

Abstract
Background
In treating major depressive disorder (MDD), we lack tests anchored in neurobiology that predict antidepressant efficacy. Cognitive impairments are a particularly disabling MDD feature. We tested whether functional connectivity during a response-inhibition task can predict response to antidepressants and whether its changes over time are correlated to symptom changes.
Methods
We analyzed data from MDD outpatients (N=124) randomized to receive escitalopram, sertraline or venlafaxine (8 weeks) and healthy controls (N=59) (ages 18–65). At pre-and post-treatment, participants were interviewed and scanned using functional MRI and functional connectivity was measured using generalized psychophysiological interaction during response inhibition (Go-NoGo task). We investigated the interaction between treatment type and response (≥50% reduction on self-reported symptoms), coupling differences between responders/non-responders at baseline, their correlation with symptom improvement and their changes in time.
Results
During response inhibition, connectivity between the dorsolateral prefrontal cortex/supramarginal gyrus and supramarginal gyrus/middle temporal gyrus was associated with response to sertraline and venlafaxine, but not escitalopram. Sertraline responders had higher functional connectivity between these regions than non-responders, whereas venlafaxine responders had lower functional connectivity. For sertraline, attenuation of connectivity in the precentral and superior temporal gyri correlated with post-treatment symptom improvement. For venlafaxine, enhancement of connectivity between the orbitofrontal cortex and subcortical regions correlated with symptom improvement.
Conclusions
Connectivity of the cognitive control circuit during response inhibition selectively and differentially predicts antidepressant treatment response and correlates with symptom improvement. These quantitative markers tied to the neurobiology of cognitive features of depression could be used translationally to predict and evaluate treatment response.
Clinical Trials Registration
Registry Name: ClinicalTrials.gov
URL: https://clinicaltrials.gov/ct2/show/NCT00693849
Registration Number: NCT00693849
Keywords: fMRI, cognitive control, Go-NoGo, major depressive disorder, treatment, biomarker
Introduction
Major depressive disorder (MDD) is the leading cause of disability worldwide (1). The lack of quantitative tests for identifying interventions for individual patients often makes treatment choice a years-long trial-and-error process (2). Tests anchored in the neurobiological mechanisms that underlie MDD are needed for predicting the efficacy of antidepressants, the most widely used MDD therapy (3). We present a predictive marker for antidepressant outcomes defined by functional coupling of the putative cognitive control circuit of the human brain.
MDD-related impairments in cognition and thought are particularly disabling and involve changes in macro-scale brain circuits that subserve these functions (4). These circuits may be probed with specifically designed tasks using functional MRI (fMRI), during which the coupling of the regions involved can be quantified as functional connectivity (5). By quantifying connectivity, symptom clusters and behavior, we can characterize “biotypes” of MDD, which might improve the reliability of diagnosis and orient treatment selection (4). Recent neuro-cognitive models of depression have hypothesized that pathology in cognitive control structures might promote biased processing of negative stimuli and blunted responses to positive stimuli (6). Therefore, we focus on the putative biotype of cognitive dyscontrol, characterized by impairment of regions activated and functionally connected during cognitive tasks (7). Studies show impairments of activation, functional connectivity and structure in these regions in MDD, which might relate to concentration deficits, ruminations, and inability to suppress negative emotions (8–12). Behaviorally, this implicates poor performance during executive function tasks, such as those requiring response inhibition, concentration to select relevant responses over time, and planning, all of which are observed in MDD and persist after treatment (13–15). Changes in activations and resting state connectivity in brain regions involved in cognitive control are also associated with higher risk of depression recurrence (16).
Despite the evidence supporting impairments of the cognitive control circuit in MDD, we know little about how its function could predict antidepressant outcomes. One foundational study observed a positive association between pre-treatment activation of the inferior frontal gyrus during a Go-NoGo response inhibition task and depressive symptom improvement with citalopram (17). More recently, in a randomized biomarker trial including 80 MDD constituting a subset of the data presented in this work, higher activation of the dorsolateral prefrontal cortex (DLPFC) during a pre-treatment Go-NoGo task was associated with subsequent symptom response to venlafaxine, escitalopram and sertraline (18). Conversely, participants showing reduced DLPFC activation did not respond to treatment (18). Participants responding to selective serotonin reuptake inhibitors (SSRIs) were also characterized by relative hypo-activation of the parietal regions of the cognitive control circuit, whereas the reverse was observed for responders to venlafaxine-extended release (venlafaxine-XR), a serotonin-norepinephrine reuptake inhibitor (SNRI) (18). Another recent study reported that functional responses to commission errors in a Go-NoGo task achieved highly accurate prediction of response to escitalopram or duloxetine (19). Taken together, these findings show promise for fMRI markers related to cognitive control in predicting who may benefit from an antidepressant, and which pharmacological class of antidepressant will be efficacious.
Few experiments have used task-evoked functional connectivity to explore whether disruptions in functional brain connectivity revert after successful therapy (20). To our knowledge, no study has investigated how connectivity during a task probing cognitive control might predict antidepressant treatment response and how its changes relate to symptom improvement longitudinally. In this study, we quantified, at baseline and after treatment, connectivity as well as activation evoked during a Go-NoGo task in MDD participants who completed both imaging and clinical assessments within a large multi-arm randomized biomarker trial (21).
Our first goal was to investigate whether pre-treatment activation and coupling during cognitive inhibition predicted clinical response following eight weeks of acute treatment with escitalopram, sertraline or venlafaxine-XR. Furthermore, we wanted to assess longitudinally the changes in these measures that correlated with changes in clinical symptoms. We aimed to identify predictors and correlates of response for each treatment and hypothesized that they would involve regions ascribed to the cognitive dyscontrol biotype of MDD, as described in Williams (4) (DLPFC, dorsal parietal cortex, precentral gyrus).
Methods and Materials
Biomarker trial design
Analyses were conducted on data collected during the International Study to Predict Optimized Treatment in Depression (iSPOT-D), a biomarker trial. All participants were assessed and scanned at baseline, after which MDD participants were randomly assigned to receive escitalopram, sertraline, or venlafaxine-XR. All participants were assessed and scanned again eight weeks later. For a complete description of the iSPOT-D trial protocol, assessments and inclusion/exclusion criteria, see Williams et al. (21).
This study was conducted according to the principles of the Declaration of Helsinki 2008. After study procedures were fully explained in accordance with the ethical guidelines of the Western Sydney Area Health Service Human Research Ethics Committee, participants provided written informed consent.
Randomization and masking
For randomization, Phase Forward’s validated, Web-based interactive response technology was used (Oracle Corporation, USA). Personnel conducting clinical interviews were blind to treatment.
Participants
Our sample consisted of 124 nonpsychotic MDD outpatients (ages 18–65) from primary or specialty care practices, and 59 age- and sex-matched healthy controls (HCs). MDD patients met DSM-IV criteria for single or recurrent nonpsychotic MDD. Comorbid diagnoses included current anxiety disorders and dysthymia. MDD participants were either antidepressant medication-naïve or underwent a washout period of at least one week (five half-lives). This study collected data only at the Sydney site, where MRI scans were conducted and included only participants who had completed the Go-NoGo task at baseline and follow-up, had complete self-reported symptom measures, and excluded participants with more than 15% (18) of volumes showing frame displacement (see Supplemental Methods, Figure S1). No harms or unintended effects occurred during the trial.
Symptom ratings and response outcomes
Depressive symptom severity was measured using the clinician-rated 17-item Hamilton Rating Scale for Depression (HRSD17) (22) and 16-item Quick Inventory of Depressive Symptomatology–Self-Rated (QIDS-SR16) (23). The primary outcome measure was a 50% decrease in QIDS-SRI6 score at follow up (treatment response). To test whether our results would hold when taking into account an observer-rated scale, we investigated them post-hoc using confirmatory one-tailed t-tests considering treatment response as a ≥50% decrease in HRSD17. We also investigated post-hoc whether our results would hold when considering remission for both scales (follow-up QIDS-SR16 ≤5 or HRSD17 ≤7).
Scanning paradigm and preprocessing
Details of all iSPOT-D imaging are outlined in Grieve et al. (24). For a detailed description of sequences, paradigm and preprocessing for the current study, see the Supplemental Methods.
