

Abstract
Citrullination is an enzyme-catalyzed post-translational modification (PTM) that is essential for a host of biological processes including gene regulation, programmed cell death, and organ development. While this PTM is required for normal cellular functions, aberrant citrullination is a hallmark of autoimmune disorders as well as cancer. Although aberrant citrullination is linked to human pathology, the exact role of citrullination in disease remains poorly characterized, in part because of the challenges associated with identifying the specific arginine residues that are citrullinated. Tandem mass spectrometry is the most precise method to uncover sites of citrullination, however, due to the small mass shift (+0.984 Da) that results from citrullination, current database search algorithms commonly misannotate spectra leading to a high number of false-positive assignments. To address this challenge, we developed an automated workflow to rigorously and rapidly mine proteomic data to unambiguously identify the sites of citrullination from complex peptide mixtures. The crux of this streamlined workflow is the ionFinder software program, which classifies citrullination sites with high confidence based on the presence of diagnostic fragment ions. These diagnostic ions include the neutral loss of isocyanic acid, which is a dissociative event that is unique to citrulline residues. Using the ionFinder program, we have mapped the sites of autocitrullination on purified protein arginine deiminases (PADs 1-4) and mapped the global citrullinome in a PAD2 over-expressing cell line. The ionFinder algorithm is a highly versatile, user-friendly, and open-source program that is agnostic to the type of instrument and mode of fragmentation that is used.
Graphical Abstract
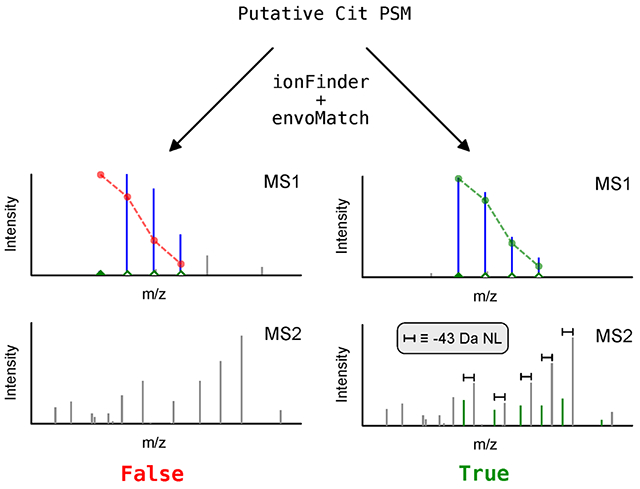
Introduction
Protein citrullination is an enzyme-catalyzed post-translational modification (PTM) that converts an arginine (Arg) residue into citrulline (Cit) (Figure 1a).1, 2 This irreversible hydrolysis reaction is catalyzed by the Protein Arginine Deiminase (PAD) family of enzymes. There are five PAD isozymes in humans and other mammals, of which, only PADs 1-4 are catalytically active. Of note, PAD activity requires high, micromolar levels of calcium.
Figure 1. Arginine citrullination and isocyanic acid dissociation.
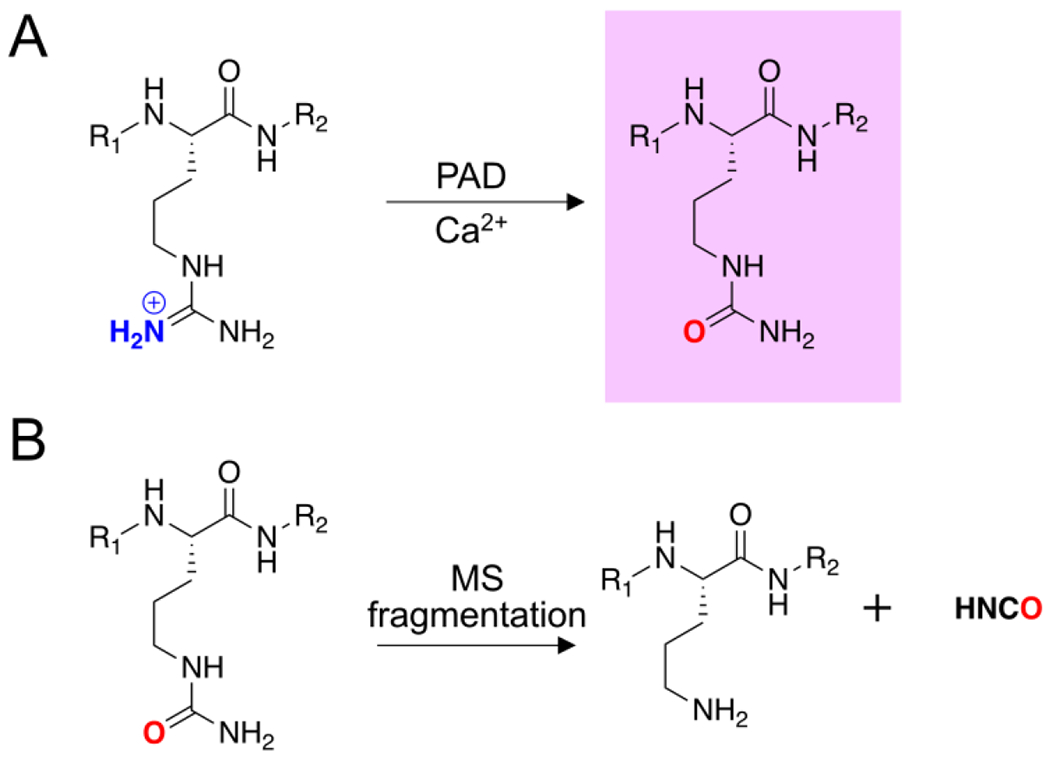
A) PAD-catalyzed conversion of an arginine residue into Cit in the presence of calcium. B) MS fragmentation results in the neutral loss of isocyanic acid from Cit.
Aberrant PAD expression and citrullination is a hallmark of rheumatoid arthritis (RA).3 In fact, patients with RA generate autoantibodies that target citrullinated proteins, and the occurrence of these anti-citrullinated protein antibodies (ACPAs) is the most specific diagnostic for RA. Antibodies targeting the citrullinated forms of vimentin, α-enolase, and fibrin correlate with different RA subtypes,4 and higher ACPA titers correlate with disease onset and severity.5 In addition to RA, dysregulated PAD activity is associated with lupus, ulcerative colitis, sepsis, and idiopathic lung fibrosis.6–11 The exact role of citrullination in driving the pathogenicity of such disparate diseases is not firmly established due to the challenges associated with identifying citrullinated proteins from complex biological samples.
Given the important role of protein citrullination in diverse disease pathologies, numerous chemical probes and mass-spectrometry (MS) strategies have been developed to enrich and identify citrullinated proteins.2, 12–14 Selective chemical derivatization of the urea group in Cit can be achieved with reagents such as 2,3-butanedione and phenylglyoxal.15 Phenylglyoxal derivatives functionalized with rhodamine and biotin have enabled the visualization and enrichment of citrullinated proteins and peptides.13, 16–18 However, assignment of the exact sites of citrullination within these proteins has been confounded by unpredictable fragmentation patterns and poor fragmentation efficiency of peptides containing the phenylglyoxal adduct. Alternatively, the presence of Cit can be detected directly by the +0.984 Da mass shift that occurs upon deimination. However, the small mass shift, coupled with the low abundance of citrullinated peptides within a proteome, necessitate the use of stringent filtering criteria to limit false-positive assignments. Specifically, false-positive identifications can result from: (1) the incorrect assignment of the monoisotopic peak within the isotopic distribution for a given peptide, and (2) the presence of a deamidated glutamine (Gln) and/or asparagine (Asn) within a peptide, which results in a mass shift that is identical to that expected for Cit.
Several strategies have been developed to minimize the number of false-positive hits resulting from database searches for citrullinated peptides. The simplest method is to use stringent precursor mass tolerances of 5 ppm to minimize the incorrect assignment of the monoisotopic species.19 However, this approach does not account for misannotations driven by the presence of deamidated Gln and Asn. A second approach utilizes a dual database searching strategy, where searches are performed with and without a +0.984 differential modification on Arg, Asn, and Gln. Peptides that score higher with the differential modification on Arg, relative to Asn and Gln, are considered true matches.19, 20 A third approach is to specifically search for diagnostic fragment ions that are unique to citrullination. These diagnostic ions include the immonium ion of Cit, and the −43.0058 Da neutral loss of isocyanic acid (HNCO) from Cit that occurs upon peptide fragmentation (Figure 1b). In particular, the isocyanic acid neutral losses can be used as diagnostic fragment ions for the unambiguous detection of a Cit-containing peptide14, 21, 22 because the fragmentation of peptides containing deamidated Gln and/or Asn do not generate similar neutral losses. Several recent studies have exploited this Cit-specific neutral loss to identify citrullinated peptides. For example, one study developed a logistic regression model to determine sites of citrullination based on the presence of predictive diagnostic ions, including the Cit-specific neutral loss species.23 In a second study by Lee et al, a library of ~2200 citrullinated peptides were chemically synthesized as reference spectra to unambiguously differentiate citrullinated peptides from their corresponding deamidated counterparts. Manual comparisons of the experimental spectra to the synthetic standards enabled the high-confidence assignment and validation of citrullinated peptides from 30 human tissues. Additionally, these studies confirmed that the neutral loss of isocyanic acid from fragment ions was a key characteristic required for assignment of sites of citrullination with high confidence.14 The requirement to detect neutral loss fragment ions was subsequently applied in a study by Salinger et al to identify sites of citrullination from neutrophil extracellular traps (NETs).24 Both the Lee et al and Salinger et al studies confirmed the importance of applying stringent criteria to minimize false-positives in citrulline assignments, but relied on labor-intensive manual assessment of fragmentation spectra to identify determining neutral loss peaks. A recent study by Chaerkady et al25 identified 833 citrullination sites in ionomycin-treated neutrophil and mast cells using neutral-loss ion-based searches in MaxQuant. Importantly, the generated data were then used to benchmark a machine-learning model to predict sites of citrullination to further facilitate sequence-based prediction of novel citrullination events.
