

Abstract
It has been suggested that new β cells can arise from specific populations of adult pancreatic progenitors or facultative stem cells. However, their existence remains controversial, and the conditions under which they would contribute to new β cell formation are not clear. Here, we use a suite of mouse models enabling dual recombinases-mediated genetic tracing to simultaneously fate map insulin+ and insulin− cells in the adult pancreas. We find that the insulin− cells, of both endocrine and exocrine origin, do not generate new β cells in the adult pancreas during homeostasis, pregnancy or injury, including partial pancreatectomy, pancreatic duct ligation or β cell ablation with streptozotocin. However, non-β cells can give rise to insulin+ cells after extreme genetic ablation of β cells, consistent with transdifferentiation. Together, our data indicate that pancreatic endocrine and exocrine progenitor cells do not contribute to new β cell formation in the adult mouse pancreas under physiological conditions.
Introduction
Insulin-secreting pancreatic β cells are central to the proper maintenance of whole-body glucose homeostasis. Normal glycemic control requires an adequate number of functional β cells; consequently β cell death is a pathological hall mark of type 1 diabetes and advanced type 2 diabetes (T2D)1,2. The ability of functional β cells to regulate real-time glycemic control, and thus the avoidance of chronic organ damage, far exceeds the capability of exogenous insulin treatment. Therefore, there is an increasing demand for therapeutic approaches that restore functional and effective β cell mass in patients with severe β cell deficiency3. Understanding how endogenous β cells are generated during homeostasis and potentially in disease conditions would provide clinically relevant information for designing new therapeutic approaches to treat diabetes. With respect to the identification of the cellular sources for the generation of new β cells in the adult pancreas, to date, two distinct mechanisms have been reported: self-replication of pre-existing β cells and generation of new β cells from non-β cells4. In support of the first mechanism, several pioneering studies using genetic lineage tracing elegantly demonstrated that β cell replication is the predominant means by which new β cells are generated after birth and in response to injury in vivo5–8. The second proposed mechanism regarding the generation of new β cells in the adult could occur by either transdifferentiation of differentiated non-β cells or by differentiation of pancreatic stem or progenitor cells (termed neogenesis).
Non-β cells of the adult human pancreas are capable of β cell differentiation when co-transplanted with fetal pancreatic cells under the mouse kidney capsule9. Adult glucagon-producing α cells and somatostatin-producing δ cells can transdifferentiate into new β cells after extreme β cell loss (> 99%) mediated by diphtheria toxin-based genetic ablation10,11. Forced over-expression of the transcription factor Pax4 or selective inhibition of Arx in mouse α cells is sufficient to promote their conversion to β cells12,13. Expression of Pdx1 and Mafa endows human α cells with β-cell characteristics14. Beyond this transdifferentiation of endocrine cells, expression of the key β-cell developmental regulators Neurogenin 3 (Ngn3), Pdx1, and Mafa, can reprogram adult mouse exocrine cells into cells that closely resemble β cells15. Similarly, the acinar cell-specific expression of Pdx1 also converts adult exocrine cells into insulin-expressing β cells16. Notably, many of the above studies supporting transdifferentiation utilized forced gene expression and employed genetic tracing to demonstrate direct conversion of differentiated cells into insulin-producing β cells. Under these specific non-physiological conditions as mentioned above, new β cells could be derived from other non-β cell lineages.
While there is a general consensus with regard to the transdifferentiation pathway for adult β cell regeneration, whether β cell neogenesis exists in the adult pancreas under physiological conditions remains controversial. Several lineage tracing studies have supported the existence of facultative β cell progenitors or stem cells that differentiate into β cells after pancreatic injury. For instance, adult ductal Ngn3-expressing cells, which have been reported as progenitors of β cells, can be reactivated autonomously to proliferate and differentiate into β cells after injury in a pancreatic duct ligation (PDL) model17. Adult carbonic anhydrase II-positive cells of the pancreatic duct may also serve as progenitors and give rise to β cells after PDL18. Similarly, a small number of adult Ptf1a+ acinar cells could undergo rapid reprogramming to generate duct cells and produce a small number of insulin+ β cells after PDL19. Additional studies have also suggested the existence of pancreatic stem cells or progenitors including CD133+ cells20, ALDH1+ cells21, Pdx1+ cells22, Sox9+ ductal cells23 and the recently reported Procr+ cells24. Contrary to these studies, a large body of evidence from lineage tracing studies that relied on a variety of exocrine and endocrine markers do not support the stem cell hypothesis for β cell neogenesis25–29. Due to the conflicting nature of these studies, there remains much debate and contention over the role of stem cells in β cell neogenesis.
In this study, we reassessed the stem cell hypothesis by developing a dual-recombinases-mediated genetic system, in which pre-existing insulin+ β cells and all insulin− non-β cells, including putative stem and progenitor cells, are simultaneously labeled by distinct surrogate markers that enable indelible tracing. With this dual fate mapping approach, we found that non-β cells generate new β cells in the adult pancreas during embryogenesis but not during homeostasis, pregnancy or after various forms of injuries, including partial pancreatectomy, pancreatic duct ligation and β cell ablation with streptozotocin. However, formation of new insulin+ cells from insulin− cells were observed after Diphtheria toxin-mediated extreme β cell loss, consistent with direct transdifferentiation from α- and δ-cells. Thus, our study supports the idea that β cell self-replication is the dominant pathway for β cell regeneration in the adult mouse pancreas; whereas there is no evidence for β cell neogenesis.
Results
Generation of fate mapping system to track β and non-β cells
To genetically label all putative β cell progenitors and stem cells without relying on prior knowledge of specific markers, we developed a dual genetic fate mapping system for simultaneous labeling of insulin-producing β cells and all non-β cells (Fig. 1a). In the construct design, we employed two orthogonal site-specific recombination systems, Cre-loxP and Dre-rox, with an interleaved reporter, IR130–33. In the IR1 reporter, two pairs of recombination recognition sites (rox and loxP) were interleaved, such that one successful recombination resulted in expression of one distinct genetic reporter and prevented any further event from another type of recombination. For example, Dre-rox recombination in β cells first removes the rox-flanked segment that contains one loxP site and ZsGreen from IR1, such that this allele would then express tdTomato and would no longer be responsive to Cre-loxP recombination (Fig. 1a). Likewise, Cre-loxP recombination in non-β cells removes the loxP-flanked segment that contains one rox site and the transcriptional stop cassette for ZsGreen expression, preventing subsequent Dre-rox-mediated recombination. Recombination is prevented even if this non-β cell (e.g. a stem cell) could later differentiate into an insulin-producing β cell. This design would thus simultaneously label β cells and non-β cells with two distinct permanent surrogate markers, tdTomato (tdT) and ZsGreen, respectively (Fig. 1a). If authentic β cell progenitors or stem cells exist and contribute to new β cells, the newly differentiated insulin-producing β cells would be genetically traced by ZsGreen expression.
Fig. 1 |. Genetic labeling of β cells and non-β cells by a Ins2-Dre;R26-iCre;IR1 strategy.
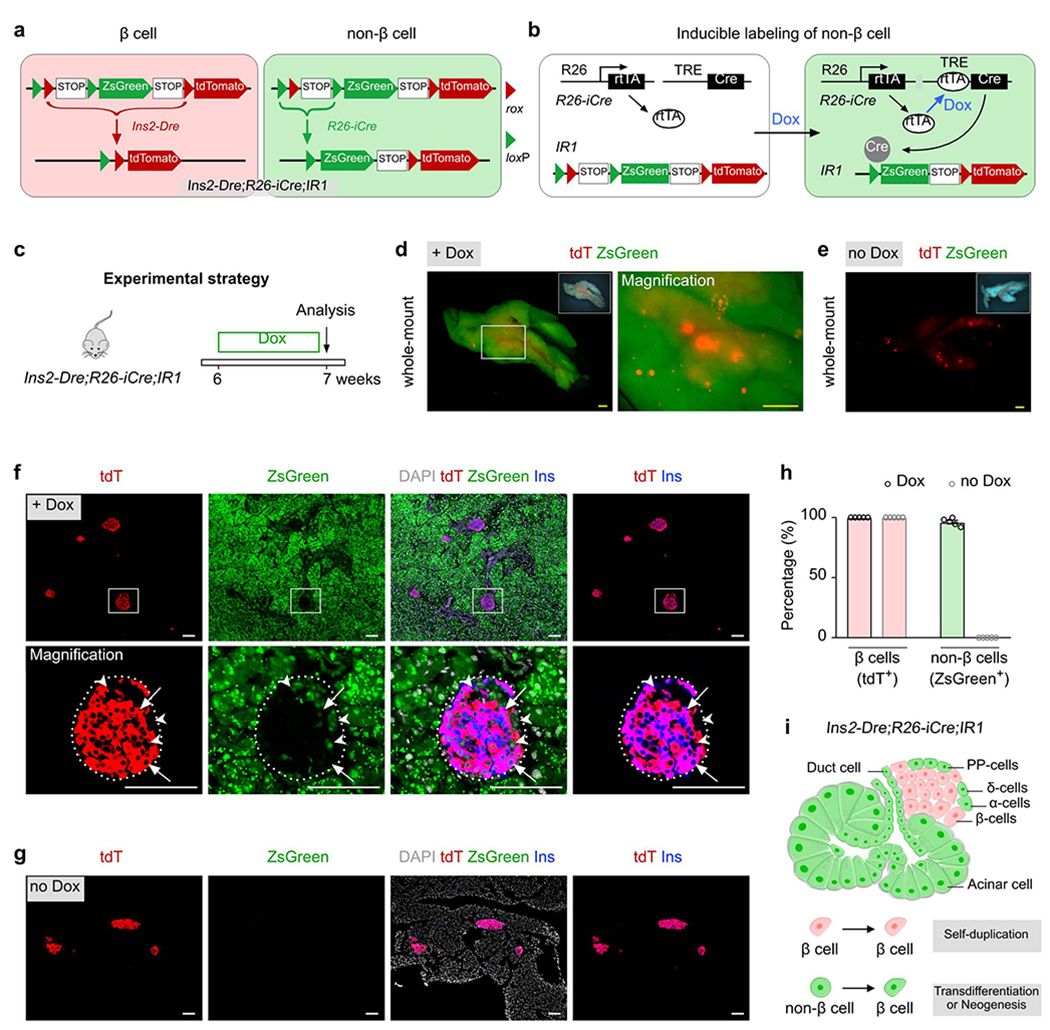
a, A Schematic diagram illustrating the strategy for labeling pancreatic β cells and non-β cells by Ins2-Dre;R26-iCre;IR1 using a dual-recombinases-based genetic approach. b, A schematic diagram illustrating the means by which doxycycline (Dox) induces the labeling of non-β cells by Cre-loxP-mediated recombination via R26-rtTA;TRE-Cre (R26-iCre). c, A schematic diagram illustrating the experimental strategy for Dox-induced lineage tracing of β cell and non-β cells. 6 weeks old mice (both male and female mice were used) were treated with either Dox (n = 5) or no Dox (n = 5) for 1 week before euthanasia. d,e, Whole-mount fluorescent images of pancreas from indicated mice with (d) or without (e) Dox treatment. Inserts, bright-field images of pancreas. f,g, Immunostaining for tdT, zsGreen, and Ins on pancreatic slides collected from Ins2-Dre;R26-iCre;IR1 mice treated with Dox (f) or no Dox (g). Arrows indicate tdT+Ins+ β cells. Arrowheads indicate zsGreen+Ins− non-β cells. h, Quantification of the percentage of β cells expressing tdT or non-β cells expressing ZsGreen from Ins2-Dre;R26-iCre;IR1 mice treated with Dox or without Dox. Numbers of investigated pancreas were as follows: Dox group, n = 5; no Dox group, n = 5, Data are mean ± SEM. i, Illustration showing the dual labeling of β cells and non-β cells by Ins2-Dre;R26-iCre;IR1 mice. Scale bars, yellow, 1 mm; white, 100 μm. Each image is representative of 5 individual biological samples.
To label insulin-producing β cells, we first generated the Ins2-Dre knock-in line by targeting the Dre recombinase cDNA located directly after the endogenous insulin gene promoter. The ATG start codon was replaced by homologous recombination using CRISPR/Cas9 (Extended Data Figure 1a). By crossing the mouse line with IR1, Ins2-Dre efficiently labeled insulin-expressing β cells, rendering virtually all β cells tdT+ in the adult islet (Extended Data Figure 1), and thus demonstrated the high efficiency of genetic targeting to insulin-expressing β cells. We next generated an inducible Cre system, R26-iCre, consisting of R26-rtTA and TRE-Cre alleles. In this system, doxycycline (Dox)-mediated rtTA binding to TRE leads to Cre expression, which recombines with the IR1 allele and yields constitutively active expression of ZsGreen (Fig. 1b).
