

Abstract
Carrageenans (CGNs) are widely used in foods and pharmaceuticals although their safety remains controversial. To investigate the effects of CGNs and CGN-degrading bacteria in the human colon, we screened for CGN degradation by human fecal microbiota, and for inflammatory response to CGNs and/or CGN-degrading bacteria in germ free mice. Thin-layer chromatography indicated that high molecular weight (MW) CGNs (≥100 kDa) remained undegraded in the presence of human fecal microbiota, whereas low MW CGNs, i.e., κ-carrageenan oligosaccharides (KCO, ~4.5 kDa) were degraded when exposed to seven of eight human fecal samples, although sulfate groups were not removed during degradation. Bacteroides xylanisolvens and Escherichia coli isolates from fecal samples apparently degraded KCO synergistically, with B. xylanisolvens serving as the primary degrader. Combined treatment of KCO with KCO-degrading bacteria led to greater pro-inflammatory effects in the colon and rectum of germ-free mice than either KCO or bacteria alone. Similarly, p-p38-, CD3-, and CD79a-positive immune cells were more abundant in combined treatment group mice than in either single treatment group. Our study shows that KCO-degrading bacteria and the low MW products of KCO can promote proinflammatory effects in mice, and represent two key markers for evaluating CGN safety in foods or medicines.
Keywords: Carrageenans, Carrageenan oligosaccharides, Oligosaccharide degrading bacteria, Intestinal inflammatory, Germ-free mouse
Introduction
Carrageenans (CGNs), extracted primarily from red algae, represent one of the three major classes of industrial algal polysaccharides (Ji, 1997). Specifically, CGNs are water soluble, linear, sulfated polysaccharides with alternating β-1–3- and α-1–4-linked galactose residues, with additional substitute residues such as xylose, glucose, methyl esters, and pyruvate groups (Ji, 1997). CGNs are classified by the types of sulfate bonds they contain, including kappa (κ-), iota (ι-), lambda (λ), and other types (Campo, 2009). K-CGNs are mainly composed of D-galactose-4-sulfate and 3,6-anhydro-D-galactose, and the sulfate content of commercial κ-CGN is approximately 22% (David et al., 2018). In addition, the gelling properties of κ-CGN have led to its wide adoption in the food and pharmaceutical industries (Li et al., 2014a; Prajapati et al., 2014). Since the human upper gastrointestinal tract lacks CGN-degrading digestive enzymes (Harmuth-Hoene and Schwerdtfeger, 1979), some CGNs are likely to remain intact at least until entry into the human colon where they inevitably encounter human colonic microbiota (Hehemann et al., 2010). Although numerous CGN-degrading bacteria have been isolated from marine environments (Ficko-Blean et al., 2017; Shen et al., 2017, 2018; Park et al., 2018; Jung et al., 2019), little is yet known about the CGN-degrading bacteria present in the human intestine.
Although CGNs have been used in the food industry for decades, and recent reports have shown that some of these polysaccharides may exhibit antiviral, immunomodulatory, anticoagulant, antioxidant, or anticancer properties, the safety of CGNs remains controversial (David et al., 2018, David et al., 2019; Jiang et al., 2021). Of particular concern are the physicochemical properties of commercial CGNs, acceptable levels of human exposure to CGNs, and the potentially adverse effects of CGNs on gut microbiome leading to dysbiosis and inflammation (David et al., 2019). Although CGNs have been classified by the Food and Drug Administration (FDA) and the European Food Safety Authority as “generally recognized as safe (GRAS)”, CGNs with molecular weight (MW) less than 100 kDa have been designated as food contaminants (Jiang et al., 2021). Furthermore, a maximum daily CGN dose of 75 mg/kg body weight is provisionally considered a safe level of average daily intake (Efsa Panel et al., 2018). However, some studies have reported maximum acceptable daily CGN exposure levels in infants, toddlers, children, adolescents, adults, and seniors (>65 years old) of 21.8–86.7, 135.3–581.4, 109.6–487, 63.5–264.6, 34.7–138.6, and 24.7–89.4 mg/kg body weight, respectively (Efsa Panel et al., 2018). Moreover, a relatively low MW CGN fraction can consistently found in CGN-containing food products, even when food-grade high-MW CGNs are used, potentially due to degradation of high-MW CGNs during food processing (David et al., 2019). It is thus analytically challenging to accurately determine the physicochemical characteristics of CGNs, such as distribution of constituent CGNs with different MWs, charge distribution, linearity, macromolecular architecture, mineral content, and impurities, during or post processing (David et al., 2019). These uncertainties, in conjunction with the lack of extensive oversight for some CGN-related products or CGN by-products have increased concerns about the safety of CGNs, especially low MW CGNs (David et al., 2019).
In this study, we investigated the degree of fermentation by human gut microbiota for three κ-CGN fragments with differing MWs, including κ-carrageenan polysaccharides (KCP, 450 kDa), mild acid degraded κ-carrageenan (SKCO, 100 kDa), and κ-carrageenan oligosaccharides (KCO, 4.5 kDa). Remarkably, we found that only KCO can be degraded by seven of eight human gut microbiota samples. We subsequently isolated and identified KCO-degrading bacteria in the human intestinal tract. Finally, we explored the proinflammatory effects of KCO and KCO-degrading bacterial strains in a germ-free mouse model. Our results showed that both KCO and KCO-degrading bacteria are promoting factors in the proinflammatory effects, indicating both of them should be considered as markers for evaluating the safety evaluation of CGNs in foods or medicines.
Results
Low MW of CGN can be degraded by human fecal microbiota
As the MW of κ-CGN may be associated with colon inflammation, we assessed the fermentability of three κ-CGN fragments (KCP: 450 kDa; SKCO: 100 kDa; and KCO: 4.5 kDa) by human colonic microbiota using an in vitro batch fermentation system. We therefore measured the degree of KCP, SKCO, and KCO degradation by fecal microbiota collected from eight healthy human volunteers using thin-layer chromatography (TLC) and found that KCP and SKCO were not degraded by any of the eight fecal microbiota samples (Fig. S1). In contrast, KCO was degraded by microbiota in seven of the eight fecal samples (K1, K2, K3, K5, K6, K7, and K8) after 24 h or 48 h of fermentation, slightly degradation of KCO by sample K4 (degradation efficiency of 4.6% at 48 h) was also observed (Figs. 1A and S1).
Fig. 1.
