

Abstract
The underlying challenge of drug delivery is the safe, controlled transport of a supply of therapeutic agent to its intended location at its effective dose. New and expanding solutions to payload delivery are being discovered in the field of hydrogels. Hydrogels are highly hydrated polymer networks that vary greatly depending on the underlying molecular structure. The subgroup of hydrogels that will be the focus of this chapter is the β-hairpin peptide hydrogel. These peptide-based materials are formed through a molecular self-assembly mechanism that only occurs after desired triggering of intramolecular peptide folding. Once folded, the β-hairpins assemble intermolecularly into a nanofibrillar network. The physical properties of the hydrogel network and its peptide foundation result in advantageous material properties which can be used for multiple biomedical applications including drug delivery. As a shear thinning solid that is easily injectable, cytocompatible, customizable, and well characterized, β-hairpin hydrogels are an exciting candidate as a drug delivery vehicle.
Graphical Abstract
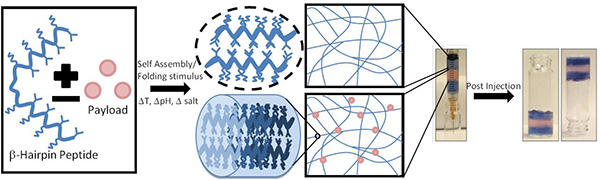
1. Introduction
Drug delivery is a challenge that requires the balance of maintaining an effective therapeutic dose in a desired location, managing and limiting side effects, and patient compliance.[1] One proposed solution to this challenge is the creation of local drug reservoirs that can release the active drug over sustained periods of time at the proper dose in a controlled manner.[2] This strategy requires a biologically compatible, easily delivered material with a well-defined payload release profile.[3] Hydrogels have emerged as an excellent candidate material for local, controlled delivery. Hydrogels, highly hydrated materials with an underlying molecular or supramolecular network, can be synthetic in nature (e.g. poly(ethylene glycol, or PEG, chemically crosslinked polymer network) or made from natural components, (e.g the natural extracellular matrix protein collagen).[4] Recently, supramolecular peptide hydrogels have become a heavily expanding segment of researched biomaterials. Peptide hydrogels are defined by a combination of intermolecularly assembled, short chain peptide molecules and high water content (generally 0.1 < 10.0 volume percent) resulting in versatile, soft-solid hydrogel materials with a wide range of applications.[5],[6] Peptide hydrogel materials can be categorized in a number of ways: the origin and type of the network molecules underlying the material (e.g. synthetic or cell-derived peptides/proteins), the type of intermolecular bonding underlying the network (e.g. physical or covalent interactions), the mechanism of crosslinking (e.g. entanglement vs. branching vs. covalent junction), or the mechanical characteristics (e.g. stiff, solid hydrogel vs. a soft solid able to exhibit some viscoelastic properties and some small amounts of flow). The use of peptides/peptidic molecules is not without precedent. For example, natural proteins such as collagen and naturally-derived proteins like Matrigel have been used to both to deliver cells and as cellular scaffolds. However, a significant hindrance to using these natural materials in quantifiable ways is their natural origin that results in materials containing proteins that are chemically/biochemically ill-defined. Collagen and Matrigel are able to promote cell proliferation and induce unique cellular phenotypes due to the presence of many growth factors and other biological molecules. However, the user is not sure of the exact soluble factors and ligands present in the materials thus making elucidation of effects on cells/drug delivery complicated or impossible. A different, finely-tuned, and well-defined strategy is the use of a synthetic, short, linear peptides, which utilize a self-assembly approach with synthetic molecules.[7–11] The assembly of short peptides almost always gives rise to fibrillar, one dimensional nanostructures resulting in mechanically and morphologically well-defined materials. Required or desired biomedical properties can be added simply through the covalent addition and/or mixing of components (e.g. desired ligands, enzyme substrates, encapsulated cells, drug compounds, etc.). Synthetic peptides tend to be easy to modify chemically although molecule synthesis and can be synthesized via solid-phase methods, bulk chemical methods, and even by recombinant biological expression techniques depending on the peptide sequence.
Peptide assemblies are growing in interest as a drug delivery vehicle. For example, micelles have been made by the Tirrell group that preferentially bind to fibrin for the targeted delivery of chemotherapeutics.[12] The Hartgerink lab recently modified a multidomain peptide to include a specific compartment on assembly that can hold hydrophobic drugs.[13] Stupp, in collaboration with Podlasek, used peptide amphiphile fibrils to deliver the protein sonic hedgehog to prevent neural degeneration.[14] While there are many types of small molecule peptide hydrogels,[7,15–18] this review will focus on self-assembling, β-hairpin peptides for encapsulation and delivery of payloads, particularly drug compounds for local, sustained delivery, of which there are many In the majority of small molecule peptides for hydrogel formation the monomer peptide unit can form a β-sheet during intermolecular assembly that is the foundation for the fibril direction and, depending on the solution assembly pathway used to form nanostructure and crosslinks, can lead to a stiff hydrogel[19,20]. In the case of a β-hairpin, the molecule undergoes a desired intramolecular folding event into a folded, β-hairpin conformation that then can assemble intermolecularly into β-sheet-rich fibrils (figure 1)[20]. Peptide hydrogels constructed from properly designed β-hairpin peptides and control of the intramolecular folding and consequent intermolecular assembly process were conceived by the Schneider and Pochan labs with thorough study of the material properties, such as control of hydrogel physical and chemical properties[21], solution assembly pathways[19], and nanostructure formation[22], as well as biomaterial properties, such as in vitro behavior with mammalian cells[23] and in vivo behavior, in support of possible biomedical applications. In this chapter, the fundamental design of the molecules for desired folding and assembly will be discussed with a focus on hydrogel material properties desired for the delivery of encapsulated molecular payloads.
Figure 1:

Transmission electron microscopy (TEM) of MAX1 β-hairpin nanofbirils. Inset schematic shows the bilayer cross-section of the nanofibril with a valine-rich hydrophobic core and hydrogen bonding along the fibril direction.[20]
2. Peptide Design
The word “peptide” generally implies one is considering a short chain of amino acids of less than ~50 amino acids. The β-hairpin peptides of focus in this chapter are all 20 amino acids in length. Also, the hydrogels to be discussed are all ≤ 2 weight percent peptide meaning ≥98% is water. The basic structure of the β-hairpin peptide is two locally amphiphilic arms of alternating hydrophobic valine and hydrophilic lysine amino acids surrounding a central turn sequence. The turn sequence forms a type II’ turn structure and promotes monomeric hairpin formation by forcing the two arms in a parallel arrangement under proper solution conditions that deprotonate lysine side chains and/or screen electrostatic interactions between lysines [22]. When folded in a parallel arrangement, the two arms undergo significant hydrogel bonding to stabilize the folded state. The originally studied peptide named MAX1 ((VK)4-VDPPT-(KV)4-NH2.) was designed to undergo the above intramolecular folding event (figure 2) [24]. While older schematics of the β-hairpin nanofibrils have shown the -VDPPT- turns opposing each other on each face of the bilayer fibril (e.g. Figure 2) recent NMR studies by Scheider and Tycko have revealed a different, regular nanostructure. In Nagy, et al., the hairpins are revealed to pack with the turn sequences all on one side of one face of the nanofibril but on the opposite side of the opposing fibril face (Figure 3). The peptides along one face of the nanofibril are also offset by one arm relative to the peptides along the opposing face instead of being directly over one another.[25] This surprising regularity in peptide packing within the fibrils provides for opportunities for specific, desired display of functionalization of the nanofibrils and overall hydrogels..
