

Abstract
Pinpointing functional noncoding DNA sequences and defining their contributions to health-related traits is a major challenge for modern genetics. We developed a high-throughput framework to map noncoding DNA functions with single-nucleotide resolution in four loci that control erythroid fetal hemoglobin (HbF) expression, a genetically determined trait that modifies sickle cell disease (SCD) phenotypes. Specifically, we used the adenine base editor ABEmax to introduce 10,156 separate A•T to G•C conversions in 307 predicted regulatory elements and quantified the effects on erythroid HbF expression. We identified numerous regulatory elements, defined their epigenomic structures, and linked them to low-frequency variants associated with HbF expression in an SCD cohort. Targeting a newly discovered γ-globin gene repressor element in SCD donor CD34+ hematopoietic progenitors raised HbF levels in the erythroid progeny, inhibiting hypoxia-induced sickling. Our findings reveal previously unappreciated genetic complexities of HbF regulation and provide potentially therapeutic insights into SCD.
Introduction
Cis-regulatory elements (CREs) are the driving force in the conversion of static genomic DNA sequences into cell-, tissue-, and developmental-stage–specific gene expression profiles1–3. Alterations of CREs can cause human diseases4–6 and influence health-related traits7. The results of genome-wide association studies (GWAS) indicate that CREs are major participants in common diseases8, as nearly 90% of disease-associated single-nucleotide variants (SNVs) are within noncoding sequences of the human genome. Global regulatory mapping performed through consortia, including the ENCODE (Encyclopedia Of DNA Elements)9 and Roadmap Epigenomics10 consortia, has identified numerous properties of CREs, including chromatin openness11, characteristic histone modifications12–14, transcription factor (TF) occupancy, and three-dimensional (3D) chromatin architecture15,16. However, the causal relation between these biochemical signals and the functions of CREs remains imprecise17,18. High-throughput systems are needed for functional validation of these putative CREs and their modulation by natural genetic variation. Ideally, such systems should interrogate CREs in their native genomic context, in biologically relevant cells, and at the single-nucleotide level.
Clustered regularly interspaced short palindromic repeats (CRISPR)–based genome-editing technologies are used to identify CREs by measuring the effects of targeted genetic perturbations on biological phenotypes7,19–21. Most high-throughput CRISPR/Cas9 mutational screening is performed by creating targeted double-stranded DNA breaks (DSBs), which are subsequently repaired in cells via nonhomologous end joining to create insertion/deletion (indel) mutations that disrupt DNA coding regions or CREs. However, Cas9-induced indels vary in size, ranging from one to hundreds of nucleotides, and are, therefore, imprecise for mapping functional CREs. This limitation may be circumvented by base editors composed of catalytically impaired Cas9, termed Cas9 nickase (Cas9n), linked to modified nucleotide deaminases22,23. Adenine base editors (ABEs) convert A•T to G•C and cytosine base editors (CBEs) convert C•G to T•A via DSB-independent mechanisms with low rates of indel formation, thereby providing nucleotide-level resolution.
Fetal hemoglobin, expressed mainly in third-trimester fetal red blood cells (RBCs), is a heterotetramer composed of two γ-globin and two α-globin subunits (α2γ2). Around birth, the γ-globin genes (HBG1 and HBG2) are transcriptionally silenced, with concomitant activation of the related β-globin gene (HBB), resulting in the production of adult hemoglobin (HbA, α2β2). Elevated levels of RBC HbF arising from residual postnatal HBG1 and HBG2 transcription alleviate β-hemoglobinopathy phenotypes and, in extreme cases, termed hereditary persistence of fetal hemoglobin (HPFH), eliminate the symptoms entirely. Postnatal HbF expression is regulated largely by genetic variation. GWAS have identified common variants associated with HbF levels at three loci: BCL11A and MYB, which encode TFs, and the β-like globin gene cluster, which includes HBG1, HBG2, and HBB coding regions and associated CREs. Rare variants in the β-like globin gene cluster and in the KLF1 erythroid TF gene have been shown to cause HPFH24–26. Genetic variants that control HbF expression act by regulating the function of CREs, some of which have been targeted in strategies aimed at inducing HbF therapeutically27–29. However, the currently identified variants account for less than half of the heritability in HbF levels30; the CREs and associated variants that account for the remainder remain unknown.
Here we present a strategy (Fig. 1a) to define the regulation of health-related traits by integrating functional genomic fine mapping with population-based studies. Specifically, we quantified the functional effects on HbF expression of thousands of ABEmax-induced point mutations targeted to computationally predicted CREs in the BCL11A, MYB-HBS1L, KLF1, and β-like globin gene loci. By combining the effects of genetic perturbation with bioinformatic predictions and computational deconvolution modeling, we identified multiple CREs with base-pair resolution, defined their genomic and epigenomic signatures, and used this information to address clinically relevant problems related to the genetics of SCD.
Figure 1. Establishment of an ABE-based system to perturb CREs.

(a) A schematic representation of the overall strategy: 1) Predicting CREs regulating HbF levels. 2) Combining a high-throughput ABEmax screen with computational deconvolution to identify CREs with base-pair resolution. 3) Applying high-resolution CRE mapping to understand functional variants and to identify potential novel therapeutic targets.
(b) ABEmax protein level in wild-type HUDEP-2 (WT) cells and HUDEP-2–ABEmax (ABEmax) cells measured by Western blot analysis. The result is representative of two independent experiments (image was cropped from source data Fig. 1).
(c) Heatmap showing the Pearson correlation of WT and ABEmax cell transcriptomes as measured by RNA-seq. The number in each cell is the correlation coefficient.
(d–i) HUDEP-2–ABEmax cells were transduced with a non-targeting control gRNA (Ctrl) or with a gRNA targeting the BCL11A enhancer (BCL11A–ENH).
(d) Flow-cytometry plots showing the expression of the RBC maturation markers Band3 and CD49d after 5 days of induced differentiation. The result is representative of three independent experiments.
(e) On-target DNA base-editing efficiencies measured by next-generation sequencing. P = 1.8 × 10−4.
(f) BCL11A mRNA measured by real-time RT-qPCR. P = 2.7 × 10−5. (g) The percentage of γ-globin mRNA (γ/γ+β) or β-globin mRNA (β/γ+β) as measured by real-time RT-qPCR.
(h) The hemoglobin (Hb) protein content measured by IE-HPLC after 5 additional days of induced erythroid maturation. The result is representative of three independent experiments.
(i) Flow-cytometry plots showing HbF immunostaining (F-cells). The result is representative of three independent experiments.
*P values were determined using two-tailed unpaired t-test. Bar plots show mean ± S.E.M. of three independent experiments.
Results
Regulatory sequence perturbation through point mutations
To assess the functional effects of CREs through precise base-pair perturbation, we stably expressed the adenine base editor ABEmax31 in HUDEP-2 cells (Fig. 1b and Extended Data Fig. 1a, b), an immortalized human erythroblast cell line that self-renews in culture and can be induced to undergo terminal erythroid maturation32. HUDEP-2 cells normally express HbA but can be induced to express HbF upon genetic disruption of CREs that repress γ-globin expression19,27,33,34. Whole-transcriptome RNA sequencing confirmed that stable expression of ABEmax did not alter gene expression in HUDEP-2 cells or the capacity of the cells to undergo terminal erythroid maturation (Fig. 1c, d and Supplementary Table 1). In contrast to reports of extensive transcriptome-wide deamination (“spurious deamination”) after base editor expression in non-erythroid cells such as HEK293T cells35, the baseline rate of adenine-to-inosine (A-to-I) conversion was low in HUDEP-2–ABEmax cells (Extended Data Fig. 1c and Supplementary Table 2). Therefore, the rates of spurious editing might be cell-type and/or ABE-dosage dependent.
To test whether ABEmax could efficiently perturb the function of CREs, we targeted a previously defined enhancer in the BCL11A gene, whose product represses HBG1/HBG2 and HbF expression7,34,36. Expression of a targeting guide RNA (gRNA) precisely converted more than 60% of the total adenines to guanines (Fig. 1e) in a core enhancer GATA1–half-Ebox element (Extended Data Fig. 1d), resulting in a 70% decrease in BCL11A mRNA (Fig. 1f), an increase in γ-globin mRNA from 12% to 60% of the total β-like (γ + β) globin transcripts (Fig. 1g), and marked induction of γ-globin protein as detected by Western blot analysis (Extended Data Fig. 1e). HbF production was increased, as evidenced by isoelectric focusing high-performance liquid chromatography (IE-HPLC) of cell lysates (Fig. 1h) and immuno-flow cytometry for cells staining for HbF (“F-cells”) (Fig. 1i).
High-throughput perturbation of putative regulatory elements
We designed 6,174 gRNAs targeting 307 putative CREs that were selected according to chromatin accessibility, erythroid occupancy of the key erythroid TF GATA1, and predictions in the literature37,38. As controls, we included 20 non-targeting gRNAs and, for the same four loci, 112 gRNAs targeting sequences that contained no adenines (Supplementary Table 3).
We generated a lentiviral vector library encoding the 6,174 gRNAs and a puromycin selection cassette, transduced HUDEP-2–ABEmax cells at a multiplicity of infection (MOI) of 0.3, selected transduced cells in puromycin for 2 days, then induced erythroid differentiation for 5 days. We next fractionated the cells by HbF immuno-flow cytometry, followed by high-throughput DNA sequencing to compare the gRNA representation in the HbFhigh and HbFlow populations (Fig. 2a).
Figure 2. High-throughput ABEmax perturbation identifies CREs that control HbF expression.
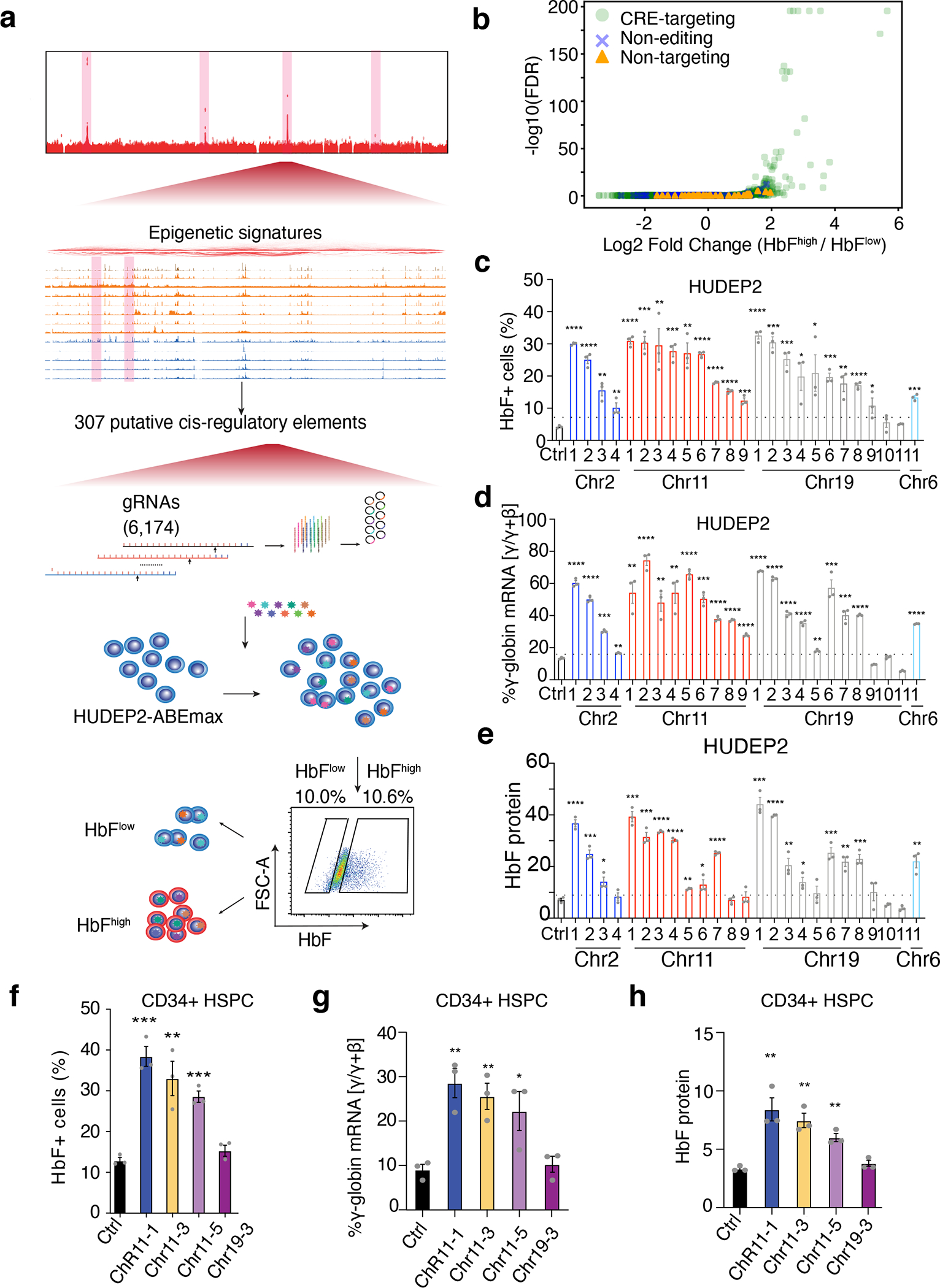
(a) Schematic representation of the workflow showing bioinformatics-informed gRNA library construction, mutagenesis, and selection for HbFhigh and HbFlow phenotypes.
(b) Differential representation of individual targeting gRNAs in HbFhigh and HbFlow populations as a volcano plot. Each dot represents a single gRNA. The x-axis shows the log fold-difference and the y-axis shows the −log10(FDR). Light green dots, CRE-targeting gRNAs; Blue crosses, non-editing control gRNAs; orange triangles, non-targeting control gRNAs.