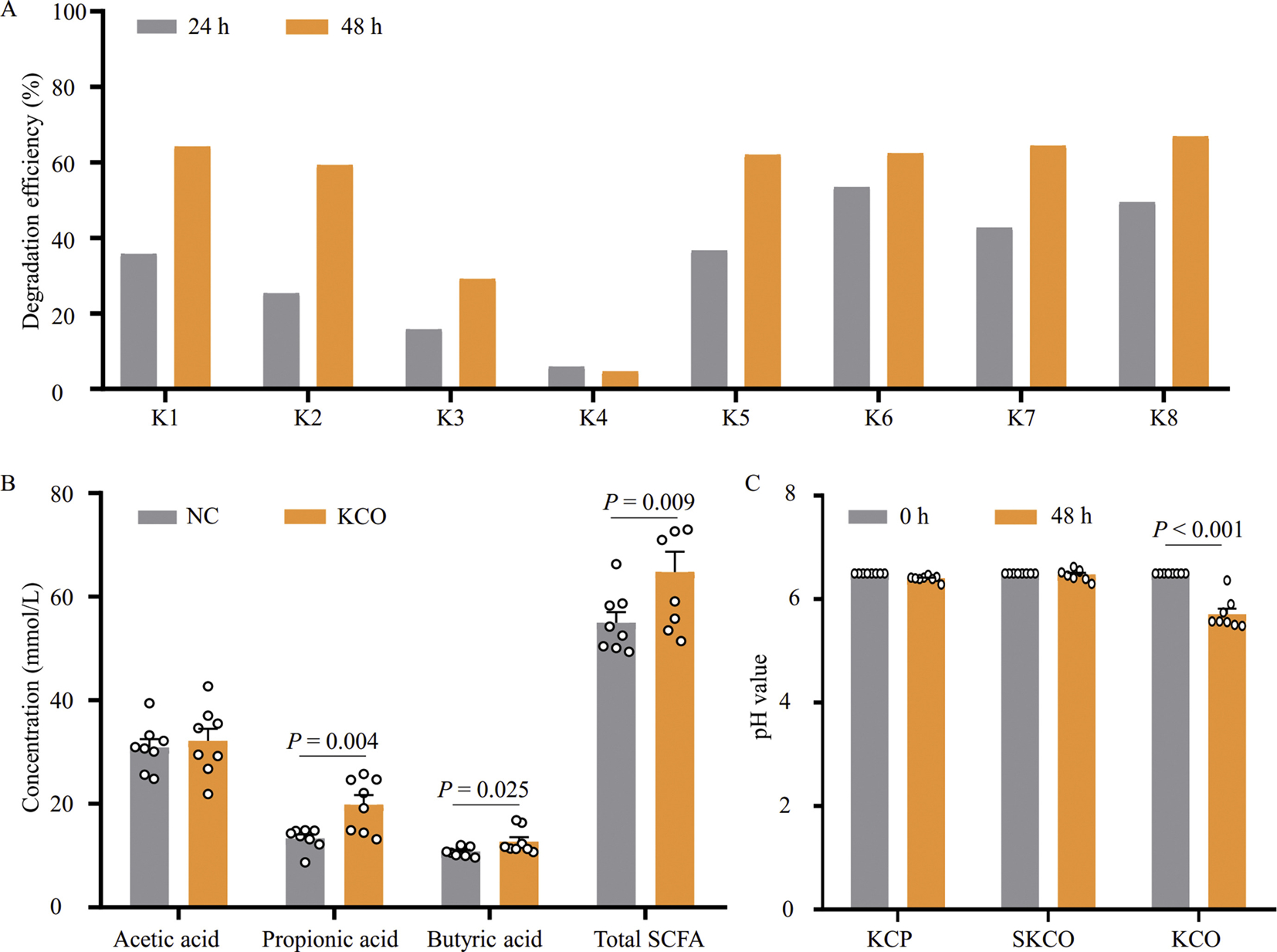
Degradation of KCO by human fecal microbiota. A: TLC measurements of KCO degradation efficiency. Degradation efficiency was calculated by quantifying changes in gray value of KCO spots in TLC using ImageJ software. Samples K1–K8 fecal microbiota were collected from healthy volunteers and subsequently inoculated into separate batch fermentations. B: HPLC detection of SCFAs produced during KCO degradation by fecal microbiota samples K1–K8 after 48 h of fermentation; total SCFAs include total contents of acetic, propionic, and butyric acids. No KCO was added to the NC. C: pH values before (0 h) and after (48 h) KCP, SKCO, or KCO fermentation. κ-carrageenan polysaccharides (KCP, 450 kDa), mild acid degraded κ-carrageenan (SKCO, 100 kDa), and κ-carrageenan oligosaccharides (KCO, 4.5 kDa); KCO, κ-carrageenan polysaccharides; TLC, thin-layer chromatography; SCFA, short chain fatty acid; NC, negative control.
Given that acetic, propionic, and butyric fatty acids are reportedly the primary short chain fatty acids (SCFAs) produced by intestinal microbiota (Nogal et al., 2021), we determined the concentrations of these SCFAs before and after in vitro fermentation using high-performance liquid chromatography (HPLC). The results showed that the concentrations of propionic and butyric acids were significantly increased after KCO fermentation (Fig. 1B). In addition, the pH values after KCP, SKCO, and KCO fermentation were also measured and, consistent with our TLC results, the results showed that the pH in KCP and SKCO fermentations remained stable, whereas the pH significantly decreased by the end of the KCO fermentation (Fig. 1C). Taken together, these results indicated that high MW CGN (KCP and SKCO) could not be degraded by any of eight human gut microbiota samples tested here. However, fecal microbiota from different individuals can degrade low MW components of CGN (KCO, 4.5 kDa), and KCO degradation results in accumulation of organic acids.
KCO was not desulfurized by human colonic microbiota
KCO, which contains sulfate groups, was degraded during fermentation, which led us to investigate whether KCO was desulfurized during this process. The results of TLC analyses indicated that the K6 and K8 fecal samples exhibited the greatest degradative effects. We analyzed the structures of the fermentation products of these two samples using gel filtration chromatography coupled with Electrospray ionization mass spectrometry (ESI-MS) and found that the spectral profiles of the K6 and K8 degradation products were similar. In both sets of degradation products, four fragments were identified: 4-O-sulfate-D-galactose (G4S, degree of polymerization [dp] = 1), κ-neocarrabiose (dp = 2), κ-carratriose (dp = 3), and κ-carrapentaose (dp = 5) (Table 1). However, no desulfation was observed during KCO degradation by human fecal microbiota based on MW analysis of the compounds detected by MS (Table 1). The sequence of κ-neocarrabiose was identified as A-G4S using electrospray ionization collision-induced-dissociation mass spectrometry (ESI-CID-MS2) and an oligosaccharide reduction method as described in our previous study (Li et al., 2017).
Table 1.
Electrospray ionization mass spectrometry and sequencing results for the fermentation broths of fecal samples K6 and K8.
| Fraction | Found ions (charge) | Calculated mol mass (Na form) | DP sequences | Theoretical mol mass (Na form) |
|---|---|---|---|---|
| F1 (dp) = 1 | 259.02 (−1) | 282.02 | G4S, salt | 282.02 |
| F2 (dp) = 2 | 403.06 (−1) | 426.06 | A-G4S | 426.06 |
| F3 (dp) = 3 | 322.03 (−2) | 690.06 | G4S-A-G4S | 690.07 |
| F4 (dp) = 5 | 343.03 (−3) | 1098.09 | G4S-A-G4S-A-G4S | 1098.12 |
G4S: D-galactose-4-sulfate; A: 3,6- D-anhydrogalactose; DP: degree of polymerization.
We next explored the effects of KCO on human fecal microbiota composition in vitro, especially bacteria which were associated with sulfation degradation. To this end, we used qPCR-based assays with taxon-specific primers to quantify changes in the relative abundances of six major groups of colonic bacteria (including Bacteroides-Prevotella, Clostridium cluster XIVab, Bifidobacterium, Lactobacillus, Enterobacteriaceae, and Desulfovibrio) after in vitro KCO fermentation. The results indicated that the relative abundances of four of the six groups (i.e., Bacteroides-Prevotella, Bifidobacterium, Clostridium cluster XIVab, and Lactobacillus) did not differ significantly from that of control fermentations lacking KCO after 48 h of fermentation (Fig. S2A). Although significant increase was observed in the relative abundance of family Enterobacteriaceae, the increase was detected both in the KCO-degrading (K1, K2, K3, K5, K6, K7, and K8) and non-degrading (K4) fermentations (Figs. S1, S2A, S2C).
Since commensal Desulfovibrio strains have been found to exhibit potent sulfate-reducing properties (Kushkevych et al., 2020), we examined its relative abundance before and after KCO fermentation. The results revealed that the abundance of Desulfovibrio increased in all the fecal samples after fermentation, regardless of the efficiency of KCO degradation (Figs. S1, S2A, S2B), which suggested that the increase in Desulfovibrio abundance was not associated with KCO degradation. In addition, ESI-MS structural analysis indicated no significant desulfation of the degradation products (Table 1). Taken together, these results suggested that KCO was not likely desulfurized during fermentation by human gut microbiota. Then we moved on to focus on investigation of the degrading bacteria of KCO.
Bacteroides xylanisolvens was identified as the primary KCO-degrading bacteria
Considering sample K8 exhibited the highest KCO degradation efficiency after 48 h of fermentation in VI medium containing KCO as the sole carbon source (Fig. 1A), bacteria from this sample were spread on KCO agar plates to screen for KCO-degrading isolates. Colonies were randomly selected from the plates and reinoculated into medium containing KCO as the sole carbon source. To characterize the ability of these isolates to degrade KCO, we then conducted TLC analysis after 110 h of incubation to analyze the KCO degradation. One isolate (38F6) that was found to degrade KCO, displayed increased degradation with fermentation time (Figs. 2A and S3A). The positive colony was purified by spreading to isolation on KCO agar plates under anaerobic conditions. Through this process, two colonies with different morphologies emerged, which we identified as Bacteroides xylanisolvens and Escherichia coli based on 16s rDNA gene sequencing. To determine which bacterium was responsible for degrading KCO, the purified strains of B. xylanisolvens and E. coli were again inoculated (separately and together) into VI medium containing KCO. We found that B. xylanisolvens significantly degraded KCO (Fig. 2B), while E. coli mainly consumed the monosaccharide 4-O-sulfate-D-galactose (the upper most spot in the TLC shown in Fig. S3B), which apparently enhanced the efficiency of B. xylanisolvens degraded KCO.