Figure 2:
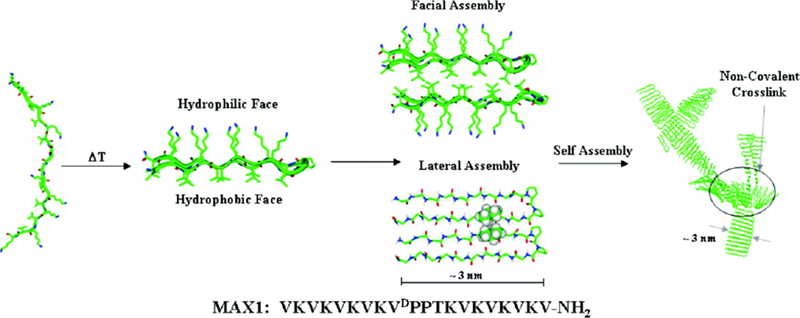
MAX1 mechanism of intramolecular folding and consequent intermolecular assembly. A desired trigger such as an increase in salt concentration, an increase in pH or an increase in temperature (as indicated in the above figure) drives the peptide to fold and undergo lateral and facial self-assembly into fibrils to form hydrogel network. [40]
Figure 3:
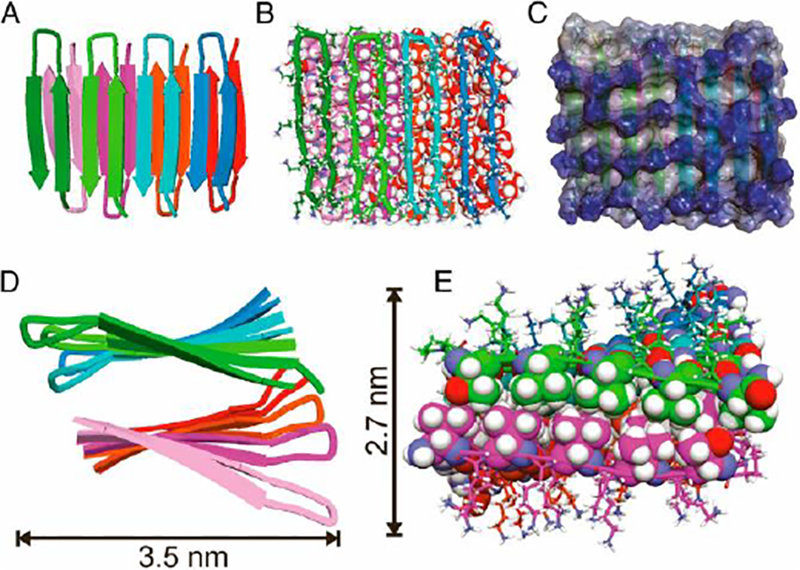
Cartoon representations of the molecular relationship between β hairpins in the MAX fibrils[25]
3. Gelation Mechanism
By controlling the folded state of the peptide molecules, one dictates gel formation and properties. As stated, if the lysine side chains are positively charged at low pH and with low salt concentration the peptide remains soluble and unfolded due to charge repulsion.[26] Therefore, folding can be triggered by a combination of an increase in the solvent pH to deprotonate the lysines, an increase in the ionic strength to introduce charge screening, and by an increase in temperature to increase the hydrophobic effect primarily between valine side chains. As will be discussed, peptide primary sequence changes from the original MAX1 sequence allows one to control the solution conditions required for molecule folding and to control the consequent rate by which the folded molecules assembly together to form a nanofibrillar hydrogel.
After folding, individual hairpins become facially amphiphilic with all valines displayed perpendicular from one face of the folded hairpins. This drives intermolecular assembly primarily through facial hydrophobic association to bury the valine-rich hydrophobic faces of the folded hairpins and define the cross-section of the eventual nanofibril as well as through lateral, intermolecular hydrogen bonding and hydrophobic interactions between the arms within one layer of the nanofibril(Figure 2, inset). The entire assembly mechanism of facial hydrophobic association and lateral intermolecular hydrogen bonding and hydrophobic association results in a fibril made of a bilayer of β-sheet hairpins with a hydrophobic core rich in valine side chains. Examples of assembly conditions include pH 9 (borate buffer), 20°C, and 50 mM added NaCl where MAX1 will undergo intramolecular folding followed by nanofibril assembly and complete hydrogel formation over the course of 20 minutes[19] or pH7.4 (phosphate buffered saline) with 150 mM NaCl, 37°C where assembly is slower due to the lower pH and more highly charged state of the lysine side chains.[19]
During molecule folding and assembly into nanofibrils and hydrogelation, the speed with which the fibrils are formed, which is controlled by the strength of the molecule folding trigger as well as peptide weight percent, has a clear effect on the stiffness of the ultimate hydrogel.[19,24] This difference in stiffness arises from the addition of crosslinks in the network during nanofibril formation. Defects during fibril formation can result from imperfect hydrophobic collapse of valine-rich faces that can become a nucleation point for the formation of a second fibril. This results in physical branch points that, consequently, lead to a stiffer network.[27] The faster a gel is formed (e.g. with a higher amount of salt/higher temperature) the more resultant hydrophobic packing defects/more resultant branch points provding a stiffer, final gel.[28] Entanglements can also form from two separate fibrils intertwining to form a physical crosslink. Both defect-induced branching and fibril entanglements result in physical crosslinks in the gel network that serve to physically stiffen the hydrogel (figure 4).[24] The branching mechanism was recently supported through the study of a new peptide that was designed to eliminate branching during assembly and significantly affect the nanofibril morphology and hydrogel properties.[29]
Figure 4:
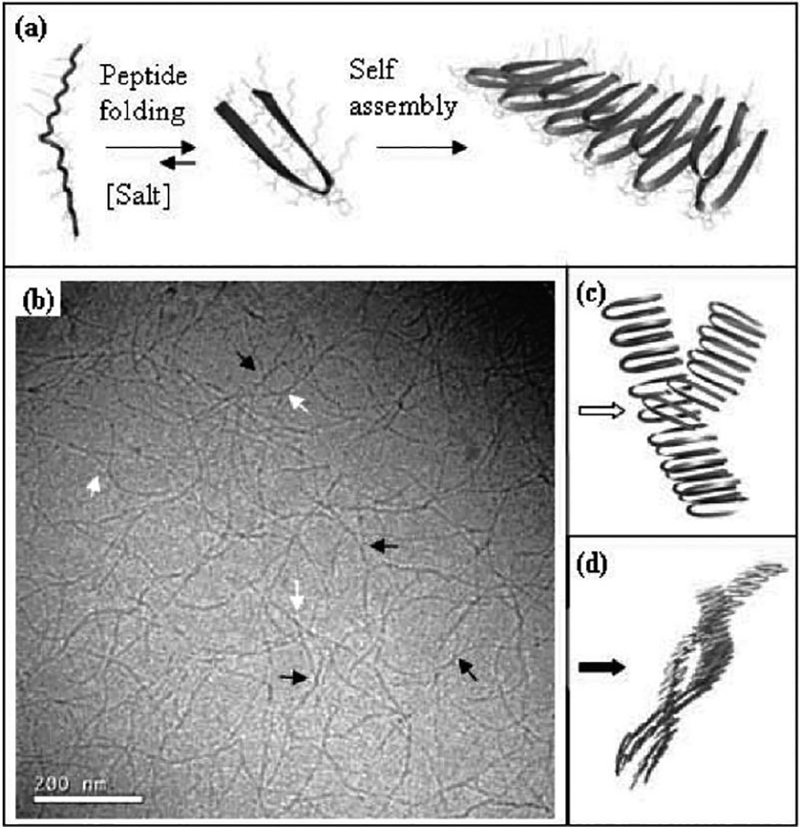
Cryogenic TEM of nanofibrillar network highlighting branch points (while arrows) and entanglements (black arrows).[24]
4. Hydrogel Properties
The above discussion reveals the mechanism for the desired formation of hydrogels from properly designed β-hairpin peptides. Practically, hydrogel formation requires simple mixing of aqueous peptide solutions with a desired buffer solution to induce molecule folding and assembly into a hydrogel. Therefore, one need only add a desired payload for encapsulation into the buffer solution in order to guarantee homogeneous encapsulation within the final, stiff hydrogel. In addition, MAX1 and other β-hairpin peptide hydrogels share several advantageous physical and chemical properties for payload delivery to a secondary site. The first advantageous property is that the materials are shear-thinning solids. The assembled hydrogel networks behave like permanent hydrogels with no flow over time despite having no covalent crosslinks.[26] However, the stiff gels can undergo shear-thinning when exposed to shear force, such as in a rheometer for gel stiffness characterization or when injected from a syringe. During shear the gel will flow with low viscosity until the shear force has ceased when it immediately heals into a solid with the same physical properties that the material exhibited prior to injection(figure 5).[30,31] Importantly, this means a preformed hydrogel can be used as a delivery vehicle via simple syringe or catheter injection in a relatively noninvasive fashion. The mechanism at work here is that following the formation of the hydrogel network a significant enough shear force can fracture the physical network into domains or layers allowing for low viscosity flow. At the cessation of shear forces the domains are able to percolate immediately into a stiff network with further recovery of stiffness through relaxation and fibrillar interpenetration between previously fractured domains or layers.[31] The recovery of hydrogel properties exhibited prior to shear-thinning delivery is critical in the strategy of using the materials as devices for payload delivery. One can encapsulate a payload and study the dispersion of the encapsulant in the material as well as the release of the payload. Importantly, the properties such as payload dispersion within the hydrogel and payload release are the same post-injection as measured prior to shear-thinning injection. Therefore, one can be confident that the payload release profile post injection, e.g. in vivo, will be the same as understood from in vitro studies as opposed to the injection of a liquid that can crosslink into a hydrogel with a payload only once in vivo. The high water content combined with well-defined nanofibril and network morphology of the gel means the payload will consistently diffuse out of the gel in a well-defined manner.
