

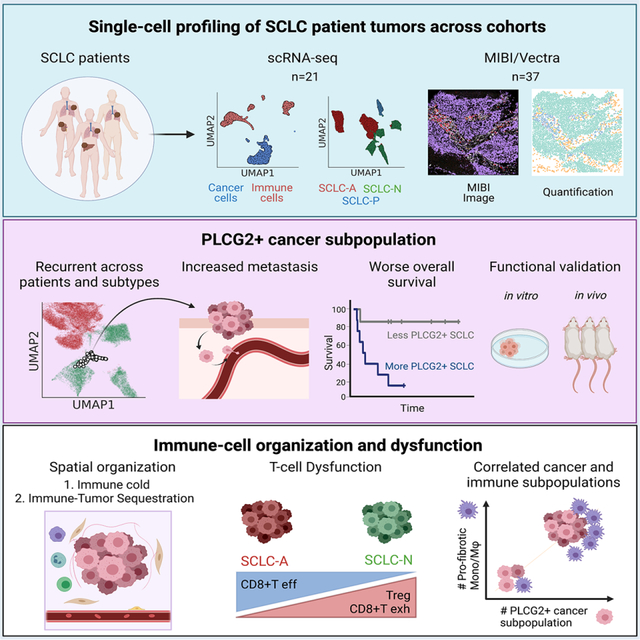
SUMMARY
Small cell lung cancer (SCLC) is an aggressive malignancy that includes subtypes defined by differential expression of ASCL1, NEUROD1, and POU2F3 (SCLC-A, -N, and -P, respectively). To define the heterogeneity of tumors and their associated microenvironments across subtypes, we sequenced 155,098 transcriptomes from 21 human biospecimens, including 54,523 SCLC transcriptomes. We observe greater tumor diversity in SCLC than lung adenocarcinoma, driven by canonical, intermediate, and admixed subtypes. We discover a PLCG2-high SCLC phenotype with stem-like, pro-metastatic features that recurs across subtypes and predicts worse overall survival. SCLC exhibits greater immune sequestration and less immune infiltration than lung adenocarcinoma, and SCLC-N shows less immune infiltrate and greater T-cell dysfunction than SCLC-A. We identify a profibrotic, immunosuppressive monocyte/macrophage population in SCLC tumors that is particularly associated with the recurrent, PLCG2-high subpopulation.
Keywords: SCLC, metastasis, scRNAseq, tumor atlas
eTOC Blurb
Chan et al. use single-cell transcriptome sequencing and imaging techniques to study the heterogeneity and tumor microenvironment of clinical small cell lung cancer specimens. This analysis identifies a PLCG2-high expressing subpopulation linked to metastasis and poor prognosis, and an enrichment of a monocyte/macrophage population with a profibrotic, immunosuppressive phenotype.
Graphical Abstract
INTRODUCTION
The prognosis for patients with small cell lung cancer (SCLC), the most aggressive lung cancer histology, remains exceptionally poor: most patients present with metastatic disease, and the recent addition of immune checkpoint blockade to first-line chemotherapy has only modestly improved median survival (Horn et al., 2018a; Rudin et al., 2021). The strong predilection for early metastasis and therapeutic resistance contribute to poor long-term outcomes, with 5-year survival of 15–30% for limited stage disease, and less than 1% for patients with extensive stage disease (Byers and Rudin, 2015; Siegel et al., 2020).
Although SCLC appears morphologically homogeneous, recent data from both murine models and human tumors suggest the existence of SCLC subtypes with distinct therapeutic vulnerabilities (Rudin et al., 2019). An emerging consensus has classified these subtypes based on differential expression of four transcription factors: ASCL1, NEUROD1, POU2F3 and YAP1 (Rudin et al., 2019). This classification has led to new questions, such as whether subtypes are associated with particular disease stages, metastatic potential or immune microenvironments, and whether there is plasticity between subtypes (Chalishazar et al., 2019; Ireland et al., 2020; Rudin et al., 2019).
Single-cell RNA sequencing (scRNA-seq) offers a unique opportunity to address these questions by dissecting the intratumoral heterogeneity of SCLC and its tumor microenvironment (TME). Multiplexed ion beam imaging (MIBI) is a complementary technology that profiles multiple protein markers simultaneously at single-cell resolution in the spatial context of tissue. Efforts to apply these technologies to human SCLC tumors have been limited, as surgical resections of primary tumors are performed in under 5% of SCLC patients(Vallières et al., 2009), and biopsied samples are not typically preserved in a manner amenable to single-cell profiling. Since resection is only clinically indicated for very early stage de novo disease, these samples fail to capture the spectrum of disease progression.
Here, we have constructed a single-cell atlas of SCLC patient tumors, with comparative lung adenocarcinoma (LUAD) and normal lung. Our analysis reveals high inter-patient transcriptomic diversity in SCLC and immune cells, largely driven by subtype-specific changes in cancer gene programs and immune dysfunction. In the midst of substantial heterogeneity, we identify a stem-like pro-metastatic tumor subpopulation marked by high PLCG2 expression that spans the full diversity of SCLC subtypes and predicts worse overall survival. Together, our analyses provide a deep characterization of the molecular features of SCLC, with clinical implications.
RESULTS
Human SCLC tumors are more heterogeneous than LUAD
We profiled the transcriptomes of 155,098 cells from 21 fresh SCLC clinical samples (Figure S1A, Table S1) obtained from 19 patients, as well as 24 LUAD and 4 tumor-adjacent normal lung samples as controls (Figures 1A and S1B). The SCLC and LUAD cohorts include treated and untreated patients (Figure 1B). Samples were obtained from primary tumors, regional lymph node metastases, and distant metastases (liver, adrenal gland, axilla, and pleural effusion) (Figure 1C).
Figure 1: The single-cell transcriptional landscape of SCLC.
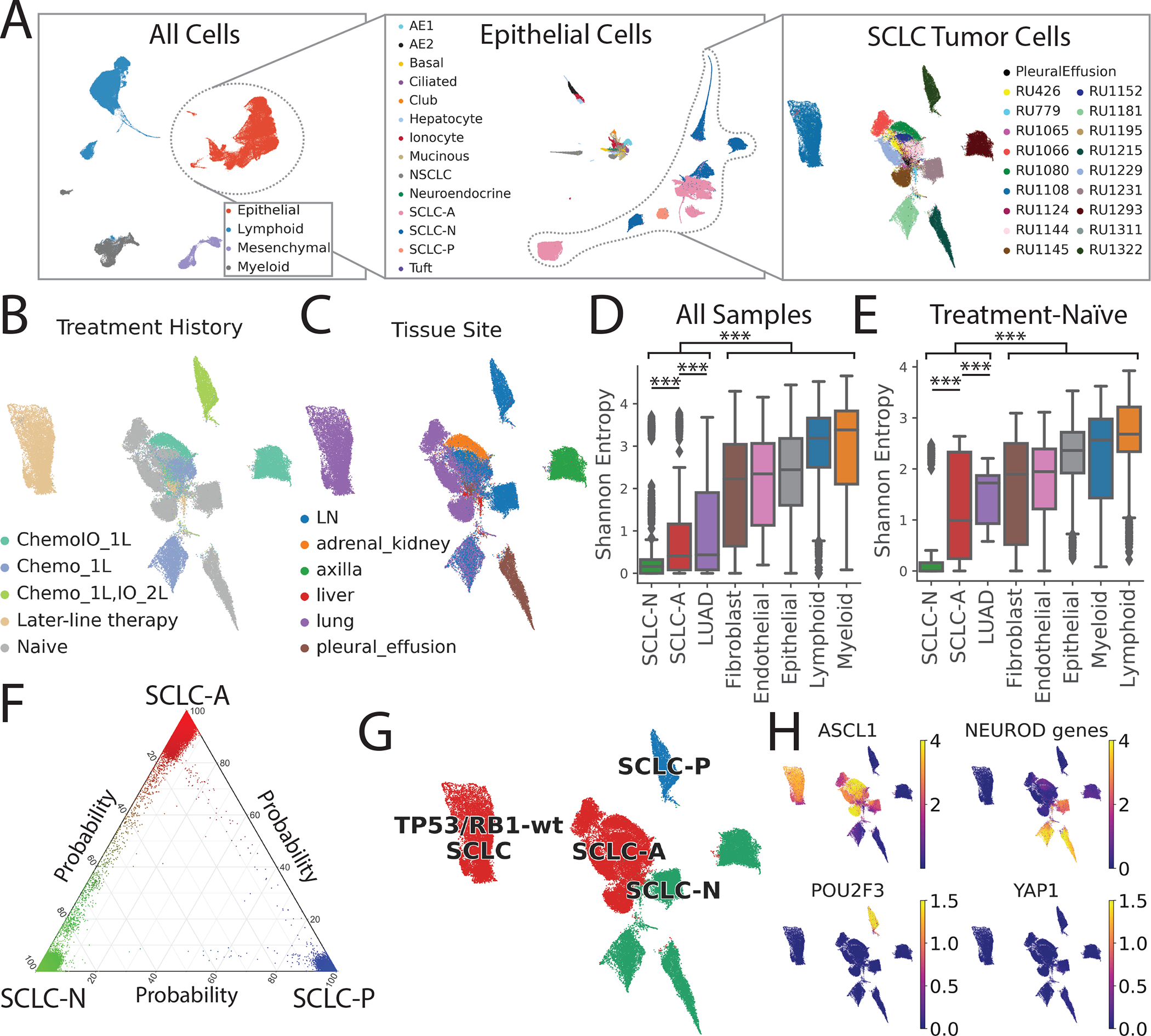
LUAD and normal adjacent lung serve as reference tissues.
(A) UMAP of iterative subsets of cells from the global level (left, n=155,098 cells) to the epithelial compartment (middle, n=64,301 cells) to SCLC cells (right, n=54,523 cells). Each dot represents a single cell colored by cell type.
UMAP of SCLC cells annotated by (B) treatment history and (C) tissue site.
Inter-patient heterogeneity within each cell type as measured by Shannon entropy for (D) all samples and (E) treatment-naïve samples (Student’s t-test, error bars: 95% confidence intervals; STAR Methods).
DC = dendritic cells, LN = lymph node, Chemo_1L = chemotherapy in first line, ChemoIO_1L = chemotherapy plus immunotherapy in first line, IO_2L = Immunotherapy in second line, later-line therapy = multiple lines of treatment. p-values: *<0.05, **<0.01, ***<0.001.
(F) UMAP of SCLC cells colored by subtype (red = SCLC-A, green = SCLC-N, blue = SCLC-P), based on maximum likelihood computed by our classifier. Sample RU1108 is labeled as a TP53/RB1 wild-type SCLC-A outlier (STAR Methods).
(G) UMAP of imputed expression of ASCL1, NEUROD1, POU2F3 and YAP1 in the SCLC cohort using MAGIC109 (k=30, t=3). Expression in units of log2(X+1) where X = normalized counts.
(H) Ternary plot of SCLC subtype probability per cell, calculated by Markov absorption probabilities (STAR Methods). Cell color is assigned by the likelihood of SCLC-A (red), SCLC-N (green), and SCLC-P (blue).
See also Figures S1–S2, and Table S1.
All scRNA-seq data were merged, normalized, batch-corrected, and clustered to identify coarse cell types, including epithelial, mesenchymal, lymphoid, and myeloid cells (Figures 1A and S1B–C; STAR Methods). Further clustering within the epithelial compartment identified cells comprising the respiratory epithelium (including alveolar epithelial types 1 and 2, ciliated, club, neuroendocrine and tuft cells) and hepatocytes derived from liver metastases.
MSK-IMPACT™ targeted sequencing (Cheng et al., 2015) of 14 SCLC samples demonstrated frequent mutation or loss of RB1 and TP53, and recurrent mutations in CREBBP and KMT2B (Figure S1D and S1E). This information facilitated the identification of cancer cells that harbor transcripts bearing characteristic variants. We also inferred single-cell copy number variation (CNV) to support cancer cell identification (STAR Methods). We detected higher CNV levels in SCLC than LUAD (Figure S1F), consistent with higher tumor mutation burden in SCLC (Yarchoan et al., 2019). Based on studies investigating cell types of origin(Ferone et al., 2020), we consider clusters of neuroendocrine and alveolar epithelial type 2-like cancer cells to represent SCLC and LUAD, respectively.
Following cell type annotation, we characterized tumor heterogeneity within our atlas. Of 38 epithelial clusters (n = 64,301 cells), we found that LUAD and SCLC clustered separately as expected; 5 LUAD clusters contain 7,635 cells from 24 tumors and 25 SCLC clusters contain 55,815 cells from 21 tumors, consistent with the higher stromal content of LUAD. To quantify the inter-patient heterogeneity of SCLC, we calculated the Shannon entropy of patients for each cluster (STAR Methods). Low Shannon entropy signifies that the cluster phenotype is rarely shared across patients, i.e., inter-patient heterogeneity is high. Malignant SCLC cells showed significantly higher inter-patient heterogeneity (lower entropy) than LUAD cells (Figure 1D), even when restricting analysis to treatment-naïve samples (Figure 1E). We observed low phenotypic diversity in stromal and immune cell populations, consistent with minimal batch effects across samples, and high diversity in neoplastic cells compared to stroma, consistent with prior studies (Azizi et al., 2018; Puram et al., 2017). Our results suggest that, despite its homogeneous histological morphology, SCLC has a high degree of transcriptional tumor heterogeneity, exceeding that of LUAD and normal stroma.
Tumor heterogeneity of canonical SCLC subtypes at single-cell resolution
Next, we considered the 54,523 SCLC cells in our dataset and characterized cell states within the canonical SCLC subtypes (Rudin et al., 2019) (STAR Methods). SCLC subtypes are typically classified by the expression of ASCL1, NEUROD1, POU2F3 and YAP1, but a single-gene strategy is unreliable for scRNA-seq, given the prevalence of gene dropout. Recent studies from our group and others have also questioned the value of YAP1 alone as a subtype marker (Baine et al., 2020; Pearsall et al., 2020).
We therefore used a neighbor-graph-based approach, which harnesses multiple genes that define the full complexity of each subtype, to calculate the probability of a given SCLC subtype per cell (Levine et al., 2015) (Figure 1F; STAR Methods). We identified the most likely subtype of each cell (Figure 1G) and used this to categorize the major subclone of each sample as SCLC-A (N = 14), SCLC-N (N = 6), or SCLC-P (N = 1). Our classification did not identify any SCLC-Y tumors, consistent with minimal expression of YAP1 in SCLC cells. This observation is supported by the relative expression of canonical transcription factors (Figure 1H), corresponding MYC family members (Figure S1G), and matched immunohistochemistry (IHC) when available (Figure S1H). Unlike single-gene expression or IHC, our strategy can classify cases with high expression of both ASCL1 and NEUROD1 (such as Ru1231, classified as SCLC-N) and those with low expression of both (such as Ru1293, classified as SCLC-N due to expression of NEUROD2 and NEUROD4). We also identified intermediate cancer cells along the SCLC-A to SCLC-N spectrum, suggesting transitional or non-canonical phenotypes, as well as tumors of admixed subtype and a non-canonical SCLC phenotype with wild-type TP53/RB1 (see Figures S2A–B, Table S2, and STAR Methods for further details).
SCLC-N exhibits a pro-metastatic neuronal and EMT phenotype
To better define the role of SCLC subtype in tumor progression, we assessed cell composition and gene expression differences across subtypes (Figure S2C). We focused on SCLC-A and -N, as our cohort only included a single SCLC-P case. Consistent with mouse models (Ireland et al., 2020; Mollaoglu et al., 2017), SCLC-A is significantly overrepresented in primary tumors, whereas SCLC-N is enriched in nodal and distant metastases (Dirichlet regression, p<3.4×10−8; Figure S2D; STAR Methods). We also observed greater interpatient diversity in SCLC-N tumors than in SCLC-A (Figure 1D). These findings are consistent with preclinical models showing that SCLC-N can derive from SCLC-A through discrete evolutionary bottlenecks.
We performed differential expression (DE) and pathway analysis to determine subtype-specific gene programs (Figures 2A and S2E; Tables S3–8). We found that SCLC-A is enriched in expression of genes regulating cell cycle progression and DNA repair, as well as EZH2 target genes implicated in SCLC cell cycle regulation (Hubaux et al., 2013; Poirier et al., 2015) (Figure S2E). In contrast, SCLC-N tumors exhibit a pro-metastatic pattern of gene expression including overexpressed markers of (1) epithelial-mesenchymal transition (EMT), (Dongre and Weinberg, 2019a), (2) TGF-β(Farabaugh et al., 2012), (3) BMP signaling (Choi et al., 2019; Dongre and Weinberg, 2019b)(Bach et al., 2018), (4) STAT (Dongre and Weinberg, 2019b); and (5) TNFα-promoted NFκB signaling (Jiang et al., 2001; Wu and Zhou, 2010) (Figures 2A, 2B and S2E).
Figure 2: Gene programs and cell-cell interactions enriched in each SCLC subtype.
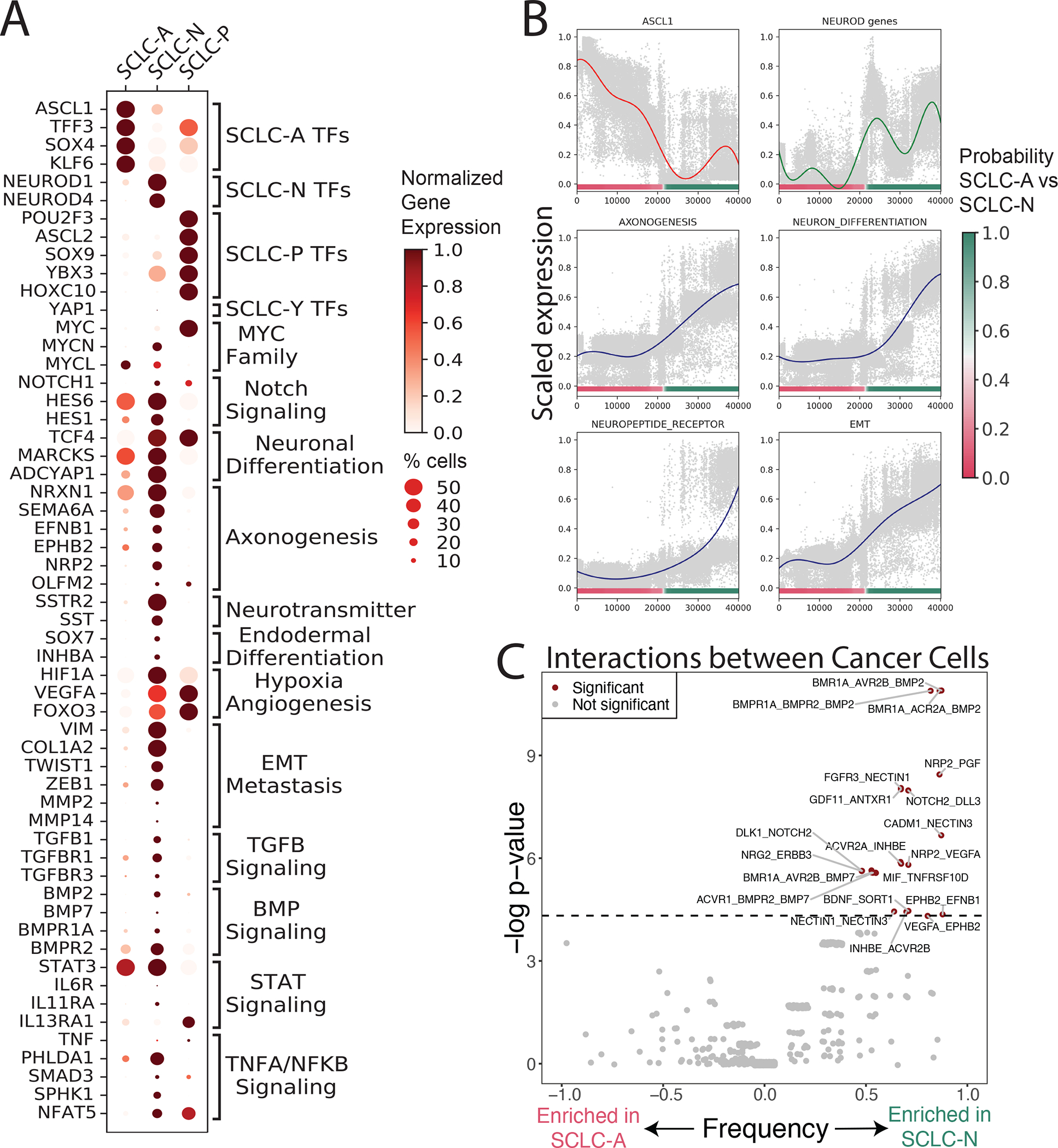
(A) Dot plot showing selected DEGs between each SCLC subtype versus the rest, as well as between SCLC-A vs SCLC-N. DEGs are grouped by enriched gene pathways as assessed by GSEA (NES > 1, FDR < 0.1) (Tables S3–8). Dot size = % cells expressing gene; dot color = mean expression scaled from 0 to 1.