Fig. 2.
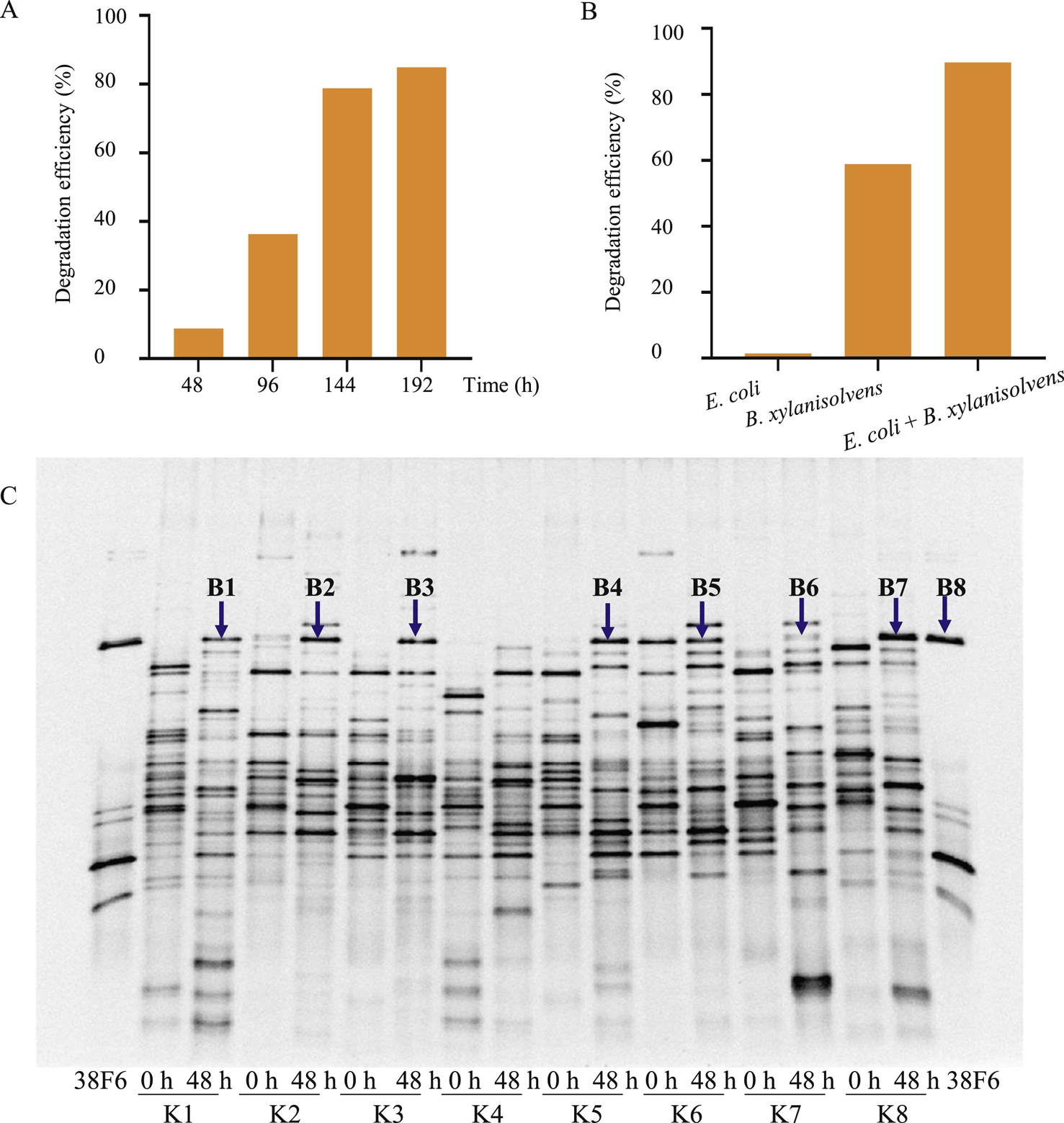
Identification of KCO-degrading bacteria. A: The KCO-degrading ability of bacterial isolate 38F6. The ability to degrade KCO by 38F6 was evaluated using TLC after 48 h, 96 h, 144 h, and 192 h of fermentation in VI medium containing 8 g/L of KCO. Degradation efficiency was calculated based on changes in gray value of the KCO spot using ImageJ. B: The KCO-degrading ability of E. coli, B. xylanisolvens, or E. coli plus B. xylanisolvens. Degradation efficiency was calculated as in (A). C: PCR-DGGE analysis of the bacterial community before (0 h) and after (48 h) KCO fermentation. Samples K1–K8 fecal microbiota samples were collected from healthy volunteers and separately inoculated into VI medium containing 8 g/L KCO as the sole carbon source. Lane 38F6 contains the PCR products generated using B. xylanisolvens genomic DNA as template. Bands B1–B8 (indicated by blue arrows) correspond to the PCR-DGGE bands which were selected and further confirmed by Sanger sequencing.
To better understand the genetic basis underlying KCO catabolism, we sequenced the B. xylanisolvens genome using next-generation sequencing. In total, 150 scaffolds and 6.09 Mb were assembled. Based on NCBI BLAST analysis, we identified two genes, designated as C0 and C1, that had 46% and 100% amino kacidsimilarity to that of B. ovatus κ-carrageenase precursors, respectively. To characterize the capacity for KCO degradation by C0 and C1, we cloned these genes separately into the pET28a plasmid and transformed the respective vector constructs into E. coli. We then tested the ability of these two strains to degrade KCO. Unfortunately, neither of the recombinant E. coli strains could degrade KCO.
To determine whether B. xylanisolvens was the primary bacterial strain that could degrade KCO in the human colon, we performed PCR-Denaturing Gradient Gel Electrophoresis (DGGE) on slurries of the eight fecal samples after KCO fermentation. The PCR products of the B. xylanisolvens isolate were used as biomarkers to test for the presence of this species in all eight of the fecal slurries. As shown in Fig. 2C, the intensities of the PCR products corresponding to B. xylanisolvens increased after KCO fermentation in all samples except for K4 and K7. Notably, K4 samples showed negligible KCO degradation. DNA sequencing of the PCR bands confirmed the presence of B. xylanisolvens in most fecal samples (Table S1), and suggested that B. xylanisolvens was the primary fecal bacterium associated with KCO degradation. However, the high levels of KCO degradation observed in samples K3 and K7 in the absence of B. xylanisolvens suggested that Firmicutes (K3) or Dialister species (K7) might also be involved in KCO degradation.
KCO and KCO-degrading bacteria could induce an inflammatory response in germ-free mice
In light of our results showing that B. xylanisolvens and E. coli co-culture resulted in greater degradative effects on KCO than B. xylanisolvens alone (Fig. 2B), we used a mixture of B. xylanisolvens and E. coli to explore the effects of KCO degradation on inflammatory response in vivo. To this end, germ-free mice were randomly divided into four groups: (i) treated with KCO (GFK), (ii) treated with B. xylanisolvens and E. coli (GN), (iii) treated with KCO, B. xylanisolvens, and E. coli (GNK), and (iv) untreated control (GF) (n = 6 per group). The experiments lasted for 16 weeks, and no mortalities were observed in any group during the experiment. Surface erosion and cryptitis were not observed in ileal, duodenal, jejunal, colonic, or rectal samples. Submucosal edema and inflammatory exudates were not detected in any ileal, duodenal, or jejunal samples (Fig. S4A), but deterioration was observed in the colonic and rectal samples (Fig. 3A). High inflammation scores were recorded in the colonic and rectal tissues of the GK, GN, and GNK groups (Figs. 3B and S4B), indicating that exposure to KCO and KCO-degrading bacteria can each lead to inflammation, independently. However, the rectal samples from the GNK group had higher scores than all other tissue samples, suggesting that the combination of a KCO-degrading bacterium (i.e., B. xylanisolvens), E. coli, and KCO could increase inflammation in mice.
Fig. 3.