Figure 5:
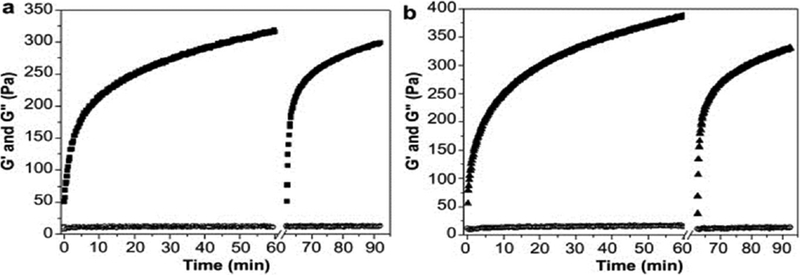
Oscillatory rheology of β-hairpin hydrogel formation during hydrogelation of A) pure peptide and B) peptide loaded with 4mM curcumin from 0–60 minutes. At 60 minutes, the hydrogels were briefly subjected to a shear-treatment to simulate injection conditions after which hydrogel recovery was observed for an additional 30 minutes. Pure peptide hydrogels behaved similarly to those with encapsulated curcumin. [58]
6. Peptide Alterations
While MAX1 is the parent, originally studied β-hairpin peptide there have been many modifications made to both investigate the fundamental nature of the material and to improve its performance for specific tasks. This design flexibility is one the advantages that comes with using a peptide hydrogel. Most of the gel physical properties can be changed by alteration of the specific amino acids. The specific temperature pH and salt concentration at which gelation will occur are all tunable with desired peptide sequence. For example, MAX2 (VKVKVK VDPPTKVKVTVKV) and MAX3 (VKVKVT VDPPTKVKVTVKV) are identical to MAX1 except for the exchange of one or two valines, respectively, for the isostructural but less hydrophobic threonine.[32] The change decreased the hydrophobicity of the new molecules causing the peptides to require a higher temperature for intramolecular folding and intermolecular assembly.
Final hydrogel stiffness can be finely controlled using hydrogel weight percent, a more complex interaction between peptide enantiomers,[33] or through modifying the hydrophobic face of the hairpin.[34] The importance of lateral hydrophobic interactions is shown through MAX4 (KVKVKVKVKDPPSVKVKVKVK) which has its valine and lysine positions on the amphiphilic arms switched so that when folded the valine side chains point to the center line of the hairpin instead of away from it.[35] The change makes lateral hydrophobic interaction more difficult and inhibits fibril growth resulting in a less stiff hydrogel compared to MAX1. More complex chemistry can be add to the peptide like a temporal UV controlled gelation trigger,[36] or UV initiated covalent crosslinking.[37] There are even simple biological cues that can be designed into the network, for example a binding ligand like RGD or a cleavable section targetable by MMP.[38]
As will be discussed later in the manuscript, MAX1 has been used successfully to culture a range of cells including fibroblasts and mesenchymal stem cells,[23,39] but because of its slow gelation time at physiological conditions it is not an ideal scaffold for cell encapsulation and delivery. Peptide alterations can be designed to lessen the overall charge of the hairpin that, in turn, makes the molecule more amenable to folding at the same solution conditions relative to MAX1. MAX8 (VKVKVK VDPPTKVEVKVKV) was designed by replacing one lysine with a glutamic acid and consequently lowering the charge of the peptide. The results is that less charge needs to be screened/obviated before hairpin folding can occur.[40] Interestingly, the location of the substitution along the peptide chain had a major effect on the intramolecular folding response to pH and temperature change. The most responsive peptide at pH 7 had a lysine exchanged for a glutamic acid at position 15. The change results in a peptide that will rapidly undergo gelation at physiological conditions (37 C, 7.4 pH). MAX8 possesses the useful mechanical properties of MAX1 and rapidly, under 1 minute, undergoes gelation at physiological conditions. The increased gelation speed creates a more physically robust network and ensures homogeneous cell distribution, or other encapsulated payload, throughout the hydrogel.[39]
7. Delivery
The use of a self-assembling, physically crosslinked, shear-thinning and immediately rehealing peptide hydrogel as a payload delivery vehicle has many advantages over other hydrogel options.[6] For example, because the hydrogel can be formed into a solid network with desired physical properties and payload delivery profile for injection as a solid, many issues associated with in vivo crosslinking can be avoided (figure 6). With in vivo crosslinking, a large amount of drug or cell payload can be lost to diffusion prior to the network forming. Additionally, the hydrogel will be diluted by other fluids and molecules, thus producing a material that is not the same as studied ex vivo. Pre-reaction monomers can interact unfavorably with local tissue. The action of kicking off chemical crosslinking using UV or heat as well as the heat from the covalent bonding can negatively affect local tissue and the encapsulated payload.[41] Following injection, the β-hairpin peptides also have the unique property of being inherently cytocompatible[42] and biocompatible[23,43] with some peptides even displaying innate anti-bacterial activity.[44] Cultures of multiple bacteria strains including E. coli, K. pneumonia, and S. aureus were grown on MAX1 and each one had their membranes physically compromised which resulted in death. Even during a co-culture between fibroblasts, A. xylosoxidans, and S. maltophilia the bacterial cells were inhibited while the mammalian cells proliferated.[44]
Figure 6:
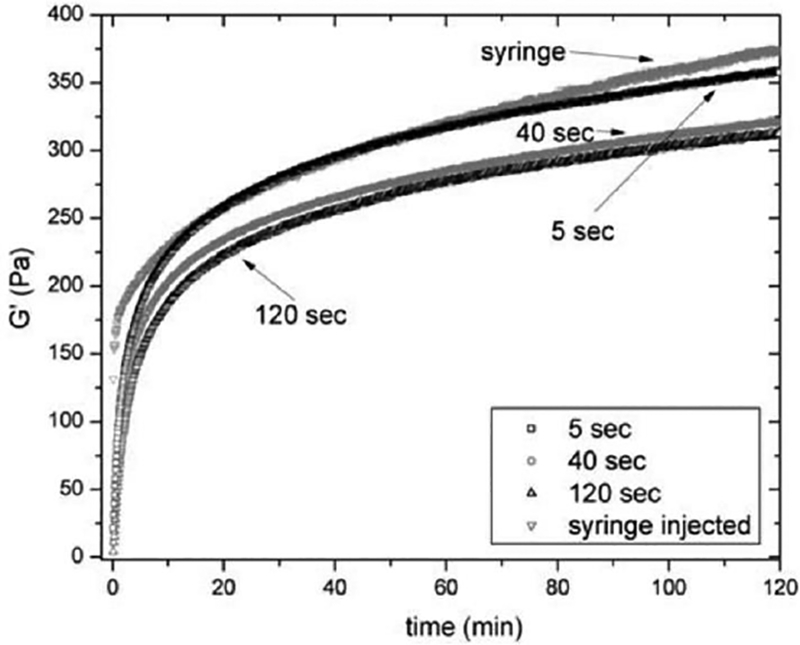
Oscillatory rheology of 0.5 wt% MAX8 (pH 7.4, 25 mM HEPES, 37 C) comparing the hydrogels storage modulus restoration response to a syringe injection or shear step [5s, 40s, 120s] (simulated injection)[31]
7.1. Cells
While cells themselves are not drugs, they are a growing section of therapeutic options that requires controlled delivery.[45] Cell-based therapies for stroke, spinal cord injury, heart attack, joint rehabilitation, and general wound healing are all in various stages of development and require the safe and effective delivery of cells.[46,47] Peptide hydrogels are particularly useful for cell delivery because of its biocompatibility as well as serving as the cell scaffold immediately following delivery. The peptide can also incorporate biologically active moieties, for example RGD a common cell binding ligand, or a matrix metalloproteinase cleavable section.[38] There are multiple options to guide and drive cell proliferation or differentiation following injection.