(B) Scaled expression of canonical markers or scaled average Z-score of select enriched pathways in SCLC-N (Y-axis), versus SCLC subtype probability (X-axis). Solid lines represent average gene/pathway trend (STAR Methods).
(C) Enrichment of interactions between cancer cells within SCLC-A vs SCLC-N. Significant interactions are assessed using CellPhoneDB102. Enrichment of interactions within SCLC-A vs SCLC-N is plotted as significance (−log2 of Fisher’s test) versus frequency. Dashed line corresponds to nominal p < 0.05.
SCLC-N is also enriched in neuronal differentiation and neuropeptide signaling, including ephrins and semaphorins, gene families involved in axonogenic signaling (Pitulescu and Adams, 2010; Yoshida, 2012) (Figures 2A, 2B and Table S3). Prior studies have shown that the axonogenesis program coordinates neuronal migration (Zhang et al., 2019a) and is implicated in SCLC metastasis (Yang et al., 2019), and ephrin and semaphorin pathway components are NEUROD1 targets (Borromeo et al., 2016) or regulators of the NEUROD1high phenotype (Wooten et al., 2019) (see STAR Methods for an in-depth characterization of enriched pathways in SCLC-A vs SCLC-N).
We further assessed differentially expressed ligand-receptor pairs within subtypes (Figure 2C; STAR Methods), and observed marked enrichment in potential homotypic interactions between cancer cells in SCLC-N compared to SCLC-A. While one cannot be certain of any individual hypothesized ligand-receptor interaction in such analysis, the difference in the number of interactions between subtypes is striking and may reflect differential interactivity between subtypes. This enrichment is consistent with how SCLC-A cell lines typically grow as loose floating aggregates and SCLC-N lines grow as a tightly adherent monolayer in cell culture (Gazdar et al., 1985; Rudin et al., 2019).
A stem-like, pro-metastatic cell cluster recurs across patients and SCLC subtypes
The transcriptomic diversity of SCLC contrasts with the uniformly poor prognosis of patients. We analyzed phenotypes spanning multiple patients to determine whether any shared cell types may account for the universal aggressiveness of SCLC. Unsupervised clustering of the SCLC malignant cell compartment identified 25 clusters. Most clusters are specific to a single tumor, but cluster 22 is strikingly recurrent across samples (Mann-Whitney p < 2.2×10−16) (Figures 3A–C and S3A; Table S1; STAR Methods), spanning a range of treatment histories, tissue sites, and predominant subtypes (Figure 3D). Cluster 22 comprises 166 cells, with 9 of 21 profiled tumors harboring at least 3% of the cluster. We confirmed that cells in the recurrent cluster have greater CNV burden than normal epithelial cells, consistent with a malignant phenotype (Figure S3B).
Figure 3: A subpopulation with metastatic, stem-like phenotype recurs broadly across SCLC tumors.
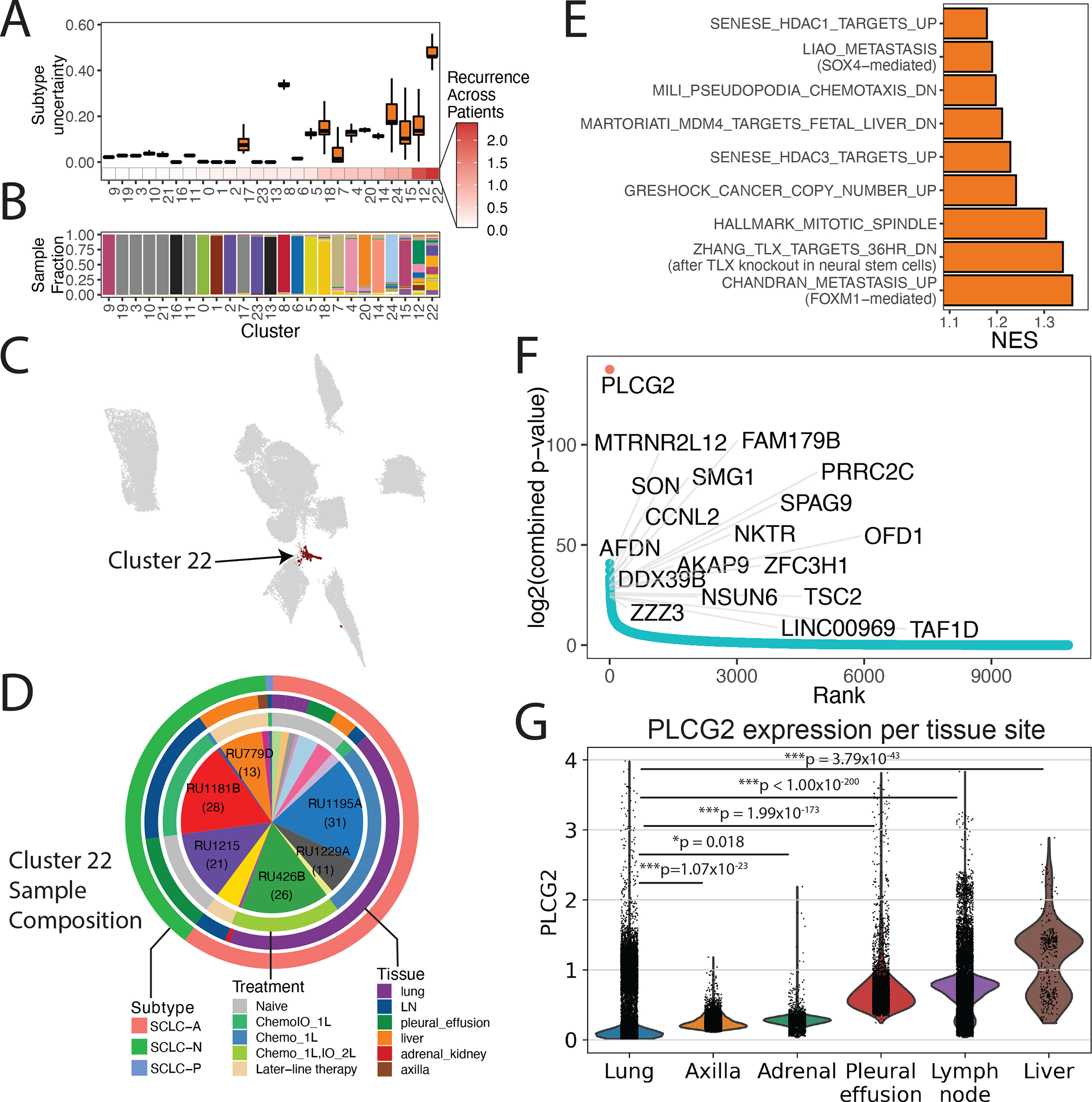
(A) Boxplot of subtype uncertainty of each SCLC cell stratfied by cluster (Y-axis; measured as entropy of subtype probabilities per cell within each cluster; error bars span 25th to 75th percentile), ordered by recurrence across patients (X-axis; measured as Shannon entropy of patients per cluster; STAR Methods).
(B) Stacked barplot of sample fraction per cluster, ordered by recurrence across patients, as in (A).
(C) UMAP of SCLC cells with recurrent cluster 22 colored in black.
(D) Proportion of samples comprising the recurrent cluster (9 of 21 profiled tumors harboring >3% of the cluster). The number of cells per sample are indicated in parentheses for samples with the greatest representation of the recurrent cluster. Outer rings indicate the major intratumoral subtype (outer), tissue site (middle), and treatment history (inner).
(E) Gene programs significantly enriched in cluster 22. Barplot of NES from GSEA for significantly enriched pathway (FDR < 0.05 and NES > 1; Table S9).
(F) Genes ordered from most to least recurrently overexpressed along the X-axis, with recurrence score plotted on the Y-axis. The recurrence score is calculated as follows. Within each sample, DEGs were assessed between the recurrent cluster vs the rest of the tumor. The adjusted p-values for differential expression within each tumor are combined using Edgington’s method. The recurrence score is the −log of the combined p-value (Table S11; STAR Methods).
(G) Violin plot with PLCG2 expression among individual cancer cells in our SCLC samples, grouped by tissue site (Bonferroni-adjusted Mann-Whitney test). Expression is plotted as log2(X+1) where X is the normalized count, imputed using MAGIC (k=30, t=3).
Cells in the recurrent cluster exhibit significantly higher uncertainty in subtype assignment than those in any other cluster (Mann-Whitney p < 2.2×10−16), suggesting a dedifferentiated phenotype (Figure 3A; STAR Methods). These cells are enriched in genes and gene programs related to metastasis and neural stem cells (Figures 3E and 3F; Table S9). In microarray data from SCLC-A and SCLC-N cell lines in the Cancer Cell Line Encyclopedia (CCLE) database (N = 54), we confirmed that the gene signature for the recurrent cluster was significantly positively correlated with many of the same pathways associated with metastasis, chemotaxis, and stemness (Figure S3C; STAR Methods).
Within Cluster 22, phospholipase C gamma 2 (PLCG2) was the top differentially upregulated gene (Figures 3F and S3D; Tables S10 and S11). PLCG2 has been previously implicated in Alzheimer’s disease (Castillo et al., 2017; van der Lee et al., 2019) and its paralog PLCG1 promotes metastasis (Kassis et al., 1999; Sala et al., 2008). We used knnDREMI (Dijk et al., 2018), which is well suited to handle data sparsity and rare cell populations, to explore the full gene program that covaries with PLCG2 (STAR Methods). We grouped results from knnDREMI into three gene modules corresponding to low (module 1), medium (module 2) and high PLCG2 expression (module 3) (Figure S3E; Table S12). Candidate genes in module 3 included FGFR1 (implicated in SCLC through frequent amplifications (Elakad et al., 2020)), and MTRNR2L8 and MTRNR2L12 (humanin family genes shown to inhibit apoptosis (Morris et al., 2020), to be neuroprotective in Alzheimer’s disease (Kusakari et al., 2018), and to promote tumor progression in triple-negative breast cancer (Moreno Ayala et al., 2020)). Among the top 5% of pathways most correlated to module 3 were those related to stemness (including OCT4 and SOX2 targets), metastatic gene signatures, and pro-metastatic signaling pathways (including Wnt and BMP signaling) (Dongre and Weinberg, 2019b) (Figures S3E and S3F; Table S13).
PLCG2 expression is associated with increased stem-like and pro-metastatic potential
Among the multiple ovexpressed genes in the recurrent SCLC cluster (Figure 3F), we began by investigating the role of PLCG2 as a potential driver of progression. Consistent with the suggested pro-metastatic phenotype of the recurrent cluster, PLCG2 is significantly upregulated in metastatic sites compared to lung, with highest levels in the liver, the most common site of SCLC metastasis (Figure 3G). These observations prompted us to test PLCG2 function directly by overexpressing the gene in SCLC cell lines with relatively low PLCG2 expression (SHP-77, SCLC-A; H82 and H446, SCLC-N) and by knocking it out in PLCG2-high SCLC cell lines (H526, SCLC-P; DMS114, SCLC-Y). Exogenous PLCG2 overexpression did not affect proliferation (data not shown) but did increase anchorage-independent growth (Figure S3G). Additionally, PLCG2 expression was associated with higher migration and invasion in vitro (Figure 4A) and with higher metastatic potential in vivo following intracardiac injection (Figures 4B and 4C), consistent with the pro-metastatic expression profile of the recurrent cluster. Western blot analyses validated key phenotypes observed in the single-cell data including (1) increased β-catenin expression, suggesting higher Wnt signaling, which was confirmed in a Wnt reporter assay (Figure S3H); (2) increased SMAD1/5 phosphorylation, consistent with higher BMP signaling; (3) increased expression of EMT/metastatic markers and (4) higher levels of stemness-related markers (Figure 4D). These results suggest that PLCG2 may be partially driving a stem-like, pro-metastatic phenotype in the recurrent cluster.
Figure 4: A role for the PLCG2+ recurrent cluster in metastasis and patient outcome associated with PLCG2 expression.
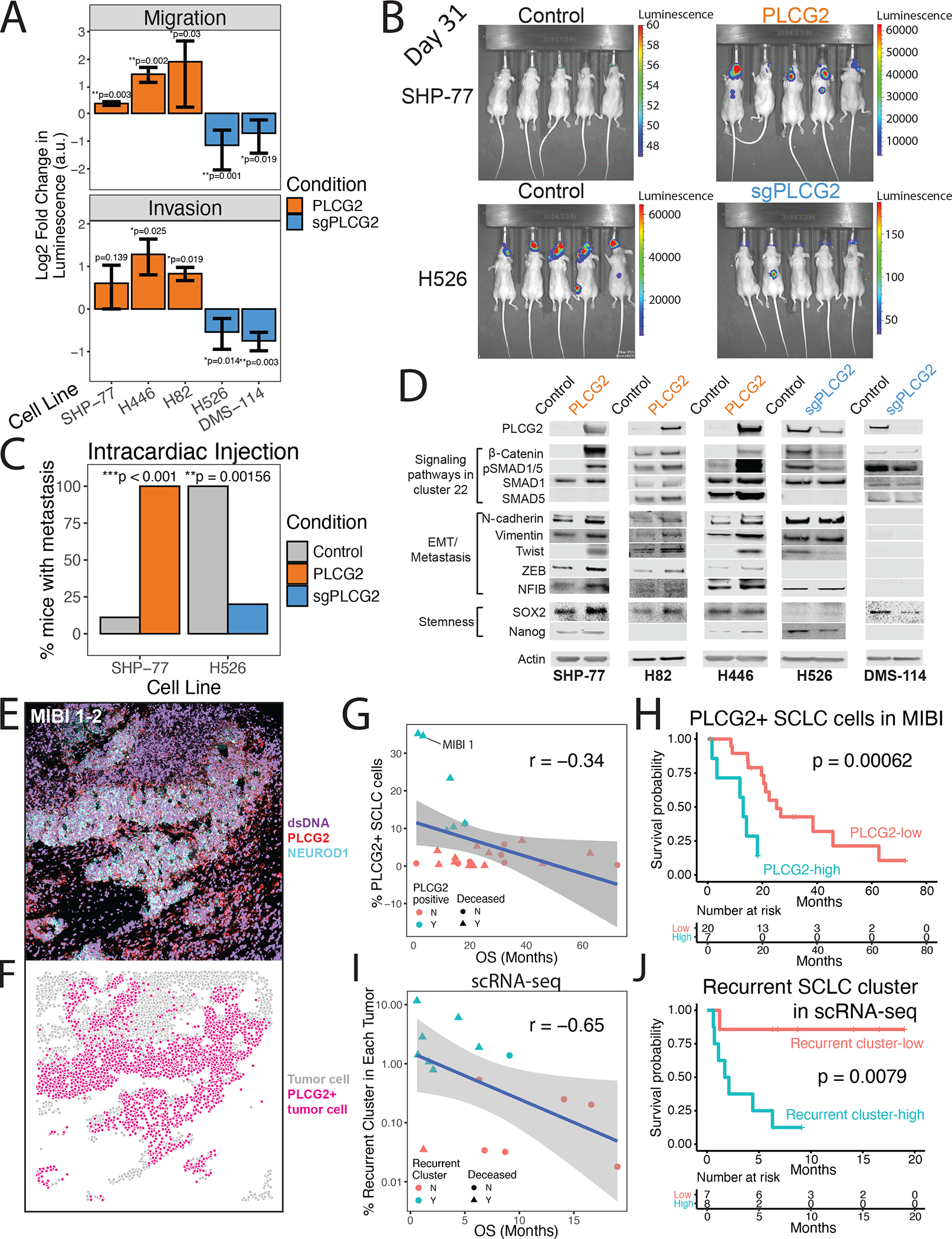
(A) Migration (top) and invasion (bottom) assays for PLCG2-overexpressing cell lines (SHP-77, H446, and H82) and PLCG2-CRISPR KO polyclonal (H526, DMS-114) cell lines, measured with a luminometric method in at least 3 independent experiments (3 technical replicates/experiment). Log2 fold change over control condition was calculated (two-tailed Student’s t-test; error bars: standard deviation).
(B) Luminescence imaging of mice at day 31 following intracardiac injection to assess metastatic capacity of PLCG2-overexpressing SHP77 cells and PLCG2-KO polyclonal H526 cells.
(C) Barplot showing the percentage of mice with metastasis in in vivo intracardiac injections of PLCG2-overexpressing SHP-77 and PLCG2-downregulated H526 cell lines in mice compared to control conditions (Fisher’s exact test).
(D) Western blots of markers associated with signaling pathways upregulated in cluster 22 (Wnt and BMP pathways), EMT/metastasis, and stemness in PLCG2-overexpressing and -KO polyclonal cell lines.
(E) Color overlay of PLCG2 (red), NEUROD1 (cyan), and dsDNA (violet) channels in SCLC tumor MIBI 1 from field of view (FoV) 2 (800 × 800 μm), illustrating high fraction of PLCG2-positive cancer cells. Error bars: 95% confidence interval.
(F) Same FoV as (E) now visualized based on segmented cancer cells using Mesmer (Greenwald et al., 2021), represented by dots colored by PLCG2 positivity. Error bars: 95% confidence interval.
(G) Scatterplot of the percent of PLCG2-positive SCLC cells per sample using MIBI-TOF vs overall survival (months) in an independent TMA cohort, annotated by percent of PLCG2+ SCLC cells >7% (cyan) and deceased patient (triangle). Spearman’s correlation r and example patient MIBI 1 from Figures 4E–F are shown.
(H) Kaplan-Meier analysis of OS in an independent cohort of SCLC patients (Table S14) with high vs low PLCG2 positivity (>7% vs ≤7% of SCLC cells with high PLCG2 staining intensity), as assessed by MIBI-TOF on a TMA. Note that the adjusted Cox proportional hazards model using the fraction of PLCG2-positive SCLC cells as a continuous rather than dichotomized covariate was also significantly predictive (p = 0.012, STAR methods).
(I) Scatterplot of the percent of the recurrent SCLC cluster per sample using scRNA-seq (log10 scale) vs overall survival (months), annotated by percent of recurrent cluster > 0.75% (cyan) and deceased patients (triangle). Spearman’s correlation r is indicated.
(J) Kaplan-Meier analysis of OS in patients with detectable PLCG2+ recurrent cluster cells by scRNA-seq (>0.75% vs ≤0.75% of SCLC cells) (Table S16). Note that the adjusted Cox proportional hazards model using the fraction of the recurrent cluster as a continuous rather than dichotomized covariate was also significantly predictive (p = 0.009, STAR methods). PLCG2 = PLCG2 overexpression, sgPLCG2 = CRISPR knockout. See also Figure S1 and Tables S1, S14, and S16.