(c–e) Validation studies of top-hit gRNAs in HUDEP-2 cells. HUDEP-2–ABEmax cells were treated with a non-targeting control (Ctrl) gRNA or with randomly chosen individual candidate gRNAs that were identified as top screening hits by their over-representation in HbFhigh cells. The chromosome locations of each gRNA are shown on the x-axis.
(c) F-cell fractions measured by immuno-flow cytometry.
(d) The percentage of γ-globin mRNA as determined by real-time RT-qPCR.
(e) The hemoglobin F fraction measured by IE-HPLC.
(f–h) Validation studies of top-hit gRNAs in normal donor CD34+ HSPC–derived erythroblasts. CD34+ cells were transfected with RNP complexes consisting of ABEmax + non-targeting control (Ctrl) gRNA or individual top-hit gRNAs, grown in culture under conditions that support erythroid differentiation, and analyzed after 14 days.
(f) F-cell fractions measured by immuno-flow cytometry.
(g) The percentage of γ-globin mRNA as determined by real-time RT-qPCR.
(h) The hemoglobin F fraction measured by IE-HPLC.
****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, were determined using two-tailed unpaired t-test (Exact P values for each test are listed in source data). Bar plots show mean ± S.E.M. of three independent experiments.
As unperturbed HUDEP-2 cells express HbF at a low level, our screen was configured to identify mutations that upregulate HbF. Lentiviral vector sequences encoding gRNAs that induced HbF were predicted to be preferentially represented in HbFhigh cells, as compared to HbFlow cells. Biological replicate screenings produced highly reproducible results (R > 0.95) (Extended Data Fig. 2a, b). We used a negative binomial model to test the difference in gRNA abundance between the two populations (Fig. 2b and Supplementary Table 4). At a false discovery rate (FDR) of <1 × 10−3, 45 gRNAs with a >2 log fold-change were significantly enriched in the HbFhigh cell population. To validate the results from the pooled library, we randomly selected 25 HbFhigh gRNAs and tested their functions individually (Supplementary Table 5). Of these candidate gRNAs, 92% (23 of 25) resulted in an increased F-cell fraction (Fig. 2c), 88% (22 of 25) resulted in an increased proportion of γ-globin mRNA (Fig. 2d), and 72% (18 of 25) led to significantly increased HbF protein levels (Fig. 2e). To validate our findings in primary erythroblasts, we electroporated mobilized peripheral blood CD34+ hematopoietic stem and progenitor cells (HSPCs) from healthy donors with ribonucleoprotein (RNP) complexes consisting of ABE and one of four separate gRNAs, after which the cells underwent in vitro erythroid differentiation. Three of the four gRNA–RNP complexes caused significant increases in the F-cell fraction (Fig. 2f), γ-globin mRNA (Fig. 2g), HbF protein as measured by IE-HPLC (Fig. 2h), and γ-globin protein as measured by Western blot analysis (Extended Data Fig. 2c). None of the ABE–gRNA RNPs altered erythroid cell differentiation, as measured using the maturation-stage markers Band3 and CD49d (Extended Data Fig. 2d).
Catalytic domain inactive Cas9 (dCas9) or Cas9n associated with base editors can affect the functions of regulatory sequences by interfering with TF-DNA interactions38. To exclude the possibility that the functional effects we observed with ABEmax were caused by the competitive binding between Cas9n and TFs to DNA, we assessed the functions of 112 gRNAs that do not contain an A/T in their sequences. The effects of gRNAs without editable adenines were similar to those of non-targeting gRNAs (P = 0.21, Wilcoxon test, Extended Data Fig. 2e). We further tested 10 gRNAs that altered HbF levels by targeting CREs that repress γ-globin gene transcription in the ABEmax system using HUDEP-2 cells stably expressing dCas9. Only 2 gRNAs modestly elevated HbF levels (Extended Data Fig. 2f). In addition, the frequencies of adenines converted by ABEmax in HbFhigh cells were consistently higher than those in HbFlow population (Extended Data Fig. 2g). Collectively, our data confirmed that ABEs disrupt CREs specifically through precise base editing.
We examined more closely the CREs with sequences targeted by HbFhigh-associated gRNAs. For BCL11A, strong signals occurred near the promoter region and in an erythroid-specific super-enhancer–like region distinguished by three DNase I hypersensitive sites (DHSs) at positions +62, +58, and +55 kb relative to the transcriptional start site (Fig. 3a, top panel). This region harbors common SNVs associated with HbF7,34. We also detected a potential CRE approximately 1 Mb downstream of the BCL11A gene (Fig. 3a, bottom panel, blue shading). Interestingly, this region, but not the BCL11A coding sequences, was eliminated by a 3.5-kb deletion in an individual with impaired erythroid BCL11A expression and elevated RBC HbF39. The current ABEmax mutagenesis screen has verified that the deleted region contains long-range CRE(s) that promote HbF expression, probably by driving erythroid BCL11A transcription. Further supporting this possibility, the CRE physically communicates with the BCL11A promoter, as revealed by H3K27ac Hi-ChIP analysis, a protein-centric chromatin conformation assay40. The KLF1 gene encodes a key erythroid TF that activates BCL11A to suppress HbF expression41,42. Rare loss-of-function variants in KLF1 cause HPFH43. We identified proximal and distal CREs of the KLF1 gene (Fig. 3b, pink shading). We also detected multiple potential HbF-regulating CREs in the nearby NFIX gene (Fig. 3b, blue shading). The NFIX gene has been reported to harbor variants associated with HbF in a Sardinian population44. Real-time qPCR showed that disrupting two identified NFIX CREs with ABEmax can decrease the expression levels of either NFIX or both NFIX and nearby KLF1 (Extended Data Fig. 3a). We also identified three functional CREs in the MYB-HBS1L region (Fig. 3c). One CRE (Fig. 3c, green shading) is within 10 and 20 bp, respectively, of two SNVs (rs4895440 and rs4895441) associated with HbF45. Another CRE (pink shading) is located in the promoter of HBS1L. A third CRE (blue shading), which showed the strongest signal, is located 20 kb upstream of HBS1L. Hi-ChIP data indicated that this element physically interacts with the HBS1L promoter and with several CREs within the MYB gene, indicating a potential multi-CRE regulatory hub. In the β-like globin gene cluster (Extended Data Fig. 3b), the ABEmax screen has verified CREs within the HBBP1 pseudogene and lncRNA BGLT346,47. In addition, we identified multiple long-range CREs in the locus control region (LCR), in the 5′ and 3′ flanking olfactory receptor (OR) genes, and immediately 3′ to the HBG1 coding region.
Figure 3. Genome browser screenshots mapping HbF-regulating CREs identified by ABE mutagenesis.
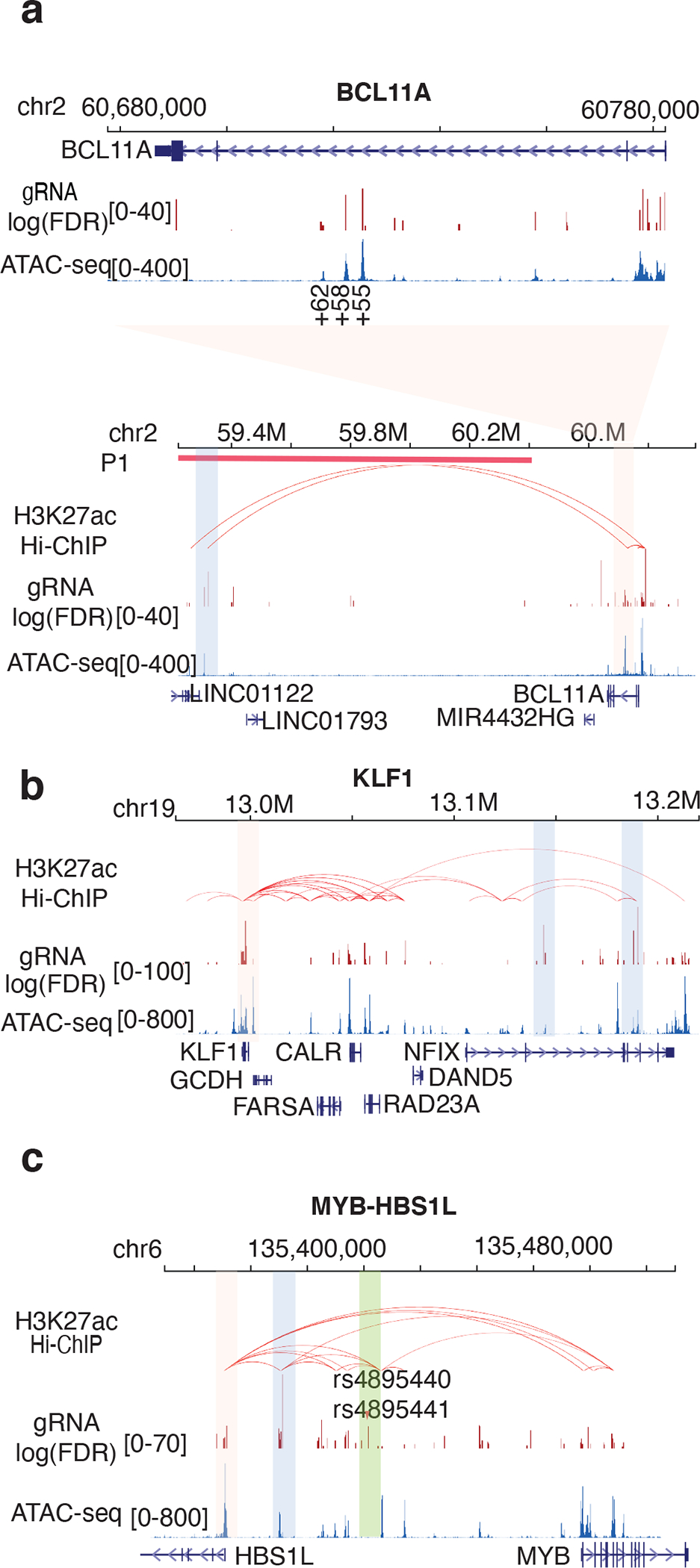
(a) BCL11A-associated HbFhigh gRNAs. The top panel shows the gene body region including an erythroid-specific super-enhancer represented by three DNase I–hypersensitive sites at positions +62, +58, and +55. gRNA log(FDR) represents the difference in gRNA abundance between the HbFhigh and HbFlow populations. ATAC-seq analysis reflects chromatin openness. The bottom panel shows a zoom-out view including a 1.8-Mb surrounding region. Chromatin interaction loops indicated by red arcs were determined by H3K27ac Hi-ChIP in HUDEP-2 cells. The thick red line (P1) represents the location of a long deletion reported in a participant with a low erythroid BCL11A level and high HbF39.
(b) KLF1-associated HbFhigh gRNAs, labeled as in panel a. Note the nearby NFIX gene, which was recently identified as regulating HbF levels.
(c) HBS1L-MYB–associated HbFhigh gRNAs, labeled as in panel a. SNVs (rs4895440, rs4895441) associated with HbF levels are labeled with red triangles.
*Specific CRE regions analyzed in further detail are color coded.
Dissection of functional regulatory sequences with base-pair resolution
One ABEmax–gRNA complex can edit multiple adenines within an approximately 5-nucleotide window23,31,48. Considering this, our pool of 6,174 gRNAs covers 10,156 editable adenines. To translate gRNA-level measurements of HbF induction to a single-adenine–level regulatory effect, we developed a computational approach to deconvolute gRNA-level measurements. To determine the editing efficiency at different distances from the protospacer adjacent motif (PAM), we first performed high-throughput amplicon sequencing in 23 ABEmax-edited target sites in HUDEP-2 cells. The amplicon sequencing data confirmed the narrow editing window of ABEmax (Extended Data Fig. 4a). We further used this information to adjust the position-dependent editing efficiency bias (Extended Data Fig. 4b). We calculated the empirical distribution of editing efficiency at different positions relative to the PAM and incorporated the adjusted position effects into our modeling. For adenines covered by multiple gRNAs, we used the empirical Brown’s method (EBM)49 to generate a combined significance value. We applied our model to all editable adenines covered by the gRNA pool library and assigned a base-pair regulation score for HbF (BPRSHbF) to each position (Fig. 4a and Supplementary Table 6).
Figure 4. Dissection of HbF-regulating CREs with base-pair resolution via ABE mutagenesis.