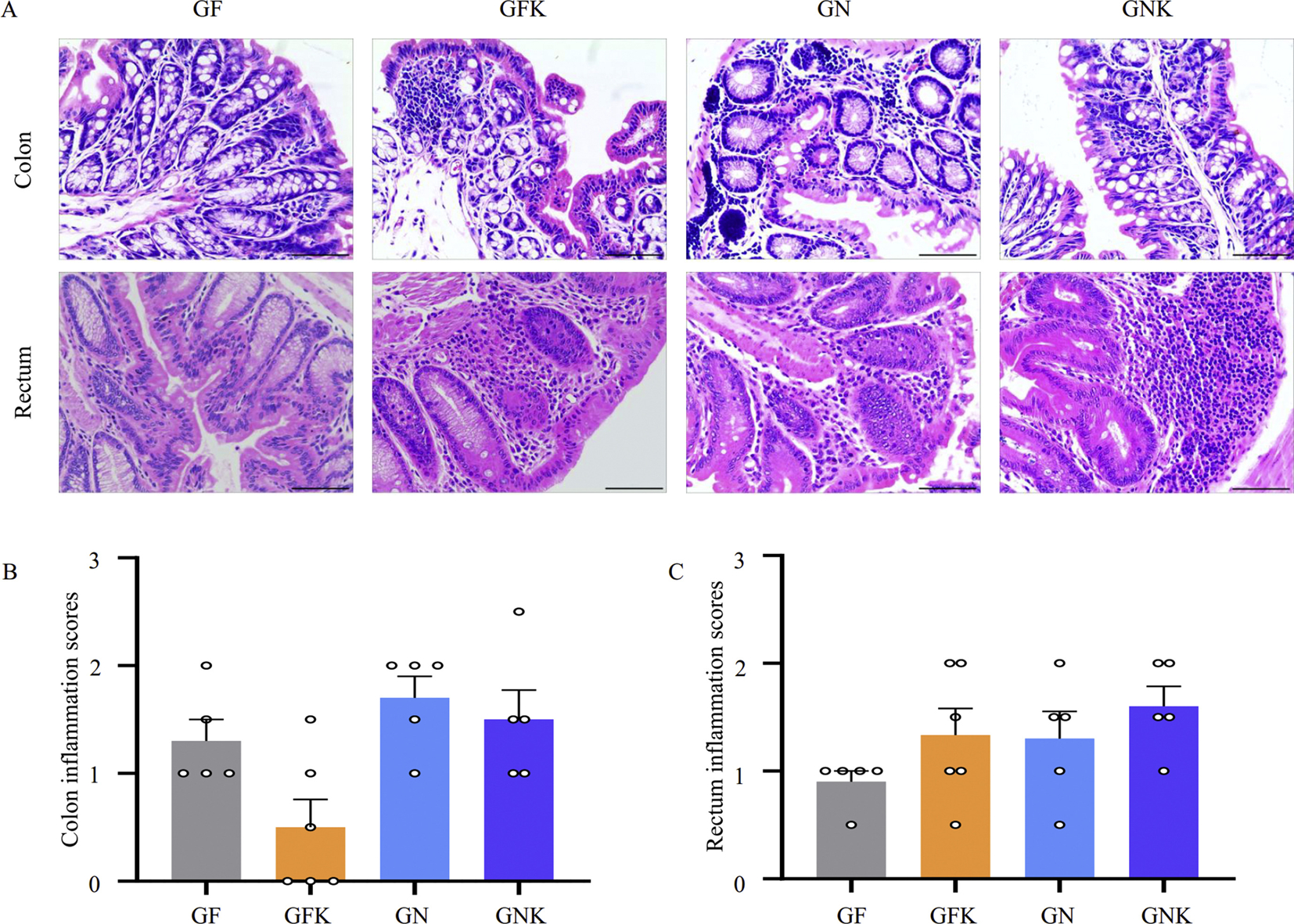
Inflammatory response to treatment with KCO and KCO-degrading bacteria in mouse colon and rectum. A: Photomicrographs of representative H&E-stained colon and rectal samples. Scale bar, 200 mm. B and C: Histopathological indexes reflect inflammation severity in the colon and rectum, respectively. Results are expressed as mean ± SEM (n = 6). Tissue samples were collected from GF, GFK (5% KCO), GN (5 × 108 CFU of B. xylanisolvens and E. coli), and GNK (5% KCO plus 5 × 108 CFU of B. xylanisolvens and E. coli) treatment groups in germ-free Kunming mice. SEM: standard error of the mean; GF, germ free control group.
To better understand which metabolic and signaling pathways in mice participated in the inflammatory response to KCO and KCO-degrading bacteria, we extracted RNA from the rectal samples to obtain the mouse transcriptome for RNA-Seq analysis. Sequencing reads covered the entire genome, and no sequencing bias was observed (Fig. S5). Expression profiling identified 434, 316, and 1454 differentially expressed genes (DEGs; log2 [fold change] ≥2 and q-value < 0.05) in groups GFK, GN, and GNK, respectively, compared to the controls (GF). The most strongly up- and down-regulated genes relative to the control were identified in the GNK group. Cluster analysis also revealed that gene expression patterns in the GNK group were distinctly different from those in the GFK and GN groups (Fig. S6).
DEGs were then subjected to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses. At the level of molecular function, 56 DEGs in the GNK group and 17 DEGs in the GN group were annotated to encode carbohydrate-binding proteins, while 35 DEGs encoding polysaccharide-binding proteins and 25 DEGs encoding heparin-binding proteins were identified in the GNK group (Table S2). At the pathway level, 28, 21, and 21 DEGs in group GNK were associated with focal adhesion, cell adhesion, and leukocyte transendothelial migration, respectively (Table S3). These pathways are reportedly associated with immune and inflammatory responses. Seven DEGs related to leukocyte transendothelial migration were significantly upregulated (PI30cas, PI3K, Cldn3, Ezrin, Mapk13, Cldn23, and Actin), and 14 were downregulated (CAMs, Pecam1, Rap1a, Rap1b, Itgam, Mmp9, Mmp2, Cxcl12, Vcam1, Itgb1, Jam2, Jam3, Cdh5, and Gnai1; Table S4); together, these 21 genes represent almost the complete leukocyte transendothelial migration pathway (Fig. 4).
Fig. 4.
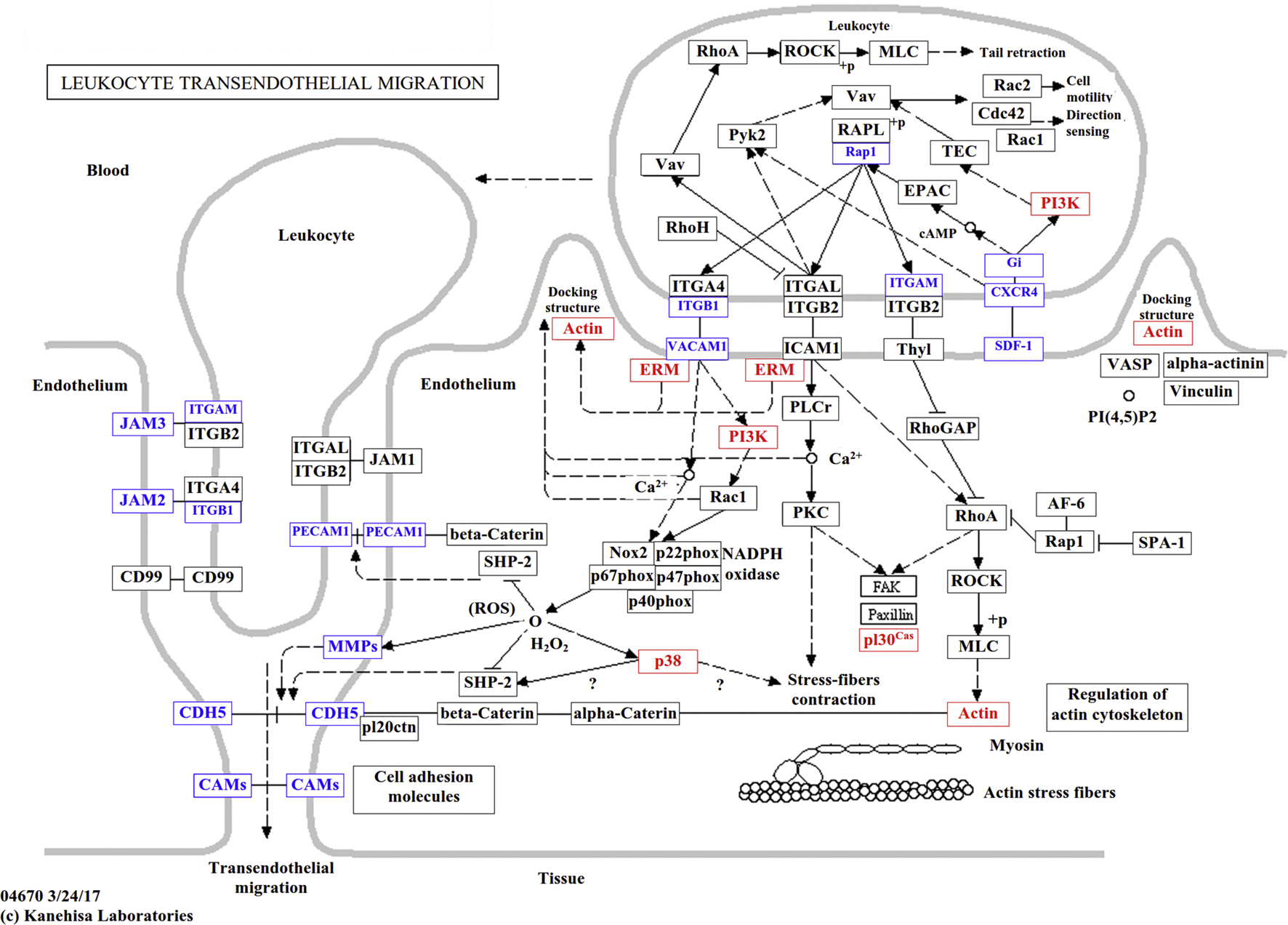
Distribution of DEGs in leukocyte transendothelial migration pathways. The nodes marked in red or blue represent genes which were up- or down-regulated, respectively, in the comparisons between the GNK vs. GF transcriptomes. Tissue samples were collected from GF (control group) and GNK (treated with 5% KCO plus 5 × 108 CFU of B. xylanisolvens and E. coli) groups.
