

Abstract
A balanced progression through mitosis and cell division is largely dependent on orderly phosphorylation and ubiquitin-mediated proteolysis of regulatory and structural proteins. These series of events ultimately secure genome stability and time-invariant cellular properties during cell proliferation. Two of the core enzymes regulating mitotic milestones in all eukaryotes are cyclin dependent kinase 1 (CDK1) with its coactivator cyclin B, and the E3 ubiquitin ligase anaphase promoting complex/cyclosome (APC/C). Discovering mechanisms and substrates for these enzymes is vital to understanding how cells move through mitosis and segregate chromosomes with high fidelity. However, the study of these enzymes has significant challenges. Purely in vitro studies discount the contributions of yet to be described regulators and misses the physiological context of cellular environment. In vivo studies are complicated by the fact that each of these enzymes, as well as many of their regulators and downstream targets, are essential. Moreover, long-term in vivo manipulations can result in cascading, indirect effects that can distort data analysis and interpretation. Many of these challenges can be circumvented using cell-free systems, which have historically played a critical role in identifying these enzymes and their contributions under quasicellular environments. Here, we describe the preparation of a newly developed human cell-free system that recapitulates an anaphase-like state of human cells. This new toolkit complements traditional cell-free systems from human cells and frog eggs and can be easily implemented in cell biology labs for direct and quantitative studies of mitotic signaling regulated by phosphorylation, APC/C-mediated proteolysis, and beyond.
Keywords: Cell-free system, Cell extracts, Ubiquitin-mediated degradation, Nondegradable cyclin B, Mitosis, Anaphase, APC/C, Cdc20, Cdh1, Cdk1
1. Introduction
The unidirectional nature of the cell cycle is first and foremost achieved by a highly regulated series of protein modifications—most notably phosphorylation, and ubiquitination-mediated proteolysis [1]. Orderly (de)phosphorylation and proteolysis is perhaps best demonstrated during mitosis and cell division. Much of the protein degradation controlling mitotic progression and exit is regulated by an E3 ubiquitin ligase, the anaphase-promoting complex/cyclosome (APC/C), and its two substrate adaptors—Cdc20 and Cdh1. Cdc20-bound APC/C (APC/CCdc20) is active from mitotic metaphase until late anaphase, mediating the proteolysis of mitotic cyclins, Securin, and other key cell cycle proteins [2–5]. Cdh1-bound APC/C (APC/CCdh1) is active from telophase through G1-phase of the cell cycle. The switch from APC/CCdc20 to APC/CCdh1 is regulated by temporal (de)phosphorylation and proteolysis, and is a central event in the overall mechanism underlying orderly cell division in all eukaryotes [6–8].
Cell-free systems are known for their capacity to recapitulate complex cellular processes in vitro while maintaining a physiologically relevant context. These systems are optimal for direct and quantitative assays, circumventing caveats associated with time-sensitive assays and long-term in vivo manipulations. Cell-free systems can either capture a certain physiological state (e.g., interphase) or reproduce biochemical and structural dynamics characterizing transitions between cell cycle phases (e.g., phosphorylation wave, chromatin condensation, and spindle formation during mitotic entry).
Much of the core machinery of the vertebrate cell cycle was originally resolved in cell-free systems derived from frog eggs and early embryos. The unmatched simplicity of obtaining near physiological protein concentration in vitro and the synchronous two-phase cell cycles during early embryogenesis provide a homogenous biochemical ‘soup’ for elucidating the cell cycle machinery [9–12]. Mitotic entry and exit, metaphase-to-anaphase transition and cytokinesis were all demonstrated in frog egg extracts [13–15]. Moreover, the APC/C and some of its key targets were both discovered in egg extracts, including Cyclin B [3], Securin [4] and Geminin [16]. With respect to Cdc20 vs. Cdh1 specificity of APC/C targets, egg extracts are optimal. Vertebrate Cdh1 is undetectable pre–mid-blastula transition, and thus, APC/C activity in mitotic egg extracts is mediated solely by Cdc20, whereas interphase egg extracts supplemented with recombinant Cdh1 recapitulate APC/CCdh1-specific activity. Systematic screens for APC/C targets in both mitotic and interphase egg extracts have shed enormous light on vertebrate cell cycle [4, 16–19].
Currently, frogs are no longer considered a popular animal model and thus, frog colonies with sufficient size are scarce. Moreover, the high cost of frog facilities is a limiting factor for many labs. In the last 20 years, human cell-free systems have been gradually integrated into cell cycle research. Extracts from synchronous cell populations provide quasi-cellular environments for analyzing orderly protein degradation, phosphorylation and other signaling events in a somatic 4-stage cell cycle context, which is lacking in egg extracts [20–23]. These environments can be manipulated either genetically in the source cells prior to extract preparation, or biochemically before, during or after extract preparation. Extracts from human cells arrested at the G1 phase exhibit APC/CCdh1-mediated proteolysis, much like interphase egg extracts. Human mitotic cell extracts are typically obtained from nocodazole arrested cells. These extracts are not equivalent to mitotic egg extracts because the activated mitotic checkpoint complex halts APC/CCdc20 activity. Removal of this checkpoint apparatus can be induced thermodynamically and/or biochemically, but then, mitotic exit is triggered and both APC/CCdc20 and Cdk1 are gradually inactivated [24].
We recently developed a human cell-free system which recapitulates an early anaphase-like state where both Cdk1 and APC/CCdc20 remain stably active [24]. The system is named ‘NDB’ and is based on HEK293 cells inducibly expressing a nondegradable mutant variant of Cyclin B1 (NDB). In this method chapter we describe the preparation of NDB extracts and source cells. The potential use of the system in studying mitotic phosphorylation and degradation and the Cdc20 vs. Cdh1 specificity of APC/C substrates is demonstrated.
2. Materials
2.1. Buffers and Reagents
2.1.1. Cell Culture Maintenance
Unless otherwise is noted, all solutions made with double deionized water (DDW); 18 MΩ.
Culture medium: Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), 2 mM L-glutamine, and 1% of X100 penicillin–streptomycin solution (10,000 U/mL penicillin, 10,000 μg/mL streptomycin).
Phosphate buffered saline (PBS), pH 7.4.
Trypsin–EDTA Solution: 0.25% Trypsin, 0.05% EDTA.
Antibiotics for cell selection: zeocin™ stock solution (100 mg/mL); blasticidin stock solution (10 mg/mL).
2.1.2. Generation of NDB Cell System
NDB cell system is based on human 293T-REx™ expressing a nondegradable mutant variant of human Cyclin B1 under Tetracycline-regulated CMV promoter.
Mammalian cell line: 293T-REx™ (Thermo Fisher Scientific; #R71007).
Mammalian expression vector pcDNA™4/TO (Thermo Fisher Scientific, #V102020).
-
A primer set for cloning human Cyclin B1 from a source vector or genomic cDNA into pcDNA™4/TO vector:
Forward— GACTGGATCCATGGCGCTCCGAGTCACCAG.
Reverse complement—GACTCTCGAGTTACAC CTTTGCCACAGCCTT.
Restriction enzymes BamHI and XhoI.
High-fidelity DNA polymerase (e.g., Agilent’s Herculase II; #600675).
T4 DNA Ligase (New England Biolabs; #M0202S).
QuikChange Lightning™ site directed mutagenesis kit (Agilent, #210519).
-
A primer set for generating nondegradable mutant Cyclin B1 using pcDNA4/TO-Cyclin B1 as a template (see Note 1).
Forward— CCAATGTCCCCAACAGCTGTTCCTGGCCTCAGTCCG.
Reverse complement— CGGACTGAGGCCAGGAACAGCTGTTGGGGACATTGG.
A plasmid DNA miniprep kit (e.g., Qiagen; #27104).
Competent bacteria DH5α™ (Thermo Fisher Scientific; #18263012).