Herein, we report on a streamlined computational workflow for assigning sites of citrullination with high confidence, based on two newly developed algorithms, termed ionFinder and envoMatch. The first algorithm, ionFinder, can rapidly identify Cit-containing peptides from tandem MS data by identifying the presence of diagnostic neutral loss ions. The second algorithm, envoMatch, automates the matching of isotopic envelopes to confirm that the monoisotopic species displays the required +0.984 Da mass shift. Both algorithms are now available as open-source programs. The ionFinder program is designed to be implemented downstream of the database-searching step of standard proteomic workflows to differentiate between high-confidence and low-confidence identifications of Cit-containing peptides. As such, ionFinder is compatible with the outputs of common database-searching algorithms, including SEQUEST, Mascot and MaxQuant, and functions on both collision-induced dissociation (CID) and high-energy collisional dissociation (HCD) fragmentation datasets. The accuracy of ionFinder was evaluated using fragmentation spectra from verified Cit-containing, and non-Cit containing, synthetic peptides generated in the study by Lee et al14 as well as proteomic data from human cells and tissues.14, 25 To demonstrate the versatility of the ionFinder and envoMatch workflow, we comprehensively mapped the sites of autocitrullination in all four active PAD isozymes. Moreover, we used this workflow to map the citrullinome of cells expressing PAD2, resulting in the identification of over 350 unique Cit-containing peptides from 220 proteins.
Materials and Methods
Expression and Purification of PAD1,2, & 3
PAD1, PAD2, and PAD3 were expressed and purified similarly to previously described methods.26, 27 Briefly, E. coli BL21(DE3) were transformed with a plasmid encoding a PAD protein with an N-terminal 10X-His tag. Single colonies were used to inoculate a 5 mL starter culture that was grown overnight in LB (0.1 mg/mL ampicillin, 0.025 mg/mL chloramphenicol) agitating at 37 °C. The starter culture (1 mL) was used to inoculate 1L of pre-warmed LB media (0.1 mg/mL ampicillin, 0.025 mg/mL chloramphenicol). Expression cultures were grown to an OD600 of 0.6-0.8. Flasks were cooled on ice for 20 min before expression was induced by the addition of 0.1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG). Cultures were agitated overnight at 16 °C, and cell pellets were harvested via centrifugation and frozen in liquid nitrogen. Frozen cells pellets were stored at −80 °C.
To lyse the cells, pellets were thawed in a water bath at room temperature. Per each g of frozen cell pellet, 1 mL of lysis buffer (20 mM Tris pH 7.6, 400 mM NaCl, 5 mM MgCl2, 5 mM imidazole, 0.5 mM TCEP, 1% Triton X-100) was added in addition to Pierce™ EDTA-free protease inhibitor tablets. The slurry was agitated for 30 min after which the mixture was spiked with universal nuclease (Promega). The mixture was briefly sonicated before clarifying the lysate of insoluble debris. Soluble lysate was applied to Ni-NTA resin via gravity. The resin was washed thoroughly with a buffer consisting of 20 mM Tris pH 7.6, 400 mM NaCl, 20 mM imidazole, 0.5 mM TCEP, and 10% (w/v) glycerol. PAD protein was eluted with wash buffer containing 250 mM imidazole. Purity was assessed via SDS-PAGE and Coomassie blue staining. The eluted protein was dialyzed overnight into a buffer of 20 mM Tris pH 7.6, 500 mM NaCl, 0.5 mM TCEP, and 10% (w/v) glycerol. Protein concentration was measured using the Bradford Assay.
Recombinant PAD4 Expression and Purification
PAD4 was expressed and purified as described previously28 with the following alterations: after the GST tag was removed with Precision Protease, PAD4 protein was dialyzed overnight against a buffer consisting of 20 mM Tris pH 7.6, 100 mM NaCl, 1 mM EDTA, 10% (w/v) glycerol, and 2 mM DTT. This low-salt buffer caused full length PAD4 to precipitate, whereas the GST tag and GST-tagged PAD4 remained in solution. The precipitate was collected via centrifugation in a 50 mL Falcon tube and washed 2 X with 20 mL of the low-salt buffer. Finally, pure PAD4 was resolubilized gently in 3 mL of buffer consisting of 20 mM Tris pH 8.1, 500 mM NaCl, 1 mM EDTA, 10% (w/v) glycerol, and 2 mM DTT. Purity was assessed via SDS-PAGE and Coomassie blue staining. Protein concentration was measured using the Bradford Assay.
Preparation of Purified Proteins for Tandem MS Analysis
Purified PADs (30 μg) were autocitrullinated for 1 h at 37 °C in 100 μL of buffer consisting of 100 mM HEPES pH 7.6, 100 mM NaCl, 1 mM TCEP, and 5 mM CaCl2. Autocitrullination was stopped by the addition of trichloroacetic acid to a final concentration of 20% (w/v). Reactions were placed on ice for 1 h to promote protein precipitation. Sample tubes were centrifuged in a 4 °C tabletop centrifuge at top speed for 15 min. The precipitant was washed with 300 μL cold acetone, which was then removed via pipette after another 10 min centrifugation at top speed. The pellets were air dried and then resuspended in a solution of 8 M urea in PBS (30 μL). Once solubilized, 100 mM ammonium bicarbonate (70 μL) was added to dilute the urea concentration to 2.4 M. The samples were then reduced by the addition of 1 M DTT (1.5 μL), and further denatured by placing them at 65 °C for 15 min. Reduced cysteines were then alkylated with iodoacetamide (12.5 mM final concentration) at 21 °C in the dark for 30 min. The urea concentration was then further diluted by the addition of PBS (120 μL). Both GluC and LysC (Promega) were used in combination (1:30 ratio, enzyme:substrate). Trypsin was used both in combination with GluC and alone (1:50, enzyme:substrate) in the presence of 1 mM CaCl2. Digests were agitated by rotating at 37 °C overnight. Protein digestion was stopped by the addition of formic acid to 5% (v/v). Peptides were then desalted using Pierce™ C18 spin columns and dried to a powder. After resuspension in water, the peptide concentration was assessed using a Pierce™ quantitative fluorometric peptide assay.
HEK-PAD2 Cell Culture and Lysate Prep
HEK293T cells that stably express full length PAD2 (HEK-PAD2 cells)13 were propagated according to previously described methods.13 Briefly, cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. The cultures were maintained in a humidified atmosphere with 5% CO2 at 37 °C. Cellular citrullination was induced according to previously described methods.13 Briefly, cells were grown to passage 5 on T-175 plates until ~90% confluency. They were then treated with Ca2+ (2 mM final concentration) and ionomycin (5 µM final concentration) for 1 h. After incubation, cells were scraped, washed with cold PBS, and then snap-frozen. Pellets were stored at −80 °C until lysis. To lyse the cells, pellets were thawed on ice in 5 mL of buffer consisting of PBS, 5 mM EDTA, 1% Triton X-100, and Pierce protease inhibitor. Lysis was achieved in an ice bath via iterative rounds of sonication using a Sonic Dismembrator (Fisher Scientific) fitted with a microtip (amplitude 10, 10x1 sec bursts with 30 sec of rest between cycles, 4 cycles total). Lysates were cleared by centrifugation, and the protein concentration assessed via the DC assay.
Preparation of HEK-PAD2 Lysate for Tandem MS Analysis
Cell lysate (50 μg) was precipitated on ice for 30 min by the addition of trichloroacetic acid to 20% (w/v). Sample tubes were centrifuged in a 4 °C tabletop centrifuge at top speed for 15 min. The precipitate was washed with 300 μL cold acetone, which was then removed via pipette after another 10 min centrifugation at top speed. The pellets were air dried and then resuspended in a solution of 8 M urea in PBS (30 μL). Once solubilized, 100 mM ammonium bicarbonate (70 μL) was added to dilute the urea concentration to 2.4 M. The samples were then reduced by the addition of 1 M DTT (1.5 μL), and further denatured by placing them at 65 °C for 15 min. Reduced cysteines were then alkylated with iodoacetamide (12.5 mM final concentration) at 21 °C in the dark for 30 min. The urea concentration was then further reduced by the addition of PBS (120 μL). Trypsin was used at a concentration of 1:50 (enzyme:substrate) in the presence of 1 mM CaCl2, and samples were proteolyzed overnight agitating by rotation at 37 °C. After digestion, tryptic peptides were separated on a ZORBAX extended C18 column (Agilent) over a 1 h, biphasic gradient from 0% Buffer A (10 mM ammonium bicarbonate) to 100% Buffer B (10 mM ammonium bicarbonate, 90% acetonitrile). Fractions (0.5 mL) were collected in a 96 well plate and pooled by column (as opposed to by row) to yield 12 samples. The 12 samples of pooled fractions were dried in a vacuum concentrator and resuspended in 5% acetonitrile/0.1% trifluoroacetic acid.