We treated 6 week-old Ins2-Dre;R26-iCre;IR1 mice with Dox for one week, and analyzed non-β cell labeling a week later (Fig. 1c). Whole-mount fluorescent imaging of the pancreas from the 7 week-old Dox-treated mice showed tdT+ islets peppered within ZsGreen+ signal (Fig. 1d), while no ZsGreen+ signal was observed in the pancreas without Dox treatment (no Dox, Fig. 1e). Immunostaining for tdT, ZsGreen and insulin (Ins) on pancreatic sections derived from the Dox-treated Ins2-Dre;R26-iCre;IR1 mice showed that virtually all Ins+ β cells were tdT+ (Fig. 1f,h), For examination of ZsGreen+ cells in dual lineage tracing models, multiple regions of immunostained pancreatic tissue sections were randomly selected for image collection and quantification of labeling percentage. In the whole pancreas sections with magnification of multiple regions, we found all regions randomly selected are highly efficient in ZsGreen labeling (Extended Data Figure 2). In examination of the different endocrine cell lineages of islets, we found broad ZsGreen expression in non-β cell lineages, such as α cells (94.95 ± 1.22%), δ cells (92.52 ± 0.86%), and PP cells (97.79 ± 0.36%, Extended Data Figure 3). In the exocrine tissues, ZsGreen were also broadly expressed in multiple non-β cells, such as acinar cells (98.58 ± 0.28%), ductal cells (97.20 ± 0.57%), endothelial cells (95.72 ± 1.08%), lymphatic endothelial cells (94.58 ± 0.86%), and fibroblasts (93.26 ± 0.73%, Extended Data Figure 4). Quantitatively, 96.30 ± 1.38% of non-β cells expressed ZsGreen (ZsGreen+tdT−DAPI+ / tdT−DAPI+), demonstrating that the vast majority of non-β cells were labeled by R26-iCre (Fig. 1f,h). In the control group lacking Dox treatment, β cells were tdT+ and non-β cells remained ZsGreen− (Fig. 1g,h). By these means, we developed a genetic system marked by tdT+ β cells and ZsGreen+ non-β cells in the mouse pancreas (Fig. 1i).
Assessment of β cell neogenesis during homeostasis
To test whether non-β cell could contribute to β cell neogenesis during homeostasis, we treated 6 week-old Ins2-Dre;R26-iCre;IR1 mice with Dox for 1 week, followed by analyses at 3- or 6-month timepoints after induction (+3m or +6m, Fig. 2a). We performed whole-mount fluorescent imaging of pancreases at these two timepoints and found a strong ZsGreen+ signal throughout the entire pancreas coupled with isolated speckles of tdT+ staining (Fig. 2b). Next, we immunostained pancreatic tissue sections at these two timepoints for tdT, ZsGreen and Ins, and found that all β cells maintained islet-specific tdT expression (Fig. 2c–e). We did not find any cells co-expressing ZsGreen and Ins in the +3m or +6m pancreatic tissues (Fig. 2c–e). All Ins+ cells exclusively expressed tdT, but none of them expressed ZsGreen (Fig. 2d,e). These fate-mapping data indicate that insulin− endocrine or exocrine cells did not give rise to new β cells in the adult pancreas under homeotic conditions (Fig. 2f).
Fig. 2 |. Non-β cells do not contribute to β cells during adult homeostasis.

a, A schematic diagram illustrating the experimental strategy to test for β cell neogenesis during homeostasis. 6 weeks old mice (both male and female mice were used) were treated with Dox for 1 week. After 3 or 6 months, mice were euthanized (3 months trace group: n = 5; 6 months trace group: n = 5). b, Whole-mount bright-field and fluorescent images showing pancreas from Ins2-Dre;R26-iCre;IR1 mice after 3 or 6 months of lineage tracing. c,d, Immunostaining for tdT, zsGreen, and Ins on pancreatic slides collected from Ins2-Dre;R26-iCre;IR1 mice after 3 (c) or 6 (d) months. Arrows indicate tdT+Ins+ β cells. Arrowheads indicate zsGreen+Ins− non-β cells. e, Quantification of the percentage of β cells expressing tdT or ZsGreen after 6 months of lineage tracing. Numbers of investigated pancreas were as follows: n = 5 (6 months trace group), Data are mean ± SEM. f, Illustration showing non-β cells do not differentiate into β cells during homeostasis. Scale bars, yellow, 1 mm; white, 100 μm. Each image is representative of 5 individual biological samples.
Prior studies have reported that multipotent endocrine progenitors exist in the embryonic and fetal pancreas, which can give rise to insulin-producing β cells25–27,34. To test if our system can track β cell generation during embryogenesis, we collected the pancreata from Ins2-Dre;R26-iCre;IR1 mice at embryonic day (E) 14.5 without Dox treatment and confirmed the presence of Ins+tdT+ cells (Extended Data Figure 5a). We then performed pulse-chase experiments by treating Ins2-Dre;R26-iCre;IR1 mice with Dox at E14.5 and collected the pancreas at E16.0 and postnatal day 21 (P21) to compare the ZsGreen labeling rate of β cells. By the genetic design, IR1 allele could be recombined by one type of recombinase only and was then permanently expressed afterwards. The pre-existing β cells are labeled by Ins2-Dre (tdTomato), and non-β cells including stem cells or progenitors would be labeled by inducible Cre after Dox treatment (ZsGreen), and their descendants including β cells would be permanently ZsGreen+. As more β cells arise from ZsGreen+ progenitors or stem cells over time during embryonic development, the tdTomato labeling rate of β cells would be expected to be diluted, while the ZsGreen labeling rate of β cells would be expected to increase over time. By examining the tissue sections stained with ZsGreen, tdTomato, and Ins, we found the zsGreen labeling rate of β cells was 9.46 ± 0.52% at E16.0 and increased to 27.14 ± 1.51% at P21 (P=1.12 ×10−4, Extended Data Figure 5b–e). However, we did not observe Ins+ZsGreen+ β cells in the islets when Dox was administered postnatally after P7 (Extended Data Figure 5f–j). These findings are consistent with prior studies that endocrine progenitors give rise to new β cells during embryogenesis, but not postnatally25–27,34. These data also provided technical controls supporting the ability of our dual genetic tracing strategy to detect non-β cell-to-β cell conversion, if any exists.
Non-β cells do not convert to β cell during PPX or pregnancy
To test whether non-β cells contribute to adult β cells under physiological conditions known to stimulate β cell proliferation, we employed a previously described partial pancreatectomy (PPX) model to induce tissue regeneration29. We treated 6 week-old Ins2-Dre;R26-iCre;IR1 mice with Dox for 1 week, performed 50% PPX after two weeks of washout and collected pancreatic tissue at 1 week, 2 weeks and 4 weeks after injury (Fig. 3a, Extended Data Figure 6a). We performed whole-mount fluorescent imaging and found readily detectable tdT+ islets in the pancreas after PPX (Fig. 3b). The newly regenerated pancreatic tissues included both islets and exocrine cells (Fig. 3c). Immunostaining for tdT, ZsGreen and Ins on pancreatic sections also confirmed that all β cells were tdT+, but none expressed ZsGreen in both the sham and PPX groups (Fig. 3d–f, Extended Data Figure 6b and Extended Data Figure 6d). In agree with previous studies5,7,35,36, we found increased incorporation of EdU in the islets of PPX pancreas compared with sham controls (1.99 ± 0.10% in PPX vs 0.45 ± 0.05% in control, P=1.22 ×10−5, Extended Data Figure 6c). Of note, we also found that a few tdT+ cells in the islets were Ins−, possibly indicating β cell dedifferentiation as previous described37–39. These data demonstrated that PPX did not lead to β cell formation from non β cells (Fig. 3g).
Fig. 3 |. Non-β cells do not contribute to β cells during partial pancreatectomy and pregnancy.
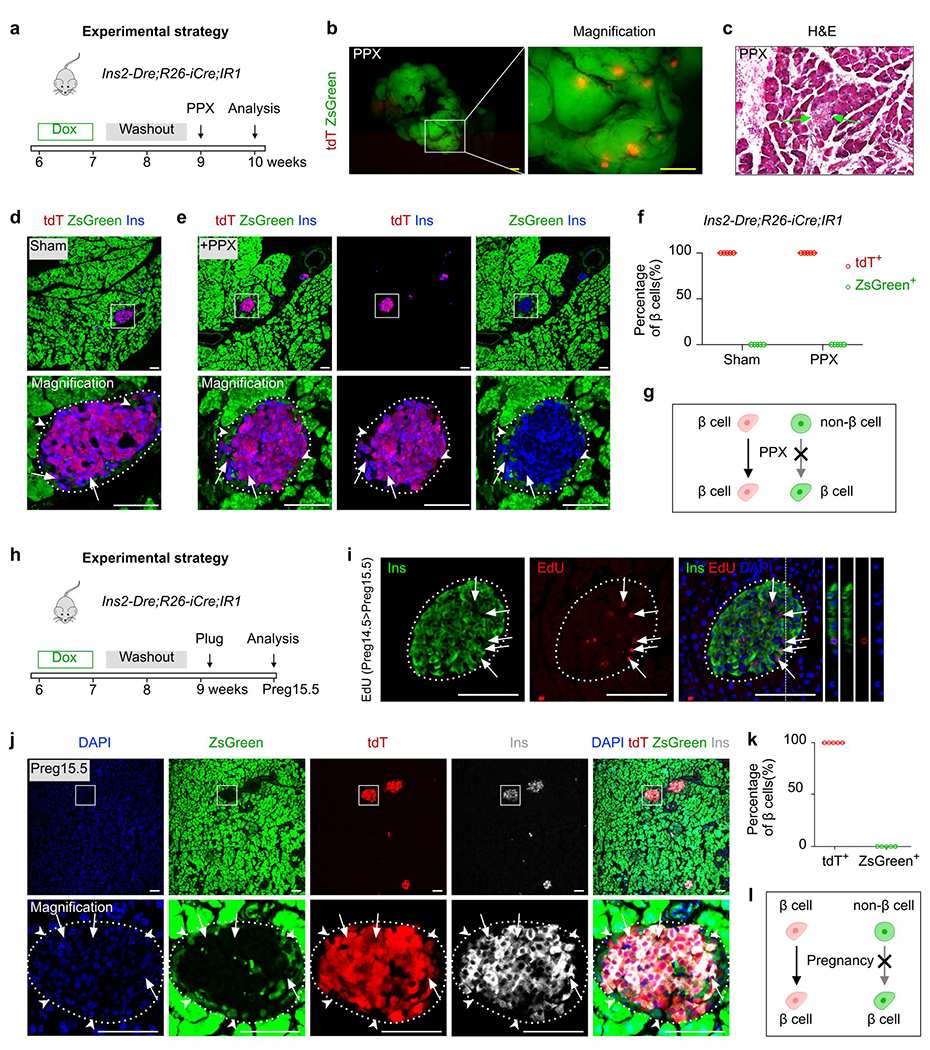
a, A schematic diagram illustrating the experimental strategy to test for β cell neogenesis after partial pancreatectomy (PPX) injury. 6 weeks old mice (both male and female mice were used) were treated with Dox for 1 week. After washout for 2 weeks and PPX injury for 1 week, mice were euthanized (PPX group: n = 5; Sham group: n = 5). b, Whole-mount fluorescent images of pancreas from PPX mice. c, H&E staining of pancreatic sections collected from PPX injured mice. Arrows, islet. d,e, Immunostaining for tdT, zsGreen, and Ins on pancreatic slides collected from Ins2-Dre;R26-iCre;IR1 Sham (d) or PPX (e) mice. Arrows indicate tdT+Ins+ β cells. Arrowheads indicate zsGreen+Ins− non-β cells. f, Quantification of the percentage of β cells expressing tdT or ZsGreen. Numbers of investigated pancreas were as follows: Sham group, n = 5; PPX group, n = 5, Data are mean ± SEM. g, Illustration showing no contribution of non-β cells to β cells after PPX. h, A schematic diagram illustrating the experimental strategy to test for β cell neogenesis during pregnancy. 6 weeks old mice (only female mice were used) were treated with Dox for 1 week. After washout for 2 weeks and Pregnancy for 15.5 days, mice were euthanized (Pregnancy group: n = 5; Sham group: n = 5). i, Immunostaining for Ins and EdU on pancreatic sections collected from pregnant mice at Preg15.5 after EdU injection at Preg14.5. Arrows indicate EdU+Ins+ β cells. j, Immunostaining for tdT, zsGreen, and Ins on pancreatic sections collected from Ins2-Dre;R26-iCre;IR1 mice after pregnancy. Arrows indicate tdT+Ins+ β cells. Arrowheads indicate zsGreen+Ins− non-β cells. k, Quantification of the percentage of β cells expressing tdT or ZsGreen. Numbers of investigated pancreas were as follows: Pregnancy group, n = 5, Data are mean ± SEM. l, Illustration showing no contribution of non-β cells to β cells during pregnancy. Scale bars, yellow, 1 mm; white, 100 μm. Each image is representative of 5 individual biological samples.
We next asked if non-β cells contribute to adult β cells during pregnancy, during which a significant increase of β cell mass occurs40. We treated 6-week old Ins2-Dre;R26-iCre;IR1 mice with Dox for 1 week, set up mating after 2 weeks of washout and examined pancreatic tissues during the course of pregnancy at Preg15.5 (Fig. 3h). We found that the islets of the pregnant dams exhibited a robust generation of new β cells as demonstrated by a large number of EdU-expressing β cells during pregnancy (Fig. 3i, Extended Data Figure 6e–f, 3.67 ± 0.14% in Pregnancy vs 0.41 ± 0.05% in control, P=3.38 ×10−6). Immunostaining for tdT, ZsGreen and Ins on pancreatic sections revealed that all β cells expressed tdT, but not ZsGreen (Fig. 3j,k). The ZsGreen+ cells detected in the pancreas were exocrine cells and non-β cells of the islets (Fig. 3j). Extrapolating from these findings, our data suggests that no β cell neogenesis occurs from progenitors or stem cells after PPX or during pregnancy (Fig. 3l).