Many cell types have been successfully encapsulated within β-hairpin hydrogels with multiple, possible delivery applications. Two areas of interest are the replacement of cartilage in joints and the repair of nerves in the spinal column.[48] Because the injection of mesenchymal stem cells suspended in media can expose the cells to shear forces, they were chosen to highlight the flow profile of the material.[49] Following encapsulation of mesenchymal stem cells in MAX8 the gel mechanical properties were tested to show that the shear thinning and rehealing ability had not been lost due to the presence of the encapsulated cells. The cells also do not alter the fibrilar nanostructure of the hydrogel. Following a test injection onto tissue culture plastic, live dead staining was done and showed minimal cell death (figure 7). To discover how the hydrogel was behaving during injection, fluorescent microbeads were used to visualize the flow profile through a capillary. The hydrogel displayed plug flow, a flow profile characterized by the central section of tube having a constant velocity. Depending on the flow rate, as much as 90% of the tube cross section experienced a constant flow, meaning the majority of encapsulated cells were protected from shear.[50] Shear stress can have many effects on cell phenotype, directing preferential growth, reorganizing the cytoskeleton, and/or promotion of proliferation. These shear effects can be good or bad depending on desired cell behavior, but the injection process must be considered when using cell-based therapy. The use of β-hairpin hydrogel as a shear-thinning sold matrix allows one to purposefully shield encapsulated cells from shear if shear is to be avoided in supporting desired cell behavior post-injection (Figure 8). As mentioned earlier, there also have been macrophage studies to show a lack of immune response to the hydrogel material in vitro.[42] While not considered dangerous necessarily, the D-proline in the turn sequence is uncommon in humans. Therefore, peptides have been designed by the Schneider group that that utilize natural turn sequences (VPDGT, VPIGT, YNGT) that can be substituted to allow bacterial expression during biological synthesis or to avoid injecting non-natural proline derivatives [51]
Figure 7:
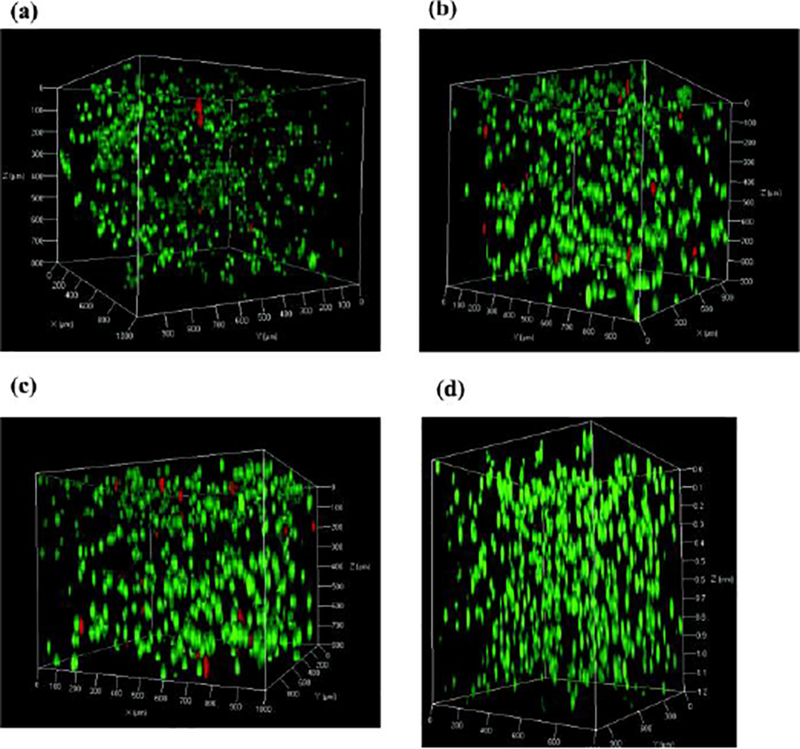
Three dimensional confocal microscopy of 5*106 cells/mL MG63 cells encapsulated in 0.5 wt% MAX8 (pH 7.4, 25 mM HEPES, 37 C) using live dead staining taken 3h following injection through a 250 μm diameter capillary at A) 4.0 mL/h B) 6.0 mL/h C) 8.0 mL/h and D) no injection for comparison.[50]
Figure 8:
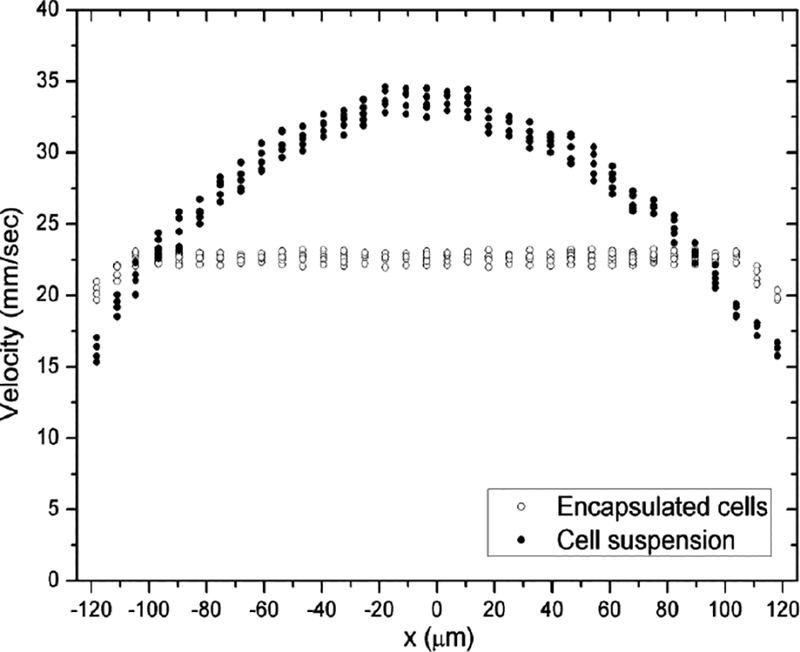
One dimensional flow profile at 4.0 mL/h of MG63 cells encapsulated in 0.75 wt% β-hairpin hydrogel (pH 7.4, 25 mM HEPES, 37 C) exhibiting plug flow with shear only at the walls compared to the parabolic flow profile of cells in aqueous media.[50]
7.2. Proteins
The use of proteins, growth factors, and enzymes as a therapeutic payload is a simple choice to make when the delivery vehicle is protein based.[52,53] The active protein can be covalently linked to the scaffold requiring enzymatic or other release or can be physically associated with the scaffold after simple mixing prior to hydrogelation.[54] To begin to investigate the effects of protein encapsulation on the bulk material properties of β-hairpin peptides and how the encapsulated protein is then released, the protein lactoferrin was encapsulated in a series of increasing weight percent hydrogels (.5, 1, 2 %) of both MAX1 and MAX8. The effect of hydrogel pore size and electrostatic interaction between network and payload was the focus of this initial study in the Schneider lab. The pore size of MAX8 hydrogels was controlled by the concentration of peptide; the higher the peptide concentration, the smaller the pore size of the nanofibrillar network. To examine the electrostatic effects of protein-network interaction, the positively charged lactoferrin was compared with a negatively charged 70 dalton dextran probe. Both molecules have a similar hydrodynamic diameter, around 8 nm. As expected, the larger pore size gels have a faster release profile. The positively charged lactoferrin was released from the hydrogel much faster when compared with the negatively charged dextran. This is due to the fact that the hydrogel network has a net-positive charge due to the preponderance of lysine residues on the surface of the nanofibrils. Of the total dextran encapsulated only 60% is released after 15 days compared with the nearly total release of lactoferrin. Two additional dextran probes were also encapsulated, one larger (150 dalton) and one smaller (20 dalton) to examine how the payload size effects the release profile. The smaller dextran probe was able to quickly and completely leave the hydrogel while the much larger probe remains largely trapped even after 30 days due to hydrogel having too small of a pore size for the large probe diffusion.[55]