To verify the RNA-Seq results, we used immunohistochemical staining (IHC) of five cytokines, including CD3, CD79a, p-p38, VCAM1, and PECAM1, to observe the inflammatory response in mouse tissue under different treatment. Because the colonic and rectal tissue sections had the highest inflammation scores in Hematoxylin and eosin (H&E) staining assays, we performed IHC analyses of colonic (Fig. S7) and rectal (Fig. 5) tissue samples from five or six mice per group. We found that p-p38-positive cells in the rectal tissues of the GNK group were more abundant and showed a stronger signal compared to the other groups, although p-p38 scores were also high in the KCO-only group (GFK). These findings suggested that inflammation occurred in the distal end of the colon, and that the combination of KCO and KCO-degrading bacteria led to more severe inflammation. A similar trend was observed in CD79a staining experiments, however the differences between groups were not statistically significant. We also noted that VCAM1 protein was downregulated in colon samples from the GFK group, compared with that in other groups, while PECAM1 was not significantly differentially expressed among groups (Fig. S7). Furthermore, IHC scores for p-p38, CD3, CD79a, VCAM1, and PECAM1 were in agreement with their mRNA expression patterns in the RNA-Seq data (Fig. S8). Taken together, these results indicated that KCO, B. xylanisolvens, and E. coli treatment lead to greater inflammation in the mouse rectum.
Fig. 5.
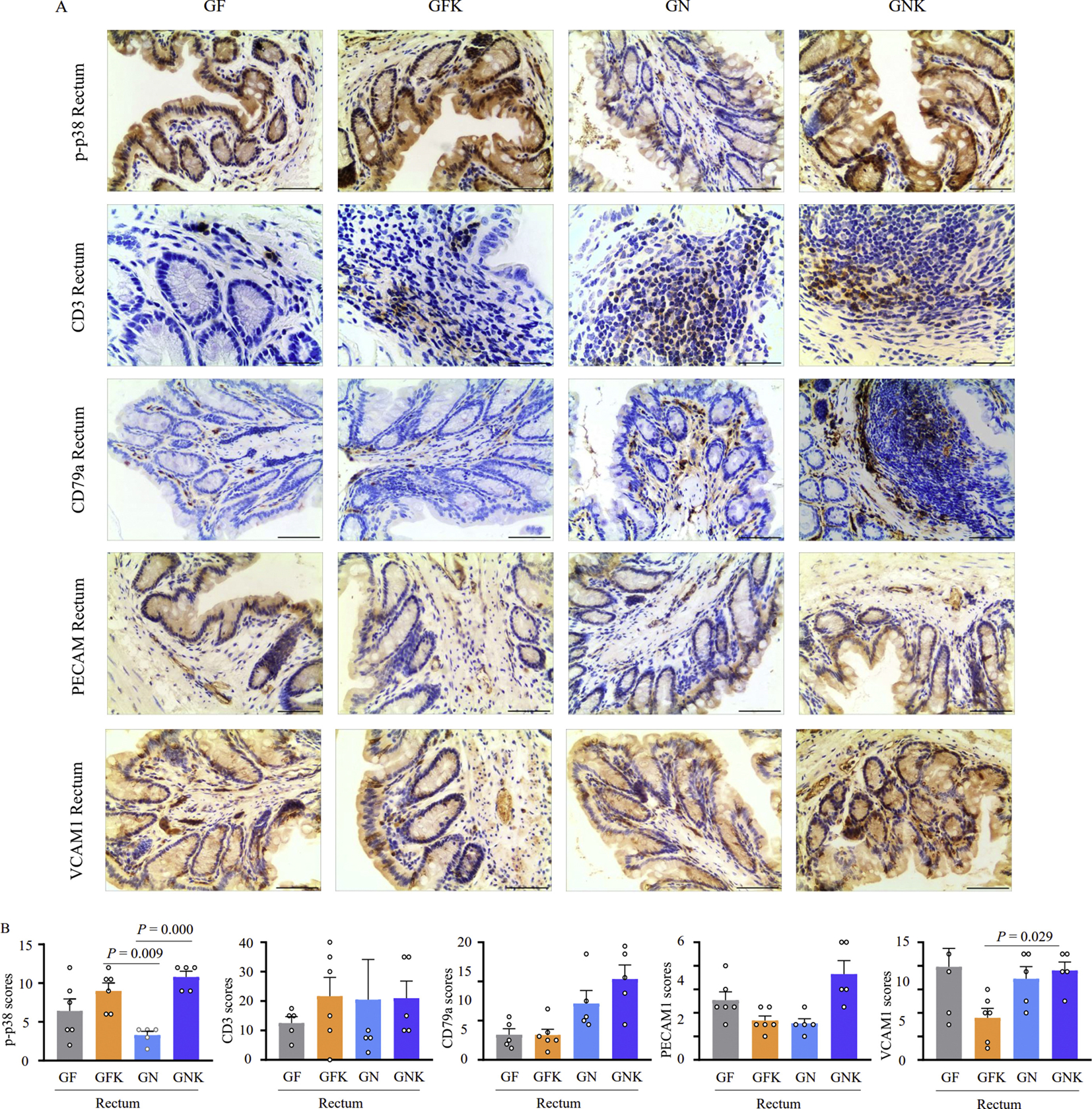
Immunohistochemical staining and inflammatory marker scoring of rectum tissue treated with KCO and KCO degrading bacteria. A: Representative images of rectal tissue samples stained for p-p38, CD3, CD79a, PECAM1, and VCAM1 inflammatory markers. Scale bar, 200 μm. B: Histopathology scores indicate inflammation severity in the rectum based on quantification of IHC staining results by ImageJ. Results are presented as mean ± SEM (n = 6). Tissue samples were collected from GF (germ free control group), GFK (5% KCO), GN (5 × 108 CFU of B. xylanisolvens and E. coli), and GNK (5% KCO plus 5 × 108 CFU of B. xylanisolvens and E. coli) treatment groups in germ-free Kunming mice. SEM, standard error of the mean.
Discussion
κ-CGNs are sulfurized seaweed polysaccharides with thermoreversible gelling properties that are widely used as food thickening agents (Ji, 1997). However, the safety of κ-CGNs remains a concern, and inconsistent detrimental effects of κ-CGN in animal model have been attributed to the varying MWs of κ-CGN compounds (David et al., 2018). Here, we propose that differences among individuals in the composition and function of their colonic microbiota may further obscure the effects of κ-CGN. To explore these possible complications, we investigated the interactions between κ-CGNs with different MWs and human colonic microbiota. The results clearly demonstrated that the capacity for κ-CGN degradation by human gut microbiota was correlated with the MW of specific κ-CGNs. More specifically, we found that κ-CGNs with an MW > 100 kDa, including both KCP and SKCO, cannot be utilized by human fecal microbiota in vitro (Fig. S1). In contrast, degradation of the low MW CGN KCO (~4.5 kDa) varied among the fecal samples tested here (Fig. 1A), suggesting that KCO degradation also depends on the presence of specific taxa in the human microbiome.