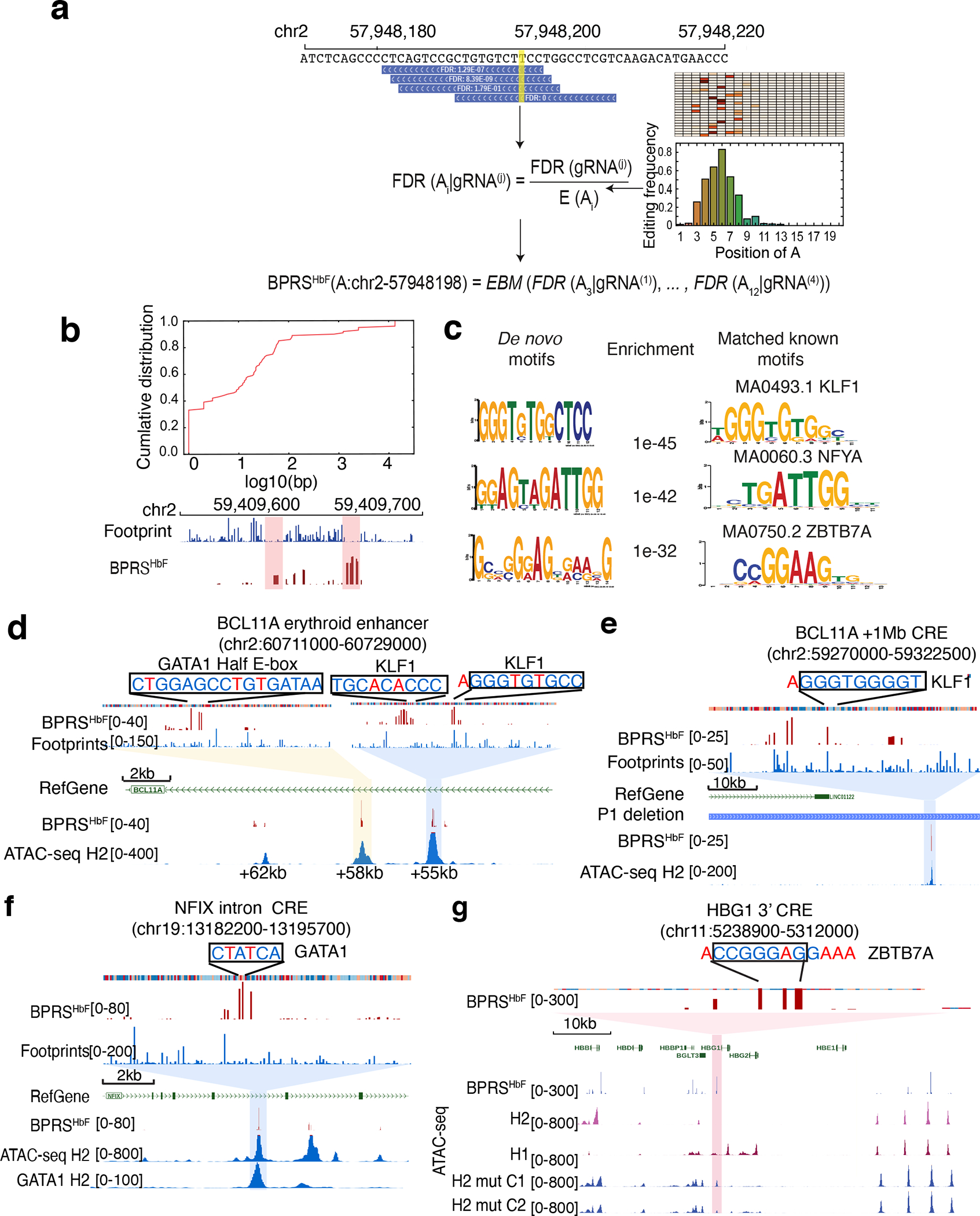
(a) Deconvoluting gRNA level measurements to base-pair regulatory scores. The left panel shows the algorithm (See the Methods for details) used to adjust for bystander editing, based on the empirical measurements obtained using amplicon sequencing. The right panel shows the frequency of A-based edits at different positions.
(b) An accumulative curve showing the distribution of distance between the 200 highest BPRSHbF adenines and their nearest TF footprints (top panel). The lower part of the panel shows high-BPRSHbF adenines (red) and TF footprints (blue) distribution within a 200-bp window in the BCL11A. Each vertical line represents one base pair.
(c) The transcription factor binding motifs most enriched in the TF footprints near adenines with high BPRSHbF.
(d–g) Fine mapping of CREs at the indicated loci. The top portion of each panel shows a zoom-in view of TF motifs disrupted. Edited adenines (or thymidines representing the opposite strand) are indicated in red font.
(d) The BCL11A intron 2 erythroid enhancer. See also Figure 3a, top panel.
(e) An HbF CRE located approximately 1 Mb downstream of the BCL11A gene. See also the blue-shaded area in Figure 3a, bottom panel.
(f) An HbF CRE located in NFIX intron. The lower track shows the GATA1 ChIP-seq signals in HUDEP-2 cells. See also the blue-shaded area in Figure 3b.
(g) An HbF CRE located immediately 3′ to HBG1. ATAC-seq signals are shown for wild-type HUDEP-1 (H1), HUDEP-2 (H2) cells and two different HUDEP-2 clones (H2_mut_C1 and H2_mut_C2) after ABEmax mutagenesis of the ZBTB7A motif.
Noncoding SNVs frequently influence health-related traits by altering interactions between TFs and their cognate motifs within CREs50. We reasoned that ABEmax mutagenesis might affect HBG1/HBG2 transcription directly by altering TF binding sites in CREs within the β-like globin gene cluster or indirectly by altering TF binding sites in CREs within the MYB, KLF1, BCL11A, or ZBTB7A (also called LRF) loci. In either case, targeted adenines with a high BPRSHbF should be enriched in DNA sequences that affect TF occupancy. To test this hypothesis, we combined chromatin accessibility patterns determined by ATAC-seq with underlying DNA sequence motifs to identify TF-specific footprints in HUDEP-2 cells (Extended Data Fig. 4c). This analysis was based on the principle that chromatin regions interacting directly with TFs exhibit unique patterns of resistance to modification by the Tn5 transposase used to detect open chromatin by ATAC-seq51. By comparing the genome-wide BPRSHbF distribution with high-resolution TF footprints, we showed that nearly 50% of the 200 targeted adenines with the highest BPRSHbF were located within 10 bp of TF footprints (Fig. 4b). In particular, the motifs of HbF regulators52–54 such as KLF1 and NFY were significantly over-represented in TF footprints close to high-BPRSHbF adenines (Fig. 4c). Another over-represented sequence matched a ZBTB7A binding motif deposited in the JASPAR database55. This motif is enriched in the binding sites of ZBTB7A, although at lower frequency than the canonical CAGGG ZBTB7A motif.
Deeper inspection of high-BPRSHbF regions resolved the modular structure of selected CREs. The BCL11A gene is regulated by an erythroid-specific exon 2 enhancer marked by three DHSs34. We observed high-BPRSHbF nucleotides in the +58 kb and +55 kb DHSs (Fig. 4d). In a prior study, Cas9-induced indels that disrupted a GATA1–half-Ebox motif in the +58 kb DHS resulted in increased erythroid HbF levels34. Our results confirmed this finding directly by showing that ABEmax-mutated adenines with high BPRSHbF occurred specifically in the GATA1–half-Ebox motif (Fig. 4d, yellow shading). Analysis of the +55 kb DHS identified high-score adenines located within or next to two core KLF1 motifs (Fig. 4d, blue shading). The ABEmax mutagenesis screen also identified a high-BPRSHbF adenine near a core KLF1 motif in a CRE located 1 Mb upstream of BCL11A (Fig. 3a, blue shading, and Fig. 4e). KLF1 is reported to regulate BCL11A gene expression directly by binding its promoter42. Our data indicate that KLF1 also regulates BCL11A through a long-range CRE. In a CRE located in intron 4 of the NFIX gene, we identified two high-BPRSHbF adenines within a GATA1 binding motif. GATA1 occupancy in this region was further confirmed by ChIP-seq (Fig. 4f).
In the β-like globin gene cluster, we identified a CRE located approximately 400 bp 3′ to the polyadenylation signal of HBG1 (Fig. 4g, pink shading). This element was previously identified as an HBG1 enhancer56,57. ATAC-seq signals reflecting open chromatin were present in HbF-expressing HUDEP-1 cells, but not in HbA-expressing HUDEP-2 cells. Motif analysis identified high-BPRSHbF adenines within a binding motif associated with the occupancy of ZBTB7A repressor protein. Analysis by ChIP-seq58 showed that the region is occupied by ZBTB7A in HUDEP-2 cells (Extended Data Fig. 5a). Mutation of the motif by ABEmax and a specific targeting gRNA in single HUDEP-2 clones disrupted ZBTB7A binding (Extended Data Fig. 5a, b), re-established the open chromatin (Fig. 4g), and induced HbF levels (Extended Data Fig. 5c, d and e). These data suggest that the HBG1 3′ CRE functions as an enhancer to drive γ-globin expression during fetal erythropoiesis and that it is inactivated by ZBTB7A repressor binding during adult erythropoiesis. ABEmax mutagenesis may interfere with ZBTB7A binding, thereby reactivating γ-globin transcription and HbF expression.
Nucleotide sequence and epigenetic determinants of HbF regulatory sequences
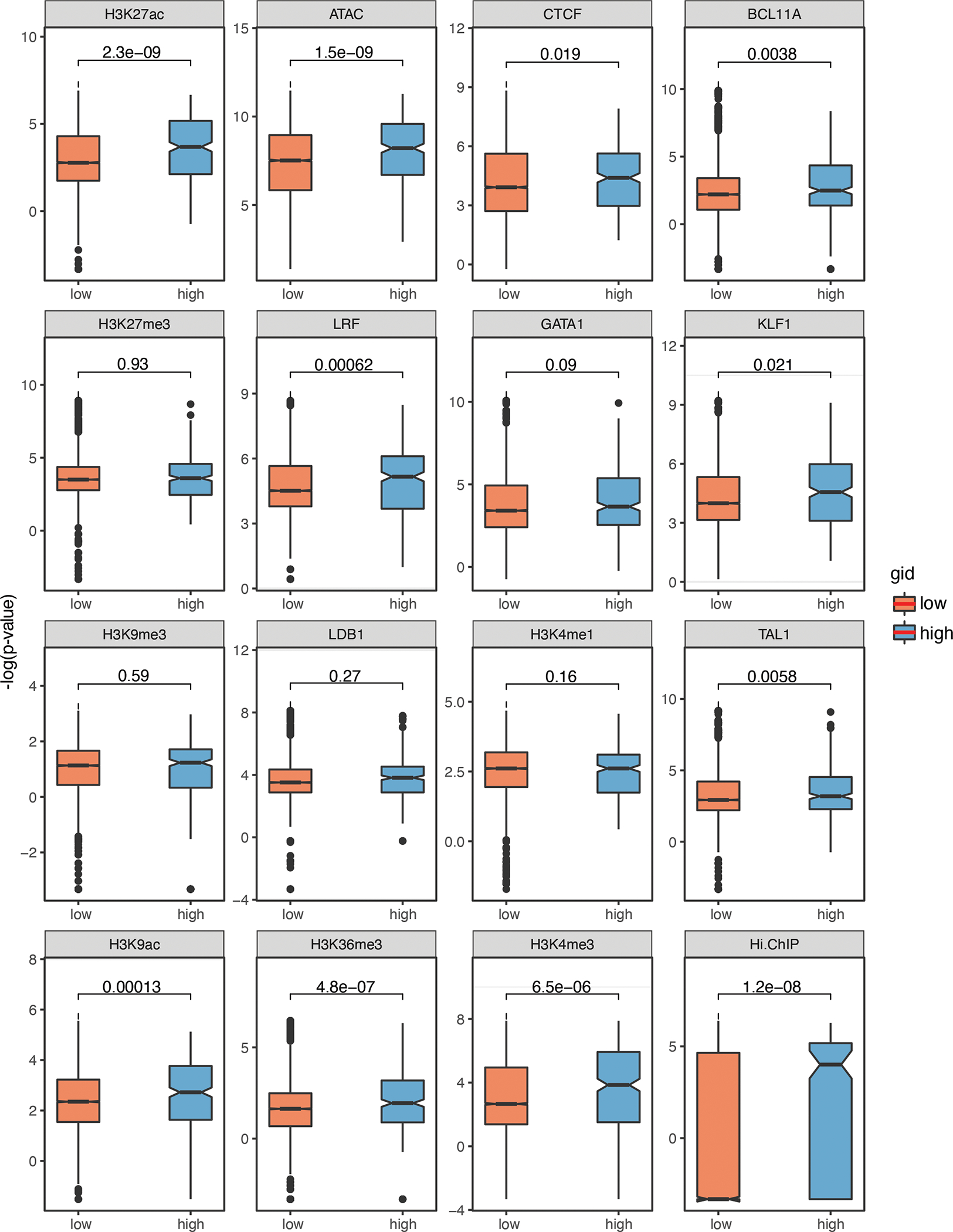
Nucleotide sequence and epigenomic features can predict functional regulatory elements, albeit imperfectly59,60. Our results provide an opportunity to investigate the determinants of the functional sequences within CREs. We analyzed wild-type HUDEP-2 cells for CRE-associated epigenetic features, including chromatin openness (by ATAC-seq), binding of key TFs (CTCF, GATA1, LDB1, TAL1, BCL11A, and ZBTB7A), and histone modifications (H3K27ac, H3K9ac, H3K4me1, H3K36me3, H3K4me3, H3K27me3, and H3K9me3) (Fig. 5a), and we performed Hi-ChIP against H3K27ac to profile 3D chromatin interactions. We divided ABEmax-edited adenines into high and low groups according to their BPRSHbF scores and compared epigenetic features within each group (Extended Data Fig. 6 and Methods). In general, adenines with strong functional effects (i.e., those with high BPRSHbF scores) showed stronger signals for chromatin accessibility, active histone modifications (H3K27ac and H3K9ac), and occupancy by key TFs (ZBTB7A, BCL11A and CTCF), and they had more 3D interactions with other regulatory elements (Fig. 5b, c and Supplementary Table 7).
Figure 5. Nucleotide sequence and epigenetic determinants of HbF regulatory sequences.

(a) Genome browser screenshot of the β-like globin gene cluster showing the distribution of BPRSHbF in relation to various epigenetic signals. The tracks are color-coded as follows: red, 3D chromatin organization; dark red, chromatin accessibility (ATAC-seq); blue, key TF occupancy; brown, histone modifications.
(b) Box plots comparing epigenetic signal distribution in ABE-mutated adenines with high (>30) (n = 313) and low (<10) BPRSHbF (n = 9,268). The y-axis represents log-transformed normalized signals. P value were determined using with two-tailed unpaired Wilcoxon test. Box depicts the interquartile range; central line indicates the median and whiskers indicate minimum/maximum values.
(c) The significance of differences in specific functional genomic features in ABE-mutated adenines with high and low BPRSHbF, ranked by −log10 (P value). P value were determined using with two-tailed Wilcoxon test.
(d) Comparison of CRE prediction models. The left panel summarizes the performance of different models for resolving high- and low-BPRSHbF adenines according to the distance between them. The y-axis represents the area under the receiver operating characteristic (AUROC) curves. The x-axis represents the threshold of minimal distance. The two panels at right show details of the prediction performance, represented by AUROC curves at different window sizes. RF refers to the random forest model that we developed. n = 400 iterations of cross-validation. Bar plot shows the mean and 95% confidence interval of the AUROC distribution.