Agarose–TAE powder blends (1%) for Gel electrophoresis (Sigma-Aldrich; #A6236–100G). Follow the manufacturer’s protocol for gel preparation.
LB Agar carbenicillin plates: 100 μg/mL carbenicillin.
2×HeBS solution: 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 1.5 mM Na2HPO4, 280 mM NaCl. Adjust pH to 7.05 with NaOH.
2.5 M CaCl2 stock solution. Dissolve 11 g of CaCl2·6H2O in a final volume of 20 mL of DDW. Sterilize the solution by passing it through a 0.22-μm filter. Store at 4 °C.
2.1.3. Validating Mitotic Arrest in NDB Cells Expressing Nondegradable Cyclin B1
Tetracycline (Tet) stock solution: 2.5 mg/mL Tet in 95% ethanol absolute and 5% DDW.
PBS.
Ethanol absolute. Store at −20 °C.
Propidium iodide (PI) staining solution: 0.02 mg/mL PI, 0.05 mg/mL RNAse A in PBS.
Anti-human Cyclin B1 antibody (Cell Signaling Technology; #4138).
Standard reagents for SDS-PAGE and Western blotting (see Subheading 2.1.8, items 6–11).
Nocodazole stock solution: 1 mM in DMSO. Store at −20 °C.
Prewarmed hypotonic solution (37 °C): 5.6 g KCl in 1000 mL DDW (0.075 M).
Fresh fixative solution: 3:1 (v/v) methanol–glacial acetic acid.
Mounting solution: Mounting medium (Richard-Allan Scientific, #4112APG) supplemented with 5 μg/mL 4′,6-diamidino-2-phenylindole (DAPI) stain.
Clear nail polish.
2.1.4. Preparation of NDB Mitotic Protein Lysate
Tet stock solution.
PBS.
0.5 M Ethylenediaminetetraacetic acid (EDTA) stock solution: Dissolve 93.05 g EDTA disodium in 300 mL DDW. Adjust pH to 8.0 with NaOH. Adjust volume to 500 mL with DDW. Store at room temperature (RT).
Cell lysis solution: 50 mM Tris–HCl (pH 7.6), 150 mM NaCl, 5 mM EDTA (pH 8.0 with NaOH), 0.5% NP-40, supplemented with a protease inhibitor cocktail (Roche; #4693159001), phosphatase inhibitor cocktails (Sigma-Aldrich; #P5726 and #P0044), 1 mM phenylmethylsulfonylfluoride (PMSF), 10 mM NaF, 20 mM β-glycerophosphate, 1 mM Na3VO4, 20 mM P-nitrophenylphosphate.
Bradford reagent (Bio-Rad #500–0006).
Bovine serum albumin (BSA).
2.1.5. Preparation of NDB Mitotic Extracts
Tet stock solution.
PBS.
Swelling Buffer: 20 mM HEPES (pH 7.5), 2 mM MgCl2, 5 mM KCl, 1 mM dithiothreitol [DTT], and protease inhibitor cocktail (Roche; #4693159001).
E-mix (Energy regeneration mixture): 20 mM ATP, 2 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 20 mM MgCl2, 150 mM creatine phosphate, 1 mg/mL creatine phosphokinase. Store 50 μL aliquots in −80 °C.
Liquid N2.
2.1.6. Degradation and Mobility-Shift Assays in NDB Mitotic Extracts
NDB mitotic extract.
SP6 expression vector carrying an ORF of interest.
TNT® SP6 coupled reticulocyte lysate system (Promega; #L2080).
35S-methionine/35S-L-cysteine mix (PerkinElmer; #NEG772002MC).
Recombinant Ubiquitin (Boston Biochem; #U-100H).
E. coli-derived recombinant UbcH10 and dominant negative UbcH10 (UbcH10DN). Commercial version of these reagents are available (Boston Biochem; #E2–650, #E2–654, #E2–652).
MG-132 proteasome inhibitor stock solution: 5 mg/mL in DMSO.
2× Laemmli sample buffer (Bio-Rad Laboratories; #161–0737).
Dithiothreitol (DTT). 1 M stock solution in DDW. Store at −20 °C.
Polyacrylamide gels (e.g., Mini-Protean TGX precast gels [Bio-Rad Laboratories]). Recommended polyacrylamide concentrations: 7.5%, 10%, and 4–15% (#456-1-23, #456-1033, #456-1083, respectively).
Destain solution: 10% methanol, 7.5% acetic acid, in DDW.
2.1.7. G1-Like NDB Extracts
NDB mitotic extracts.
Cdk1 inhibitor stock solution: 10 mM RO-3306 (Cdk1 inhibitor) in DMSO. Store −20 °C.
DMSO (dimethyl sulfoxide).
2.1.8. Immuno-precipitation of APC/C from NDB Extracts and Immunoblot of Cdc20/Cdh1
Wash buffer I: 150 mM NaCl, 20 mM Tris–HCI (pH 7.5), 10% glycerol, 0.1% Triton, 1 mM EDTA.
Wash buffer II: 75 mM NaCl, 20 mM Tris-HCI (pH 7.5), 10% glycerol, 0.1% Triton, 1 mM EDTA.
Agarose-conjugated monoclonal anti-Cdc27 antibody: clone AF3.1 (Santa Cruz Biotechnology; SC-9972AC).
Anti-Cdc20 antibody (Santa Cruz Biotechnology; #SC-8358).
Anti-Cdh1 antibody (MilliporeSigma™; #CC43100UG [Clone DH01]).
Secondary antibodies conjugated to horseradish peroxidase (HRP) from Jackson ImmunoResearch: goat anti-rabbit IgG (#111-035-144), goat anti-Mouse IgG (#115-035-003), mouse anti-rabbit IgG, light chain specific (#211-032-171), goat anti-mouse IgG light chain specific (#115-035-174).
TBST buffer: 150 mM NaCl, 50 mM Tris–HCl, 0.1% Tween 20, in DDW. pH adjusted to 7.6 with HCl.
Transfer buffer: 25 mM Tris, 192 mM glycine, 20% methanol.
Running buffer: 25 mM Tris, 192 mM glycine, 0.1% sodium dodecyl sulfate (SDS).
Antibody solution: 5% BSA, 0.05% sodium azide in TBST (see item 7 above).
Reagents for Enhanced Chemiluminescence (ECL) Western detection (e.g., Thermo Fisher Scientific; #34095).
2.2. Equipment
2.2.1. General Use
Pipettes for 2, 10, 200, and 1000 μL, and matching pipette tips.
Pipette-aid and serological pipettes: 5, 10, and 25 mL.
Tubes: 0.2, 1.5, 2, 15, and 50 mL.
Refrigerated centrifuges and adaptors for 1.5 and 2 mL tubes, and 15, 50, and 500 mL conical tubes.
PCR machine.
Temperature controlled water bath.
Thermomixer or dry block incubator for 1.5 mL tubes.
Spectrophotometer for DNA quantification and Bradford protein assay.
4 °C environment.
Freezers: −20 and −80 °C.
Apparatuses and power supplies for preparing and running agarose and acrylamide gels.
Gel loading tips, 1–200 μL (e.g., Fisherbrand™ H01096RSFIS).
Autoclave.
2.2.2. Cell Culture Maintenance
37 °C, 5% CO2 controlled incubator.
Laminar flow hood.
Tissue culture plates: 60, 100, and 150 mm/diameter.
Multiwell tissue culture dishes: 24- and 96-well dishes.
2.2.3. Microscopy
Inverted phase contrast microscope equipped with 20× and 40× objectives.
Epifluorescence microscope equipped with a light source and a filter set for imaging DAPI-stained chromosomes, and ×40-× 100 oil objectives.
Glass slides (1 mm thick, 25 mm × 75 mm).
Coverslips (No. 1 thickness, 22 mm × 22 mm).
2.2.4. Flow Cytometry
Optical flow cytometer with a 488 nm laser and filter/detector for Phycoerythrin (PE).