Non-β cells do not contribute to adult β cells after PDL
As previous studies have reported that partial duct ligation (PDL) triggers Ins− progenitor cells to differentiate into new β cells17–19, we next induced PDL in 9-week old Ins2-Dre;R26-iCre;IR1 mice after 1 week of Dox treatment that started at 6 weeks of age and was followed by a 2-week washout period (Fig. 4a). Loss of acinar cells in the ligated tail of the pancreas gave the region a more translucent appearance within 1 week after PDL (Fig. 4b). The islets in the tail section of the pancreas could be seen clearly with the naked eye (Fig. 4b). H&E staining also showed severe inflammation and atrophy in the exocrine tissues of the tail section, but not the head, of the pancreas (Fig. 4c), with a marked increase of CK19+ ductal cells in the injured tail (Fig. 4d). Immunostaining for tdT, ZsGreen and Ins revealed that ZsGreen+ cells did not express Ins, and all Ins+ β cells in both the head and tail of the pancreas were tdT+ within 1 week, 2 weeks and 4 weeks after PDL (Fig. 4e, Extended Data Figure 7). In the model of pancreatic duct ligation-induced injuries, we did not detect significant increase in β cell generation compared with those of controls (Extended Data Figure 7c, P=0.63). Of note, the expression levels of Ins among these tdT+ cells were not homogenous, as some cells expressed a weak level of insulin in the islets (Fig. 4e). We did not detect any ZsGreen+ cell expressing Ins in the sham group, similar with that in PDL group (Fig. 4f,g). Altogether, our results showed no obvious β cell neogenesis after PDL, consistent with previous works27,36 (Fig. 4h).
Fig. 4 |. Non-β cells do not give rise to new β cells after partial duct ligation-induced injury.

a, A schematic diagram illustrating the experimental strategy to test for β cell neogenesis after partial-duct ligation (PDL). 6 weeks old mice (both male and female mice were used) were treated with Dox for 1 week. After washout for 2 weeks and PDL injury for 1 week, mice were euthanized (PDL group: n = 5; Sham group: n = 5). b, Whole-mount fluorescent images of pancreas from indicated mice 1 week later after PDL injury. Insert, bright field image. c, H&E staining of pancreatic sections from the tail (top) and the head (bottom) of the pancreas after PDL. d, Immunostaining of pancreatic sections for CK19 from the tail (top) and the head (bottom) of the pancreas after PDL. e, Immunostaining on pancreatic tail (top) and head (bottom) sections for tdT, ZsGreen, and Ins collected after PDL. Arrows indicate tdT+Ins+ β cells. Arrowheads indicate zsGreen+Ins− non-β cells. f, Immunostaining of pancreatic tail sections for tdT, ZsGreen, and Ins collected from sham mice. Arrows indicate tdT+Ins+ β cells. Arrowheads indicate zsGreen+Ins− non-β cells. g, Quantification of the percentage of β cells expressing tdT or ZsGreen in pancreas from sham and PDL mice. Numbers of investigated pancreas were as follows: Sham group, n = 5; PDL-Tail, n = 5; PDL-Head, n = 5, Data are mean ± SEM. h, Illustration showing no contribution of non-β cells to β cells after PDL injury. Scale bars, yellow, 1 mm; white, 100 μm. Each image is representative of 5 individual biological samples.
No evidence of β cell neogenesis after STZ-mediated injury
To test whether β cell loss as a result of diabetes may trigger non-β cell-to-β cell conversion, we treated Ins2-Dre;R26-iCre;IR1 mice with a single injection of β cell-specific toxin streptozotocin (STZ) intraperitoneally following 1 week of Dox administration and then analyzed the pancreases from these mice at either 1, 2 or 4 weeks after depletion (Fig. 5a). As expected, STZ-treated mice exhibited significantly higher fasting blood glucose (5.50 ± 0.43 mmol/L vs. 12.14 ± 0.33 mmol/L, P=3.32 ×10−6, Fig. 5b), and they displayed reduced tdT expression in the islets as demonstrated by whole-mount fluorescent imaging compared with controls (Fig. 5c). Immunostaining for tdT, ZsGreen and Ins on the pancreatic sections showed a striking loss of Ins+ β cells one week after STZ treatment compared to the controls (Fig. 5d,e). In samples collected 2 and 4 weeks after STZ treatment, we observed more Ins+ β cells in the islets compared with the level present at 1 week post-STZ treatment (Fig. 5g,h), possibly indicating a slow recovery of β cells after a severe degree of β cell death(Fig. 5f, P=0.69). However, these Ins+ cells did not express ZsGreen (Fig. 5i), excluding their non-β cell origins. These data showed lack of β cell neogenesis in the adult pancreas following STZ-induced β cell injury (Fig. 5j).
Fig. 5 |. Non-β cells do not generate new β cells during streptozocin-induced β cell loss.

a, A schematic diagram illustrating the experimental strategy to test for β cell neogenesis after streptozocin (STZ)-induced injury. 6 weeks old mice (both male and female mice were used) were treated with Dox for 1 week. After washout for 2 weeks and STZ injury for 1 week, 2 weeks and 4 weeks, mice were euthanized (Sham group, n = 5; +1 week group, n = 5; +2 weeks group, n = 5; +4 weeks group, n = 5). b, Detection of fasting blood glucose level of STZ group and PBS group after 1 week. Numbers of investigated mice were as follows: PBS group, n = 5; STZ group, n = 5, Data are mean ± SEM, P=3.32×10−6, *P<0.05 (unpaired, two-sided, Student’s t-test). c, Whole-mount fluorescent images of pancreatic tissue for tdT+ islets after STZ treatment, compared with that from PBS group. d-e, Immunostaining of pancreatic sections for tdT, zsGreen, and Ins on pancreatic slides collected from Ins2-Dre;R26-iCre;IR1 after treated with PBS (d) and STZ 1 week (e). Arrows indicate tdT+Ins+ β cells. Arrowheads indicate zsGreen+Ins− non-β cells. f, Immunostaining on pancreatic sections for tdT, ZsGreen, and EdU collected after STZ 2 weeks. Arrows indicate EdU+tdT+cells. EdU was injected 12 hours before sacrifice. Right panel shows quantification of the percentage of tdT+ cells with incorporated EdU. Numbers of investigated mice were as follows: PBS group, n = 5; STZ group, n = 5, Data are mean ± SEM; P=0.69, n.s., not significant (unpaired, two-sided, Student’s t-test). g-h, Immunostaining of pancreatic sections for tdT, zsGreen, and Ins on pancreatic slides collected from Ins2-Dre;R26-iCre;IR1 after treated with STZ 2 weeks (g) and 4 weeks (h). Arrows indicate tdT+Ins+ β cells. Arrowheads indicate zsGreen+Ins− non-β cells. i, Quantification of the percentage of β cells expressing tdT or ZsGreen. Numbers of investigated mice were as follows: Sham group, n = 5; +1 week group, n = 5; +2 weeks group, n = 5; +4 weeks group, n = 5, Data are mean ± SEM. j, Illustration showing no contribution of non-β cells to β cells during STZ-induced injury. Scale bars, yellow, 1 mm; white, 100 μm. Each image is representative of 5 individual biological samples.
Non-β cells generate Ins+ cells after extreme β cell loss
To provide a further technical control to demonstrate the capability of our dual-tracing system in detecting the conversion of non-β cells to β cells, we developed an iterated version of IR1 mouse line with an inclusion of the diphtheria toxin receptor (DTR), IR1-DTR (Extended Data Figure 8a). Immunostaining on pancreatic sections of Ins2-Dre;IR1-DTR mice for ZsGreen, tdT, Ins and DTR showed specific expression of tdT and DTR, but not ZsGreen, in Ins+ β cells (Extended Data Figure 8b–d). DT (10 μg/g /day*2) treatment led to a significant reduction of tdT+ β cells compared with the control group without DT (Extended Data Figure 8e–h). However, there are some cells that express DTR but not tdTomato, which could be due to the possibility that the protein degradation rate of tdTomato and DTR may vary in these cells within 2 days after DT treatment. We confirmed that the tdTomato and DTR are co-localized well in β cells without DT treatment (Extended Data Figure 8g,h). Having successfully generated the IR1-DTR line, we crossed it with Ins2-Dre and R26-iCre mice to trace β cells and non-β cells after genetic ablation of β cells by treating with DT (10 μg/g/day*5) after Dox-mediated labeling for 1 week followed by a washout period of 2 weeks (Fig. 6a,b). Immunostaining for ZsGreen, tdT and Ins of pancreatic sections showed a significantly lower number of tdT+ β cells in the DT treatment group compared to the control (Fig. 6c). Over 99% of tdT+ β cells were genetically ablated, which was also confirmed by immunostaining for DTR+ β cells (Fig. 6d). The remaining cells of the islets were largely non-β cells, such as glucagon-producing α cells and somatostatin-producing δ cells (Fig. 6e).
Fig. 6 |. Non-β cells contribute to insulin+ cells after genetic ablation of β cells.
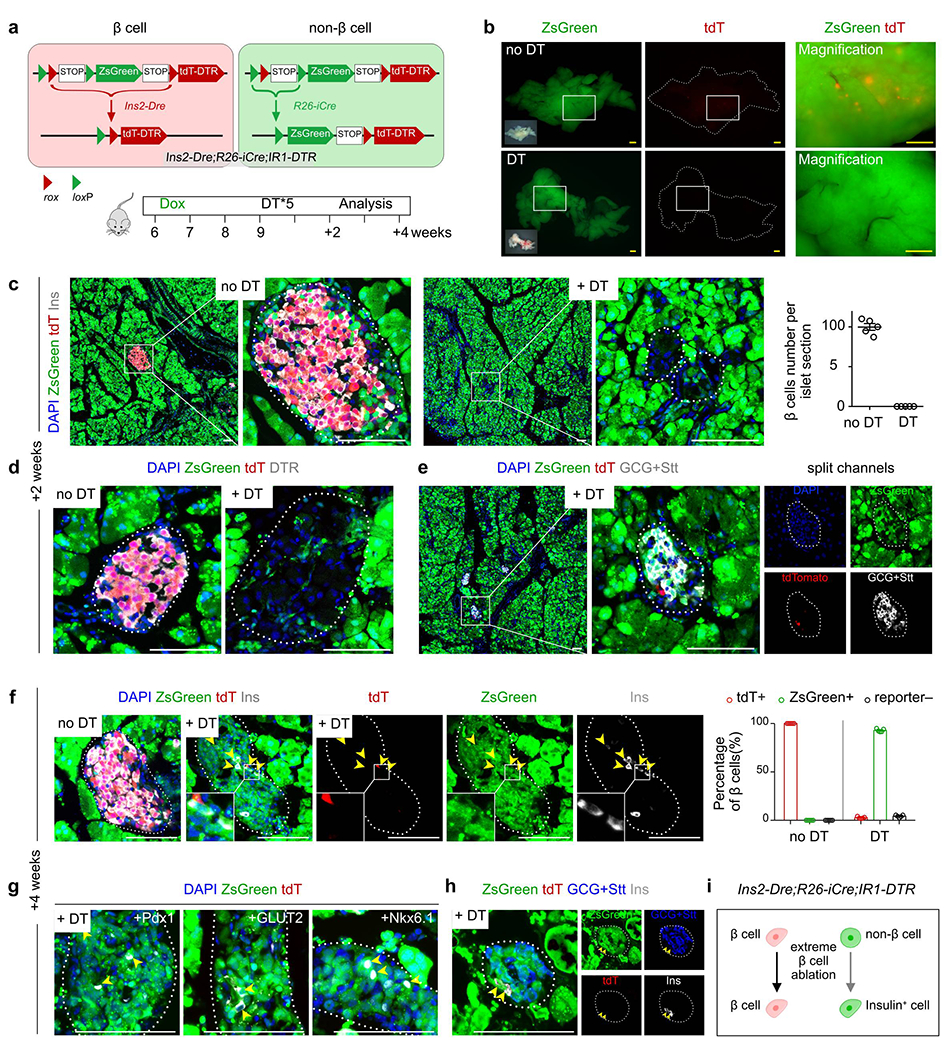
a, A schematic diagram illustrating the experimental strategy to test for β cell neogenesis after severe loss of β cell mass. 6 weeks old mice (both male and female mice were used) were treated with Dox for 1 week. After washout for 2 weeks and Diphtheria toxin (DT) injury for 2 weeks and 4 weeks, mice were euthanized (+2 weeks group, n = 5; +2 weeks Sham group, n = 5; +4 weeks group, n = 5; +4 weeks Sham group, n = 5). b, Whole-mount bright-field and fluorescent images of pancreas from Ins2-Dre;R26-iCre;IR1-DTR mice at 2 weeks post-DT or no DT. c, Immunostaining of pancreatic sections for tdT, zsGreen and Ins at 2 weeks post-DT or no DT. Right panel shows quantification of Insulin+ β cell number per islet sections. Numbers of investigated pancreas were as follows: no DT group, n = 5; DT group, n = 5, Data are mean ± SEM. d, Immunostaining of pancreatic sections for tdT, zsGreen and DTR at 2 weeks post-DT or no DT. e, Immunostaining ofs pancreatic sections for tdT, zsGreen, Glucagon (GCG) and Somatostatin (Stt) at 2 weeks post-DT or no DT. f, Immunostaining of pancreatic sections for tdT, zsGreen and Ins at 4 weeks post-DT or no DT. Right panel shows quantification of the percentage of Insulin+ cells expressing tdT or ZsGreen in DT or no DT groups. Numbers of investigated pancreas were as follows: no DT group, n = 5; DT group, n = 5, Data are mean ± SEM. Arrowheads indicate zsGreen+Ins+ β cells. g, Immunostaining for tdT, zsGreen, Pdx1, GLUT2 and Nkx6.1 on pancreatic sections at 4 weeks post-DT. h, Immunostaining for tdT, zsGreen, GCG, Stt and Ins on pancreatic sections at 4 weeks post-DT. i, Schematic figure showing non-β cells can contribute to Ins+ cells after genetic ablation of β cells. Scale bars, yellow, 1 mm; white, 100 μm. Each image is representative of 5 individual samples.