We built a random forest–based prediction model incorporating epigenetic profiles and DNA sequence features (see Methods) and tested its ability to predict functionally important adenines identified by ABEmax mutagenesis. The model discriminated between high- and low-BPRSHbF adenines most accurately when they were separated by more than 500 bp (area under the receiver operating characteristic curve, AUROC = 0.7) (Fig. 5d). The performance declined with decreasing distance to an AUROC of approximately 0.58 for immediately adjacent mutated adenines (in the 0-bp window). Features with the highest contribution in our model were TF binding and TF-motif footprints, which contributed 55% of predictive power at >500-bp distances and 76% at 0 bp. In contrast, histone modifications contributed 21% in the >500-bp window and 10% at 0 bp. This analysis demonstrates that epigenetic and DNA sequence features can predict CREs, albeit imperfectly and with low resolution. Therefore, functional screens, such as the one described here, are necessary to define regulatory regions with greater certainty. In contrast to our model, two state-of-the-art computational models that were developed to predict deleterious variants in various human cell types (DeepSEA61 and CADD62) could not discriminate between HbF regulatory and nonregulatory adenines identified in the ABEmax screen. This discrepancy indicates that 1) cell type–specific regulatory features might be necessary to predict some functional CREs and 2) programs designed to identify deleterious mutations might be less effective at analyzing variants associated with benign traits.
Applications of high-resolution CREs mapping in SCD research
We investigated whether high-resolution mapping of CREs that regulate HbF levels could provide insight into two active concerns of SCD genetic research: 1) the failure of common variants identified by GWAS to explain most of the genetic variation in HbF expression30; and 2) the identification of CREs that can be targeted therapeutically to induce HbF27,29,63.
We performed whole-genome sequencing and RBC HbF determinations on 454 individuals with SCD (genotype HbSS or HbS-β0-thalassemia) who were older than 2 years and not receiving hydroxyurea therapy (https://sickle-cell.stjude.cloud). We validated the established associations of HbF with common SNVs in BCL11A, HBS1L-MYB, and the β-like globin cluster, which together accounted for approximately 25% of the heritability in HbF variation. The remaining HbF heritability could be partly explained by multiple rare variants in established HbF modifier loci. Identifying such variants by GWAS requires large cohorts to achieve statistical power. However, insights into this problem might be gained by combining human genome sequencing data with the ABEmax-generated high-resolution functional map of HbF regulatory elements. In this case, naturally occurring SNVs that modulate HbF expression by altering CREs should be preferentially located near ABEmax-mutated adenosines with high BPRSHbF, as compared to SNVs that do not affect HbF expression. Therefore, we compared the proximity of SNVs to high-BPRSHbF adenines in individuals with high and normal HbF levels (Fig. 6a and Extended Data Fig. 7a). Compared to participants with SCD with normal HbF levels (n = 357), those with high HbF levels (n = 97) showed an increased burden of SNVs near functional high-BPRSHbF adenines. This difference was most significant within a 10-bp window centered at high-BPRSHbF adenines (Empirical P = 0.0001), gradually decreased with increasing window sizes, and became insignificant for windows greater than 100 bp (Fig. 6b). To validate these findings, we designated a “known set” of 18 common variants that are reported to be associated with HbF levels and a “novel set” of 56 variants within 20 bp of high-BPRSHbF adenines (Supplementary Table 8). We developed a random forest–based model to discriminate between participants with SCD with high HbF and those with normal HbF by using the mutation burden of known and novel variants, conducted three-fold cross-validation 400 times, and averaged the model performance. Using the known set as a positive control, the area under the receiver operating characteristic curve (AUROC) and the area under the precision-recall curve (AUPRC) were 0.71 and 0.46, respectively (Fig. 6c and Extended Data Fig. 7b). Adding the novel variant set improved the model significantly: the AUROC increased from 0.71 to 0.78 (P = 1.08 × 10−100) (Fig. 6d), and the AUPRC increased from 0.46 to 0.51 (P = 1.47 × 10−52) (Extended Data Fig. 7b, c). Using a threshold corresponding to 75% recall, the false-positive rate of approximately 50% for the known set was reduced to 32% after incorporating the novel set (Extended Data Fig. 7b). These findings indicate that rare SNVs contribute to HbF variability in an SCD population by modulating the function of novel CREs identified by the high-throughput ABEmax screen.
Figure 6. Novel HbF CREs identified by ABE mutagenesis are associated with high HbF levels in an SCD cohort.

(a) Workflow for the Analysis of SNV burden in SCD participants with differing HbF levels (see Methods).
(b) Comparison of normalized mutation burden (y-axis) within different window sizes (x-axis) centered on adenines with high BPRSHbF(see Methods). The cyan area indicates the 95% confidence interval for the distribution of participants with normal HbF based on 1,000 samplings. The median values plotted as the center line.
(c) The ROC curve of the classification model that discriminates between subjects with high and normal HbF levels. The green curve represents the model including only known GWAS variants (n = 18), and the red curve represents the models including both known GWAS variants and variants within 20 bp of adenines with high BPRSHBF(n = 56).
(d) Box plot showing a pair-wise performance comparison of the two models described in (c). n = 400 random samplings. P value is determined using paired two-tailed t-test. Box depicts the interquartile range; central line indicates the median and whiskers indicate minimum/maximum values. Data beyond the end of the whiskers are outlying points that are plotted individually.
(e−g) CD34+ HSPCs from donors with SCD were transfected with RNP consisting of ABE and Chr11–1 gRNA targeting the 3′ HBG1 enhancer or non-targeting control gRNA (Ctrl). The results are representative of two donors.
(e) F-cell fractions measured by immuno-flow cytometry.
(f) The hemoglobin F fraction measured by IE-HPLC.
(g) The fraction of sickled cells among reticulocytes from the edited and control populations incubated for 8 hours in 2% oxygen.
Base editing to disrupt CREs that negatively regulate HBG1/HBG2 expression is a promising approach to raising HbF levels to treat SCD or β-thalassemia63. Approximately 30% of adenines with high BPRSHbF scores in the ABEmax screen (62 of 200) are predicted by their chromatin accessibility pattern to be erythroid specific64 (Extended Data Fig. 8a and Supplementary table 9). Manipulating these CREs in hematopoietic stem cells can potentially regulate erythroid HbF levels without affecting other blood lineages. To test the therapeutic potential of CREs discovered by ABEmax mutagenesis, we transfected SCD donor CD34+ HSPCs with RNP consisting of ABE and the gRNA (Chr11–1), thereby disrupting the 3′ HBG1 enhancer identified by the ABEmax screen (see Fig. 4g), and examined the erythroid progeny generated by in vitro differentiation. Base editing led to a more than two-fold increase in the proportion of F cells (Fig. 6e), increased levels of HbF (Fig. 6f), and an increased proportion of γ-globin mRNA (Extended Data Fig. 8b) without affecting erythroid maturation (Extended Data Fig. 8c, d). Moreover, the RBCs derived from ABE-edited HSPCs exhibited reduced sickling after exposure to hypoxia (2% O2) when compared to RBCs generated from control HSPCs (Fig. 6g and Extended Data Fig. 8e). Therefore, high-throughput ABEmax mutagenesis combined with biologically meaningful phenotype assessment can be used to identify specific nucleotides as potential therapeutic targets.
Discussion
Our findings have elucidated the complex regulation of developmental globin gene expression, defined further the epigenetic features of CREs, and identified potential targets for therapeutic induction of HbF. More generally, our approach has defined a powerful paradigm for defining CREs with unprecedented resolution across a chromosomal region. Of particular importance is our demonstration that point mutagenesis by ABEs is superior to conventional Cas9 for functional dissection of CREs.
High-throughput CRISPR/Cas9-based perturbation has advanced our capacity to study coding and noncoding DNA by creating targeted loss-of-function mutations that can be correlated with altered cellular properties34,65. Most assays employ alterations in gRNA abundance (i.e., “dropout” or enrichment) to associate genotypes with phenotypes. However, individual gRNAs usually generate mixtures of variably sized indels in unpredictable patterns that can differ across cell types66, thereby limiting the sensitivity, specificity, and reproducibility of CRISPR-based screens. In contrast, base editing usually produces precise, single-nucleotide mutations within a 4−8-bp window67. Considering the average distance between two adjacent TF footprints is about 20 bp68, base editing can provide high-resolution information on CREs and their functional DNA elements compared to conventional Cas9-induced indels.
Our data confirmed the functions of important HbF regulatory elements identified through decades of effort using in vitro, cellular, mouse and human genetic studies30,69–71. In addition, we identified numerous previously unappreciated CREs. The complex genetic regulatory network controlling the RBC HbF level, in particular the numerous CREs controlling the expression of BCL11A, KLF1 and MYB that repress γ-globin gene expression, probably represents what has been referred to as a “regulatory archipelago”72 of genetic control elements with distinct, redundant, and cooperative functions that may be influenced by the developmental stage and environmental conditions during erythropoiesis21,73. Our findings identify high-throughput base editor screening as a powerful tool to elucidate the genetics of HbF expression, and more generally, to define complex transcriptional regulatory networks underpinning other biologically important processes.
Numerous variants in the BCL11A, HBS1L-MYB, and the β-like globin gene loci have been defined, but these account for less than half of the inter-individual HbF variation30. The CREs newly identified in our study were significantly associated with low-frequency SNVs in participants with SCD with high HbF. Therefore, multiple rare variants in KLF1, BCL11A, HBS1L-MYB, and the β-like globin gene cluster probably account for some of the unexplained HbF heritability. Additionally, several erythroid-specific CREs that repress HBG1 and HBG2 expression are under investigation as potential therapeutic targets for genome editing–mediated induction of HbF77. The optimal therapeutic CRE target is unknown, and there are additional candidates to explore among the CREs identified in our study. In this regard, base editing might be used therapeutically to induce HbF by disrupting CREs that negatively regulate HBG1 and HBG2 transcription or to create new binding sites for transcriptional activators in these genes27,28,74.
The genomic coverage of our screen was not comprehensive because of several technical issues. ABEmax requires an NGG PAM site in genomic DNA, which limits the number of regions that can be targeted. Additionally, only adenine (and opposite-strand thymidine) is perturbed by ABEs. Therefore, the overlap between mutations introduced by ABEmax and naturally occurring HbF-associated SNVs was low (<0.6%). For the same reason, it is likely that not all HbF-associated CREs were detected. The sensitivity of ABE screening will be improved by several approaches, including the parallel use of CBEs, newly developed ABEs that expand coverage by recognizing NG instead of NGG PAM sites75–77, and dual base editor systems78–80. Another approach for targeted mutagenesis, prime editing81, can theoretically produce a virtually unlimited mutational repertoire.
Methods
HUDEP cell culture.
HUDEP clone 2 (HUDEP-2) cells were grown in culture as described previously32. They were expanded in StemSpan serum-free expansion medium (SFEM; Stem Cell Technologies, cat. #09650) supplemented with 1 M dexamethasone (Sigma, cat. #D4902), 1 g/ml doxycycline (Sigma, cat. #D9891), 50 ng/ml human stem cell factor (hSCF, PeproTech, cat. #300–07), 3 units/ml erythropoietin (EPO Amgen, cat. #55513–144-10), and 1% penicillin–streptomycin (ThermoFisher, cat. #15140122). Differentiation was achieved by growing cells for 5 days (with one medium change at day 3) in Iscove’s modified Dulbecco’s medium (IMDM) (Mediatech, cat. #MT10016CV) supplemented with 50 ng/ml hSCF, 3 units/ml erythropoietin, 2.5% fetal bovine serum, 250 μg/ml holo-transferrin (Sigma, cat. #T4132), 1% penicillin–streptomycin, 10 ng/ml heparin (Sigma, cat. #H3149), 10 μg/ml insulin (Sigma, cat. #I9278), and 1 μg/ml doxycycline.
CD34+ HSPC cultures and in vitro differentiation.
Circulating granulocyte colony-stimulating factor (G-CSF)–mobilized human CD34+ cells were enriched by immunomagnetic bead selection using an AutoMACS instrument (Miltenyi Biotec). CD34+ HSPCs were grown in culture via a three-phase erythroid differentiation protocol82. The basic medium consisted of IMDM (Gibco) supplemented with 2% human blood type AB plasma (Sera Care), 3% human AB serum (Atlanta Biologicals), 1% penicillin–streptomycin (Gibco), 3 units/ml heparin, 10 g/ml insulin, and 3 units/ml erythropoietin (EPO) (Amgen). In phase 1 of the culture (days 1–7), the medium also included 200 g/ml holo-transferrin (Sigma-Aldrich), 10 ng/ml stem cell factor (SCF) (PeproTech, Inc.), and 1 ng/ml interleukin 3 (IL-3) (PeproTech, Inc.). In phase 2 (days 8–12), the medium included the same cytokines as in phase 1, except that IL-3 was withdrawn. During phase 3 (day 13 and beyond), the concentration of holo-transferrin was increased to 1 mg/ml and SCF was removed. Samples for Western blot analysis and HbF FACS were harvested on day 12 of culture. Samples for HPLC were harvested on day 15 of culture. Erythroid differentiation and maturation were monitored by flow cytometry using a fluorescein isothiocyanate (FITC)–conjugated anti-CD235 antibody (BD Biosciences, clone GA-R2), an allophycocyanin (APC)-conjugated anti-Band3 antibody (a gift from Xiuli An, NY Blood Center), and a VioBlue-conjugated anti–a4-integrin antibody (Miltenyi, clone MZ18–24A9), gating strategies were presented on Extended Data Figure 9.
Establishment of HUDEP-2–ABEmax cell lines.