Falcon® round-bottom polypropylene 5 mL tubes with cell strainer cap.
2.2.5. Extract Preparation
Liquid N2 dewar.
250 or 500 mL conical tubes.
10 mL beaker.
21 G needle.
3 or 5 mL syringe.
Cooled centrifuge for 15–500 mL conical tubes, and a swing bucket rotor.
Cooled centrifuge for 1.5 to 2 mL tubes.
2.2.6. Degradation/Phosphorylation Assays
Whatman™ paper (46 cm × 57 cm; Whatman grade: 3MM).
Gel drying machine (e.g., Bio-Rad, model #583).
Phosphoimager.
Phosphor screen and cassette.
A phosphorscreen eraser.
Image analysis software for signal quantification (e.g., ImageJ).
2.2.7. Immunoblot and Immunoprecipitation
Rocking shaker.
Tube rotator.
Western blot imaging system.
3. Methods
3.1. Generation of NDB Cell Line
Culture 293T-REx™ cells in a medium containing 5 μg/mL Blasticidin (see Notes 2–4).
Amplify human Cyclin B1 ORF using (a) a source plasmid or genomic cDNA as a template; (b) a primer set (see Subheading 2.1.2); (c) restriction enzymes BamHI and XhoI; and (d) a PCR reaction with a high-fidelity DNA polymerase (see Subheading 2.1.2, items 4 and 5).
Clone human Cyclin B1 ORF into pcDNA™4/TO expression vector using a standard DNA ligation protocol.
Generate a nondegradable mutant variant of Cyclin B1 by introducing two point mutations for substituting Arg 42 and Leu 45 at the destruction box of Cyclin B1 with Gly and Val, respectively [5, 25]. QuikChange Lightning™ site-directed mutagenesis kit is optimal for this application. Use the pcDNA™4/TO-Cyclin B1 plasmid as a template. A primer set for mutagenesis is indicated in Subheading 2.1.2.
Digest the PCR product with DpnI restriction enzyme (DpnI is supplied with the site-directed mutagenesis kit) and transform into DH5α E. coli. Select transformants bacteria on agar plates containing 100 μg/mL Carbenicillin.
Select a transformed colony for plasmid preparation using a standard miniprep protocol.
Validate mutagenesis by sequencing the miniprep product.
Transfect 293T-REx™ cells with pcDNA™4/TO-Cyclin B1-DM plasmid using a standard transfection method (see Note 5).
Forty-eight hours post transfection split and reculture cells at 25% confluency. Start cell selection by adding 200 μg/mL Zeocin™ in addition to 5 μg/mL blasticidin.
Change selection medium every 3–4 days until single colonies are formed.
Pick at least 24 colonies and expand them separately (see Note 6).
Culture colonies in 24-well dishes.
Once cells reach 50% confluency, treat cells with 1 μg/mL Tet for 18–24 h (see Note 7).
Assess synchronization quality by phase contrast microscopy. Cells in which the ORF encoding nondegradable cyclin B1 was integrated successfully into the genome and expression is properly induced arrest in mitosis, become round, form aggregates and partly detach from the surface (Fig. 1). From this point onward, maintain about five positive colonies for further validation. In addition, cryofreeze two vials for each colony.
In addition, assess synchronization quality by DNA quantification before and after 22 h treatment with 1 μg/mL Tet (see Subheading 3.2). Maintain colonies in which G2/M index following mitotic arrest reaches ~95% (Fig. 1).
Validate expression of nondegradable Cyclin B1 by Western blotting with anti-Cyclin B1 before and 22 h after induction by Tet (see Note 8).
Validate sister chromatid separation in mitotic-arrested NDB cells. Tet-induced NDB cells arrest in mitosis post metaphase-to-anaphase transition. At this anaphase-like stage, full or partial sister chromatid separation has already taken place [24]. A standard protocol for chromosome spreads can be used to validate this feature of the arrested cells (see Subheading 3.3 and refs. 24, 26, 27). As a control, nocodazole-arrested NDB cells should be analyzed in parallel for visualizing unseparated sister chromatids for comparison. To this end, treat NDB cells with 100 nM nocodazole for ~16 h to enrich for prometaphase cells. Chromosomes can be visualized by DAPI staining on glass slides using an epifluorescence microscope equipped with a 405 nm light source and 40–100× oil-immersion objectives. NDB cell colonies exhibiting a high fraction of Tet-induced cells with separated sister chromatids should be prioritized for further experiments.
For all further experiments, select a cell colony whose proliferating time, DNA distribution in steady state culture conditions, overall shape and size range are most similar to the source cell line (293T-REx™) [24].
Fig. 1.
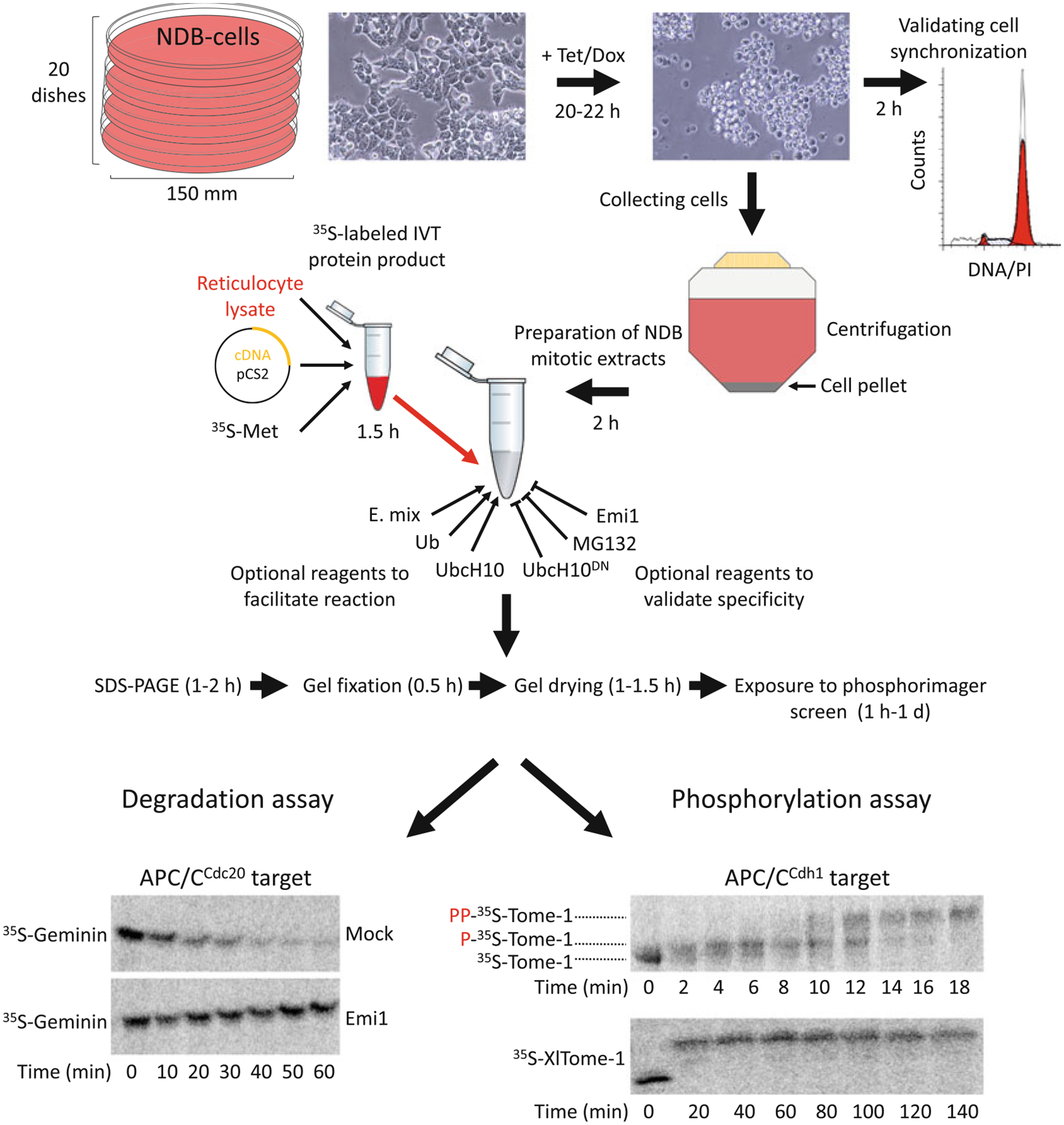
NDB cell- and cell-free systems for elucidating human mitosis. (a) NDB cell system is based on human 293-T-REx™ cells stably expressing nondegradable (ND) mutant of Cyclin B1 under a tetracycline (Tet)/doxycycline (Dox)-regulated CMV promoter. Expression of ND-Cyclin B1 induces mitotic arrest in an anaphase-like state. The constant activity of the Cdk1–Cyclin B1 complex prevents mitotic exit and maintains a peak level of APC/CCdc20 activity for hours. NDB extracts are typically prepared from 20 plates of 150 mm/diameter. Recommended cell confluency for Tet-induced expression is 75–80%. Following 20–22 h treatment with Tet, cells acquire a spherical shape. This archetypal morphology indicates mitotic arrest. Cell synchronization should be further validated by standard DNA quantification. The arrested cells will be loosely attached to the surface, if at all. Thus, cells can be collected directly by gentle pipetting (without trypsinization) into a single 500 mL conical tube. Once cells are pelleted and washed, they are ready for extract preparation. In vitro translated (IVT) protein products are generated in reticulocyte lysate supplemented with radiolabeled methio-nine (35S-Met). pCS2-based expression vectors and reticulocyte lysates supporting transcription from SP6 promoter are