It has been established that H2S produced by sulfate reducing bacteria leads to a signal cascade resulting in detrimental effects to colonocytes (Libiad et al., 2019). However, it remains unknown whether free sulfate can be released from CGNs by human colonic microbiota. Our structural analysis of KCO fragments after fermentation by human fecal microbiota identified the major derivatives as 4-O-sulfate-D-galactose, κ-neocarrabiose, κ-carratriose, and κ-carrapentaose, suggesting that sulfate is not hydrolyzed from KCO. Our qPCR results showed that the total abundance of Desulfovibrio increased at a generally constant rate, irrespective of KCO degradation rate. These results indicated that human colonic microbiota could degrade KCO, but do not remove sulfate from KCO or its derivatives, which is consistent with a previous study (Shang et al., 2017). Therefore, the enzymes produced by human colonic microbiota that mediate degradation of marine sulfated polysaccharides may differ from those that degrade sulfurized glycans in the human gut.
Although several strains of marine CGN-degrading bacteria have been previously isolated (Ficko-Blean et al., 2017; Shen et al., 2017, 2018; Park et al., 2018; Jung et al., 2019), few studies have investigated CGN-degrading bacteria derived from animal or human intestines. Using in vitro batch fermentation with KCO as the substrate to screen for degradation, we identified two closely associated KCO-degrading species, B. xylanisolvens, and E. coli which degraded KCO in a synergistic manner. Consistent with the findings of a previous study (Li et al., 2014b), the presence of E. coli improved the KCO-degrading ability of B. xylanisolvens, suggesting that cross-feeding may occur between the KCO degrader and E. coli. Based on these findings, we hypothesize that E. coli can utilize metabolites generated by B. xylanisolvens, particularly galactose, thereby reducing the negative feedback effects of the monosaccharide degradation products on carrageenase synthesis in B. xylanisolvens, ultimately improving the capacity for KCO degradation by this bacterium. Moreover, PCR-DGGE analysis and DGGE band sequencing showed that treatment with KCO results in higher growth of B. xylanisolvens in five of the eight fecal microbiota that could degrade KCO (Fig. 2C and Table S1), confirming that B. xylanisolvens plays a major role in KCO utilization by human colonic microbiota. Two genes homologous to κ-carrageenase were identified using bioinformatic analyses, but no KCO degradation activity was detected after transformation of these two genes into E. coli. This result may be attributable to unsuitability of the E. coli expression system for the functional degradation of KCO by the C0 and C1 genes. Alternatively, other genes in B. xylanisolvens may be necessary to facilitate KCO degradation. Thus, the mechanism by which B. xylanisolvens degrades KCO requires further study.
Although the impact of CGNs on the colonic immune response has been previously investigated (David et al., 2018), to date, the effects on host inflammatory response by CGN-degrading bacteria and the associated CGN degradation products have not been explored. To address this gap in knowledge, we treated germ-free mice with (i) KCO alone; (ii) B. xylanisolvens and E. coli; (iii) KCO, B. xylanisolvens, and E. coli; or (iv) water (control). Interestingly, H&E staining showed significantly higher inflammation in the colon and rectum than in the duodenum, jejunum, and ileum. In addition, the combination of KCO with KCO-degrading bacteria resulted in the strongest proinflammatory effect. It was previously shown that the administration of CGN into the mouse pleural cavity induces pleurisy by activating the p38 mitogen-activated protein kinases (MAPK) pathway (Vigil de Mello et al., 2016). Here, p38 and p-p38 were upregulated in the mouse rectal samples, especially in samples from mice treated with a combination of KCO, B. xylanisolvens, and E. coli. IHC using p-p38 revealed a low degree of inflammation in the groups treated with B. xylanisolvens and E. coli or KCO alone, but a significant increase in inflammation in the KCO, B. xylanisolvens, and E. coli treatment group, indicating that KCO-degradation products lead to tissue inflammation in the mouse rectum.
As KCO treatment alone could cause low grade inflammation compared with conditions in GF mice, in addition to the significant exacerbation of inflammation observed in the KCO plus KCO-degrading bacteria treatment group, we inferred that the increased inflammatory response was associated with the KCO derivatives produced during the bacterial KCO degradation process. Indeed, CGNs with low MWs have higher reported rates of diffusion into the intestinal epithelium through the mucus layer (David et al., 2018). In addition, as KCO was only degraded by colonic microbiota after at least 48 h of incubation, KCO degradation most likely occurred in the colon and rectum.
Cell-to-cell and cell-to-matrix adherence are essential for the maintenance of proper epithelial tissue homeostasis (Witherden et al., 2014). KEGG analysis showed that the DEGs detected under KCO plus KCO-degrading bacteria treatment were mainly involved in leukocyte transendothelial migration. It warrants mention that the DEGs Vcam1, Jam2, Jam3, Pecam1, and Cdh5 encode proteins critical for diapedesis, and that the mRNA levels of all six genes were decreased in mice treated with KCO, B. xylanisolvens, and E. coli. This finding suggests that the metabolites generated by KCO degradation could potentially reduce the adherence of tight junctions among epithelial and endothelial cells (Bazzoni, 2003). Moreover, JAM2 and JAM3 are consistently differentially expressed in tumors and adjacent tissues (Hajjari et al., 2013), implying that the proinflammatory effects of KCO and KCO-degrading bacteria might contribute to increases in gut permeability.
In summary, high-MW κ-CGNs (>100 kDa) are not apparently degraded by human intestinal microflora in vitro, whereas 7 of the 8 gut microbiotas tested in this study could degrade 4.5 kDa KCO and use it as a sole carbon source in vitro. B. xylanisolvens and E. coli operate synergistically to degrade KCO. However, these bacteria are unable to remove the sulfate group from KCO. Experiments with germ-free mice suggest that KCO and the KCO-degrading strain B. xylanisolvens together exert proinflammatory effects on the mouse colon and rectum. Therefore, our results suggest that KCO-degrading bacteria, in addition to low MW CGNs, can act as proinflammatory agents and may affect the function and viability of cells in the lower intestine. These findings provide a basic framework for future considerations of CGN safety.
Material and methods
Preparation of CGN polysaccharides and oligosaccharides
KCP was obtained from Yantai Runlong Marine Biological Products Co., Ltd., China. To prepare SKCO, 10 mg/ml CGN was adjusted to pH 1.26 with 0.1 M HCl and then heated to 37°C for 3 h. To prepare KCO, 30 mg/ml CGN was dissolved at 70°C and treated with 732 cation-exchange resin (Sinopharm Chemical Reagent Co., Ltd, China) for 4 h. The hydrolytic product was neutralized with 1 M NaOH and transferred to a dialysis bag (MW cutoff of 200–500 Da) to remove the salt. The final product was obtained using rotary evaporation and lyophilization. The MWs of KCP, SKCO, and KCO were measured using HPLC (Agilent 1260, USA) with a Shodex OH pak SB-804 HQ column (Shodex, Japan) and detected with a refractive index detector (Agilent 1260, USA) using multiangle laser light scattering (Wyatt, USA). The average MWs of KCP, SKCO, and KCO were 450 kDa, 100 kDa, and 4.5 kDa, respectively.
ESI-MS for sugar structure analysis
After removing the bacteria by centrifugation, each supernatant was separated on a Superdex Peptide 10/300 column (GE, USA). The sequence of each fraction was determined using a Thermo LTQ Orbitrap XL (Thermo Finnigan Corp, USA). Samples were then dissolved in CH3CN/H2O (1:1, v/v) at a concentration of 10 pmol/μl, and 5 ml aliquots were injected. Solvent volatilization and capillary temperatures were 275°C. The sheath gas flow rate was 8 arb. The flow rate was 8 μL/min during negative-ion ESI-MS. Helium was used as the collision gas, with a collision energy of 20–25 eV.
Origin of human fecal samples
Eight healthy human volunteers from Hangzhou, China (24–27 years old) were recruited for the current study. The donors had not taken anti-, pro-, or prebiotics for at least three months prior to sample collection. All volunteers provided informed, written consent, and the study was approved by the Ethics Committee of the Zhejiang Academy of Agricultural Sciences (Hangzhou, China).