To further elucidate the effects of hydrodynamic diameter and charge on the release of proteins from β-hairpin peptides multiple proteins (lactalbumin, lysozyme, myoglobin, albumin, lactoferrin, and immunoglobulin g) were encapsulated and their release profiles were examined by the Schneider group. As with the lactoferrin and dextran study, neutral and positively charged proteins were able to release fully from the gel, but negatively charged proteins remained trapped, physically releasing less than 10% of original load.[55] The result suggests that the negatively charged proteins adsorb to the positively charged fibrils retarding release. Rheology determined that the protein loading could slightly decrease the hydrogel stiffness up to 12% depending on the protein loaded, but for other encapsulated proteins there was no effect. Importantly, none of the loaded proteins interfered with the hairpin fibril formation or shear thinning and rehealing properties. Importantly, syringe delivery of the protein-loaded hydrogels did not cause a significant change to the protein release profiles as compared to pre-syringe injection. This result supports the earlier discussed network fracture mechanism of shear-thinning and flow and that there is not a complete disruption of the network even during shear-thinning injection.
To study the therapeutic potential of loaded growth factor proteins, nerve growth factor and brain-derived neurotrophic factor were encapsulated in MAX8 and delivered in vitro to PC12 cells, a cell line derived from a neuroendocrine tumor of the adrenal glands. The presence of NGF or BDNF promotes cell growth and neurite extensions in the cell line, providing a metric to determine the presence of therapeutically active levels of growth factor. The growth factors were encapsulated in 0.5, 1.0, and 1.5 weight percent MAX8 hydrogel at multiple growth factor concentrations. The higher the initial amount of growth factor encapsulated and the lower the weight percent of the hydrogel, the larger amount of growth factor that was released.[48] The release was consistent over the initial two-week encapsulation, and there is a negligible amount of loaded growth factor released immediately following injection (figure 9). Both results are useful for a drug delivery vehicle, but the most interesting result was the protection the peptide gel seemed to afford the growth factors. Both growth factors have an activity half-life of 3.5 days in solution, but following encapsulation for 1 month with regular media sink changes the eluted growth factors were still able to have a phenotypic effect on the cells causing the neurite extensions. This result indicates that the growth factors are able to associate with the hydrogel in a way that prevents degradation and opens up possible long-term therapy options.
Figure 9:
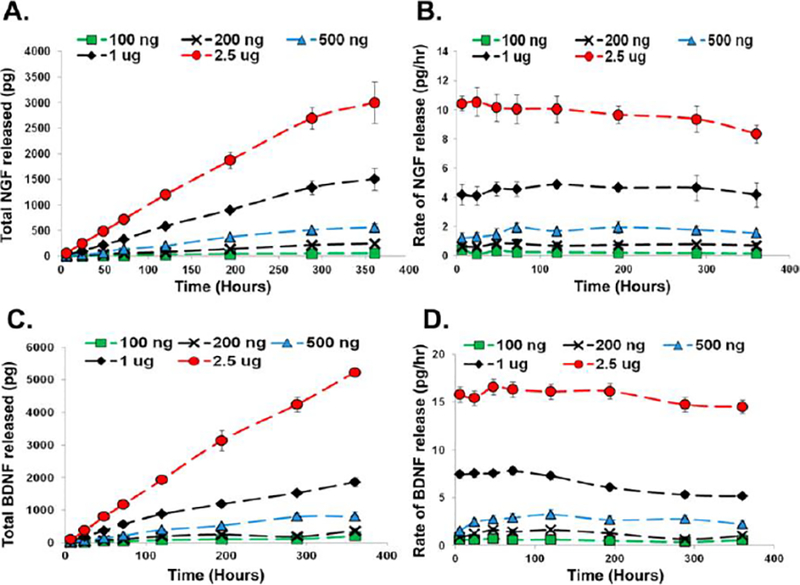
Total release and rate of release of 100ng through 2.5 μg of NGF (A, B) or BDNF (B, C) from 1.0 wt% MAX8 (pH 7.4, 25 mM HEPES, 37 C) over two weeks measured by ELISA[48]
7.3. Chemotherapeutics
The concept of long-term, controlled release of a therapeutic that normally has a very short half-life in bulk solution is also critical when considering chemotherapy delivery. When designing the delivery of chemotherapeutics, harmful side effects should be minimized while the cancer should be effectively treated for a positive patient outcome.[56] The ability to deliver a long-term controlled, local release of chemotherapeutics would cut down on the exposure of healthy tissue to chemotherapy drugs, decreasing side effects and would require fewer treatments to keep the drug in its therapeutic range. This situation is well suited for β-hairpin hydrogels. An example of small molecule therapeutic delivery was shown with curcumin, a hydrophobic molecule extracted from the turmeric root. It has anti-inflammatory and cytotoxic effects, but because it is hydrophobic and has a half-life of 8 hours it can be challenging to properly deploy as a therapeutic intervention.[57] For these challenging reasons it was chosen as an ideal candidate for delivery via an amphiphilic β-hairpin peptide. Curcumin (0 mM, 2 mM, 4 mM) was encapsulated in MAX8 (0.5, 1.0, 2.0 weight percent) and shown to release micromolar amounts of curcumin up to two weeks following encapsulation. (figure 11)[58] The released compound was shown to have a cytotoxic effect at levels lower than those reported in the literature supporting the idea that a continuous, low-dose treatment is preferable to a single bolus of drug. Rheology shows the loaded hydrogel is stiffer than unloaded, this suggests that the curcumin is buried in the fibril core stiffening them or creating additional crosslinking between the fibrils.