Go-NoGo paradigm
For a detailed description of this paradigm, see Supplemental Methods and Figure S2. The utility of the Go-NoGo task for probing poor cognitive control in MDD has been previously reported (9, 18). Due to malfunctions in custom-designed software and hardware, the behavioral data of 53 HC and 25 MDD participants were lost.
To investigate behavioral markers of treatment response, we built two general linear models within the MDD group, predicting false positives and mean reaction time with independent variables: treatment type, response, age, sex, baseline QIDS-SR16. Each model included all main effects and interactions between treatment type and response. Behavioral data is presented in Table S1 (baseline) and Table S2 (follow-up).
Preprocessing
Preprocessing was conducted using the same protocols as previous studies using data from iSPOT-D (for example, see Goldstein-Piekarski et al. (25)). For a detailed description of preprocessing, see the Supplemental Methods.
Within-participant analyses
See the Supplemental Methods for a detailed description of task activation quantification, rationale, creation of regions of interest (ROIs), and definition of generalized psychophysiological interaction (gPPI) models for the connectivity analysis.
In summary, for activation, contrast maps for NoGo>Go were created at pre-treatment baseline and follow-up using one-tailed t-tests. An additional contrast map (ΔNoGo>Go) was created for each participant by subtracting the follow-up from the baseline contrast map. To create seed ROIs for the connectivity analysis, first we identified 10 areas that were comparably active in the NoGo>Go condition in both MDD participants and HCs (Figure S3). Then, spheres with a 6 mm diameter were built around peak voxels of these clusters. This yielded 10 ROIs: left postcentral gyrus, right supramarginal gyrus, left orbitofrontal cortex, left external cerebellum, right frontal pole, left globus pallidus, right middle temporal gyrus, and three located in the right middle frontal gyrus (Table S3; see Figure S4 for an illustration of the placement of these ROIs in relation to the original clusters; see Figure S5 for their placement in relation to a meta-analysis of cognitive control). To assess task-based functional connectivity between each ROI and the rest of the brain, we used the gPPI toolbox (5). The contrast of the gPPI terms for NoGo>Go was computed in all participants at baseline and follow-up using a one-sided t-test. As with the activation, we created an additional contrast map for each participant representing the difference between the follow-up and baseline contrast maps (ΔNoGogPPI>GogPPI).
Group-level analyses
See the Supplemental Methods for a detailed breakdown of all group-level fMRI models and the contrasts included in each.
In summary, we assessed differences between HC and MDD for both activation and functional connectivity at baseline and their changes in time. Within the MDD group, we tested the interaction between response and treatment arm, then compared responders to non-responders within each treatment arm.
All group-level models included age and sex as covariates and were masked using a grey matter mask (average of grey matter segmented data from all participants, thresholded at >0.10 intensity). To detect significant results within each model, a whole-brain uncorrected cluster-forming threshold of p<0.001 was set, followed by family-wise error (FWE) cluster-level correction at p<0.05. Data smoothness was checked for all group-level models using the SPM results window output to assess compatibility with the assumptions of random field theory (29). To account for testing of multiple models, we used two strategies. Our main approach was to apply a second step of FWE correction by multiplying the pFWE cluster-level values of the 18 surviving clusters by 18 and considering as significant only those with resulting p<0.05 (pFWE analysis-level). As an alternative approach, we used an experiment-wide correction, and considered significant clusters for which the uncorrected p-value was less than 0.05 divided by the number of group-level tests (0.05/110=p<0.00045). When we found functional differences between responders and non-responders, we ran post-hoc Spearman correlations using change in clinical scores across both groups to assess whether results would be applicable regardless of the arbitrary cut-off of 50% symptom reduction.
Predictive models of treatment response
To provide an example of how our functional markers could be helpful in a treatment decision and to inform future studies, we ran three cross-validated classification analyses predicting treatment response. As features, we first used mean contrast values extracted from the result clusters. Then, to assess generalizability to regions independent from the specific areas we detected in our participants, we reran our gPPI analyses using anatomically and meta-analytically defined ROIs in proximity of our results (Table S4). For details, see the Supplemental Methods.
Results
Sample characteristics
The HC and MDD groups did not differ regarding age and sex (all tests p>0.05). Most treatment arm groups did not differ regarding age, sex, QIDS-SR16 (baseline and follow-up) or HRSD17 (baseline and follow-up) (all tests p>0.05). Sertraline responders were younger than non-responders (t=2.48, p=0.02, df=40, d=0.78). Escitalopram responders had higher baseline QIDS-SR16 scores than non-responders (U=327.50, p=0.02, d=0.77) (Table 1).
Table 1.
Demographics and clinical information of our sample.
| Feature | HC (N=59) |
Escitalopram | Sertraline | Venlafaxine-XR | HC vs. MDD | Treatment | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NR (N=22) |
R (N=21) |
NR vs R | NR (N=19) |
R (N=23) |
NR vs R | NR (N=21) |
R (N=18) |
NR vs R | ||||
| Sex (F/M) | 29/30 | 11/11 | 9/12 | χ2=0.22 p=0.64 | 7/12 | 10/13 | χ2=0.19 p=0.66 |
14/7 | 10/8 | χ2=0.51 p=0.48 | χ2<0.01 p=1 | χ2=3.78 p=0.15 |
| Age | 30.37 (13.30) |
35.59 (12.91) |
29.36 (7.30) |
t=1.93 p=0.06 |
37.67 (14.16) |
29.32 (7.07) |
t=2.48 p=0.02 |
35.52 (13.40) |
30.98 (10.72) |
t=1.15 p=0.26 |
t=−1.19 p=0.23 | F=0.06 p=0.94 |
| QIDSBL | 13 (6–23) |
17 (8–22) |
U=327.50 p=0.02 |
16 (3–20) |
15 (6–20) |
U=194.0 p=0.53 |
14 (8–19) |
14 (7–19) |
U=195.50 p=0.86 |
KW=1.07 p=0.59 |
||
| QIDSFU | 10 (5–19) |
5 (1–7) |
U=38 p<0.01 |
11 (2–19) |
5 (1–9) |
U=25.50 p<0.01 |
10 (6–17) |
3.5 (1–7) |
U=4 p<0.01 |
KW=0.24 p=0.88 |
||
| HRSDBL | 22.50 (17–29) |
23 (16–34) |
U=269.50 p=0.35 |
21 (16–30) |
21 (16–26) |
U=207.50 p=0.78 |
21 (16–31) |
21.50 (17–28) |
U=224.50 p=0.32 |
KW=4.11 p=0.13 |
||
| HRSDFU | 13 (3–20) |
7 (2–14) |
U=5.00 p<0.01 |
12 (5–21) |
7 (2–16) |
U=88.50 p<0.01 |
13 (6–25) |
5 (3–16) |
U=33.50 p<0.01 |
KW<0.01 p=1 |
||
| Dysthymia | 12 | 7 | χ2=1.96 p=0.16 |
9 | 11 | χ2<0.01 p=0.98 |
7 | 8 | χ2=0.51 p=0.47 |
χ2=0.70 p=0.40 |
||
| Panic | 5 | 2 | χ2=1.37 p=0.24 |
3 | 1 | χ2=1.58 p=0.21 |
1 | 2 | χ2=0.55 p=0.46 |
χ2=1.70 p=0.19 |
||
| Agoraphobia | 4 | 6 | χ2=0.65 p=0.42 |
6 | 4 | χ2=1.15 p=0.28 |
4 | 2 | χ2=0.47 p=0.49 |
χ2=1.07 p=0.30 |
||
| Social phobia | 8 | 8 | χ2=0.01 p=0.92 |
6 | 11 | χ2=1.14 p=0.29 |
9 | 9 | χ2=0.20 p=0.65 |
χ2=0.69 p=0.41 |
||
| Specific phobia | 4 | 3 | χ2=0.12 p=0.73 |
3 | 5 | χ2=0.24 p=0.62 |
5 | 0 | χ2=4.92 p=0.03 |
χ2=0.58 p=0.47 |
||
| Generalized anxiety | 0 | 2 | χ2=2.20 p=0.14 |
1 | 0 | χ2=1.24 p=0.26 |
2 | 0 | χ2=1.81 p=0.18 |
χ2=0.46 p=0.50 |
||
Factors and comorbidities are reported as counts, parametric variables as mean (standard deviation), non-parametric variables as median (minimum-maximum). We also report results of statistical tests comparing NR and R within each treatment arm, comparing MDD and HC, assessing differences between treatment arms at baseline. Response was defined as 50% reduction of baseline QIDS. When defined based on 50% reduction of HRSD instead, remission rates were: escitalopram: 24/43, sertraline: 24/42, venlafaxine: 21/39.