We next analyzed the pancreas 4 weeks after DT treatment to examine whether new β cells could be regenerated from non-β cells over time. Immunostaining pancreatic sections for ZsGreen, tdT and Ins showed reappearance of Ins+ β cells at 4 weeks after DT (Fig. 6f). Most of these newly formed Ins+ β cells were ZsGreen+, indicating that they originated from pre-labeled ZsGreen+ non-β cells (917 Ins+ β cells from 191 islets in sections, n = 5; Fig. 6f). Quantification of the percentage of Ins+ β cells expressing tdT or ZsGreen showed that while all Ins+ β cells in the control mice were tdT+ZsGreen−, the majority of Ins+ β cells were ZsGreen+ with a minority as tdT+ after DT (Fig. 6f). To further evaluate these newly generated insulin+ cells, we performed immunostaining of other β cell makers with tdT and ZsGreen, we found that some ZsGreen+tdT− cells in the islet expressed other hallmarks of β cells including Pdx1, GLUT2, and Nkx6.1 (Fig. 6g). While ~80% of Ins+tdT−ZsGreen+ cells expressed glucagon and somatostatin, ~20% of Ins+tdT−ZsGreen+ cells have lost glucagon and somatostatin expression at 4 weeks after diphtheria treatment, suggesting both existence of bi-hormonal cells and formation of mono-hormonal β cells in this genetic system. (Fig. 6h). The results from the mice after 4 weeks post-DT treatment mice demonstrated that our dual-tracing genetic system was able to detect conversion of non-β cells to β cells in the adult pancreas, if any exists (Fig. 6i).
Inducible saturated tracing of β cells reveals no neogenesis
It is possible that Ins2-Dre could unexpectedly label very few putative stem cells that transiently express insulin at the fetal stage. In this case, these rare putative stem cells would be labeled as tdT+ by Ins2-Dre;IR1, escaping non-β cell labeling as ZsGreen+ by R26-iCre;IR1. Labeling β cells at a specific time window with an inducible method avoids this caveat caused by the constitutive activation of Dre. We adopted the elegant strategy of lineage tracing at saturation of cell labeling, as reported previously41, to re-assess β cell regeneration in the adult pancreas. In this design, almost every single Ins+ β cell is genetically labeled by Ins-inducible recombinase at a given time after tamoxifen treatment. When Ins+ β cells express a reporter, such as tdT, other non-β cells that contain the putative β cell progenitors do not express insulin and become tdT−. After the pulse-chase period, if tdT− stem cells differentiate into new β cells, they would start to express Ins. However, these newly formed Ins+ β cells would remain as tdT−, as there is no tamoxifen at this stage to activate the Ins-specific inducible recombinase. The efficacy of this strategy in addressing β cell sources largely depends on the efficiency of labeling β cells by the inducible recombinase. In theory, every pre-existing β cell is labeled if the efficiency is 100%, and, therefore, any new β cell differentiated from a putative stem cell would be observed as Ins+tdT−. If the efficiency of β cell labeling is not 100% but very close to 100% (e.g. >99.9%), the newly formed Ins+tdT− β cells differentiated from putative stem cells could be recorded when the percentage of tdT− β cells increases significantly over 0.1%. Therefore, the accuracy in interpretation using this system requires an Ins-specific inducible recombinase line with super efficiency.
To address this requirement, we first tested the labeling efficiency of a previously reported inducible Cre line, Rip-CreER5, and found that it labels a substantial portion of β cells after tamoxifen induction. Although efficient, we wished to reach even higher labeling saturation (i.e. >99.9% to 100%). Consequently, we generated two new mouse lines: an Ins-specific inducible recombinase mouse line and a new responding reporter line, as both recombinase and reporter alleles significantly influence the recombination efficiency42. We first generated a new Ins2-DreER knock-in line by inserting DreER into the insulin gene, replacing the endogenous start codon by a homologous recombination using CRISPR/Cas9 (Fig. 7a). We also generated a new rox knock-in reporter, H11-rox-tdTomato (Fig. 7a) by targeting the rox-stop-rox-tdT cassette on the intergenic Hipp11 (H11) locus of mouse chromosome 1143. By crossing Ins2-DreER with H11-rox-tdT, tamoxifen-induced Dre-rox recombination removes the Stop cassette, resulting in constitutively active expression of tdT in Ins+ cells (Fig. 7b). New β cells derived from stem cells at the chasing stage would not express tdT due to the nature of the pulse-chase strategy. We injected 3 doses of tamoxifen, then analyzed tissue samples at 1 (+1W) or 25 weeks (+25W) afterwards (Fig. 7c). To our surprise, the adult Ins2-Dre;H11-rox-tdTomato mice exhibited an unprecedentedly high labeling efficiency for β cells, albeit with the use of an inducible recombinase. We examined Ins+ β cells for >1000 islets and found that virtually every Ins+ β cell was tdT+ (Fig. 7d and Extended Data Figure 9). These tdT+ cells were exclusively Ins+ β cells (Fig. 7d,e and Extended Data Figure 9), demonstrating the technique’s specificity for lineage tracing of β cells. Examination of the +25W pancreatic tissues revealed that virtually all Ins+ β cells were tdT+ (>99.9%) (Fig. 7f). This saturation of β cell labeling was not due to leakiness of DreER activity, as all Ins+ β cells are tdT− in the pancreas without tamoxifen treatment (Fig. 7g). It should be noted that we found no detectable dilution of tdT+ labeling in β cells from +1W to +25W (Fig. 7d,f), further suggesting that non-β cells did not contribute to any detectable adult β cell biogenesis during tissue homeostasis.
Fig. 7 |. Genetic tracing of β cells by inducible Ins2-DreER;H11-rox-tdT system during homeostasis and after injury.

a, A schematic diagram illustrating generation of Ins2-DreER and H11-rox-tdT knock-in mouse lines by homologous recombination. b, A schematic diagram illustrating genetic lineage tracing of Ins+ β cells by Dre-rox recombination. c, A schematic diagram illustrating the experimental strategy. 6–7 weeks old mice (both male and female mice were used) were treated with Tamoxifen for 3 times. After 1 week and 25 weeks, mice were euthanized (+1 week Tam group, n = 5; +1 week no Tam group, n = 5; +25 weeks Tam group, n = 5; +25 weeks no Tam group, n = 5). d, Whole-mount fluorescent images of pancreas and immunostaining for tdT and Insulin on sections from Ins2-DreER;H11-rox-tdT mice at 1 week after Tam. Right panel shows quantification of labeling efficiency and specificity for β cells. Data are mean ± SEM, n = 5 (Tam group). e, Immunostaining of pancreatic sections for tdT, Glu, Stt, or PP, and the quantification of each (right) indicating the percentage of tdT+ cells expressing these markers. Data are mean ± SEM, n = 5 (Tam group). f,g, Immunostaining for tdT and Insulin on pancreatic sections collected at 25 weeks after Tam (f) or no Tam (g). Quantification data of labeling efficiency for β cells. Data are mean ± SEM, n = 5 (Tam group). h, A schematic diagram illustrating the experimental design to test for β cell neogenesis after PPX in the inducible system. 6 weeks old mice (both male and female mice were used) were treated with Tamoxifen for 3 times. After washout for 2 weeks and PPX injury for 2 weeks, mice were euthanized (PPX group, n = 5; no Tam group, n = 5). i, Whole-mount fluorescence image of the regenerated pancreas (right side of the dotted line) after PPX. j, Immunostaining of pancreatic sections for tdT and Insulin after PPX (left) and quantification of β cells expressing tdT (right) in the Tam-treated and no Tam groups. Numbers of investigated pancreas were as follows: Tam group, n = 5; no Tam group, n = 5, Data are mean ± SEM. k, Illustration showing that non-β cells do not contribute to β cells during homeostasis and after injuries. Scale bars, yellow, 1 mm; white, 100 μm. Each image is representative of 5 individual samples.
Given the super-efficient and highly specific properties of the Ins2-DreER line, we next used it to re-assess the possible differentiation of putative tdT− stem cells into β cells after PPX (Fig. 7h). Whole-mount fluorescent images of the regenerated part of pancreas collected from Ins2-DreER;H11-rox-tdT mice showed tdT+ islets (Fig. 7i). Immunostaining of pancreatic sections for tdT and Ins showed virtually all Ins+ cells were tdT+ after PPX (Fig. 7j). Indeed, quantification data revealed that >99.9% Ins+ cells remained as tdT+ (Fig. 7j), indicating no β cell neogenesis at a detectable level after PPX. We also performed other models including pregnancy, PDL and STZ-induced injuries on Ins2-DreER;H11-rox-tdT and didn’t find β-cell neogenesis (Extended Data Figure 10). Together, the above results based on tracing at saturation argue against β cell neogenesis in the adult pancreas during homeostasis or after injury (Fig. 7k).
Discussion
In this study, we developed a novel genetic lineage tracing system to simultaneously label both the β and non-β cell populations with two distinct and irreversible genetic markers in the adult mouse pancreas. Using non-genetically manipulated injury models for inducing β cell death, cell growth, regeneration and inflammation, we found that newly formed β cells during repair and regeneration were derived from pre-existing β cells, but not non-β cells. Additionally, we adopted a strategy of lineage tracing at saturation by generating a super-efficient β cell labeling tool, and we confirmed that non-β cells did not contribute to any new β cells during homeostasis, pregnancy and after various injuries. These results suggest that stem cell differentiation is unlikely to be the mechanism for β cell regeneration, at least in the injury models tested in this study.
The controversial conclusions regarding the existence of endogenous stem cells or progenitors for β cell conversion in the adult pancreas have also been drawn from genetic lineage tracing studies. The main caveat of these lineage tracing methods is the nonspecific labeling by reporters that are supposed to be activated through Cre recombinase driven under the control of “cell-specific” markers44,45. The discrepancy between studies supporting or refuting the stem cell hypothesis is at least partly due to the specificity and stability of the proposed stem cell markers used in these studies. For example, the evidence in support of Ngn3+ stem cells generating new β cells is predicated on the notion that Ngn3 is not expressed in the pre-existing β cells17. Lack of Ngn3 expression in β cells was the linchpin of this study; however, subsequent follow-up studies have revealed that Ngn3 is actually expressed by both proliferating ducts and pre-existing β cells29, possibly leading to data misinterpretation. Similarly, a handful of previous studies have recorded the cell fate commitment of facultative stem cells to β cells by lineage tracing in mice, in which the data interpretation heavily relies on the specificity of the stem cell markers17–24. In theory, these “stem cell markers” should not be expressed by the pre-existing Ins+ β cells. It should also be noted that a weak expression of stem cell marker genes driving Cre below the sensitivity limit for antibody detection might still induce Cre-loxP recombination in some β cells. The antecedent weak Cre expression in β cells could thus result in unexpected labeling of pre-existing β cells in subsequent studies, giving rise to false positive results and misinterpretation. Therefore, the previous conclusions about the contribution of stem cells in β cell neogenesis require independent re-assessments to more closely scrutinize whether a low-level of β cells were pre-labeled.
The previous studies against progenitor or stem cells mainly used inducible lineage tracing of β cells5, and found no statistically significant dilution of β cell labeling percentage in pulse-chase experiments (~30%-40%), suggesting no differentiation of β cell progenitors. However, this strategy based on statistical evaluation could not exclude small contributions of β cells from putative stem or progenitor cells. Another line of evidence used constitutive Cre driven by insulin promoter and took advantage of mTmG reporter for capturing the transient co-expression of GFP and mTomato during β cell neogenesis, and showed no double fluorescent cells in adult pancreas after various models of β cell loss, growth, and regeneration29. However, it is technically difficult to capture the transient stage during β cell differentiation, especially when very few β cells are derived from progenitors or stem cells. Due to the above reasons, there are possibilities that some undifferentiated stem cells or progenitors, beyond the detection by previous lineage tracing studies, contribute to a small number of β cells during homeostasis or injuries. In this study, we developed a dual recombinases-mediated genetic system to label those non-β cells and examine if they could contribute to β cells during pancreas homeostasis and injuries. Our work is not merely confirmatory. First, this dual genetic system differs significantly from previous studies in that it tracks non-β cells permanently with a fluorescence marker, and evaluation of non-β cells to β cell contribution does not rely on statistical significance, but at single cell basis. Even with very few contributions from stem cells, if this happens minimally (e.g. < 1%), we could readily detect it with positive fluorescence lineage tracing. Secondly, our genetic system enables random labeling of non-β cells using ubiquitously active promoter, thus avoiding relying on one particular stem cell marker. This is important as there may be putative progenitors with unknown markers residing in islets. Our genetic system enables us to examine non-β cell contribution without knowing markers for stem cells. The fate mapping results showed that non-β cells do not generate new β cells in the pancreas during homeostasis, pregnancy or injury. Of note, if most β cells were genetically ablated, we could detect a small number of new β cells arising from non-β cells, indicating that our dual recombinases-mediated genetic tracing system is capable of detecting β cell neogenesis, even if it occurs at low frequency.