Tandem MS
Peptide mixtures were separated on a NanoAcquity UPLC (Waters Corporation, Milford, MA) using an in-house packed pre-column (C18, 200A, 5µm, 2cm) and an in-house packed analytical column (C18, 100A, 3 µm, 25 cm) using the aqueous mobile phase of water + 0.1 % formic acid (A) and an organic mobile phase of acetonitrile + 0.1 % formic acid (B). Peptide trapping was operated at 4 min at 4 µL/min and 5% B. Then the peptides were transferred to the analytical column at the flow rate of 300 nL/min using the gradient of 5% to 35% B over 60 minutes, then 35% to 60% B for 30 min, followed by 15 min of high organic wash of 90% B and 18 min of 5% B for re-equilibration.
Ions were then introduced to a Q Exactive hybrid quadrupole-Orbitrap (Thermo Fisher Scientific Inc., Waltham, MA) mass spectrometer, performing at positive electrospray ionization (ESI+) with the ionization voltage set at 1.4 kV. The full MS (MS1) data scan was acquired in a m/z scan range of 300-1750 Da, using an AGC target of 1e6, the maximum injection time of 30 ms and a resolution of 70000 at m/z 200. The MS/MS (MS2) data were acquired in data-dependent acquisition mode, performing MS/MS on the top 10 most abundant precursor ions using an AGC target of 1e5, a maximum injection time of 110 ms, an isolation width of 1.6 Da, a resolution of 17500 at m/z 200, and a collision energy of 27 volts using higher-energy collisional dissociation fragmentation (HCD).
MS Data Analysis
The LC-MS/MS raw data were processed using Thermo Proteome Discoverer (PD) 2.1.1.21 (Thermo Fisher Scientific Inc.). The data obtained for the autocitrullinated PADs was searched against an E. coli SwissProt database FASTA file that included the sequences corresponding to the PAD proteins, and the HEK-PAD2 samples were searched against the Human SwissProt database. Both searches were performed using Mascot Server 2.6.2 (Matrix Science Ltd). The search parameters included searching specific protease cleavage sites with 2 maximum missed cleavages. Carbamidomethyl cysteine modifications were set as a fixed modification, while variable modifications included: peptide N-terminal acetylation, methionine oxidation, N-terminal glutamine to pyroglutamate, and citrullination of arginine. In addition to these modifications, the deamidation of Asn and Gln was set as variable modifications. A 10 ppm m/z cutoff was employed for the precursor mass and 0.05 Da for the fragment ion mass tolerance.
Protein identification and validation was done using Scaffold 4.10.0 (Proteome Software Inc.), employing 1% FDR threshold for peptides, and a 99% probability threshold for protein identification, using Peptide Prophet and Protein Prophet algorithms.29, 30 The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE31 partner repository with the dataset identifier PXD027358.
Automated assignment of “true” citrullinated residues using ionFinder
ionFinder was written in c++, utilizing routines from the MSToolkit library for mzXML and mzML file parsing.32 Results were exported from Scaffold in the form of a spectrum report and converted into a format suitable for ionFinder using a custom python script. After reading the required input files, ionFinder analyzes peptide-spectrum matches (PSM) in 3 major steps. In the first step, PSM assigned by PD are re-searched by ionFinder for a customized list of theoretical fragment ions. For each peptide, a list of masses for theoretical b and y ions are calculated. In addition, b and y −43.0058 Da neutral loss (NL) ion masses are calculated for citrullination up to the multiplicity of citrulline residues on the peptide. In this work, the MS2 spectra were charge deconvoluted prior to searching with PD, so that only singly charged fragment ions were considered. Each MS2 spectrum was searched for theoretical fragment ions within a 10 ppm m/z tolerance. If multiple ions are found in the specified range, ties are broken by intensity. In cases where multiple fragments have the same predicted mass, all possible fragments are considered found if the ion is found in the MS2 spectrum.
In the second analysis step, fragment ions which were identified in the spectrum are classified into 1 of 5 categories with respect to how they provide evidence supporting or contradicting the citrullination of a given peptide. These assignments were based on the decision tree shown in Figure 2a. Unmodified b or y ions which do not contain any citrullines are classified as ambiguous (Amb); fragment ions which are citrullinated, but also contain N or Q will also be classified as Amb; b or y ions which contain Cit are classified as Cit determining (Det). NL ions which unambiguously belong to a Cit containing fragment are classified as Cit determining NL (DetNL); NL ions on multiply modified peptides which cannot unambiguously be assigned to 1 or both modifications are classified as ambiguous (Amb); if a NL is observed for an ion which does not contain Cit it is classified as artifact NL (ArtNL). ArtNL ions in effect function as decoy fragment ions because they would not be expected to be observed in the fragmentation spectrum in significant abundance. Once each fragment ion has been classified, a dynamic ion intensity cutoff is set such that the percentage of ArtNL ion intensities in the spectrum is less than 1%. The sum of intensities for ion type t, above cutoff k is defined as:
| (1) |
where ai,t is the intensity for an ion i of type t (out of n ions of type t). The fraction of intensity from ArtNL ions out of all ion types is defined as:
| (2) |
k is set such that PArtNL ≤ 0.01. If PArtNL is already less than 1% at a k of 0, no intensity cutoff is applied. If PArtNL > 0.01, the value of k is determined by iterating through the intensities of ArtNL ions in ascending order.
Figure 2. Decision trees and filtering criteria for ionFinder.
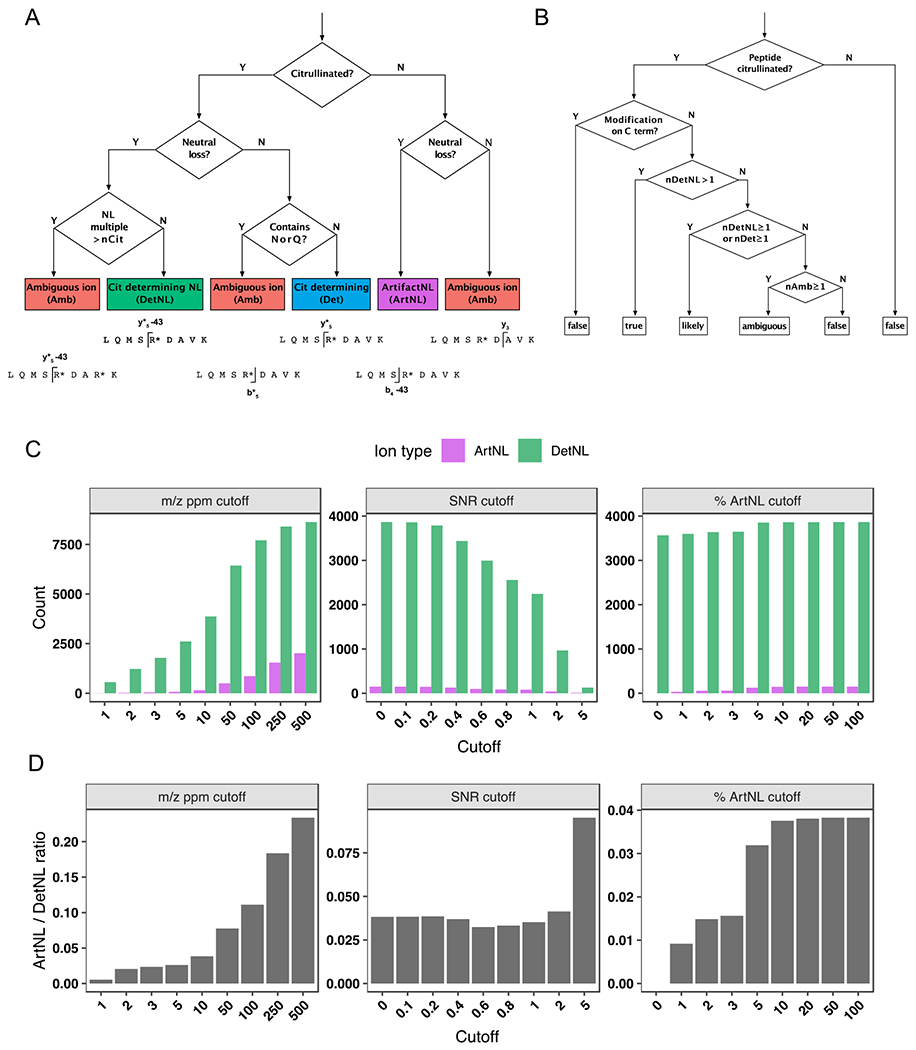
A) A decision tree depicting how each fragment ion within an MS/MS spectrum is binned into one of five categories: a citrulline-determining fragment (Det); (2) a citrulline-determining neutral loss fragment (DetNL); (3) an artifact neutral loss fragment (ArtNL); and, (4) an ambiguous fragment (Amb). B) A decision tree depicting how each PSM is assigned categories of “true”, “likely”, “ambiguous” and “false”, based on the number of DetNL, Det, and Amb fragments. C) Bar graphs depicting DetNL and ArtNL fragments upon varying the m/z ppm cutoff value (left panels), signal-to-noise cutoff (middle panels), and a dynamic % ArtNL cutoff (right panel). The data are depicted as total counts of fragments. D) Bar graphs depicting DetNL and ArtNL fragments upon varying the m/z ppm cutoff value (left panels), signal-to-noise cutoff (middle panels), and a dynamic % ArtNL cutoff (right panel). The data are depicted as ArtNL/DetNL ratios (bottom panels).
In the third analysis step, the number of ions observed in each of the 5 classes are used to assign each site of citrullination as “true”, “likely”, “ambiguous”, or “false” according to the decision tree in (Figure 2b). A classification of true, likely, ambiguous, or false is assigned to each site on multiply-citrullinated peptides individually, then the value assigned to the peptide is the lowest classification of all sites; i.e. a doubly citrullinated peptide will only be classified as “true”, if both sites are classified as “true”.