PLCG2 and the recurrent cluster are associated with reduced overall survival in patients
To determine the clinical significance of PLCG2 expression, we performed MIBI imaging on a tissue microarray (TMA) representing an independent cohort of SCLC tumor specimens (N = 37; Table S14). We optimized cell-type-specific antibodies (Table S15) in combination with kernel density estimation of cells (STAR Methods) to identify SCLC, immune, and stromal cell types (Figure S4A), which were consistent with IHC review of an adjacent TMA section by a pathologist (data not shown). Using a monoclonal PLCG2 antibody, we identified a subset of patient tumors with high fraction of cancer cells expressing PLCG2, as exemplified by patient MIBI 1 (Figures 4E–F, STAR Methods). Considering only tumors that were ever extensive-stage (either at initial diagnosis or upon relapse; N = 27 passing quality control, see STAR Methods), we found that the presence of PLCG2-expressing cancer cells is negatively correlated with overall survival (Spearman’s correlation r = −0.34; t-test p = 0.04; Figure 4G). Kaplan-Meier analysis revealed worse overall survival in patients with tumors exhibiting high PLCG2 expression (>7% of SCLC cells with high PLCG2 intensity; p = 0.00062; Figure 4H). An adjusted Cox proportional hazards model confirmed decreased overall survival (p = 0.041) and showed furthermore that high PLCG2 positivity is a stronger predictor of worse outcome than treatment history, presence of metastatic disease, or SCLC subtype (Figure S4B). The same model, using the fraction of PLCG2-positive SCLC cells as a continuous rather than dichotomized covariate, was also significantly predictive (p = 0.012), indicating that the analysis does not depend on selecting a threshold for PLCG2-positive SCLC cells.
PLCG2 overexpression is just one feature of the recurrent cluster phenotype. We also assessed whether the prevalence of this subpopulation has prognostic significance, and found that the fractional representation of recurrent cluster cells (log fraction out of all cancer cells in each tumor) is negatively correlated with overall survival (Spearman’s correlation r = −0.65; asymptotic t-test = 0.009; Figure 4I). Patients for whom this subpopulation represents >0.75% of total cancer cells had significantly decreased overall survival relative to others (p = 0.008; Figure 4J; Table S16). An adjusted Cox proportional hazards model confirmed worse overall survival and greater hazard ratio than PLCG2 positivity in the MIBI analysis (44.4 vs 5.47); PLCG2 positivity is a strong predictor, but less so than the full transcriptional phenotype of the recurrent cluster (Figure S4C). We repeated this analysis using recurrent cluster fraction as a continuous covariate and confirmed significantly worse survival without pre-selecting a threshold (p = 0.009). Taken together, these data support that a small stem-like, pro-metastatic subpopulation with high PLCG2 expression has a remarkably large prognostic impact across SCLC subtypes.
Immune cells in the SCLC TME are fewer and more sequestered
SCLC is recognized as a particularly immune-cold cancer46, and the addition of immune checkpoint blockade to standard-of-care chemotherapy only modestly improves median survival (Horn et al., 2018b; Paz-Ares et al., 2019). However, recent findings suggest some subtype-dependent heterogeneity in immunogenicity (Best et al., 2020a; Dora et al., 2020; Gay et al., 2021a; Owonikoko et al., 2021), including a non-NE inflamed SCLC subtype (Gay et al., 2021b). Understanding the role of subtype in shaping the immune environment will be key to developing effective interventions. However, a comprehensive characterization of the SCLC immune compartment has not been feasible due to limited biospecimen availability and the poor accuracy of low-abundance cell type deconvolution from bulk RNA-seq data.
We aimed to assess the influence of SCLC subtype on the immune TME. Our scRNA-seq dataset could not be used to assess total immune cell abundance, since we enriched for non-immune (CD45−) cells by sorting (STAR Methods). Instead, we analyzed flow cytometry data from this cohort, as well as an independent SCLC cohort (N = 11, Table S17). Focusing on SCLC-A and SCLC-N, we confirmed fewer CD45+ cells than LUAD, and found further reduction specifically in SCLC-N and NEUROD1-positive tumors (Figures S4D and S4E), consistent with prior bulk RNA-seq data suggesting that NEUROD1-positive tumors express lower levels of immune-related genes (Best et al., 2020b).
We next sought to characterize the spatial architecture of the immune TME using MIBI on an independent cohort with available NEUROD1 staining (N = 33). Following a prior definition of immune-hot tumors as harboring > 250 immune cells in an 800 × 800 μm field of view (FoV) (Keren et al., 2018a), we found that the majority of SCLC tumors (20 out of 33) in this cohort were immune cold. Moreover, significantly more NEUROD1+ SCLC tumors are immune cold (univariate test - Fisher’s exact p = 0.0066; Figures 5A, 5B and S4F). To account for possible confounders, we modeled immune infiltration (hot vs. cold) as a logistic regression that incorporates clinical covariates including NEUROD1 positivity, histology (single vs. admixed with adenocarcinoma), treatment (treated vs. naive) and location (primary vs. metastatic). The regression model found that tumor location separates predictions of immune infiltration, with immune-cold tumors represented by all 5 metastases and 15 of 28 primaries. Upon adjusting for all clinical covariates, including tumor location, only NEUROD1 positivity is a significant predictor of immune-cold status (Student’s t-test p = 0.037; Figure S4G).
Figure 5: Analysis of therapy and subtype-specific changes in immune phenotype indicate suppressed T-cell activity in SCLC-N.
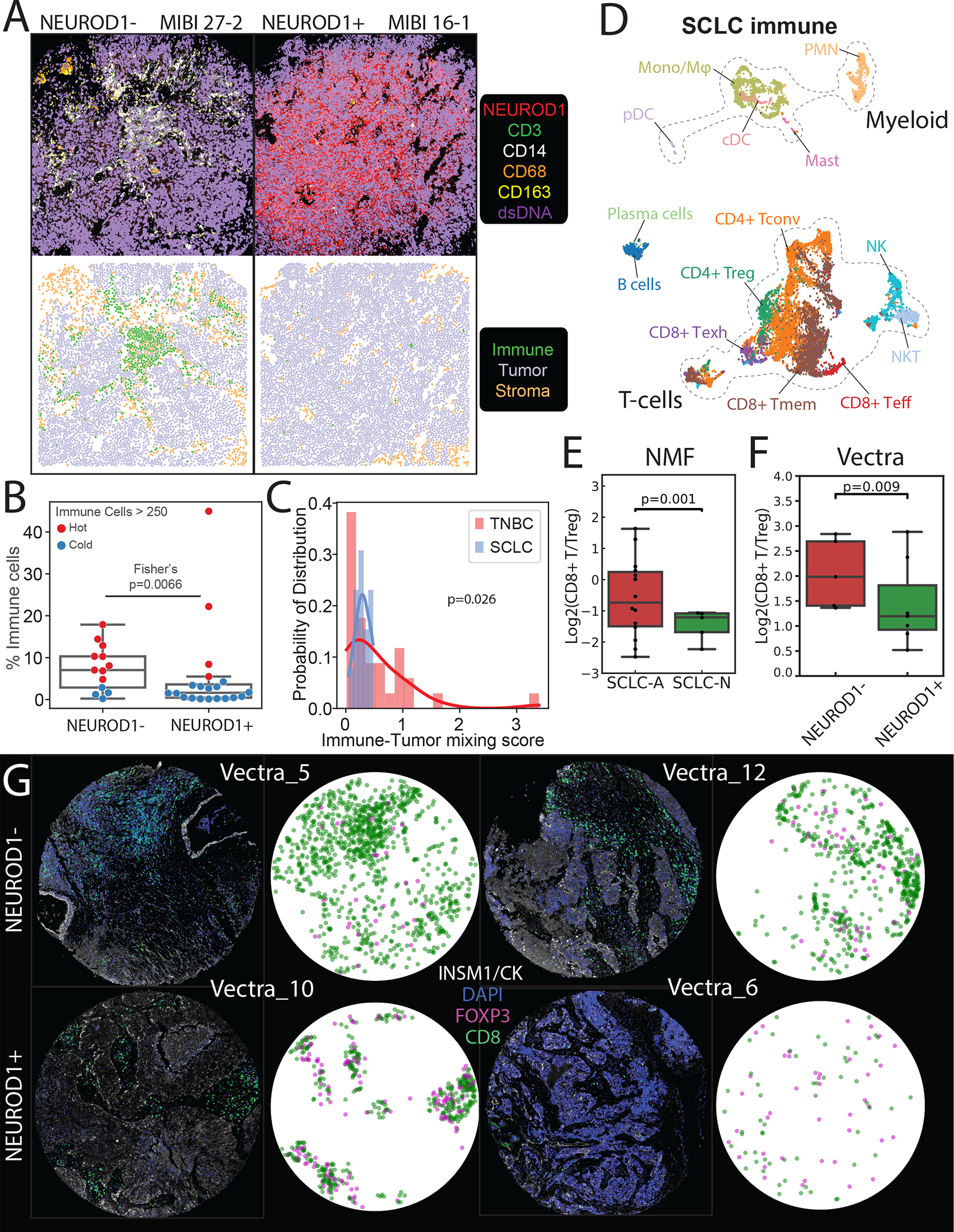
(A) Comparison of MIBI images depicting NEUROD1− SCLC tumor MIBI 27 from FoV 2 (left) and NEUROD1+ SCLC tumor MIBI 16 from FoV 1 (right) (each FoV 800 × 800 μm), illustrating differences in immune abundance and sequestration. Top: Color overlay of NEUROD1 (red), CD3 (green), CD14 (white), CD68 (orange), CD163 (yellow), and dsDNA (violet) channels. Bottom: FoV from the top panel now visualized with segmented cancer cells using Mesmer(Greenwald et al., 2021), represented by dots colored by cell type (immune, tumor, and stroma).
(B) Boxplot comparing the percent of immune out of total cells between NEUROD1− vs NEUROD1+ SCLC cells. The overlying swarmplot is colored by hot (red) vs cold (blue) where hot is defined as number of immune cells > 250 in an 800 × 800 μm FoV (N=33, Fisher’s exact test; error bars: 95% confidence intervals).
(C) The probability distribution of the immune-tumor mixing score in SCLC vs TNBC, defined as the number of interactions between immune and cancer cells divided by the number of interactions between immune and non-cancer cells (N=47, Welch’s t-test).
(D) UMAPs of SCLC immune subsets. Tconv = conventional T-cell; Treg = regulatory T-cell; Teff = effector T-cell; Tmem = memory T-cell; Tgd = γδ T-cell; Mono/Mφ = monocyte/macrophage; PMN = neutrophil; cDC = conventional dendritic cell; pDC = plasmacytoid dendritic cell.
(E) Barplot comparing CD8+ Teff/Treg log ratio based on NMF cell loadings associated with T-cell phenotype in SCLC-A vs SCLC-N in our single-cell cohort (N=19), adjusted for treatment and tissue site (weighted t-test; error bars: 95% confidence interval).
(F) Barplot comparing CD8+ T/Treg log ratio in NEUROD1− vs NEUROD1+ SCLC in an independent cohort with Vectra imaging (N=12; weighted t-test; error bars: 95% confidence interval).
(G) Select Vectra imaging of NEUROD1− vs NEUROD1+ SCLC (2 samples each). Fluorescent markers include CD8 (cytotoxic T-cells), Foxp3 (Tregs), INSM1/CK7 (epithelial and cancer cells), and DAPI (DNA). CD8 (green) or Foxp3 (pink) positivity of segmented cells are shown.
See also Figures S4–S5 and Tables S14, S17, and S18.
To gain potential insights into the extent of immune interactions in SCLC, we quantified the degree of compartmentalization between immune and tumor cells among immune-hot tumors (> 250 immune cells/FoV). We defined an immune-tumor mixing score as the ratio of immune-cancer-cell interactions to immune-stromal-cell interactions (defining stroma as all non-cancer cells), whereby a larger ratio corresponds to greater mixing (STAR Methods). To benchmark this metric, we leveraged a published triple-negative breast cancer (TNBC) dataset (Keren et al., 2018a), as no MIBI comparator is available in LUAD. We found that the distribution of the immune-tumor mixing score among immune-hot SCLC tumors (N = 13) was significantly lower than in immune-hot TNBC tumors (N = 34) (Welch’s t-test p = 0.026; Figures 5A, 5C and S4F). Collectively, we find evidence of 1) decreased immune infiltrate in SCLC and particularly SCLC-N subtype, and 2) immune sequestration in SCLC cases that do contain more immune cells.
SCLC-N exhibits greater T-cell dysfunction
We next wanted to assess differences in immune subsets within the SCLC TME at the single-cell level. To this end, we pooled immune cells across the 21 SCLC samples in our cohort (N = 16,475 cells), using immune cells from LUAD (N = 45,535 cells) and normal adjacent lung (N = 10,934 cells) as a reference (Figure S5A). We analyzed the myeloid and T-cell compartments separately to facilitate cell type annotation (Figures 5D, S5A–E, S6C–F and S7C; Table S18; STAR Methods). Our cohort is well-balanced with respect to treatment history (7 untreated, 6 treated with chemotherapy and 8 with chemotherapy and immunotherapy) (Figure S1A).
To assess how SCLC subtype impacts T-cell phenotype, we applied non-negative matrix factorization (NMF) (Chung et al., 2017; Puram et al., 2017; Stein-O’Brien et al., 2018), which excels in settings of continuous phenotypes with uncertain cluster boundaries, and identified 30 factors that facilitate cell-type annotation (Figures S6A and S6B; STAR Methods). Of these factors, 7 correspond to T-cell phenotypes: CD4+ regulatory (Tregs, factor 4), CD4+ conventional (Tconv, factors 19 and 23), CD8+ exhausted (Texh, factor 7), CD8+ memory (Tmem, factor 12), CD8+ effector (Teff, factor 28), and CD8+ gamma delta T-cells (Tgd, factor 29) (Figure S6C; STAR Methods). A parallel cluster-based phenotyping approach confirmed the annotation of discrete T-cell phenotypes (Figures S6D–F; STAR Methods). To assess whether any T-cell phenotypes are enriched by subtype, we compared factor loadings between SCLC-A and SCLC-N while adjusting for treatment and tissue. SCLC-N exhibits significantly higher Treg factor 4 and CD8+ exhausted factor 7, as well as significantly lower CD8+ effector-like factor 28 and Tgd factor 29 compared with SCLC-A (Figure S6G). A low ratio of CD8+ effector to Treg cells has been correlated with poor prognosis in cancer patients in a variety of contexts (Baras et al., 2016; Preston et al., 2013; Shang et al., 2015). The ratio of CD8+ effector to Treg factor loadings is significantly lower in SCLC-N than SCLC-A (p = 0.001; Figure 5E; STAR Methods) and is robust to the number of factors (Figure S6H). This measure of immunosuppression is consistent with a parallel cluster-based CD8+ effector/Treg ratio (p = 0.001; Figure S6I; STAR Methods).
We sought to validate these findings by imaging the independent SCLC cohort (N = 35 passing quality control). Given the relatively low T-cell representation in SCLC (mean 1.7% of cells ± s.d. 4.2% across samples, estimated by MIBI-TOF), we chose to use Vectra rather than MIBI imaging to assess T-cell abundance, as Vectra has (1) a substantially larger FoV, (2) greater sensitivity for FOXP3 staining, and (3) access to more treatment-naive tumors that pass quality control. As a proxy for SCLC subtype, we divided samples according to NEUROD1 positivity in IHC due to the near absence of any ASCL1− samples in this cohort. We found a similarly reduced ratio of CD8+ T-cells to Tregs in NEUROD1+ samples (p = 0.009; Figures 5F and 5G; Table S14; STAR Methods). Our findings identify compositional differences between SCLC-A and SCLC-N T-cell populations, including relative depletion of cytotoxic T-cells and increase in Tregs in SCLC-N.
Populations resembling fibrosis-associated macrophages are enriched in SCLC metastases
To examine the myeloid compartment, we reclustered these cells from SCLC samples in our scRNA-seq dataset (N = 2,951 cells), resulting in 7 monocyte/macrophage (Mono/Mφ), 4 neutrophil, and 2 dendritic clusters (Figure 6A; STAR Methods; see Figures S7A–B for mapping to the combined SCLC, LUAD, and normal lung myeloid dataset). SCLC myeloid clusters 1, 7, 9, and 12 represent a subset of THBS1+ VCAN+ Mono/Mφ cells that overexpress genes related to the extracellular matrix (ECM), including VCAN, FCN1, S100A4, S100A6, S100A8 and S100A9 (Figures 6A and S7C; Table S19; STAR Methods). This phenotype resembles monocytic myeloid-derived suppressor cells (MDSCs) in mice (Gao et al., 2012) and MDSC-like Mφ expressing THBS1+ S100 proteins in human hepatocellular carcinoma (Zhang et al., 2019b).
Figure 6: SCLC tumors are associated with a pro-fibrotic, immunosuppressive Mono/Mφ subset.

(A) UMAP of SCLC myeloid cells (N=2,951 cells) annotated by myeloid cell type (left) and clusters within the SCLC compartment only (right).
(B) Heatmap showing normalized mean expression of select markers from the IPF-associated profibrotic macrophage gene signature(Adams et al., 2020) (N=143 genes with log fold change > 0.3) per Mono/Mφ subsets. Expression is imputed using MAGIC (k=30, t=3) and scaled from 0 to 1 across clusters. Left barplot shows average z-scored gene expression across the entire gene signature per cluster. Clusters (rows) ordered by signature score.
(C) UMAP of SCLC myeloid cells showing gene signature scores for IPF-associated pro-fibrotic macrophages (left) and monocytes (right). Scores are calculated by taking the average Z-score of imputed expression of a given gene set, taken from(Adams et al., 2020).
(D) Heatmaps showing normalized mean imputed expression of IPF-associated pro-fibrotic macrophage (left) and monocytic (right) gene signatures(Adams et al., 2020) per SCLC Mono/Mφ cluster, as described in (B).
(E) Boxplot showing the proportion of pro-fibrotic Mono/Mφ in each sample of the combined LUAD and SCLC myeloid compartment (combined myeloid cluster 6, which includes SCLC clusters 1 and 7) in different histologies for all samples (N=48) and treatment-naive samples (N=23). We also denote in the overlying swarmplot which samples are matched to the same patient (Mann-Whitney test; error bars: 95% confidence interval). p-values: *<0.05, **<0.01, ***<0.001.
See also Figures S6–S7 and Tables S18–S19.
Given that clusters 1, 7, 9, and 12 belong to a Mono/Mφ subset known to secrete ECM-related proteins, we compared these to myeloid populations in idiopathic pulmonary fibrosis (IPF)(Adams et al., 2020). This subset, and clusters 1 and 7 in particular, closely resemble previously defined IPF-associated macrophage populations (Figure 6B). Cluster 1 scores highest for a profibrotic macrophage signature within IPF, and cluster 7 scores highest for a monocytic signature within IPF (Figures 6C and 6D).
Unsupervised clustering of the combined myeloid compartment of SCLC, LUAD, and normal lung identified a single cluster (‘combined cluster 6’), which is comprised of Mono/Mφ from SCLC clusters 1 and 7 (N = 514 cells from 14 SCLC samples) and from LUAD samples (N = 467 cells from 6 LUAD samples), but none from normal lung (Figures S7A–B). We found that the fraction of combined cluster 6 cells out of all Mono/Mφ cells is significantly higher in primary and metastatic SCLC samples compared to primary LUAD, whereas these cells are undetected in normal lung and metastatic LUAD (Figure 6E). The enrichment in SCLC is even more striking in untreated samples. Combined cluster 6 cells are also enriched, but not significantly, in metastatic compared to primary SCLC.
We sought to characterize the transcriptional profile of SCLC Mono/Mφ cluster 1 resembling profibrotic IPF-associated macrophages. Differential expression (Figure S7D; Table S19) identified cluster 1 as a CD14+ CD16+ (FCGR3A) CD81+ ITGAX+ CSF1R+ subpopulation that secretes specific pro-fibrotic, pro-metastatic growth factors involved in ECM deposition and remodeling(Winkler et al., 2020), including fibronectin 1 (FN1) (Park and Helfman, 2019; Wang and Hielscher, 2017), cathepsins (CTSB and CTSD) (Egeblad and Werb, 2002; Guo et al., 2002), and osteopontin (SPP1) (Giopanou et al., 2017; Pang et al., 2019). In addition, cluster 1 upregulates genes related to immune inhibition, including (1) SPP1, implicated in T-cell suppression and tumor immune evasion in colon cancer (Shurin, 2018) and NSCLC (Lin et al., 2015); (2) CD74, implicated in both immune suppression in metastatic melanoma (Figueiredo et al., 2018) and migration inhibitory factor-induced pulmonary inflammation (Takahashi et al., 2009); and (3) VSIG4, implicated in macrophage suppression (Li et al., 2017). Collectively, these findings suggest that cluster 1 is a subpopulation with a pro-fibrotic and immunosuppressive Mono/Mφ phenotype that is selectively increased in SCLC. Further functional analyses would be required to assess whether this population contributes to SCLC tumorigenesis or metastasis.