Abbreviations: BL=baseline, F=female, FU=follow-up, HC=healthy controls, HRSD=Hamilton Rating Scale for Depression, KW=Kruskall-Wallis test, M=male, MDD=depressed patients, NR=non-responders, QIDS=Quick Inventory of Depressive Symptomatology scale, R=responders, t=independent samples t-test, U=Mann-Whitney U test, χ2=chi-square test, XR=extended release.
Task performance
We found no effect of the interaction between treatment type and response toward predicting number of false positives or average reaction time. Performance did not differ between treatment responders and non-responders, MDD and HC, or overall treatment arms (Tables S1–2, all tests p>0.05).
fMRI
Table 2 shows our primary imaging results. See the Supplemental Results for those surviving our secondary strategy for multiple comparisons correction.
Table 2.
Results of our gPPI analyses.
| Group contrast | Subject contrast | ROI | punc | pFWE (cluster) |
pFWE (analysis) |
Size (voxels) |
Test | Z | Peak (x y z) |
Region | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MDD vs HC | ||||||||||||
| NoGo>Go | - | - | - | - | - | - | - | - | - | |||
| ΔNoGo>Go | - | - | - | - | - | - | - | - | - | |||
| NoGogPPl>GogPPl | All | - | - | - | - | - | - | - | - | - | ||
| ΔNoGogPPl>GogPPl | Rt SMG | 0.002 | 0.012 | 0.216 | 589 | 4.74 | 4.60 | 44 | −58 | 34 | Rt angular | |
| Others | - | - | - | - | - | - | - | - | - | |||
| Treatment × Response | ||||||||||||
| NoGo>Go | - | - | - | - | - | - | - | - | - | |||
| ΔNoGo>Go | - | - | - | - | - | - | - | - | - | |||
| NoGogPPl>GogPPl | Rt MTG | 0.007 | 0.048 | 0.864 | 331 | 13.40 | 4.39 | −18 | −70 | 42 | L superior parietal | |
| Rt MFG1 | 6.28*10−5 | 4.89*10−4 | 8.80*10−3 | 778 | 14.89 | 4.63 | 68 | −30 | 28 | Rt supramarginal | ||
| Rt SMG | 3.47*10−8 | 2.68*10−7 | 4.83*10−6 | 1854 | 19.11 | 5.27 | 52 | −52 | −4 | Rt middle temporal | ||
| 0.003 | 0.024 | 0.432 | 360 | 15.50 | 4.74 | −40 | −68 | 0 | L inferior occipital | |||
| Cerebellum | 0.001 | 0.008 | 0.144 | 413 | 14.84 | 4.63 | −16 | −62 | 54 | L superior parietal | ||
| Others | - | - | - | - | - | - | - | - | - | |||
| ΔNoGogPPl>GogPPl | All | - | - | - | - | - | - | - | - | - | ||
| EscR>EscNR | ||||||||||||
| NoGo>Go | - | - | - | - | - | - | - | - | - | |||
| ΔNoGo>Go | - | - | - | - | - | - | - | - | - | |||
| NoGogPPl>GogPPl | All | - | - | - | - | - | - | - | - | - | ||
| ΔNoGogPPl>GogPPl | Rt SMG | 0.003 | 0.015 | 0.270 | 495 | 4.13 | 3.98 | 16 | −38 | −4 | Rt lingual | |
| Others | - | - | - | - | - | - | - | - | - | |||
| SerR>SerNR | ||||||||||||
| NoGo>Go | - | - | - | - | - | - | - | - | - | |||
| ΔNoGo>Go | - | - | - | - | - | - | - | - | - | |||
| NoGogPPl>GogPPl | Rt MFG1 | 0.002 | 0.013 | 0.234 | 549 | 4.70 | 4.48 | −24 | 42 | 42 | L superior frontal | |
| 0.001 | 0.005 | 0.090 | 692 | 4.34 | 4.17 | −8 | −42 | 42 | L precuneus | |||
| 0.002 | 0.011 | 0.198 | 571 | 4.29 | 4.13 | 64 | −32 | 32 | Rt supramarginal | |||
| Rt SMG | 3.97*10−5 | 2.22*10−4 | 0.004 | 1197 | 5.33 | 5.03 | 52 | −50 | −6 | Rt middle temporal | ||
| 0.001 | 0.005 | 0.090 | 692 | 4.65 | 4.45 | −40 | −68 | 0 | L inferior occipital | |||
| Cerebellum | 1.59*10−4 | 0.001 | 0.018 | 829 | 5.02 | 4.76 | −42 | 2 | −6 | L anterior insula | ||
| 0.005 | 0.033 | 0.594 | 385 | 5.01 | 4.75 | −14 | −76 | 36 | L precuneus | |||
| Others | - | - | - | - | - | - | - | - | - | |||
| ΔNoGogPPl>GogPPl | L PCG | 3.88*10−4 | 0.002 | 0.036 | 745 | 4.85 | 4.62 | −62 | −10 | −10 | L middle temporal | |
| Rt MFG2 | 0.001 | 0.007 | 0.126 | 642 | 5.02 | 4.77 | −52 | −42 | −2 | L middle temporal | ||
| Others | - | - | - | - | - | - | - | - | - | |||
| VenR>VenNR | ||||||||||||
| NoGo>Go | - | - | - | - | - | - | - | - | - | |||
| ΔNoGo>Go | - | - | - | - | - | - | - | - | - | |||
| NoGogPPl>GogPPl | All | - | - | - | - | - | - | - | - | - | ||
| ΔNoGogPPl>GogPPl | L OFC | 6.01*10−6 | 3.21*10−5 | 0.001 | 1548 | 5.16 | 4.89 | 6 | −2 | −6 | Rt ventral diencephalon | |
| 0.006 | 0.029 | 0.522 | 440 | 4.34 | 4.17 | 10 | 26 | 2 | Rt caudate | |||
| Others | - | - | - | - | - | - | - | - | - | |||
Results from our analyses comparing activation or functional connectivity in the NoGo versus the Go condition and the longitudinal changes (Δ) in this contrast. For brevity, we show only results surviving a cluster-level correction within their respective general linear models of pFWE<0.05. To account for testing of multiple models, after this step, another family-wise error correction was applied on the 18 surviving clusters across all models. Results surviving the analysis-level correction are highlighted in bold and are our main findings. We also show which models did not return any findings surviving the within-model cluster-level correction step (dashes). Coordinates of peak values are in MNI space. ROI abbreviations match the ones reported in Table 1. Test values are F for the Treatment × Response interaction and T for all other contrasts.
Abbreviations: gPPI=generalized psychophysiological interaction, L=left, MFG=middle frontal gyrus, MTG=middle temporal gyrus, NR=non-responders, OFC= orbitofrontal cortex, PCG=postcentral gyrus, pFWE=family-wise error corrected p, pUNC=uncorrected p, R=responders, Rt=right, ROI=region of interest, Ser=sertraline, SMG=supramarginal gyrus, Ven=venlafaxine.
Functional connectivity predictors of differential response between treatments
We found an interaction effect between treatment type and response for the connectivity between the right DLPFC and right supramarginal gyrus (peak F=14.84, pFWE=0.009, model df=116) and for the coupling between the right supramarginal gyrus and middle temporal gyrus (peak F=19.11, pFWE<0.001, model df=116) (Figure 1).
Figure 1: Functional connectivity of the DLPFC and supramarginal gyrus predicting differential treatment response to sertraline and venlafaxine.