Lenti-ABEmax plasmids were derived from previously described plasmids31 and cloned by the Gibson assembly method. Plasmids expressing sgRNAs were constructed by ligating annealed oligonucleotides into a BsmBI-digested acceptor vector (Addgene plasmid no. 65777). All vectors for mammalian cell experiments were purified using Plasmid Plus Midiprep Kits (Qiagen). We generated stable HUDEP-2 cell lines with different ABEmax concentrations, quantified by Western blot analysis. We further tested the relation between ABEmax concentration and editing efficiency by using a known gRNA at the HBG promoter region27. We found a nearly linear correlation between ABE concentration and editing efficiency when the ABEmax concentration was low. However, above a certain concentration, increased ABEmax concentrations no longer increased the editing efficiency, suggesting that over-expression of ABEmax is toxic. From among the HUDEP-2–ABEmax cell lines with high editing efficiency, we chose the one with the lowest ABEmax concentration for our high-throughput screen (Extended Data Fig. 1b). We routinely used low-passage HUDEP-2-ABEmax cells to avoid potentially genetically unstable cells and those with chromosomal abnormalities. In addition, we frequently validated the erythroid differentiation capacities of the cells and their responsiveness to HbF expression induction.
RNA-seq and analysis.
For RNA-seq, RNA was extracted from 1 million HUDEP-2 cells (at least three biological replicates each), using the RNeasy Mini Kit (Qiagen). The TruSeq Stranded mRNA Library Prep Kit (Illumina) was used to enrich for polyA+ RNA and to create libraries for HiSeq2000 sequencing (Illumina). Gene expression profiles were quantified using Kallisto (v0.43.1)83 on hg19 Ensembl v75 cDNA annotation. Differential expression analysis was performed using Sleuth(v0.30.0)84. Gene expression correlation was calculated based on TPM values.
ATAC-seq.
The ATAC-seq library was prepared according to the published omni-ATAC protocol, using 50,000 live cells per sample85. Libraries were sequenced using an Illumina HiSeq 4000 system (100-bp pair-end sequencing). Biological replicates for HUDEP-2 and HUDEP-1 were merged. Raw reads were trimmed to remove Tn5 adaptor sequence using skewer (v0.2.2) and then mapped to hg19 by using BWA mem (v0.7.16a). Duplicated multi-mapped reads were removed with samtools (v0.17). ATAC-seq peaks were called using MACS2 (v2.1.1) with the following parameters: macs2 callpeak --nomodel --shift −-100 --extsize 200. BigWiggle files were generated using DeepTools (v3.2.0). Footprinting analysis was performed using HINT-ATAC (v0.12.1)86 with correction of strand-specific Tn5 cleavage bias (--atac-seq --paired-end). ATAC-seq footprints that were within 100 bp of the top adenosines were searched for de novo motifs by using Homer (v4.9.1)87.
ChIP-seq.
ChIP experiments were performed as previously described38 with the following modifications. Briefly, approximately 2.5 × 107 cells were used for each immunoprecipitation. Cells were cross-linked with 1% formaldehyde for 10 min at room temperature with rotation, and the reaction was quenched with glycine at a final concentration of 125 mM. Cross-linked cells were then lysed and resuspended in 2 ml of RIPA buffer and sonicated for 12 cycles with a Branson 250 sonifier (10 s on, 90 s off for a total of 2 min of pulses with 20% output from the micro-tip) to obtain fragments of chromatin approximately 200–300 bp in size. Supernatants were precleared by incubation with 200 μl of protein A/G agarose bead slurry (Thermo Fisher Scientific, cat. #15918014) overnight at 4°C with rotation. Meanwhile, 12.5 μg of IP antibody was incubated with 50 μl of protein A/G agarose bead slurry in 1 ml of PBS overnight at 4°C with rotation. Saved precleared chromatin (20 μl) was used as the input sample. Precleared chromatin was incubated with the antibody–bead complex for 7 h at 4°C with rotation. Cross-linking of DNA was reversed by incubation with RNase A (1 μg/μl), proteinase K (0.2 mg/ml), and 0.25 M NaCl overnight at 65°C. Immunoprecipitated DNA was purified using the Qiagen PCR Extraction Kit and eluted with 20 μl of EB elution buffer. Sequencing libraries were prepared using the NEBNext Ultra II DNA Library Prep Kit (NEB, cat. #E7645) with homemade TruSeq adaptors. Libraries were sequenced using an Illumina HiSeq 4000 system. FASTQ files were mapped to hg19 by using BWA mem (v0.7.16a). Reads that could not be uniquely mapped to the human genome were removed by samtools (v0.17, samtools view -q 1). ChIP-seq peaks were called by using MACS2 (v2.1.1), and peaks identified by two biological replicates were kept.
Hi-ChIP.
Hi-ChIP libraries were prepared as previously described88. After proximity ligation, the nuclear lysates were sonicated with a Branson 250 sonicator. Reverse cross-linked ChIP DNAs were purified with the DNA Clean & Concentrator-5 purification kit (Zymo Research) and quantified with Qubit (Life Technologies). The samples were then end-repaired, dATP-labeled, and adaptor-ligated with an NEBNext Ultra II DNA library prep kit, followed by streptavidin pull-down and PCR amplification of the library. PCR products (300–1,000 bp) were size selected by using E-gel (Invitrogen). Libraries were sequenced with an Illumina HiSeq 4000. Reads were mapped to hg19 with HiC-Pro (v2.11.1) with default parameters. The outputs were further processed with hichipper (v0.7.7) to identify significant loops with default parameters.
Transcriptome-wide deamination analysis.
To avoid the bias potentially introduced by differences in sequencing depth during A-to-G conversion quantification, we first down-sampled RNA-seq with greater sequencing depth to match the experiment with the lowest depth (30 million uniquely mapped reads) using seqtk (v1.2). Down-sampled FASTQ files were mapped to the hg19 reference genome by using STAR (v2.5.3a)89 with parameters of --twopassMode Basic --outSAMtype BAM SortedByCoordinate --outFilterMultimapNmax 20 --alignSJoverhangMin 8 --alignSJDBoverhangMin 1 --outFilterMismatchNmax 999 --outFilterMismatchNoverReadLmax 0.04 --alignIntronMin 20 --alignIntronMax 1000000 --alignMatesGapMax 1000000 --outSAMattributes Standard --sjdbScore 1. Next, duplicated reads were removed using picard (v2.9.4) and indel realignment and base recalibration were performed based on known variants from the GATK(v3.5)90 bundle (i.e., Mills_and_1000G_gold_standard.indels.hg19.vcf, 1000G_phase1.indels.hg19.vcf, and dbsnp_138.hg19.vcf). Variants were called using GATK HaplotypeCaller and were filtered using “-window 35 -cluster 3 -filterName FS -filter “FS > 30.0” -filterName QD -filter “QD < 2.0”.” Variants were further filtered to exclude loci without a high-confidence reference in the control group. For HUDEP-2–ABEmax cells, HUDEP-2 wild-type cells were used as controls; for HEK293T-HEKsite2-ABEmax (GSM3724238) and HEK293T-GFP (GSM3724258), HEK293T-HEKsite2-control (GSM3724237) was used as a control. High-confidence references were determined using bam_count (v0.8.0) (https://github.com/genome/bam-readcount) with read depth ≥ 10 and reference allele frequency ≥ 0.99.
ABE screen for CREs regulating RBC HbF levels.
An sgRNA library was constructed according to the published protocol91. Oligonucleotide PCR products were gel-purified and cloned into a BsmBI-digested and gel-purified pLentiGuide-Puro5 vector (Addgene cat. #52963), which encodes a puromycin resistance cassette, by Gibson assembly using NEBuilder HiFi DNA Assembly Master Mix (NEB). The assembly products were transformed into NEB stable competent cells, then the plasmids were extracted. Lentiviral particles were generated by transfecting HEK 293T cells with the library plasmid, pVSVG, pRevtat, and pGagpol, using Lipofectamine 2000 (Life Technologies). Viral vector titers were determined by serial dilution. HUDEP-2–ABEmax cells were transduced with the library at a low multiplicity of infection (MOI) of 0.3 to minimize the transduction of cells with more than one vector particle and to achieve an approximately 1,000-fold coverage of the sgRNA library. After 24 h, transduced cells were selected in puromycin (1 μg/ml) for 2 additional days and then maintained in the expansion medium for 5 days. Erythroid maturation was induced in differentiation medium for 5 days. Cells were then stained for HbF as indicated above and sorted into HbFhigh and HbFlow populations. Genomic DNA was extracted with the PureLink Genomic DNA Mini Kit (Life Technologies). sgRNAs were amplified for 27 cycles, using barcoded lentiGuide-Puro–specific primers with 400 ng of genomic DNA per reaction. The PCR products were then pooled for each sample. The barcoded libraries were pooled at an equimolar ratio, gel purified, and subjected to massively parallel sequencing with a MiSeq instrument (Illumina), using 75-bp paired-end sequencing. For data analysis, FASTQ files obtained after MiSeq sequencing were demultiplexed using the MiSeq Reporter software (Illumina). The reads were subsequently mapped to the reference sgRNA library and normalized to control sgRNAs by using MAGeCK (v0.5.9) count. The significance (FDR) of the difference in gRNA abundance between the HbFhigh and HbFlow populations was calculated with the MAGeCK RRA test with default parameters92.
Base-pair–HbF regulation score (BPRSHbF).
To assign regulatory scores to each editable adenine (A) based on the gRNA FDRs, we first calculated an average editing frequency at each gRNA position based on the on-target amplicon sequencing data in HUDEP-2 cells, and we identified an editing window from the first nucleotide to the thirteenth nucleotide (e.g., positions with at least 1% observed editing efficiency). Then, for an A at position i in gRNA j, denoted by Ai|gRNAj, an adjusted FDR was calculated by dividing the original FDR by the editing frequency, namely,
where i denotes the ith position in gRNA j, and FDR* denotes the adjusted FDR. Lastly, if a particular A was covered by multiple gRNAs, the empirical Brown’s method93 was used to combine multiple adjusted FDRs. We called the adjusted FDR values for each adenine as BPRSHbF. The source code and analyzed data are available at https://github.com/YichaoOU/ABE_NonCoding_functional_score.
Analysis of SNV burden in participants with SCD with differing HbF levels.
Participants with SCD were classified into three groups based on their HbF levels: the low-HbF group (< mean − SD), the normal-HbF group (mean ± SD), and the high-HbF group (> mean + SD). Only the participants in the normal-HbF group (n = 357) and the high-HbF group (n = 93) were used in our analysis. The top 200 BPRSHbF genomic loci were used as target sites to calculate the mutation burden. The mutation burden was calculated based on the following equation:
(Total no. of SNVs / Total no. of participants / Total length of targeted loci) × 1000
Total no. of SNVs were calculated along the lengths of targeted loci. To control for the imbalance between high-HbF participants and the low-HbF participants, 93 participants from the normal-HbF group were selected for comparison. This random sampling was repeated 1,000 times. Variants with different minor allele frequency (MAF) were extracted from the VCF files generated from a previous study61.
Machine learning classification of participants with SCD.
Binary values representing the presence or absence of genetic variants in the participants with SCD were used as machine learning features. NA values, caused by incomplete variant calling, were treated as absence. Next, 18 known GWAS variants that were associated with HbF were used as the “known set”. Fifty-six variants (MAF < 0.2) within ±10 bp of the top 200 BPRSHbF genomic loci were used as the “novel set.” We tested the hypothesis that using the known set + the novel set could provide more power than using only the known set in discriminating participants with SCD from those with normal HbF (an HbF level within 1 standard deviation) or high HbF (an HbF level above 1 standard deviation). Random forest models were implemented using sklearn (v0.22). Stratified three-fold cross-validation was repeated 400 times. The AUROC (area under the receiver operating characteristics curve) and AUPRC (area under the precision-recall curve) were calculated. Welch’s t-test was performed to determine the significance of adding the novel set.
Predicting BPRSHbF with sequence and epigenetic features.
To evaluate the prediction of BPRSHbF scores using the available sequence and epigenetic features, we developed a random forest model to classify variants with high BPRSHbF or low BPRSHbF scores. Genomic loci with high BPRSHbF (≥30, positive data, n = 313) or low BPRSHbF (≤10, negative data, n = 9,268) were used as a training set. The training and testing were conducted at six different resolutions: 0, 5, 10, 50, 100, and 500 bp. At each resolution, any adenines in the negative data that were within ± the window size (i.e., the resolution bp) of adenines in the positive set were removed. For machine learning feature representation, two types of feature were included: (1) Epigenetic features were represented using data from seven histone ChIP-seq experiments, seven TF ChIP-seq experiments, ATAC-seq, and H3K27ac Hi-ChIP. For 1D signal data (i.e., ChIP-seq or ATAC-seq data), an average signal over ±5 bp from the A sites was used. For 2D interaction data (i.e., Hi-ChIP data), the number of interactions overlapping with the A sites was used. (2) TF binding site features were generated using known motifs for key erythroid TFs (i.e., GATA1, ZBTB7A, KLF1, BCL11A, NFIX, E2F, NFYA, and NFE2) and the chromatin organization factor CTCF from four motif databases: JASPAR, Homer, CIS-BP, and ENCODE-motif. Motif PWM scanning was performed using FIMO (v5.1.0) on sequences corresponding to ±100 bp from each A site. The top five matched PWM scores were retained and were then multiplied by the ATAC-seq footprint score. The footprint score was calculated as the difference between the flanking cutting frequency and the core cutting frequency, formulated as follows: let S = {s1,s2,…,si,…,si+m,…,sn} be the vector of the Tn5 cutting frequency, m the motif length, and i the start position of the motif match, then the flanking (± 2 bp) cutting frequency is FLANK = mean (si−2,si−1,si+m+2,si+m+1) and the core cutting frequency is CORE = mean (si,…si+m). The footprint score for the motif match is FLANK™lCOR. For evaluation, we used a leave-one-chromosome-out cross-validation strategy (e.g., training on chr2, chr11, chr6, and testing on chr19) and repeated the CV 100 times. Random forest models were implemented using sklearn (v0.22). The AUROC and AUPRC were calculated for each resolution.