In our system, most of the non-β cells are labeled with ZsGreen (>96%), which is more efficient than any other inducible tracing strategy that we know of. The promoter of Rosa26 used in our system is widely employed in genetic lineage tracing and has been shown to be ubiquitously active in tissues. The genetic labeling strategy is therefore entirely random and does not exclude any particular cell populations. Thus, any potential stem cell population, if it exists, will be labeled and detected. We used the Ins2-Dre knock-in line to label all Ins+ β cells as tdT+, and non-β cells as ZsGreen+ by the exclusive dual reporter IR1. Although all tdT+ cells were Ins+ in the initial stage, we did observe a subset of tdT+ cells that expressed weak or non-existent levels of insulin after severe injuries. This was likely due to dedifferentiation of β cells, which has been reported as a mechanism to compensate for the loss of proper β cell mass in different models of diabetes in mice37. Another possibility is that very few non-β cells at the adult stage were labeled as tdT+ as they may have transiently expressed insulin and as such were labeled during early development by constitutive Dre. To address this issue of constitutive Dre, we took advantage of a strategy called inducible lineage tracing at saturation41. We unexpectedly found an inducible Ins2-DreER mouse line that worked super-efficiently after it was crossed with H11-rox-tdTomato to label virtually all Ins+ β cells at the adult stage. We did not see any flux or dilution of the tdT surrogate marker in β cell labeling during pancreatic homeostasis or after injuries, demonstrating that there was no β cell progenitor accounting for any detectable β cell neogenesis. Furthermore, transdifferentiation of non-β cells such as α, δ, or exocrine cells, did not contribute to adult β cells in our non-genetically manipulated injury models. Taken together, our findings leverage the recent advances in the dual-tracing genetic technology coupled with the inducible lineage tracing at saturation approach to show that adult β cell regeneration is predominantly mediated by self-replication, rather than by stem or progenitor cell differentiation.
Methods
Mice.
All mouse experiments were carried out in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the Institute of Biochemistry and Cell Biology, and the Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Science. Mice were maintained on a C57BL6/ICR mixed background, housed in standard cages and were maintained on a 12-hour light/dark cycle and fed a normal diet (Jiangsu Xietong Pharmaceutical Bioengineering Co. Ltd, 1010085). Unless otherwise stated, both male and female heterozygote mice (6–8 weeks old) were randomly allocated to control and experimental groups. Littermate controls were used when appropriate. Only female mice were used in the Pregnancy group and the sham group. R26-rtTA, TRE-Cre and IR1 mouse lines were described previously5,33,46–50. Ins2-Dre and Ins2-DreER were generated using the CRISPR/Cas9 technology by Shanghai Biomodel Organism Co., Ltd. A cDNA encoding Dre or DreER was targeted to the translational start codon ATG of Ins gene by homologous recombination, replacing the endogenous translational start codon ATG, followed by Woodchuck post-transcriptional regulatory element (WPRE) and polyA. PCR primers, spanning the genomic DNA, were designed to test the correctly targeted allele (Ins2-Dre 5’mut-F: AAGATACTAGGTCCCCAACTGCAAC, 5’mut-R: TTCAGCATGGCGTAGTGCTTGTCG, 5’ wt-F: AAACAGCAAAGTCCAGGGGG, 5’wt-R: ATGGGTGTGTAGAAGAAGCCACGC; Ins2-DreER 5’mut-F: GATCCGCTACAATCAAAAACCATCAGC, 5’mut-R: TCCCGGGCCTCCACC, 5’ wt-F: CCTCTCTTACGTGAAACTTTTGCTATCCTC, 5’wt-R: TCAGACAGAGGAGGCAGGCCA). H11-rox-tdTomato was generated using the CRISPR/Cas9 technology by Shanghai Biomodel Organism Corporation. A cDNA encoding rox-Stop-rox-tdTomato was targeted to the Hipp11 site by homologous recombination, followed by WPRE and polyA. PCR primers spanning the genomic DNA were designed to test the correctly targeted allele (H11-rox-tdTomato 5’mut-F: CACTCTACTGGAGGAGGACAAACTG, 5’mut-R: ACAACGAGGACTACACCATCGTG, 5’ wt-F: TGGAGGAGGACAAACTGGTCAC, 5’wt-R: TTCCCTTTCTGCTTCATCTTGC). IR1-DTR was generated using the CRISPR/Cas9 technology by Shanghai Biomodel Organism Corporation. A cDNA encoding CAG-loxP-rox-Stop-loxP-ZsGreen-Stop-rox-tdTomato-P2A-DTR cassette was targeted to the Rosa26 site by homologous recombination. PCR primers spanning the genomic DNA were designed to test the correctly targeted allele (IR1-DTR, 3’mut-F: GGCACGCTGATCTACAAGGTGAA, 3’mut-R: GTGCTTGTGGCTTGGAGGATAA, 5’ wt-F: TTGGAGGCAGGAAGCACTTG, 5’wt-R: CCGACAAAACCGAAAATCTGTG). These new mouse lines will be deposited on commercial animal company and also be available upon reasonable request directly. Genomic DNA was extracted from a mouse tail snip and prepared for genotyping by Proteinase K lysis, isopropanol precipitation and 70% ethanol washing. The start points for each experiment involved 6-8 weeks old adult mice. For adult mouse treatment, doxycycline (Dox) was dissolved in ddH2O (2 mg/ml) and to feed mice for 1 week. For embryo mouse treatment, doxycycline was dissolved in ddH2O (20 mg/ml) and introduced by gavage (0.1 mg/g) to pregnant mice 2 times in 3 days. For postnatal mouse treatment, doxycycline (Dox) was dissolved in ddH2O (20mg/ml) and introduced by gavage (0.1 mg/g) 2 times. To assess cell proliferation, mice were injected subcutaneously with EdU (Invitrogen, C10338, 10 μg/g) dissolved in PBS 24 hours or 12 hours before sacrifice. Tamoxifen (Tam) was dissolved in corn oil (20 mg/ml) and introduced by gavage (0.1 mg/g) at the indicated time points. The β cell toxin Streptozotocin (STZ) was dissolved as previously described3 and introduced by intraperitoneal injection (150mg/kg body weight). Blood glucose levels of different groups of mice were measured after a 12-hour fasting period. Diphtheria toxin (DT) was dissolved in PBS (1 μg/μl) and introduced by intraperitoneal injection (10 μg/g) per mouse, 2 times each, to induce some β cell loss and 5 times to induce extreme β cell loss. The newly generated mice lines in this study would be deposited in the commercial animal company, and would also be provided upon reasonable request directly (contact Dr. Bin Zhou, zhoubin@sibs.ac.cn).
Partial pancreatectomy (PPX).
PPX was performed as previously described29,35. Briefly, adult mice were anesthetized with isoflurane gas and a midline abdominal incision was made. The mesenteric connections to the stomach, small intestine, and retroperitoneum were partially broken. The splenic lobe of the pancreas was moved and then the pancreatic tissue, bordered by the spleen and stomach, but not including the small flap of pancreas attached to the pylorus, was removed. The sham group underwent laparotomy. All mice received a standard laboratory chow diet and tap water postoperatively.
Pancreatic duct ligation (PDL).
PDL was performed as previously described29. Briefly, adult mice were anesthetized with isoflurane gas and a midline abdominal incision was made. The duodenum and the head of the pancreas were lifted off the retroperitoneum. The pancreatic duct at the left side of the portal vein, which separates the splenic and gastro-duodenal lobes were carefully ligated. Sham group underwent laparotomy. All mice received a standard laboratory chow diet and tap water postoperatively.
Whole-mount microscopy.
The collected tissues at the indicated time points were washed in phosphate-buffered saline (PBS, pH=7.4) twice and set down gently on an agar gel in a petri dish. The whole-mount bright field and fluorescent images were taken using the Zeiss stereo microscope (AxioZoom V16), and automated Z-stack images were acquired to determine magnification of the boxed region.
Immunofluorescent staining.
Immunofluorescent staining was performed as previously described51. Briefly, tissues were collected in PBS, fixed in 4% paraformaldehyde (PFA) for 0.5-1 hour at 4°C, washed in PBS 3 times at room temperature and dehydrated in 30% sucrose overnight at 4°C. The tissues were then embedded in optimum cutting tissue (OCT) for 1h at 4°C, and frozen in block with OCT and sectioned (Thermo HM525 cryosection machine) or stored at −80°C. 10 μm-thick cryosections were collected on slides, washed in PBS, incubated in 5% normal donkey serum (NDS) in PBST (PBS with 0.1% Triton X-100) for 0.5 hours at room temperature and incubated with primary antibodies diluted in 2.5% NDS diluted in PBST overnight at 4°C. All primary antibodies were commercially available reagents: ZsGreen (Clontech, 632474, 1:1000), Insulin (Dako, A0564, 1:500), Glucagon (Sigma, SAB4501137, 1:500), Somatostatin (Santa Cruz, sc-47707, 1:500), Somatostatin (Abcam, ab-111912, 1:500), Pancreatic polypeptide (Abcam, ab77192, 1:500), Pancreatic polypeptide (Abcam, ab255827, 1:500), E-cadherin (Cell Signaling, 3195, 1:500), Amylase (Sigma, A8273, 1:500), Cytokeratin 19 (Developmental Studies Hybridoma Bank, TROMA-III, 1:200), PDGFRa (R & D, AF1062, 1:500), VE-cadherin (R & D, AF1002, 1:100), Lymphatic vessel endothelial hyaluronan receptor 1 (Abcam, ab14917, 1:500), EdU (Invitrogen, C10338, 1:400), DTR (R & D, AF-259-NA, 1:200), GLUT2 (Abcam, ab54460, 1:500), Nkx6.1 (Abcam, ab221549, 1:500), Pdx1 (Abcam, ab47267, 1:500). After washing, signals were developed with the secondary Alexa fluorescent conjugated antibodies (Invitrogen, 1:1000) for 0.5 hours at room temperature and the cell nuclei were counterstained with DAPI. After, slides were rinsed with PBS, and they were mounted with fluorescence-protecting mounting medium (Vector Lab). Images were obtained by a Nikon confocal microscopy system (Nikon A1) and image data were analyzed by the ImageJ (NIH) software.
H&E staining.
H&E staining was performed as previously described52. Briefly, 10-μm-thick cryosections were incubated in Hematoxylin A solution for 3 mins, washed with running tap water for 3 times, rinsed in 1% concentrated hydrochloric acid diluted in 70% ethanol for 1 min and washed with water 3 times. The slides were then incubated in 1% ammonia water for 1 min, washed 3 times, stained with Eosin-Y solution for 8-10 s, dehydrated in a series of ethanol and xylene, and lastly, mounted with neural balsam. All Image data were acquired using an Olympus microscope (Olympus, DP72).
Statistical analysis.
All mice were randomly assigned to different groups and all data were obtained from 5 individual samples, as indicated in each experiment. For all experimental data, calculations were performed by Prism (GraphPad software, La Jolla, CA). Unpaired two-tailed Student’s t-tests were performed to analyze the P value for single comparison between two groups. Significance was accepted when P < 0.05. All data are presented as mean ± SEM. All experiments were done in blinded fashion, and no predetermination was done for sample size.
Extended Data
Extended Data figure 1.