Automated verification of precursor isotopic envelopes with envoMatch
envoMatch is written in Python and utilizes routines from the Pyteomics33 package for calculation of isotopic envelopes and mzXML and mzML file parsing. envoMatch compares the observed isotopic envelope for the precursor peptide with the theoretical envelope of the PSM peptide. For a peptide with n citrulline residues, theoretical envelopes of a peptide with {n, n − 1, …, 0} citrulline residues are calculated. An envelope similarity score comparing the observed envelope to each theoretical isotopic envelope is generated using a Pearson correlation. A peptide is considered to have a “true” site of citrullination if the best match was for the envelope with n cit residues and if the similarity score is greater than 0.8.
Software availability
ionFinder and envoMatch are open-source software and are distributed under MIT licenses. Source code, installation instructions, and software manuals with instructions for customizing the search settings including the mass of the neutral loss, can be found at their respective GitHub repositories. ionFinder (https://github.com/weerapana-lab/ionFinder), and envoMatch (https://github.com/weerapana-lab/envoMatch).
Results
Development and validation of ionFinder.
To facilitate the unambiguous detection of citrulline and automate the process of validating peptide-spectrum matches (PSMs) of citrullinated peptides assigned by database-searching algorithms, we developed a software package named ionFinder. As a starting point for ionFinder, we adapted the empirically determined rules developed by Lee et al,14 where the first step in the workflow was the elimination of peptides annotated as having a C-terminal Cit, because citrullinated Arg residues no longer serve as a recognition element for trypsin. In subsequent filtering steps, a requirement for diagnostic neutral loss (NL) peaks in the fragmentation spectra was instituted to unambiguously assign sites of citrullination. As a first step to automating the identification of NL species, we developed a decision tree to categorize diagnostic fragment ions for peptides annotated to contain Cit. For a given MS/MS spectra, each fragment ion is classified into 1 of 4 categories (Figure 2a): (1) a Cit-determining fragment ion (Det), where a +0.984 Da mass shift from the unmodified Arg peptide can be assigned to a Cit-containing peptide fragment that does not contain Asn or Gln; (2) a Cit-determining neutral loss fragment ion (DetNL), where a Cit-containing peptide fragment shows a −43.0058 Da neutral loss; (3) an artifact neutral loss fragment ion (ArtNL), where a −43.0058 Da neutral loss is observed for a peptide fragment that does not contain Cit; and, (4) an ambiguous fragment ion (Amb), which lacks diagnostic peptide fragments to confidently assign the presence or absence of Cit.
After classifying the fragment ions from each MS/MS scan into the different categories, the number of each type of fragment ion (Det, DetNL, ArtNL, and Amb) is then used to assign a value of “true”, “likely”, “ambiguous”, or “false” to each peptide identification according to a second decision tree (Figure 2b). Spectra containing two or more DetNL fragment ions are classified as “true”, whereas spectra with no Det or DetNL fragment ions are classified as “false”. Spectra containing a single DetNL fragment ion, or any number of Det fragment ions, are classified as “likely”. Lastly, spectra containing no Det or DetNL, and only Amb fragment ions, are listed as “ambiguous”. For example, if a peptide is annotated as containing a single Cit, ionFinder rapidly searches the fragmentation data and automates the categorization of each PSM into the “true”, “likely”, “ambiguous”, or “false” categories. For those peptides that contain multiple annotated Cit residues, the categorization of “true”, “likely”, “ambiguous”, or “false” is given to each individual site, and then the value assigned to the peptide as a whole is the lowest classification of all sites. For example, a doubly citrullinated peptide will only be classified as “true” if both individual Cit sites are categorized as “true”.
To validate the utility of ionFinder, we first applied this algorithm to a previously described dataset14 that includes Cit-containing synthetic peptides, as well as non-Cit containing peptide controls. Specifically, we randomly selected fragmentation spectra from 100 Cit-containing peptides (validated “true” hits), and 100 non-Cit containing peptides that contain one or more deamidated Asn or Gln residues (validated “false” hits). Upon initial application of ionFinder to this dataset, we observed numerous ArtNL fragments, where a −43.0058 Da neutral loss was observed for fragments that did not contain a Cit residue. Upon manual inspection, many of these ArtNL fragment peaks displayed relatively low intensity, compared to the majority of DetNL peaks. Moreover, these ArtNL fragments were a very small fraction of the total NL species, where DetNL fragments clearly dominated (Figure 2c, 2d). Nevertheless, we sought to eliminate these ArtNL fragments because the presence of a NL species is a critical component of our Cit assignment. Initially, we evaluated the use of m/z ppm cutoffs, as well as signal-to-noise cutoffs, to minimize the number of ArtNL fragments and provide greater confidence in the assignment of DetNL fragments. However, the use of more stringent ppm and signal-to-noise cutoffs not only reduced the ArtNL fragment counts, but also led to a corresponding decrease in total fragment counts (Figure 2c, d). Consequently, we evaluated the use of a dynamic ion-intensity cutoff, wherein the cutoffs are set to self-adjust to provide an abundance of ArtNL fragments below a specified percentage of the total abundance of all other fragment types. We tested several potential cutoffs before establishing that a 1% threshold reduced the number of ArtNL fragments significantly, without altering the number of Det and DetNL fragments (Figure 2c, d). In summary, the use of these criteria to minimize ArtNL fragments provides higher confidence of NL peak assignments that are used as diagnostic species for the presence of Cit residues.
When this optimized version of ionFinder was applied to the verified “true” and “false” spectra from the study by Lee et al,14 77 of the 100 “true” spectra were automatically assigned as “true” by ionFinder (Figure 3a; Table S1). By contrast, only 11 of the 100 “false” spectra were assigned as “true” by ionFinder. Examination of the annotated fragmentation spectra confirmed that the “true” spectra contained high-intensity NL peaks, whereas the annotated NL peaks in the “likely” assignments were of low intensity and precluded high-confident assignment of that PSM as a definitive Cit-containing peptide (Figure 3b; Figures S1–S7).
Figure 3. Verifying the accuracy of ionFinder and envoMatch using an established “true” and “false” dataset.
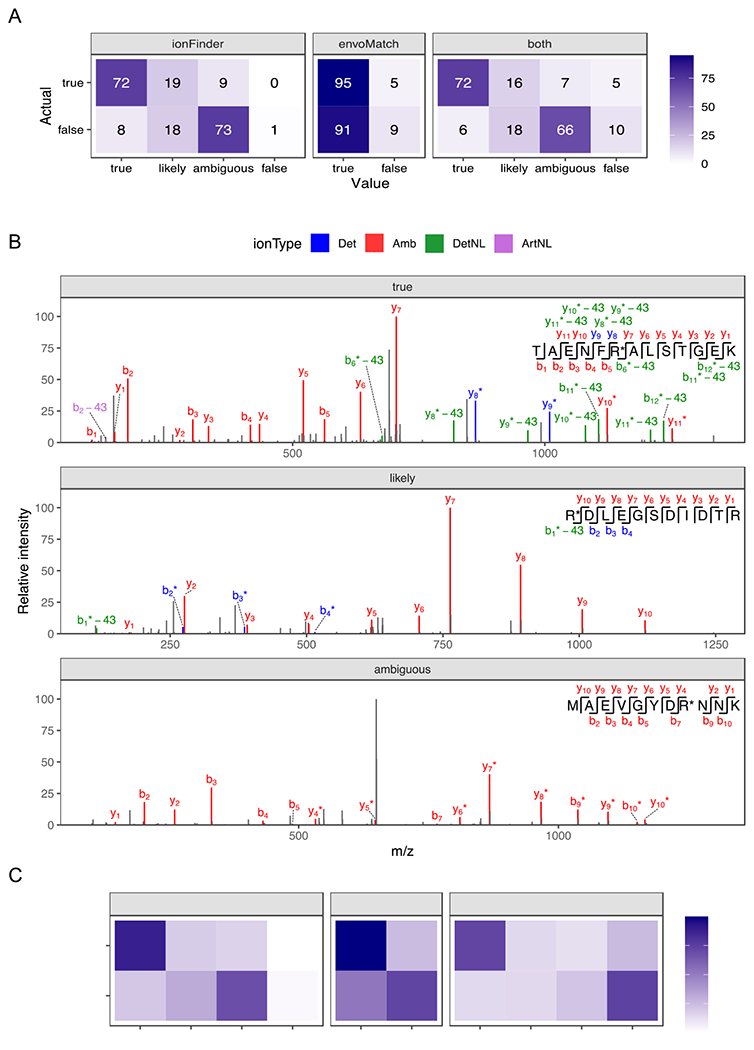
A) Fragmentation spectra from 100 Cit-containing and 100 non-Cit containing synthetic peptides that contain confirmed deamidation events from Lee et al,14 were analyzed by ionFinder, envoMatch, or both. The number of verified “true” and “false” hits that fell into each category by ionFinder and envoMatch are shown. B) Representative fragmentation spectra from peptides classified as “true”, “likely”, and “ambiguous” by ionFinder. C) 2644 PSMs from a complex proteomic analysis were analyzed by ionFinder, envoMatch, or both. The number of verified “true” and “false” hits that fell into each category by ionFinder and envoMatch are shown.
We then expanded our test dataset to include deep proteomic data from human tissues used in the study by Lee et al14, where Cit-containing peptides were assigned by comparing to synthetic standards. We analyzed 2644 PSMs from these proteomic datasets, of which 456 and 2188 were classified as true and false, respectively. Analysis of these 2644 PSMs with ionFinder resulted in ~71% of the verified hits being classified as “true”, while only ~17% of the false hits were annotated to be “true” (Figure 3c).