Erythroid-specific score.
ATAC-seq data for different blood lineages were collected from the GSE115672 and GSE75384 data sets. Mean ATAC-seq signals were extracted over ±10 bp from all 13,147 adenine sites by using bigWigAverageOverBed (http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/). They were then divided by the sum of all adenine sites to generate normalized chromatin openness signals. The same analysis was conducted for each cell type (BM_CD34, B cell, CB_CD34, CB_Ery, CD4 T cell, CD8 T cell, CLP, CMP, GMP, HSC, LMPP, MEP, MPP, Mono, MyP, and NK cell). The erythroid-specific score was calculated as , where xi was the normalized signal at position xi (i.e., some A) in the CB_Ery cells and μi and σi were, respectively, the mean signal and the standard deviation over all cell types for position i.
Flow cytometry for F-cells.
Intracellular staining was performed as described previously. Cells were fixed with 0.05% glutaraldehyde (Sigma) for 10 min at room temperature, washed three times with PBS/0.1%BSA (Sigma, cat. #A7906), and permeabilized with 0.1% Triton X-100 (Life Technologies) for 5 min at room temperature. After one wash with PBS/0.1% BSA, the cells were stained with an antibody against human HbF (clone HbF-1 with FITC; Life Technologies) for 20 min in the dark. The cells were washed to remove unbound antibody before FACS analysis was performed. Control cells without staining were used as a negative control, gating strategies were presented on Extended Data Figure 10.
Hemoglobin HPLC.
Hemolysates were prepared from erythroid cells after differentiation by using Hemolysate Reagent (Helena Laboratories, Beaumont, TX, cat. #5125). The hemolysates were cleared by centrifugation and analyzed for identity and levels of hemoglobin variants (HbF and HbA) by cation-exchange high-performance liquid chromatography (HPLC) (Primus Diagnostics). Hemoglobin was quantified directly by integrating the areas under the HbF and HbA peaks. A commercial standard containing approximately equal amounts of HbF, HbA, HbS, and HbC (Helena Laboratories) was used as an isotype reference.
Sickling assay.
Human SCD CD34+ HSPCs (HBB genotype SS) were purified from de-identified, discarded whole blood from partial-exchange RBC transfusions (this was not considered human subject research by the St. Jude Children’s Hospital Institutional Review Board), differentiated into erythroblasts, and grown in culture in 2% oxygen. Between days 14 and 16 of culture, the percentage of sickled cells was determined by manual counting, with the counter blinded to the sample genotype.
Amplicon deep sequencing.
For editing type analysis with deep sequencing, DNA was extracted from cells with the PureLink Genomic DNA Mini Kit (Life Technologies). First-round PCR amplification using 2× Phusion High-Fidelity PCR Master Mix was performed with the corresponding primers. An additional PCR amplification was performed with primers containing sample-specific adapters. This was followed by sequencing on a MiSeq platform (Illumina) with 2 × 150-bp paired-end reads. Sequence alignment and mutation detection were performed using CRISPResso2 software. The first-round PCR primers are listed in Supplementary Table 10.
Real-time RT-qPCR.
Total RNA was extracted from cells with RNeasy Kits (Invitrogen) according to the manufacturer’s protocol. Reverse-transcription reactions were performed with the SuperScript VILO cDNA Synthesis Kit. qPCR reactions were prepared with Power SYBR Green (ThermoFisher Scientific) and were run with the default cycle parameters of the Applied Biosystems 7500 Fast Real-Time PCR System. The expression levels of genes of interest were normalized against β-actin levels. The real-time PCR primers are listed in Supplementary Table 11.
Plasmids.
The ABE expression vector was constructed by inserting the human codon–optimized ABEmax cDNA (Addgene, cat. #112101) into a blasticidin-expressed lentiviral backbone. All gRNA-encoding oligonucleotides were inserted into a lentiviral U6-puro-guide expression vector via a BsmBI restriction site.
RNP electroporation.
Electroporation was performed using a Lonza 4D Nucleofector (V4XP-3032 for 20-μl Nucleocuvette Strips) in accordance with the manufacturer’s instructions. The modified synthetic gRNA (with 2′-O-methyl-3′-phosphorothioate modifications in the first and last three nucleotides) was purchased from Synthego. CD34+ HSPCs were thawed 24 h before electroporation. The RNP complex (20 μl) was prepared by mixing ABE protein (20 μmol) and gRNA (60 μmol) and incubating the mixture for 15 min at room temperature immediately before electroporation. HSPCs (2 × 105) resuspended in 5 μl of P3 solution were mixed with RNP and transferred to a cuvette for electroporation with program DS-130. The electroporated cells were resuspended in StemSpan II medium with cytokines, which was replaced after 24 h by phase I medium for in vitro differentiation.
Virus preparation and infections.
Virus was produced in HEK293T cells grown in DMEM supplemented with 10% fetal bovine serum and 2% penicillin–streptomycin. HEK293Ts were plated in 10-cm plates 24 h before transfection such that the cells were approximately 90% confluent by the time of transfection. Expression vectors were mixed with pGagpol, pVSVG, and pRevtat plasmids in a 6:3:1:1 ratio in 500 μl of OPTI-MEM (Thermo Fisher Scientific, cat. #31985070) and added to 500 μl of OPTI-MEM containing 50 μl of Lipofectamine 2000 for precipitation. Plasmid precipitations were added to HEK293T cells, and transfections were incubated at 37°C in 5% CO2 for 6 h, after which fresh medium was placed on the cells. Viral supernatants were harvested at 48 h post transfection and pooled. One million HUDEP-2 cells were infected with ABE or single-gRNA–containing virus. HUDEP-2–ABE cells were selected, starting 24 h post infection, for 5 days in 1 μg/ml blasticidin. For individual gRNA infections, HUDEP-2–ABE cells were transduced with lentiviral vectors encoding individual gRNAs, with selection for 2 days, starting 24 h post infection, in 1 μg/ml puromycin. To generate single-gene–edited clones, pooled edited cells were sorted into 96-well plates and expanded for 14–21 days. gDNA was extracted with the PureLink Genomic DNA Mini Kit (Life Technologies). The editing type was characterized by PCR followed by next-generation sequencing.
Western blot analysis.
Western blotting was performed using standard procedures. Cells were suspended in 1× radioimmunoprecipitation assay (RIPA) buffer (Sigma–Aldrich) to which was added 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, and 1:500 protease inhibitor cocktail (Sigma–Aldrich). Proteins were loaded onto polyacrylamide gels (BioRad), transferred to a PVDF membrane, and incubated in blocking buffer (PBS) from Odyssey (cat. #927–40000). Antibody staining was visualized using the Odyssey CLx Imaging System. Rainbow protein standards were loaded on each gel for size estimation.
Cytocentrifuge preparations.
Approximately 100,000–200,000 cells were washed and resuspended in 200 μl of PBS then deposited onto poly-L-lysine–coated microscope slides by centrifugation in a Shandon Cytospin 4 Cytocentrifuge (Thermo Fisher Scientific) at 250 rpm for 5 min. Dried slides were stained with May–Grünwald solution (Sigma–Aldrich) for 5 min, rinsed in deionized water, and stained in Giemsa solution (Sigma–Aldrich) for 15 min. Slides were washed in water and dried, and coverslips were mounted. All images were acquired with AxioVision software (Zeiss) at 60× magnification.
Statistics and reproducibility.
Details of statistic test are indicated in the figure legends. Sample sizes (n) for each experiment refers to the number independent experiments or experiments with different SCD participants, as indicated in the legends. Independent experiments are defined as cells were treated multiple times, harvested and analyzed independently, as in the case of pooled gRNA library screen, RNA-seq, ChIP-seq and ATAC-seq. P values were not calculated for datasets with n < 3. SD stands for standard deviation. S.E.M. stands for standard error of the mean.
Data Availability Statement
Raw and processed sequencing data generated in this study are available from the Gene Expression Omnibus under accession GSE157311. All unprocessed Western Blotting for Figure 1b, Extended Data Figures 1a,e and 2c can be found in Source data provided with this paper.
Code Availability Statement
Custom source code used in this paper can be downloaded from https://github.com/YichaoOU/ABE_NonCoding_functional_score
Extended Data
Extended Data Fig. 1. Establishment of an ABEmax-based system to perturb regulatory sequences.

(a) ABEmax-Cas9 protein levels measured by Western blot analysis in wild-type HUDEP-2 cells (WT) and HUDEP-2 cells infected with different dosages of ABEmax lentivirus. β-Actin was used as a loading control. The result is representative of three independent experiments (Image was cropped from source data Fig. 2).
(b) HUDEP-2 cells with different levels of ABEmax expression were transduced with the same amount of gRNA targeting the HBG promoter. The graphs show the hemoglobin (Hb) protein content, as measured by isoelectric focusing high-performance liquid chromatography (IE-HPLC) in HUDEP-2 cells after 5 additional days of induced erythroid maturation. The result is representative of three independent experiments.
(c) Jitter plots showing the percentage of adenosine-to-inosine RNA modification by ABEmax in wild-type HUDEP-2 cells (WT), HUDEP-2 cells stably expressing an ABE (ABEmax), and HEK293T cells. The y-axis represents the efficiency of A-to-I RNA editing. n = total number of modified adenines observed.
(d) Targeted deep-sequencing analysis of the BCL11A CRE after editing with ABEmax and BCL11A_ENH gRNA. The mutations are indicated in bold. The red arrowhead indicates the targeted nucleotide.
(e) Western blot analysis with the indicated antibodies in undifferentiated (Day 0) and differentiated (Day 5) HUDEP-2 cells transduced with non-targeting control gRNA (Ctrl) or with BCL11A-ENH gRNAs. The result is representative of three independent experiments. (Image was cropped from source data Fig. 3)
Extended Data Fig. 2. High-throughput mapping of CREs regulating HbF in HUDEP-2 cells and single-gRNA validation in CD34+ HSPCs.
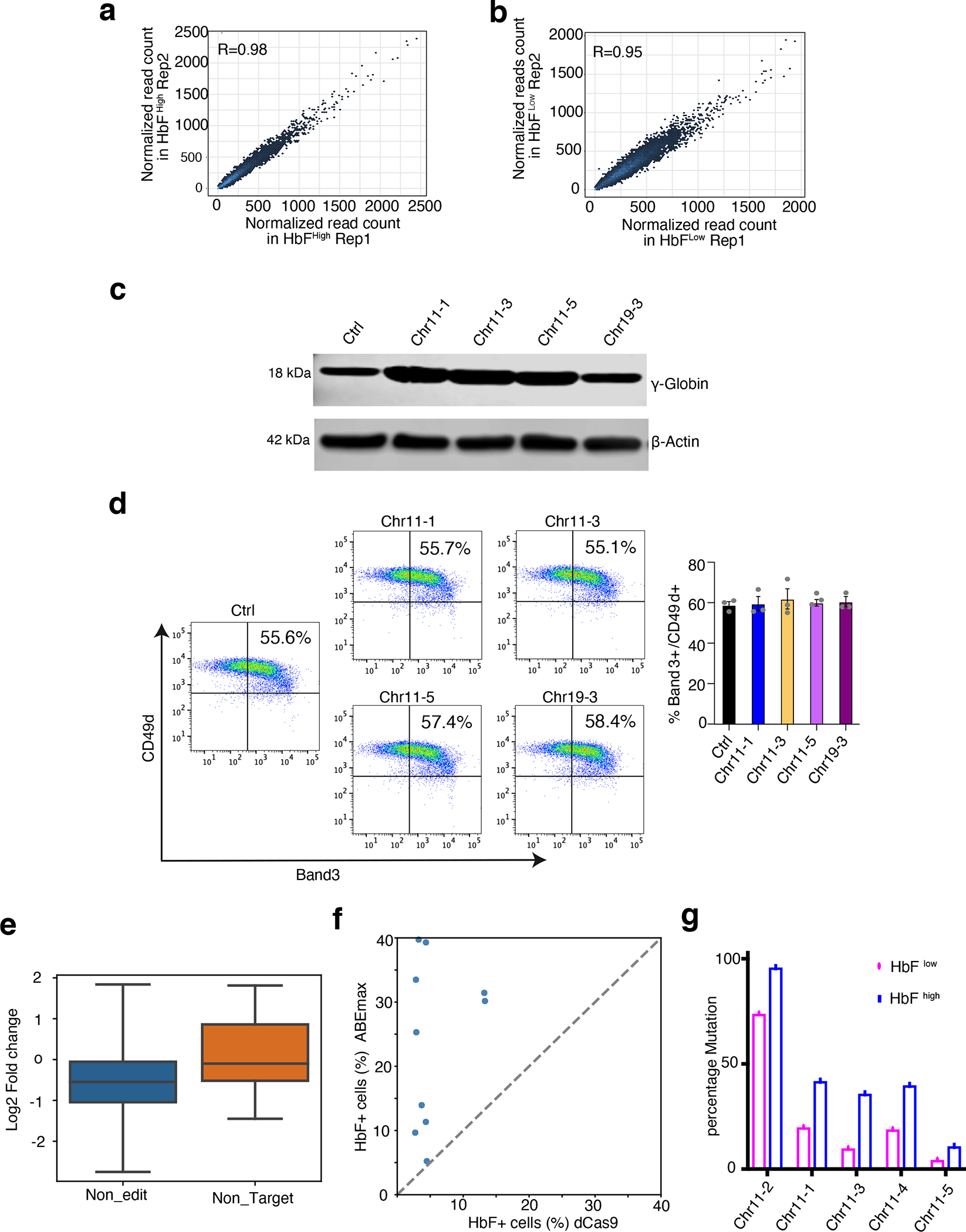
(a,b) Dot plots showing the correlation between two biological replicates of ABE screens for the HbFhigh (a) and HbFlow (b) cell populations. Each dot represents one gRNA; the x- and y-axes represent the normalized read counts.