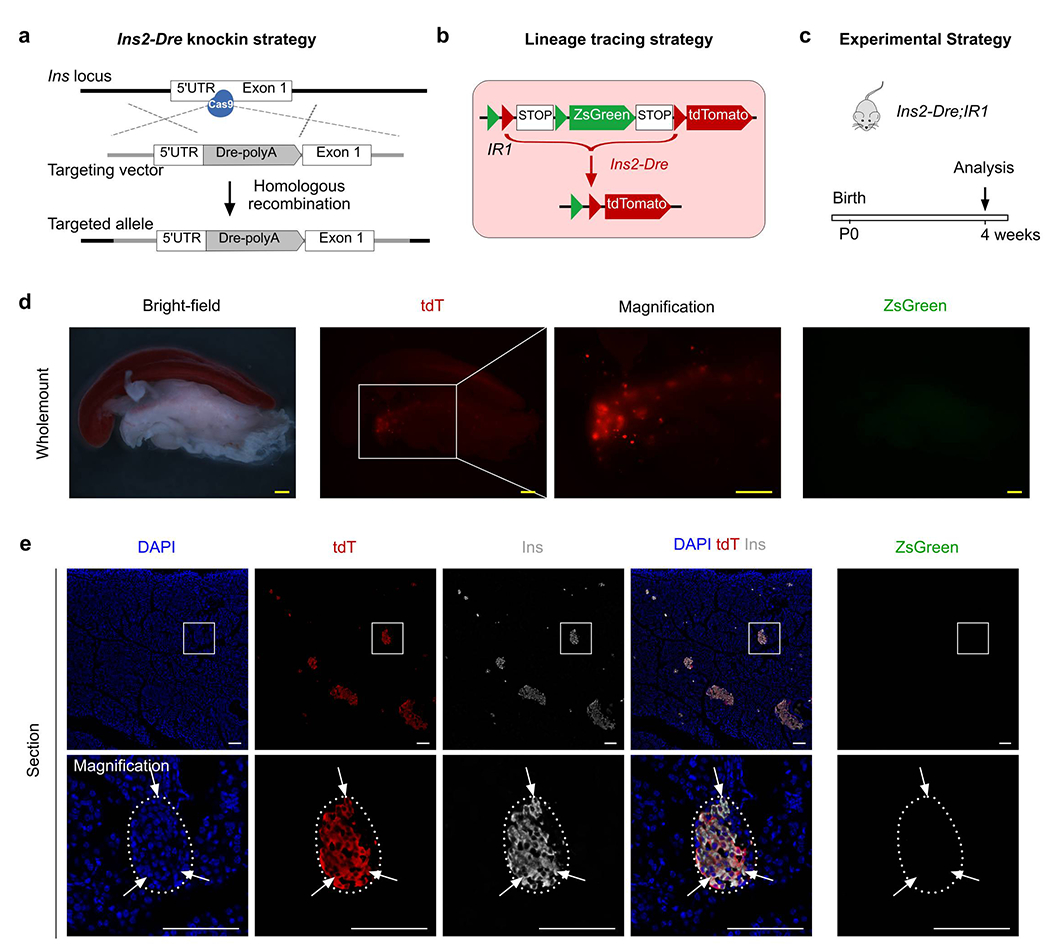
Generation and characterization of Ins2-Dre mouse allele.
(a) A Schematic diagram illustrating knock-in strategy for Ins2-Dre by homologous recombination using CRISPR/Cas9. (b) A Schematic diagram illustrating the strategy for labeling pancreatic β cells by Ins2-Dre using interleaved reporter 1 (IR1). (c) A Schematic diagram illustrating the experimental strategy. (d) Whole-mount bright-field and fluorescent images of pancreas from Ins2-Dre;IR1. (e) Immunostaining for tdT, zsGreen, and Ins on pancreatic slides collected from Ins2-Dre;IR1. Arrows indicate tdT+Ins+ β cells. Scale bars, yellow, 1 mm; white, 100 μm. Each image is representative of 5 individual biological samples.
Extended Data Figure 2.

Ins2-Dre;R26-iCre;IR1 efficiently labels most of cells in pancreas.
(a-b) Immunostaining for tdT and zsGreen on slides collected from Ins2-Dre;R26-iCre;IR1 mice at 7 weeks old after Dox treatment. Boxed regions in (a) are magnified in (b). Scale bars, yellow, 1mm; white, 100 μm. Each image is representative of 5 individual biological samples.
Extended Data Figure 3.
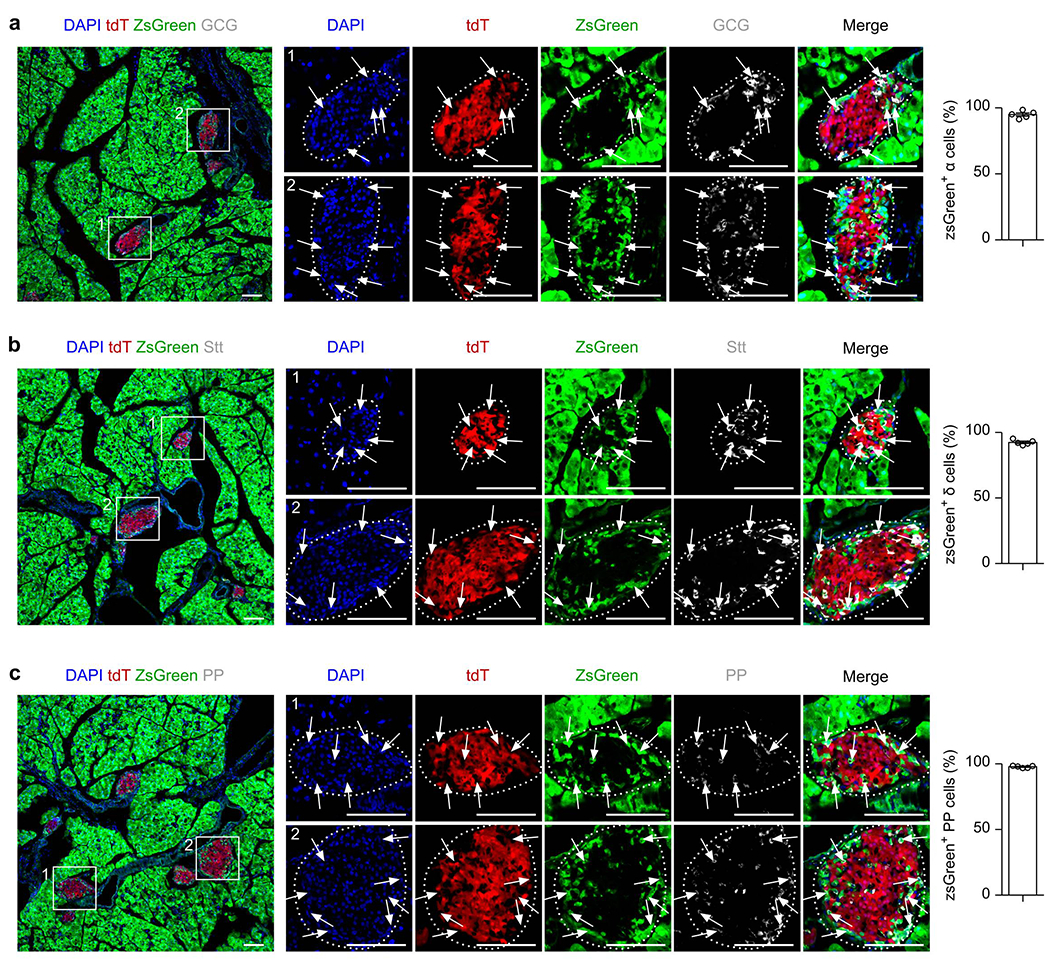
R26-iCre efficiently labels most of pancreatic endocrine glands.
(a-c) Immunostaining for tdT, zsGreen, Glucagon (GCG, a), Somatostatin (Stt, b) and Pancreatic Polypeptide (PP, c) on slides collected from Ins2-Dre;R26-iCre;IR1 mice at 7 weeks old after Dox treatment. Arrows indicate zsGreen+ cell lineages. Right panel is the quantification of the percentage of indicated non-β cell lineages expressing zsGreen. Data are mean ± SEM, n = 5. Scale bars, 100 μm. Each image is representative of 5 individual biological samples.
Extended Data Figure 4.

R26-iCre efficiently labels most of cell lineages in pancreatic exocrine gland.
(a-f) Immunostaining for tdT, zsGreen and E-cadherin (E-cad), or Amylase (Amy), Cytokeratin 19 (CK19), Vascular endothelial cadherin (VE-cad), Lymphatic vessel endothelial hyaluronan receptor 1 (Lyve1) and Platelet derived growth factor receptor a (Pdgfra) on slides collected from Ins2-Dre;R26-iCre;IR1 mice at 7 weeks old after Dox treatment. Right panel is the quantification of the percentage of indicated non-β cell lineages expressing zsGreen. Data are mean ± SEM, n = 5 (Dox group). Scale bars, 100 μm. Each image is representative of 5 individual biological samples.
Extended Data Figure 5.
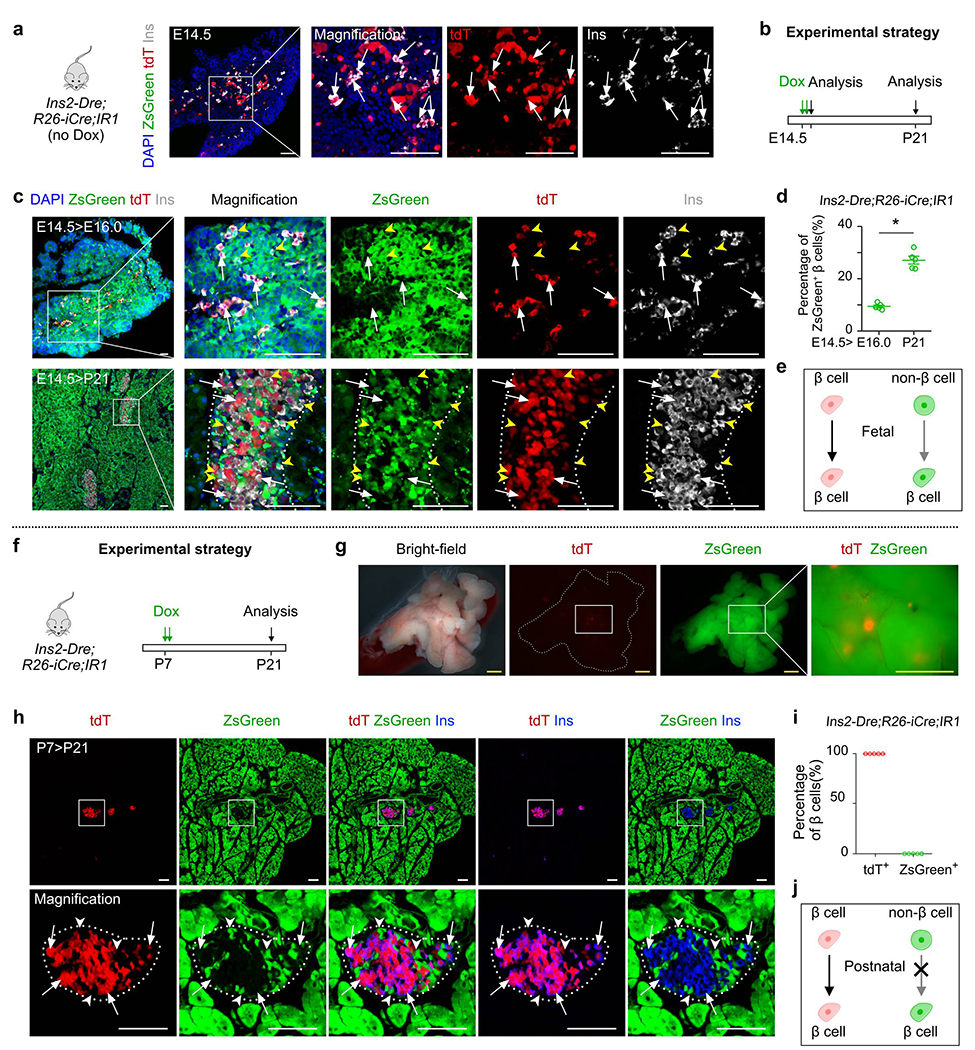
Non-β cells contribute to β cells in embryonic stage.
(a) Immunostaining for tdT, zsGreen, and Ins on pancreatic sections of Ins-Dre;R26-iCre;IR1 mice without Dox treatment at E14.5. (b) A schematic figure showing experimental strategy of Dox injection and analysis. (c) Immunostaining for tdT, zsGreen, and Ins on pancreatic sections of Ins-Dre;R26-iCre;IR1 mice at E16.0 (top) and P21 (bottom) treated with Dox at E14.5. White arrows indicate tdT+Ins+ β cells. Yellow arrowheads indicate zsGreen+Ins+ β cells. (d) Quantification of the percentage of zsGreen+ β cells from indicated mice. Numbers of investigated mice were as follows: E14.5>E16.0 group, n = 5; E14.5>P21 group, n = 5, Data are mean ± SEM, P=1.12 ×10−4, *P<0.05 (unpaired, two-sided, Student’s t-test). (e) Illustration showing contribution of non-β cells to β cells in embryonic stage. (f) A schematic figure showing experimental strategy of Dox injection and analysis. (g) Whole-mount bright-field and fluorescent images of pancreas from Ins-Dre;R26-iCre;IR1 mice with Dox treatment. (h) Immunostaining for tdT, zsGreen, and Ins on pancreatic sections of Ins-Dre;R26-iCre;IR1 mice at P21 with Dox treatment at P7. Arrows indicate tdTIns+ β cells. Arrowheads indicate zsGreen+Ins− non-β cells. (i) Quantification of the percentage of β cells expressing tdT or zsGreen from indicated mice. Data are mean ± SEM, n=5 (P7>P21 group). (j) Illustration showing no contribution of non-β cells to β cells in postnatal stage. Scale bars, yellow, 1 mm; white, 100 μm. Each image is representative of 5 individual samples.
Extended Data Figure 6.