To further improve the reliability of our assignments, we developed an isotopic envelope matching algorithm, termed envoMatch, that would verify the proper monoisotopic peak identification of Cit-containing peptides. Note that isotopic envelope matching was excluded from the analysis by Lee et al.,14 but successfully employed by Salinger et al.24 to identify Cit-containing peptides from NETs. In this previous study, isotopic envelope matching was performed by using commercial software to predict MS1 chromatograms for both Arg- and Cit-containing peptides. Then, subsequent manual comparisons of the experimental and predicted envelopes were performed to determine an appropriate fit. The envoMatch program automates this process by comparing experimental isotopic envelopes to predicted Arg- and Cit-containing isotopic envelopes. The program then assigns a value of “true” or “false” quantitatively based on a Pearson correlation between the experimental and predicted isotopic envelopes. When the synthetic sample datasets were analyzed, envoMatch correctly classified the Cit-containing isotopic envelopes with ≥95% accuracy (Figure 3A). As expected, 95% of the “false” dataset was also classified as “true”, because these “false” peptides were all deaminated, and therefore have the same isotopic distribution as the citrullinated peptide. Isotopic envelope verification through envoMatch becomes more important when analyzing complex proteomic samples where there is a higher likelihood of observing co-eluting isobaric species. When the 2644 PSMs from the complex proteomic sample were analyzed by both ionFinder and envoMatch, the number of verified true positives decreased from 70% to 58% (Figure 3c). We also observed an ~5% decrease in the false-positive rate, where the detNL-containing species did not display the required isotopic distribution. We therefore recommend a workflow by which spectra are analyzed by both ionFinder and envoMatch to generate high-confidence assignments of sites of citrullination, i.e., those that receive annotations of ‘true’ based on an analysis of both MS1 and MS2 spectra.
Sites of autocitrullination in PADs 1, 2, 3, and 4
To evaluate the utility of ionFinder in a ‘real world’ situation, we examined the autocitrullination of all four active PADs, i.e. PADs 1-4. Note that extensive research has established that the PADs autocitrullinate in biological systems, and once modified, protein-protein interactions are impacted.34–36 Notably, recombinant, purified PADs readily undergo autocitrullination under reducing conditions in the presence of Ca2+. As such, PADs 1-4 were incubated with Ca2+ (5 mM) and TCEP (1 mM) at 37 ºC for 1 h to induce autocitrullination. The proteins were processed for tandem mass spectrometry using different combinations of proteases (LysC/GluC, Trypsin/GluC, and Trypsin) to increase the depth of coverage and mitigate the lower efficiency of trypsin towards Cit-containing proteins. After processing the resulting data using envoMatch and ionFinder, we found that a large percentage of arginine residues from each PAD were autcitrullinated. Specifically, ionFinder unambiguously mapped 28 citrullinated residues on PAD1 (76% of arginine residues), 20 on PAD2 (61% of arginine residues), 19 on PAD3 (49% of arginine residues), and 21 on PAD4 (78% of arginine residues) (Figure 4). We compared the PAD4 autocitrullination sites obtained from ionFinder to a previously published dataset where sites of autocitrullination of PAD4 were evaluated in a time-dependent manner using tandem mass tag (TMT) labeling.36 The sites identified by ionFinder displayed significant overlap with the previously determined sites of PAD4 autocitrullination.
Figure 4. Autocitrullination sites in PADs 1, 2, 3, and 4.
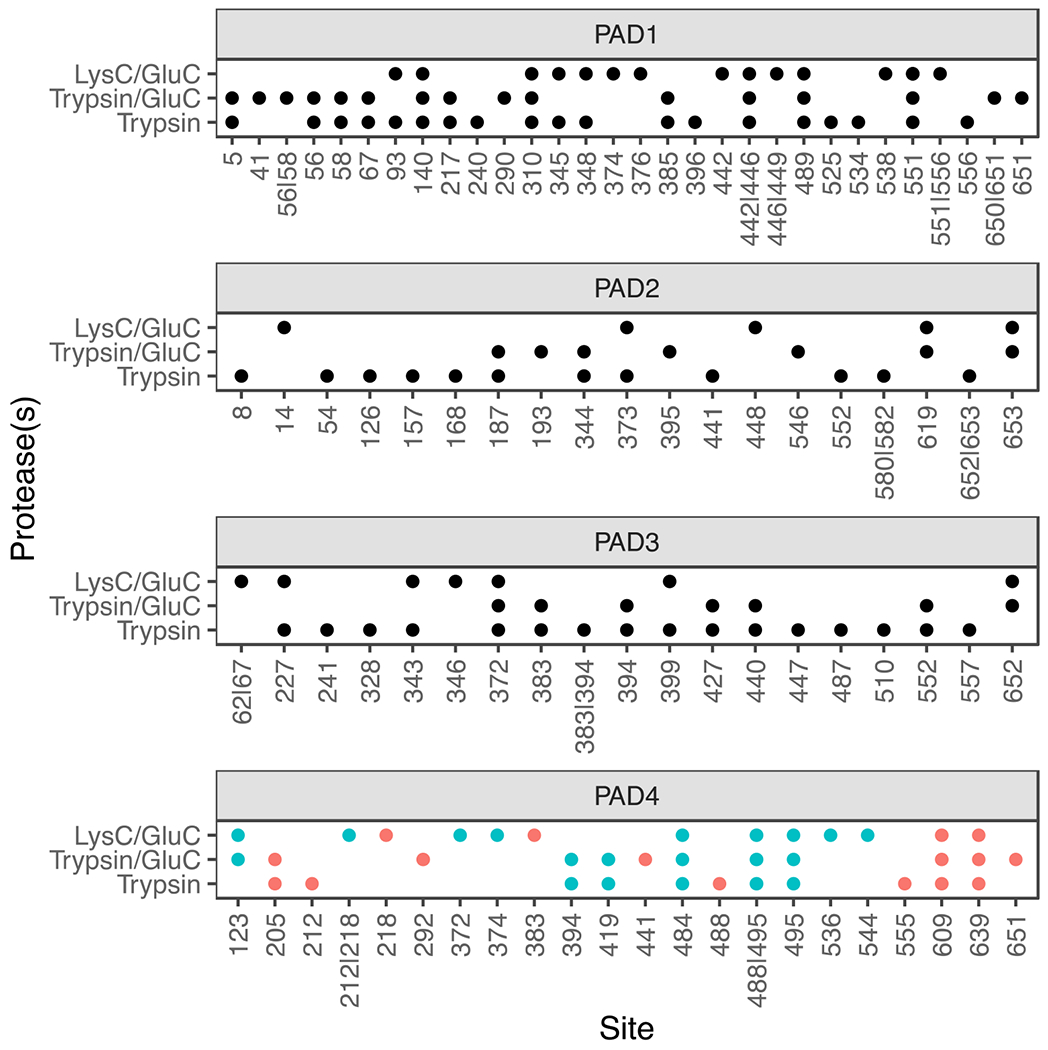
The residues that are identified as citrullinated for each individual PAD under three different digestion conditions; LysC/GluC, Trypsin/GluC, and Trypsin are shown. The PAD4 data were compared to a previous study.36 Citrullination sites identified in that Mondal et al36 are shown in blue. Unique sites are shown in red.
Since these autocitrullination sites were obtained using purified, recombinant PADs, we cannot conclude that they all occur endogenously. However, we can use these data to search for patterns in PAD substrate preferences. First, we examined the citrullination data for each PAD to establish the existence of a consensus amino acid sequence flanking the site of citrullination (Figure 5a).37 Upon inspection, the results indicate that there are no obvious consensus sequences for PAD autocitrullination. For example, PADs 1, 2, and 4 show no discernable preference for a particular residue, or type of residue, in positions that span a 3-residue window N-terminal and C-terminal to the site of citrullination. PAD3, however, does show a small preference for hydrophobic residues at the R+2 and R+3 positions.
Figure 5. Specificity of autocitrullination.
