specific locations of HIV-infected and uninfected areas were recorded to perform post hoc correlative electron microscopy of the same area analyzed by confocal microscopy. Beclin and actin staining were used as a positive control for inter-organelle interactions because HIV increases autophagy and actin aggregation, as described (Bertrand and Toborek, 2015; Campbell and Spector, 2019; Dever et al., 2015; Dinkins et al., 2015; Santos-Llamas et al., 2018).
Mitochondria staining using mitotracker Orange CMTMRos indicated that mean fluorescence intensity was minimally decreased only after infection of U87CD4CCR5 cells, but not primary astrocytes (Fig. 2J). Upon HIV infection, primary astrocytes lost 11.89±5.69 % of the staining associated with mitochondrial membrane potential 7 days post-infection. U87CD4CCR5 cultures exposed to HIV lost 20.43±8.46 % of mitochondrial staining 2 days post-infection. Staining for ER and Golgi markers increased, probably due to the disorganization of these structures and their movement into the plasma membrane in response to HIV infection (Fig. 2 K and L, respectively). Interestingly, changes in the distribution of mitochondria, ER, and the Golgi apparatus were present in HIV and neighboring uninfected astrocytes up to a distance of 300 μm following the pattern of HIV-induced bystander damage. The addition of raltegravir (5 μM, Ral) or dolutegravir (5 μM, Dol) to the cultures prevented the small peak in HIV replication induced by HIV infection alone; however, both integrase inhibitors further increased the disorganization of the inter-organelles and compromised OCR determinations. This indicates that although these ART drugs decreased HIV replication, they also had a toxic effect on the organelles examined (see Supplemental Fig. 3). Overall, we propose that dysregulation of the complexes among Golgi-ER-mitochondria-PM induced by HIV infection (HIV-p24 positive cells) profoundly impacts inter-organelle interactions that could participate in the formation of viral reservoirs and bystander apoptosis induced by HIV-infected astrocytes.
HIV infection compromises mitochondrial function in astrocytes.
To examine the functional consequences of the dysregulation of the Golgi-ER-mitochondria-PM complex induced by HIV-compromised mitochondrial function, we determined the oxygen consumption rate (OCR) by SeaHorse Analyzer (Agilent Technologies, Santa Clara, CA). The system enables us to determine basal and maximal mitochondrial respiration, spare capacity, ATP production, maximal respiration, spare respiratory capacity, and proton leak (see details, https://www.agilent.com/en-us/products/cell-analysis-(seahorse)/mitochondrial-respiration-the-xf-cell-mito-stress-test) (Fig. 3A), using different drugs to target specific pathways of the mitochondrial energy process (Fig. 3B). A representative example of SeaHorse analysis of primary astrocyte cultures, uninfected and HIV-infected, is shown in Fig. 3C (uninfected, blue line; HIV-infected, green line). We used three astrocyte models: human primary astrocytes and the U87 astrocytoma cell lines, U87MG (resistant to HIV infection but used as a control), and U87CD4CCR5 (sensitive to HIV infection).
Figure 3. HIV infection of astrocytes compromises mitochondrial function.
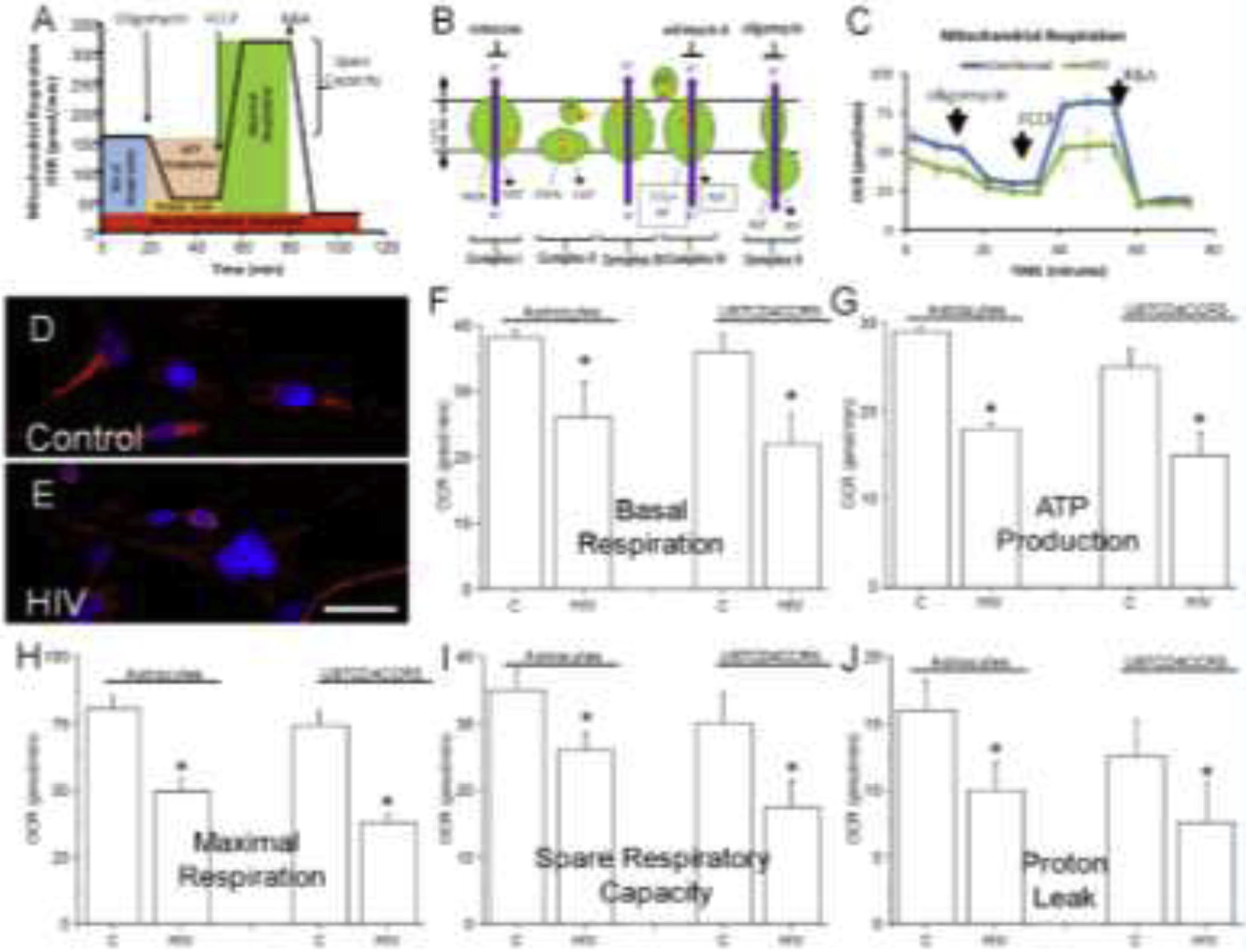
To assess mitochondrial function in HIV-infected cultures of human primary astrocytes and U87CD4CCR5 cells, the oxygen consumption rate (OCR) was determined using the Seahorse System. (A) Schematic representing the different measures obtained by OCR. (B) Schematic indicating the molecular target of each drug used in (A). (C) A representative example of mitochondrial respiration using human primary astrocytes, uninfected and HIV-infected, as indicated, after 7 days in culture. (D and E) Correspond to mitochondrial staining and distribution in control and HIV-infected conditions. (F) Corresponds to Basal Respiration, (G) ATP production, (H) maximal respiration, (I) spare respiratory capacity, and (J) proton leak in primary astrocytes (indicated as astrocytes), and U87 cells transfected with CD4 and CCR5 in the absence and presence of HIV infection. (*p≤0.004 as compared to control conditions, n=8–11 independent experiments). All data points are represented as mean ± SD. Micro-photograph bar: 5 μm.
HIV-infected cultures (primary astrocytes and U87CD4CCR5) were analyzed 7 days post-infection. In primary astrocytes, viral replication was minimal to undetectable (see Fig. 1B), and for U87CD4CCR5 cells, replication was high (258.3±26.9 pg/ml of HIV-p24). Mitochondria staining in control and HIV-infected conditions denotes the loss of mitochondrial membrane potential in HIV conditions (Fig. 3 D and E, control and HIV, respectively). In both cell types, basal respiration (Fig. 3F), ATP production (Fig. 3G), Maximal Respiration (Fig. 3H), Spare Respiratory Capacity (Fig. 3I), and Proton Leak (Fig. 3J), resulted in a reduction in all mitochondrial parameters by ~20–30% as compared to control uninfected conditions in both cell types (Fig. 3F–J) despite the low infectivity and minimal viral replication.
None of the mitochondrial changes observed in response to HIV infection were associated with alterations in cell number, viral replication, or cell death. No changes in OCR function were detected in U87MG cells exposed to HIV at any time point analyzed (0, 3, and 7 days post-exposure, data not shown). In addition, changes in OCR induced by HIV infection of primary astrocyte and U87CD4CCR5 cultures were not replicated when astrocytes were exposed to glutamate (100 μM, −5.4±2.4%), HIV-gp120 (Hx and Bal, 300 μM, −8.0±5%), IFN-γ or TNF-α (100 ng/ml, −0.16±0.03%). Furthermore, the mitochondrial compromise and OCR parameters induced by HIV infection were further increased by 14.5±6.1 % by the addition of either of the anti-retroviral drugs, raltegravir (5 μM, Ral) or dolutegravir (5 μM, Dol) 24 h post-infection (see Supplemental Fig. 3A–F). These data indicate that ART drugs have intrinsic mitochondrial toxicity associated with bystander damage induced by the few HIV-infected astrocytes. In conclusion, HIV infection of only a few astrocytes resulted in significant changes in mitochondrial metabolism.
Free intracellular levels of IP3 are elevated in HIV-infected astrocyte cultures.
As described above, bystander apoptosis and stability of the interactions among mitochondria-ER-Golgi-PM are dependent on lipid and inositol metabolism (Baskin et al., 2016; Chung et al., 2015; Dong et al., 2016; Giordano et al., 2013; Kumar et al., 2018; Saheki and De Camilli, 2017). Inositol metabolites, including IP3, are essential components of the mammalian cell-to-cell communication system; however, proper signaling requires functional communication among mitochondria/ER/Golgi/PM. PLC-β enzymatically cleaves the membrane phospholipid, phosphatidylinositol-4,5-biphosphate (PIP2), into DAG and IP3. Upon IP3 generation, several enzymes process IP3 into other IP isoforms to prevent cell overactivation (Fig. 4A). DAG remains in the plasma membrane, and IP3 diffuses into the cytoplasm to activate the opening of IP3-dependent calcium channels on the ER to mediate several physiological and pathological conditions (Litosch, 2015; Zhu et al., 2018). To examine if IP3 and the associated enzymes are affected by HIV infection, we quantified the amount of these second messengers and enzymes involved in the processing of IP3.
Figure 4. IP3 and inositol-related enzymes are compromised in HIV-infected astrocytes.