recommended. A typical reaction mix contain 10–20 μL NDB mitotic extracts and 0.5–1.5 μL IVT product. Extracts already contain energy-regeneration mix (E-mix), as well as endogenous Ubiquitin (Ub) and the E2 enzyme UbcH10. As such, extracts are highly active. Yet adding 0.5–1 μL of ×20 E. mix, recombinant Ub, and UncH10 into the reaction mix can facilitate proteolysis mediated by APC/CCdc20. Conversely, proteasome inhibitors (e.g., MG132), dominant negative UbcH10 (UbcH10DN), or APC/C specific inhibitors (e.g., Emi1 or TAME) can be added for validating the potency and specificity of the assay. Assays are typically performed in a temperature range of 23–30 °C. During degradation/mobility-shift assays, 3–5 μL of the reaction mix is sampled each time point, mixed with Laemmli buffer, boiled and frozen at 80 °C. Protein sampled are resolved by SDS-PAGE. Gels are then soaked in a distain solution, heat/vacuum dried, and exposed to a Phosphorimager screen for 1 h to 1 day. Longer exposure might be helpful in case of weak signals. The potency and specificity of NDB mitotic extracts is demonstrated; Geminin, an APC/CCdc20 substrate, but not Tome-1, an APC/CCdh1 substrate, is degraded. This degradation is blocked by Emi1. Orderly phosphorylation of Tome-1 is evident in minute-scale resolution by a gradual mobility shift. The high mobility shift of Tome-1 observed after 140 min indicates for the stability and potency of this cell-free system
3.2. PI Staining Protocol for Quantifying DNA Content by Flow Cytometry
Harvest about one million cells and transfer to a 10 mL conical tube.
Centrifuge cells for 5 min (220 × g, 4 °C), discard media.
Resuspend cells with 10 mL cold PBS. Centrifuge cells for 5 min (220 × g, 4 °C), discard supernatant.
Resuspend cells with 250 μL cold PBS.
Fix cells by adding 750 μL absolute Ethanol (−20 °C), drop-by-drop, while agitating tube with your finger. Store in −20 °C for 2 h or longer.
For PI staining, centrifuge fixed cells for 5 min (220 × g, 4 °C), discard media.
Resuspend cells with 1 mL PI staining solution, mix and incubate for 30 min at RT in the dark.
Transfer cells through a cell strainer cap into a round-bottom polypropylene 5 mL tubes.
Analyze cells using a flow cytometer equipped with a 488 nm laser.
3.3. Chromosome Spreads
As a control, nocodazole-arrested NDB cells should be analyzed in parallel for visualizing unseparated sister chromatids for comparison. To this end, treat NDB cells with 100 nM nocodazole for ~16 h to enrich for prometaphase cells. NDB cell colonies exhibiting a high fraction of Tet-induced cells with separated sister chromatids should be prioritized for further experiments.
Harvest Tet-induced and nocodazole-arrested NDB cells from a 10 cm/diameter dish by gentle pipetting.
Transfer cell to a 15 mL conical tube. Centrifuge cells for 5 min (220 × g, RT). Discard media.
Resuspend cells with 10 mL PBS, centrifuge cells for 5 min (220 × g, RT).
Aspirate PBS, leaving 0.5 mL of buffer in the tube. Gently resuspend the pellet by flicking the tube with your fingers.
Add 2 mL of prewarmed (37 °C) hypotonic solution (0.56% KCl), drop-by-drop, while agitating tube.
Add additional 10 mL hypotonic solution and incubate for 10 min in 37 °C. Centrifuge for 5 min (220 × g, RT) and aspirate supernatant, leaving 0.5 mL solution. Gently resuspend pellet.
Prepare fresh fixative solution (3:1 v/v methanol–glacial acetic acid).
Add 5 mL fixative solution (3:1 v/v methanol–glacial acetic acid) from the side of the tube. Mix by gently inverting the tube. Centrifuge cells for 5 min (220 × g, RT) and discard supernatant. Repeat step 8.
Add 500 μL fixative solution, mix well by flicking tube. Let large clumps settle.
Immerse precleaned slides in fixative solution for 10 min. Air-dry the slide.
Drop cell droplets on tilted glass slides from 1-m height.
Air-dry slides, mount with a mounting/DAPI solution, place a coverslip, and seal with clear nail polish.
Image chromosomes using an epifluorescence microscope equipped with a 405 nm light source, a filter set for imaging DAPI, and ×40–×100 oil objectives.
3.4. Harvesting Mitotic NDB Cells for Whole-Cell Protein Lysate Preparation and Immunoblot Assays
Split NDB cells into a desired number of plates. One 10 cm plate is sufficient for ~1 mg total protein.
The following day, when cells reach ~75% confluency, add 1 μg/μL Tet for 22 h.
Tet-induced NDB cells will be fully or partially detached from the surface. Collect medium with a 10 mL serological pipette. Pipet up and down gently across the plate in a serpentine manner to facilitate cell detachment.
Transfer cells to a 15 mL conical tube. Rewash the plate with 5 mL fresh media and collect remaining cells.
Centrifuge cells for 5 min (220 × g, 4 °C). Remove supernatant.
Resuspend cell pellet gently with 10 mL cold PBS and centrifuge again. Remove supernatant.
Resuspend cell pellet gently with 1 mL cold PBS, transfer cells to a 1.5 mL tube and centrifuge for 5 min (220 × g, 4 °C). Remove supernatant.
Resuspend pellet with 150–200 μL cell lysis solution and incubate 30 min on ice.
Centrifuge for 40 min (20,000 × g, 4 °C) to remove cell debris. Collect protein lysate (supernatant).
Transfer protein lysate into a new precooled tube.
Determine protein concentration by a standard Bradford assay.
Store protein lysate in −80 °C. For immunoblot applications, storing protein lysate in aliquots of 10–20 μL is recommended.
3.5. Preparation of NDB Mitotic Extracts
-
Split NDB cells into 21 plates of 150 mm/diameter (by trypsinization). We recommend preparing extracts from 20 plates. Expected extract yield is 2–3 mL. Keep at least one extra dish for backup or maintenance. We typically split cells from seven plates of ~90% cell confluency in a 1:3 ratio 24 h before adding Tet (see Note 9).