(c,d) Validation studies of top-hit gRNAs in normal donor CD34+ HSPC–derived erythroblasts. CD34+ cells were transfected with RNP complexes consisting of ABEmax + non-targeting control (Ctrl) gRNA or individual top-hit gRNAs and analyzed after 12 days of erythroid differentiation.
(c) HbF protein levels measured by Western blot analysis. The result is representative of three independent experiments. (Image was cropped from source data Fig. 4)
(d) Flow-cytometry plots showing the expression of the RBC maturation markers Band3 and CD49d after 12 days of differentiation (left) and a bar chart summarizing the results from three replicates (right). Error bars represent the mean +/− S.E.M from three independent experiments.
(e) Boxplot comparing the HbF effects of gRNAs without editable adenines (n=112) and none targeting control gRNAs (n=20). Y-axis is log2 ratio of gRNA reads counts between HbFhigh and HbFlow cells. P-value was determined by unpaired two-tailed Wilcoxon test. Box depicts the interquartile range; central line indicates the median and whiskers indicate minimum/maximum values.
(f) Scatterplot showing the F-cell fractions measured by immune-flow cytometry in HUDEP-2- ABEmax and HUDEP-2-dCas9 cells transfected with 10 gRNAs. Each dot represents one gRNA.
(g) Comparison of target site mutation frequencies in HbFhigh and HbFlow cells. Cells were treated with ABEmax and 5 different gRNAs and then sorted based on HbF levels after 5 days differentiation. The frequencies are calculated based on one argeted deep-sequencing result.
Extended Data Fig. 3. ABE mutagenesis at different genomic loci.

(a) Bar plot showing the effects of NFIX CREs on the expression levels of NFIX and KLF1. Y-axis shows relative mRNA expression measured by real-time RT-qPCR in HUDEP-2 cells edited with ABEmax and the two indicated gRNAs. The expression levels were normalized by those from HUDEP-2 cells treated with ABEmax and non-targeting control gRNA (Ctrl) (n=3 independent experiments).
(b) β-Like globin gene cluster–associated HbFhigh gRNAs: Chromatin interaction loops, indicated by red arcs, were determined by H3K27ac Hi-ChIP in HUDEP-2 cells. gRNA -log(FDR) represents the difference in gRNA abundance between the HbFhigh and HbFlow populations. ATAC-seq analysis reflects chromatin openness.
Extended Data Fig. 4. Empirical distribution of ABE editing efficiency and DNA sequence motifs measured by ATAC-seq.
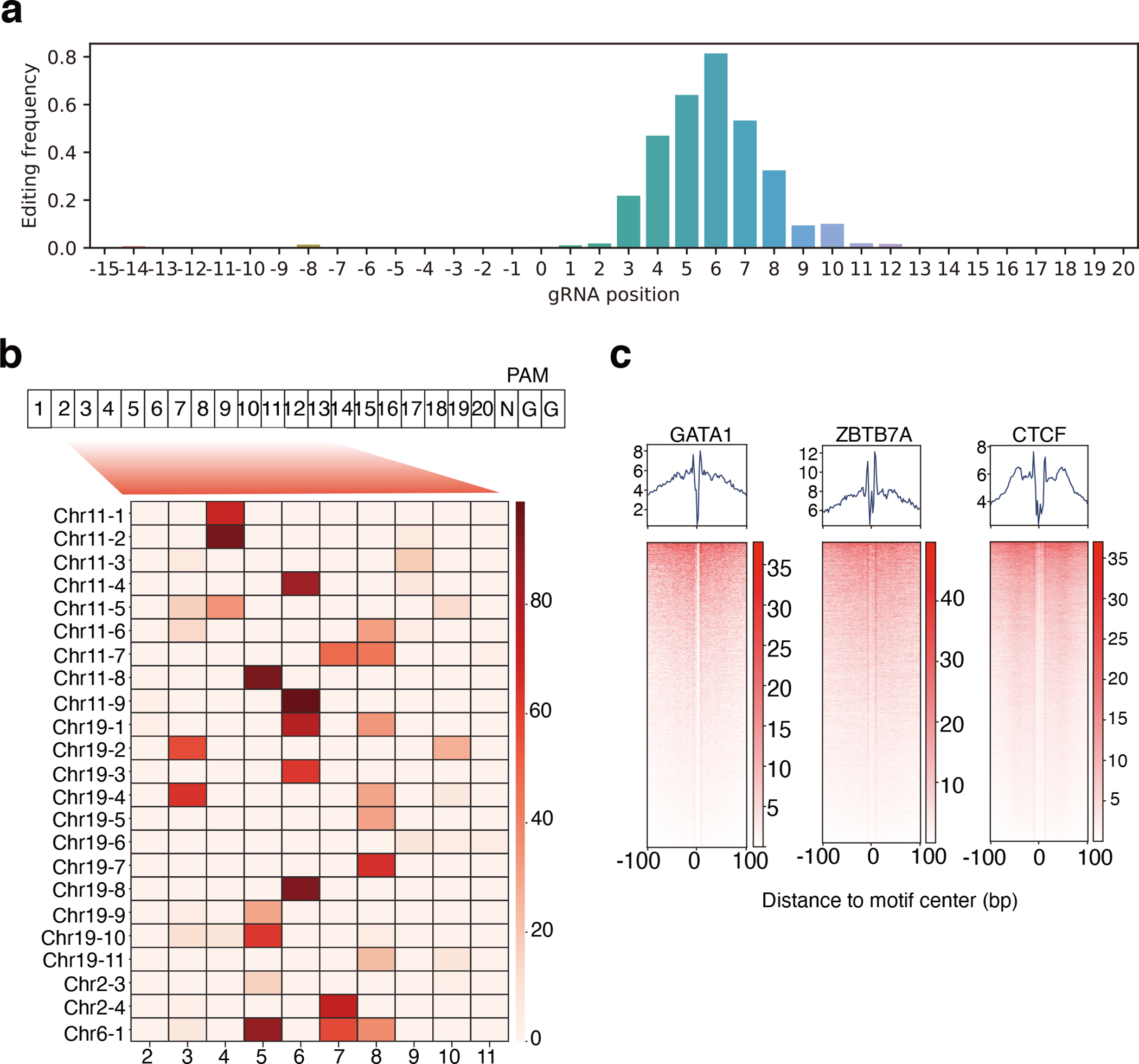
(a) Empirical distribution of ABEmax editing activities in HUDEP-2 cells. Bar plot shows the average editing activity at different positions among 23 different ABEmax edited loci. X-axis denotes positions relative to protospacer start (position 1). Y-axis shows the A to G conversion rate.
(b) A heatmap of on-target base-editing efficiencies of ABEmax as measured by targeted amplicon sequencing of 23 different edited genomic loci (row). Each cell represents one nucleotide. The cell number indicates the relative position of the nucleotide relative to the PAM sequence. The editing efficiency was measured by determining the percentage of nucleotide converted by ABEmax.
(c) The footprint profiles of GATA1, ZBTB7A, and CTCF binding sites derived from deep sequencing (ATAC-seq). The heatmap represents the ATAC-seq signals within a ±100-bp window for the top 1000 binding sites for each TF. Each row represents one binding site. Aggregated signals are plotted in the top panels.
Extended Data Fig. 5. 3′ HBG1 enhancer–edited clones in HUDEP-2 cells.
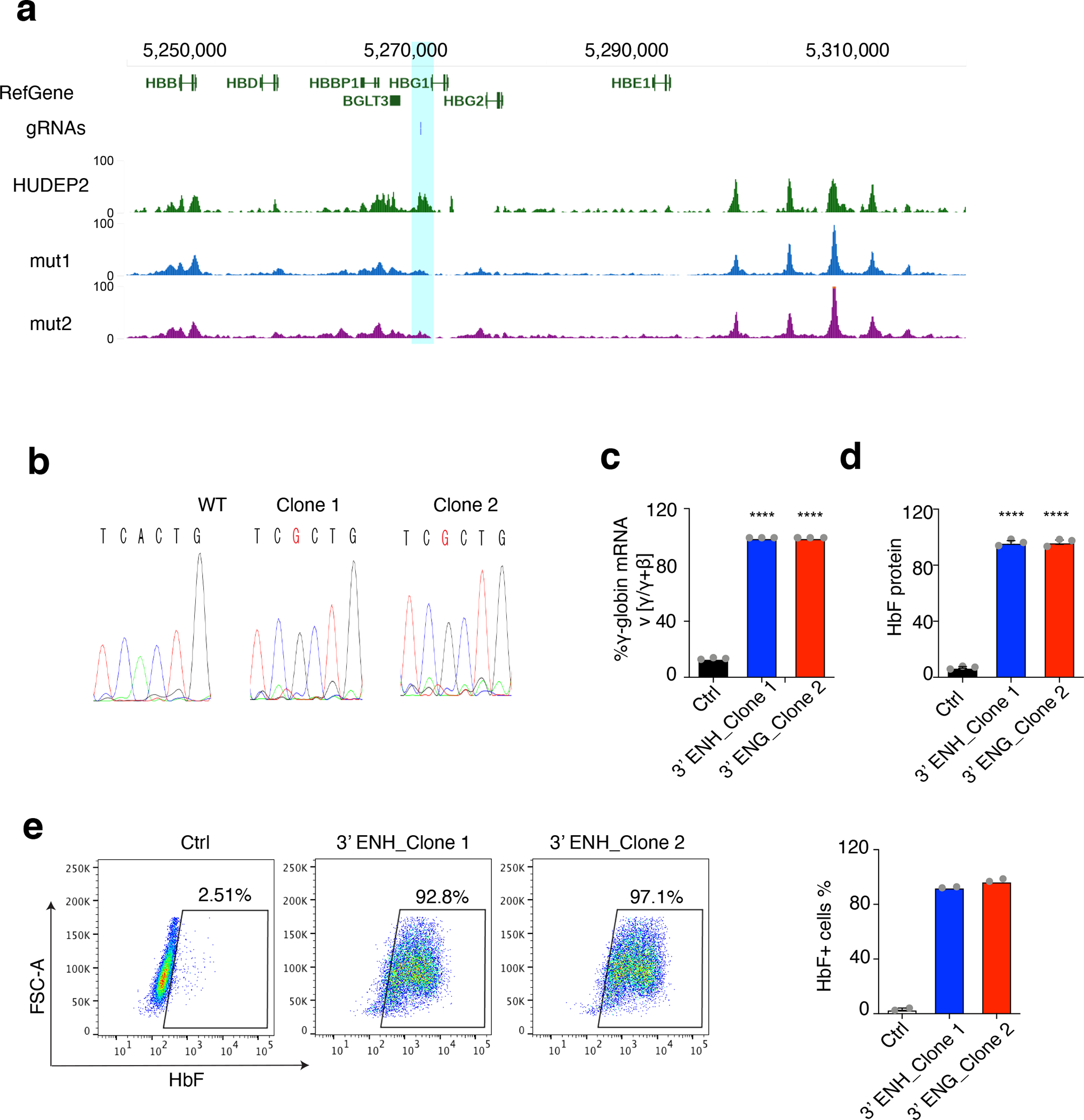
(a) Genome browser screenshot of ZBTB7A occupancy profiles in HBG1 locus. gRNA track showing the location of the gRNA. Wild type HUDEP-2 ChIP-seq was downloaded from GSE103445. Two mutated clones (designated H2_mut_C1 and H2_mut_C2) were generated using ABEmax and gRNAs targeting 3’ HBG1 CRE. The position of the CRE was highlighted in blue.
(b) Amplicon sequencing confirming the mutations in HUDEP-2 cells derived from single clones after treatment with the Chr11–3 gRNA. Edited adenines are marked in red box.
(c–e) Validation studies of two HUDEP-2 single clones with mutations in the 3′ HBG1 enhancer.
(c) The percentage of γ-globin mRNA as determined by real-time RT-qPCR. The error bars represent the ± S.E.M from three independent experiments. **** P = 4X10−7; unpaired t-test, two side.
(d) The hemoglobin F fraction measured by IE-HPLC. The values represent the mean ± S.E.M from three independent experiments. ****P = 6×10−7; unpaired t-test, two side.
(e) F-cell fractions measured by immuno-flow cytometry (left). The bar chart (right) shows the values from two independent experiments.
Extended Data Fig. 6. Epigenetic signals of CREs regulating HbF levels.
Box plots showing the epigenetic signal distribution among adenines with high (>30) (n=313) and low (<10) BPRSHbF (n=9268). (P-value were determined using with two-tailed Wilcoxon test. Box depicts the interquartile range; central line indicates the median and whiskers indicate minimum/maximum values.
Extended Data Fig. 7. Functional noncoding sequences and SNVs associated with HbF levels in patients with SCD.

(a) The ratio of the mutation burden in patients with SCD with high HbF to that in patients with SCD with normal HbF at genomic loci with high BPRSHbF (the top 200). The x-axis represents the threshold of minor allele frequency (MAF) that was used to filter variants. The y-axis represents the different window sizes centered on genomic loci with high BPRSHbF. The number in each cell represents the ratio of the normalized mutation burden (see Methods) in patients with SCD with high RBC HbF levels to that in patients with SCD with normal HbF levels.
(b) The precision-recall curve representing the performance of a random forest model that predicts HbF levels by using the mutation burden within two groups of genomic loci. The green curve represents the model including only 18 common GWAS variants, and the red curve represents the model including the common GWAS variants plus 56 variants with high BPRSHbF. Dashed lines represent the precision at 75% recall rate.
(c) A box plot showing a pair-wise performance comparison of the two models. n= 400 random samplings. P-value is determined using paired two-tailed t-test. Box depicts the interquartile range; central line indicates the median and whiskers indicate minimum/maximum values.
Extended Data Fig. 8. Targeting erythroid-specific regulatory elements to increase HbF levels in erythroid progeny derived from HSPCs from donors with SCD.