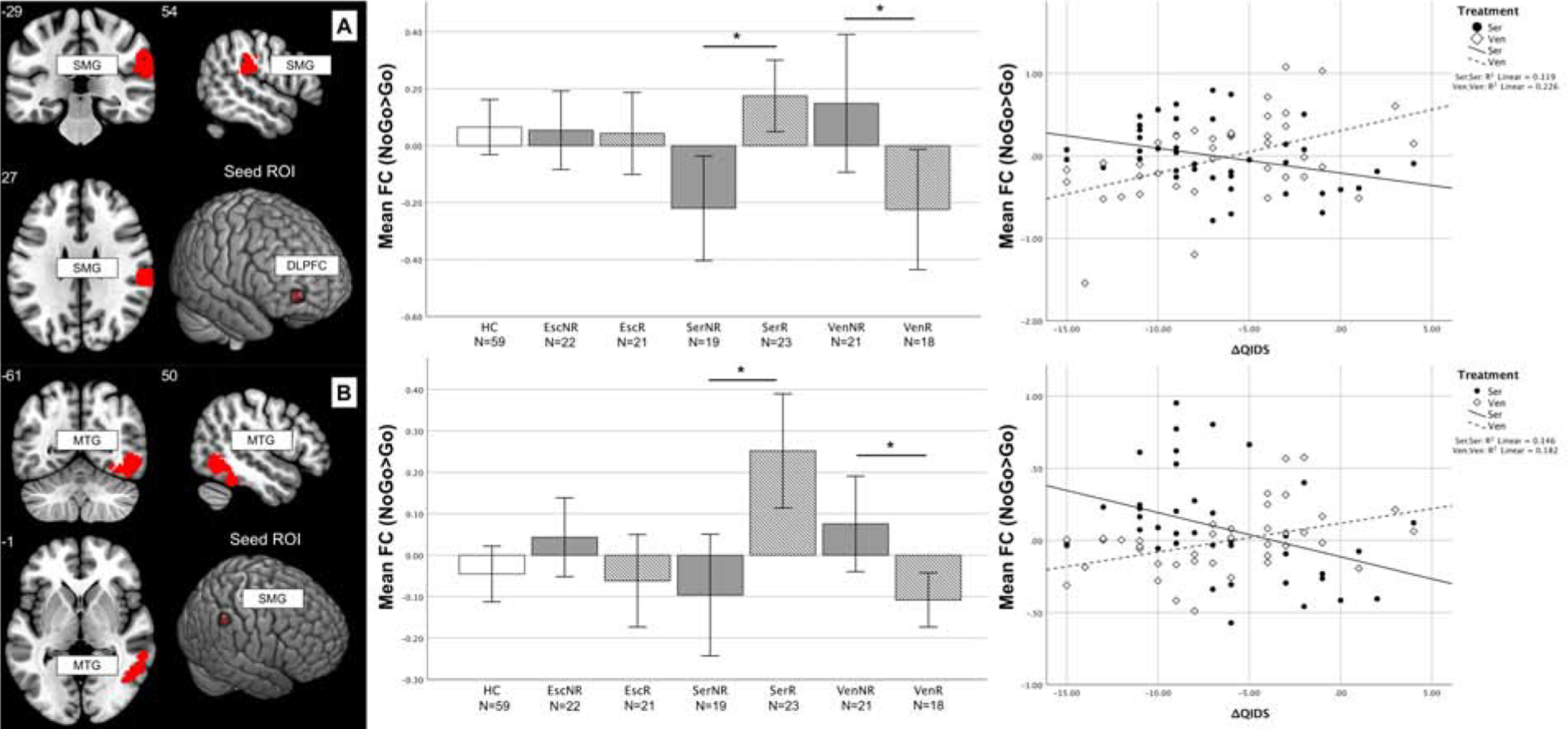
A: Responders to sertraline showed higher FC between the right DLPFC and supramarginal gyrus (t=3.809, p<0.001) whereas, in the venlafaxine-XR group, the opposite was true (t=−2.391, p=0.020). Higher coupling at baseline was associated with greater QIDS-SR16 decrease after treatment in the sertraline group (rho=−0.566, p<0.001) and the opposite was true for venlafaxine-XR (rho=0.441, p=0.005). B: The coupling between right supramarginal gyrus and middle temporal gyrus was higher in responders to sertraline compared to non-responders (t=3.591, p=0.001), whereas in venlafaxine-XR responders connectivity was lower compared to non-responders (t=−2.770, p=0.009). Higher coupling at baseline was associated with greater QIDS-SR16 decrease after treatment in the sertraline group (rho=−0.560, p<0.001) and the opposite was true in the venlafaxine-XR group (rho=0.457, p=0.003). Numbers indicate slice coordinates in MNI space. Stars mark significant comparisons between groups (t-test p<0.05). For correlations, a linear fit line calculated based on the data is shown. ROI coordinates in MNI space: DLPFC=(34 58 0), SMG=(52 −44 46). FC is expressed as difference of the parameter estimates of the hemodynamic response function (beta values) for the NoGo and Go conditions. Abbreviations: DLPFC=dorsolateral prefrontal cortex (MFG1 in Table 2), Esc=escitalopram, FC=functional connectivity, HC=healthy controls, MNI=Montreal neurological institute MTG=middle temporal gyrus, NR=non-responders, QIDS=Quick Inventory of Depressive Symptomatology scale, R=responders, ROI=region of interest, Ser=sertraline, SMG=supramarginal gyrus, Ven=venlafaxine, XR=extended-release, Δ=difference between follow-up and baseline.
Sertraline responders had higher functional connectivity during response inhibition compared to non-responders both between DLPFC/supramarginal gyrus (mean difference=0.421, CI95%=0.224–0.617, t=4.326, p<0.001, df=40, d=1.368) and supramarginal gyrus/middle temporal gyrus (mean difference=0.431, CI95%=0.265–0.596, t=3.591, p=0.001, df=40, 1.136). By comparing connectivity during the NoGo and Go conditions separately, we found that responders had higher connectivity specifically during NoGo trials, whereas non-responders showed lower connectivity (Figure S7 and S8). Follow-up correlations confirmed that in the sertraline group, higher coupling at baseline was associated with greater post-treatment QIDS-SR16 improvement for both region pairs (respectively: rho=−0.566, CI95%=−0.742 – −0.316, p<0.001, df=40; rho=−0.560, CI95%=−0.739 – −0.309, p<0.001, df=40).
In the venlafaxine-XR group, the opposite was observed such that responders had lower functional connectivity compared to non-responders for both DLPFC/supramarginal gyrus (mean difference=−0.517, CI95%=−0.807 – −0.227, t=−3.611, p=0.001, df=37, d=−1.187) and supramarginal gyrus/middle temporal gyrus (mean difference=−0.238, CI95%=−0.368 – −0.109, t=−3.721, p=0.009, df=37, d=−1.223). Responders had lower connectivity specifically during NoGo trials, whereas non-responders showed higher connectivity (Figure S7 and S8). Follow-up correlations confirmed that lower coupling at baseline was associated with greater QIDS-SR16 improvement in the venlafaxine-XR group for both region pairs (respectively: rho=0.441, CI95%=0.146–0.664, p=0.005, df=37 and rho=0.457, CI95%=0.165–0.675, p=0.003, df=37).
This opposing pattern of functional connectivity was reflected in comparisons to HCs. Compared to HCs, functional connectivity was lower in non-responders to sertraline for DLPFC/supramarginal gyrus (mean difference=−0.333, CI95%=−0.524 – −0.142, t=−3.477, p<0.001, df=76, d=−0.797) and supramarginal gyrus/middle temporal gyrus (mean difference=−0.122, CI95%=−0.251 – −0.008, t=−2.081, p=0.044, df=76, d=0.477), and higher in responders to sertraline for supramarginal gyrus/middle temporal gyrus (mean difference=0.309, CI95%=0.163–0.454, t=4.315, p<0.001, df=80, d=0.965).
Conversely, responders to venlafaxine-XR showed lower functional connectivity compared to HCs for DLPFC/supramarginal gyrus (mean difference=−0.348, CI95%=−0.562–0.133, t=−3.234, p=0.01, df=75, d=0.747), and non-responders to venlafaxine-XR had higher coupling than HCs for supramarginal gyrus/middle temporal gyrus (mean difference=0.150, CI95%=0.028–0.271, t=2.489, p=0.017, df=78, d=0.564).