The following day, at ~75% cell confluency, add freshly prepared 1 μg/mL Tet for 22 h.
Assess synchronization quality by phase microscopy. Nearly all cells should be rounded, in clusters, and either fully or partially detached from the surface (Fig. 1).
Collect cells from each plate by pipetting the medium up-and down 2–3 times across the plate in a serpentine manner using 25–50 mL serological pipettes. Transfer all media into a single 500 mL conical tube (see Note 10).
Rewash all plates with 25 mL medium by transferring medium from plate to plate. Collect remaining cells.
Place the 500 mL tube in a swing bucket rotor with a matching adaptor, balance weight, and centrifuge 5 min (220 × g, 4 °C).
Discard supernatant by pouring the medium gently into a chilled beaker in case the cell pellet is inadvertently detached.
Resuspend cell pellet with 40 mL ice-cold PBS and transfer into 50 mL conical tube. Centrifuge for 5 min (220 × g, 4 °C). Discard supernatant. Repeat step 7.
Resuspend cell pellet with 2–3 mL ice-cold PBS and transfer into precooled 2 mL tubes. Tube number should be minimized. Centrifuge for 5 min (220 × g, 4 °C). Discard supernatant. Pellet volume should be approximately 1 mL per tube.
For each 1 mL cell pellet, resuspend with 750 μL swelling buffer and 50 μL E-mix. Incubate 30 min on ice. Every 5 min mix by inverting the tube three times.
To break cells, flash-freeze the tubes in liquid N2 and thaw quickly in 30 °C water bath. Minimize thawing time. Repeat step 10.
Combine the contents of all tubes into a 10 mL precooled beaker. To facilitate lysis, pass liquid ten times through a 21 G needle using a precooled 3–5 mL syringe.
Divide liquid into precooled 2 mL tubes and centrifuge for 10 min at 20,000 × g (4 °C).
Collect supernatant from all tubes with a 200 μL pipette and transfer to new precooled 2 mL tubes. Fill tubes all the way up. Centrifuge for 40 min at 20,000 × g (4 °C). Expect to see (a) cell pellet at the bottom of the tube; (b) a clear phase of cell extracts; and (c) a thin layer of floating lipids.
Collect supernatant gently with a 200 μL pipette. Place the tip well above the pellet phase. Aspirate protein extracts slowly and with minimal mixing. Avoid taking up pellet or lipids floating at the top. Typically, 1–1.2 mL extracts can be safely collected per full 2 mL tube.
Combine cell extracts in new precooled tubes. Set aside a few μL of the extracts to measure protein concentration by a Bradford assay. Extract concentration typically ranges from 18 to 25 mg/mL (see Note 11).
Aliquot extracts in 0.2 mL tubes. Strips of thin-wall PRC tubes are recommended. Aliquots of 45–65 μL are convenient.
Snap-freeze tubes in liquid N2, and store at −80 °C (see Notes 12 and 13).
3.6. Degradation and Mobility Shift Assays in NDB Extracts
Express protein of interest in reticulocyte lysate following manufacturer’s protocol. We recommend having the desired ORF in a pCS2-based vector and using an SP6 premixed version of Promega’s TNT-coupled transcription–translation reaction kit (see Note 14). Add 35S-Met to reaction mix to generate a radiolabeled in vitro translated (IVT) protein product (Fig. 1; see Note 15). Degradation and mobility shift assays are typically performed with 0.5–1.5 μL IVT product. Based on our experience, IVT products can be refrozen five times without noticeable damage.
Prepare reaction mix on ice (Fig. 1). A default reaction mix for degradation assay contains (a) 20 μL NBD extract; (b) 1 μL radiolabeled IVT product; (c) 10 μg ubiquitin; and (d) 1 μL E-mix (see Note 16).
Reaction mix can contain additional reagents that either facilitate or block APC/CCdc20-mediated proteolysis (Fig. 1). These supplements are critical controls for testing the potency and specificity of the assay. For example, APC/CCdc20-mediated proteolysis is facilitated by recombinant UbcH10 but blocked by UbcH10DN, Emi1 (APC/C inhibitor), Securin (competitive inhibitor), or TAME (small molecule drug) [28]. Control experiments with proteasome inhibitors (e.g., 20 μM MG-132) are recommended.
For phosphorylation-mediated mobility shift assays, ubiquitin is not required. Freshly added E-mix is also dispensable in assays shorter than 1 h. Instead, control assays can contain the Cdk1 inhibitor RO-3306 (15 μM) to couple mobility shift of a tested protein to Cdk1-Cyclin B1-dependent phosphorylation [22–24]. Small molecule inhibitors of other mitotic kinases (e.g., Plk1) can also be used.
Mix all reagents in a 0.2 mL PCR tube on ice or in a precooled metal rack on ice. Add IVT product last.
Homogenize reaction mix by pipetting up and down 4–5 times with a 10 μL pipette. Avoid foaming.
Prepare 1.5 mL tubes with 10 μL 2× Laemmli sample buffer to stop the reaction; one tube per time point. The volume allocated for each time point may vary according to the desired number of samples and the overall volume of the reaction mix (see Note 17).
While reaction mix is on ice, take the first sample and mix with 2× Laemmli buffer. Snap-freeze in liquid N2. This sample represents time point 0.
Place the reaction mix tube in a prewarmed PCR machine. Recommended temperature range for the assay is 23–30 °C (see Fig. 2 and Notes 18 and 19).
For each time point, transfer 3–5 μL reaction mix into a tube containing 10 μL 2× Laemmli buffer and snap-freeze in liquid N2 (see Note 17).
Store all samples in −80 °C.
Denature proteins in 95 °C for 10 min just before gel loading. Quick-spin samples and load the entire sample on a gel.
Resolve sample by SDS-PAGE. We typically use fresh gels of 1 mm thick and 8% or 10% acrylamide and a Tris-Glycine running buffer.
Soak gel in destain solution for 20 min.
Lay gels on a wet Whatman paper. Add a piece of dry Whatman paper underneath and a piece of saran wrap on top (the gel should remain unwrapped). Dry the gel using a vacuum/heat gel drying system (90 min, 80 °C).
Expose the dry gel to a phosphor screen for 1 day and scan by phosphorimager (see Note 20).
Quantify 35S-signal by ImageJ software.
Fig. 2.