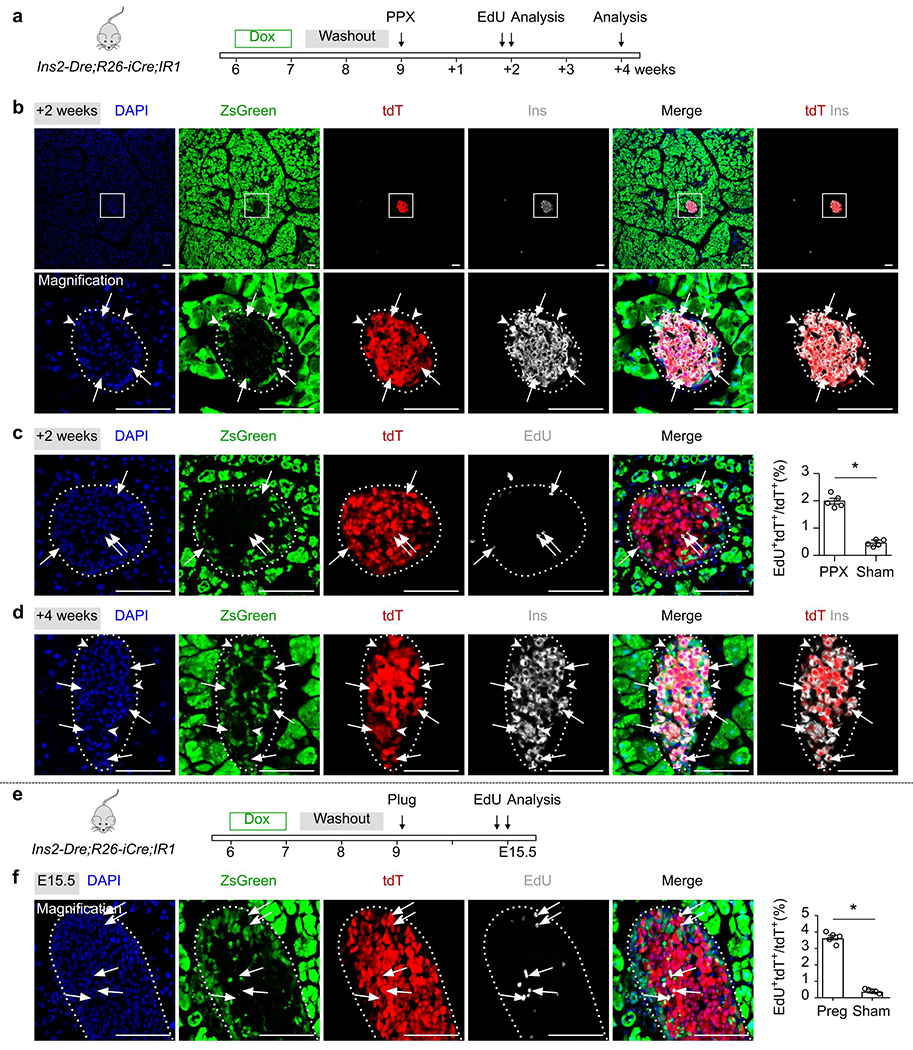
β cells proliferated after partial pancreatectomy and during pregnancy.
(a) A schematic diagram illustrating the experimental strategy for Dox induction, partial pancreatectomy (PPX) and analysis. (b) Immunostaining for tdT, ZsGreen and Ins on pancreatic sections collected after PPX 2 weeks. Arrows indicate tdT+Ins+ β cells. Arrowheads indicate zsGreen+Ins− non-β cells. (c) Immunostaining on pancreatic sections for tdT, ZsGreen and EdU collected after PPX 2 weeks. Arrows indicate EdU+tdT+cells. EdU was injected 12 hours before sacrifice. Right panel shows quantification of the percentage of tdT+ cells with incorporated EdU. Numbers of investigated mice were as follows: PPX group, n = 5; Sham group, n = 5, Data are mean ± SEM, P=1.22 ×10−5, *P<0.05 (unpaired, two-sided, Student’s t-test). (d) Immunostaining for tdT, ZsGreen and Ins on pancreatic sections collected after PPX 4 weeks. Arrows indicate tdT+Ins+ β cells. Arrowheads indicate zsGreen+Ins− non-β cells. (e) A schematic diagram illustrating experimental strategy of Dox induction, pregnancy and analysis. (f) Immunostaining for tdT, ZsGreen and EdU on pancreatic sections collected after pregnancy. Arrows indicate EdU+tdT+ cells. EdU was injected 12 hours before sacrifice. Right panel shows quantification of the percentage of tdT+ cells with incorporated EdU. Numbers of investigated mice were as follows: Pregnancy group, n = 5; Sham group, n = 5, Data are mean ± SEM, P=3.38 ×10−6, *P<0.05 (unpaired, two-sided, Student’s t-test). Scale bars, 100 μm. Each image is representative of 5 individual samples.
Extended Data Figure 7.
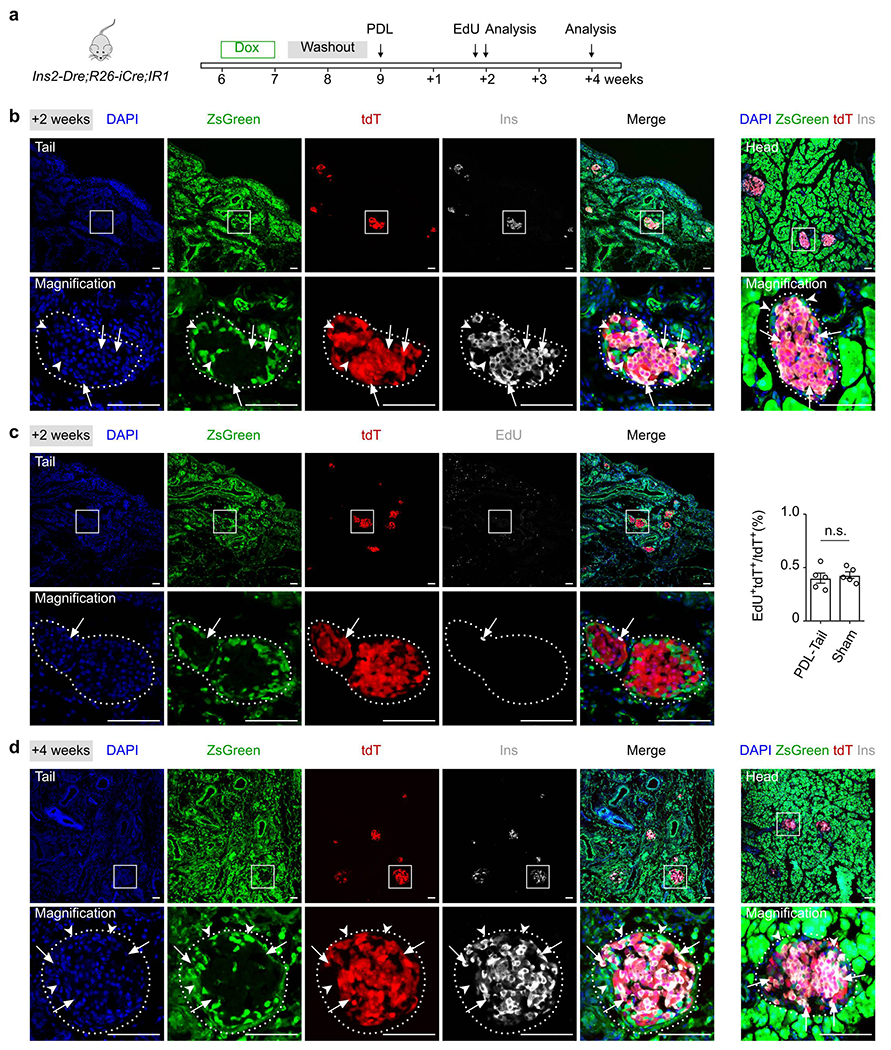
β cells are not generated from non-β cells after pancreatic ductal ligation.
(a) A schematic diagram illustrating experimental strategy of Dox induction, pancreatic ductal ligation (PDL) and analysis. (b) Immunostaining for tdT, ZsGreen and Ins on pancreatic tail (left) and head (right) sections collected after PDL 2 weeks. Arrows indicate tdT+Ins+ β cells. Arrowheads indicate zsGreen+Ins− non-β cells. (c) Immunostaining for tdT, ZsGreen and EdU collected after PDL. Arrowheads indicate EdU+tdT+ cells. EdU was injected 12 hours before sacrifice. Right panel shows quantification of the percentage of tdT+ cells with incorporated EdU. Numbers of investigated mice were as follows: PDL group, n = 5; Sham group, n = 5, Data are mean ± SEM, n.s., not significant (unpaired, two-sided, Student’s t-test). (d) Immunostaining for tdT, ZsGreen and Ins on pancreatic tail (left) and head (right) sections collected after PDL 4 weeks. Arrows indicate tdT+Ins+ β cells. Arrowheads indicate zsGreen+Ins− non-β cells. Scale bars, 100 μm. Each image is representative of 5 individual samples.
Extended Data Figure 8.

Generation and characterization of IR1-DTR.
(a) A schematic diagram illustrating knock-in strategy for generation of IR1-DTR by homologous recombination using CRISPR/Cas9. (b) Strategy for labeling pancreatic β cells by Ins2-Dre;IR1-DTR mouse line. (c) Immunostaining for tdT, zsGreen and Ins on pancreatic slides collected from 8 weeks old Ins2-Dre;IR1-DTR. (d) Immunostaining for tdT, zsGreen, and DTR on pancreatic slides collected from 8 weeks old Ins2-Dre;IR1-DTR. (e) Schematic figure showing experimental strategy of DT injection and analysis. (f) Whole-mount bright-field and fluorescent images of pancreas from Ins2-Dre;IR1-DTR mice. (g) Immunostaining for tdT, zsGreen, and Ins on pancreatic sections of Ins2-Dre;IR1-DTR mice. (h) Immunostaining for tdT, zsGreen, and DTR on pancreatic sections of Ins2-Dre;IR1-DTR mice. Scale bars, yellow, 1 mm; white, 100 μm. Each image is representative of 5 individual biological samples.
Extended Data Figure 9.
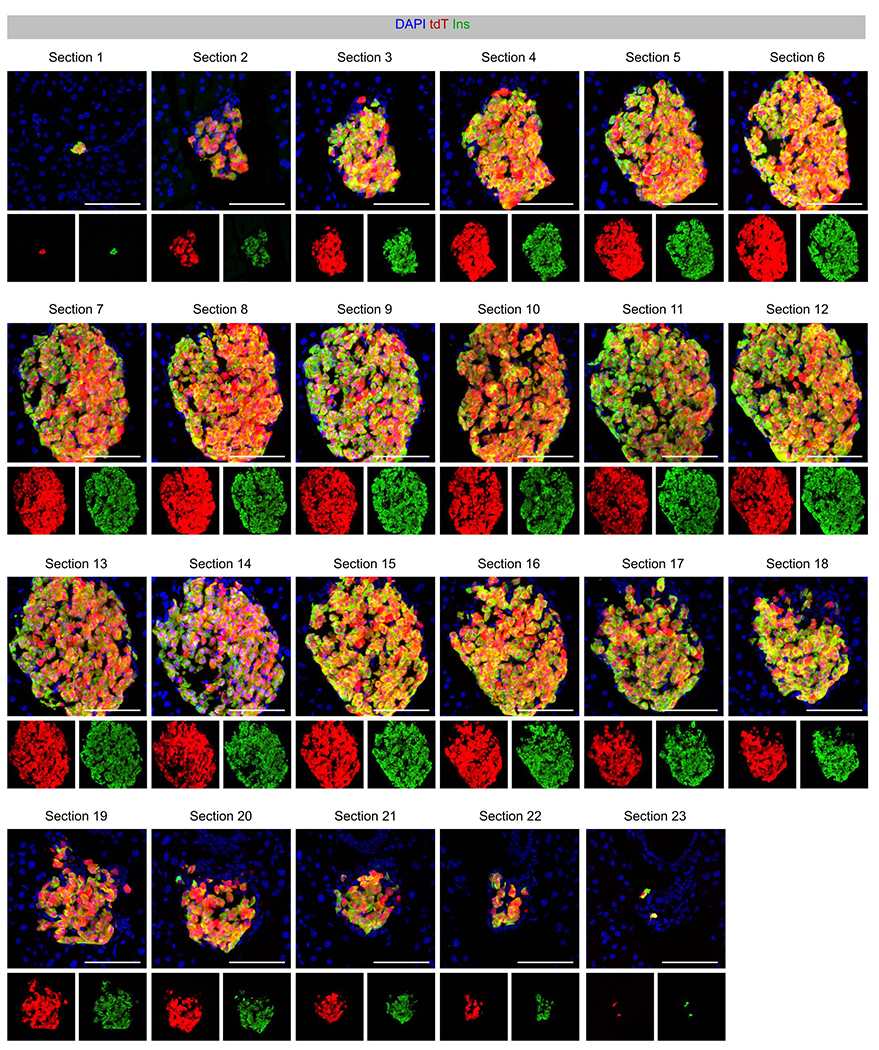
Ins2-DreER specifically and efficiently labels β cells in the adult pancreas.
Immunostaining for tdT and Insulin on consecutive sections (section 1-23) of pancreas islets of Ins2-DreER;H11-rox-tdT mice treated with Tam. Scale bars, 100 μm. This data is representative of 5 individual biological samples.
Extended Data Figure 10.