Figure 11:
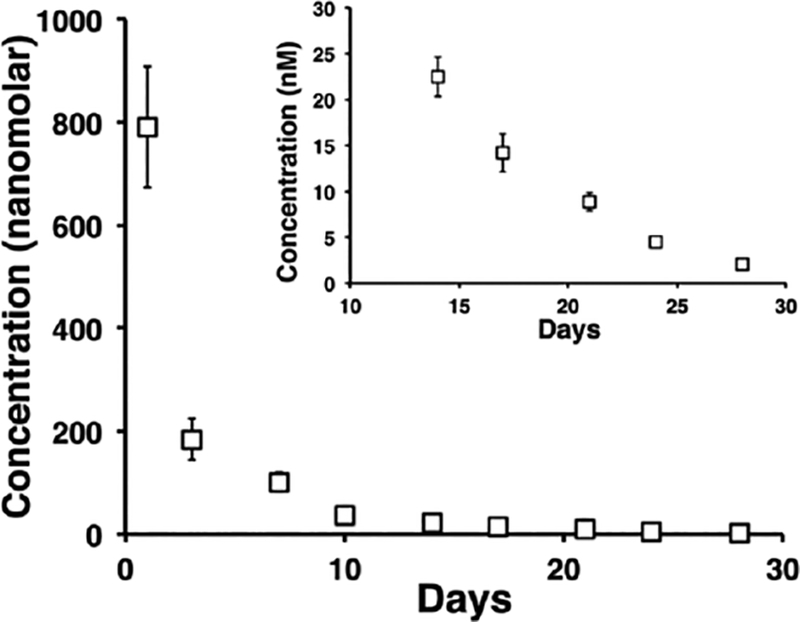
The released profile of tritiated vincristine over 4 weeks from 0.5 wt% MAX8 (pH 7.4, 25 mM HEPES, 37 C) with an insert of the final two weeks showing the released amounts are nonzero and are still able to have a therapeutic effect on the cells.[62]
Vincristine is a more traditional chemotherapeutic that is approved for use treating multiple types of cancer including leukemias and lymphomas.[59] Vincristine is also hydrophobic and has a half-life of 35 hours in bulk solution. It works by indiscriminately preventing cell division through tubulin binding, and, as a result, has severe side effects. Vincristine (0 uM, 10 uM, 500 uM) was encapsulated in 0.5 weight % MAX8 for 30 days. The hydrogel was shown to release nanomolar amounts of drug during the entire 30 days with the drug still actively able to kill cells even on the last day of the release study. Using rheology, no physical difference was seen between the loaded and unloaded gels, and the shear-thinning and rehealing behavior was unaffected. Neutron scattering was done to investigate the loaded hydrogel nanostructure to find the location of the vincristine in the nanostructure. Because the scattering was largely unaffected it suggests that the vincristine is intimately, physically bound to the peptide fibrils (figure 10). This result also supports the ability of the hydrogel to protect the vincristine from degradation.
Figure 10:
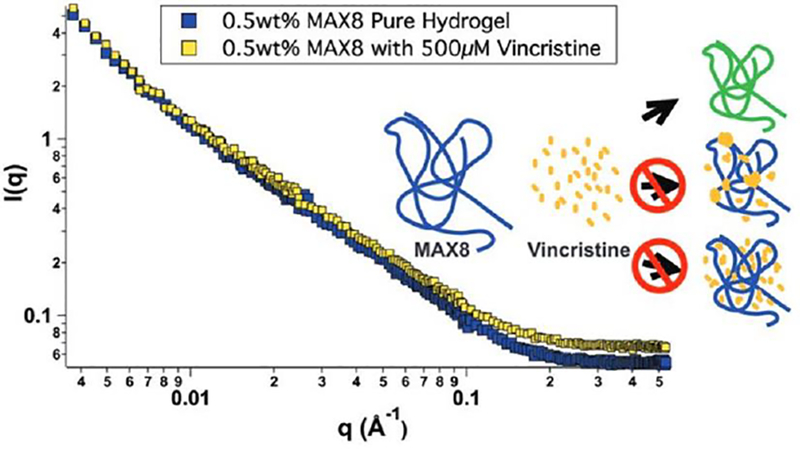
Small angle neutron scattering of 0.5 wt% MAX8 (pH 7.4, 25 mM HEPES, 37 C) with 500 μM vincristine (yellow) and without vincristine (blue). The similar shape and slope of the lines indicate the encapsulation of vincristine does not alter the structure or folding of MAX8, and the cartoon indicates possible drug-gel combinations.[62]
7.3. DNA
DNA is another fragile therapeutic agent that can require special considerations to ensure a successful delivery.[60] Plasmid DNA can force cells to express specific tumor antigens that will create a heightened immune response. MAX1, MAX8, and HLT2 were loaded with plasmid DNA that encodes for a melanoma antigen by the Schneider group.[43] HLT2 was designed by altering MAX8 with the goal to lower the peptide charge to +5 making the hydrogel gentler on cells during encapsulation but without changing the physical properties.[61] HLT2 was created by exchanging two lysines for leuceines and two valines for threonines. The loaded gels were injected into mice and the immune response was measured. The MAX8 and HLT2 mice showed evidence of plasmid uptake by the cells via increased lymphocyte proliferation. The HLT2 gel showed the most evidence of gel resorbtion, cell infiltration, and tissue growth. The mouse’s immune response was also found to be driven specifically by its reaction to the melanoma specific antigen which the encapsulated plasmid encoded for and promoted macrophage infiltration. Before injection each hydrogel was tested for plasmid retention over 2 weeks and less than 10% was released. The results show the gel is safely delivering the plasmid in a way that promotes cellular uptake and therapeutic effect.
8. Conclusion
β-hairpin peptide hydrogels provide several unique material advantages that make them well in vivo delivery. The peptide hydrogels can be preformed into solids prior to injection with physical and payload delivery characteristics. Delivery can then occur with shear-thinning immediate rehealing through syringe or catheter injection. The hydrogels can locally release a drug or protein in a defined and continuous manner with a release profile that is dependent on design of the peptide and hydrogel network. The materials can deliver shear-sensitive payloads and able to protect therapeutic agents delivering compounds that are still active long after they would have broken down in bulk solution. Current research efforts work to engineer more desired properties the peptide hydrogel to fine-tune payload delivery. For example, compared to covalently hydrogels, physical gels are considerably weaker/less stiff. This prevents injections in high stress/strain tissues if the goal is for the scaffold to remain in place. Current work also is addressing how the peptide scaffolds affect cellular growth and phenotype through both mechanical stiffness, morphology chemical or biological cues built into the scaffold. Finer mechanical control including new degradation pathways or, conversely, the prevention of degradation would be useful for controlling drug release surrounding cell behavior. The advantages of β-hairpin peptide hydrogels for drug delivery warrant their continued study.