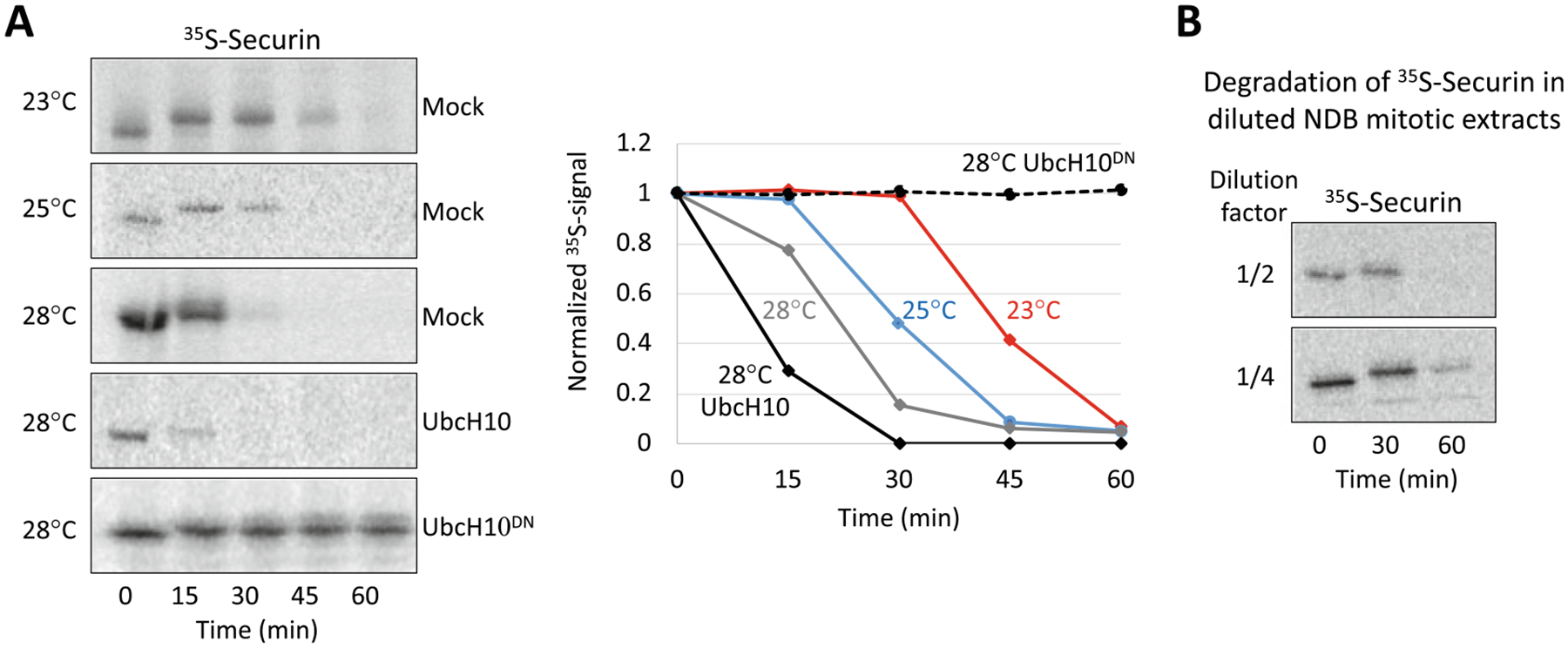
Characterization of APC/CCdc20-mediated degradation in NDB mitotic extracts. (a) Time-dependent degradation of Securin (35S-labeled, IVT product) in mitotic NDB extracts was assayed in three different temperatures. Extracts were supplemented with E-mix and Ub (0.5 mg/mL). The impact of adding recombinant UbcH10 or UbcH10DN (0.5 mg/mL) on the degradation of Securin also tested at 28 °C. Degradation was assayed by SDS–PAGE and autoradiography. Quantification of the depicted raw data (left) are plotted on the right. Data are normalized to max signal at t = 0. (b) Time-dependent degradation of Securin in diluted NDB mitotic extracts are shown (28 °C). See (a) for details. NDB extracts were diluted two- or fourfold in swelling buffer
3.7. Recapitulating a G1-Like State in NDB Extracts
Both NDB cells and extracts override mitotic arrest in the presence of small molecule inhibitors of Cdk1 (e.g., RO-3306) (Fig. 3). Inactivation of Cdk1 effectively generates a synchronous population of NDB cells in a quasi G1 state. In vitro, this treatment signals mitotic exit into a G1-like state (see Notes 21 and 22). The end result is a complementary extract system in which mitotic vs. G1 signaling of target proteins can be tested, including the Cdc20 vs. Cdh1 specificity of APC/C targets [24].
Fig. 3.
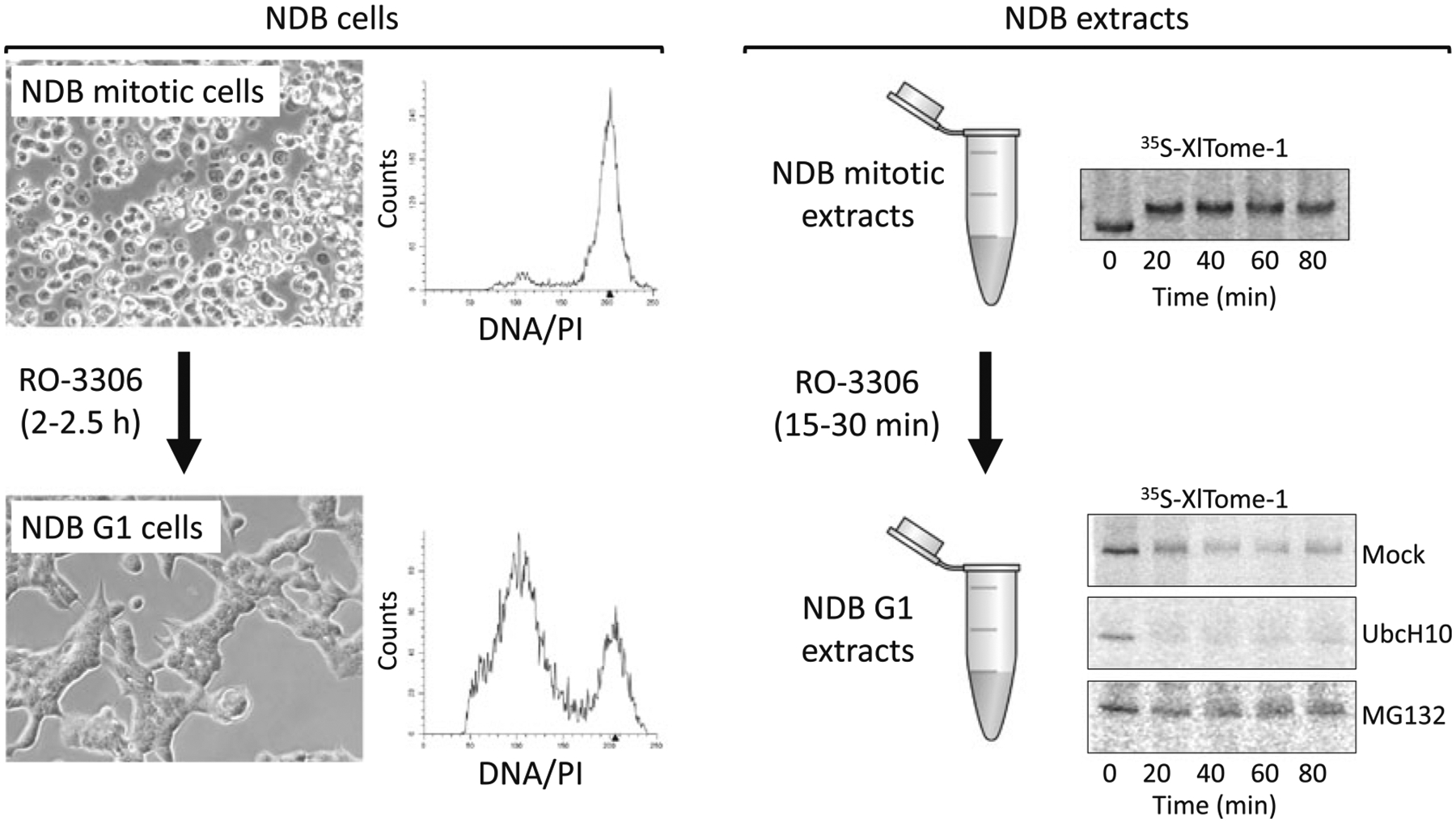
Mitotic NDB cells and extracts can exit mitosis by Cdk1 inhibition. NDB cells induced by Tet are static in an anaphase-like state. These cells can, however, progress into G1 following treatment with the Cdk1 inhibitor RO-3306. Phase images and DNA distributions are shown before and after 2.5 h treatment with RO-3306. Similarly, RO-3306 induces mitotic exit in NDB extracts. The resulting G1-like extracts support the degradation of Tome-1 (APC/CCdh1 substrate). Moreover, the mitotic—Cdk1-dependent—mobility shift of this protein (top right panel) is no longer apparent (bottom right panels). Tome-1 degradation is G1-like NDB extracts is facilitated by UbcH10 and blocked by MG132. These results demonstrate the shift from APC/CCdc20- to APC/CCdh1-specific activity in mitotic vs. G1-like NDB extracts. Degradation of Tome-1 (35S-labeled, IVT product) was assayed in 28 °C, and resolved by SDS-PAGE and autoradiography
3.7.1. Mitotic Exit in Tet-Induced NDB Cells
Culture NDB cells in plates until they reach 75–80% confluency.