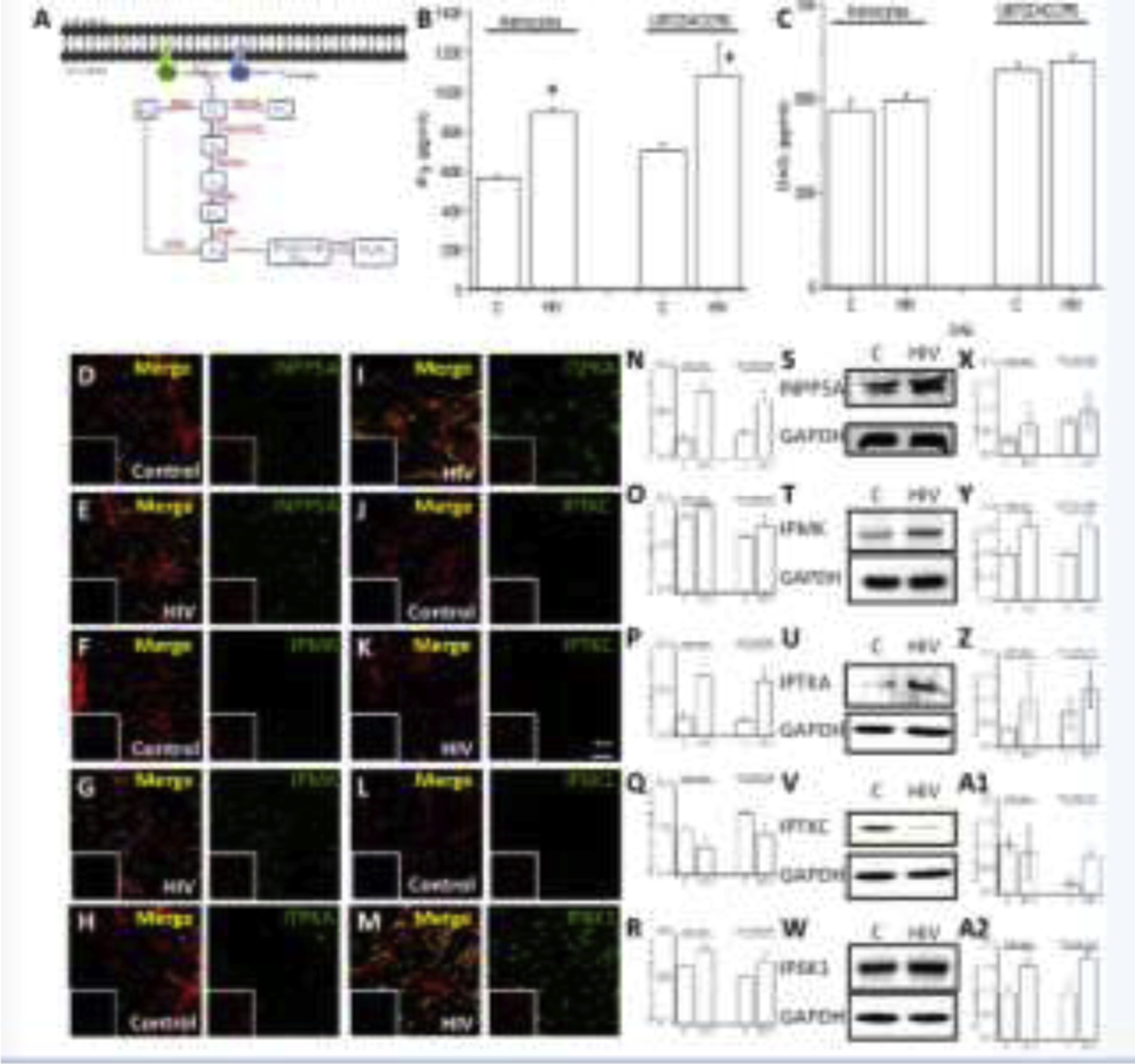
To examine the mechanism of IP3/calcium dependency of the bystander apoptosis in response to HIV infection, we examined the intracellular levels of IP3 by ELISA and the expression and distribution of key enzymes involved in IP3 metabolism using western blot and immunostaining, as described in the schematic in C. (A) Cartoon denoting the different steps examined in this figure. (B) Intracellular IP3 levels determined by ELISA in uninfected control (C) and HIV-infected conditions (HIV) in primary (astrocytes) and U87CD4CCR5 astrocytoma cells (U87CD4CCR5). (C) quantification of DAG in uninfected control (C) and HIV-infected conditions (HIV) in primary (astrocytes) and U87CD4CCR5 astrocytoma cells (U87CD4CCR5). (D and E) Correspond to the staining of astrocytes with INPP5 antibodies, DAPI, and phalloidin (detecting actin) in control and HIV conditions. (F and G) Correspond to astrocytes staining with IPMK antibodies, DAPI, and phalloidin in control and HIV conditions. (H and I) Correspond to the staining of astrocytes with ITPKA antibodies, DAPI, and phalloidin in control and HIV conditions. (J and K) Correspond to the staining of astrocytes with IPTKC antibodies, DAPI, and phalloidin in control and HIV conditions. (L and M) Correspond to the staining of astrocytes with IP6K1 antibodies, DAPI, and phalloidin in control and HIV conditions. (N to R) Quantification of the total fluorescence intensity of the confocal images using NIS Elements for INPP5A, IPMK, IPTKA, IPTKC, and IP6K1. (S to W) Representative western blot for INPP5A, IPMK, IPTKA, IPTKC, and IP6K1 for uninfected control cultures and HIV-infected conditions (n=4). (X to A2) Corresponds to the quantification of the western blots (n=3 independent experiments). (*p≤0.004 as compared to control conditions, n=5 independent experiments). All data points are represented as mean ± SD.
Quantification of the intracellular levels of IP3 and DAG by ELISA in uninfected and HIV-infected astrocyte cultures indicate that IP3, but not DAG, was elevated in human primary astrocytes and the U87CD4CCR5 astrocytoma cell line upon HIV infection. These data indicate that IP3 metabolism is compromised in HIV-infected cultures despite the low to undetectable HIV replication (Fig. 4B and C, *p≤0.004 as compared to control conditions, C, n=5). This finding is exciting because the IP3 half-life is normally around a few seconds-to-minutes in several multicellular systems to prevent cell overactivation by a calcium-mediated mechanism (Chatton et al., 1998; Huque et al., 1992; Sims and Allbritton, 1998). Thus, HIV is either increasing the stability of IP3 or reducing IP3 degradation.
To determine the mechanisms of IP3 accumulation observed in HIV-infected astrocyte cultures (primary and the U87CD4CCR5 cell line), we examined the five potential enzymes involved in IP3 metabolism, Inositol Polyphosphate-5-Phosphatase A (INPP5A), inositol polyphosphate multikinase (IPMK), Inositol-trisphosphate 3-kinase A (IPTKA), inositol 1,4,5-trisphosphate 3-kinase C (IPTKC), and inositol hexakisphosphate kinase 1 (IP6K1) by confocal microscopy and western blot (Fig. 4A). The analysis of these enzymes included all possible metabolite pathways involved in IP3 degradation. First, we examined the intracellular distribution of these enzymes in primary cultures of astrocytes and the U87CD4CCR5 cell line in the presence and absence of HIV infection (Fig. 4D–M, the first panel correspond to the merge of DAPI, blue staining, actin, red staining, and the enzyme described in green). These enzymes were localized in the cytoplasm and nucleus, as described in other cell types (Kim et al., 2013; Lee et al., 2012), without significant changes in the nucleus-cytoplasm ratio for the five enzymes examined in the presence of HIV infection (Fig. 4 D–M). Quantification of the total fluorescence intensity shows an increase in the total expression of INPP5A (Fig. 4N, catalyze IP3 to IP2, Fig. 4A), IPMK (Fig. 4O, catalyze IP4 to IP5, Fig. 4A), IPTKA (Fig. 4P, catalyze IP3 to IP4, Fig. 4A), and IP6K1 (Fig. 4R, *p=0.0023 as compared to control uninfected conditions, n=7) in response to HIV infection. However, IPTKC expression decreased as compared to uninfected conditions (Fig. 4Q, *p=0.0023, n=4). These data indicate that, despite low astrocyte infectivity and minimal-to-undetectable viral replication, significant lipid and IP3 metabolism dysregulation occurs. Further, western blot analysis of uninfected and HIV-infected cultures also supported our confocal and image analysis data: HIV infection increases the expression of INPP5A (Fig. 4S and X), IPMK (Fig. 4T and Y), IPTKA (Fig. 4U and Z), and IP6K1 (Fig. 4W and A2). In contrast, IPTKC was maintained or reduced depending on the cell type analyzed (Fig. 4V and A1). In conclusion, HIV infection of astrocytes, despite being minimal, highly localized, and poorly replicative, has profound consequences on IP3 accumulation and its metabolism.
HIV infection of astrocytes did not alter the expression or localization of IP3 receptors 1, 2, or 3.
We examined the mechanism by which HIV infection of astrocyte cultures compromises the expression and distribution of IP3 receptors; we used qRT-PCR, western blot, and immunofluorescence. As described above, changes in inter-organelle interactions, IP3 levels, mitochondrial function, and IP metabolism were detected upon HIV infection. However, whether IP3 receptor expression or distribution was altered in response to HIV infection is unknown. qRT-PCR for IP3 receptor 1, 2, and 3 at 3, 7- and 14-days post-HIV infection of primary and U87CD4CCR5 did not show significant changes in mRNA expression (Fig. 5A) despite the differences in organelle interaction, IP3 intracellular levels, and mitochondrial function as described above. In agreement, western blots of uninfected (Control, C) and HIV-infected cultures (HIV, H) of primary astrocytes (Fig. 5B) and U87CD4CCR5 cells (data not shown) did not show differences in protein expression as quantified by densitometric analysis (Fig. 5C). Controls using individual, purified proteins (IP3R1, R2, and R3) were used as a control to ensure proper band detection (data not shown). Immunofluorescence for DAPI (blue staining), mitotracker (red staining), and IP3 receptor 1 (Fig. 5D, green staining), receptor 2 and receptor 3 (See Supplemental Fig. 4 and 5 for uninfected and HIV-infected human primary astrocytes and the astrocytoma U87CD4CCR5 cultures) indicate that IP3 receptors did not change in expression or distribution upon HIV infection (Fig. 5D and supplemental Fig. 4 and 5). Overall, mRNA, protein expression, and distribution of IP3 receptors (R1, 2, and 3) were not altered by HIV infection.
Figure 5. IP3 receptor expression and distribution are not affected by HIV infection.