Batch culture fermentation of KCP, SKCO, and KCO with human fecal slurries
Batch culture fermentation was performed as described by Lei et al. (2012). KCP, SKCO, or KCO were used as the sole carbon source in each culture. The mass of each compound added to the growth medium varied based on its viscosity; we added 1.0 g of KCP, 5.0 g of SKCO, or 8.0 g of KCO to basic growth medium VI, and adjusted each culture to pH 6.5 before autoclaving. Fresh fecal samples were separately homogenized in Stomacher bags with 0.1 M anaerobic phosphate-buffered saline (PBS) (pH 7.0) to generate 10% (wt/vol) slurries. Large food residues were removed by passing each mixture through a 0.4 mm sieve. Each human fecal slurry (7 mL) was inoculated into a bottle containing 63 ml of growth medium, and the bottles were incubated at 37°C for 48 h in an anaerobic chamber (anaerobic workstation AW 500, Electrotek Ltd., UK). Fermentation products were collected at various time points for further analysis. The pH values after 48 h of fermentation were measured using a pH probe (Eutech, Singapore).
TLC analysis
To perform TLC, 0.2 μL of each sample was loaded on a pre-coated silica gel-60 TLC aluminum plate (Merck, Germany). After development with a solvent system consisting of formic acid, n-butanol, and water (6:4:1, v:v:v), the plate was soaked in orcinol reagent, and carbohydrates were visualized by heating at 120 C for 3 min.
To calculate the degradation rates of these carbohydrates, IntDen of each dot at the spot site was measured using ImageJ software (https://imagej.nih.gov/ij; National Institutes of Health, Bethesda, MD, USA). Then the degradation rates were calculated by comparing to control (0 h).
DNA extraction and PCR-DGGE profiling of the gut microbiota
Bacterial genomic DNA was extracted using a QIAamp DNA Stool Mini Kit (QIAGEN, Germany), following the manufacturer’s instructions. To characterize the microbial communities in the samples, the V3 region of the bacterial 16S rRNA gene was amplified and analyzed using PCR-DGGE, as described previously (Yin et al., 2010). PCR product was obtained by excision of selected DGGE bands followed by elution in water at 4°C overnight. Eluted DNA was reamplified, cloned into pMD18-T vector and sequenced. DNA sequences were taxonomically classified based on BLAST searches for aligned sequences (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Quantitation of bacterial groups using real-time PCR
Bacterial groups from fermentations were quantitated by real-time PCR. Each real-time PCR (20 μL) contained 10 μL Thunderbird SYBR qPCR Mix (Toyobo Co., Ltd, Japan), 0.04 μL 50× ROX reference dye, 0.5 mM of each primer, 1 μL DNA template (20 ng/μL), and distilled water. Amplifications were performed using an ABI PRISM 7500 Real-Time PCR Detection System (Applied Biosystems, USA), with the following PCR program: one cycle of 95°C for 1 min, followed by 40 cycles of 95°C for 15 s, an appropriate annealing temperature for 35 s, and 72°C for 35 s. Six primer pairs were selected to quantify the abundances of the following bacterial groups: Bacteroides-Prevotella, Bifidobacterium, Clostridium cluster XI, Enterobacteriaceae, Lactobacillus, and Desulfovibrio. Primer sequences and primer-specific annealing temperatures are shown in Table S5. Unknown samples were quantified using standard curves generated based on known concentrations of plasmid DNA containing the amplicons produced by each set of primers.
SCFA analysis
The production of SCFAs was measured using HPLC (Agilent Technologies, USA) with an Aminex HPX-87H Exclusion Column (Bio-Rad, USA). In brief, fermentation products were centrifuged at 14,000 rpm for 15 min, and each supernatant was analyzed. H2SO4 (5 mM) was used as the mobile phase, with a flow rate of 0.6 mL/min. The column temperature was 50°C, and a refractive index detector (Agilent Technologies, USA) was used at a wavelength of 215 nm. Acetic, propionic, and butyric acid standards were purchased from Sigma-Aldrich (USA).
Isolation and identification of KCO-degrading bacteria
Human sample No. 8 was collected after 48 h of fermentation and spread on a KCO agar plate (basic growth medium VI supplemented with 8 g/L KCO and 12 g/L agar) using a 10-fold dilution method. Colonies obtained from the plates were randomly selected and reinoculated into VI medium with KCO as the sole carbon source. KCO degradation was assessed based on TLC analysis of the supernatant and confirmed using gel filtration chromatography and ESI-MS. Positive colonies were further purified by repeating the 10-fold dilution.
Isolates capable of degrading KCO were identified based on 16S rRNA gene sequences. In brief, genomic DNA was extracted from each isolate using an OMEGA Bacterial Genome DNA extraction kit (OMEGA, USA), and the 16S rRNA gene was PCR-amplified using the primers 27F (5'-CAG AGT TTG ATC CTG GCT-3') and 1492R (5'-AGG AGG TGA TCC AGC CGC A-3')(Frank et al., 2008). DNA sequencing was performed by Shanghai Sangon Biotech Co., Ltd. (Shanghai, China). Bacterial species were identified based on BLAST searches for aligned 16S rRNA gene sequences (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Animal experiments
Twenty-four three-week-old germ-free Kunming mice (both males and females) were randomly divided into four groups. KCO-degrading bacteria (5 × 108 CFU in 0.5 mL) were inoculated intragastrically into mice in the GN and GNK groups. Fecal pellets were collected from each group four weeks after inoculation, and bacterial community composition was assessed using 16s rRNA gene clone library sequencing (Table S6). Bacterial colonization was allowed to progress for a further four weeks, and then mice in the GNK and GK groups were given 5% KCO in their drinking water for an additional 8 weeks. Control mice (group GF) were untreated. Mice were killed via cervical dislocation, and serum, liver, cecal content, and gut tissue samples were collected as shown in Fig. S9. All procedures involving mice were approved by the Zhejiang Academy of Agricultural Science and Third Military Medical University (Hangzhou & Chongqing, China).
RNA-Seq analysis
Samples with RNA integrity ≥8 were prepared using the TruSeq RNA Sample Prep Kit (Illumina, USA). cDNA was prepared via multiplex amplification using an Illumina HiSeq 2000 (Illumina, USA), with a paired-end read length of 100 base pairs. RNA-Seq analyses were performed by Shanghai Personal Biotechnology Co., Ltd., (Shanghai, China) (http://www.personalbio.cn/en/).
The RNA-Seq data were quality controlled using the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and analyzed using the DNABOX suite (http://www.dnabox.cn/). The NCBI reference mouse genome (Mus_musculus.GRCm38.75) was used to identify known transcripts. DEGs (P < 0.05) were identified and subjected to GO and KEGG analyses using DAVID (https://david.ncifcrf.gov/).
H&E staining and IHC analysis
Samples of the duodenum, jejunum, ileum, colon, and rectum were immediately removed, immersed in 4% paraformaldehyde in 0.1 M PBS (pH 7.4) for 24 h, paraffin-embedded, and sectioned. Each 4-mm section was either stained with H&E or immunostained with a primary antibody: anti-CD3 antibody, ab16669, Abcam; anti-VCAM1 antibody, ab134047, Abcam; anti-PECAM1 antibody, ab182981, Abcam; Phospho-p38 (p-p38) MAPK (Thr180/Tyr182) Antibody, Cell Signaling Technology, followed by the Horseradish peroxidase (HRP) polymer and diaminobenzidine (DAB). Sections were observed by a medical pathologist using the double-blind method. For IHC analysis, cells in five randomly selected fields were selected to determine the relative abundance of positive cells. H&E-stained sections were observed under a microscope (Leica DM 2500, Germany) to assess changes in ulcer size and the infiltration of inflammatory cells. Injuries to these tissues were scored as previously described (Chiu Cj Fau -McArdle et al., 1970).
Statistical analyses
All values are presented as mean ± standard error of the mean (SEM). Statistical analyses, including independent-samples t tests and one-way ANOVAs, were performed using SPSS v20.0 (SPSS Inc., USA).
Data availability
The raw mouse RNA-Seq data and the draft genome of the KCO-degrading bacterium have been submitted to the NCBI database (accession numbers PRJNA589115 and PRJNA588896, respectively).