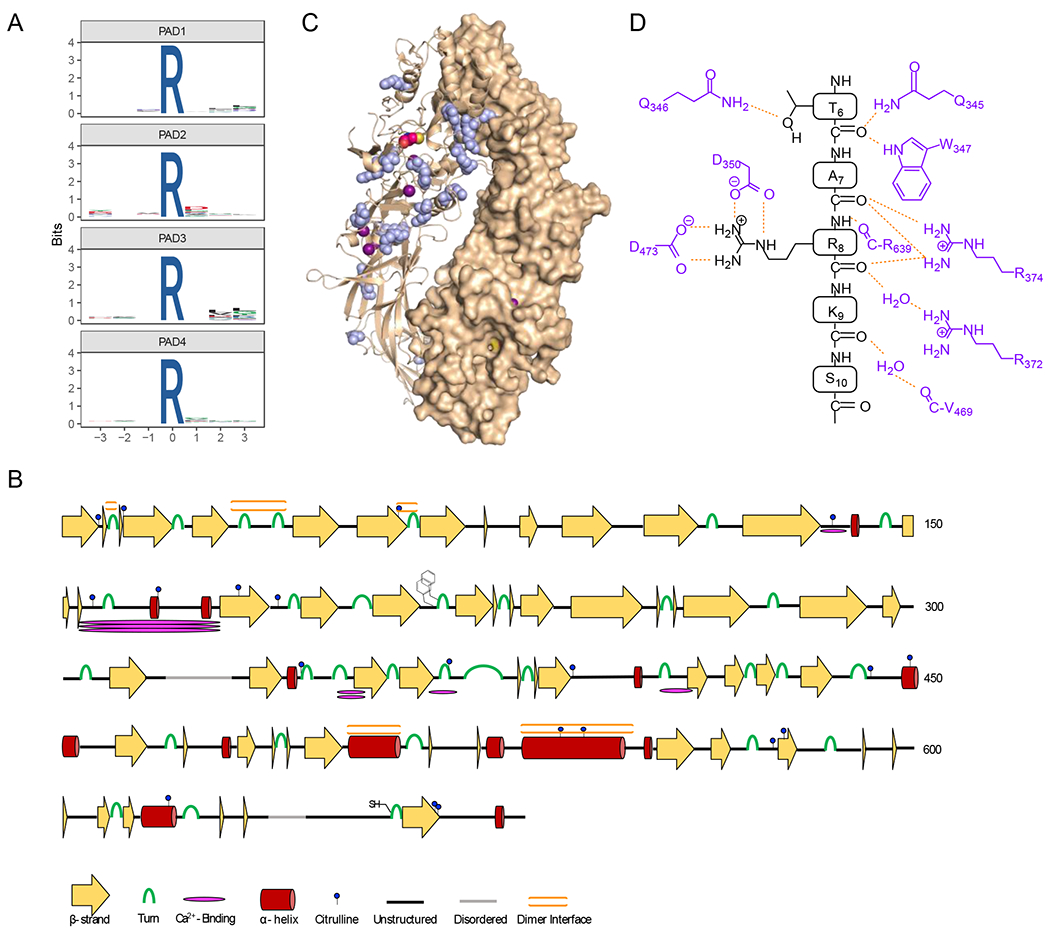
A) WebLogos depicting the substrate specificity determinants of autocitrullination by PAD1, PAD2, PAD3, and PAD4 (top to bottom). B) Secondary structural analysis of PAD2 autocitrullination shows that many of the sites of citrullination do not fall on canonical secondary structural regions. The cartoon highlights the sites of citrullination, different secondary structures, the FF-motif which plays a role in nuclear localization, and the location of the active site cysteine. C) Structure of the holo PAD2 dimer (PDBID: 4N2C) with citrullinated Arg residues shown in blue. D) Cartoon showing how histone H3 binds in the active site of PAD4 (PDB ID: 2DEW). The interactions are primarily between the enzyme and the substrate backbone, indicating that specific side chains do not make a major contribution to substrate specificity.
Next, we mapped the sites of PAD2 autocitrullination onto high-resolution structures of PAD2 available in the protein databank.26 These structural analyses revealed that 9 of the 20 citrullinated residues are not on strands or helices, and are instead located on looped or intermediate regions of the enzyme (Figure 5b). The Cit residues that lie in regions of canonical secondary structure seem to be on the cap region, such as the C-termini of α-helices, and both termini of β-strands. Finally, we found that a quarter of the citrullinated residues were present at, or adjacent to, the dimer interface in PAD2 (Figure 5c). These data indicate that PADs preferentially citrullinate Arg residues that are accessible and lie on flexible regions of proteins. Consistent with this notion is the fact that the PAD active site consists of a narrow ~21 Å long channel that interacts with the substrate guanidinium and main chain carbonyls of a substrate. While this organization confers steric selectivity for peptidyl-arginine over free arginine, active site residues only contact water and the backbone of the substrate and not key side chains in the R-3 to R+3 positions, as revealed from structures of PAD4 bound to histone tail analogues (Figure 5d).38 As such, PADs preferentially citrullinate protein domains that are flexible enough to provide access to the enzyme, rendering looped regions, or caps of helices and strands, ideal locations for citrullination. Based on the MS data presented here, and the strong homology amongst PAD isozymes, we hypothesize that this explanation likely holds true for all PADs.
The HEK-PAD2 citrullinome
Having established the utility of envoMatch and ionFinder for identifying Cit-containing peptides from a simple purified protein system, we next sought to evaluate the assignment of Cit residues within a complex proteome. For these studies, we used a previously described HEK293T-PAD2 cell line13 that stably overexpresses PAD2. Addition of Ca2+ and ionomycin to the cell growth medium leads to robust citrullination of hundreds of proteins. After quenching with EDTA, cells were harvested, and proteomes were extracted and proteolytically digested for MS. Based on our prior work identifying citrullinated peptides in NETs,24 we expected that a two-dimensional peptide fractionation step prior to MS analysis, would provide improved coverage of low-abundant Cit peptides. Therefore, samples were segmented offline into 12 fractions on a C18 column at high pH, followed by on-line reverse-phase separation at neutral pH. The resulting MS data were analyzed with envoMatch and ionFinder, resulting in the identification of ~350 unique Cit-containing PSMs on over 220 proteins (Table S3).
We previously described the development of a suite of phenyl glyoxal(PG)-based probes that can be used to chemically modify citrullines, thereby enabling the visualization (rhodamine-PG), and enrichment (biotin-PG), of citrullinated proteins.13,16 Employing biotin-PG, we previously identified proteins that are citrullinated in the HEK-PAD2 cell line upon treatment with Ca2+ and ionomycin.13 Comparing the data from the biotin-PG-enriched lysates (500 μg), and the unenriched lysate analyzed with envoMatch and ionFinder (50 μg), we found 53 common proteins amongst the two groups. Notably, ionFinder identified an additional 171 citrullinated proteins that were distinct from the biotin-PG dataset (Figure 6a). Differences between the two datasets are expected given the numerous variations in the analysis workflows. For example, the protein hits generated using biotin-PG are determined by the stoichiometry of the chemical reaction of the probe with citrullinated proteins. In addition, biotin-PG does not directly identify the sites of citrullination, instead protein identification is based on non-citrullinated tryptic peptides from citrullinated proteins that were modified by biotin-PG. Therefore, the abundance of MS-amenable tryptic peptides can further skew the protein identifications. By contrast, envoMatch and ionFinder directly identify the exact site of citrullination, without the need for biotin-PG labeling and enrichment. Here, peptide identifications are dependent on the ionization and fragmentation characterizations of the Cit-containing peptide. Additionally, due to the lack of an enrichment step, Cit identifications are also biased by the relative abundance of the citrullinated protein. In fact, the proteins identified as being citrullinated by ionFinder were, on average, over the 70th percentile in protein abundance (as assessed with cumulative intensity-based absolute quantification (iBAQ) values) (Figure 6b). We also compared the Cit identifications from the Lee et al14 and the Chaerkady et al25 studies to our HEK-PAD2 data. These three studies were similar in that there was no specific enrichment of Cit peptides, but differed in the data analysis workflows used for Cit identification. There was little overlap across the three studies (Figure S8), likely due to the significant differences in the proteomes that were analyzed in these three studies (various human tissues, neutrophils, and HEK cells, respectively).
Figure 6. Global analysis of the HEK293T-PAD2 citrullinome.
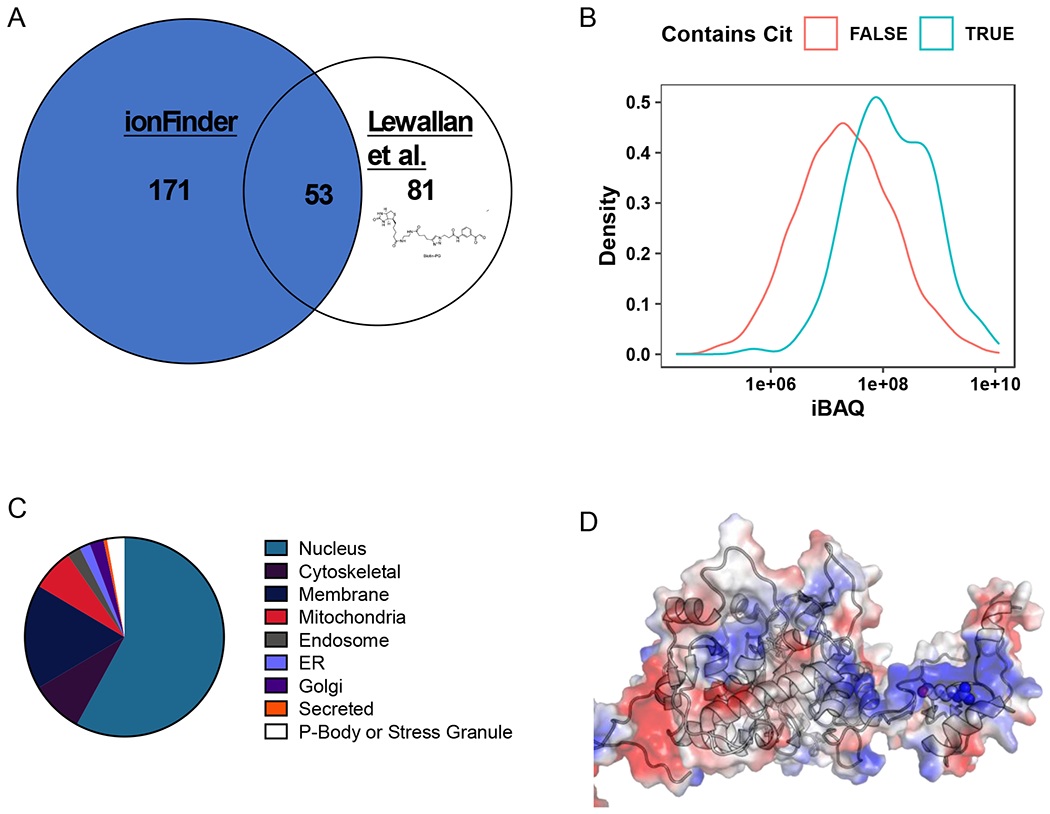
A) Comparison of the citrullinome identified by ionFinder and Lewallen et al.13 B) Kernel density estimate plot depicting the probability distribution of protein families, TRUE and FALSE, abundance based on cumulative iBAQ values from fractionated samples. The iBAQ at the maximum density in the plot was 7.45e+07 for Cit-containing proteins and 1.94e+07 for non-Cit-containing proteins. Thus, the maximum probability density of protein iBAQ values was 3.8 times greater for Cit-containing proteins than non-Cit containing proteins. C) Cellular localization of the citrullinated proteins identified by ionFinder, demonstrating that many of the proteins are, as expected, nuclear. D) Crystallographic depiction of IMPDH2 electrostatic distribution (PDB 1NF7) showing the location of Arg224 in the apo state of the enzyme.44 Arg 224 is in an exposed, and positively charged region, making it a target for citrullination by PAD2. When GTP is bound, Arg224 is no longer as accessible, as it lies in the nucleotide binding pocket near the dimer interface.