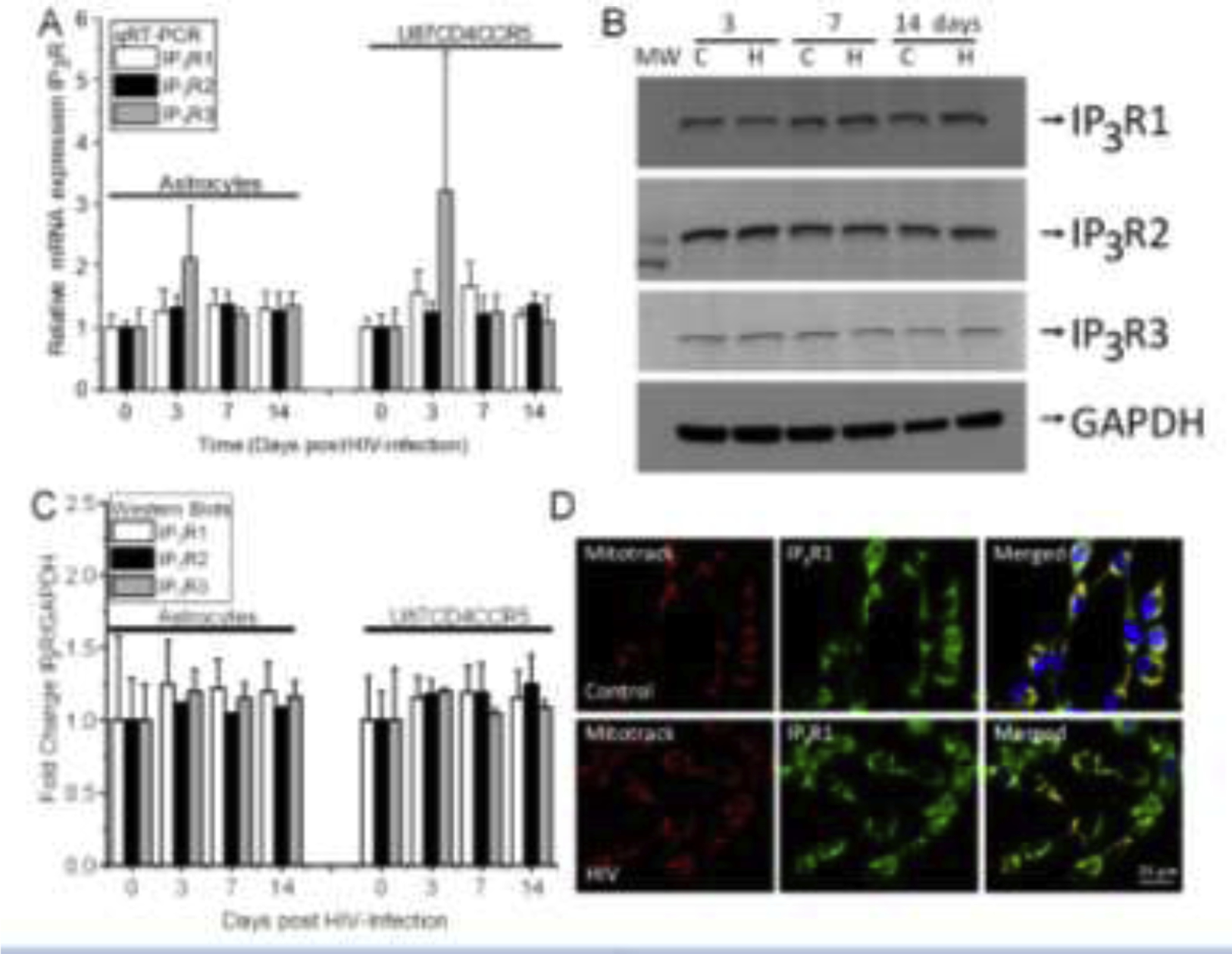
To examine whether IP3 receptors were dysregulated in response to HIV infection, we determined mRNA, protein expression, and distribution in primary (indicated as astrocytes) and the U87 astrocytoma cell line transfected with CD4 and CCR5 (U87CD4CCR5). (A) IP3 receptor 1, 2, and 3 mRNA expression in primary and U87CD4CCR5 cells at 0, 3, 7- and 14-days post-infection. (B) Representative western blot for IP3 receptors 1, 2, and 3 in control (C) and HIV-conditions (H) at 3, 7, and 14 days post-infection. GAPDH was used as a loading control for the quantification shown in the panel. (C) Quantification of the IP3 receptor 1, 2, and 3 protein expression in primary and U87CD4CCR5 cells at 0, 3, 7- and 14-days post-infection. (D) Distribution of IP3 receptor 1 and mitotracker in control and HIV conditions. (n=4–5 independent experiments). All data points are represented as mean ± SD. Micro-photograph bar: 25 μm.
HIV-infected astrocytes survive infection by controlling IP3/calcium-related signaling and spread toxicity into neighboring uninfected astrocytes in a GJ-dependent manner.
To perform these experiments, we used the astrocytoma cell line U87 for several reasons: first, better control of infectivity and bystander damage between U87CD4CCR5 and U87MG cells; second, a more reliable number of HIV-infected cells and uninfected cells with spatial distribution; third, excellent control of extracellular infectivity because U87MG cells cannot be infected, or affected, by an extracellular virus; fourth, better separation and characterization of HIV-infected and uninfected cells to perform laser capture and subsequent analyses, and lastly, any potential contamination can be examined by CD4 and CCR5 expression.
Briefly, as indicated above, U87MG cells are resistant to HIV infection, even at high viral titers (0.1 to 100 MOI, 1 MOI, 50 ng/ml is shown in Fig. 1). However, upon stable transfection with CD4 and CCR5 (U87CD4CCR5), the cells become highly sensitive to HIV infection. Using these differences in HIV infectivity, we co-cultured U87MG (negative for CD4 and CCR5) and U87CD4CCR5 (uninfected and HIVADA-infected, see Fig. 6A and B) in WillCo dishes for subsequent laser capture microdissection, in which both cell populations were separated by a silicon ring 10–20 μm wide. Upon removing the silicon ring, both cell types established gap junction communication between U87MG and U87CD4CCR5 cells. After 24 h in co-culture, the plates were subjected to calcium imaging, confocal microscopy, or laser capture microdissection with the equipment shown in Fig. 6C. As indicated at the bottom of Fig. 6H, the U87CD4CCR5 cells were labeled as HIV-infected astrocytes and corresponded to the cell type indicated as 0 μm, and diffusion of the factors examined in the U87MG area of the culture is indicated with positive numbers (Fig. 6D–H). The location of the cells on the plate was recorded to facilitate spatial localization of the functional data with co-immunoprecipitation and IP3 ELISA using laser captured material, as described in Fig. 6D–H. HIV infection of U87CD4CCR5 (HIV-infected astrocytes) for two days resulted in 83±9.5 % of cells expressing HIV-p24 protein and without appreciable apoptosis, 4.1±2.1 % (Fig. 6D, yellow area). However, U87MG cells (uninfected astrocytes) are resistant to HIV infection. Still, upon forming functional GJ channels with the U87CD4CCR5 HIV-infected cells, U87MG cells underwent apoptosis up to 240 μm away from the silicon ring (Fig. 6D).
Figure 6. Gap junctions are essential to amplify inflammation and bystander apoptosis induced by the few HIV-infected astrocytes.
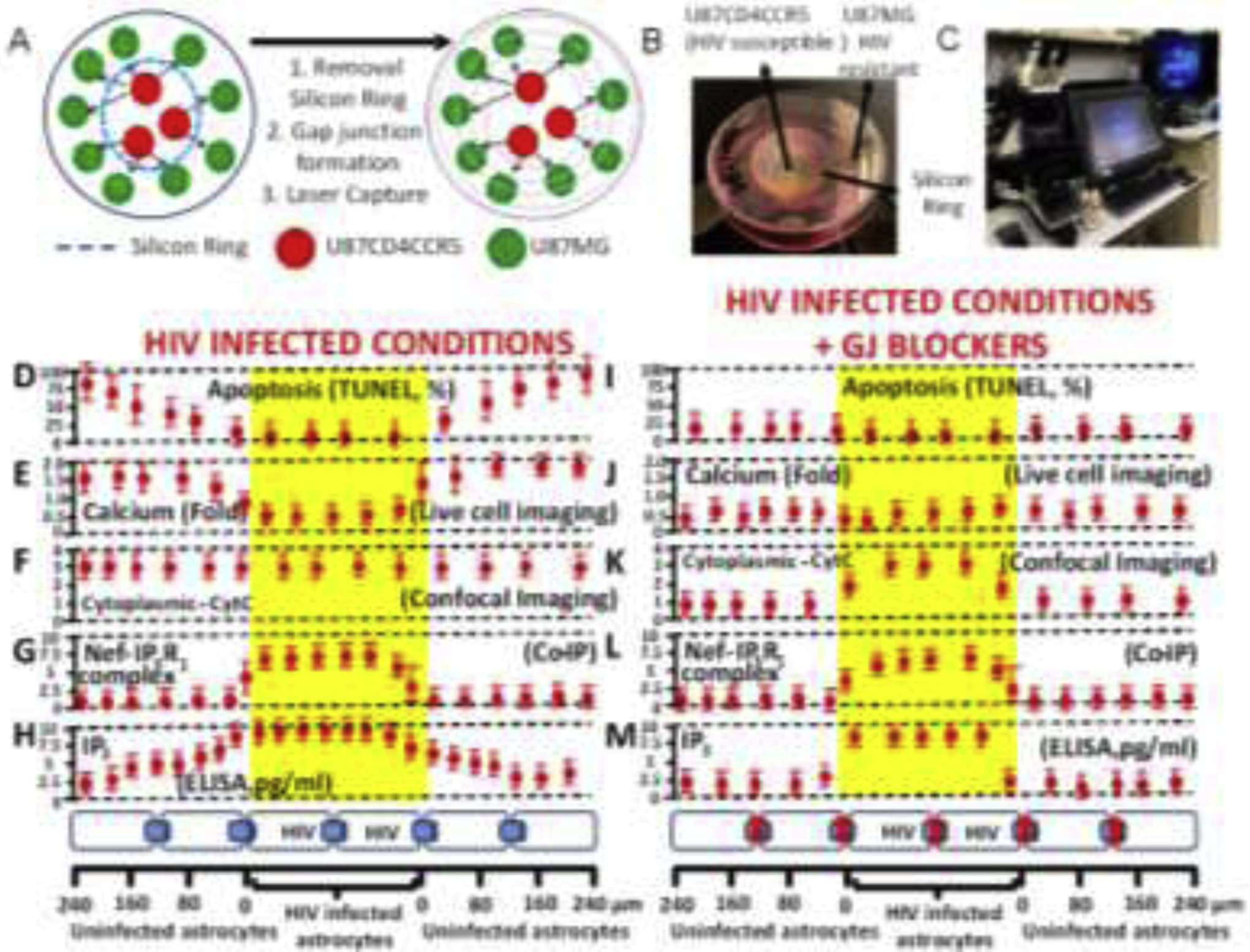
(A) A co-culture model between U87MG (resistant to HIV infection) and U87CD4CCR5 (sensitive to HIV infection) was used in the following experiments. The cell types are divided by a silicon ring that, upon removal, enables both cell types to establish gap junctional communication. This model provided the necessary spatial resolution to examine apoptosis and distance from HIV-infected into uninfected cells. (B) Representative picture of the model used. (C) Equipment used for the concentric laser capture is described below. (D to H) represent cocultures of both cell types in HIV-infected conditions with functional GJ communication (see bottom of the figure to see cell distribution and distances examined). (D) Quantification of apoptosis versus distance from HIV-infected cells. Apoptosis was only detected in uninfected cells (primary or U87MG cells). (E) Levels of intracellular calcium as determined by calcium imaging using fura-2. Data are expressed in folds as compared to control conditions. Calcium increased mostly in uninfected cells. (F) Quantification of CytC in the cytoplasm or “leak” of CytC from the mitochondria into the cytoplasm using confocal microscopy for mitotracker, DAPI, and CytC. CytC was present in the cytoplasm of HIV-infected and uninfected cells. (G) Co-immunoprecipitation of the laser captured material to examine the interaction between Nef and IP3 receptors 1, 2, or 3. Only IP3R1/3 interacted with Nef, but only in the HIV-infected astrocytes. (H) ELISA of unfixed laser captured material to quantify the amount of IP3. IP3 was high in HIV-infected astrocytes, and IP3 concentration was reduced in a distance-dependent manner in uninfected astrocytes. (I–M) correspond to the same set of experiments shown in D to H in the presence of the gap junction blocker, AGA. Functional GJ communication is required for bystander apoptosis of uninfected cells, calcium increase in uninfected cells (compare E versus J), the release of CytC into the cytoplasm in uninfected cells (compare F versus K), the formation of the complex between Nef and IP3R1 was not affected in HIV-infected astrocytes (compare G versus L), and IP3 did not diffuse into uninfected astrocytes (compare H versus M). Overall, these data indicate that HIV-infected astrocytes are protected from apoptosis by altering the calcium/IP3 axis and probably by generating specific interactions between viral proteins and the ER receptors. In future work, we will expand our study of this critical point in the survival of HIV-infected astrocytes. All data points are represented as mean ± SD. Each point corresponds to the data integration of an area of 25±8.98 μm. n=4 independent experiments.