Analysis of β cells after injuries by inducible Ins2-DreER;H11-rox-tdT system.
(a) A schematic diagram illustrating the experimental design to test for β cell neogenesis during pregnancy in the inducible system. (b) Immunostaining for tdT and Ins on pancreatic slides collected from Ins2-DreER;H11-rox-tdT during pregnancy (Preg15.5). (c) Quantification of β cells expressing tdT in the indicated mice. Numbers of investigated mice were as follows: Pregnancy group, n = 5; Sham group, n = 5, Data are mean ± SEM, n.s., not significant (unpaired, two-sided, Student’s t-test). (d) A schematic diagram illustrating the experimental design to test for β cell neogenesis after pancreatic ductal ligation (PDL). (e) Immunostaining for tdT and Ins on pancreatic slides collected from Ins2-DreER;H11-rox-tdT after PDL. (f) Quantification of β cells expressing tdT in the indicated mice. Numbers of investigated mice were as follows: PDL group, n = 5; Sham group, n = 5, Data are mean ± SEM, n.s., not significant (unpaired, two-sided, Student’s t-test). (g) A schematic diagram illustrating the experimental design to test for β cell neogenesis after streptozocin (STZ)-induced injury. (h) Immunostaining for tdT and Ins on pancreatic slides collected from Ins2-DreER;H11-rox-tdT after streptozocin (STZ)-induced injury. (i) Quantification of β cells expressing tdT in the indicated mice. Numbers of investigated mice were as follows: STZ group, n = 5; Sham group, n = 5, Data are mean ± SEM, n.s., not significant (unpaired, two-sided, Student’s t-test). Scale bars, yellow, 1 mm; white, 100 μm. Each image is representative of 5 individual biological samples.
Fig. 8 |. Non-β cells do not generate new β cells in adults under pathological conditions.
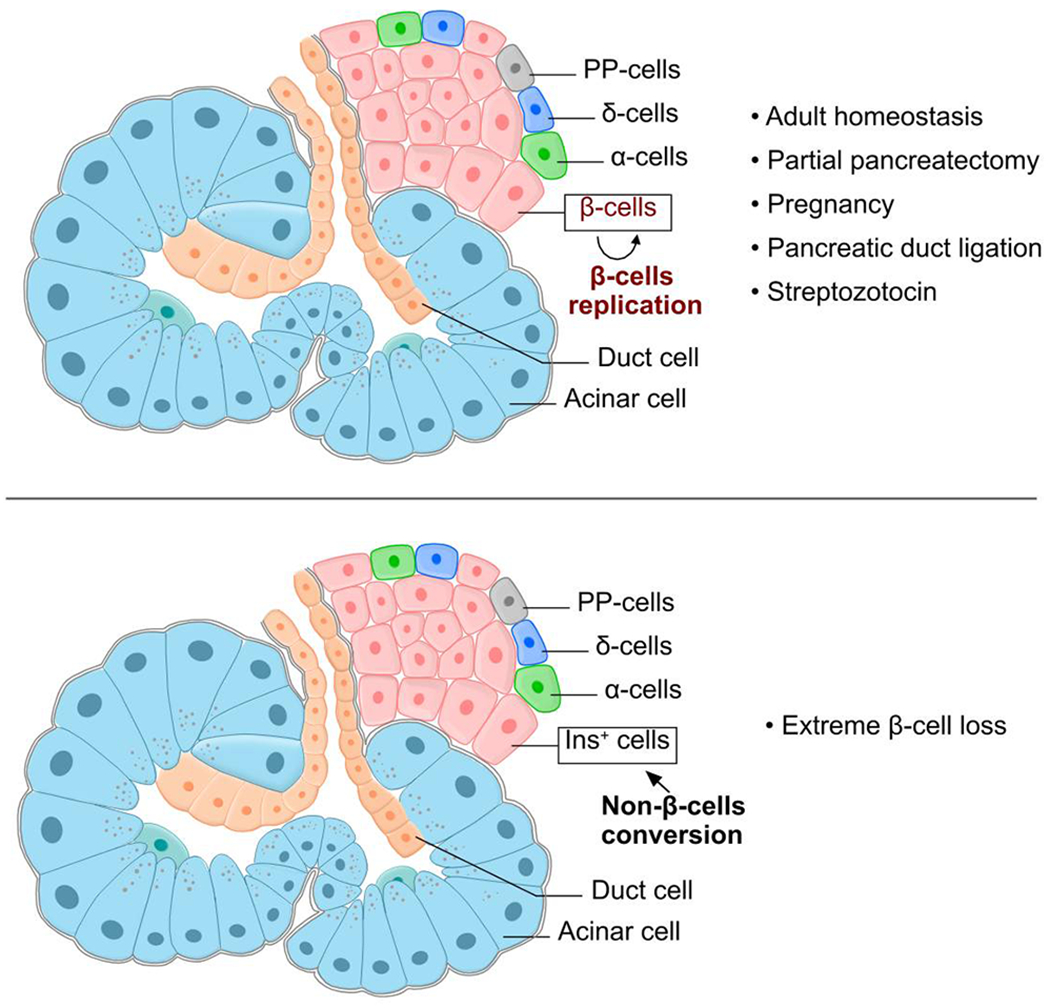
Cartoon image showing labeling of β cells and non-β cells simultaneously. Non-β cells, including α-cells, δ-cells, PP-cells, acinar cell, duct cells and et al., do not generate new β cells under homeostatic and pathological conditions (top). Non-β cells do generate new Ins+ cells in adults after extreme loss of existing β cells (bottom).
Acknowledgments
We thank Shanghai Biomodel Organism Co., Ltd. for mouse generation, and Hongkui Zeng for reporter mice. This study was supported by the National key Research & Development Program of China (2019YFA080200, 2019YFA011040, 2018YFA010810, and 2017YFC1001303), Strategic Priority Research Program of the Chinese Academy of Sciences (CAS, XDB19000000 and XDA16010507), National Science Foundation of China (31730112, 82088101, 32050087, 91849202, 31625019, 31922032, 81872241, and 31900625), Key Project of Frontier Sciences of CAS (QYZDB-SSW-SMC003), International Cooperation Fund of CAS, Shanghai Science and Technology Commission (20QC1401000, 19JC1415700, 19YF1455300, and 19ZR1479800), China Postdoctoral Science Foundation, National Postdoctoral Program for Innovative Talents (BX20190343), the Pearl River Talent Recruitment Program of Guangdong Province (2017ZT07S347), Royal Society-Newton Advanced Fellowship, AstraZeneca, Boehringer-Ingelheim, Sanofi-SIBS Fellowship, SIBS President Fund, and the support from the XPLORER PRIZE.
Footnotes
Competing interests
The authors declare no competing financial interests.
Data availability
The data that support the plots within this paper and other findings of this study are available from the corresponding author upon reasonable request.
References
- 1.McCarthy MI Genomics, type 2 diabetes, and obesity. N Engl J Med 363, 2339–2350 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Butler AE et al. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52, 102–110 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Zhou Q & Melton DA Pancreas regeneration. Nature 557, 351–358 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aguayo-Mazzucato C & Bonner-Weir S Pancreatic β Cell Regeneration as a Possible Therapy for Diabetes. Cell Metab 27, 57–67 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dor Y, Brown J, Martinez OI & Melton DA Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429, 41–46 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Georgia S & Bhushan A Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest 114, 963–968 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teta M, Rankin MM, Long SY, Stein GM & Kushner JA Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell 12, 817–826 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Nir T, Melton DA & Dor Y Recovery from diabetes in mice by beta cell regeneration. J Clin Invest 117, 2553–2561 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hao E et al. Beta-cell differentiation from nonendocrine epithelial cells of the adult human pancreas. Nat Med 12, 310–316 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Thorel F et al. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 464, 1149–1154 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chera S et al. Diabetes recovery by age-dependent conversion of pancreatic delta-cells into insulin producers. Nature (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Hasani K et al. Adult duct-lining cells can reprogram into β-like cells able to counter repeated cycles of toxin-induced diabetes. Dev Cell 26, 86–100 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Courtney M et al. The inactivation of Arx in pancreatic α-cells triggers their neogenesis and conversion into functional β-like cells. PLoS Genet 9, e1003934 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furuyama K et al. Diabetes relief in mice by glucose-sensing insulin-secreting human α-cells. Nature 567, 43–48 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Q, Brown J, Kanarek A, Rajagopal J & Melton DA In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 455, 627–632 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyazaki S, Tashiro F & Miyazaki J Transgenic Expression of a Single Transcription Factor Pdx1 Induces Transdifferentiation of Pancreatic Acinar Cells to Endocrine Cells in Adult Mice. PLoS One 11, e0161190 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu X et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 132, 197–207 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Inada A et al. Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc Natl Acad Sci U S A 105, 19915–19919 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan FC et al. Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development 140, 751–764 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin L et al. Cells with surface expression of CD133highCD71low are enriched for tripotent colony-forming progenitor cells in the adult murine pancreas. Stem Cell Res 16, 40–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rovira M et al. Isolation and characterization of centroacinar/terminal ductal progenitor cells in adult mouse pancreas. Proc Natl Acad Sci U S A 107, 75–80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Criscimanna A et al. Duct cells contribute to regeneration of endocrine and acinar cells following pancreatic damage in adult mice. Gastroenterology 141, 1451–62, 1462.e1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El-Gohary Y et al. Intraislet Pancreatic Ducts Can Give Rise to Insulin-Positive Cells. Endocrinology 157, 166–175 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang D et al. Long-Term Expansion of Pancreatic Islet Organoids from Resident Procr+ Progenitors. Cell 180, 1198–1211.e19 (2020). [DOI] [PubMed] [Google Scholar]
- 25.Solar M et al. Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev Cell 17, 849–860 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Kopinke D & Murtaugh LC Exocrine-to-endocrine differentiation is detectable only prior to birth in the uninjured mouse pancreas. BMC Dev Biol 10, 38 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kopp JL et al. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development 138, 653–665 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kopinke D et al. Lineage tracing reveals the dynamic contribution of Hes1+ cells to the developing and adult pancreas. Development 138, 431–441 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao X et al. No evidence for beta cell neogenesis in murine adult pancreas. J Clin Invest 123, 2207–2217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sauer B & McDermott J DNA recombination with a heterospecific Cre homolog identified from comparison of the pac-c1 regions of P1-related phages. Nucleic Acids Res 32, 6086–6095 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anastassiadis K et al. Dre recombinase, like Cre, is a highly efficient site-specific recombinase in E. coli, mammalian cells and mice. Dis Model Mech 2, 508–515 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Hermann M et al. Binary recombinase systems for high-resolution conditional mutagenesis. Nucleic Acids Res 42, 3894–3907 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He L et al. Enhancing the precision of genetic lineage tracing using dual recombinases. Nat Med 23, 1488–1498 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Q et al. A multipotent progenitor domain guides pancreatic organogenesis. Dev Cell 13, 103–114 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Xiao X et al. TGFβ receptor signaling is essential for inflammation-induced but not β-cell workload-induced β-cell proliferation. Diabetes 62, 1217–1226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rankin MM et al. β-Cells are not generated in pancreatic duct ligation-induced injury in adult mice. Diabetes 62, 1634–1645 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Talchai C, Xuan S, Lin HV, Sussel L & Accili D Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 150, 1223–1234 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rahier J, Guiot Y, Goebbels RM, Sempoux C & Henquin JC Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 10 Suppl 4, 32–42 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Butler AE et al. β-Cell Deficit in Obese Type 2 Diabetes, a Minor Role of β-Cell Dedifferentiation and Degranulation. J Clin Endocrinol Metab 101, 523–532 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim H et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med 16, 804–808 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wuidart A et al. Quantitative lineage tracing strategies to resolve multipotency in tissue-specific stem cells. Genes Dev 30, 1261–1277 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma Q, Zhou B & Pu WT Reassessment of Isl1 and Nkx2-5 cardiac fate maps using a Gata4-based reporter of Cre activity. Dev Biol 323, 98–104 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tasic B et al. Site-specific integrase-mediated transgenesis in mice via pronuclear injection. Proc Natl Acad Sci U S A 108, 7902–7907 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tian X, Pu WT & Zhou B Cellular Origin and Developmental Program of Coronary Angiogenesis. Circ Res 116, 515–530 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao H & Zhou B Dual genetic approaches for deciphering cell fate plasticity in vivo: more than double. Current Opinion in Cell Biology 61, 101–109 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Hochedlinger K, Yamada Y, Beard C & Jaenisch R Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell 121, 465–477 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Perl AK, Wert SE, Nagy A, Lobe CG & Whitsett JA Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc Natl Acad Sci U S A 99, 10482–10487 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Y et al. Genetic Lineage Tracing of Non-Myocyte Population by Dual Recombinases. Circulation 138, 793–805 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Madisen L et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang H et al. Endocardium Minimally Contributes to Coronary Endothelium in the Embryonic Ventricular Free Walls. Circ Res 118, 1880–1893 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Tian X et al. De novo formation of a distinct coronary vascular population in neonatal heart. Science 345, 90–94 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao H et al. Apj+ Vessels Drive Tumor Growth and Represent a Tractable Therapeutic Target. Cell Rep 25, 1241–1254.e5 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the plots within this paper and other findings of this study are available from the corresponding author upon reasonable request.