The recurrent PLCG2-high SCLC population is associated with a pro-fibrotic, immunosuppressive Mono/Mφ subpopulation and CD8+ T-cell exhaustion
We hypothesized that the subset of pro-fibrotic, immunosuppressive Mono/Mφ cells might interact with specific cancer subpopulations to facilitate progression. We found that SCLC-A is significantly correlated with Mono/Mφ clusters 2 and 12, whereas SCLC-N is significantly correlated with clusters 1 and 9 (p < 0.01 and p < 0.01; Figure 7A; STAR Methods). We asked whether these myeloid clusters are correlated with cancer phenotypes associated with SCLC-N and found that clusters 1, 7, and 9—which most closely resemble IPF-associated Mono/Mφ (Figure 6C)—are significantly correlated with EMT in SCLC cells (p < 0.001, p < 0.01 and p < 0.001, respectively; Figure 7A). Beyond canonical SCLC subtypes, we tested for associations with the recurrent PLCG2-high SCLC subpopulation and found a significant correlation with Mono/Mφ clusters 1 and 7 (p < 0.01 and p < 0.01, respectively; Figure 7A). Separately, we found that Mono/Mφ cluster 1 is enriched in samples harboring the recurrent SCLC cluster and that this enrichment is robust to sampling (p = 0.018; Figure S7E and S7F; STAR Methods). We also confirmed that the profibrotic Mono/Mφ population is significantly correlated with PLCG2 and EMT gene signatures in independent bulk RNA-seq datasets (N = 81; Figure S7G) (George et al., 2015; Rudin et al., 2012).
Figure 7: The recurrent PLCG2-high SCLC phenotype is associated with the pro-fibrotic, immunosuppressive Mono/Mφ subset and exhausted CD8+ T-cells.
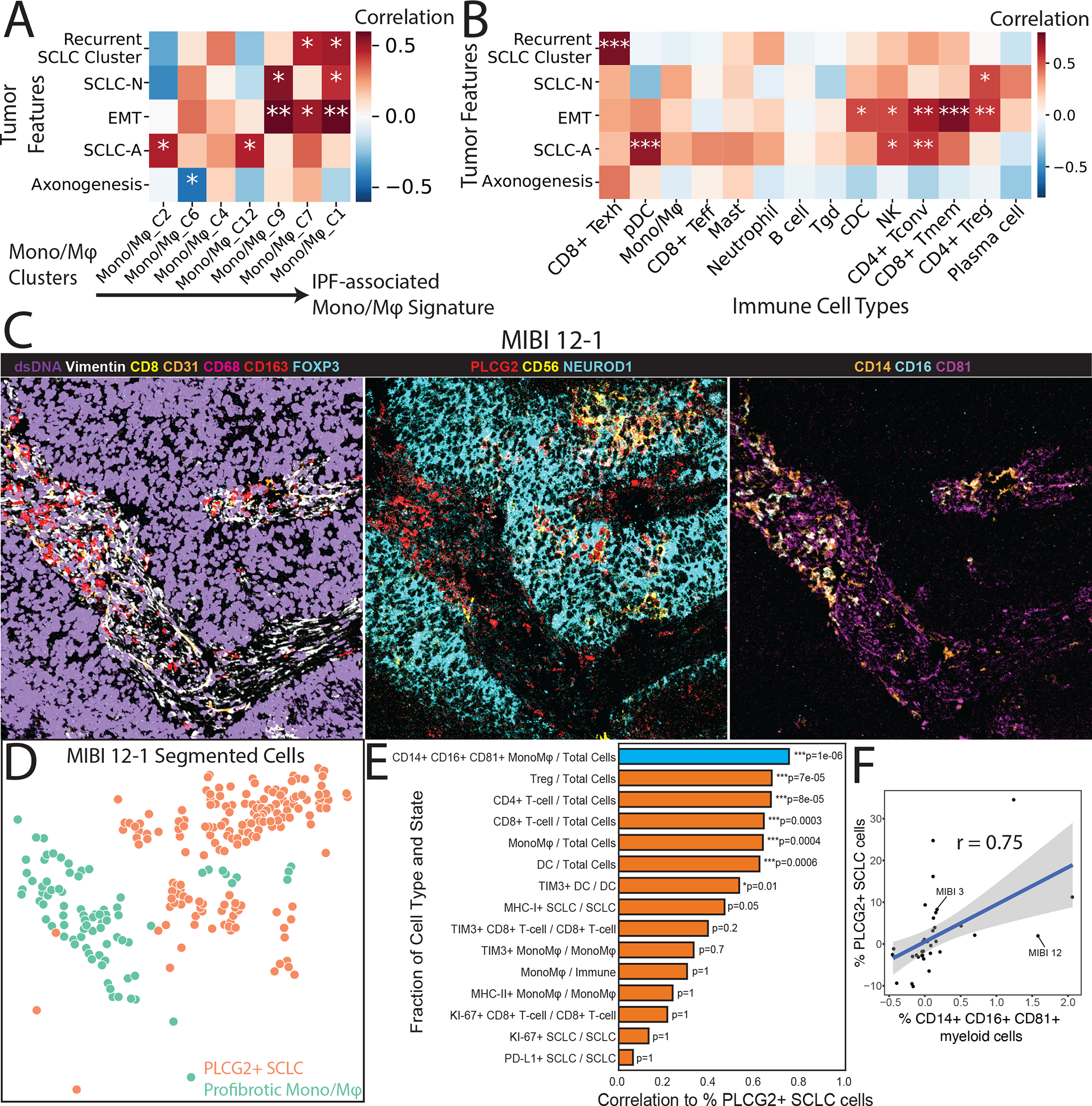
Heatmaps showing covariate-adjusted Spearman’s correlation of SCLC phenotypes with (A) Mono/Mφ subsets and (B) coarse immune cell types. Mono/Mφ in (A) are arranged along columns from low to high score for IPF-associated Mono/Mφ, as in Figure 6C. Treatment and tissue covariates were adjusted (STAR Methods). Tumor features in (A) are arranged by hierarchical clustering using Euclidean distance and average linkage. Tumor features in (B) follow the ordering in (A) for readability. p-values: *<0.05, **<0.01, ***<0.001.
(C) Color overlay of SCLC tumor MIBI 12 at FoV 1 (500 × 500 μm) showing the co-occurrence of the PLCG2-positive SCLC cells and the putative profibrotic Mono/Mφ. Left: Channels dsDNA (violet), Vimentin (white), CD8 (yellow), CD31 (orange), CD68 (red), CD163 (red), and FOXP3 (cyan) illustrate the global tumor environment structure. Middle: Channels PLCG2 (red), CD56 (yellow), and NEUROD1 (cyan) identify PLCG2+ tumor. Right: Channels CD14 (orange), CD16 (cyan), and CD81 (magenta) identify the profibrotic Mono/Mφ.
(D) FoV from the (C) now visualized with segmented cancer cells using Mesmer(Greenwald et al., 2021), represented by dots colored by PLCG2+ SCLC cells vs profibrotic Mono/Mφ.
(E) Barplot of covariate-adjusted Spearman’s correlation between the percent of PLCG2+ SCLC cells and the fraction of different cell types/states in MIBI-TOF of an independent TMA cohort. The following covariates were adjusted: SCLC subtype (NEUROD1+/−), treated vs naive, combined vs single histology and distant metastasis vs primary (Student’s t-test; STAR Methods). PLCG2+ SCLC cells had the highest correlation with CD14+ CD16+ CD81+ Mono/Mφ, shown in blue (r=0.75, N=37, Bonferroni-adjusted p = 1 × 10−6; STAR methods).
(F) Scatterplot of residuals for the fraction of CD14+ CD16+ CD81+ myeloid cells out of all myeloid cells (representing the profibrotic Mono/Mφ) vs the fraction of PLCG2+ SCLC cells out of all SCLC cells (representing the recurrent PLCG2-high SCLC phenotype). Residuals correspond to the partial correlation described in (E). Example patient MIBI 12 from Figures 7C is indicated.
We also assessed whether SCLC phenotypes are correlated with non-myeloid immune subpopulations. Notably, the PLCG2-high subpopulation is the only SCLC phenotype that correlates significantly with exhausted CD8+ T cells (p < 0.0001, Figures 7B; STAR Methods). We confirmed significant correlation of CD8+ T-cell exhaustion to profibrotic Mono/Mφ and PLCG2 in published bulk RNA-seq datasets (N = 81; Figure S7G).
Finally, we sought to validate the association of PLCG2-positive SCLC cells with the profibrotic Mono/Mφ population using MIBI-TOF on an independent cohort of SCLC tumors (N = 37). We used the specific combination of CD14, CD16, and CD81 markers to differentiate the putative profibrotic Mono/Mφ cells from other myeloid cells (STAR Methods) and found that PLCG2-positive SCLC cells coexist with this population in a number of patients. For example, in patient MIBI 12, we find a subset of NEUROD1+ SCLC cells with PLCG2 positivity adjacent to a population of profibrotic Mono/Mφ cells (Figures 7C and 7D). We see a similar association of NEUROD1+ SCLC cells with PLCG2 positivity and profibrotic Mono/Mφ in patient MIBI 3 (Figures S7H and S7I). Across the MIBI-TOF cohort, we found that the fraction of CD14+ CD16+ CD81+ myeloid cells is better correlated with the fraction of PLCG2+ SCLC cells than all other tumor and immune cell types and states (adjusted partial Spearman’s r = 0.75, Bonferroni-adjusted p = 6.71 × 10−8; Figure 7E–F; STAR Methods). Together, our findings suggest that this recurrent SCLC subpopulation may exist in an immunosuppressed TME characterized by exhausted CD8+ T-cells and a pro-fibrotic, immunosuppressive Mono/Mφ population that may be associated with EMT.
DISCUSSION
SCLC was classically considered a homogeneous disease based on its highly consistent histology, but more recent analyses have revealed distinct transcriptomic subtypes (Rudin et al., 2019) with potential prognostic and therapeutic implications (Mollaoglu et al., 2017; Saunders et al., 2015). Here, we expose a level of biological complexity that cannot be described by bulk-level subtyping, demonstrating that SCLC tumors—particularly SCLC-N—are more heterogeneous than LUAD and that SCLC has the potential for plasticity and interconversion between subtypes, particularly SCLC-A and -N.
Despite substantial clinical heterogeneity in patients with SCLC, we detected a subpopulation that was shared among tumors across subtypes, treatments, and tissue locations, pointing to a potentially universal characteristic of this malignancy. This subpopulation demonstrates a pro-metastatic, stem-like phenotype marked by profound PLCG2 overexpression. Signaling by the related phospholipase PLCG1 has been implicated in promoting metastasis in other tumor types (Kassis et al., 1999; Sala et al., 2008). Direct genetic manipulation validated that PLCG2 expression promotes pro-metastatic and stem-like features. We further found that PLCG2 expression is significantly higher in metastases and correlates with worse overall survival. While these results demonstrate the utility of PLCG2 as a single prognostic marker, tracking the recurrent PLCG2-high subpopulation in our scRNA-seq data demonstrated an even greater hazard ratio than PLCG2 expression alone, suggesting that additional factors determine the full phenotype of the recurrent cluster. Even though the recurrent PLCG2-high SCLC cluster constitutes a minor fraction of the malignant cells comprising the tumors under study, this small subpopulation has a strong correlation with survival, illustrating its prognostic importance and the value of single-cell analysis.
Analysis of the TME in our data confirmed an immune-cold phenotype in SCLC, particularly in SCLC-N, notable for more Tregs and fewer CD8+ T-cells than SCLC-A. Analysis of the myeloid milieu revealed that SCLC is enriched for a subset of Mono/Mφ with an immunosuppressive phenotype resembling IPF-associated macrophages. One specific Mono/Mφ cluster displayed a pro-fibrotic, immunosuppressive phenotype. Among the SCLC cohort, we identified a constellation of immune and tumor phenotypes (exhausted CD8+ T-cells and pro-fibrotic Mono/Mφ) associated with SCLC-N, EMT, and the recurrent PLCG2-high phenotype. These associations raise the possibility that CD8+ T-cells in the TME of the PLCG2+ SCLC subpopulation are impeded by immunosuppressive Mono/Mφ cells. This same Mono/Mφ cluster may also provide the fibrotic substrate that facilitates mobility of the pro-metastatic PLCG2-high subset of cancer cells. Further investigation into these immune populations may reveal novel tumor-immune interactions that enable metastasis.
Our findings were facilitated by unbiased, high-throughput profiling of SCLC phenotypes and the surrounding immune microenvironment across multiple modalities (scRNA-seq, MIBI, Vectra, FACS and published bulk RNA-seq) and independent cohorts; yet there are important limitations. A portion of our samples come from small biopsies, which may not fully represent the biology of the entire tumor. Additionally, our cohort includes a diversity of treatment histories, tissue locations and SCLC subtypes. Nonetheless, we detected the consistent presence of tumoral and immune subpopulations with potential implications for SCLC metastasis. Further analyses of expanded cohorts with more tightly restricted clinical variables will contribute to further deciphering the role of intratumoral heterogeneity in SCLC.
The picture of SCLC that emerges from our atlas is that a spectrum of subtypes and a PLCG2-high recurrent population enlist diverse gene programs to define pronounced heterogeneity and facilitate metastasis in a profoundly immunosuppressed TME. Our dataset has potential implications for the design of novel targeted therapies and immunotherapeutic approaches.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information should be directed to and will be addressed by the Lead Contact, Charles Rudin (rudinc@mskcc.org).
Materials Availability
Requests for resources should be directed to and will be addressed by the Lead Contact.
Data and Code Availability
Software and tools used for the enclosed data analysis will be provided open source at http://github.com/dpeerlab. In collaboration with the NIH-funded HTAN Data Coordinating Center (U24), single-cell analysis at time of publication will be made available as an interactive, online platform for independent visualization and analysis. MIBI-TOF data will be made available at https://mskcc.ionpath.com/tracker.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient cohorts
Patients with LUAD or SCLC undergoing a surgical resection or tissue biopsy at Memorial Sloan Kettering Cancer Center (MSKCC) were identified and biospecimens collected prospectively from 2017 to 2019. All patients from whom biospecimens were obtained provided informed consent through an Institutional Review Board-approved biospecimen collection and analysis protocol. Clinical, demographic, pathologic, and molecular data using MSK-IMPACT were identified by retrospective review of the electronic medical record. Single cell RNA-seq were performed on 21 clinical specimens with SCLC, 24 clinical specimens with lung adenocarcinoma, and 4 tumor-adjacent normal lung tissue samples (Table S1). IHC for subtyping TFs was performed on the SCLC samples as previously described (Baine et al., 2020) and reviewed by a pathologist at MSKCC.
MIBI and Vectra analyses were performed on a TMA constructed with additional independent SCLC cohorts. IHC was also performed on the same TMA for benchmarking. 26 cases were amenable for IHC evaluation and 12 for Vectra analysis (Table S14). For TMA construction, archival formalin-fixed, paraffin-embedded (FFPE) samples were identified and collected retrospectively from SCLC and NSCLC cases between 2007 and 2017. Human kidney samples were used as a positive control in both TMAs.
Flow cytometry analysis of CD45 positive cells was performed on an independent cohort of 11 SCLC patients (Table S17) collected prospectively from 2017 to 2019.
Cell lines
H82 (male), SHP-77 (male), H526 (male), H446 (male) and DMS-114 (male) were purchased from ATCC, authenticated through the STR characterization method and regularly tested for Mycoplasma. Both cell lines were cultured in RPMI 1640 supplemented with 10% FBS and cultured according to ATCC guidelines.
METHOD DETAILS
Sample handling
Clinical samples were received in the lab immediately after extraction (Median delivery time±SEM, 0.75±0.72 hours) and processed rapidly (Median±SEM processing time from delivery until 10x protocol started, 1.75±0.27 hours) to ensure high sample viability and quality for single cell RNA-seq.
Sample processing: Resection and small biopsies dissociation
Upon delivery to the lab, samples were mechanically/enzymatically dissociated using the tumor dissociation kit (#130–095-929, Miltenyi) and the GentleMACS Octo Dissociator with Heaters (Miltenyi, # 130–096-427). Resection samples were chopped and added to 7.5 mL of enzyme mix in the GentleMACS tube, while core needle biopsies/fine needle aspiration samples were added to 2.5 mL of enzyme mix in the GentleMACS tube. After 15–30 minutes dissociation, depending on sample size and consistency, bigger size samples were filtered with MACS SmartStrainers (70 μm) (Miltenyi, #130–098-462) into 50 mL tubes, and smaller samples were filtered with 35 uM stainer cap FACS tube (Corning # 352235). Then, samples were centrifuged (800g, 1 minute) and supernatant was discarded. Pelleted cells were then stained as indicated below.
Sample processing: Pleural effusions cell collection
Upon delivery to the lab, samples were centrifuged at 800g, 10 minutes. The supernatant was discarded, and the pellet resuspended in 40 mL of 1X PBS containing 2.5% FBS. Next, 15 mL of Ficoll Paque (GE healthcare, #17–1440-03) was added per tube to two SepMate tubes (STEMCELL Technologies, #85450). Then, 20 mL of pleural fluid was added onto each SepMate tube, slowly, drop by drop, to avoid mixing of the sample and Ficoll, followed by centrifugation at 1200g for 20 minutes at RT. After centrifugation, 15 mL of the upper fluid layer were discarded, and the remaining 5 mL above the dividing plastic surface in the tube were collected, resuspending the cells located in it. Finally, cells were pelleted by centrifugation at 800g, 2 minutes and stained with anti-CD45 antibody and calcein dye as indicated below.
Sample processing: staining for sorting and CD45+ composition analyses
Cell pellet was resuspended in 200–3000 uL of Red Blood Cell Lysis Solution (ACK lysis buffer), depending on the pellet size. After incubation for 2 minutes at room temperature the ACK buffer was diluted 10-times with 1X PBS containing 2.5% FBS and pelleted again. Cell pellet was resuspended in 100 uL of 1X PBS + 2.5% FBS, mixed with 5 uL of Human TruStain FcX (Biolegend #422302), 3 uL of PE CD45 antibody (Biolegend # 368510 and 0.1 uL of calcein (1μg/μL, Calcein (Biolegend #425201)), and left for 15 minutes on ice. Stained samples were washed twice with 2 ml of 1X PBS + 2.5% FBS, and finally resuspended in the same buffer supplemented with DAPI dye. Using BD FACSAria (BD Biosciences) or Sony MA900 (Sony) flow cytometers, cells were sorted on DAPI-, Calcein+ (FITC+) to select for live cells. In addition, we sorted CD45+ (immune cells) and CD45− (cell population enriched in cancer cells) populations into separate tubes, and mixed them back in an artificial ratio to balance the compartmental representation (1:5–1:10 ratio, depending on cell availability). To define the percentage of immune cells in each sample, we registered the fraction of CD45+ and CD452210032 in the live cell (DAPI-, Calcein+) population.
Sample processing: single-cell RNA-seq
FACS-sorted cells were subjected to scRNA-seq protocol using Chromium (10X genomics) instrument and Single Cell 3’ Reagent Kit (v3). Each sample, containing approximately 3000–8000 cells was encapsulated and barcoded following the manual (CG000183 Rev B). The viability of samples varied between 58–98% (median 80%), as confirmed with 0.2% (w/v) Trypan Blue staining. The final sequencing libraries were double-size purified (0.6–0.8X) with SPRI beads and sequenced on Illumina Nova-Seq platform (R1 – 26 cycles, i7 – 8 cycles, R2 – 70 cycles or higher). On average, 3,330 cells per clinical sample (N = 62) were sequenced at a depth of ~42.000 reads per cell (195 million reads per sample). The unique mapping was high, between 79–88%, and a median number of unique transcripts per cell was 4.393.
PLCG2 overexpression/CRISPR knock out
H82 (male), SHP-77 (male), H526 (male), H446 (male) and DMS-114 (male) were purchased from ATCC, authenticated through the STR characterization method and regularly tested for Mycoplasma. Both cell lines were cultured in RPMI 1640 supplemented with 10% FBS and cultured according to ATCC guidelines.