Acknowledgements
This work is supported by NIST UD CNS 70NANB12H239, NIH grant P30114736, and funds from the DO Believe and Nemours Foundations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
9. References
- [1].Goldberg M, Langer R, Jia XQ, Nanostructured materials for applications in drug delivery and tissue engineering, J. Biomater. Sci. Ed 18 (2007) 241–268. doi: 10.1163/156856207779996931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jiang Y, Chen J, Deng C, Suuronen EJ, Zhong Z, Click hydrogels, microgels and nanogels: Emerging platforms for drug delivery and tissue engineering, Biomaterials. 35 (2014) 4969–4985. doi: 10.1016/j.biomaterials.2014.03.001. [DOI] [PubMed] [Google Scholar]
- [3].Branco MC, Schneider JP, Self-assembling materials for therapeutic delivery, Acta Biomater. 5 (2009) 817–831. doi: 10.1016/j.actbio.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nguyen QV, Huynh DP, Park JH, Lee DS, Injectable polymeric hydrogels for the delivery of therapeutic agents: A review, Eur. Polym. J 72 (2015) 602–619. doi: 10.1016/j.eurpolymj.2015.03.016. [DOI] [Google Scholar]
- [5].Guvendiren M, Lu HD, Burdick J. a., Shear-thinning hydrogels for biomedical applications, Soft Matter. 8 (2012) 260. doi: 10.1039/c1sm06513k. [DOI] [Google Scholar]
- [6].Yan C, Pochan DJ, Rheological properties of peptide-based hydrogels for biomedical and other applications., Chem. Soc. Rev 39 (2010) 3528–40. doi: 10.1039/b919449p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kang MK, Colombo JS, D’Souza RN, Hartgerink JD, Sequence effects of self-assembling multidomain peptide hydrogels on encapsulated SHED cells., Biomacromolecules. 15 (2014) 2004–11. doi: 10.1021/bm500075r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bakota EL, Sensoy O, Ozgur B, Sayar M, Hartgerink JD, Self-assembling multidomain peptide fibers with aromatic cores., Biomacromolecules. 14 (2013) 1370–8. doi: 10.1021/bm4000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Maslovskis A, Guilbaud J, Grillo I, Hodson N, Miller AF, Saiani A, Self-assembling peptide/thermoresponsive polymer composite hydrogels: effect of peptide-polymer interactions on hydrogel properties., Langmuir. 30 (2014) 10471–80. doi: 10.1021/la502358b. [DOI] [PubMed] [Google Scholar]
- [10].Zhou M, Ulijn RV, Gough JE, Extracellular matrix formation in self-assembled minimalistic bioactive hydrogels based on aromatic peptide amphiphiles., J. Tissue Eng 5 (2014) 2041731414531593. doi: 10.1177/2041731414531593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Song B, Song J, Zhang S, Anderson M. a, Ao Y, Yang C-Y, Deming TJ, Sofroniew MV, Sustained local delivery of bioactive nerve growth factor in the central nervous system via tunable diblock copolypeptide hydrogel depots., Biomaterials. 33 (2012) 9105–16. doi: 10.1016/j.biomaterials.2012.08.060. [DOI] [PubMed] [Google Scholar]
- [12].Ji E, Cheng Y, Morshed R, Nord K, Han Y, Wegscheid ML, Auf B, Wainwright DA, Lesniak MS, Tirrell MV, Biomaterials Fibrin-binding, peptide amphiphile micelles for targeting glioblastoma, 35 (2014). doi: 10.1016/j.biomaterials.2013.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li I, Moore AN, Hartgerink D, “ Missing Tooth “ Multidomain Peptide Nano fi bers for Delivery of Small Molecule Drugs, (2016). doi: 10.1021/acs.biomac.6b00309. [DOI] [PubMed]
- [14].Choe S, Bond CW, Harrington DA, Stupp SI, Mcvary KT, Podlasek CA, Peptide amphiphile nanofiber hydrogel delivery of sonic hedgehog protein to the cavernous nerve to promote regeneration and prevent erectile dysfunction, Nanomedicine Nanotechnology, Biol. Med 13 (2017) 95–101. doi: 10.1016/j.nano.2016.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].King PJS, Giovanna Lizio M, Booth A, Collins RF, Gough JE, Miller AF, Webb SJ, A modular self-assembly approach to functionalised β-sheet peptide hydrogel biomaterials, Soft Matter. 12 (2016) 1915–1923. doi: 10.1039/C5SM02039E. [DOI] [PubMed] [Google Scholar]
- [16].Glassman MJ, Avery RK, Khademhosseini A, Olsen BD, Toughening of Thermoresponsive Arrested Networks of Elastin-Like Polypeptides to Engineer Cytocompatible Tissue Scaffolds, Biomacromolecules. 17 (2016) 415–426. doi: 10.1021/acs.biomac.5b01210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cardoso AZ, Mears LLE, Cattoz BN, Griffiths PC, Schweins R, Adams DJ, Linking micellar structures to hydrogelation for salt-triggered dipeptide gelators, Soft Matter. 12 (2016) 3612–3621. doi: 10.1039/C5SM03072B. [DOI] [PubMed] [Google Scholar]
- [18].Foster AA, Marquardt LM, Heilshorn SC, The diverse roles of hydrogel mechanics in injectable stem cell transplantation, Curr. Opin. Chem. Eng 15 (2017) 15–23. doi: 10.1016/j.coche.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ozbas B, Kretsinger J, Rajagopal K, Schneider JP, Pochan DJ, Salt-triggered peptide folding and consequent self-assembly into hydrogels with tunable modulus, Macromolecules. 37 (2004) 7331–7337. doi: 10.1021/ma0491762. [DOI] [Google Scholar]
- [20].Ozbas B, Rajagopal K, Schneider JP, Pochan DJ, Semiflexible chain networks formed via self-assembly of Beta-hairpin molecules, Phys. Rev. Lett 93 (2004) 1–4. doi: 10.1103/PhysRevLett.93.268106. [DOI] [PubMed] [Google Scholar]
- [21].Hule RA, Nagarkar RP, Hammouda B, Schneider JP, Pochan DJ, Dependence of self-assembled peptide hydrogel network structure on local fibril nanostructure, Macromolecules. 42 (2009) 7137–7145. doi: 10.1021/ma9003242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lamm MS, Rajagopal K, Schneider JP, Pochan DJ, Laminated morphology of nontwisting β-sheet fibrils constructed via peptide self-assembly, J. Am. Chem. Soc 127 (2005) 16692–16700. doi: 10.1021/ja054721f. [DOI] [PubMed] [Google Scholar]
- [23].Kretsinger JK, Haines LA, Ozbas B, Pochan DJ, Schneider JP, Cytocompatibility of self-assembled β-hairpin peptide hydrogel surfaces, Biomaterials. 26 (2005) 5177–5186. doi: 10.1016/j.biomaterials.2005.01.029. [DOI] [PubMed] [Google Scholar]
- [24].Yucel T, Micklitsch CM, Schneider JP, Pochan DJ, Direct observation of early-time hydrogelation in β-hairpin peptide self-assembly, Macromolecules. 41 (2008) 5763–5772. doi: 10.1021/ma702840q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nagy-smith K, Moore E, Schneider J, Tycko R, Molecular structure of monomorphic peptide fibrils within a kinetically trapped hydrogel network, (2015). doi: 10.1073/pnas.1509313112. [DOI] [PMC free article] [PubMed]
- [26].Veerman C, Rajagopal K, Palla CS, Pochan DJ, Schneider JP, Furst EM, Gelation kinetics of β-hairpin peptide hydrogel networks, Macromolecules. 39 (2006) 6608–6614. doi: 10.1021/ma0609331. [DOI] [Google Scholar]
- [27].Hule R. a., Nagarkar RP, Altunbas A, Ramay HR, Branco MC, Schneider JP, Pochan DJ, Correlations between structure, material properties and bioproperties in self-assembled β-hairpin peptide hydrogels, Faraday Discuss. 139 (2008) 251. doi: 10.1039/b717616c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Branco MC, Nettesheim F, Pochan DJ, Schneider JP, Wagner NJ, Fast dynamics of semiflexible chain networks of self-assembled peptides., Biomacromolecules. 10 (2009) 1374–80. doi: 10.1021/bm801396e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sathaye S, Zhang H, Sonmez C, Schneider JP, Macdermaid CM, Von Bargen CD, Saven JG, Pochan DJ, Engineering complementary hydrophobic interactions to control β-hairpin peptide self-assembly, network branching, and hydrogel properties, Biomacromolecules. 15 (2014) 3891–3900. doi: 10.1021/bm500874t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sathaye S, Mbi A, Sonmez C, Chen Y, Blair DL, Schneider JP, Pochan DJ, Rheology of peptide- and protein-based physical hydrogels: Are everyday measurements just scratching the surface?, Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology 7 (2015) 34–68. doi: 10.1002/wnan.1299. [DOI] [PubMed] [Google Scholar]