Supplementary Material
Acknowledgments
Xin Wang was supported by National Natural Science Foundation of China (NSFC, 31870106), Key Research & Development of Zhejiang Province (2018C02048), and State Key Laboratory for Managing Biotic and Chemical Threats to the Quality and Safety of Agro-products (2010DS0024-ZZ006). Guangli Yu was supported by National Natural Science Foundation of China (NSFC, 81991522), the National Science and Technology Major Project for Significant New Drug Development (2018ZX09735004), and Taishan Scholar Climbing Project (TSPD20210304). Yeshi Yin was supported by Distinguished Young Scholars of Hunan Natural Science Foundation (2020JJ2016). HDY was supported by NIGMS R44GM113545 and P20GM103434. DAP is supported by NIGMS WV-INBRE P20GM103434.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jgg.2021.08.001.
References
- Bazzoni G, 2003. The JAM family of junctional adhesion molecules. Curr. Opin. Cell Biol. 15, 525–530. [DOI] [PubMed] [Google Scholar]
- Campo VL, K DF, Silva DB, da Carvalho I, 2009. Carrageenans: Biological properties, chemical modifications and structural analysis - a review. Carbohydr. Polym. 77, 167–180. [Google Scholar]
- Chiu CJ, McArdle AH, Brown R, Scott HJ, Gurd FN, 1970. Intestinal mucosal lesion in low-flow states. I. A morphological, hemodynamic, and metabolic reappraisal. Arch. Surg. 101, 478–483. [DOI] [PubMed] [Google Scholar]
- David S, Fahoum L Fau, Rozen G, Rozen G Fau, Shaoul R, Shaoul R Fau, Shpigelman A, Shpigelman A Fau, Meyron-Holtz EG, Meyron-Holtz Eg Fau, et al. , 2019. Reply to the comment on “Revisiting the carrageenan controversy: do we really understand the digestive fate and safety of carrageenan in our foods?” by M. Weiner and J. McKim. Food. Funct 10, 1763–1766. [DOI] [PubMed] [Google Scholar]
- David S, Shani Levi C, Fahoum L, Ungar Y, Meyron-Holtz EG, Shpigelman A, Lesmes U, 2018. Revisiting the carrageenan controversy: do we really understand the digestive fate and safety of carrageenan in our foods? Food. Funct. 9, 1344–1352. [DOI] [PubMed] [Google Scholar]
- Efsa Panel on Food ANS, Younes M, Aggett P, Aguilar F, Crebelli R, Filipič M, Frutos MJ, Galtier P, Gott D, et al. , 2018. Re-evaluation of carrageenan (E 407) and processed Eucheuma seaweed (E 407a) as food additives. EFSA J. 16, e05238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficko-Blean E, Prechoux A, Thomas F, Rochat T, Larocque R, Zhu Y, Stam M, Genicot S, Jam M, Calteau A, et al. , 2017. Carrageenan catabolism is encoded by a complex regulon in marine heterotrophic bacteria. Nat. Commun. 8, 1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank JA, Reich CI, Sharma S, Weisbaum JS, Wilson BA, Olsen GJ, 2008. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl. Environ. Microbiol. 74, 2461–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjari M, Behmanesh M, Sadeghizadeh M, Zeinoddini M, 2013. Junctional adhesion molecules 2 and 3 may potentially be involved in progression of gastric adenocarcinoma tumors. Med. Oncol. 30, 380. [DOI] [PubMed] [Google Scholar]
- Harmuth-Hoene AE, Schwerdtfeger E, 1979. Effect of indigestible polysaccharides on protein digestibility and nitrogen retention in growing rats. Nutr. Metab. 23, 399–407. [DOI] [PubMed] [Google Scholar]
- Hehemann JH, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G, 2010. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature 464, 908–912. [DOI] [PubMed] [Google Scholar]
- Ji M, 1997. Seaweed Chemistry. Science Press, Beijing. [Google Scholar]
- Jiang J, Zhang W, Ni W, Shao J, 2021. Insight on structure-property relationships of carrageenan from marine red algal: a review. Carbohydr. Polym. 257, 117642. [DOI] [PubMed] [Google Scholar]
- Jung J, Bae SS, Chung D, Baek K, 2019. Tamlana carrageenivorans sp. Nov., a carrageenan-degrading bacterium isolated from seawater. Int. J. Syst. Evol. Microbiol. 69, 1355–1360. [DOI] [PubMed] [Google Scholar]
- Kushkevych I, Cejnar J, Treml J, Dordevic D, Kollar P, Vítězová M, 2020. Recent advances in metabolic pathways of sulfate reduction in intestinal bacteria. Cells 9, 698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei F, Yin Y, Wang Y, Deng B, Yu HD, Li L, Xiang C, Wang S, Zhu B, Wang X, 2012. Higher-level production of volatile fatty acids in vitro by chicken gut microbiotas than by human gut microbiotas as determined by functional analyses. Appl. Environ. Microbiol. 78, 5763–5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Ni R, Shao Y, Mao S, 2014a. Carrageenan and its applications in drug delivery. Carbohydr. Polym. 103, 1–11. [DOI] [PubMed] [Google Scholar]
- Li M, Li G, Zhu L, Yin Y, Zhao X, Xiang C, Yu G, Wang X, 2014b. Isolation and characterization of an agaro-oligosaccharide (AO)-hydrolyzing bacterium from the gut microflora of Chinese individuals. PLoS One 9, e91106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Shang Q, Li G, Wang X, Yu G, 2017. Degradation of marine algae-derived carbohydrates by bacteroidetes isolated from human gut microbiota. Mar. Drugs 15, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libiad M, Vitvitsky V, Bostelaar T, Bak DW, Lee HJ, Sakamoto N, Fearon E, Lyssiotis CA, Weerapana E, Banerjee R, 2019. Hydrogen sulfide perturbs mitochondrial bioenergetics and triggers metabolic reprogramming in colon cells. J. Biol. Chem. 294, 12077–12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogal A, Valdes AA-O, Menni C, 2021. The role of short-chain fatty acids in the interplay between gut microbiota and diet in cardio-metabolic health. Gut Microb. 13, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Won SM, Yoon JH, 2018. Dokdonia ponticola sp. nov., a carrageenan-degrading bacterium of the family flavobacteriaceae isolated from seawater. Curr. Microbiol. 75, 1126–1132. [DOI] [PubMed] [Google Scholar]
- Prajapati VD, Maheriya PM, Jani GK, Solanki HK, 2014. Carrageenan: a natural seaweed polysaccharide and its applications. Carbohydr. Polym. 105, 97–112. [DOI] [PubMed] [Google Scholar]
- Shang Q, Sun W, Shan X, Jiang H, Cai C, Hao J, Li G, Yu G, 2017. Carrageenan-induced colitis is associated with decreased population of anti-inflammatory bacterium, Akkermansia muciniphila, in the gut microbiota of C57BL/ 6J mice. Toxicol. Lett. 279, 87–95. [DOI] [PubMed] [Google Scholar]
- Shen J, Chang Y, Chen F, Dong S, 2018. Expression and characterization of a κ-carrageenase from marine bacterium Wenyingzhuangia aestuarii OF219: a biotechnological tool for the depolymerization of κ-carrageenan. Int. J. Biol. Macromol. 112, 93–100. [DOI] [PubMed] [Google Scholar]
- Shen J, Chang Y, Dong S, Chen F, 2017. Cloning, expression and characterization of a ι-carrageenase from marine bacterium Wenyingzhuangia fucanilytica: a biocatalyst for producing ι-carrageenan oligosaccharides. J. Biotechnol. 259, 103–109. [DOI] [PubMed] [Google Scholar]
- Vigil de Mello SV, da Rosa JS, Facchin BM, Luz AB, Vicente G, Faqueti LG, Rosa DW, Biavatti MW, Fröde TS, 2016. Beneficial effect of Ageratum conyzoides Linn (Asteraceae) upon inflammatory response induced by carrageenan into the mice pleural cavity. J. Ethnopharmacol. 194, 337–347. [DOI] [PubMed] [Google Scholar]
- Witherden DA, Ramirez K, Havran WL, 2014. Multiple receptor-ligand interactions direct tissue-resident gd T cell activation. Front. Immunol. 5, 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Lei F, Zhu L, Li S, Wu Z, Zhang R, Gao GF, Zhu B, Wang X, 2010. Exposure of different bacterial inocula to newborn chicken affects gut microbiota development and ileum gene expression. ISME J. 4, 367–376. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw mouse RNA-Seq data and the draft genome of the KCO-degrading bacterium have been submitted to the NCBI database (accession numbers PRJNA589115 and PRJNA588896, respectively).