Lentiviral plasmids were used for PLCG2 overexpression (GeneCopoeia, #EX-A8643-Lv201) and for PLCG2 CRISPR knock out (Sigma-Aldrich, #HSPD0000031727). Lentiviral particles were produced by standard protocols, transfecting HEK293T cells using JetPrime reagent (Polyplus, #114–15) and concentrated viruses using Lenti-X Concentrator (Takara Bio, #631232) and SCLC cells were transduced at high multiplicity of infection in a spin transduction protocol (Centrifugation of cells at 800×g, 30 minutes with 8ug/mL polybrene).
Immunoblotting
Protein extraction was performed by pelleting cells and resuspending in cold RIPA buffer (ThermoFisher, #89901) supplemented with phosphatase/protease inhibitors (ThermoFisher, #78446) and incubating for 1 hour on ice. Then, protein extracts were clarified at 14,000 rpm for 10 min in a refrigerated benchtop centrifuge (Eppendorf, #5340 R). Protein lysates were quantified using a micro BCA protein assay kit (Pierce, #23235) and then diluted with extraction buffer, NuPAGE® LDS sample buffer and reducing reagent (Life Technologies) prior to resolving on 4–12% Bis-Tris gradient gels. Gels were wet-transferred to 0.45 μm Immobilon-FL PVDF membrane (Millipore, #IPFL00010). All primary antibodies were incubated overnight with membranes in TBS Odyssey blocking buffer supplemented with 0.1% Tween-20 (LI-COR, #927–50000), while secondary antibodies (donkey anti-rabbit IRDye 800CW (LI-COR, #926–32213) and donkey anti-mouse IRDye 680LT (LI-COR, #926–68022) were incubated at room temperature with agitation for 1 hour in primary blocking buffer supplemented with 0.01% SDS. Membranes were dried at 37°C and protected from light before imaging (LI-COR; Odyssey Sa). Antibodies for PLCG2 (#3872, Cell Signaling Technology), Beta-catenin (#8480, Cell Signaling Technology), pSMAD1/5 (#9576, Cell Signaling Technology), SMAD1 (#6944, Cell Signaling Technology), SMAD5 (#12534, Cell Signaling Technology), N-cadherin (#14215, Cell Signaling Technology), Vimentin (#5741, Cell Signaling Technology), Twist (#46702, Cell Signaling Technology), ZEB (#70512, Cell Signaling Technology), NFIB (#ab186738, Abcam), SOX2 (#3579, Cell Signaling Technology), Nanog (#4903, Cell Signaling Technology) and actin (#3700, Cell Signaling Technology) were used. Immunohistochemistry was performed as previously described16, using antibodies for ASCL1 (#556604, BD), NEUROD1 (#ab205300, Abcam), POU2F3 (Santa Cruz, #6D1) and PLCG2 (#HPA020100, Sigma-Aldrich).
In vitro tumorigenic surrogate analyses and reporter assay
Surrogate assays were performed as indicated in (Quintanal-Villalonga et al., 2019). For growth curves, multiple 96-well plates were seeded with 3,000 cells/well and cell density was quantified using a luminescent assay (CellTiter-Glo 2.0 assay, #G9242, Promega). Cell proliferation was determined by normalizing to the day 0 cell density measurement. For agar assays, the number of colonies was counted after a period of 2 weeks to 1 month after seeding. Wnt signaling reporter assay (#60500, BPS Biosciences) was performed following manufacturer instructions with a Firefly/Renilla luciferase assay kit (#SCT152, Sigma Millipore). Three biological replicates (independent experiments) were performed for each assay. For each biological replicate, three technical replicates per condition were carried out.
In vitro metastasis surrogate analyses
Migration and invasion assays were performed using Cultrex BME Cell invasion assay kit (#3455–096-K, R&D Systems), following manufacturer’s instructions. 50.000 cells were seeded per chamber on day 0 on 0% FBS media, with 10% FBS media in the bottom well, and results were collected on day 4 using a luminescent assay (CellTiter-Glo 2.0 assay, #G9242, Promega). Each experiment was replicated a minimum of three times in independent assays, and the experimental condition was normalized to control condition, which was assigned a value of 1. Analysis of invasion/migration capacity was performed by averaging values in the independent replicates and by performing a two-tailed Student’s t-test to assess for statistical significance.
In vivo intracardiac injections
All mice were kept in specific pathogen-free animal facilities at Memorial Sloan Kettering Cancer Center (MSK), and procedures were performed in accord with the guidelines of MSK Institutional Animal Care and Use Committee under an approved protocol. A total of 0.5 million cells were injected in the left ventricle of anesthetized 6–8 week old athymic female mice (Envigo). Immediately after surgery, and then weekly, animals were injected with D-luciferin (# LUCK-5G, GoldBiotechnology) at 15 mg/Kg retro-orbitally and photonic emission was imaged using the In Vivo Imaging System (IVIS, Perkin Elmer) with a collection time of 1 minute. Tumor bioluminescence was quantified by integrating the photonic flux (photons per second) through a region encircling each tumor as determined by the LIVING IMAGES software package per manufacturer’s instructions (Perkin Elmer). At day 31 after injection, we counted the number of mice with or without metastasis and represented the percentages of each group for conditions under assay, for each cell line. A Chi-Square test was performed to measure statistical significance of the differences in percentage for each cell line tested.
Tissue microarray construction for imaging
Tissue microarrays (TMAs) were constructed in the pathology core lab of Precision Pathology Center using an automated TMA Grand Master (3DHistech) and TMA Control software (Version 2.4). TMAs were designed and constructed using archival paraffin-embedded lung cancer tissue samples (N=54) retrieved from the files of the Department of Pathology, Memorial Sloan Kettering Cancer, New York, NY. Histology sections were reviewed by a pathologist and most representative areas to be cored were selected and marked on the H&E slides. To obtain better representation of the tumor, 1 mm diameter donor cores were sampled from three tumor regions.
MIBI-TOF imaging
Antibody conjugation
BSA and protein carrier free antibodies were obtained and optimized using standard immunohistochemical staining on the Leica Bond RX automated research stainer with DAB detection kit (Leica Bond Polymer Refine Detection DS9800). Using 4 μm formalin-fixed, paraffin-embedded multi-tissue control sections and serial antibody titrations, the optimal antibody concentration was determined by MSKCC pathologist (TH). All primary antibodies (except Foxp3) were conjugated using the Ionpath MIBItag kit per manufacturer’s instructions. Conjugated antibody was diluted in Candor PBS Antibody Stabilizer (Candor Bioscience GmbH, #131125) to 0.5mg/ml final concentration and stored long-term at 4°C. Prior to the assay, post conjugation antibody concentrations were determined by testing serial dilutions on the MIBIscope to obtain equivalency with standard DAB sensitivity. For FOXP3 protein detection, tissue staining was performed using a Foxp3-AF488 primary antibody with detection using anti-AF488 secondary antibody conjugated with Nd146.
A summary of antibodies, MIBItag, and concentrations can be found in Table S15.
IHC Staining
Tissue sections (4 μm) were cut from FFPE tissue blocks of the non-small cell carcinoma tissue microarray (TMA) or multi-tissue control block using a Leica microtome (Leica, RM2255), mounted on Ionpath slides (Ionpath, #567001) for MIBI SCOPE staining. Slide-tissue sections were baked at 62°C for 1 hour. Slide sections were deparaffinized with xylene(2x) (Sigma-Aldrich, #534056–4L) and then rehydrated with successive incubated with ethanol 100% (2x) (Sigma-Aldrich, # R8382), 95% (2x) (Sigma-Aldrich, # R3404), 70% (1x) (Sigma-Aldrich, # R3154), and distilled water. The sections were then immersed in epitope retrieval buffer (Target Retrieval Solution, pH 9, DAKO Agilent, Santa Clara, CA) and incubated at 97°C for 40 min and cooled down to 65°C using Lab vision PT module (Thermofisher Scientific, Waltham, MA). Slides were washed with a wash buffer made with TBS (pH 7.4) (Ionpath, # 567005). 5%(v/v) Donkey serum (Sigma-Aldrich, #D9663) with TBS wash buffer was applied to block the nonspecific staining for 30min. A metal-conjugated antibody cocktail including the Foxp3-AF488 antibody was prepared in 5% (v/v) donkey serum with TBS wash buffer and filtered using centrifugal filter, 0.1 μm PVDF membrane (Ultrafree-MC, Merck Millipore, Tullagreen Carrigtowhill, Ireland). The antibody cocktail was incubated overnight at 4°C in humid chamber. After overnight incubation, slides were washed with TBS wash buffer three times, 2min for each. Nd146 conjugated anti-AF488 secondary antibody was prepared in 5% (v/v) donkey serum with TBS wash buffer and filtered using centrifugal filter, 0.1 μm PVDF membrane as before, applied on the slide and incubated 2 hours at RT. Slides were then washed twice 5 min in TBS wash buffer and fixed for 5 min in diluted 2% glutaraldehyde solution (Electron Microscopy Sciences, Hatfield, PA) in PBS-low barium(Ionpath, #567004). Slides were then rinsed briefly in TBS wash buffer and then dehydrated with successive washes of Tris 0.1 M (pH 8.5), (3x) (Ionpath, #567003 ), distilled water (2x), and ethanol 70% (1x), 80%(1x), 95% (2x), 100% (2x). Slides were immediately dried in a vacuum chamber for at least 1 h prior to imaging.
Imaging Acquired with MIBI SCOPE
Quantitative imaging was performed using MIBI SCOPE (Ionpath) with the following settings: 2048×2048 resolution, 1ms Dowell time, “fine” mode for imaging acquisition, and 800μm FOV size.
Vectra Imaging
Multiplex Tissue Staining and Imaging
Primary antibody staining conditions were optimized using standard immunohistochemical staining on the Leica Bond RX automated research stainer with DAB detection (Leica Bond Polymer Refine Detection DS9800). Using 4 μm formalin-fixed, paraffin-embedded tissue sections and serial antibody titrations, the optimal antibody concentration was determined by a pathologist (TH) followed by transition to a seven-color multiplex assay with equivalency. Multiplex assay antibodies and conditions are described in the following table:
| Antigen | Antibody Clone | Manufacturer | Concentration | Detection Dye (cycle) |
|---|---|---|---|---|
| CD8 | C8/114B | Cell Signaling | 0.125 μg/ml | Opal 520 (1) |
| FoxP3 | 236A/E7 | Biocare | μg/ml | Opal 540 (2) |
| CTLA4 | BSB88 | BioSB | 1.34 μg/ml | Opal 570 (3) |
| Perforin | 5B10 | Leica | 0.267 μg/ml | Opal 620 (4) |
| CD56 | MRQ-42 | Cell Marque | 0.110 μg/ml | Opal 650 (5) |
| INSM1 | A-8 | Santa Cruz | 0.800 μg/ml | Opal 690 (6) |
| CK7 | OV-TL-12/30 | Abcam | 0.250 μg/ml | Opal 690 (6) |
Seven-color multiplex imaging assay
4 μm FFPE tissue sections were baked for 3 hrs. at 62 degrees Celsius in vertical slide orientation with subsequent deparaffinization performed on the Leica Bond RX followed by 30 minutes of antigen retrieval with Leica Bond ER2 followed by 6 sequential cycles of staining with each round including a 30-minute combined block and primary antibody incubation (PerkinElmer antibody diluent/block ARD1001).
Detection of all primary antibodies was performed using a goat anti-mouse Poly HRP secondary antibody or goat anti-rabbit Poly HRP secondary antibody (Invitrogen B40961/2; 10-minute incubation). The HRP-conjugated secondary antibody polymer was detected using fluorescent tyramide signal amplification using Opal dyes 520, 540, 570, 620, 650 and 690 (Akoya FP1487001KT, FP1494001KT, FP1488001KT, FP1495001KT, FP1496001KT, FP1497001KT). The covalent tyramide reaction was followed by heat induced stripping of the primary/secondary antibody complex using Perkin Elmer AR9 buffer (AR900250ML) and Leica Bond ER2 (90% ER2 and 10% AR9) at 100 degrees Celsius for 20 minutes preceding the next cycle. After 6 sequential rounds of staining, sections were stained with Hoechst (Invitrogen 33342) to visualize nuclei and mounted with ProLong Gold antifade reagent mounting medium (Invitrogen P36930).
Multispectral imaging, spectral unmixing and cell segmentation
Seven color multiplex-stained slides were imaged using the Vectra Multispectral Imaging System version 3 (Perkin Elmer). Scanning was performed at 20X (200X final magnification). Filter cubes used for multispectral imaging were DAPI, FITC, Cy3, Texas Red and Cy5. A spectral library containing the emitted spectral peaks of the fluorophores in this study was created using the Vectra image analysis software (Perkin Elmer). Using multispectral images from single-stained slides for each marker, the spectral library was used to separate each multispectral cube into individual components (spectral unmixing) allowing for identification of the seven marker channels of interest using Inform 2.4 image analysis software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis of in vitro and in vivo experiments
Quantitative analyses of in vitro and in vivo experiments include two-tailed Student’s t test with unequal variance. When possible, all experiments were reproduced a minimum of three times (independent biological replicates). Sample sizes were in line with previous literature and our laboratories’ standard practices. Statistical parameters can be found in the figure legends and/or METHOD DETAILS section above.
Pre-processing of scRNA-seq data
Pre-processing steps of scRNA-seq are illustrated in Figure S1C and detailed as follows. Fastq files from patient samples were individually processed using the SEQC pipeline (Azizi et al., 2018) based on the hg38 human genome reference and default parameters for the 10x single-cell 3’ library. The SEQC pipeline performs read alignment, multi-mapping read resolution, as well as cell barcode and UMI correction to generate a count matrix (cells × genes). The pipeline then performs the following initial cell filtering steps: 1) true cells are distinguished from empty droplets based on the cumulative distribution of total molecule counts; 2) cells with a high fraction of mitochondrial molecules are filtered (> 20%); and 3) cells with low library complexity are filtered (cells that express very few unique genes). In addition, we perform additional filtering of empty droplets using the CB2 package with parameter “lower” set at 100 to estimate the background distribution of ambient RNA and an FDR threshold of 0.01 for calling real cells (Ni et al., 2020). Putative doublets were removed using the DoubletDetection package (DOI 10.5281/zenodo.2658729). Genes that were expressed in more than 10 cells were retained for further analysis. Combining samples in the entire cohort of samples from SCLC, LUAD, and normal adjacent lung yielded a filtered count matrix of 155,098 cells by 23,628 genes, with a median of 5,654 molecules per cell and a median of 3,041 cells per sample. The count matrix was then normalized by library size, scaled by median library size, and log2-transformed with a pseudocount of 0.1 for analysis of the combined dataset. Principal component analysis (PCA) was performed with the top 50 principal components (PCs) retained with 42% variance explained.
Batch correction of the combined dataset
We performed batch correction in the combined dataset of clinical samples--including SCLC, LUAD, and normal adjacent lung--using fastMNN with cosine distance applied to the log2 transform of the library-size normalized count matrix with pseudocount of 1, reduced to the top 50 PCs. We favored fastMNN due to the ability to perform hierarchical merging among samples first from the same patient, then from the same histology, with samples containing a greater number of cells merged first. To evaluate the effect of batch correction, we used an entropy-based measure that quantifies how much normalized expression mixes across patients (Azizi et al., 2018). We constructed a k-nearest neighbors graph (k=30) from the normalized dataset using Euclidean distance and computed the fraction of cells qT derived from each tumor sample T in the neighborhood of each cell j. We then calculated the Shannon entropy Hj of sample frequencies within each cell’s neighborhood as:
High entropy indicates that the most similar cells come from a well-mixed set of tumors, whereas low entropy indicates that most similar cells derive from the same tumor. This sample entropy was projected on the UMAP (Figure S1B). As expected, immune cells generally had the highest entropy consistent with shared phenotypes across tumors, whereas SCLC and LUAD cells had the lowest entropy consistent with increased inter-tumoral diversity. These results indicate a good trade-off that corrects for batch effect while maintaining true biological heterogeneity. Importantly, we did not perform downstream batch correction in subsetted compartments of coarse cell types out of concern of over-correcting tumor phenotypes.
Gene imputation
Given the sparse nature of scRNA-seq that arises from gene dropout, we used gene imputation using MAGIC (knn = 30, t=3)(Dijk et al., 2018) when performing knnDREMI calculations (described in section “Identifying the PLCG2-related gene module”) and for visualizing gene expression on both UMAPs and heatmaps (Figures 1G, S1G, S2B, S2C, S3E).
Visualization of single-cell RNA-seq
Visualization of different cell type compartments
To visualize single cells of the global atlas as well as epithelial, SCLC, immune, T-cell, and myeloid subsets, we used UMAP projections(McInnes et al., 2018) to generate lower dimensional representations using knn = 15, min_dist = 0.3–0.5, and init_pos = ‘paga’ (Figure 1A–C, 1G–H, 3C, 5D, 6A, 6C, S1B, S1G, S2B, S3A, S5A–B, S7A–B). The initialization for the UMAPs were based on partition-based graph abstraction (PAGA) implemented in the scanpy package using Phenograph clusters (except for cell type annotation in the T-cell compartment).
Visualizing phenotypic changes along the SCLC-A vs SCLC-N spectrum
For better visualization of cancer cells along the SCLC-A vs SCLC-N spectrum (Figure 2B), we excluded SCLC-P cells and renormalized the Markov absorption probabilities of SCLC-A and SCLC-N (described in section “Subtype classification and deconvolution in the SCLC tumor compartment”). We ordered the cells by these probabilities from SCLC-A to SCLC-N along the X-axis and colored the corresponding subtype probability on the horizontal color bar. We rescaled marker expression or pathway scores from 0 to 1 along the Y-axis and plotted this value for each cell (grey dots) as subtype probability along the X-axis increasing from SCLC-A to SCLC-N. We calculated pathway scores as the average of Z-scored expression of genes belonging to a pathway. The average trend for each gene marker or pathway was computed by a generalized additive model of 8 splines with spline order 3 using the python package pyGAM (DOI 10.5281/zenodo.1208723).
Differential expression
Differential expression in bulk reference datasets
To facilitate annotation of our single cells by tumor histology and SCLC subtype, we used available reference RNA-sequencing of bulk tumors. These datasets included SCLC subtypes (SCLC-A, SCLC-N, SCLC-P, and SCLC-Y from George et al. (George et al., 2015) and Rudin et al. (Rudin et al., 2012). We performed differential expression using limma(Ritchie et al., 2015) based on log transcripts per million (TPM) counts (Tables S20–23). We considered only DEGs with absolute value of log2 fold-change > 1.5 and Benjamini-Hochberg adjusted p-values < 0.05.
Differential expression of tumor and immune subsets in scRNA-seq
We performed differential expression for the following comparisons: 1) each SCLC subtype vs rest (Tables S4–6), 2) SCLC-A vs SCLC-N cells (Table S3), and 3) each unsupervised cluster vs rest (Tables S10 and S24). All differential expression was performed using MAST (version 1.8.2) (Finak et al., 2015), which provides a flexible framework for fitting a hierarchical generalized linear model to the expression data. We used a regression model that adjusts not only for cellular detection rate (cngeneson, or number of genes detected per sample), but also tissue status (primary vs LN vs distant metastasis) and treatment status (naive vs most recently chemo-treated vs most recently immunotherapy-treated):
where condition represents the condition of interest and Yi is the expression level of gene i in cells in cluster j, transformed by natural logarithm with a pseudocount of 1. To homogenize cell sampling per batch, we downsampled such that the cell complexity (i.e. the number of genes per cell) was evenly matched across groups. In particular, we partitioned cells from each cluster into 10 equally-sized bins based on cell complexity and subsampled from each bin to match cell complexity distribution across samples. We downsampled to at most k cells per sample, where k is the median sample size. We verified that the mean expression levels from the full and downsampled datasets were strongly correlated. We considered genes to be significantly differentially expressed for Bonferroni-adjusted p-value < 0.05 and absolute log fold-change > 0.3.