Cocultures of U87CD4CCR5 cells with surrounding U87MG cells and subjected to calcium imaging using fura-2 indicate that free calcium intracellular levels were high in uninfected U87MG cells in contact with HIV-infected U87CD4CCR5 cells (Fig. 6E). However, free calcium in HIV-infected U87CD4CCR5 cells was low (Fig. 6E, each point recorder was every 25±9 μm). No changes in free calcium levels were detected in pure cultures of U87MG cells exposed to HIV, suggesting that calcium dysregulation in U87MG cells depends on GJ communication with HIV-infected U87CD4CCR5 cells.
As we previously described, changes in intracellular calcium and bystander apoptosis were associated with cytochrome C (CytC) release from the mitochondria into the cytoplasm to trigger apoptosis (Berman et al., 2016; Castellano et al., 2017; Castellano et al., 2019; Eugenin and Berman, 2013; Orellana et al., 2014). Quantification of cytoplasmic CytC by confocal microscopy and subsequent image analysis indicated that CytC leak into the cytoplasm from the mitochondria was present in both cell types upon HIV infection of U87MG, U87CD4CCR5, and upon HIV exposure (Fig. 4F). However, apoptosis only occurs in uninfected U87MG cells. Normally, in most cell types, including U87 cells, 100% of cytochrome C is sequestered inside the mitochondria to prevent apoptosis (Castellano et al., 2017; Eugenin and Berman, 2013). Thus, HIV infection dysregulates mitochondria to induce the release of CytC into the cytoplasm. However, apoptosis was only detected in uninfected cells. HIV-infected cells were protected from apoptosis despite mitochondrial dysfunction and the initial release of CytC to induce apoptosome formation.
To examine the role of IP3 receptors in bystander apoptosis and associated dysfunction, we used laser captured material of IP3 receptors 1, 2, and 3 to determine their spatial expression by western blot, without significant expression changes, supporting our data in Fig 5 using primary and U87 astrocyte cultures (data not shown). However, we determined that HIV-Nef, but not other HIV proteins such as Gp120 or Tat, can directly interact with the IP3R1, as determined by co-immune precipitation (Fig. 6G). Interestingly, HIV-Nef only interacted with IP3R1 in HIV-infected cells but not in uninfected surrounding cells (Fig. 6G). This result that Nef can interact with IP3 receptor 1 was confirmatory of the work of others (De and Marsh, 1994; Manninen and Saksela, 2002). We propose that Nef-IP3R1 interaction, in addition to localized inter-organelle interactions, calcium, and apoptosis, perfectly correlated with the survival pattern of HIV-infected cells and bystander apoptosis of neighboring uninfected cells by a GJ dependent mechanism.
To further examine whether IP3 intracellular levels correlated with bystander apoptosis, we quantified the amount of IP3 in the cell fractions of HIV-infected cells and the concentric laser-captured areas of uninfected cells (Fig. 6H). Intracellular levels of IP3 were elevated mostly in HIV-infected astrocytes and spread into uninfected cells, most likely in a GJ-dependent mechanism, as previously described in hepatocytes (Clair et al., 2001; Gaspers and Thomas, 2005; Saez et al., 1989). Despite the increase in intracellular IP3 levels in HIV-infected cells (Fig. 6H), neither calcium nor apoptosis correlated with the levels of this master second messenger. These data correlate with the increased overall levels of IP3 in the primary cultures of human astrocytes (Fig. 4B). We propose that HIV-infected cells survive infection due to the interaction of Nef with the IP3 receptor at the ER (Fig. 6G and Fig. 2). Overall, our data indicate that survival of HIV-infected astrocytes correlates with a unique mechanism of calcium regulation involving free calcium, the release of Cytochrome C into the cytoplasm (to induce apoptosis), and a large accumulation of IP3 in the cytoplasm, but without apoptosis. We believe that Nef binding to the IP3R1 prevents the calcium release and formation of the apoptosome even when cytoplasmic Cytochrome C is available to form the apoptosome. In contrast, uninfected cells lack the HIV protective mechanism, Nef binding to IP3R1, resulting in calcium release, leak of CytC, and subsequent apoptosis.
Blocking gap junctional communication between uninfected and HIV-infected astrocytes prevents bystander apoptosis, calcium, cytochrome C, and IP3 dysregulation.
As described above, we used the co-culture system in Fig. 6A and B to examine the mechanism of HIV-mediated bystander apoptosis. The addition of the GJ blocker, 18-α-glycyrrhetinic acid (AGA, 35 μM), or the Cx43 blocking peptide, gap26 (300 μM), after 24–36 h post removal of the silicon ring prevented functional GJ communication between HIV-infected cells and uninfected surrounding cells as well as bystander apoptosis (Fig. 6I). We demonstrated that blocking GJ communication prevented the diffusion of IP3 (Fig. 6M) and calcium (Fig. 6J) from HIV-infected cells into uninfected neighboring cells reducing mitochondrial compromise (Fig. 6K) and bystander apoptosis (Fig. 6I). However, blocking GJ did not prevent Nef interaction with the IP3 receptor 1 in HIV-infected cells (Fig. 6L). Thus, using a combination of calcium/live cell/confocal imaging, spatial correlative microscopy, biochemistry, and laser capture microdissection, we identified that HIV-infected cells were protected from apoptosis by a Nef, cytochrome C, and IP3 dependent mechanism. Furthermore, functional GJs containing Cx43 protein were essential in spreading bystander apoptosis and damage into neighboring uninfected cells.
Discussion
Astrocyte networks are the most extensive communication system in the CNS, playing critical roles in many CNS functions, including neuronal excitability, diffusion of metabolites, short and long-range communication/signaling, immune activation, inflammation, memory, and BBB maintenance (Carmignoto and Haydon, 2012; Plog and Nedergaard, 2018; Santello et al., 2019). Due to the potential misuse of GJ communication for pathogens, the body responds by a total shutdown of Cx expression and GJ communication (Eugenin, 2014; Valdebenito et al., 2018). However, our work demonstrates that HIV infection is different in this regard. No other pathogen has been found to follow this pattern. Here, we provide the first evidence that HIV uses Cx-containing channels, GJ, and HC to spread inflammation and bystander apoptosis even in the absence of significant HIV replication. Furthermore, a low level of expression of viral proteins such as HIV-Tat contributes to maintain and enhance Cx43 expression at the promotor level (Berman et al., 2016). In agreement with data from other laboratories (De and Marsh, 1994; Manninen and Saksela, 2002), we have identified that HIV-Nef binds to the IP3 receptor 1 in association with impairment of IP3-IP3 receptor signaling, compromised ER structure, and impaired inter-organelle interactions, which could explain the survival of HIV-infected astrocytes and the bystander damage and inflammation observed in at least half of the HIV-infected population even in the current ART era.
Although intra-organelle communication among plasma membrane, endoplasmic reticulum, Golgi apparatus, mitochondria, lysosomes/endosomes, lipid droplet, and peroxisomes is an area in development (see review by (Saheki and De Camilli, 2017)), our data indicate a strong correlation between HIV survival and bystander damage. The results indicate a reorganization of the interactions between the different organelles and sometimes the membranes’ fusion, especially in glial viral reservoirs. However, we cannot discard the possibility that our inter-organelle interaction can be better defined using more sensitive techniques such as focused-ion-beam scanning electron microscopes (FIB-SEM). We propose that these interactions are essential for cell-to-cell signaling and the survival of latently HIV-infected cells. In a different context, proper membrane contact interactions are crucial to maintaining low concentrations of free intracellular calcium (nM range) compared to the extracellular space and the endoplasmic reticulum lumen, preventing overactivation and apoptosis (Cali et al., 2012; Cuchillo-Ibanez et al., 2002; McCarron et al., 2018; Stefan, 2018). Mutations affecting these membrane contacts can result in several neurological diseases, including amyotrophic lateral sclerosis and Huntington and Alzheimer’s-like diseases (Landstrom et al., 2014; Wu et al., 2017a; Wu et al., 2017b) – all features observed in HIV-infected individuals with cognitive disease.
Our data that HIV compromises inter-organelle interactions to promote survival of HIV-infected astrocytes and, simultaneously, promotes apoptosis in the surrounding uninfected cells by a GJ-, IP3R1-, and a calcium-dependent mechanism is novel and has not been previously described. Normally, the ER network extends into all cell compartments, even those most distant, including the plasma membrane and the nucleus. For example, the membrane contact sites between ER and the mitochondria can cover up to 4% of this organelle’s surface, as seen as membrane tubules and shown by electron microscopy (Wu et al., 2017a). Membrane junctions between the ER and the mitochondria are essential for transferring Ca+2 from the ER lumen to the mitochondria matrix (Dickson et al., 2016). Also, protein-mediated transport is particularly important for lipid exchange between the ER and mitochondria as these two organelles are not connected by membrane traffic. Yet, mitochondria use precursors imported from the ER to produce most of their lipids. The ER-mitochondria membrane contact sites are required for lipid biosynthesis. It has been proposed that the ERMES complex transfers phosphatidylserine and phosphatidylcholine to the mitochondrial membrane (Wu et al., 2018). It is also well known that these interactions can regulate calcium handling, mitochondrial division, and metabolism. In fact, contacts with the ER have been implicated in mitochondria fission, as it has been shown that human VPS13A and VPS13C bind to the ER, tethering it to mitochondria (VPS13A) (Kumar et al., 2018). As a lipid transport between the ER and other organelles such as mitochondria, VPS13 is related to membrane lipid homeostasis, and its dysregulation is associated with different neuronal diseases. The mitochondrial fission mechanism involves VPS13A, and VPS13C binds to the ER and strangles the mitochondria to mediate fission (Kumar et al., 2018; Munoz-Braceras et al., 2019; Yeshaw et al., 2019). Compromise of these proteins at the ER and the mitochondria has been associated with fusion/fission problems as well as signaling compromise (Madreiter-Sokolowski et al., 2019; Rasul et al., 2021). However, the mechanism involved has only recently been uncovered. Interestingly, all these phenotypes are observed in the primary HIV-infected astrocytes, including the strangled and enlarged mitochondria, but without clear fission of mitochondria affecting not only the size but also the interaction with the ER, lipid-containing vesicles, and lack of efficient signaling among the organelles, including calcium and IP3 as well as the viral protein Nef binding directly to IP3R1. Thus, a potential mechanism for HIV to survive is to prevent mitochondrial fission, metabolism changes, and signaling among organelles.