Notably, many of the proteins we identified as being citrullinated with envoMatch and ionFinder were nuclear proteins, with an overrepresentation of DNA and RNA-binding proteins (Figure 6c). In fact, 58% of the citrullinated proteins are annotated as localized to the nucleus based on information provided by Uniprot. In addition, 21% of citrullinated proteins are classified as nucleotide binding proteins (PANTHER: PC00171). This is unsurprising when one considers that Arg-rich proteins are often responsible for binding to the net negatively charged backbone of DNA and RNA.39 One interesting example is the splicing factorU1 70K snRNP, which is citrullinated at Arg222. Arg222 is in a S/R/E rich region adjacent to the N-terminal RNA-binding motif, and many studies have shown that the surrounding Ser residues can be phosphorylated to regulate alternative splicing, cell death, and autoantigenicity.40–43 Another intriguing example is eEF1, an EF-Tu homologue, which acts as an elongation factor that facilitates aminoacyl-tRNA binding into the A-site of ribosomes during translation. The region that is citrullinated (Arg96, 427, and 430) is part of a positively charged cluster of amino acids involved in binding tRNA. Lastly, the enzyme Inosine-5’-monophosphate dehydratase 2 (IMPDH2) is citrullinated at Arg224. This region of IMPDH2 is involved in nucleotide binding, where Arg224 contributes to GTP binding in the holo conformation.44 In the apo form, Arg224 is on a positively charged, readily accessible loop (Figure 6d) where this residue is far more exposed and available to act as a substrate for a PAD. Fascinatingly, mutations of this arginine residue in IMPDH1 are associated with retinopathy.45
Conclusions
Citrullination remains an understudied area of research. While many advances have been made over the last several decades, site-specific decoding of the citrullinome has remained challenging. Identifying sites of citrullination by mass spectrometry is plagued by false-positive annotations due to the small mass shift that results from the modification, as well as confounding isobaric Asn and Gln deamidations. Accurate assignment of Cit residues within peptides requires extensive mining of fragmentation spectra for the presence of neutral loss species that are unique to Cit. Previous studies used manual spectral matching to confirm sites of citrullination. Here, we automated this process through the development of a suite of computational tools, envoMatch and ionFinder, which can mine fragmentation data to unambiguously map the sites of citrullination. These tools are intended to be incorporated downstream of database-search algorithms, such as SEQUEST, Mascot, or MaxQuant, to further parse for high-confident assignments of Cit-containing peptides. In the recent Chaerkady et al study25 a similar approach was taken, where MaxQuant data was parsed for hits that specifically displayed Cit-specific NL species. Our described workflow incorporates more stringent filtering than that available through MaxQuant by setting a dynamic NL cutoff to differentiate determining from artifactual NL species. Additionally, MaxQuant does not implement the isotopic envelope matching performed by envoMatch, which serves to further decrease false positives from the output and increase confidence in the identified hits.
EnvoMatch and ionFinder were used to comprehensively map of the sites of PAD autocitrullination. Our data indicate that PADs prefer substrate Arg residues on flexible, surface-exposed regions of the protein, supporting the hypothesis that PADs are opportunistic in their substrate selection. Furthermore, many of these resulting Cit residues were in the dimer interface of the protein, suggesting that autocitrullination may therefore affect PAD dimerization. With these citrullination maps at our disposal, our future work is focused on discovering the molecular consequences of autocitrullination on enzyme structure, activity, and subcellular localization.
In addition to sites of autocitrullination on purified PAD proteins, envoMatch and ionFinder also identified a long list of cellular targets for PAD2. Upon activating cells with calcium and ionomycin, PAD2 re-localizes to the nucleus.46 Our data confirms the nuclear localization of PAD2 because the majority of the identified sites of citrullination were found on nuclear proteins. Furthermore, many of the targets, nuclear or otherwise, are involved in nucleotide binding. Proteins which bind to nucleotides are often arginine-rich and have higher pI values to promote a favorable interface with the negatively charged sugar-phosphate backbone. These qualities make this family of proteins prime targets for the PADs. Future work entails mechanistic investigation of these targets to understand the functional consequences of citrullination in vitro and in cells using our recently described approach to site-specifically incorporate citrulline into proteins.36
In summary, ionFinder and envoMatch, automate the process of verifying the assignment of Cit annotations by standard database-search algorithms, primarily through the automated assignment of diagnostic neutral loss species. The primary strength of ionFinder and envoMatch is the speed of analysis, which far exceeds the throughput amenable by manual spectral matching. Additionally, the programs are flexible in the types of instruments, fragmentations modes, and database-search algorithms that are used to generate the input data. The primary limitation in the described ionFinder workflow is the limited coverage of citrullination events on low abundance proteins. Since the input data for ionFinder is generated from cell lysates that have not specifically been enriched for Cit-containing proteins or peptides, there is an expected bias toward identification of sites of citrullination from abundant proteins within the proteome. Further advances in peptide fractionation methodology, and the advent of instruments with faster scan times, will likely help to overcome this abundance bias, and allow for a deeper interrogation of citrullination events from complex biological samples. Lastly, although specifically designed for analysis of sites of citrullination, ionFinder and envoMatch are easily adapted to other PTMs that show diagnostic NL species. We believe that the customizable nature of ionFinder and envoMatch will provide a useful tool for proteomic analyses in general, by setting more stringent criteria for assignment of modified peptides.
Supplementary Material
Funding Sources
This work was supported in part by NIH grants R35 GM118112 (P.R.T.) and R35GM134964 (E.W.).
Footnotes
Supporting information. The Supporting Information is available free of charge on the ACS Publications website at DOI: Supporting information includes annotated fragmentation spectra (Figures S1–S7), comparison to other proteomic datasets (Figure S8), and complete mass spectrometry data (Tables S1–S3).
The authors declare the following competing financial interest(s): P.R.T. is a consultant for Related Sciences.
Accession Codes.
| Protein | Uniprot ID | Link |
|---|---|---|
| PAD1 | Q9ULC6 | https://www.uniprot.org/uniprot/Q9ULC6 |
| PAD2 | Q9Y2J8 | https://www.uniprot.org/uniprot/Q9Y2J8 |
| PAD3 | Q9ULW8 | https://www.uniprot.org/uniprot/Q9ULW8 |
| PAD4 | Q9UM07 | https://www.uniprot.org/uniprot/Q9UM07 |
References
- (1).Fuhrmann J, and Thompson PR (2016) Protein Arginine Methylation and Citrullination in Epigenetic Regulation, ACS Chem Biol 11, 654–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Tilvawala R, and Thompson PR (2019) Peptidyl arginine deiminases: detection and functional analysis of protein citrullination, Curr Opin Struct Biol 59, 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Carmona-Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, Liu Y, Bicker KL, Wahamaa H, Hoffmann V, Catrina AI, Thompson P, Buckner JH, Robinson WH, Fox DA, and Kaplan MJ (2017) Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis, Sci Immunol 2, eaaz9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, and van Venrooij WJ (1998) Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies, J Clin Invest 101, 273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).van Boekel MA, Vossenaar ER, van den Hoogen FH, and van Venrooij WJ (2002) Autoantibody systems in rheumatoid arthritis: specificity, sensitivity and diagnostic value, Arthritis Res 4, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Li FJ, Surolia R, Li H, Wang Z, Liu G, Kulkarni T, Massicano AVF, Mobley JA, Mondal S, de Andrade JA, Coonrod SA, Thompson PR, Wille K, Lapi SE, Athar M, Thannickal VJ, Carter AB, and Antony VB (2021) Citrullinated vimentin mediates development and progression of lung fibrosis, Sci Transl Med 13, eaba2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Jones JE, Causey CP, Knuckley B, Slack-Noyes JL, and Thompson PR (2009) Protein arginine deiminase 4 (PAD4): Current understanding and future therapeutic potential, Curr Opin Drug Discov Devel 12, 616–627. [PMC free article] [PubMed] [Google Scholar]