(a) A heatmap showing the distribution of chromatin accessibility, as measured by ATAC-seq, near edited adenines for 15 different blood cell types. Representative adenines with high (top) and low (bottom) erythroid-specific scores (Z-scores) were selected for plotting. The cell types for each track are shown at the bottom.
(b–e) CD34+ HSPCs from two donors with SCD were transfected with RNP consisting of ABE and Chr11–1 gRNA targeting the 3′ HBG1 enhancer or a non-targeting control gRNA (Ctrl), then grown in culture under conditions that support erythroid differentiation. Hemoglobinized erythroblasts were analyzed at day 12.
(b) The percentage of γ-globin mRNA as determined by real-time RT-qPCR (n=2 different SCD participants).
(c) Representative flow-cytometry plots showing the expression of the RBC maturation markers Band3 and CD49d (n=2 different SCD participants).
(d) May–Grünwald–Giemsa–stained erythroblasts. Scale bar, 20 μM. This is representative results of 2 SCD participants.
(e) Images of sickled erythroid cells. Arrowheads mark cells with sickle-like morphology. This is representative results of 2 SCD participants. Original picture was visualized by phase-contrast microscopy using the IncuCyte S3 Live-Cell Analysis System (Sartorius) with a 20X objective; Size bars, 20 μM.
Extended Data Fig. 9. Gating strategies used for cell sorting during RBC maturation.

(a) Gating strategy to determine the percentage of RBC maturation markers Band3 and CD49d after 5 additional days of induced differentiation in WT and HUDEP-2-ABEmax cells presented on Fig. 1d.
(b) Gating strategy to determine the percentage of the RBC maturation markers Band3 and CD49d after 12 days of differentiation of normal CD34+ HSPCs (transfected by 5 gRNAs, respectively.) presented on Extended data Fig. 2d.
(c) Gating strategy to determine the percentage of the RBC maturation markers Band3 and CD49d after 12 days of differentiation of SCD derived CD34+ HSPCs (transfected by 2 gRNAs, respectively.) presented on Extended data Fig. 8c.
Extended Data Fig. 10. Gating strategies used for F cells sorting.

(a) Gating strategy to determine the percentage of F cells in Ctrl or BCL11A-ENH gRNA transfected HUDEP-2 cells presented on Fig. 1i.
(b) Gating strategy to determine the percentage of F cells after 12 days of differentiation of SCD derived CD34+ HSPCs (transfected by 2 gRNAs, respectively.) presented on Fig. 6e.
(c) Gating strategy to determine the percentage of F cells presented on Extended data Fig. 5e.
Supplementary Material
Acknowledgments
Ryo Kurita and Yukio Nakamura (Cell Engineering Division, RIKEN BioResource Center, Tsukuba, Japan) provided the HUDEP-2 cells. Xiuli An (Laboratory of Membrane Biology, New York Blood Center) provided the anti-Band3 antibody. We thank the St. Jude Children’s Research Hospital Flow Cytometry core facility for performing the cell sorting, the Hartwell Center core facility for performing the high-throughput sequencing, and the Center for Advanced Genome Engineering for performing the targeted deep sequencing. We thank Keith A. Laycock for scientific editing of the manuscript. This work was supported by St. Jude Children’s Research Hospital and ALSAC and by NIH grants R35GM133614 (Y.C.), P01HL053749 (M.J.W.), the St. Jude Collaborative Research Consortium (M.J.W. and Y.C.), the Doris Duke Foundation grant 2017093 (M.J.W.) and R24DK106766 (M.J.W, R.C.H., and Y.C.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing Interests Statement
M.J.W. is a consultant for Cellarity Inc. and Novartis and has equity in Beam Therapeutics (a base editing company).
A.S. is the St. Jude Children’s Research Hospital site principal investigator of clinical trials for genome editing of SCD sponsored by Vertex Pharmaceuticals/CRISPR Therapeutics (NCT03745287) and by Novartis (NCT04443907). The industry sponsors provide funding for the clinical trial, which includes salary support paid to A.S.’s institution. A.S. also is a consultant for Spotlight Therapeutics.
References
- 1.Agrawal P, Heimbruch KE & Rao S Comprehensive Physiology. Compr Physiol 9, 439–455 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rickels R & Shilatifard A Enhancer Logic and Mechanics in Development and Disease. Trends Cell Biol 28, 608–630 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Bolt CC & Duboule D The regulatory landscapes of developmental genes. Development 147, dev171736 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Driscoll MC, Dobkin CS & Alter BP Gamma delta beta-thalassemia due to a de novo mutation deleting the 5’ beta-globin gene activation-region hypersensitive sites. Proc National Acad Sci 86, 7470–7474 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kioussis D, Vanin E, deLange T, Flavell RA & Grosveld FG β-Globin gene inactivation by DNA translocation in γβ-thalassaemi. Nature 306, 662–666 (1983). [DOI] [PubMed] [Google Scholar]
- 6.Lettice LA et al. Disruption of a long-range cis-acting regulator for Shh causes preaxial polydactyly. Proc National Acad Sci 99, 7548–7553 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauer DE et al. An Erythroid Enhancer of BCL11A Subject to Genetic Variation Determines Fetal Hemoglobin Level. Science 342, 253–257 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chatterjee S & Ahituv N Gene Regulatory Elements, Major Drivers of Human Disease. Annu Rev Genom Hum G 18, 1–19 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Dunham I et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kundaje A et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thurman RE et al. The accessible chromatin landscape of the human genome. Nature 489, 75–82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heintzman ND et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heintzman ND et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39, 311–318 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Bulger M & Groudine M Functional and Mechanistic Diversity of Distal Transcription Enhancers. Cell 144, 327–339 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng H & Xie W The role of 3D genome organization in development and cell differentiation. Nat Rev Mol Cell Bio 20, 535–550 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Schoenfelder S & Fraser P Long-range enhancer–promoter contacts in gene expression control. Nat Rev Genet 20, 437–455 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Henikoff S & Shilatifard A Histone modification: cause or cog? Trends Genet 27, 389–396 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Cheng J et al. A Role for H3K4 Monomethylation in Gene Repression and Partitioning of Chromatin Readers. Mol Cell 53, 979–992 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Canver MC et al. Variant-aware saturating mutagenesis using multiple Cas9 nucleases identifies regulatory elements at trait-associated loci. Nat Genet 49, 625–634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diao Y et al. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat Methods 14, 629–635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diao Y et al. A new class of temporarily phenotypic enhancers identified by CRISPR/Cas9-mediated genetic screening. Genome Res 26, 397–405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komor AC, Badran AH & Liu DR CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 168, 20–36 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaudelli NM et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wienert B et al. KLF1 drives the expression of fetal hemoglobin in British HPFH. Blood 130, 803–807 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Wienert B, Martyn GE, Funnell APW, Quinlan KGR & Crossley M Wake-up Sleepy Gene: Reactivating Fetal Globin for β-Hemoglobinopathies. Trends Genet 34, 927–940 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Perkins A et al. Krüppeling erythropoiesis: an unexpected broad spectrum of human red blood cell disorders due to KLF1 variants. Blood 127, 1856–1862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Traxler EA et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med 22, 987–990 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Y et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med 25, 776–783 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Métais J-Y et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv 3, 3379–3392 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galarneau G et al. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet 42, 1049–1051 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koblan LW et al. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat Biotechnol 36, 843–846 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurita R et al. Establishment of Immortalized Human Erythroid Progenitor Cell Lines Able to Produce Enucleated Red Blood Cells. Plos One 8, e59890 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grevet JD et al. Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells. Science 361, 285–290 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Canver MC et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grünewald J et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569, 433–437 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu N et al. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 173, 430–442.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dogan N et al. Occupancy by key transcription factors is a more accurate predictor of enhancer activity than histone modifications or chromatin accessibility. Epigenet Chromatin 8, 16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng Y et al. Principles of regulatory information conservation between mouse and human. Nature 515, 371–375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Funnell APW et al. 2p15-p16.1 microdeletions encompassing and proximal to BCL11A are associated with elevated HbF in addition to neurologic impairment. Blood 126, 89–93 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mumbach MR et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods 13, 919–922 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borg J et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet 42, 801–805 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou D, Liu K, Sun C-W, Pawlik KM & Townes TM KLF1 regulates BCL11A expression and γ- to β-globin gene switching. Nat Genet 42, 742–744 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Natiq A et al. Hereditary persistence of fetal hemoglobin in two patients with KLF1 haploinsufficiency due to 19p13.2–p13.12/13 deletion. Am J Hematol 92, E2–E3 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Danjou F et al. Genome-wide association analyses based on whole-genome sequencing in Sardinia provide insights into regulation of hemoglobin levels. Nat Genet 47, 1264–1271 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thein SL Genetic association studies in β-hemoglobinopathies. Hematology 2013, 354–361 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Huang P et al. Comparative analysis of three-dimensional chromosomal architecture identifies a novel fetal hemoglobin regulatory element. Gene Dev 31, 1704–1713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ivaldi MS et al. Fetal γ-globin genes are regulated by the BGLT3 long noncoding RNA locus. Blood 132, 1963–1973 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rees HA & Liu DR Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet 19, 770–788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poole W, Gibbs DL, Shmulevich I, Bernard B & Knijnenburg TA Combining dependent P- values with an empirical adaptation of Brown’s method. Bioinformatics 32, i430–i436 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gusev A et al. Partitioning Heritability of Regulatory and Cell-Type-Specific Variants across 11 Common Diseases. Am J Hum Genetics 95, 535–552 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Masuda T et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science 351, 285–289 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mantovani R et al. The effects of HPFH mutations in the human γ-globin promoter on binding of ubiquitous and erythroid specific nuclear factors. Nucleic Acids Res 16, 7783–7797 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ronchi AE, Bottardi S, Mazzucchelli C, Ottolenghi S & Santoro C Differential Binding of the NFE3 and CP1/NFY Transcription Factors to the Human γ- and ∊-Globin CCAAT Boxes (∗). J Biol Chem 270, 21934–21941 (1995). [DOI] [PubMed] [Google Scholar]
- 55.Fornes O et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 48, D87–D92 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bodine DM & Ley TJ An enhancer element lies 3’ to the human A gamma globin gene. Embo J 6, 2997–3004 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Purucker M, Bodine D, Lin H, McDonagh K & Nienhuis AW Structure and function of the enhancer 3′ to the human A γ, globin gene. Nucleic Acids Res 18, 7407–7415 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martyn GE et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat Genet 50, 498–503 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Degner JF et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 482, 390–394 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang F & Lupski JR Non-coding genetic variants in human disease. Hum Mol Genet 24, R102–R110 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou J & Troyanskaya OG Predicting effects of noncoding variants with deep learning–based sequence model. Nat Methods 12, 931–934 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rentzsch P, Witten D, Cooper GM, Shendure J & Kircher M CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47, D886–D894 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zeng J et al. Therapeutic base editing of human hematopoietic stem cells. Nat Med 26, 535–541 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Corces MR et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet 48, 1193–1203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sanjana NE et al. High-resolution interrogation of functional elements in the noncoding genome. Science 353, 1545–1549 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Allen F et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat Biotechnol 37, 64–72 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anzalone AV, Koblan LW & Liu DR Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 38, 824–844 (2020). [DOI] [PubMed] [Google Scholar]
- 68.Vierstra J et al. Global reference mapping of human transcription factor footprints. Nature 583, 729–736 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Menzel S et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet 39, 1197–1199 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Stadhouders R et al. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. J Clin Invest 124, 1699–1710 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vinjamur DS, Bauer DE & Orkin SH Recent progress in understanding and manipulating haemoglobin switching for the haemoglobinopathies. Brit J Haematol 180, 630–643 (2018). [DOI] [PubMed] [Google Scholar]
- 72.Montavon T et al. A Regulatory Archipelago Controls Hox Genes Transcription in Digits. Cell 147, 1132–1145 (2011). [DOI] [PubMed] [Google Scholar]
- 73.Snetkova V & Skok JA Enhancer talk. Epigenomics-uk 10, 483–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jeong J et al. High-efficiency CRISPR induction of t(9;11) chromosomal translocations and acute leukemias in human blood stem cells. Blood Adv 3, 2825–2835 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walton RT, Christie KA, Whittaker MN & Kleinstiver BP Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 368, 290–296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nishimasu H et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361, eaas9129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hu JH et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang X et al. Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat Biotechnol 38, 856–860 (2020). [DOI] [PubMed] [Google Scholar]
- 79.Grünewald J et al. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat Biotechnol 38, 861–864 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sakata RC et al. Base editors for simultaneous introduction of C-to-T and A-to-G mutations. Nat Biotechnol 38, 865–869 (2020). [DOI] [PubMed] [Google Scholar]
- 81.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-Only References
- 82.Hu J et al. Isolation and functional characterization of human erythroblasts at distinct stages: implications for understanding of normal and disordered erythropoiesis in vivo. Blood 121, 3246–3253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bray NL, Pimentel H, Melsted P & Pachter L Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 84.Pimentel H, Bray NL, Puente S, Melsted P & Pachter L Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods 14, 687–690 (2017). [DOI] [PubMed] [Google Scholar]
- 85.Corces MR et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li Z et al. Identification of transcription factor binding sites using ATAC-seq. Genome Biol 20, 45 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Heinz S et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Qi Q et al. Dynamic CTCF binding directly mediates interactions among cis-regulatory elements essential for hematopoiesis. Blood (2020) doi: 10.1182/blood.2020005780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McKenna A et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sanjana NE, Shalem O & Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li W et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Landau W, Niemi J & Nettleton D Fully Bayesian analysis of RNA-seq counts for the detection of gene expression heterosis. J Am Stat Assoc 114, 0–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed sequencing data generated in this study are available from the Gene Expression Omnibus under accession GSE157311. All unprocessed Western Blotting for Figure 1b, Extended Data Figures 1a,e and 2c can be found in Source data provided with this paper.