Interestingly, there are several genetic diseases in which protein VPS13 loses its function, resulting in multiple diseases such as chorea acanthocytosis, a Huntington-like syndrome associated with red-cell shape abnormalities and improper development (Rampoldi et al., 2001; Vonk et al., 2017), mental disability (Kolehmainen et al., 2003), early onset of Parkinson’s disease (Lesage et al., 2016), and a recently described form of ataxia with spasticity (Gauthier et al., 2018; Seong et al., 2018). However, these diseases are genetic. Furthermore, HIV, an infectious acquired disease, exhibits a similar phenotype quickly after infection. Overall, our study identifies several mechanisms compromised by HIV infection but not replication; however, we must acknowledge that we do not directly demonstrate that inter-organelle interactions control survival and apoptosis of neighboring uninfected cells in a GJ-dependent manner.
A critical second messenger transferred by GJs is IP3 (Charles et al., 1992; Leybaert and Sanderson, 2012; Sanderson et al., 1990; Scemes et al., 1998; Simard et al., 2003; Stout and Charles, 2003; Verkhratsky and Kettenmann, 1996). IP3 is a high-energy molecule, usually with a short life span to prevent cell overactivation and consequent cell death (Ando et al., 2018; Egorova and Bezprozvanny, 2018; Ivanova et al., 2014; Tada et al., 2016). It has been calculated that different active forms of inositol are metabolized in milliseconds to minutes; thus, these molecules are highly unstable (Ivanova et al., 2014; Vermassen et al., 2004; Xia and Yang, 2005). However, our data indicate that HIV infection either increase production or prevents degradation of IP3 in HIV-infected astrocytes, which may be due to the increased expression of INPP5A, IPMK, IPTKA, and IP6K1 and the decrease in levels of IPTKC enzymes. These studies open the possibility that other forms of inositol, such as IP7, may be involved in the survival of HIV reservoirs due to the extensive literature on the role of IP7 and IP8 in cancer, insulin sensitivity, viral immunity, apoptosis, and proliferation (see review by (Kim et al., 2017; Park et al., 2019; Rajadurai et al., 2016)). The inositol enzymes involved in IP metabolism also have an active role in regulating several protein targets non-catalytically via protein-protein interactions, including Akt, mammalian target of rapamycin complex 1 (mTORC1), and the energy-sensing kinase, AMPK (Dailey and Kim, 2012; Kim et al., 2011). AMPK amplifies inflammation by being an essential regulator of Toll-like receptor innate immunity due to its interaction with tumor necrosis factor receptor-associated factor 6 (TRAF6) (Kim et al., 2017). A critical result is the increased expression of the enzymes INPP5A, IPMK, IPTKA, and IP6K due to HIV infection. However, a decrease in IPTKC is also observed in HIV-infected cultures. Despite the complexity of IP3 stability, it is remarkable that a low level of HIV infection, few infected cells, and poor replication compromise IP metabolism. Changes in expression of these enzymes have been associated with the development and progression of several cancers and metastasis (Cumsky et al., 2019; Maly et al., 2019; Patel et al., 2018; Yang et al., 2015; Yang et al., 2018). These enzymes’ loss or gain has been associated with cerebral degeneration and apoptosis (Yang et al., 2015; Yang et al., 2018), two clear features of viral reservoirs within the brain. Thus, inositol metabolites play a role in ER-Golgi-mitochondria-plasma membrane interaction, in addition to apoptosis, proliferation, and signaling. Furthermore, we propose that chronic HIV-infected individuals have problems processing high-energy phosphates. Our data indicate that ATP circulates in the bloodstream for extended periods in addition to the inositol metabolites, and this high ATP concentration correlates with changes in cognitive impairment in the HIV-infected population (Velasquez et al., 2019). Thus, some of these high-energy phosphor metabolites could be used as biomarkers of HIV-associated disease and perhaps to detect long-lasting viral reservoirs.
A critical and controversial question is how HIV entry occurs in astrocytes and whether astrocytes are viral reservoirs. HIV entry into astrocytes is CD4 independent. As described by others (Hao et al., 1997; Hao and Lyman, 1999; Peudenier et al., 1991; Schweighardt et al., 2001) and us (Eugenin and Berman, 2007, 2013; Gajardo-Gomez et al., 2020), HIV infection of astrocytes is not by a CD4-mediated mechanism, but several alternative receptors have been described. Our human primary astrocytes are CD4 negative by mRNA and protein analysis (data not shown). But several groups that quantified large numbers of cells demonstrated integrated HIV DNA in a few astrocytes suggesting an alternative mechanism of entry and infection to CD4+ T lymphocytes (Churchill et al., 2006; Edara et al., 2020; Li et al., 2020; McCarthy et al., 1998; Narasipura et al., 2014). Now, we demonstrate that the high survival of HIV-infected astrocytes is associated with inter-organelle dysfunction and signaling dysregulation and the anti-apoptotic action of viral proteins such as Nef.
Despite the exciting data, we must be careful with the interpretation of Fig. 6 that was generated using an astrocytoma cell line, U87MG or U87CD4CCR5, which has multiple genetic deficiencies (Furnari et al., 1997). However, several mechanisms have been demonstrated in astrocyte primary cultures, such as mitochondrial dysregulation, CytC release, lack of apoptosome formation, and calcium deficiencies (Eugenin and Berman, 2007, 2013; Gajardo-Gomez et al., 2020; Malik et al., 2017; Orellana et al., 2014; Valdebenito et al., 2021). More importantly, these bystander damage mechanisms have been observed in vivo supporting our findings in the cell line described (Eugenin et al., 2011; Malik et al., 2017). Thus, the laser capture experiments denote the exquisite mechanism of HIV-astrocyte survival and the GJ/HC bystander mechanism of toxicity; however, several questions remain, including a better direct demonstration that inter-organelle dysfunction is a direct link to latency and viral reservoir survival. In the same direction, inter-organelle dysfunction will compromise lipid metabolism and protein trafficking, as we can observe in our electron microcopy data as well as mass spectrometry determination. We propose that nuclei, endosomes, lysosomes, and peroxisomes may be involved because inter-organelle interactions corresponded to interdependent networks; thus, identifying where and how HIV compromises these networks is essential to understanding its physiology and pathology. Further, our data that Nef and Tat strongly alter the mechanism of viral reservoir survival and calcium signaling could provide tools to target other genetic and neurodegenerative diseases.
Interestingly, HIV infection of astrocytes does not depend on GJ or HC activation. Instead, we propose that HIV uses Cx43-containing channels after the initial infection to amplify inflammation and thus achieve several points, including an early amplification of apoptosis into uninfected surrounding cells to induce localized inflammation to recruit immune cells to infectious sites generating hyperreactive astrocytes. More importantly, in the long run, this also generates an inflammatory/fibrotic environment or niche to transfer infection into lymphoid, myeloid, and glial cells. Our in vivo data demonstrate that these sanctuaries within the human brain and in monkey and mouse models operate in a highly localized manner (Berman et al., 2016; Eugenin and Berman, 2007, 2013; Eugenin et al., 2011; Malik and Eugenin, 2016; Orellana et al., 2014). Further, viral replication is not required for the survival of the sparse HIV-infected astrocytes, supporting the theory that HIV-infected astrocytes are reservoirs of the virus. However, other groups indicate that HIV positivity corresponds to engulfed infected material (Ko et al., 2019; Russell et al., 2017). Further, viral proteins are not enough to induce bystander damage, but HIV-Tat alone induces expression of Cx43 and maintains GJ communication (Berman et al., 2016). Now, we report that Nef can bind to the IP3R1, thereby potentially preventing apoptosis.
In conclusion, HIV-infected astrocytes, despite their low number and undetectable replication, are viral reservoirs, survive the infection, and spread toxicity into neighboring uninfected cells via a GJ-mediated mechanism. Considering astrocytes as a long-term reservoir and identifying the unique mechanism of survival and bystander toxicity will provide a new mechanism to target to eliminate these residual cells in the current ART era, thus achieving a functional cure.
Supplementary Material
Acknowledgments:
This work was funded by The National Institute of Mental Health grant, MH096625 and MH128082, the National Institute of Neurological Disorders and Stroke, NS105584, and UTMB internal funding (to E.A.E). We want to thank the Albert Einstein College of Medicine Imaging Facilities for their assistance with SEM and TEM. Heather Lander, Dr. Richard Coggeshall, and Dr. Shelly Buffington for editing the manuscript.
Abbreviations:
- AGA
18-α-glycyrrhetinic acid
- Akt
protein kinase B
- AMPK
5’ adenosine monophosphate-activated protein kinase
- APAF-1
apoptotic protease activating factor 1
- 2-APB
2-aminoethoxydiphenyl borate
- ART
anti-retroviral therapy
- ATP
adenosine triphosphate
- BAPTA-AM
glycine, N,N’-[1,2-ethanediylbis(oxy-2,1-phenylene)]bis[N-[2-[(acetyloxy)methoxy]-2-oxoethyl]]-, bis[(acetyloxy)methyl] ester
- BBB
blood brain barrier
- Ca+2
Calcium
- cDNA
complementary DNA
- CD4
Cluster of differentiation-4
- CD40L
Cluster of differentiation-40 ligand
- CNS
central nervous system
- CO2
carbon dioxide
- Cx
connexin
- CryoEM
cryogenic electron microscopy
- CytC
cytochrome C
- Cy3
cyanine 3
- DAG
diacylglycerol
- DAPI
2-[4-(Aminoiminomethyl)phenyl]-1H-Indole-6-carboximidamide hydrochloride
- DMSO
dimethyl sulfoxide
- DOL
dolutegravir
- ECAR
extracellular acidification rate
- EDTA
ethylenediaminetetraacetic acid
- EM
electron microscopy
- ER
endoplasmic reticulum
- FBS
fetal bovine serum
- FIB-SEM
focused ion beam scanning electron microscopy
- FITC
Fluorescein isothiocyanate
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- GAP26
Gap blocking peptide 26
- GFAP
Glial fibrillar acid protein
- GJ
gap junction
- GOLPH3
Golgi phosphor protein 3
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HIV
human immunodeficiency virus-1
- H2O2
hydrogen peroxide
- INF-γ
interferon gamma
- IP6K1
Inositol hexakisphosphate kinase 1
- INPP5
Inositol polyphosphate 5-phosphatase A
- IPMK
Inositol polyphosphate multikinase
- IPTKA
Inositol-trisphosphate 3-kinase A
- IPTKC
Inositol-trisphosphate 3-kinase C
- IP3
inositol triphosphate
- IP3 pep
inositol triphosphate receptor release-blocking peptide
- IP3R
inositol triphosphate receptor
- mRNA
messenger ribonucleotide acid
- MOI
multiplicity of infection
- mTORC1
rapamycin complex 1
- OCR
oxygen consumption rate
- PBS
phosphate buffer solution
- PDI
protein disulfide isomerase
- PM
plasma membrane
- PLC-β
phospholipase C
- PIP2
phospholipid phosphatidylinositol-4,5-biphosphate
- P/S
penicillin/streptomycin
- RAL
raltegravir
- TEM
transmission electron microscopy
- TNF-α
Tumor necrosis factor-alpha
- TRAF6
TNF Receptor Associated Factor 6
- TUNEL
Terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling
- VPS13A
Vacuolar Protein Sorting 13 Homolog A
- VPS13C
Vacuolar Protein Sorting 13 Homolog C
- Xestos
Xestospongin B or C
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors declare no competing interest.