Treat cells with 1 μg/mL Tet for 22 h.
Collect and transfer mitotic cells into a 50 mL tube. Centrifuge for 5 min (room temperature [RT], 220 × g) and discard supernatant.
Resuspend with fresh, Tet-free, prewarmed media and reculture cells on plates.
Treat cells with 15 μM RO-3306 FOR 2–2.5 h.
Validate mitotic exit by monitoring cell morphology (phase contrast microscopy) and DNA quantification (see Subheading 3.2).
Harvest cells for immunoblot or other assays.
3.7.2. Mitotic Exit in NDB Extracts
Prepare fresh mitotic NDB extracts or thaw premade extracts on ice (see Subheading 3.5 for details).
Incubate NDB mitotic extracts with 15–30 μM RO-3306 for 15–30 min at 28 °C. Place tube on ice.
For Western blotting or protein immunoprecipitation assays one can either use fresh RO-3306-activated NDB extracts or snap-freeze extracts and conduct the assay later.
For Western blotting, we typically mix 50–80 μg of the extracts with 2× Laemmli buffer, denature samples for 10 min in 95 °C, and resolve by SDS-PAGE.
For degradation or mobility-shift assays, assemble a reaction mix on ice and conduct the assay (see Subheading 3.6).
3.7.3. Immuno-precipitation of APC/C in Mitotic vs. G1-Like NDB Extracts
Wash 15 μL agarose-conjugated anti-Cdc27 monoclonal antibodies (AF3.1 clone) with 500 μL PBS.
Centrifuge beads for 1 min (250 × g, 4 °C). Aspirate supernatant using a fine protein-loading tip while tilting the tube sideways to minimize contact with the beads.
Wash agarose-conjugated antibodies with 500 μL wash buffer I (150 mM NaCl). Centrifuge beads for 1 min (250 × g, 4 °C). Aspirate supernatant.
Prepare or thaw 200–250 μL of mitotic and/or G1-like NDB extracts (see Subheading 3.5).
Put aside 10 μL of the extracts for input sample. Mix this input sample with 2× Laemmli buffer, denature 10 min at 95 °C and store at −80 °C.
Mix cell extracts with (a) 100 μL Wash buffer I; (b) protease and phosphatase inhibitor cocktails; and (c) the prewashed agarose-conjugated antibodies. Rotate tube for 4 h at 4 °C.
Spin-down agarose-conjugated antibodies (1 min, 220 × g, 4 °C), aspirate supernatant, resuspend with 500 μL wash buffer I, rotate tube 5 min at 4 °C, and centrifuge for 1 min (250 × g, 4 °C). Repeat step 7.
Resuspend agarose-conjugated antibodies with 500 μL wash buffer II (75 mM NaCl), Rotate tube 5 min at 4 °C, and centrifuge for 1 min (250 × g, 4 °C).
Resuspend agarose-conjugated antibodies with 15 μL 2× Laemmli buffer, denature 10 min at 95 °C and resolve by SDS-PAGE.
Process the gel for Western blotting (see Note 23).
4. Notes
Primers for generating nondegradable mutant of human Cyclin B1 were designed using the Agilent QuikChange Primer Design tool. Two missense mutations are introduced for substituting Arg 42 to Gly and Leu 44 to Val.
NDB cell system were generated based on 293T-REx™. Flp-In™ T-Rex™−293 cell system can be used instead (Thermo Fisher Scientific; #R78007).
Repetitive attempts to generate NDB cells based on T-REx™--HeLa cell line failed.
Either Tet or doxycycline can be used for activating expression of nondegradable Cyclin B1.
Derivates of HEK293 cells are easy to transfect. We used standard HeBSx2 CaCl2 transfection protocol for generating NDB cells. However, we expect any other transfection reagent to be successful.
Colony picking can be performed manually using cloning cylinder (e.g., PYREX® 6 × 8 mm Cloning Cylinders; # 3166–6). To this end, aspirate media, wash the plate with PBS, and dip the end of the cloning cylinder into sterile silicone grease before pressing to the bottom of a culture dish to create an isolated well around the colony. Trypsinize the cell colony with 2–3 μL Trypsin solution. After 1–2 min, collect cells and transfer into a 24-well-dish filled with 0.5 mL media and selection antibiotics. Alternatively, trypsinize cell colonies and utilize single cell sorting pipeline for sorting individual cells into a 96 well-dish filled with conditioned media (generated from 293T-REx™ cell culture) and selection antibiotics.
Tet solution is ethanol based. To minimize ethanol volume in the media, prepare a stock solution of 2.5–5 mg/mL. Higher Tet concentration might be more challenging to dissolve. After mixing Tet with 95% absolute ethanol, pipette vigorously or vortex. Centrifuge tube for 30 s (>250 × g, RT) to assure there is no pellet of undissolved material. If so, incubate in 37 °C water bath for 5 min before repeating the process until no pellet is detected. Note, Tet is considered a light-sensitive material. For preparation of NDB mitotic extract, we recommend using a freshly made Tet solution. As a default, we add 10 μL of 2.5 mg/mL Tet solution into a 150 mm/diameter plate containing 25 mL medium.
Tet-induced NDB cells arrest with high APC/CCdc20 activity. Consequently, the level of endogenous Cyclin B1 is low and the overall signal of Cyclin B1 at this stage results almost exclusively from the exogenous variant. Cell colonies exhibiting highest levels of nondegradable Cyclin B1 following mitotic arrest should be prioritized for further characterization.
NDB cells are always maintained in the presence of blasticidin and Zeocin™. However, to avoid unnecessary additional costs, there is no need to add these antibiotics during culture expansion for extract preparation.
Harvest mitotic arrested NDB cells by gentle pipetting using 25 or 50 mL pipette. Pipettes with smaller orifices may damage the cells. After centrifugation, resuspend pellet by pipetting slowly 2–3 times using 25 or 50 mL pipette.
During extract preparation, cells are shredded in a 10 mL beaker on ice. To minimize foaming: (a) tilt the beaker while shredding the cells; (b) pull syringe plunger back gently; (c) always leave a quarter of the liquid in the beaker; (d) maintain needle tip in liquid during the entire process; and (e) use blunt tip needles.
NDB extract cannot be reused once defrosted. We recommend storing extracts in aliquots of 45–65 μL in 0.2 mL PCR tubes.
NDB extracts remain active up to 6 months in −80 °C.
For expressing protein substrates in vitro we recommend using pCS2 or pCS-based vectors and the TNT® SP6 coupled reticulocyte lysate system. Protein translation was satisfactory in both premixed and non-premixed lysate systems (Promega; #L4600). Expression by T3 or T7 promoters is possible by matching TNT® systems from Promega.
Degradation and mobility shift assays can be performed with tagged IVT products (e.g., Myc or FLAG). Then, detection is based on immunoblot and primary antibodies coupled to a fluorophore, rather than autoradiography. A designated imaging system is required for signal quantification (e.g., Odyssaey®; LI-COR).
Mitotic NDB extracts are highly active. We often find recombinant ubiquitin and freshly added E-mix superfluous. Moreover, degradation assays in 10–15 μL extracts (instead of 20 μL) are almost equally informative (data not shown). In fact, extracts diluted two and even fourfold in swelling buffer can still support targeted proteolysis, though with slower kinetics (Fig. 2). These qualities save cell extracts.
For degradation and mobility shift assays we recommend sampling 3–5 μL for each time point. Lower amount saves reagents but with the cost of a fainter signal. On the flip side, higher sample volume may damage protein separation by SDS-PAGE due to overloading (5 μL reaction mix is about 100 μg total protein).