- [31].Yan C, Altunbas A, Yucel T, Nagarkar RP, Schneider P, Pochan DJ, Schneider JP, Pochan DJ, Injectable solid hydrogel: mechanism of shear-thinning and immediate recovery of injectable β-hairpin peptide hydrogels., Soft Matter. 6 (2010) 5143–5156. doi: 10.1039/C0SM00642D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pochan DJ, Schneider JP, Kretsinger J, Ozbas B, Rajagopal K, Haines L, Thermally reversible hydrogels via intramolecular folding and consequent self-assembly of a de novo designed peptide, J. Am. Chem. Soc 125 (2003) 11802–11803. doi: 10.1021/ja0353154. [DOI] [PubMed] [Google Scholar]
- [33].Nagy KJ, Giano MC, Jin A, Pochan DJ, Schneider JP, Enhanced mechanical rigidity of hydrogels formed from enantiomeric peptide assemblies, J. Am. Chem. Soc 133 (2011) 14975–14977. doi: 10.1021/ja206742m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Micklitsch CM, Medina SH, Yucel T, Nagy-Smith KJ, Pochan DJ, Schneider JP, Influence of hydrophobic face amino acids on the hydrogelation of -hairpin peptide amphiphiles, Macromolecules. 48 (2015) 1281–1288. doi: 10.1021/ma5024796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rajagopal K, Ozbas B, Pochan DJ, Schneider JP, Probing the importance of lateral hydrophobic association in self-assembling peptide hydrogelators, Eur. Biophys. J 35 (2006) 162–169. doi: 10.1007/s00249-005-0017-7. [DOI] [PubMed] [Google Scholar]
- [36].Haines LA, Rajagopal K, Ozbas B, Salick DA, Pochan DJ, Schneider JP, Light-activated hydrogel formation via the triggered folding and self-assembly of a designed peptide, J. Am. Chem. Soc 127 (2005) 17025–17029. doi: 10.1021/ja054719o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rughani RV, Branco MC, Pochan DJ, Schneider JP, De novo design of a shear-thin recoverable peptide-based hydrogel capable of intrafibrillar photopolymerization, Macromolecules. 43 (2010) 7924–7930. doi: 10.1021/ma1014808. [DOI] [Google Scholar]
- [38].Giano MC, Pochan DJ, Schneider JP, Controlled biodegradation of Self-assembling Beta-hairpin Peptide hydrogels by proteolysis with matrix metalloproteinase-13, Biomaterials. 32 (2011) 6471–6477. doi: 10.1016/j.biomaterials.2011.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Haines-Butterick L, Rajagopal K, Branco M, Salick D, Rughani R, Pilarz M, Lamm MS, Pochan DJ, Schneider JP, Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells., Proc. Natl. Acad. Sci. U. S. A 104 (2007) 7791–6. doi: 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Rajagopal K, Lamm MS, Haines-Butterick LA, Pochan DJ, Schneider JP, Tuning the pH responsiveness of β-hairpin peptide folding, self-assembly, and hydrogel material formation, Biomacromolecules. 10 (2009) 2619–2625. doi: 10.1021/bm900544e. [DOI] [PubMed] [Google Scholar]
- [41].Patenaude M, Smeets NMB, Hoare T, Designing injectable, covalently cross-linked hydrogels for biomedical applications, Macromol. Rapid Commun 35 (2014) 598–617. doi: 10.1002/marc.201300818. [DOI] [PubMed] [Google Scholar]
- [42].Haines-Butterick L.a, Salick D.a, Pochan DJ, Schneider JP, In vitro assessment of the pro-inflammatory potential of beta-hairpin peptide hydrogels., Biomaterials. 29 (2008) 4164–9. doi: 10.1016/j.biomaterials.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Medina SH, Li S, Howard OMZ, Dunlap M, Trivett A, Schneider JP, Oppenheim JJ, Enhanced immunostimulatory effects of DNA-encapsulated peptide hydrogels, Biomaterials. 53 (2015) 545–553. doi: 10.1016/j.biomaterials.2015.02.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Salick D.a, Kretsinger JK, Pochan DJ, Schneider JP, Inherent antibacterial activity of a peptide-based beta-hairpin hydrogel., J. Am. Chem. Soc 129 (2007) 14793–9. doi: 10.1021/ja076300z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kabu S, Gao Y, Kwon BK, Labhasetwar V, Drug delivery, cell-based therapies, and tissue engineering approaches for spinal cord injury, J. Control. Release 219 (2015) 141–154. doi: 10.1016/j.jconrel.2015.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Canazza A, Minati L, Boffano C, Parati E, Binks S, Experimental models of brain ischemia: A review of techniques, magnetic resonance imaging, and investigational cell-based therapies, Front. Neurol 5 February (2014) 1–15. doi: 10.3389/fneur.2014.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Stuckey DW, Shah K, Stem cell-based therapies for cancer treatment: separating hope from hype., Nat. Rev. Cancer 14 (2014) 683–691. doi: 10.1038/nrc3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lindsey S, Piatt JH, Worthington P, S??nmez C, Satheye S, Schneider JP, Pochan DJ, Langhans SA, Beta hairpin peptide hydrogels as an injectable solid vehicle for neurotrophic growth factor delivery, Biomacromolecules. 16 (2015) 2672–2683. doi: 10.1021/acs.biomac.5b00541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yourek G, McCormick SM, Mao JJ, Reilly GC, Shear stress induces osteogenic differentiation of human mesenchymal stem cells., Regen. Med 5 (2010) 713–24. doi: 10.2217/rme.10.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yan C, Mackay ME, Czymmek K, Nagarkar RP, Schneider JP, Pochan DJ, Injectable solid peptide hydrogel as a cell carrier: effects of shear flow on hydrogels and cell payload., Langmuir. 28 (2012) 6076–87. doi: 10.1021/la2041746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sonmez C, Nagy KJ, Schneider JP, Design of self-assembling peptide hydrogelators amenable to bacterial expression, Biomaterials. 37 (2015) 62–72. doi: 10.1016/j.biomaterials.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Aenlle KK, Curtis KM, Roos B.a, Howard G.a, Hepatocyte growth factor and p38 promote osteogenic differentiation of human mesenchymal stem cells., Mol. Endocrinol 28 (2014) me20131286. doi: 10.1210/me.2013-1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Nagahara a H., Mateling M, Kovacs I, Wang L, Eggert S, Rockenstein E, Koo EH, Masliah E, Tuszynski MH, Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice, J Neurosci 33 (2013) 15596–15602. doi: 10.1523/JNEUROSCI.5195-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Qiu Y, Park K, Environment-sensitive hydrogels for drug delivery, Adv. Drug Deliv. Rev 64 (2012) 49–60. doi: 10.1016/j.addr.2012.09.024. [DOI] [PubMed] [Google Scholar]
- [55].Branco MC, Pochan DJ, Wagner NJ, Schneider JP, The effect of protein structure on their controlled release from an injectable peptide hydrogel., Biomaterials. 31 (2010) 9527–34. doi: 10.1016/j.biomaterials.2010.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Steichen SD, Caldorera-Moore M, Peppas NA, A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics, Eur. J. Pharm. Sci 48 (2013) 416–427. doi: 10.1016/j.ejps.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Salem M, Rohani S, Gillies ER, Curcumin, a promising anti-cancer therapeutic: a review of its chemical properties, bioactivity and approaches to cancer cell delivery, RSC Adv. 4 (2014) 10815. doi: 10.1039/c3ra46396f. [DOI] [Google Scholar]
- [58].Altunbas A, Lee SJ, Rajasekaran S.a, Schneider JP, Pochan DJ, Encapsulation of curcumin in self-assembling peptide hydrogels as injectable drug delivery vehicles., Biomaterials. 32 (2011) 5906–14. doi: 10.1016/j.biomaterials.2011.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Iwamoto T, Clinical Application of Drug Delivery Systems in Cancer Chemotherapy : Review of the Efficacy and Side Effects of Approved Drugs, Biol. Pharm. Bulliten 36 (2013) 715–718. [DOI] [PubMed] [Google Scholar]
- [60].Scholz C, Wagner E, Therapeutic plasmid DNA versus siRNA delivery: Common and different tasks for synthetic carriers, J. Control. Release 161 (2012) 554–565. doi: 10.1016/j.jconrel.2011.11.014. [DOI] [PubMed] [Google Scholar]
- [61].Sinthuvanich C, Haines-Butterick LA, Nagy KJ, Schneider JP, Iterative design of peptide-based hydrogels and the effect of network electrostatics on primary chondrocyte behavior, Biomaterials. 33 (2012) 7478–7488. doi: 10.1016/j.biomaterials.2012.06.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sun JEP, Stewart B, Litan A, Lee SJ, Schneider JP, Langhans SA, Pochan DJ, Sustained release of active chemotherapeutics from injectable-solid β-hairpin peptide hydrogel, Biomater. Sci (2016). doi: 10.1039/C5BM00538H. [DOI] [PMC free article] [PubMed] [Google Scholar]