- (8).Chumanevich AA, Causey CP, Knuckley BA, Jones JE, Poudyal D, Chumanevich AP, Davis T, Matesic LE, Thompson PR, and Hofseth LJ (2011) Suppression of Colitis in Mice by Cl-Amidine: A Novel Peptidylarginine Deiminase (Pad) Inhibitor, Am J Physiol Gastrointest Liver Physiol 300, G929–G938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tian Y, Qu S, Alam HB, Williams AM, Wu Z, Deng Q, Pan B, Zhou J, Liu B, Duan X, Ma J, Mondal S, Thompson PR, Stringer KA, Standiford TJ, and Li Y (2020) Peptidylarginine deiminase 2 has potential as both a biomarker and therapeutic target of sepsis, JCI insight 5, e138873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Knight JS, Subramanian V, O'Dell AA, Yalavarthi S, Zhao W, Smith CK, Hodgin JB, Thompson PR, and Kaplan MJ (2015) Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice, Ann Rheum Dis 74, 2199–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Liu Y, Lightfoot YL, Seto N, Carmona-Rivera C, Moore E, Goel R, O'Neil L, Mistry P, Hoffmann V, Mondal S, Premnath PN, Gribbons K, Dell'Orso S, Jiang K, Thompson PR, Sun HW, Coonrod SA, and Kaplan MJ (2018) Peptidylarginine deiminases 2 and 4 modulate innate and adaptive immune responses in TLR-7-dependent lupus, JCI insight 3, e124729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Clancy KW, Weerapana E, and Thompson PR (2015) Detection and identification of protein citrullination in complex biological systems, Curr Opin Chem Biol 30, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lewallen DM, Bicker KL, Subramanian V, Clancy KW, Slade DJ, Martell J, Dreyton CJ, Sokolove J, Weerapana E, and Thompson PR (2015) Chemical Proteomic Platform To Identify Citrullinated Proteins, ACS Chem Biol 10, 2520–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lee CY, Wang D, Wilhelm M, Zolg DP, Schmidt T, Schnatbaum K, Reimer U, Ponten F, Uhlen M, Hahne H, and Kuster B (2018) Mining the Human Tissue Proteome for Protein Citrullination, Mol Cell Proteomics 17, 1378–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Holm A, Rise F, Sessler N, Sollid LM, Undheim K, and Fleckenstein B (2006) Specific modification of peptide-bound citrulline residues, Anal Biochem 352, 68–76. [DOI] [PubMed] [Google Scholar]
- (16).Bicker KL, Subramanian V, Chumanevich AA, Hofseth LJ, and Thompson PR (2012) Seeing citrulline: development of a phenylglyoxal-based probe to visualize protein citrullination, J Am Chem Soc 134, 17015–17018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tutturen AE, Holm A, Jorgensen M, Stadtmuller P, Rise F, and Fleckenstein B (2010) A technique for the specific enrichment of citrulline-containing peptides, Anal Biochem 403, 43–51. [DOI] [PubMed] [Google Scholar]
- (18).Tilvawala R, Nguyen SH, Maurais AJ, Nemmara VV, Nagar M, Salinger AJ, Nagpal S, Weerapana E, and Thompson PR (2018) The Rheumatoid Arthritis-Associated Citrullinome, Cell Chem Biol 25, 691–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Nepomuceno AI, Gibson RJ, Randall SM, and Muddiman DC (2014) Accurate Identification of Deamidated Peptides in Global Proteomics Using a Quadrupole Orbitrap Mass Spectrometer, J Proteome Res 13, 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wang X, Swensen AC, Zhang T, Piehowski PD, Gaffrey MJ, Monroe ME, Zhu Y, Dong HL, and Qian WJ (2020) Accurate Identification of Deamidation and Citrullination from Global Shotgun Proteomics Data Using a Dual-Search Delta Score Strategy, J Proteome Res 19, 1863–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hao G, Wang D, Gu J, Shen Q, Gross SS, and Wang Y (2009) Neutral loss of isocyanic acid in peptide CID spectra: a novel diagnostic marker for mass spectrometric identification of protein citrullination, J Am Soc Mass Spectrom 20, 723–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Jin Z, Fu Z, Yang J, Troncosco J, Everett AD, and Van Eyk JE (2013) Identification and characterization of citrulline-modified brain proteins by combining HCD and CID fragmentation, Proteomics 13, 2682–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Huh S, Hwang D, and Kim MS (2020) Statistical Modeling for Enhancing the Discovery Power of Citrullination from Tandem Mass Spectrometry Data, Anal Chem 92, 12975–12986. [DOI] [PubMed] [Google Scholar]
- (24).Salinger AJ, Dubuke ML, Carmona-Rivera C, Maurais AJ, Shaffer SA, Weerapana E, Thompson PR, and Kaplan MJ (2020) Technical comment on "Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis", Sci Immunol 5, eaax5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chaerkady R, Zhou Y, Delmar JA, Weng SHS, Wang J, Awasthi S, Sims D, Bowen MA, Yu W, Cazares LH, Sims GP, and Hess S (2021) Characterization of Citrullination Sites in Neutrophils and Mast Cells Activated by Ionomycin via Integration of Mass Spectrometry and Machine Learning, J Proteome Res 20, 3150–3164. [DOI] [PubMed] [Google Scholar]
- (26).Slade DJ, Fang P, Dreyton CJ, Zhang Y, Fuhrmann J, Rempel D, Bax BD, Coonrod SA, Lewis HD, Guo M, Gross ML, and Thompson PR (2015) Protein arginine deiminase 2 binds calcium in an ordered fashion: implications for inhibitor design, ACS Chem Biol 10, 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Knuckley B, Causey CP, Jones JE, Bhatia M, Dreyton CJ, Osborne TC, Takahara H, and Thompson PR (2010) Substrate specificity and kinetic studies of PADs 1, 3, and 4 identify potent and selective inhibitors of protein arginine deiminase 3, Biochemistry 49, 4852–4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kearney PL, Bhatia M, Jones NG, Luo Y, Glascock MC, Catchings KL, Yamada M, and Thompson PR (2005) Kinetic characterization of protein arginine deiminase 4: a transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis, Biochemistry 44, 10570–10582. [DOI] [PubMed] [Google Scholar]
- (29).Keller A, Nesvizhskii AI, Kolker E, and Aebersold R (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search, Anal Chem 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- (30).Nesvizhskii AI, Keller A, Kolker E, and Aebersold R (2003) A statistical model for identifying proteins by tandem mass spectrometry, Anal Chem 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- (31).Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, Perez E, Uszkoreit J, Pfeuffer J, Sachsenberg T, Yilmaz S, Tiwary S, Cox J, Audain E, Walzer M, Jarnuczak AF, Ternent T, Brazma A, and Vizcaino JA (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data, Nucleic Acids Res 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hoopmann M (2020) MSToolkit, GithHub Repository. [Google Scholar]
- (33).Levitsky LI, Klein JA, Ivanov MV, and Gorshkov MV (2019) Pyteomics 4.0: Five Years of Development of a Python Proteomics Framework, J Proteome Res 18, 709–714. [DOI] [PubMed] [Google Scholar]
- (34).Andrade F, Darrah E, Gucek M, Cole RN, Rosen A, and Zhu X (2010) Autocitrullination of human peptidyl arginine deiminase type 4 regulates protein citrullination during cell activation, Arthritis Rheum 62, 1630–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Slack JL, Jones LE Jr., Bhatia MM, and Thompson PR (2011) Autodeimination of protein arginine deiminase 4 alters protein-protein interactions but not activity, Biochemistry 50, 3997–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Mondal S, Wang S, Zheng Y, Sen S, Chatterjee A, and Thompson PR (2021) Site-specific incorporation of citrulline into proteins in mammalian cells, Nat Commun 12, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004) WebLogo: a sequence logo generator, Genome Res 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Arita K, Shimizu T, Hashimoto H, Hidaka Y, Yamada M, and Sato M (2006) Structural basis for histone N-terminal recognition by human peptidylarginine deiminase 4, Proc Natl Acad Sci U S A 103, 5291–5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lafarga V, Sirozh O, Diaz-Lopez I, Galarreta A, Hisaoka M, Zarzuela E, Boskovic J, Jovanovic B, Fernandez-Leiro R, Munoz J, Stoecklin G, Ventoso I, and Fernandez-Capetillo O (2021) Widespread displacement of DNA- and RNA-binding factors underlies toxicity of arginine-rich cell-penetrating peptides, EMBO J, e103311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Krämer A (1996) The structure and function of proteins involved in mammalian pre-mRNA splicing, Annu Rev Biochem 65, 367–409. [DOI] [PubMed] [Google Scholar]
- (41).Smith CW, and Valcárcel J (2000) Alternative pre-mRNA splicing: the logic of combinatorial control, Trends Biochem Sci 25, 381–388. [DOI] [PubMed] [Google Scholar]
- (42).Utz PJ, Hottelet M, van Venrooij WJ, and Anderson P (1998) Association of phosphorylated serine/arginine (SR) splicing factors with the U1-small ribonucleoprotein (snRNP) autoantigen complex accompanies apoptotic cell death, J Exp Med 187, 547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Hof D, Raats JM, and Pruijn GJ (2005) Apoptotic modifications affect the autoreactivity of the U1 snRNP autoantigen, Autoimmun Rev 4, 380–388. [DOI] [PubMed] [Google Scholar]
- (44).Fernández-Justel D, Núñez R, Martín-Benito J, Jimeno D, González-López A, Soriano EM, Revuelta JL, and Buey RM (2019) A Nucleotide-Dependent Conformational Switch Controls the Polymerization of Human IMP Dehydrogenases to Modulate their Catalytic Activity, J Mol Biol 431, 956–969. [DOI] [PubMed] [Google Scholar]
- (45).Aherne A, Kennan A, Kenna PF, McNally N, Lloyd DG, Alberts IL, Kiang AS, Humphries MM, Ayuso C, Engel PC, Gu JJ, Mitchell BS, Farrar GJ, and Humphries P (2004) On the molecular pathology of neurodegeneration in IMPDH1-based retinitis pigmentosa, Hum Mol Genet 13, 641–650. [DOI] [PubMed] [Google Scholar]
- (46).Zheng L, Nagar M, Maurais AJ, Slade DJ, Parelkar SS, Coonrod SA, Weerapana E, and Thompson PR (2019) Calcium Regulates the Nuclear Localization of Protein Arginine Deiminase 2, Biochemistry 58, 3042–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.