Enzymatic assays in cell free systems are highly sensitive to temperature shifts (Fig. 2). Higher temperature will shorten the assay but may lower the resolution of protein dynamics (Fig. 2). This can be critical in phosphorylation-mediated mobility shift assays (Fig. 1). On the flip side, increased temperature may compensate for lower activity associated with imperfect batch preparation, diluted extracts, or the lack of facilitating reagents (e.g., ubiquitin or E-mix).
We find mitotic NDB extracts to be more active than equivalent mitotic extracts from HeLa S3. In optimal assay conditions (see above), there is no real reason to extend degradation assays beyond 1 h. Within this time frame, time-intervals of 15 min are recommended (Figs. 1 and 2). Phosphorylation-mediated mobility shifts are considerably faster than proteolysis; a maximal mobility shift can be obtained within 15–30 min, if not faster. Oversampling during this time period is recommended for monitoring orderly phosphorylation (Fig. 1).
The default exposure time for autoradiography is one day. In case of a faint signal, increasing exposure time to 3 days is helpful. For the most part, short exposure for 1–2 h is informative though provide a less esthetic image.
Mitotic exit in mitotic arrested NDB cells and extracts is induced by RO-3306, that is, the most selective Cdk inhibitor to date. However, we expect roscovitine and flavopiridol to be equally effective for this purpose [5, 29].
Incubation time and RO-3306 concentration must be calibrated per extract batch by testing (a) the level of endogenous Cdc20 (an APC/CCdh1 target) in extracts before and after Cdk1 inactivation by RO-3306 using immunoblot. Cdc20 levels should drop during mitotic exit as APC/CCdh1 becomes active; (b) the potency of the extracts to support APC/CCdh1-mediated degradation of Tome-1 IVT product following mitotic exit (see Subheading 3.6); and (c) the switch between Cdc20 bound APC/C to Cdh1 bound APC/C by immunoprecipitation of Cdc27 (a core APC/C subunit) followed by immunoblot with anti-Cdc20/Cdh1 antibodies (see Subheading 3.7.3).
We recommend over-night incubation (4 °C) with anti-Cdc20 and anti-Cdh1 primary antibodies, and the use of IgG light chain-specific HRP-coupled secondary antibodies.
Acknowledgments
This study was by the Israel Cancer Research Fund (ICRF), Grant no. RCDA00102, and the Israel Science Foundation (ISF) Grant no. 659/16 and 2038/19. The Emanuele lab is supported by funds from the UNC University Cancer Research Fund, National Institutes of Health (R01GM120309, R01GM134231), American Cancer Society (RSG-18-220-01-TBG) and donations from the Brookside Foundation.
References
- 1.Morgan DO (2007) The Cell Cycle, Principles of Control. New Science Press, 2007. 297 pp. ISBN: 978-0-9539181-2-6 [Google Scholar]
- 2.Sudakin V, Ganoth D, Dahan A et al. (1995) The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell 6:185–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.King RW, Peters JM, Tugendreich S et al. (1995) A 20s complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell 81:279–288 [DOI] [PubMed] [Google Scholar]
- 4.Zou H, McGarry TJ, Bernal T et al. (1999) Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science 285:418–422 [DOI] [PubMed] [Google Scholar]
- 5.Zur A (2001) Securin degradation is mediated by fzy and fzr, and is required for complete chromatid separation but not for cytokinesis. EMBO J 20:792–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kernan J, Bonacci T, Emanuele MJ (2018) Who guards the guardian? Mechanisms that restrain APC/C during the cell cycle. Biochim Biophys Acta Mol Cell Res 1865(12):1924–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barford D (2020) Structural interconversions of the anaphase-promoting complex/cyclosome (APC/C) regulate cell cycle transitions. Curr Opin Struct Biol 61:86–97 [DOI] [PubMed] [Google Scholar]
- 8.Yamano H (2019) APC/C: current understanding and future perspectives. F1000Res 8: F1000 Faculty Rev-725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vorlaufer E, Peters JM (1998) Regulation of the cyclin B degradation system by an inhibitor of mitotic proteolysis. Mol Biol Cell 9:1817–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buendia B, Draetta G, Karsenti E (1992) Regulation of the microtubule nucleating activity of centrosomes in Xenopus egg extracts: Role of cyclin A-associated protein kinase. J Cell Biol 116:1431–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shamu CE, Murray AW (1992) Sister chromatid separation in frog egg extracts requires DNA topoisomerase II activity during anaphase. J Cell Biol 117:921–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Masui Y (1992) Towards understanding the control of the division cycle in animal cells. Biochem Cell Biol 70(10–11):920–945 [DOI] [PubMed] [Google Scholar]
- 13.Funabiki H, Murray AW (2000) The Xenopus chromokinesin Xkid is essential for metaphase chromosome alignment and must be degraded to allow anaphase chromosome movement. Cell 102:411–424 [DOI] [PubMed] [Google Scholar]
- 14.Murray AW, Desai AB, Salmon ED (1996) Real time observation of anaphase in vitro. Proc Natl Acad Sci U S A 93:12327–12332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen PA, Groen AC, Loose M et al. (2014) Spatial organization of cytokinesis signaling reconstituted in a cell-free system. Science (80) 346:244–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGarry TJ, Kirschner MW (1998) Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell 93:1043–1053 [DOI] [PubMed] [Google Scholar]
- 17.Ayad NG, Rankin S, Ooi D, et al. (2005) Identification of ubiquitin ligase substrates by in vitro expression cloning. Methods Enzymol. 399:404–414 [DOI] [PubMed] [Google Scholar]
- 18.Ayad NG, Rankin S, Murakami M et al. (2003) Tome-1, a trigger of mitotic entry, is degraded during G1 via the APC. Cell 113:101–113 [DOI] [PubMed] [Google Scholar]
- 19.Rankin S, Ayad NG, Kirschner MW (2005) Sororin, a substrate of the anaphase-promoting complex, is required for sister chromatid cohesion in vertebrates. Mol Cell 18:185–200 [DOI] [PubMed] [Google Scholar]
- 20.Nguyen H, Gitig DM, Koff A (1999) Cell-free degradation of p27 kip1, a G 1 cyclin-dependent kinase inhibitor, is dependent on CDK2 activity and the proteasome. Mol Cell Biol 19:1190–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rape M, Kirschner MW (2004) Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature 432:588–595 [DOI] [PubMed] [Google Scholar]
- 22.Pe’er T, Lahmi R, Sharaby Y et al. (2013) Gas2l3, a novel constriction site-associated protein whose regulation is mediated by the APC/CCdh1 complex. PLoS One 8(2): e57532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen M, Vecsler M, Liberzon A et al. (2013) Unbiased transcriptome signature of in vivo cell proliferation reveals pro- and antiproliferative gene networks. Cell Cycle 12:2992–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wasserman D, Nachum S, Cohen M et al. (2020) Cell cycle oscillators underlying orderly proteolysis of E2F8. Mol Biol Cell 2020: mbcE19120725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glotzer M, Murray AW, Kirschner MW (1991) Cyclin is degraded by the ubiquitin pathway. Nature 349:132–138 [DOI] [PubMed] [Google Scholar]
- 26.Zur A, Brandeis M (2002) Timing of APC/C substrate degradation is determined by fzy/fzr specificity of destruction boxes. EMBO J 21:4500–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Panet E, Ozer E, Mashriki T et al. (2015) Purifying cytokinetic cells from an asynchronous population. Sci Rep 5:13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng X, King RW (2012) An APC/C inhibitor stabilizes cyclin B1 by prematurely terminating ubiquitination. Nat Chem Biol 8:383–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Potapova TA, Daum JR, Pittman BD et al. (2006) The reversibility of mitotic exit in vertebrate cells. Nature 440:954–958 [DOI] [PMC free article] [PubMed] [Google Scholar]