Functional connectivity predictors of response within individual treatment arms
Responders to sertraline also showed higher functional connectivity between the right supramarginal gyrus and the right middle temporal cortex (right: peak t=5.33, pFWE=0.004, model df=116) (Figure 2), and greater functional connectivity between the left external cerebellum and the left transverse temporal gyrus and insula (peak t=4.76, pFWE=0.018, model df=116). Responders had higher connectivity specifically during NoGo trials (Figure S9). Follow-up correlations confirmed that higher coupling at baseline was associated with greater QIDS-SR16 decrease after treatment (respectively rho=−0.527, CI95%=0.266–0.716, p<0.001; rho=−0.462, CI95%=0.184–0.672, p<0.001, all df=40). In all cases, when compared to HCs, non-responders had lower functional connectivity (all p<0.05) and responders had higher coupling (all p<0.05).
Figure 2: Functional connectivity of the cerebellum and SMG predicting sertraline response.
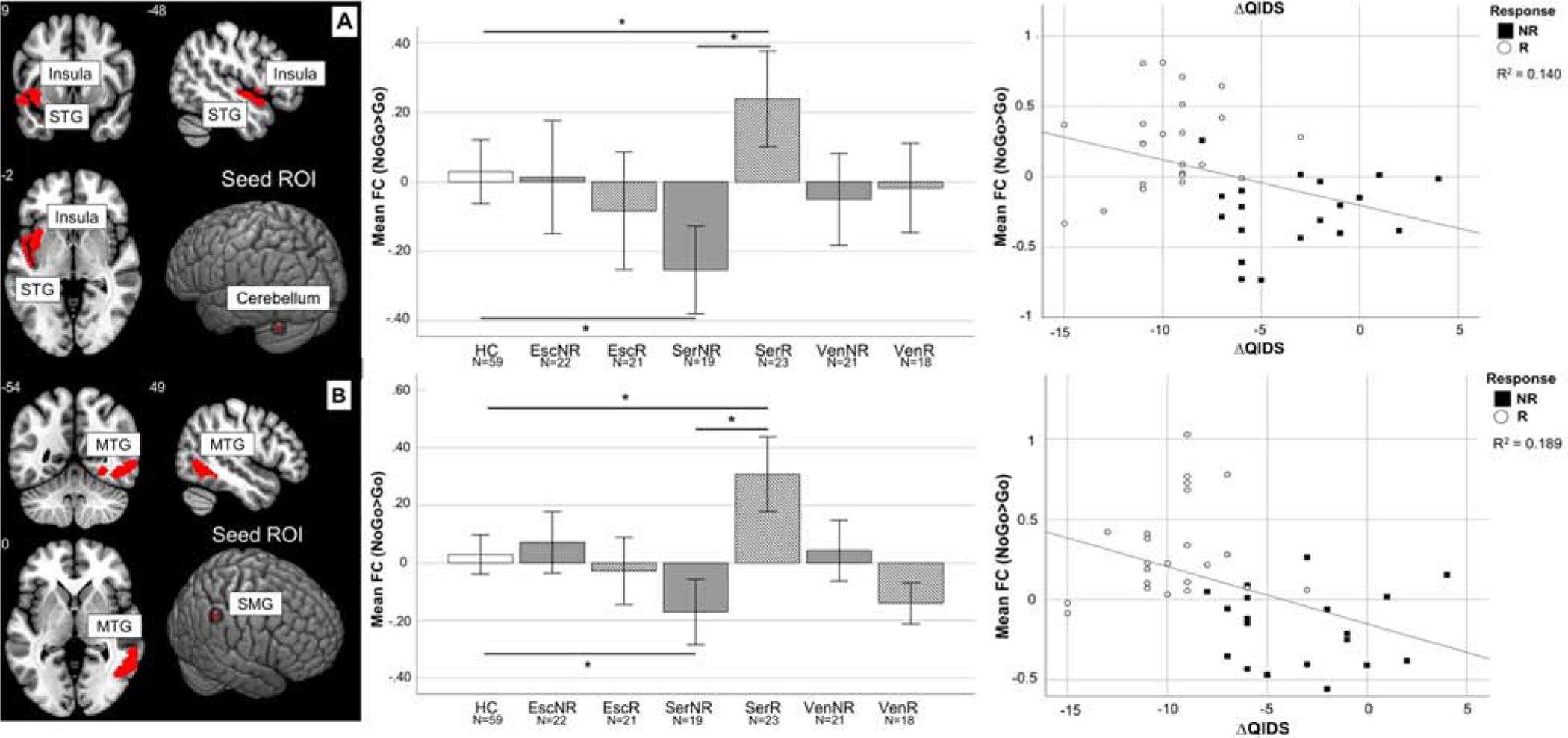
A: Responders showed higher FC between the left external cerebellum and a cluster encompassing the left transverse temporal gyrus and insula during response inhibition compared to non-responders (t=5.33, p<0.001) and HC (t=2.48, p=0.01). Non-responders showed reduced FC compared to HC (t=−3.20, p=0.002). Higher FC at baseline was also associated with greater QIDS decrease after treatment (rho=−0.462, p<0.001). B: Responders showed higher FC between the right supramarginal gyrus and right middle temporal gyrus during response inhibition compared to non-responders (t=5.33, p<0.001) and HC t=2.64, p=0.01). Non-responders showed reduced FC compared to HC (t=−3.27, p=0.002). Higher FC at baseline was also associated with greater QIDS decrease after treatment (rho=−0.460, p<0.001). Numbers indicate slice coordinates in MNI space. Stars mark significant comparisons between groups (t-test p<0.05). For correlations, a linear fit line calculated based on the data is shown. ROI coordinates in MNI space: Cerebellum=(−40 −66 −30), SMG=(52 −44 46). FC is expressed as difference of the parameter estimates of the hemodynamic response function (beta values) for the NoGo and Go conditions.
Abbreviations: Esc=escitalopram, FC=functional connectivity, HC=healthy controls, MNI=Montreal neurological institute, MTG=middle temporal gyrus, NR=non-responders, PCU=precuneus, QIDS= Quick Inventory of Depressive Symptomatology scale, R=responders, ROI=region of interest, Ser=sertraline, SMG=supramarginal gyrus, STG=superior temporal gyrus, Ven=venlafaxine, Δ=difference between follow-up and baseline.
Functional connectivity changes related to treatment response
Compared to non-responders, sertraline responders showed a significant change in functional connectivity between the left precentral gyrus and left superior temporal gyrus (peak t=4.85, pFWE=0.036, model df=116) (Figure 3). Responders showed a decrease in coupling between these regions after treatment (mean difference=−0.391, CI95%=−0.624 – −0.160, t=−3.476, p=0.002, df=22, d=1.482), whereas non-responders showed an increase (mean difference=0.712, CI95%=0.366–1.058, t=4.535, p<0.001, df=18, d=2.137). These changes were driven specifically by NoGo trials (Figure S10). Follow-up correlations confirmed that a greater functional connectivity decrease was associated with greater QIDS-SR16 reduction after treatment (rho=0.588, CI95%=0.346–0.756, p<0.001, df=40). Changes in responders and non-responders were greater than changes in HCs (respectively t=3.567, p=0.001 and t=−3.066, p=0.004).
Figure 3: Functional connectivity correlates of antidepressant treatment response.