Data Availability Statement.
The data presented in the manuscript is fully available upon reasonable request to the authors.
References
- Aktas O, Schulze-Topphoff U, Zipp F, 2007. The role of TRAIL/TRAIL receptors in central nervous system pathology. Frontiers in bioscience : a journal and virtual library 12, 2912–2921. [DOI] [PubMed] [Google Scholar]
- Ando H, Kawaai K, Bonneau B, Mikoshiba K, 2018. Remodeling of Ca(2+) signaling in cancer: Regulation of inositol 1,4,5-trisphosphate receptors through oncogenes and tumor suppressors. Adv Biol Regul 68, 64–76. [DOI] [PubMed] [Google Scholar]
- Bai D, del Corsso C, Srinivas M, Spray DC, 2006. Block of specific gap junction channel subtypes by 2-aminoethoxydiphenyl borate (2-APB). The Journal of pharmacology and experimental therapeutics 319, 1452–1458. [DOI] [PubMed] [Google Scholar]
- Baskin JM, Wu X, Christiano R, Oh MS, Schauder CM, Gazzerro E, Messa M, Baldassari S, Assereto S, Biancheri R, Zara F, Minetti C, Raimondi A, Simons M, Walther TC, Reinisch KM, De Camilli P, 2016. The leukodystrophy protein FAM126A (hyccin) regulates PtdIns(4)P synthesis at the plasma membrane. Nat Cell Biol 18, 132–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher B, Prat A, Antel JP, 2000. Brain-immune connection: immuno-regulatory properties of CNS-resident cells. Glia 29, 293–304. [PubMed] [Google Scholar]
- Berman JW, Carvallo L, Buckner CM, Luers A, Prevedel L, Bennett MV, Eugenin EA, 2016. HIV-tat alters Connexin43 expression and trafficking in human astrocytes: role in NeuroAIDS. J Neuroinflammation 13, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand L, Toborek M, 2015. Dysregulation of Endoplasmic Reticulum Stress and Autophagic Responses by the Antiretroviral Drug Efavirenz. Mol Pharmacol 88, 304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc EM, Bruce-Keller AJ, Mattson MP, 1998. Astrocytic gap junctional communication decreases neuronal vulnerability to oxidative stress-induced disruption of Ca2+ homeostasis and cell death. J Neurochem 70, 958–970. [DOI] [PubMed] [Google Scholar]
- Boehning D, van Rossum DB, Patterson RL, Snyder SH, 2005. A peptide inhibitor of cytochrome c/inositol 1,4,5-trisphosphate receptor binding blocks intrinsic and extrinsic cell death pathways. Proc Natl Acad Sci U S A 102, 1466–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cali T, Ottolini D, Brini M, 2012. Mitochondrial Ca(2+) as a key regulator of mitochondrial activities. Adv Exp Med Biol 942, 53–73. [DOI] [PubMed] [Google Scholar]
- Campbell GR, Spector SA, 2019. DIABLO/SMAC mimetics selectively kill HIV-1-infected resting memory CD4(+) T cells: a potential role in a cure strategy for HIV-1 infection. Autophagy 15, 744–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmignoto G, Haydon PG, 2012. Astrocyte calcium signaling and epilepsy. Glia 60, 1227–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano P, Prevedel L, Eugenin EA, 2017. HIV-infected macrophages and microglia that survive acute infection become viral reservoirs by a mechanism involving Bim. Sci Rep 7, 12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano P, Prevedel L, Valdebenito S, Eugenin EA, 2019. HIV infection and latency induce a unique metabolic signature in human macrophages. Sci Rep 9, 3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P, Hellmuth J, Spudich S, Valcour V, 2016. Cognitive Impairment and Persistent CNS Injury in Treated HIV. Curr HIV/AIDS Rep 13, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YC, Xue WJ, Ji W, Wang Y, Wang YP, Shi WY, Shan LY, Zhang L, Tan CY, Ma KT, Li L, Si JQ, 2018. The Protective Effect of Propofol Against Ischemia-Reperfusion Injury in the Interlobar Arteries: Reduction of Abnormal Cx43 Expression as a Possible Mechanism. Kidney Blood Press Res 43, 1607–1622. [DOI] [PubMed] [Google Scholar]
- Charles AC, Naus CC, Zhu D, Kidder GM, Dirksen ER, Sanderson MJ, 1992. Intercellular calcium signaling via gap junctions in glioma cells. The Journal of cell biology 118, 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatton JY, Cao Y, Stucki JW, 1998. Perturbation of myo-inositol-1,4,5-trisphosphate levels during agonist-induced Ca2+ oscillations. Biophysical journal 74, 523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB, Narayanaswamy P, Wenk MR, Nakatsu F, De Camilli P, 2015. INTRACELLULAR TRANSPORT. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science 349, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill MJ, Gorry PR, Cowley D, Lal L, Sonza S, Purcell DF, Thompson KA, Gabuzda D, McArthur JC, Pardo CA, Wesselingh SL, 2006. Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J Neurovirol 12, 146–152. [DOI] [PubMed] [Google Scholar]
- Clair C, Chalumeau C, Tordjmann T, Poggioli J, Erneux C, Dupont G, Combettes L, 2001. Investigation of the roles of Ca(2+) and InsP(3) diffusion in the coordination of Ca(2+) signals between connected hepatocytes. J Cell Sci 114, 1999–2007. [DOI] [PubMed] [Google Scholar]
- Cotrina ML, Kang J, Lin JH, Bueno E, Hansen TW, He L, Liu Y, Nedergaard M, 1998. Astrocytic gap junctions remain open during ischemic conditions. J Neurosci 18, 2520–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuchillo-Ibanez I, Albillos A, Aldea M, Arroyo G, Fuentealba J, Garcia AG, 2002. Calcium entry, calcium redistribution, and exocytosis. Ann N Y Acad Sci 971, 108–116. [DOI] [PubMed] [Google Scholar]
- Cumsky HJL, Costello CM, Zhang N, Butterfield R, Buras MR, Schmidt JE, Drenner K, Nelson SA, Ochoa SA, Baum CL, Pittelkow MR, DiCaudo DJ, Sekulic A, Mangold AR, 2019. The prognostic value of inositol polyphosphate 5-phosphatase in cutaneous squamous cell carcinoma. J Am Acad Dermatol 80, 626–632 e621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey MJ, Kim S, 2012. Inositol polyphosphate multikinase: an emerging player for the central action of AMP-activated protein kinase. Biochem Biophys Res Commun 421, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De SK, Marsh JW, 1994. HIV-1 Nef inhibits a common activation pathway in NIH-3T3 cells. J Biol Chem 269, 6656–6660. [PubMed] [Google Scholar]
- Dever SM, Rodriguez M, Lapierre J, Costin BN, El-Hage N, 2015. Differing roles of autophagy in HIV-associated neurocognitive impairment and encephalitis with implications for morphine co-exposure. Front Microbiol 6, 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson EJ, Jensen JB, Hille B, 2016. Regulation of calcium and phosphoinositides at endoplasmic reticulum-membrane junctions. Biochem Soc Trans 44, 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkins C, Pilli M, Kehrl JH, 2015. Roles of autophagy in HIV infection. Immunol Cell Biol 93, 11–17. [DOI] [PubMed] [Google Scholar]
- Doengi M, Hirnet D, Coulon P, Pape HC, Deitmer JW, Lohr C, 2009. GABA uptake-dependent Ca(2+) signaling in developing olfactory bulb astrocytes. Proc Natl Acad Sci U S A 106, 17570–17575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong R, Saheki Y, Swarup S, Lucast L, Harper JW, De Camilli P, 2016. Endosome-ER Contacts Control Actin Nucleation and Retromer Function through VAP-Dependent Regulation of PI4P. Cell 166, 408–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edara VV, Ghorpade A, Borgmann K, 2020. Insights into the Gene Expression Profiles of Active and Restricted Red/Green-HIV(+) Human Astrocytes: Implications for Shock or Lock Therapies in the Brain. Journal of virology 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorova PA, Bezprozvanny IB, 2018. Inositol 1,4,5-trisphosphate receptors and neurodegenerative disorders. FEBS J 285, 3547–3565. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, 2014. Role of connexin/pannexin containing channels in infectious diseases. FEBS Lett 588, 1389–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, Berman JW, 2007. Gap junctions mediate human immunodeficiency virus-bystander killing in astrocytes. J Neurosci 27, 12844–12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, Berman JW, 2013. Cytochrome C dysregulation induced by HIV infection of astrocytes results in bystander apoptosis of uninfected astrocytes by an IP3 and calcium-dependent mechanism. J Neurochem 127, 644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, Clements JE, Zink MC, Berman JW, 2011. Human immunodeficiency virus infection of human astrocytes disrupts blood-brain barrier integrity by a gap junction-dependent mechanism. J Neurosci 31, 9456–9465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, D’Aversa TG, Lopez L, Calderon TM, Berman JW, 2003. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J Neurochem 85, 1299–1311. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, King JE, Nath A, Calderon TM, Zukin RS, Bennett MV, Berman JW, 2007. HIV-tat induces formation of an LRP-PSD-95- NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci U S A 104, 3438–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantseva MV, Kokarovtseva L, Naus CG, Carlen PL, MacFabe D, Perez Velazquez JL, 2002. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J Neurosci 22, 644–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich F, Christiano R, Olson DK, Alcazar-Roman A, DeCamilli P, Walther TC, 2014. A role for eisosomes in maintenance of plasma membrane phosphoinositide levels. Mol Biol Cell 25, 2797–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari FB, Lin H, Huang HS, Cavenee WK, 1997. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc Natl Acad Sci U S A 94, 12479–12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajardo-Gomez R, Santibanez CA, Labra VC, Gomez GI, Eugenin EA, Orellana JA, 2020. HIV gp120 Protein Increases the Function of Connexin 43 Hemichannels and Pannexin-1 Channels in Astrocytes: Repercussions on Astroglial Function. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganor Y, Real F, Sennepin A, Dutertre CA, Prevedel L, Xu L, Tudor D, Charmeteau B, Couedel-Courteille A, Marion S, Zenak AR, Jourdain JP, Zhou Z, Schmitt A, Capron C, Eugenin EA, Cheynier R, Revol M, Cristofari S, Hosmalin A, Bomsel M, 2019. HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat Microbiol 4, 633–644. [DOI] [PubMed] [Google Scholar]