Filtering ambient RNA from differential expression in scRNA-seq
Following the approach first demonstrated in (Smillie et al., 2019) to remove candidate DEGs that likely represent ambient RNA, we follow a stepwise, regression-based approach that identifies likely contaminant genes per cell type (Smillie et al., 2019). For each general cell type (ingroup), expression of each gene is plotted against the expression of that gene in all other cells (outgroup). An initial Loess regression is fitted to the entire dataset. Genes are then binned by expression (number of bins = 25), and the 50 genes with the most negative residuals per bin are then assessed. A second linear regression is fit to genes with negative residuals. Finally, those genes with residuals for the second regression that are < 2 are considered ambient RNA. Likely ambient RNA is colored in red, with known specific markers of other cell types highlighted in red boxes. For instance, PTPRC detected in epithelial cells is highly likely to be contaminant RNA from lysed immune cells. We excluded any genes representing ambient RNA from DEGs per cluster or SCLC subtype.
Identifying enriched gene pathways in single-cell data
Enriched gene pathways were identified using pre-ranked GSEA, as implemented by the R package fGSEA (Korotkevich et al., 2016) using 10,000 permutations. Gene ranks were calculated using −log(p-value)*log fold change based on MAST(Finak et al., 2015) differential expression (described in section “Differential expression of tumor and immune subsets in scRNA-seq”). To assess enriched pathways in SCLC subtypes and clusters, we used a curated set of pathways from MSigDB v 7.1 (Data S1) (Subramanian et al., 2005). To assess enriched pathways in myeloid clusters, we used IPF-related gene sets (see Data S2) in addition to HALLMARK and KEGG subset of Canonical Pathways in MSigDB v 7.1 (Subramanian et al., 2005). Using the same cutoff as in the original GSEA paper, we considered pathways with Benjamini-Hochberg adjusted p-values < 0.1 to be significant.
Cell type annotation
Coarse cell type identification and subsetting
We used a hierarchical strategy to identify cell types, starting at coarse resolution (epithelial versus immune) and then fine resolution (basal versus NE cell). At the global level, we first performed unsupervised clustering on the batch-corrected count matrix (described in section “Batch correction of the combined dataset”) to identify 58 clusters. Similar to other single-cell studies in lung(Travaglini et al., 2019), we annotated clusters by coarse cell type based on expression of tissue compartment markers (for example, PTPRC for immune cells, EPCAM for epithelial cells, COL1A1 for fibroblasts, and CLDN5 for endothelial cells) (Figure 1A, Data S2). We subsetted the data based on these coarse cell types for downstream analysis (Figure S1C).
Cell type annotation of the epithelial compartment
We subsetted the EPCAM+ epithelial cells (N=64,301 cells). We projected normalized counts without log transform onto the first 45 PCs selected by detecting the knee-point (minimum radius of curvature in eigenvalues), corresponding to 85.3% variance explained. We identified 38 Phenograph clusters. We considered a cell cluster to be neuroendocrine based on expression of canonical markers (CHGA, CHGB, NCAM1, SYP, ASCL1, ASCL2, BEX1, also see Data S2). Using this classification, we further divided the epithelial compartment into a neuroendocrine subset restricted to SCLC tumors (N=54,523 cells) and a non-neuroendocrine subset (N=9,778 cells). As expected, samples with the highest abundance of cells expressing neuroendocrine markers were those tumors identified as SCLC tumors on pathology.
Cell type annotation of the non-neuroendocrine epithelial compartment
We subsetted the non-neuroendocrine epithelial cells. We projected the normalized counts without log transform onto the first 30 PCs selected by knee-point detection, corresponding to 90.5% variance explained. We then curated multiple recent publications for specific canonical markers for a range of cell types, including epithelial lineages in the lung (Laughney et al., 2020; Montoro et al., 2018; Travaglini et al., 2019), and liver (Aizarani et al., 2019) (see Data S2). Using these cell type-specific gene sets, we first transformed the data by z-score and calculated the average expression of each curated gene set per cell type subtracted from the average expression of a reference set of genes using the score_genes function in scanpy. The subsequent cell type scores were transformed again by z-score, with cell types ultimately annotated by maximum cell type score (Figure 1A). These cell types include alveolar epithelial cells type 1 (AE1) and type 2 (AE2) cells, basal cells, ciliated cells, club cells, hepatocytes, ionocytes, mucinous cells, and tuft cells.
Cancer cell identification using single-cell SNV and CNV calls
We identify cancer cells in the epithelial compartment by applying several criteria:
First, we ensure that all putative cancer subpopulations cluster separately from cells derived from normal lung samples.
Additionally, we identify cancer cells harboring genomic mutations including single nucleotide variants (SNVs) and copy number variants (CNVs) based on matched bulk DNA-sequencing from MSK IMPACT, downloaded from cBioPortal.
To account for the sparsity of scRNA-seq, as well as confounding gene fragments from lysed cancer cells that contaminate normal single-cell droplets, we consider cell clusters to be cancer if they are enriched in reads calling SNVs compared to immune and mesenchymal cells as a negative control, based on Fisher’s p-value adjusted by Bonferroni calculation for multiplicity with a threshold of < 0.05. We reasoned that any cluster with a significant enrichment of variant alleles above a null distribution of normal immune and mesenchymal cells likely represents a cluster of cancer cells.
We also identify CNVs at the single-cell level using InferCNV (Anoop P. Patel,*1, 2, 3, 4 Itay Tirosh,*3 John J. Trombetta, 3 Alex K. Shalek, 3 Shawn M. Gillespie, 2, 3, 4 Hiroaki Wakimoto, 1 Daniel P. Cahill, 1 Brian V. Nahed, 1 William T. Curry, 1 Robert L. Martuza, 1 David N. Louis, 2 Orit Rozenblatt-Rosen, 3 Mari and Human, 2014) using a sliding window of 200 genes, with a diploid mean and standard deviation determined by available normal adjacent tumor samples. We considered any deviations from the diploid mean of at least two standard deviations to be a copy number change.
We noted that the fraction of the genome altered by CNV followed a bimodal distribution across cells, consistent with normal and malignant cells having low and high CNV burden, respectively. We noted that CNV burden was higher in SCLC tumors compared to LUAD (Figures S1F), consistent with SCLC having a higher tumor mutation burden (Yarchoan et al., 2019). We use two different measures of CNV burden: fraction of the genome changed and Pearson’s correlation between single-cell and bulk CNV profiles, both of which have a bimodal distribution in tumor samples, with a lower peak corresponding to normal stromal cells and a higher peak corresponding to mutated cancer cells. On the other hand, the normal samples have a unimodal distribution that coincides with the normal stromal peak in tumor samples. Based on the bimodal distribution, we identify cancer cells using a threshold of >10% fraction of genome altered and Pearson’s correlation to bulk CNV profile rho >0.2. Of the epithelial cell compartment (N=64,301 cells), clusters that were identified as both tumor and neuroendocrine were therefore subsetted as the SCLC tumor compartment (N=54,523 cells). Epithelial cell clusters identified as tumor but not neuroendocrine (N=7,635 cells) were considered LUAD. These tumor-type calls were consistent with the histology read of the tumor by clinical pathology.
Cell type annotation in the immune compartment
We subsetted the CD45+ immune cells from all SCLC patients (N=16,098 cells). We projected the log2-transformed, normalized counts onto the first 40 PCs based on knee-point detection, corresponding to 26% variance explained. Using Phenograph with k = 30 nearest neighbors, we identified 21 clusters, annotated as B/plasma, T, Myeloid and NK cells using marker genes curated from multiple publications for canonical markers for major immune cell types (including CD79A, CD3D, CD3E, CD14, ITGAM, ITGAX, MS4A2, SDC1, FCGR3A; also see Data S2). Using these cell type-specific gene sets, we transformed the data by z-score and calculated the average expression of each curated gene set per cell type subtracted from the average expression of a reference set of genes using the score_genes function in scanpy. The subsequent cell type scores were transformed again by z-score and cells annotated by maximum cell type score. Cell type labels were smoothed by cluster after manual inspection to ensure accurate separation of cells (Figure S5D).
Cell type annotation in the T-cell compartment
Defining SCLC T-cell subsets was complicated by the relatively lower T-cell infiltrate in SCLC and lower average library size of T-cells in general, both of which can prevent clean separation of subsets based on poorly captured markers like CD4 and CD8. First, to gain more power by boosting the number of T-cells in our analysis, we added the T-cells from LUAD and normal lung samples, resulting in n=46,140 cells. Second, to enhance the gene-based signal, we z-scored the log2-transformed, normalized counts of each gene, projected onto the first 65 PCs based on knee-point detection, corresponding to 7% variance explained (the relatively lower explained variance is expected given the z-score and log transformation). We then performed annotation of T-cell phenotypes using two following parallel approaches, which converged to similar annotation.
Non-negative matrix factorization
Gene factor analysis via matrix factorization has been previously used in single-cell analysis (Levitin et al., 2019; Puram et al., 2017) and excels in settings of continuous phenotypes which are less amenable for robust partitioning by clustering. In this class of methods, cells and genes are projected into the same lower-dimensional space. The resulting latent factors are associated with weights or loadings for each cell and each gene. These cell and gene loadings can be used to associate gene programs to different cells.
We used non-negative matrix factorization (NMF) implemented in scikit-learn (version 20.0) with default parameters except for tolerance for stopping condition 10−4, maximum number of iterations 500, and number of factors k = 30, as selected by calculating the kneepoint of the log2 reconstruction error over the number of factors (Figure S6A). We ensured that NMF factors were robust over a range of k=24–36 based on correlation (Figure S6B, described in section “Robustness analysis of clustering and factor analysis”). To facilitate comparison across factors, gene loadings were first scaled by standard deviation across genes, then z-scored across factors. Each factor was then annotated by genes with the highest loadings. By comparing to a reference set of gene markers (Data S2), we annotated 7 factors with T-cell phenotypes (2 Tconv, 1 Treg, 1 effector-like, 1 memory-like, 1 exhausted, and 1 Tgd factor). The complete set of NMF loadings are provided in an adata file made available for download at https://data.humantumoratlas.org/.
Cluster-based approach
In parallel to our factor-based approach, we also performed a cluster-based approach to annotating T-cell phenotypes, similar to our strategy in other cell type compartments. However, given the challenges of T-cell clustering, we performed an additional test of robustness. In addition to confirming robustness of clusterings by adjusted Rand index (described in section “Robustness analysis of clustering and factor analysis”), we also ensured that clustering was not driven by individual samples. To this end, we repeated clustering with each sample left out and confirmed that the ultimate clustering was robust to dropping samples based on rand index. With these steps, we used Phenograph with k=40 (selected based on section “Robustness analysis of clustering and factor analysis”), which identified 34 clusters of T-cells pooled from SCLC, LUAD, and normal lung.
We then performed differential expression between each cluster and the rest (described in section “Differential expression of tumor and immune subsets in scRNA-seq”) and compared DEGs to curated markers of T-cell phenotype (Data S2) (Figure S6C–D). Finally, we confirmed agreement of our cluster-based cell typing with NMF factors, by calculating the mean cell loadings of each T-cell annotated factor within each cluster-based cell type (Figure S6E). Having successfully identified T-cell subsets at the combined level, we confirmed that these annotations restricted to SCLC were also consistent with known gene markers (Data S2).
Cell type annotation in the myeloid compartment
We subsetted the myeloid cells from SCLC patients (N=2,951 cells). We projected the log2-transformed, normalized counts onto the first 50 PCs based on knee-point detection, corresponding to 30% variance explained. We identified 13 clusters, including 7 clusters of monocyte-derived myeloid cells, 4 clusters of granulocyte-derived myeloid cells, and 2 clusters of dendritic cells (Figure 6A). To annotate myeloid subsets, we identified DEGs between each cluster vs the rest and compared these genes to curated markers of each myeloid subset (Data S2). We show select DEGs that characterize each of the myeloid clusters in Figure S7C.
Robustness analysis of clustering and factor analysis
In all cell type compartments, we performed Phenograph clustering (Levine et al., 2015) over a range of values for the parameter k (number of neighbors in the knn-graph) to ensure that subsequent cell typing is consistent. To ensure robustness, we used the adjusted Rand index to evaluate the consistency of clusterings across different k (from 5 up to 100). We then chose k from the window where the Rand index is consistently highest, indicating stable clusterings. Ultimately, we chose k = 30 for clustering in all cell compartments, with the exception of the T-cell compartment where we used k = 40 (described in section “Cell type annotation in the T-cell compartment”).
For T-cells, we also performed NMF for cell typing (described in section “Cell type annotation in the T-cell compartment”). We performed NMF over a range of k number of components from 5 to 100 and selected k=30 based on the kneepoint of the reconstruction error, defined as the Frobenius norm of the matrix difference between the observed gene expression matrix and the reconstructed matrix (Figure S6A). We ensured the robustness of our NMF factors to the choice of k by generating NMF factors over a range of k = 24–36 and computing the Pearson’s correlation between the cell loadings of each factor with those obtained using k=30 (Figure S6B). In general, for each comparison between cell loadings based on ka and our final choice of k=30, we were able to identify a subset of min(ka,30) factors showing 1-to-1 correspondence across ka and k based on max correlation. We further validated the robustness of our T-cell analysis over values of k. Specifically, we performed a weighted t-test on the ratio of CD8+ T-cells/Treg factors in SCLC-A vs SCLC-N over a range of k = 24–36 and ensured that the ratio was significantly higher in SCLC-A for all values of k.
Measuring inter-patient heterogeneity per cell type
We used an entropy-based measure of inter-patient diversity for each cell type. Here, we use the PhenoGraph clusters within each coarse cell type compartment created without batch correction (described in “Cell Type Annotation”), where each cluster C represents a discrete phenotype of a given cell type, including epithelial, myeloid, lymphoid, fibroblast, endothelial, LUAD, SCLC-A and SCLC-N. We did not consider SCLC-P, as we cannot quantify inter-patient heterogeneity for a single sample. To account for differences in the number of cells per cluster and cell type, we subsampled 100 cells from each cluster 100 times with replacement and calculated the Shannon entropy of patient frequencies P in each subsample HC as:
We then compared the distribution of Shannon entropies bootstrapped from clusters between cell types using Bonferroni-adjusted two-sample t-test (Figures 1D, 1E).
Characterizing canonical SCLC subtypes
Subtype classification and deconvolution in the SCLC tumor compartment
We aimed to characterize inter-patient tumor heterogeneity of the SCLC tumor compartment within the context of canonical and non-canonical subtypes. To focus our analysis on the features that would best discriminate known SCLC subtypes, we considered a limited set of biologically relevant genes (feature selection). We performed feature selection on bulk DEGs between each SCLC-subtype (SCLC-A, SCLC-N, SCLC-P, SCLC-Y) vs rest (described in section “Differential expression in bulk reference datasets”, Tables S20–23), and excluded genes from cell cycle, hypoxia, and apoptosis pathways that are non-specific to SCLC subtype and might confound classification. These filtered genes included pathways from REACTOME_CELL_CYCLE_MITOTIC, REACTOME_MITOTIC_G1_G1_S_PHASES, HALLMARK_G2M_CHECKPOINT, HALLMARK_HYPOXIA, HALLMARK_APOPTOSIS downloaded from MSigDB. We used these features to subset the count matrix and then projected the normalized counts without log transform onto the first 56 PCs selected by knee-point detection, corresponding to 78.8% variance explained.
We then consider the following semi-supervised classification problem of assigning SCLC subtype. For N cells where a subset of L cells has known subtype (training data), we must assign the remaining N-L cells (test data) the probability of represents subtype S ϵ {s1,s2,s3} = {SCLC-A, SCLC-N, and SCLC-P}. We excluded SCLC-Y, as we did not identify any YAP1-expressing cancer cells in our SCLC cohort (Figure 1H). We want an approach that not only assigns probabilities of each subtype per cell, but is able to deconvolve the phenotype of cancer cells residing on a continuum between different SCLC subtypes.
We solve this problem by using the probabilistic knn graph-based Phenograph classifier as implemented in the Phenograph package80 and has been shown to be highly successful in cases of mixed phenotypes which are frequently observed in cancer (Laughney et al., 2020). As input, we provide representative labeled cells for each known cell type. As output, each unlabeled cell is assigned a probability of belonging to each known cell type (termed Markov absorption probability). In cases where the probability for a given cell type is close to 1, we can annotate the cell with that cell type. In cases where the cell-type probability distribution is spread evenly across all cell types, we can consider these cells to represent a mixed phenotype.
To implement this method, we first must have labeled training data available. To this end, we identify cells that can be confidently assigned to each subtype prior to calculating Markov absorption probabilities. Using reference RNA-sequencing of bulk tumors comparing SCLC subtypes (described in section “Differential expression in bulk reference datasets”), we used the top 30 overexpressed DEGs per SCLC subtype and calculated the average Z-score over this gene set for each cell. The top 100 highest scoring cells were then used as training examples for each cell type.
Next, we constructed a Markov graph from the dataset. We first constructed a diffusion map based on the first 56 PCs to obtain the first 15 diffusion components (DCs) retained by eigengap. Using the Phenograph package, we transformed this diffusion graph additionally into a Jaccard graph between k-neighborhoods, which has been shown to be more robust to noise. The resulting graph represents a Markov chain where we can therefore calculate the Markov absorption probabilities for each unlabeled cell to reach a labeled cell of a given subtype. Based on the resulting probabilities for each subtype, we can then perform a hard classification of SCLC subtype by maximum likelihood, or consider the per-cell probabilities of SCLC-A, SCLC-N, and SCLC-P to be a deconvolution of mixed phenotype that can be readily represented by a 3-coordinate ternary graph, as implemented in the ggtern package (Hamilton and Ferry, 2018)(Figure 1F).
Of note, hard classification of SCLC subtypes on the UMAP shows that our feature selection facilitates a visualization that shows separation of cells based on canonical SCLC subtype while demonstrating inter-patient diversity (Figure 1A, 1G). This visualization stands in contrast to a previously published visualization of SCLC circulating tumor cell-derived xenograft cells where discrete clusters of cells represent different patients consistent with inter-patient diversity but without demonstrating clear relationship between patients (Stewart et al., 2020).
Continuity of mixed phenotypes between SCLC-A and SCLC-N
The vast majority of tumors were predominantly composed of a single SCLC subtype (Figure S2A). However, among predominantly SCLC-A or SCLC-N tumors, we observed that while most cells were strongly associated with either SCLC-A or SCLC-N, a substantial minority of cells comprised a relatively continuous spectrum of cells from SCLC-A to SCLC-N (Figure 1F). This minority (8.9% of cells drawn from 20 samples have <95% probability of representing SCLC-A and <95% probability of representing SCLC-N) comprised a relatively uniform continuum of mixed cell-states with almost any proportion of SCLC-A/N probability. In comparison, cells from our single SCLC-P did not contain any such mixed phenotypes with either SCLC-A or SCLC-N (0.37% of cells). Our analysis indicates cells in apparent transition between SCLC-A and SCLC-N, which may represent non-canonical phenotypes or intermediate subtype states. These findings are consistent with our previous report of ASCL1+/NEUROD1+ cells in SCLC clinical samples (Baine et al., 2020; Ireland et al., 2020).
Establishing clonality in the biphenotypic tumor with SCLC-A and SCLC-N components
One sample (Ru1215) harbored two discrete SCLC-A and SCLC-N subpopulations within the same tumor (Figure S2B). We sought to establish the clonality between the SCLC-A and SCLC-N subpopulations of the biphenotypic tumor Ru1215. Similar to our method of calling cancer cells using SNV detection, we leveraged bulk targeted DNA sequencing using the MSK IMPACT platform (Cheng et al., 2015) that was previously performed on a patient-derived xenograft (PDX) derived from a different tumor sample from the same patient. We leveraged this mutational information to assess for variants detected within the SCLC-A and SCLC-N subpopulations. We considered only variant calls that significantly exceeded the rate of variant detection in ambient RNA, estimated in normal non-epithelial cells within Ru1215. We used a Poisson model with k = number of variant calls in the tumor subpopulation and λ = expected number of variant calls given ambient rate of detection a. Here, we define ambient rate a of detecting variants to be the number of variant calls in the normal non-epithelial compartment divided by the number of normal non-epithelial cells. We considered only variants with a Benjamini-Hochberg adjusted FDR < 0.05. Of the 16 SNVs detected in SCLC-A and the 123 variants detected in SCLC-N, we found 15 variants shared between the SCLC-A and SCLC-N subpopulations (Table S2), confirming shared ancestry.