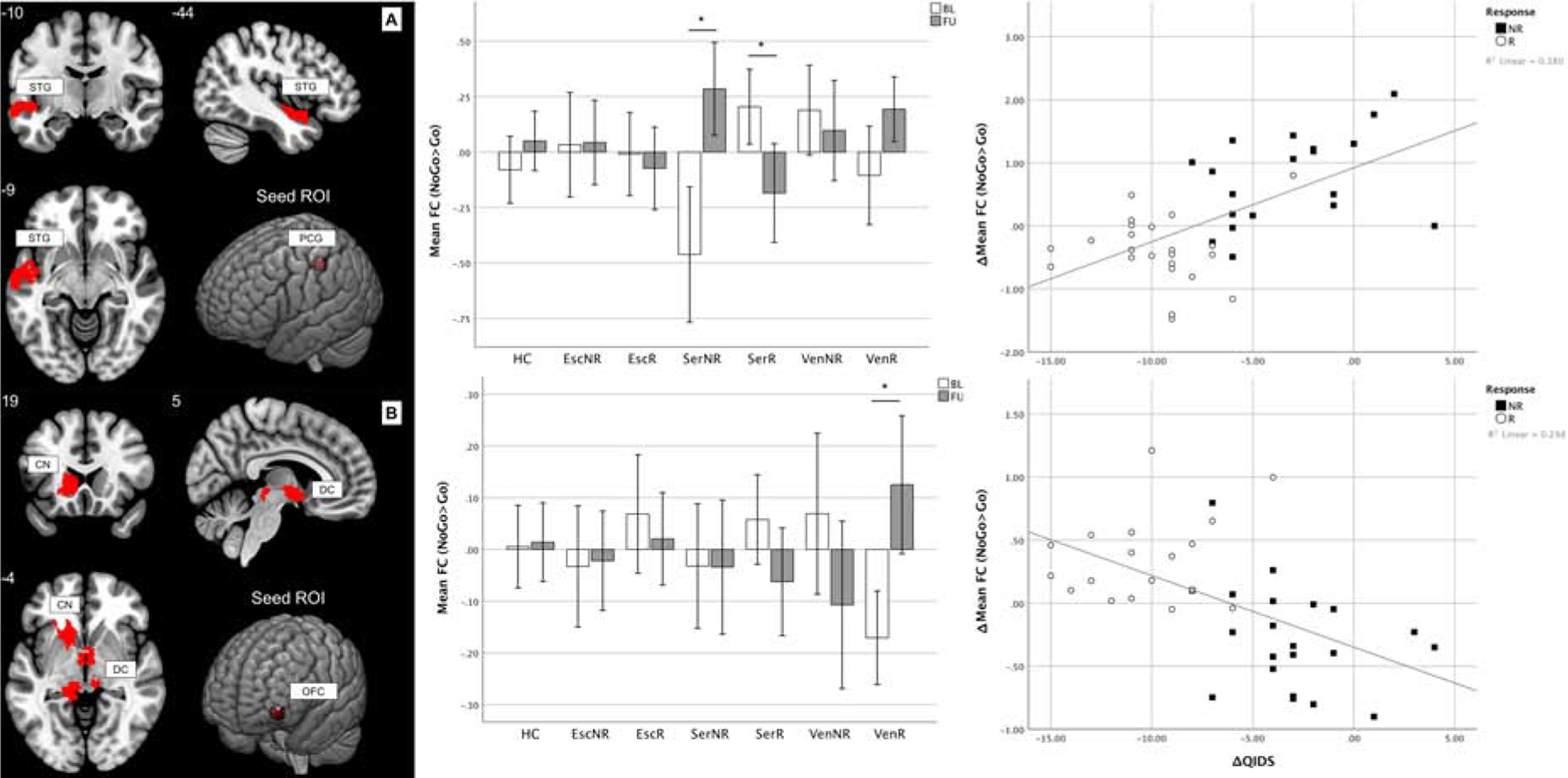
A: Participants responding to sertraline showed a decrease of FC between the left postcentral gyrus and left superior temporal gyrus (t=−3.476, p=0.002), whereas non-responders showed an increase (t=4.535, p<0.001). Also, changes in responders and non-responders were greater than the changes in HC (respectively t=−3.567, p=0.001 and t=3.066, p=0.004). Across all MDD participants, FC decrease was correlated with QIDS reduction after treatment (rho=0.588, p<0.001). B: MDD participants responding to venlafaxine-XR showed a decrease of FC between the left orbitofrontal cortex and the brainstem as well as caudate nucleus (t=3.853, p=0.001). Also, changes in responders were greater than the changes in HC (t=3.127, p=0.004). Across all MDD participants, FC decrease was correlated with QIDS reduction after treatment (rho=−0.658, p<0.001). Numbers indicate slice coordinates in MNI space. Stars mark significant comparisons (t-test p<0.05). For correlations, a linear fit line calculated based on the data is shown. ROI coordinates in MNI space: PCG=(−42 −30 46), OFC=(−40 54 −8). FC is expressed as difference of the parameter estimates of the hemodynamic response function (beta values) for the NoGo and Go conditions.
Abbreviations: BL=baseline, CN=caudate nucleus, DC=diencephalon, Esc=escitalopram, FC=functional connectivity, FU=follow-up, HC=healthy controls, MNI=Montreal neurological institute NR=non-responders, OFC=orbitofrontal cortex, PCG=postcentral gyrus, QIDS=Quick Inventory of Depressive Symptomatology scale, R=responders, ROI=region of interest, Ser=sertraline, STG=superior temporal gyrus, Ven=venlafaxine, Δ=difference between follow-up and baseline.
Compared to non-responders, venlafaxine-XR responders showed a greater change in coupling between the left orbitofrontal cortex and the brainstem and left caudate nucleus (peak t=5.16, pFWE<0.001, model df=116). Functional connectivity between these regions increased in responders (mean difference=0.296, CI95%=0.134–0.458, t=3.853, p=0.001, df=17, d=1.868). These changes were driven specifically by NoGo trials (Figure S11). Follow-up correlations confirmed that a greater connectivity increase was associated with greater QIDS-SR16 reduction after treatment (rho=−0.658, p<0.001, df=37). Changes in responders were greater than changes in HCs (mean difference=0.287, CI95%=0.100–0.474, t=3.127, p=0.004, df=75, d=0.722).
Consideration of other potential contributing factors
We first considered whether the findings were attributable to MDD diagnosis. We found no activation or functional connectivity differences at baseline or follow-up and no difference in the longitudinal changes of these measures between HCs and MDD (Table 2). Second, we found no significant difference in activation between responders and non-responders within each treatment arm (Table 2). Third, we determined that results presented in the preceding sections remained significant when accounting for baseline symptom severity assessed by the QIDS-SR16 (Figure S6). Fourth, we showed that our findings based on the QIDS-SR16 were comparable when measuring response by clinical observation using the HRSD17 and for remission on both scales (Table S5). Correlations were mostly replicated, which was not always the case when comparing groups defined by a cut-off in either scale (Table S5). Fifth, we considered our results within the context of known resting state functional connectivity networks as defined by Yeo et al. (30) and found they were mostly localized in the frontoparietal, ventral and dorsal attention networks (Figures S12–S14).
Predictive models of treatment response
We note the caveat that post-hoc descriptive models, such as those implemented here, may be prone to over-represent predictive accuracy because of overfitting. In the sertraline arm, imaging provided a better classification (accuracy 83%, p<0.001) than baseline QIDS-SR16 (accuracy 64%, p=0.139). These results also generalized well to anatomically-defined regions (accuracy 80%, p<0.001), but not to meta-analysis-derived ones (accuracy 36%, p=0.995). The performance of models predicting venlafaxine-XR response based on our clusters was slightly lower (accuracy 77%, p=0.002), but still higher than the one based on baseline QIDS-SR16 (accuracy 64%, p=0.130) and did not generalize to anatomically defined regions (accuracy 49%, p=0.789), or meta-analysis-derived ones (accuracy 15%, p=1). Details are shown in Supplemental Results and Tables S6–S7.
Discussion
We used data from a controlled biomarker trial to demonstrate that functional connectivity of cognitive control regions probed by a Go-NoGo inhibition task predicts clinical response to specific antidepressants in MDD, and that it matches the changes in symptoms after treatment.
First of all, an improvement of ≥50% in subjectively-reported symptoms after sertraline treatment was associated with higher connectivity at baseline between the DLPFC and supramarginal gyri, corresponding to higher connectivity of the frontoparietal with the ventral and dorsal attention resting state networks (Table S8, Figure S12) (30). Participants who showed reduced connectivity between the same regions responded to venlafaxine-XR.
Sertraline responders and non-responders were differentiated from HCs by comparatively higher and lower functional connectivity of the cerebellum and portions of the temporal lobe and insula, indicating coupling changes between the dorsal and ventral attention resting-state networks (Table S8, Figure S13) (30). These opposing differences from HCs highlight that when averaging across features in case-control studies, we potentially conflate distinct underlying neurobiological biotypes relevant to clinical response.