- Garcia-Dorado D, Inserte J, Ruiz-Meana M, Gonzalez MA, Solares J, Julia M, Barrabes JA, Soler-Soler J, 1997. Gap junction uncoupler heptanol prevents cell-to-cell progression of hypercontracture and limits necrosis during myocardial reperfusion. Circulation 96, 3579–3586. [DOI] [PubMed] [Google Scholar]
- Gaspers LD, Thomas AP, 2005. Calcium signaling in liver. Cell Calcium 38, 329–342. [DOI] [PubMed] [Google Scholar]
- Gauthier J, Meijer IA, Lessel D, Mencacci NE, Krainc D, Hempel M, Tsiakas K, Prokisch H, Rossignol E, Helm MH, Rodan LH, Karamchandani J, Carecchio M, Lubbe SJ, Telegrafi A, Henderson LB, Lorenzo K, Wallace SE, Glass IA, Hamdan FF, Michaud JL, Rouleau GA, Campeau PM, 2018. Recessive mutations in VPS13D cause childhood onset movement disorders. Ann Neurol 83, 1089–1095. [DOI] [PubMed] [Google Scholar]
- Giordano F, Saheki Y, Idevall-Hagren O, Colombo SF, Pirruccello M, Milosevic I, Gracheva EO, Bagriantsev SN, Borgese N, De Camilli P, 2013. PI(4,5)P(2)-dependent and Ca(2+)-regulated ER-PM interactions mediated by the extended synaptotagmins. Cell 153, 1494–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao HN, Chiu FC, Losev L, Weidenheim KM, Rashbaum WK, Lyman WD, 1997. HIV infection of human fetal neural cells is mediated by gp120 binding to a cell membrane-associated molecule that is not CD4 nor galactocerebroside. Brain research 764, 149–157. [DOI] [PubMed] [Google Scholar]
- Hao HN, Lyman WD, 1999. HIV infection of fetal human astrocytes: the potential role of a receptor-mediated endocytic pathway. Brain research 823, 24–32. [DOI] [PubMed] [Google Scholar]
- Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I, 2011. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. Journal of neurovirology 17, 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huque T, Brand JG, Rabinowitz JL, 1992. Metabolism of inositol-1,4,5-trisphosphate in the taste organ of the channel catfish, Ictalurus punctatus. Comp Biochem Physiol B 102, 833–839. [DOI] [PubMed] [Google Scholar]
- Ivanova H, Vervliet T, Missiaen L, Parys JB, De Smedt H, Bultynck G, 2014. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim Biophys Acta 1843, 2164–2183. [DOI] [PubMed] [Google Scholar]
- Kaur N, Dendukuri N, Fellows LK, Brouillette MJ, Mayo N, 2019. Association between cognitive reserve and cognitive performance in people with HIV: a systematic review and meta-analysis. AIDS Care, 1–11. [DOI] [PubMed] [Google Scholar]
- Kielian T, 2008. Glial connexins and gap junctions in CNS inflammation and disease. J Neurochem 106, 1000–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Beon J, Lee S, Park SJ, Ahn H, Kim MG, Park JE, Kim W, Yuk JM, Kang SJ, Lee SH, Jo EK, Seong RH, Kim S, 2017. Inositol polyphosphate multikinase promotes Toll-like receptor-induced inflammation by stabilizing TRAF6. Sci Adv 3, e1602296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Tyagi R, Lee JY, Park J, Kim YR, Beon J, Chen PY, Cha JY, Snyder SH, Kim S, 2013. Inositol polyphosphate multikinase is a coactivator for serum response factor-dependent induction of immediate early genes. Proc Natl Acad Sci U S A 110, 19938–19943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim SF, Maag D, Maxwell MJ, Resnick AC, Juluri KR, Chakraborty A, Koldobskiy MA, Cha SH, Barrow R, Snowman AM, Snyder SH, 2011. Amino acid signaling to mTOR mediated by inositol polyphosphate multikinase. Cell Metab 13, 215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko A, Kang G, Hattler JB, Galadima HI, Zhang J, Li Q, Kim WK, 2019. Macrophages but not Astrocytes Harbor HIV DNA in the Brains of HIV-1-Infected Aviremic Individuals on Suppressive Antiretroviral Therapy. J Neuroimmune Pharmacol 14, 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolehmainen J, Black GC, Saarinen A, Chandler K, Clayton-Smith J, Traskelin AL, Perveen R, Kivitie-Kallio S, Norio R, Warburg M, Fryns JP, de la Chapelle A, Lehesjoki AE, 2003. Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet 72, 1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N, Leonzino M, Hancock-Cerutti W, Horenkamp FA, Li P, Lees JA, Wheeler H, Reinisch KM, De Camilli P, 2018. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J Cell Biol 217, 3625–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landstrom AP, Beavers DL, Wehrens XH, 2014. The junctophilin family of proteins: from bench to bedside. Trends Mol Med 20, 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Kim YR, Park J, Kim S, 2012. Inositol polyphosphate multikinase signaling in the regulation of metabolism. Ann N Y Acad Sci 1271, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, Cormier-Dequaire F, Hassoun SM, Pujol C, Ciura S, Erpapazoglou Z, Usenko T, Maurage CA, Sahbatou M, Liebau S, Ding J, Bilgic B, Emre M, Erginel-Unaltuna N, Guven G, Tison F, Tranchant C, Vidailhet M, Corvol JC, Krack P, Leutenegger AL, Nalls MA, Hernandez DG, Heutink P, Gibbs JR, Hardy J, Wood NW, Gasser T, Durr A, Deleuze JF, Tazir M, Destee A, Lohmann E, Kabashi E, Singleton A, Corti O, Brice A, 2016. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am J Hum Genet 98, 500–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letendre S, 2011. Central nervous system complications in HIV disease: HIV-associated neurocognitive disorder. Top Antivir Med 19, 137–142. [PMC free article] [PubMed] [Google Scholar]
- Leybaert L, Sanderson MJ, 2012. Intercellular Ca(2+) waves: mechanisms and function. Physiological reviews 92, 1359–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GH, Maric D, Major EO, Nath A, 2020. Productive HIV infection in astrocytes can be established via a non-classical mechanism. AIDS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litosch I, 2015. Regulating G protein activity by lipase-independent functions of phospholipase C. Life Sci 137, 116–124. [DOI] [PubMed] [Google Scholar]
- Lutgen V, Narasipura SD, Barbian HJ, Richards M, Wallace J, Razmpour R, Buzhdygan T, Ramirez SH, Prevedel L, Eugenin EA, Al-Harthi L, 2020. HIV infects astrocytes in vivo and egresses from the brain to the periphery. PLoS Pathog 16, e1008381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madreiter-Sokolowski CT, Ramadani-Muja J, Ziomek G, Burgstaller S, Bischof H, Koshenov Z, Gottschalk B, Malli R, Graier WF, 2019. Tracking intra- and inter-organelle signaling of mitochondria. Febs J 286, 4378–4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, Eugenin EA, 2016. Mechanisms of HIV Neuropathogenesis: Role of Cellular Communication Systems. Curr HIV Res 14, 400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, Eugenin EA, 2019. Role of Connexin and Pannexin containing channels in HIV infection and NeuroAIDS. Neurosci Lett 695, 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, Theis M, Eugenin EA, 2017. Connexin43 Containing Gap Junction Channels Facilitate HIV Bystander Toxicity: Implications in NeuroHIV. Front Mol Neurosci 10, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maly CJ, Cumsky HJL, Costello CM, Schmidt JE, Butterfield RJ, Zhang N, DiCaudo DJ, Nelson SA, Smith ML, Ochoa SA, Baum CL, Nagel TH, Pittelkow MR, Sekulic A, Mangold AR, 2019. Prognostic value of inositol polyphosphate-5-phosphatase expression in recurrent and metastatic cutaneous squamous cell carcinoma. J Am Acad Dermatol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manninen A, Saksela K, 2002. HIV-1 Nef interacts with inositol trisphosphate receptor to activate calcium signaling in T cells. J Exp Med 195, 1023–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarron JG, Saunter C, Wilson C, Girkin JM, Chalmers S, 2018. Mitochondria Structure and Position in the Local Control of Calcium Signals in Smooth Muscle Cells. 173–190. [PubMed]
- McCarthy M, He J, Wood C, 1998. HIV-1 strain-associated variability in infection of primary neuroglia. Journal of neurovirology 4, 80–89. [DOI] [PubMed] [Google Scholar]
- Meini A, Sticozzi C, Massai L, Palmi M, 2008. A nitric oxide/Ca(2+)/calmodulin/ERK1/2 mitogen-activated protein kinase pathway is involved in the mitogenic effect of IL-1beta in human astrocytoma cells. Br J Pharmacol 153, 1706–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Braceras S, Tornero-Ecija AR, Vincent O, Escalante R, 2019. VPS13A is closely associated with mitochondria and is required for efficient lysosomal degradation. Disease models & mechanisms 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasipura SD, Kim S, Al-Harthi L, 2014. Epigenetic regulation of HIV-1 latency in astrocytes. J Virol 88, 3031–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor JJ, 2013. Targeting tumour necrosis factor-alpha in hypoxia and synaptic signalling. Ir J Med Sci 182, 157–162. [DOI] [PubMed] [Google Scholar]
- Ohagen A, Ghosh S, He J, Huang K, Chen Y, Yuan M, Osathanondh R, Gartner S, Shi B, Shaw G, Gabuzda D, 1999. Apoptosis induced by infection of primary brain cultures with diverse human immunodeficiency virus type 1 isolates: evidence for a role of the envelope. J Virol 73, 897–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okafo G, Prevedel L, Eugenin E, 2017. Tunneling nanotubes (TNT) mediate long-range gap junctional communication: Implications for HIV cell to cell spread. Sci Rep 7, 16660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okafo G, Valdebenito S, Donoso M, Luu R, Ajasin D, Prideaux B, Gorantla S, Eugenin EA, 2020. Role of Tunneling Nanotube-like Structures during the Early Events of HIV Infection: Novel Features of Tissue Compartmentalization and Mechanism of HIV Spread. Journal of immunology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellana JA, Saez JC, Bennett MV, Berman JW, Morgello S, Eugenin EA, 2014. HIV increases the release of dickkopf-1 protein from human astrocytes by a Cx43 hemichannel-dependent mechanism. J Neurochem 128, 752–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Longo F, Park SJ, Lee S, Bae M, Tyagi R, Han JH, Kim S, Santini E, Klann E, Snyder SH, 2019. Inositol polyphosphate multikinase mediates extinction of fear memory. Proc Natl Acad Sci U S A 116, 2707–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AB, Mangold AR, Costello CM, Nagel TH, Smith ML, Hayden RE, Sekulic A, 2018. Frequent loss of inositol polyphosphate-5-phosphatase in oropharyngeal squamous cell carcinoma. J Eur Acad Dermatol Venereol 32, e36–e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pender MP, Rist MJ, 2001. Apoptosis of inflammatory cells in immune control of the nervous system: role of glia. Glia 36, 137–144. [DOI] [PubMed] [Google Scholar]