There are several caveats to this analysis. First, there is decreased sensitivity of scRNA-seq for calling variants due to read coverage dependent on gene expression and variant position near the 3’ end. Second, because we need to boost read depth by pooling cells within the same cluster, our sensitivity to call variants is dependent on cluster size and therefore cell sampling. The latter caveat suggests that we observe a greater number of SNVs in SCLC-N due to a greater number of captured cells (61 SCLC-A cells vs 3,862 SCLC-N cells), though increased genetic diversity in SCLC-N cannot be excluded. Regardless, any overlapping variants called in both SCLC-A and SCLC-N is sufficient to establish clonality. These findings are consistent with other reports showing SCLC-A to SCLC-N transitions upon disease progression in SCLC preclinical models (Ireland et al., 2020; Mollaoglu et al., 2017).
Molecular Characterization of Canonical Subtypes in the SCLC cohort
To characterize the canonical subtypes in our SCLC cohort, we performed DE analysis between each subtype vs the rest, as well as between the predominant subtypes in our cohort SCLC-A vs SCLC-N, using MAST on the non-imputed count matrix (Tables S3–6). We then performed pathway analysis using GSEA to determine subtype-specific gene programs (Figures 2A and S2E; and Tables S7–8). To visualize the gene signatures characterizing each subtype, we plotted the heatmap, following hierarchical clustering of imputed gene expression (Figure S2C). Expression values are imputed using MAGIC (k=30, t=3). We found typical markers for SCLC-A (ASCL1, SOX4, STMN2, DOC, STMN2), SCLC-N (NEUROD1, ADCYAP1, NRXN1, SSTR2, ID1, ID3, SST, DLK1), and the one SCLC-P sample (POU2F3, ASCL2, CD44, MYC, KIT, YBX1).
We found that SCLC-A is enriched in expression of genes regulating cell cycle progression and DNA repair, as well as EZH2 target genes implicated in SCLC cell cycle regulation(Hubaux et al., 2013; Poirier et al., 2015) (Figure S2E). In contrast, SCLC-N tumors exhibit a pro-metastatic pattern of gene expression including overexpressed markers of epithelial-mesenchymal transition (EMT) (VIM, ZEB1 and TWIST1)(Dongre and Weinberg, 2019a) and hypoxia and angiogenesis (HIF1A, VEGFA or FOXO3) (Figures 2A, 2B and S2E). SCLC-N also overexpressed metastasis-related signaling pathways, including (1) TGF-β (Farabaugh et al., 2012) (upregulation of TGFB1 and TFGBR1/3); (2) BMP signaling (Choi et al., 2019; Dongre and Weinberg, 2019b) (upregulation of ligands BMP2/7 and receptors BMPR1A/2) (Bach et al., 2018); (3) STAT signaling (upregulation of STAT3, IL6R, IL11RA) (Dongre and Weinberg, 2019b); and (4) TNFα-promoted NFκB signaling (upregulation of TNF, SMAD3, PHLDA1) (Jiang et al., 2001; Wu and Zhou, 2010) (Figures 2A, 2B and S2E).
SCLC-N displayed a neuronal differentiation phenotype, with high expression of the key neurogenesis factor TCF4( Chen et al., 2016; Schmidt-Edelkraut et al., 2014) involved in BMP signaling and metastasis (Hrckulak et al., 2018; Zhao et al., 2004), as well as a neuropeptide signaling signature (SSTR2, SST and MARCKS) (Figures 2A, 2B and Table S3). SCLC-N was enriched in two main axonogenic signaling pathways: ephrin (EFNB1 and EPHB2, among others) (Pitulescu and Adams, 2010) and semaphorin (SEMA6A and NRP2, among others) (Yoshida, 2012). Consistent with these results, prior studies have shown that the axonogenesis program coordinates cell polarity with neuronal migration (Zhang et al., 2019a) and is implicated in SCLC metastasis (Yang et al., 2019), and ephrin and semaphorin pathway components are NEUROD1 targets (Borromeo et al., 2016) or regulators of the NEUROD1high phenotype (Wooten et al., 2019). We have shown that LUAD hijacks endodermal developmental pathways in metastasis (Laughney et al., 2020); similarly, our findings here suggest that SCLC-N may adopt a neuronal developmental phenotype to achieve a metastatic state.
Modeling cell fraction of SCLC subtypes in primary vs metastatic sites
We used several approaches to compare the fraction of cancer cells of different SCLC subtypes in primary lung vs lymph node vs distant metastasis (Figure S2D). We performed Dirichlet regression using the DirichletReg R package using common parameterization to adjust for treatment status (naive vs most recently chemo-treated vs most recently immunotherapy-treated) and tissue status (primary vs regional lymph node vs distant metastasis). This method tests for differences in cell type composition between groups while accounting for proportions of all other cell subsets. In addition to the multivariate Dirichlet regression, we also used univariate Mann-Whitney as a parallel statistical test to ensure consistency.
Characterizing non-canonical SCLC phenotypes
Identifying the recurrent PLCG2+ tumor subclone
Beyond canonical SCLC subtypes, a central question is whether any novel tumor phenotypes are shared across patients. We identified 25 clusters corresponding to distinct SCLC phenotypes. We first assessed whether any of these clusters poorly matched canonical SCLC subtypes and could therefore represent a novel tumor phenotype. Having assigned probabilities for each SCLC subtype s for each cell j using Markov absorption probabilities psj (described in section “Subtype classification and deconvolution in the SCLC tumor compartment”), we identified cells with high uncertainty for any SCLC subtype by calculating the entropy over the cell probabilities for each subtype Uj = Σs pj(s) log pj(s). Cells that have high entropy do not bear obvious similarity to any SCLC subtype. We compared the distribution of subtype uncertainties per cluster and found that cluster 22 had significantly higher subtype uncertainties than all other clusters by Mann-Whitney U test, suggesting a non-canonical subtype.
Having identified a possibly non-canonical SCLC phenotype, we next assessed if it arose beyond a single patient. We used a similar approach to assessing inter-patient diversity per cell type (described in section “Measuring inter-patient heterogeneity per cell type”), but instead of stratifying the bootstrapped entropies of patient labels from each cluster by cell type, we directly compared the bootstrapped entropies of each cluster versus the rest using Bonferroni-adjusted Mann-Whitney U test. We again identified cluster 22 as the most highly recurrent cluster across patients (Figures 3A, 3B).
Recurrent gene markers of the PLCG2+ tumor subclone
To assess the gene program of the recurrent PLCG2+ tumor subclone, we performed differential expression of cluster 22 vs the rest of the cancer cells using MAST (Table S10). To assess for recurrence of overexpressed genes across samples harboring the recurrent subclone, we consider 7 samples that have an unsupervised cluster, at least 10% of which belongs to the recurrent subclone. For each of these samples, we perform differential expression between the cluster and the outgroup. For each gene, we have an adjusted FDR of differential expression, and we calculate a combined p-value p by the Edgington’s method to score the recurrence of each gene. In this way, we can avoid pseudoreplication bias that emerges from a variably sequenced number of cells per sample (Sungnak et al., 2020; Zimmerman et al., 2020). We rank the recurrence of each gene by significance −log(p) and find PLCG2 to be the most highly recurrent DEG (Table S11).
Identifying the PLCG2-related gene module
To better characterize the PLCG2 pathway in the context of SCLC, we used knnDREMI (conditional-Density Resampled Estimate of Mutual Information) (Dijk et al., 2018) to estimate the functional relationship of PLCG2 expression to other genes across the dynamic range of expression. To this end, knnDREMI estimates mutual information between two genes by using conditional density instead of joint density. The key feature of knnDREMI is replacing the heat diffusion based kernel-density estimator (KDE) (Botev et al., 2010) with a knn-based density estimator (Sricharan et al., 2012), which is robust and scales well in sparse, high-dimensional data. For two genes x and y, knnDREMI performs a coarse-grained mutual information calculation on a KDE of p(x,y).
First, the KDE is calculated by constructing a knn graph from a fine-grained grid of points. The density at each grid point is computed as:
Where N is the total number of data points, k is the number of nearest neighbors, and r is the distance to the kth nearest neighbor. V(r,d) is then the volume of a d-dimensional ball of radius r:
Here, we use d = 2 for considering pairwise relationships between genes and k = 10 to be robust against outliers.
Second, we coarse-grain the KDE to calculate discrete mutual information. While KDE is calculated at fine resolution to smooth and fill in gaps in sparse data, mutual information is calculated over a coarse scale for robustness to noise and any irregularities in partitioning. The conditional density estimate, which is a column-normalized joint density estimate, better captures the functional relationship across the entire dynamic range of expression robust to density sampling.
Finally, we calculate mutual information for gene expression x and y based on the conditional density estimate. In general, mutual information is defined as
where H(y) is Shannon entropy:
and H(y|x) is conditional Shannon entropy:
On the other hand, knnDREMI uses the conditional density estimate to calculate mutual information above, which effectively adds another level of conditioning:
In the SCLC cohort, we identify genes functionally related to PLCG2 by calculating knnDREMI of each gene y conditioned on x fixed as PLCG2 expression. knnDREMI is best applied on imputed data. We therefore used MAGIC (Dijk et al., 2017) using parameters t=3 and k=10 to impute a count matrix. We applied knnDREMI to the imputed count matrix and identified genes with the highest knnDREMI > 1. We plotted the z-scored expression of the genes with the highest knnDREMI on a heatmap, ordering columns by PLCG2 expression (top row) (Figure S3E). We then performed hierarchical clustering to find three gene modules corresponding to low, intermediate, and high PLCG2 (Table S12).
To identify other pathways associated with the PLCG2-high gene module m, we calculated for each cell x a score Zm, which is the average Z-score of expression for all genes within the PLCG2-high gene module. We similarly calculated for each cell a score Zn the average Z-score of expression for all genes in each pathway n from a curated set of MSigDB. We then calculated Pearson’s correlation between Zm and each Zn to identify gene pathways that correlate with the PLCG2-high gene module. We considered pathways among the top 5% correlated, corresponding to a minimum correlation threshold of ρ = 0.341 (Figure S3F, Table S13). The remaining set therefore represents candidate gene pathways that are also increased in cells that have increased expression of the PLCG2-high gene module.
Molecular characterization of the RB1/TP53-wildtype SCLC sample
Interestingly, sample Ru1108 had a strong subtype probability for SCLC-A but was transcriptionally distinct from the rest of the SCLC-A group (Figure 1G, S2C). This sample with wild type TP53 and RB1 had high expression of ASCL1, DLL3 and neuroendocrine markers consistent with SCLC-A subtype, but also overexpressed CDK4 consistent with a previous report (Sonkin et al., 2019) and a NSCLC gene signature (average Z-score of the differentially overexpressed genes in NSCLC vs SCLC cell lines from the CCLE database, not shown). Together, our subtype classification demonstrated tumor diversity in canonical SCLC subtypes, but also identified additional non-canonical phenotypes in our cohort, including this TP53/RB1 wild-type SCLC.
Survival analysis
To assess the prognostic impact of the recurrent PLCG2+ subpopulation, we performed survival analyses in our single-cell SCLC cohort and validated these findings in an independent cohort with MIBI-TOF staining for PLCG2. Both cohorts were balanced for different covariates, including treatment history and tissue type (Tables S14 and S16). For both analyses, we considered samples with extensive-stage ES-SCLC or limited-stage LS-SCLC that recurred (ever had extensive-stage disease). OS was defined as the time of biopsy to death or censoring. For our validation cohort with MIBI-TOF, samples were divided based on NEUROD1 protein expression into ASCL1+ NEUROD1− and ASCL1(+/−) NEUROD1+ subgroups, due to the minimal number of ASCL1− NEUROD1+ samples and no ASCL1− NEUROD1− samples in the cohort.
We then performed Kaplan-Meier (univariate) and Cox proportional hazards (multivariate) survival analysis using the survival R package (Therneau and Grambsch, 2000). We separated cohorts under analyses into two subgroups using thresholds on the predictor variable using maximally selected rank statistics as determined by the surv_cutpoint function in the survminer R package (https://cran.r-project.org/web/packages/survminer/index.html). We used a threshold of (1) at least 0.75% of SCLC cells comprising the recurrent PLCG2+ subpopulation as assessed by scRNAseq, (2) >7% of SCLC cells exhibiting positive PLCG2 protein expression on MIBI-TOF (see below) or (3) >15% of SCLC cells exhibiting high PLCG2 protein expression (Intensity 3) on PLCG2 as assessed by a pathologist. To ensure that our results are robust to threshold selection, we also performed a Cox regression using a continuous predictor variable that confirmed similar significant results. In the Cox proportional hazards model, we adjusted for presence of classical vs variant SCLC subtype, treatment, and distant metastasis vs primary/regional lymph node. For the MIBI-TOF data, we considered treated vs naive as a covariate, as all treated patients received chemotherapy alone. In our scRNA-seq dataset on the other hand, treated patients received either chemotherapy alone or chemotherapy with immunotherapy added either in first or second line. Three patients were treated with later-line chemotherapies including temozolomide (Figure S1A and Table S1). We therefore considered most recently chemo-treated without immunotherapy and most recently immunotherapy-treated covariates. Our adjusted covariates were dichotomized to ensure a stable fit for the adjusted Cox regression. In general, the corresponding Schoenfeld residuals were invariant to time, but for completeness, our Kaplan-Meier univariate analysis is independent of the proportional hazards assumption. P-values were calculated using Wald test and were also consistent with bootstrapped p-values.
Cell-cell interaction analysis
We sought to identify cell-cell interactions among tumor subclones of the same SCLC subtype and between tumor subclones of different subtype. For this analysis, we used CellPhoneDB (Efremova et al., 2020), which efficiently identifies outlying co-expression of ligand-receptor (L-R) pairs compared to a null distribution generated from permuted cell type labels. While this method in no way indicates the existence of the interaction, it does identify candidate interactions in the data. We first considered whether tumor-tumor L-R interactions are enriched in SCLC-A vs SCLC-N. Given a list of significant interactions based on CellPhenoDB, we assessed enrichment of interactions using Fisher’s exact test and found that all significant interactions were found in SCLC-N rather than SCLC-A (Figure 2C). While we have limited confidence in any individual interaction represented in this figure, the sheer difference between 20 significant interactions found in SCLC-N versus no significant interaction SCLC-A suggests interactions between SCLC-N cells, consistent morphological descriptions of SCLC-N as tightly adherent cells in contrast to SCLC-A.
Comparing T-cell phenotype between SCLC-A vs SCLC-N
Assessing changes in NMF loadings between SCLC subtypes
To analyze the phenotypic shifts in T cell compartment across SCLC subtypes, we considered NMF factors associated with T-cell phenotype (described in section “Cell type annotation in the T-cell compartment”). Using NMF, we compared the distribution of factor loadings across T cells in SCLC-A and SCLC-N. To ensure that factors are assessed on the same scale, we first log2-transformed cell loadings with a pseudocount of 0.0001, shifted the minimum of each factor to 0, and scaled each factor by standard deviation across cells. We accounted for the effect of treatment and tissue site by fitting a linear model between the factor loadings and the treatment and tissue status of cells. We then performed a Bonferroni-adjusted two-sample t-test on the residuals of the factor loadings (Figure S6G). We used tissue status (primary vs LN vs distant metastasis) and treatment status (naive vs most recently chemo-treated vs most recently immunotherapy-treated) as covariates in the model.
Analysis of CD8+ T cell/Treg ratio in SCLC subtypes
As a measure of immune response in tumor-infiltrating lymphocytes that can be readily calculated from both scRNA-seq and Vectra imaging platforms and has demonstrated prognostic value in a variety of contexts in cancer (Baras et al., 2016; Preston et al., 2013; Shang et al., 2015), we used the ratio of CD8+ T-cells to Tregs in SCLC-A versus SCLC-N. We first compared the ratio of CD8+ T effector/Tregs phenotypes using NMF factors (described in section “Assessing changes in NMF loadings between SCLC subtypes”). Specifically, we compared the ratio of the averaged loadings of factor 28 (effector-like) and factor 4 (Tregs) across T cells per sample in SCLC-A and SCLC-N. We accounted for the effect of treatment and tissue site by fitting a linear model between the ratio of CD8+ T effector factor loading/Treg factor loading and the treatment status and tissue site of the samples (similar to correlation analysis described in section “Correlation analysis of immune subset abundance and tumor phenotypes”) and comparing the model residuals. We accounted for the difference in numbers of cells collected per sample using a weighted one-sided t-test (as implemented by ttest_ind in the python library statsmodels (Seabold and Perktold, 2010)). Within each SCLC subtype, the weight of the i-th sample was given by:
With nj denoting the total number of T cells in patient i and P being the total number of patients in that group (SCLC-A or SCLC-N). We calculated FDR by generating a null distribution using a permutation test on cell type labels. We also performed Goodman-Kruskal’s test as a parallel statistical test to ensure consistency. To ensure the results are not driven by individual samples, we performed leave-one-sample-out cross-validation and verified that the result remains significant for every case. To ensure the results are not driven by the choice of k for NMF, we also verified that the result remains significant over a range of k=24–36 (Figure S6H).
We verified the same difference in factor-based ratio of CD8+ T-cell/Treg abundances between SCLC-A vs SCLC-N using several approaches. We first performed the same analysis by using cells labeled with cluster-based T-cell phenotyping (described in section “Cell type annotation in the T-cell compartment”), which also showed decreased CD8+ T-cell/Treg ratio in SCLC-N.
Finally, we used Vectra imaging (see “Vectra analysis”) to validate these findings. We restricted analysis to 12 treatment-naive, primary SCLC samples. We then compared the ratio of CD8+ T cells/Tregs in NEUROD1− and NEUROD1+ subtypes to quantify the immune response of tumor-infiltrating lymphocytes.
Detailed characterization of pro-fibrotic Mono/Mφ cluster 1
We noted high expression of ECM-related genes in Mono/Mφ cluster 1, we compared our dataset to gene signatures from a single-cell atlas of IPF (Adams et al., 2020) and found that cluster 1 stood out as having an outlying pro-fibrotic signature as well as increased inflammatory macrophage signature (Figures 6C–E). Differential expression using MAST (see “Differential expression of tumor and immune subsets in scRNA-seq”) (Figures S7D) identifies cluster 1 as a CD14+ ITGAX+ CSF1R+ subpopulation. Cluster 1 expressed monocytic features that include VCAN, FCN1, and S100 proteins. At the same time, it also overexpressed scavenger receptor (MARCO, MSR1, CD36, CD68, CD163) and scavenger binding protein (APOE, APOC1) genes, suggesting that cluster 1 represents a monocyte-derived but tissue-enriched myeloid subset. In addition, cells from this cluster express secrete pro-fibrotic, pro-metastatic growth factors involved in ECM deposition and remodeling (Winkler et al., 2020), including FN1 (Park and Helfman, 2019; Wang and Hielscher, 2017), cathepsins (CTSB and CTSD) (Egeblad and Werb, 2002; Guo et al., 2002), and SPP1(Giopanou et al., 2017; Pang et al., 2019), suggesting a role in promoting metastasis. In addition, cluster 1 overexpressed genes related to immune inhibition, including (1) SPP1 (Shurin, 2018) and NSCLC (Lin et al., 2015)]; (2) CD74 (Figueiredo et al., 2018; Takahashi et al., 2009); and (3) VSIG4(Li et al., 2017).
Correlation analysis of immune subset abundance and tumor phenotypes
We aimed to identify significant partial correlations between any immune subset and tumor phenotype in SCLC while adjusting for any clinical covariates. To this end, we first consider cell abundance X and cell abundance Y of interest, as well as clinical covariates Z. We fit separate multivariate linear regression models between X and Z, and between Y and Z using the numpy.linalg package (Harris et al., 2020). We then compute the Spearman’s rank correlation between model residuals of X and Y. For this analysis, we adjusted for tissue status (primary vs lymph node vs distant metastasis) and treatment status (naive vs most recently chemo-treated vs most recently immunotherapy-treated). We verified the false discovery rate (FDR) remains significant by generating a null distribution using a permutation test on the cell type labels for 2000 times. To test robustness, we performed a leave-one-sample-out validation and confirmed that the result remains significant even after excluding any sample.