Connectivity between cognitive control regions also changed from pre- to post-treatment. Sertraline responders showed a decrease in connectivity between the left postcentral gyrus and left superior temporal gyrus, correlating with their symptom improvement, whereas non-responders showed an increase in connectivity and little change in symptoms. When considering resting state patterns, this corresponded to higher connectivity between the dorsal attention and default mode networks (Table S8, Figure S14) (30). In the venlafaxine-XR group, a connectivity increase from pre- to post-treatment between the left orbitofrontal cortex (belonging to the frontoparietal network (30); Table S8, Figure S14) and the brainstem as well as the caudate nucleus mirrored the improvement in symptoms. HCs showed no change in connectivity.
Our results involve areas which are commonly activated (32) and functionally connected (33) during Go/NoGo paradigms. Such regions might regulate limbic areas, leading to biased processing of negative and positive stimuli as well as ruminations when their connectivity is impaired (6, 34). Also, when relating our findings to established functional networks, we show that during cognitive inhibition, MDD have altered coupling especially between the frontoparietal, default mode, dorsal attention and ventral attention networks, consistently with resting-state studies (11).
The opposing directionality of predictors and correlates of sertraline compared to venlafaxine-XR response could be related to their underlying pharmacological mechanisms. Similarly, a previous study conducted on a subset of this sample showed that inferior parietal activation was greater pretreatment in SSRI remitters compared to non-remitters, with SNRIs showing the opposite (18). Animal studies have shown that functional connections correspond to neurotransmitter pathways (35) and a recent study reported a decrease of resting state connectivity in most cortical and subcortical areas after a single dose of SSRI versus placebo (36). In our study, it was indeed the participants with higher connectivity compared to controls who benefitted from an SSRI, and for these participants a decrease in connectivity was correlated with clinical improvement. Our findings in the venlafaxine-XR arm are consistent with its role as an inhibitor of the reuptake of serotonin, dopamine and noradrenaline (37). The caudate nucleus is a primary site where dopaminergic neurons are located, and most noradrenergic neurons are situated in the brainstem (locus coeruleus) from where they project to cortical regions (38). Overall, the mechanistic relationship between the pharmacological action of these drugs and functional connectivity is still relatively underexplored. Further studies are needed to elucidate the underlying mechanisms of these effects.
Our null findings in the escitalopram group contrast with previous evidence (19). This difference may be due to escitalopram participants being undistinguishable from HCs regarding functional connectivity at baseline. Indeed, both responders and non-responders to sertraline and venlafaxine-XR differed from HCs. Alternatively, this difference could be due to the action of the two drugs at the receptor level, in particular considering that sertraline, unlike other SSRIs, inhibits dopamine (39, 40). Future studies with targeted designs could assess whether our findings are specific for sertraline or apply to SSRIs in general and use cross-over designs to disentangle whether individuals are more likely to respond to one drug than another.
Classification models indicated high accuracy for classifying responders versus non-responders to both sertraline and venlafaxine-XR, based on baseline connectivity. This was not surprising, since we defined our features based on between-group comparisons, but we still sought to provide an operational example of how such measures could be used in clinical practice. Using anatomical ROIs, accuracy remained high only for sertraline response. ROIs derived by meta-analysis did not predict response to any treatment. In our Supplement, we provide coordinates for these ROIs to encourage replication of our findings in new samples and as a foundation of future studies using connectivity of the putative cognitive control network as a predictor of treatment outcomes.
Study limitations include a finding that whole-brain activations during response inhibition did not predict treatment outcome. This effect may be too small to survive whole-brain statistical correction and given our focus on connectivity, we did not investigate it further. Behavioral task information for a large number of participants was lost due to technical issues, which warrants further investigation of the relationship of our findings with behavioral markers of cognitive dyscontrol. Also, some results may have been confounded by a higher number of motor responses during the NoGo condition in some participants. Using anatomically-derived ROIs in proximity of our results predicted response only to sertraline, and ROIs derived from a meta-analytic map of cognitive control did not predict response to any treatment. This suggests that defining ROIs in regions that are maximally active might perform better compared to Neurosynth maps, which aggregate a large number of heterogeneous studies. Alternatively, it may suggest overfitting within out models that may limit generalization to other samples, or that there are subtle differences in normalization across samples that result in overly expansive Neurosynth masks. These limitations both amplify the importance of within and across sample validation of results, before any application to clinical populations could be fruitful. Future studies might consider using a functional localizer to identify the areas of peak activation for individuals to improve classification. Regarding participants undergoing medication washout before participation, one week is sufficient to clear the drug from the bloodstream (five half-lives), but downstream effects tied to the drug’s efficacy might persist. We cannot exclude that these might have influenced baseline measures.
In sum, we show that in depressed patients, connectivity of the cognitive control circuit during a response inhibition task predicts response to antidepressant treatment and correlates with symptom improvement over time. Therefore, we present quantitative markers tied to the neurobiology of depression which could be used for prospective prediction of who is likely to benefit from an antidepressant and for ongoing assessment of response.
Supplementary Material
KEY RESOURCES TABLE
| Resource Type | Specific Reagent or Resource | Source or Reference | Identifiers | Additional Information |
|---|---|---|---|---|
| Add additional rows as needed for each resource type | Include species and sex when applicable. | Include name of manufacturer, company, repository, individual, or research lab. Include PMID or DOI for references; use “this paper” if new. | Include catalog numbers, stock numbers, database IDs or accession numbers, and/or RRIDs. RRIDs are highly encouraged; search for RRIDs at https://scicrunch.org/resources. | Include any additional information or notes if necessary. |
| Software; Algorithm | Statistical parametric mapping | The FIL Methods group | RRID:SCR_007037 | |
| Software; Algorithm | Matlab | Mathworks | RRID:SCR_001622 | |
| Software; Algorithm | Generalized psychophysiological interaction toolbox | doi: 10.1016/j.neuroimage.2012.03.068 | RRID:SCR_009489 | |
| Software; Algorithm | SPSS | IBM | RRID:SCR_002865 | |
| Software; Algorithm | R | R project for statistical computing | RRID:SCR_001905 | |
| Software; Algorithm | TSDiffAna | Freiburg Brain Imaging | RRID:SCR_016656 | Only used in preprocessing |
| Software; Algorithm | FMRIB Software Library | Analysis Group, FMRIB, Oxford, UK. | RRID:SCR_002823 | Only used in preprocessing |
Acknowledgments
This work was supported by the National Institutes of Health [grant numbers R01MH101496, F32MH108299, T32MH019938, U01MH109985] and Brain Resource Company Operations Pty Ltd. The funder of this study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. LMW was the academic Principal Investigator for iSPOT-D from 2008–2013. Claire Day, Ph.D. was the global trial manager for iSPOT-D from 2009–2015. Sheryl Foster was the senior MRI Research Coordinator at Westmead Hospital and specialist radiographer who oversaw the scanning of participants for iSPOT-D from 2009–2015. LT is supported by the National Institutes of Health [grant numbers U01MH109985]. We acknowledge the editorial support of Jon Kilner, MS, MA (Pittsburgh, PA, USA) and acknowledge Scott Lanyon Fleming (Stanford University, Stanford, USA) in the setup of our predictive models.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data sharing statement
Data collected for the study, including individual participant data and a data dictionary defining each field in the set, will be made available after approval of a proposal with investigator support. Protocols for the current clinical trial are published online at https://trialsjournal.biomedcentral.com/articles/10.1186/1745-6215-12-4.