- Peudenier S, Hery C, Ng KH, Tardieu M, 1991. HIV receptors within the brain: a study of CD4 and MHC-II on human neurons, astrocytes and microglial cells. Res Virol 142, 145–149. [DOI] [PubMed] [Google Scholar]
- Plog BA, Nedergaard M, 2018. The Glymphatic System in Central Nervous System Health and Disease: Past, Present, and Future. Annu Rev Pathol 13, 379–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevedel L, Morocho C, Bennett MVL, Eugenin EA, 2017. HIV-Associated Cardiovascular Disease: Role of Connexin 43. Am J Pathol 187, 1960–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajadurai CV, Havrylov S, Coelho PP, Ratcliffe CD, Zaoui K, Huang BH, Monast A, Chughtai N, Sangwan V, Gertler FB, Siegel PM, Park M, 2016. 5’-Inositol phosphatase SHIP2 recruits Mena to stabilize invadopodia for cancer cell invasion. J Cell Biol 214, 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampoldi L, Dobson-Stone C, Rubio JP, Danek A, Chalmers RM, Wood NW, Verellen C, Ferrer X, Malandrini A, Fabrizi GM, Brown R, Vance J, Pericak-Vance M, Rudolf G, Carre S, Alonso E, Manfredi M, Nemeth AH, Monaco AP, 2001. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat Genet 28, 119–120. [DOI] [PubMed] [Google Scholar]
- Rasul F, Zheng F, Dong F, He J, Liu L, Liu W, Cheema JY, Wei W, Fu C, 2021. Emr1 regulates the number of foci of the endoplasmic reticulum-mitochondria encounter structure complex. Nat Commun 12, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawanduzy A, Hansen A, Hansen TW, Nedergaard M, 1997. Effective reduction of infarct volume by gap junction blockade in a rodent model of stroke. J Neurosurg 87, 916–920. [DOI] [PubMed] [Google Scholar]
- Russell RA, Chojnacki J, Jones DM, Johnson E, Do T, Eggeling C, Padilla-Parra S, Sattentau QJ, 2017. Astrocytes Resist HIV-1 Fusion but Engulf Infected Macrophage Material. Cell Rep 18, 1473–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC, 2003. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev 83, 1359–1400. [DOI] [PubMed] [Google Scholar]
- Saez JC, Connor JA, Spray DC, Bennett MV, 1989. Hepatocyte gap junctions are permeable to the second messenger, inositol 1,4,5-trisphosphate, and to calcium ions. Proc Natl Acad Sci U S A 86, 2708–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saheki Y, De Camilli P, 2017. Endoplasmic Reticulum-Plasma Membrane Contact Sites. Annu Rev Biochem 86, 659–684. [DOI] [PubMed] [Google Scholar]
- Sanderson MJ, Charles AC, Dirksen ER, 1990. Mechanical stimulation and intercellular communication increases intracellular Ca2+ in epithelial cells. Cell Regul 1, 585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santello M, Toni N, Volterra A, 2019. Astrocyte function from information processing to cognition and cognitive impairment. Nat Neurosci 22, 154–166. [DOI] [PubMed] [Google Scholar]
- Santos-Llamas A, Monte MJ, Marin JJG, Perez MJ, 2018. Dysregulation of autophagy in rat liver with mitochondrial DNA depletion induced by the nucleoside analogue zidovudine. Arch Toxicol 92, 2109–2118. [DOI] [PubMed] [Google Scholar]
- Scemes E, Dermietzel R, Spray DC, 1998. Calcium waves between astrocytes from Cx43 knockout mice. Glia 24, 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweighardt B, Shieh JT, Atwood WJ, 2001. CD4/CXCR4-independent infection of human astrocytes by a T-tropic strain of HIV-1. Journal of neurovirology 7, 155–162. [DOI] [PubMed] [Google Scholar]
- Seong E, Insolera R, Dulovic M, Kamsteeg EJ, Trinh J, Bruggemann N, Sandford E, Li S, Ozel AB, Li JZ, Jewett T, Kievit AJA, Munchau A, Shakkottai V, Klein C, Collins CA, Lohmann K, van de Warrenburg BP, Burmeister M, 2018. Mutations in VPS13D lead to a new recessive ataxia with spasticity and mitochondrial defects. Ann Neurol 83, 1075–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M, 2003. Signaling at the gliovascular interface. The Journal of neuroscience : the official journal of the Society for Neuroscience 23, 9254–9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims CE, Allbritton NL, 1998. Metabolism of inositol 1,4,5-trisphosphate and inositol 1,3,4,5-tetrakisphosphate by the oocytes of Xenopus laevis. The Journal of biological chemistry 273, 4052–4058. [DOI] [PubMed] [Google Scholar]
- Siushansian R, Bechberger JF, Cechetto DF, Hachinski VC, Naus CC, 2001. Connexin43 null mutation increases infarct size after stroke. J Comp Neurol 440, 387–394. [DOI] [PubMed] [Google Scholar]
- Stefan CJ, 2018. Building ER-PM contacts: keeping calm and ready on alarm. Curr Opin Cell Biol 53, 1–8. [DOI] [PubMed] [Google Scholar]
- Sticozzi C, Belmonte G, Meini A, Carbotti P, Grasso G, Palmi M, 2013. IL-1beta induces GFAP expression in vitro and in vivo and protects neurons from traumatic injury-associated apoptosis in rat brain striatum via NFkappaB/Ca(2)(+)-calmodulin/ERK mitogen-activated protein kinase signaling pathway. Neuroscience 252, 367–383. [DOI] [PubMed] [Google Scholar]
- Stout C, Charles A, 2003. Modulation of intercellular calcium signaling in astrocytes by extracellular calcium and magnesium. Glia 43, 265–273. [DOI] [PubMed] [Google Scholar]
- Sun EW, Guillen-Samander A, Bian X, Wu Y, Cai Y, Messa M, De Camilli P, 2019. Lipid transporter TMEM24/C2CD2L is a Ca(2+)-regulated component of ER-plasma membrane contacts in mammalian neurons. Proc Natl Acad Sci U S A 116, 5775–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada M, Nishizawa M, Onodera O, 2016. Roles of inositol 1,4,5-trisphosphate receptors in spinocerebellar ataxias. Neurochem Int 94, 1–8. [DOI] [PubMed] [Google Scholar]
- Tedaldi EM, Minniti NL, Fischer T, 2015. HIV-associated neurocognitive disorders: the relationship of HIV infection with physical and social comorbidities. Biomed Res Int 2015, 641913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdebenito S, Barreto A, Eugenin EA, 2018. The role of connexin and pannexin containing channels in the innate and acquired immune response. Biochim Biophys Acta Biomembr 1860, 154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdebenito S, Castellano P, Ajasin D, Eugenin EA, 2021. Astrocytes are HIV reservoirs in the brain: A cell type with poor HIV infectivity and replication but efficient cell-to-cell viral transfer. J Neurochem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Meide PH, Schellekens H, 1996. Cytokines and the immune response. Biotherapy 8, 243–249. [DOI] [PubMed] [Google Scholar]
- Vejar S, Oyarzun JE, Retamal MA, Ortiz FC, Orellana JA, 2019. Connexin and Pannexin-Based Channels in Oligodendrocytes: Implications in Brain Health and Disease. Front Cell Neurosci 13, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasquez S, Prevedel L, Valdebenito S, Gorska AM, Golovko M, Khan N, Geiger J, Eugenin EA, 2019. Circulating levels of ATP is a biomarker of HIV cognitive impairment. EBioMedicine, 102503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Kettenmann H, 1996. Calcium signalling in glial cells. Trends in neurosciences 19, 346–352. [DOI] [PubMed] [Google Scholar]
- Vermassen E, Parys JB, Mauger JP, 2004. Subcellular distribution of the inositol 1,4,5-trisphosphate receptors: functional relevance and molecular determinants. Biol Cell 96, 3–17. [DOI] [PubMed] [Google Scholar]
- Vonk JJ, Yeshaw WM, Pinto F, Faber AI, Lahaye LL, Kanon B, van der Zwaag M, Velayos-Baeza A, Freire R, van ISC, Grzeschik NA, Sibon OC, 2017. Drosophila Vps13 Is Required for Protein Homeostasis in the Brain. PLoS One 12, e0170106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TF, Zhou C, Tang AH, Wang SQ, Chai Z, 2006. Cellular mechanism for spontaneous calcium oscillations in astrocytes. Acta Pharmacol Sin 27, 861–868. [DOI] [PubMed] [Google Scholar]
- Wu H, Carvalho P, Voeltz GK, 2018. Here, there, and everywhere: The importance of ER membrane contact sites. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Whiteus C, Xu CS, Hayworth KJ, Weinberg RJ, Hess HF, De Camilli P, 2017a. Contacts between the endoplasmic reticulum and other membranes in neurons. Proc Natl Acad Sci U S A 114, E4859–E4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Thiyagarajan S, O’Shaughnessy B, Karatekin E, 2017b. Regulation of Exocytotic Fusion Pores by SNARE Protein Transmembrane Domains. Front Mol Neurosci 10, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia HJ, Yang G, 2005. Inositol 1,4,5-trisphosphate 3-kinases: functions and regulations. Cell Res 15, 83–91. [DOI] [PubMed] [Google Scholar]
- Xing L, Yang T, Cui S, Chen G, 2019. Connexin Hemichannels in Astrocytes: Role in CNS Disorders. Front Mol Neurosci 12, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AW, Sachs AJ, Nystuen AM, 2015. Deletion of Inpp5a causes ataxia and cerebellar degeneration in mice. Neurogenetics 16, 277–285. [DOI] [PubMed] [Google Scholar]
- Yang M, Zhai X, Ge T, Yang C, Lou G, 2018. miR-181a-5p Promotes Proliferation and Invasion and Inhibits Apoptosis of Cervical Cancer Cells via Regulating Inositol Polyphosphate-5-Phosphatase A (INPP5A). Oncol Res 26, 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yeshaw WM, van der Zwaag M, Pinto F, Lahaye LL, Faber AI, Gomez-Sanchez R, Dolga AM, Poland C, Monaco AP, van ISC, Grzeschik NA, Velayos-Baeza A, Sibon OC, 2019. Human VPS13A is associated with multiple organelles and influences mitochondrial morphology and lipid droplet motility. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Jones C, Zhang G, 2018. The Role of Phospholipase C Signaling in Macrophage-Mediated Inflammatory Response. J Immunol Res 2018, 5201759. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in the manuscript is fully available upon reasonable request to the authors.