Disclosures
LMW received non-salary direct research costs as an iSPOT-D investigator from Brain Resource Pty Ltd.; and fees as a scientific advisor for Psyberguide and as a consultant for BlackThorn Therapeutics. All other authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Friedrich MJ (2017): Depression is the leading cause of disability around the world. JAMA. 317: 1517–1517. [DOI] [PubMed] [Google Scholar]
- 2.Cipriani A, Furukawa TA, Salanti G, Chaimani A, Atkinson LZ, Ogawa Y, et al. (2018): Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Lancet Lond Engl. 391: 1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trivedi MH, McGrath PJ, Fava M, Parsey RV, Kurian BT, Phillips ML, et al. (2016): Establishing moderators and biosignatures of antidepressant response in clinical care (EMBARC): Rationale and design. J Psychiatr Res. 78: 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams LM (2016): Precision psychiatry: a neural circuit taxonomy for depression and anxiety. Lancet Psychiatry. 3: 472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLaren DG, Ries ML, Xu G, Johnson SC (2012): A generalized form of context-dependent psychophysiological interactions (gPPI): a comparison to standard approaches. Neuroimage. 61: 1277–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith R, Alkozei A, Killgore WDS, Lane RD (2018): Nested positive feedback loops in the maintenance of major depression: An integration and extension of previous models. Brain Behav Immun. 67: 374–397. [DOI] [PubMed] [Google Scholar]
- 7.Niendam TA, Laird AR, Ray KL, Dean YM, Glahn DC, Carter CS (2012): Meta-analytic evidence for a superordinate cognitive control network subserving diverse executive functions. Cogn Affect Behav Neurosci. 12: 241–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drevets WC, Price JL, Furey ML (2008): Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct. 213: 93–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korgaonkar MS, Grieve SM, Etkin A, Koslow SH, Williams LM (2013): Using standardized fMRI protocols to identify patterns of prefrontal circuit dysregulation that are common and specific to cognitive and emotional tasks in major depressive disorder: first wave results from the iSPOT-D study. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 38: 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bora E, Fornito A, Pantelis C, Yücel M (2012): Gray matter abnormalities in Major Depressive Disorder: A meta-analysis of voxel based morphometry studies. J Affect Disord. 138: 9–18. [DOI] [PubMed] [Google Scholar]
- 11.Kaiser RH, Andrews-Hanna JR, Wager TD, Pizzagalli DA (2015): Large-scale network dysfunction in major depressive disorder: a meta-analysis of resting-state functional connectivity. JAMA Psychiatry. 72: 603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lam RW, Kennedy SH, McIntyre RS, Khullar A (2014): Cognitive Dysfunction in Major Depressive Disorder: Effects on Psychosocial Functioning and Implications for Treatment. Can J Psychiatry Rev Can Psychiatr. 59: 649–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Snyder HR (2013): Major depressive disorder is associated with broad impairments on neuropsychological measures of executive function: a meta-analysis and review. Psychol Bull. 139: 81–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Day CV, Gatt JM, Etkin A, DeBattista C, Schatzberg AF, Williams LM (2015): Cognitive and emotional biomarkers of melancholic depression: An iSPOT-D report. J Affect Disord. 176: 141–150. [DOI] [PubMed] [Google Scholar]
- 15.Shilyansky C, Williams LM, Gyurak A, Harris A, Usherwood T, Etkin A (2016): Effect of antidepressant treatment on cognitive impairments associated with depression: a randomised longitudinal study. Lancet Psychiatry. 3: 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langenecker SA, Jenkins LM, Stange JP, Chang Y-S, DelDonno SR, Bessette KL, et al. (2018): Cognitive control neuroimaging measures differentiate between those with and without future recurrence of depression. NeuroImage Clin. 20: 1001–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langenecker SA, Kennedy SE, Guidotti LM, Briceno EM, Own LS, Hooven T, et al. (2007): Frontal and limbic activation during inhibitory control predicts treatment response in major depressive disorder. Biol Psychiatry. 62: 1272–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gyurak A, Patenaude B, Korgaonkar MS, Grieve SM, Williams LM, Etkin A (2016): Frontoparietal activation during response inhibition predicts remission to antidepressants in patients with major depression. Biol Psychiatry. 79: 274–281. [DOI] [PubMed] [Google Scholar]
- 19.Crane NA, Jenkins LM, Bhaumik R, Dion C, Gowins JR, Mickey BJ, et al. (2017): Multidimensional prediction of treatment response to antidepressants with cognitive control and functional MRI. Brain J Neurol. 140: 472–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gudayol-Ferré E, Peró-Cebollero M, González-Garrido AA, Guàrdia-Olmos J (2015): Changes in brain connectivity related to the treatment of depression measured through fMRI: a systematic review. Front Hum Neurosci. 9. doi: 10.3389/fnhum.2015.00582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams LM, Rush AJ, Koslow SH, Wisniewski SR, Cooper NJ, Nemeroff CB, et al. (2011): International Study to Predict Optimized Treatment for Depression (iSPOT-D), a randomized clinical trial: rationale and protocol. Trials. 12: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamilton M (1986): The Hamilton Rating Scale for Depression. In: Sartorius DN, Ban DTA, editors. Assess Depress. Springer Berlin Heidelberg, pp 143–152. [Google Scholar]
- 23.Rush AJ, Trivedi MH, Ibrahim HM, Carmody TJ, Arnow B, Klein DN, et al. (2003): The 16-Item quick inventory of depressive symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry. 54: 573–583. [DOI] [PubMed] [Google Scholar]
- 24.Grieve SM, Korgaonkar MS, Etkin A, Harris A, Koslow SH, Wisniewski S, et al. (2013): Brain imaging predictors and the international study to predict optimized treatment for depression: study protocol for a randomized controlled trial. Trials. 14: 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldstein-Piekarski AN, Korgaonkar MS, Green E, Suppes T, Schatzberg AF, Hastie T, et al. (2016): Human amygdala engagement moderated by early life stress exposure is a biobehavioral target for predicting recovery on antidepressants. Proc Natl Acad Sci. 113: 11955–11960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Power JD, Mitra A, Laumann TO, Snyder AZ, Schlaggar BL, Petersen SE (2014): Methods to detect, characterize, and remove motion artifact in resting state fMRI. NeuroImage. 84: 320–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siegel JS, Power JD, Dubis JW, Vogel AC, Church JA, Schlaggar BL, Petersen SE (2014): Statistical improvements in functional magnetic resonance imaging analyses produced by censoring high-motion data points. Hum Brain Mapp. 35: 1981–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Power JD, Barnes KA, Snyder AZ, Schlaggar BL, Petersen SE (2012): Spurious but systematic correlations in functional connectivity MRI networks arise from subject motion. NeuroImage. 59: 2142–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chumbley J, Worsley K, Flandin G, Friston K (2010): Topological FDR for neuroimaging. NeuroImage. 49: 3057–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas Yeo BT, Krienen FM, Sepulcre J, Sabuncu MR, Lashkari D, Hollinshead M, et al. (2011): The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J Neurophysiol. 106: 1125–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.FDA-NIH Biomarker Working Group (2016): BEST (Biomarkers, EndpointS, and other Tools) Resource. Silver Spring (MD): Food and Drug Administration (US). Retrieved October 26, 2018, from http://www.ncbi.nlm.nih.gov/books/NBK326791/. [PubMed] [Google Scholar]
- 32.Zhang R, Geng X, Lee TMC (2017): Large-scale functional neural network correlates of response inhibition: an fMRI meta-analysis. Brain Struct Funct. 222: 3973–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dambacher F, Sack AT, Lobbestael J, Arntz A, Brugman S, Schuhmann T (2014): A network approach to response inhibition: dissociating functional connectivity of neural components involved in action restraint and action cancellation. Eur J Neurosci. 39: 821–831. [DOI] [PubMed] [Google Scholar]
- 34.Disner SG, Beevers CG, Haigh EAP, Beck AT (2011): Neural mechanisms of the cognitive model of depression. Nat Rev Neurosci. 12: 467–477. [DOI] [PubMed] [Google Scholar]
- 35.Schwarz AJ, Gozzi A, Reese T, Bifone A (2007): In vivo mapping of functional connectivity in neurotransmitter systems using pharmacological MRI. NeuroImage. 34: 1627–1636. [DOI] [PubMed] [Google Scholar]
- 36.Schaefer A, Burmann I, Regenthal R, Arélin K, Barth C, Pampel A, et al. (2014): Serotonergic Modulation of Intrinsic Functional Connectivity. Curr Biol. 24: 2314–2318. [DOI] [PubMed] [Google Scholar]
- 37.Wellington K, Perry CM (2001): Venlafaxine Extended-Release. CNS Drugs. 15: 643–669. [DOI] [PubMed] [Google Scholar]
- 38.Delgado PL, Moreno FA (2000): Role of norepinephrine in depression. J Clin Psychiatry. 61 Suppl 1: 5–12. [PubMed] [Google Scholar]
- 39.Sanchez C, Reines EH, Montgomery SA (2014): A comparative review of escitalopram, paroxetine, and sertraline: are they all alike? Int Clin Psychopharmacol. 29: 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nemeroff CB, Owens MJ (2004): Pharmacologic differences among the SSRIs: focus on monoamine transporters and the HPA axis. CNS Spectr. 9: 23–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.