Validation using independent bulk datasets
Validating enriched pathways in the recurrent PLCG2-high cluster in the microarray dataset of SCLC cell lines
We validate gene signatures enriched in the recurrent cluster using a bulk microarray dataset of 54 SCLC-A and SCLC-N cell lines from the Cancer Cell Line Encyclopedia (Barretina et al., 2012). We consider the bulk expression matrix of dimension S × G, where S is the set of SCLC-A and SCLC-N cell lines and G is the set of genes. For each sample s ∈ S, we calculate a score Zm for the gene signature of the recurrent PLCG2-high SCLC cluster. To calculate this score Zm, we consider the set of recurrent DEGs m of the PLCG2-high SCLC cluster identified in our scRNA-seq analysis, where m ⊂ G (Table S11). We then calculate the average Z-score of expression for these DEGs across samples. Similarly, we calculated for each sample a score Zn for the gene signature of each pathway found to be significantly enriched in the PLCG2-high cluster using GSEA in our scRNA-seq analysis, detailed in section “Identifying enriched gene pathways in single-cell data”. To calculate score Zn for each pathway, we consider the leading-edge genes n ⊂ G identified by GSEA for each pathway. We then calculate the average Z-score expression for the leading-edge genes in each pathway n. We then computed the Spearman’s correlation between Zm and each Zn to identify gene pathways that correlate with the PLCG2-high recurrent cluster in the bulk samples.
Validating tumor phenotypes correlated with the profibrotic Mono/Mφ subset in bulk RNA-seq
Similarly, we validate gene signatures correlated with the profibrotic myeloid population using an independent bulk-RNA seq cohort of 81 SCLC-A and SCLC-N patient tumors collected from George et al. (George et al., 2015) and Rudin et al. (Rudin et al., 2012). For each sample s, we calculate a score Zx, which is the average Z-score of expression for DEGs in the profibrotic myeloid population (Table S19). We then calculate Zy for each pathway enriched in the profibrotic myeloid population based on GSEA. We calculate the average Z-scored expression for the leading-edge genes in each pathway y. We then compute the Spearman’s correlation between Zx and each Zy to identify gene pathways that correlate with the profibrotic myeloid signature in bulk samples.
Marker selection for MIBI-TOF panel
To validate the recurrent PLCG2-high SCLC cluster and its association with the profibrotic Mono/Mφ subset, we built a validation experiment based on applying MIBI-TOF on fresh frozen paraffin-embedded (FFPE) samples. We took this direction because (1) SCLC is rare, and we had more SCLC samples available in FFPE, which is not amenable to scRNA-seq. (2) This approach provides a spatial context to assess the statistical correlations found in Figures 7A and 7B.
To detect the recurrent PLCG2-high SCLC cluster, we optimized a monoclonal antibody for PLCG2 to be used in MIBI-TOF, which was consistent with both monoclonal and polyclonal antibodies for PLCG2 previously used in IHC for the same TMA. We sought to obtain a specific set of 3 markers to identify the profibrotic myeloid subset. We considered all combinations of 3 genes from the list of DEGs between cluster 1 vs other myeloid cells in our scRNA-seq dataset. We then used support vector classification using the sklearn.svm package to calculate for each combination of markers the F1 score for differentiating cluster 1 vs other Mono/Mφ. Among the highest ranking F1 scores, the combination of CD14, CD16, and CD81 was highly specific for the profibrotic Mono/Mφ. We had a previously optimized antibody for CD14 by Ionpath but optimized antibodies for CD16 and CD81. A full table of the MIBI-TOF marker panel is included in Table S15, including those that were not included in final analysis due to failure to pass quality control.
MIBI-TOF analysis
Image segmentation
Single-cell segmentation was performed with Mesmer (Greenwald et al., 2021), a deep learning algorithm pretrained on TissueNet (Basha et al., 2017). We used the dsDNA channel as a nuclear marker. To define the membrane, we used the combination of LAP2, CD45, CD3, CD14 channels with each scaled by the 80th percentile in non-zero intensity values with a cap at 10. We constrained the minimum area of a cell to be 25 pixels, with an additional one-pixel expansion to account for cells whose membrane marker is not included in the input membrane channel. We excluded any calls for segmented cells greater than 1600 pixels.
Expression pre-processing
We normalized the sum of the total marker expression within each segmented cell by the cell area to obtain the average marker intensity per cell. We further scaled marker expression per cell by clipping the 1st and 99th percentile and performed min-max normalization. Finally, following (Greenwald et al., 2021) all expression values underwent arcsinh transformation and were normalized by standard deviation.
Cluster-based cell typing
To differentiate cell types, we first performed coarse clustering of the arcsinh-transformed expression of immune markers (CD11c, CD14, CD16, CD163, CD3, CD4, CD45, CD68, CD8, FoxP3, HLA-DR), epithelial markers (Keratin, NeuroD1, CD56), and endothelial markers (CD31). We then performed Leiden clustering (Traag et al., 2019) with knn=30 and resolution=1. Of note, IHC of the adjacent slice of the TMA confirmed that no epithelial stromal cells were present, and therefore any cells positive for epithelial markers represent SCLC cells. Cells with total summed expression below 0.1 were removed prior to clustering. Based on marker expression, we therefore classified cells into immune, endothelial, SCLC (with positive epithelial markers), and others (without any positive markers).
To subtype the immune population, we used the average intensity expression matrix using markers CD11c, CD14, CD16, CD163, CD3, CD4, CD45, CD68, CD8, and FoxP3. After subsetting to the immune population, we used Phenograph with k=30 to identify 23 clusters. Based on the marker expression, we merged fine clusters into DC (CD11c+), Mono/Mφ (CD14+, CD16+, CD68+, or CD163+), T-cells (CD3+) subdivided into CD4+ and CD8+ T-cell, Tregs (FoxP3+), Other Immune, and Others (clusters negative for any immune marker). We show the relative frequency of each cell type per sample in Figure S4A. Our cell typing was consistent with cell morphology and subcellular marker distribution.
Tumor region detection
Because SCLC typically presents as large sheets of tightly packed small cells, we used cell density as a feature in addition to cluster-based cell typing (described above in section “Cluster-based cell typing”) to increase sensitivity for identifying SCLC cells. In an adjacent cut of the TMA that underwent IHC, we confirmed by pathological review that all epithelial cells present were cancer cells. Additionally, we confirmed that the TMA did not include any NSCLC even in samples of combined histology, and that any putative cancer cells are unlikely to be of any other lineage than SCLC. We therefore created a mask of the tumor region by calculating the kernel density of non-immune and non-endothelial cells in each core with scipy gaussian_kde function (Virtanen et al., 2020) over a grid of 500×500 pixels and bw_method=0.05. We then set the threshold density as 2 × 10–7 and excluded disconnected putative tumor regions of small area <2500 pixels. Finally, we considered SCLC cells to be 1) non-immune and non-endothelial cells captured in the mask region of high cell density, or 2) cells belonging to clusters positive for Keratin, NeuroD1, or CD56 that had been previously assigned to SCLC cell type in the section “Cluster-based cell typing”.
Batch normalization
To account for systematic differences in intensity due to periodic ion detector adjustments, we performed batch correction using LAP2 expression, which is universally expressed across all cell types but at different levels depending on cell type. We therefore normalized all marker expressions in each core based on the median LAP2 expression within cancer cells for subsequent analysis.
Cell state assignment
To identify an appropriate threshold for calling PLCG2 positivity in cancer cells, we compared PLCG2 expression in cancer cells using MIBI-TOF to IHC staining PLCG2 in an adjacent cut of the same TMA. We found that a minimum threshold of 0.2 for batch-corrected PLCG2 average intensity matched the parallel IHC assessment best. Similarly, to define pro-fibrotic Mono/Mφ, we used markers CD14, CD16, and CD81, and set the minimum threshold as 0.2 for the batch-corrected average intensity.
Accounting for boundary-dependent intensity dropoff
In our tissue microarray, we noted an edge-dependent dropoff in signal intensity for lowly expressed markers like PLCG2 at the edges of each field of view across samples. We sought to create a mask of these low-intensity regions to exclude from analysis. First, we applied radial basis function interpolation to PLCG2 intensity based on the scipy.interpolate.Rbf package (Virtanen et al., 2020) using multiquadric functions with smooth = 100 and epsilon = 100. Any region with PLCG2 intensity less than 0.07 was removed for quantifying PLCG2+ SCLC cells. Additionally, we excluded any cell within 50 pixels from any edge of the FoV.
Assessing immune hot vs cold tumors and immune compartmentalization vs mixing
We sought to study the immune spatial architecture in relation to the tumor in our SCLC cohort. Following Keren et al. (Keren et al., 2018b), we considered samples with less than 250 immune cells in a 800 μm by 800 μm FoV to be immune-cold tumors.
For tumors that were not considered immune-cold, we considered the degree that immune cells were compartmentalized from vs intermixed with the SCLC cells. To quantify the degree of immune-tumor mixing, we calculated a score as follows. First, we built a cell-cell neighborhood graph from Delaunay triangulation and assessed interaction between tumor, immune, and other stromal cells with Squidpy (Palla et al., 2021). We then calculated the immune-tumor mixing score as the number of immune-tumor interactions divided by the number of immune-stromal interactions, where we consider the stroma to correspond to all non-cancer cells, including immune. In calculating the immune-tumor mixing score, we excluded small contiguous tumor cell regions from analysis, as we sought to quantify the extent of immune cell infiltration of the main SCLC tumor region. Moreover, we reasoned that small SCLC tumor regions may be 1) the product of an oblique slice of a larger tumor region or 2) budding SCLC cells in transit that should not contribute to a mixing score measuring the extent of immune infiltration. We therefore excluded any contiguous tumor cell regions less than 2500 pixels based on the tumor cell density mask (see section “Tumor region detection”). To provide a baseline comparison for our measure of immune-tumor mixing in SCLC, we performed the same analysis on TNBC MIBI data, downloaded from https://www.angelolab.com/mibi-data. Cell segmentation, cell type annotation from the original dataset were used for our analysis.
Correlation analysis
Using a similar approach to the section “Correlation analysis of immune subset abundance and tumor phenotypes,” we calculated the partial Spearman’s correlation between PLCG2+ SCLC cells and other immune and SCLC subpopulations (Figure 7E). We adjusted for the following clinical covariates: distant metastasis vs primary, chemotherapy-treated vs untreated, combined vs single histology, and SCLC subtype based on NEUROD1 positivity as estimated from IHC (Table S14).
Vectra analysis
To assess differences in T-cell subsets between SCLC-A vs SCLC-N using spatial imaging, we opted to use the Vectra platform over MIBI-TOF because Vectra has (1) a substantially larger field of view, (2) increased sensitivity for FOXP3 staining, and (3) a greater number of treatment-naive tumors available following quality control. The first two points were particularly important because of the relatively lower T-cell abundance in SCLC. To study the result from Vectra images, we developed a pipeline for multiplexed imaging quality control and processing (https://github.com/dpeerlab/Vectra_Imaging_pipeline). We describe the analysis process below and code, notebook tutorials are available in the GitHub.
Batch normalization
To compare different markers across samples, we normalized intensity values of each marker. We first applied a Gaussian kernel with σ=3 to smooth intensity over the target image. We considered the maximum intensity value M of a marker in a given sample to be an initial value for intensity normalization. We then assessed the distribution of maximum intensity values of each marker across samples, which generally follows a bimodal distribution. This bimodal distribution allows for an intensity threshold that readily separates signal from noise. We therefore considered the filtered distribution of intensities greater than this threshold. Finally, we constrained the value for intensity normalization M to be greater than the minimum but less than the maximum of the filtered intensity values across samples.
Noise removal
We used the following procedure to remove noise introduced by non-specific staining in our fluorescence multiplexed imaging data. First, we applied a median filter with size 2 to remove outliers, and then a Gaussian kernel with σ=1 was applied to smooth the image. We automated remaining noise removal using either Otsu or Triangle thresholding. For a specific channel, if the 80th percentile intensity is ⋧5, we use the Otsu method. Otherwise we used the Triangle method. To guide automatic noise removal, we manually set a lower boundary (to remove obvious noise) and an upper boundary (to retain obvious signal) per sample. We then combine batch normalization and noise removal to generate a quality check report to further guide preprocessing. This initial automation facilitates manual correction of parameters for image processing.
Single-cell instance segmentation
To obtain single-cell information, we developed a segmentation toolbox based on Mask R-CNN (https://github.com/dpeerlab/MaskRCNN_cell), a deep learning framework for object instance segmentation to perform cell instance segmentation on our multiplexed imaging data. This model generates bounding boxes and segmentation masks for each instance of an object in the image. We optimized the parameters of this framework for the single-cell segmentation task, characterized by high object density, small but consistent object size. To avoid cropping TMA images into small pieces and cutting cells overlying boundaries into two, we developed seamless stitching features that allow segmentation on very large images. To generate the training data, we manually annotated 24 sample images with nuclear and cell membrane markers (DAPI, CD8, FOXP3, INSM1 et al). Training images were augmented by random horizontal flips, random vertical flips, random rotation, random gaussian blur, random zoom in and zoom out, random brightness changes, and random shear. Training was performed using a step per epoch of 1000 and was run for 10 epochs for heads layers and 30 epochs on all layers. To segment images of interests, we visualize the images with the same color pattern that was used in training.
Cell typing
Segmentation, normalization, and noise-removal of the image dataset as described above yielded a 7-dimensional single-cell protein marker expression profile with sum of marker expression, expression area, cell size et al information. Cells with low nuclear area (lower than 16 pixels) were removed prior to analysis. A marker was considered positive when the average expression (total expression divided by cell size) is above 0.1 (0.06 only for FOXP3, which s lowly expressed) and expression area is above 4 pixels. For markers that do not co-express, we classified cells into double-negative, 1 marker positive only and 2 markers positive only, based on the distribution of average expression.
ADDITIONAL RESOURCES
Raw and processed data from this paper are publicly available on the Human Tumor Atlas Network (HTAN) data portal at https://data.humantumoratlas.org/.
Supplementary Material
Table S1. Clinical characteristics of samples analyzed by single-cell RNA-seq, Related to Figures 1 and S1
Table S2. Shared mutations detected in SCLC-A and SCLC-N subpopulations of the biphenotypic sample Ru1215, Related to Figure S2
Table S3. Differentially expressed genes comparing SCLC-A versus SCLC-N in single-cell RNA-seq of SCLC cells using MAST, Related to Figures 2 and S2
Table S4. Differentially expressed genes comparing SCLC-A versus rest in single-cell RNA-seq of SCLC cells using MAST, Related to Figures 2 and S2
Table S5. Differentially expressed genes comparing SCLC-N versus rest in single-cell RNA-seq of SCLC cells using MAST, Related to Figures 2 and S22
Table S6. Differentially expressed genes comparing SCLC-P versus rest in single-cell RNA-seq of SCLC cells using MAST, Related to Figures 2 and S2
Table S7. Pathway enrichment of SCLC-A versus SCLC-N in SCLC cells using GSEA, Related to Figures 2 and S2
Table S8. Pathway enrichment of SCLC-P versus rest in SCLC cells using GSEA, Related to Figures 2 and S2
Table S9. Pathway enrichment in the recurrent, PLCG2-high SCLC cluster versus rest of SCLC cells using GSEA, Related to Figures 3 and S3
Table S10. Differentially expressed genes of SCLC recurrent subclone (cluster 22) versus rest in single-cell RNA-seq using MAST, Related to Figures 3 and S3
Table S11. Differentially expressed genes that are recurrently overexpressed in the recurrent, PLCG2-high SCLC cluster across samples, ranked by the Bonferroni-adjusted Edgington’s combined p-value, Related to Figures 3 and S3
Table S12. Gene modules with high knnDREMI conditioned on PLCG2, divided by low, medium, and high PLCG2 expression, Related to Figure S3
Table S13. Pathways with average z-scores of gene expression correlated with the average z-score of gene expression in the high-PLCG2 gene module, Related to Figure S3
Table S14. Clinical characteristics and ASCL1/NEUROD1 positivity on immunohistochemistry for an independent SCLC cohort analyzed by Vectra and MIBI, Related to Figures 4, 5, 7, S4, S7
Table S15. A summary of antibodies, MIBItag, and concentrations used in MIBI-TOF, Related to Figures 4, 5, 7, S4, S7
Table S16. Survival data and clinical covariates of samples analyzed by single-cell RNA-seq and stratified by fraction of the recurrent, PLCG2-high SCLC cluster, Related to Figures 4 and S4
Table S17. Clinical characteristics, CD45+ percentage, and ASCL1/NEUROD1 positivity on immunohistochemistry for an independent SCLC cohort analyzed by flow cytometry, Related to Figure S4
Table S18. Number of immune cell types per sample in scRNA-seq, Related to Figure S5
Table S19. Differentially expressed genes of Mono/Mφ cluster 1 vs other Mono/Mφ subsets in single-cell RNA-seq using MAST, Related to Figure S7
Table S20. Differentially expressed genes of SCLC-A versus rest in bulk RNA-seq data from (George et al., 2015) and (Rudin et al., 2012) using limma, Related to STAR Methods
Table S21. Differentially expressed genes of SCLC-N versus rest in bulk RNA-seq data from (George et al., 2015) and (Rudin et al., 2012) using limma, Related to STAR Methods
Table S22. Differentially expressed genes of SCLC-P versus rest in bulk RNA-seq data from (George et al., 2015) and (Rudin et al., 2012) using limma, Related to STAR Methods
Table S23. Differentially expressed genes of SCLC-Y versus rest in bulk RNA-seq data from (George et al., 2015) and (Rudin et al., 2012) using limma, Related to STAR Methods
Table S24. Pathway enrichment in Mono/Mφ cluster 1 vs other Mono/ Mφ subsets using GSEA, Related to STAR Methods
Data S1. Gene sets curated from MSigDB and literature, used for pathway enrichment analysis in SCLC subtypes and clusters, Related to STAR Methods
Data S2. Markers used for cell type annotation, curated from literature, Related to STAR Methods
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PLCG2 (Western blot) | Cell Signaling Technology | #3872 |
| Beta-catenin (Western blot) | Cell Signaling Technology | #8480 |
| pSMAD1/5 (Western blot) | Cell Signaling Technology | #9576 |
| SMAD1 (Western blot) | Cell Signaling Technology | #6944 |
| SMAD5 (Western blot) | Cell Signaling Technology | #12534 |
| N-cadherin (Western blot) | Cell Signaling Technology | #14215 |
| Vimentin (Western blot) | Cell Signaling Technology | #5741 |
| Twist (Western blot) | Cell Signaling Technology | #46702 |
| ZEB (Western blot) | Cell Signaling Technology | #70512 |
| NFIB (Western blot) | Abcam | #ab186738 |
| SOX2 (Western blot) | Cell Signaling Technology | #3579 |
| Nanog (Western blot) | Cell Signaling Technology | #4903 |
| Actin (Western blot) | Cell Signaling Technology | #3700 |
| donkey anti-rabbit IRDye 800CW | LI-COR | #926–32213 |
| donkey anti-mouse IRDye 680LT | LI-COR | #926–68022 |
| dsDNA (MIBI) | Ionpath | 708901–100 |
| LAP2 (MIBI) | BD Biosciences | 611000 |
| PLCG2 (MIBI) | CST | 55512BF |
| CD163 (MIBI) | Bio-Rad | MCA1853 |
| CD4 (MIBI) | Ionpath | 714301–100 |
| CD11c (MIBI) | Ionpath | 714401–100 |
| FoxP3-AF488 (MIBI) | BD Pharmingen | 561181 |
| Anti-Alexa Fluor 488 (MIBI) | Invitrogen | A11094 |
| CD81 (MIBI) | Abcam | ab233692 |
| PD-1 (MIBI) | Ionpath | 714801–100 |
| PD-L1 (MIBI) | Abcam | ab226766 |
| CD56 (MIBI) | Ionpath | 715101–100 |
| CD31 (MIBI) | Ionpath | 715201–100 |
| ki-67 (MIBI) | Ionpath | 715302–100 |
| CD68 (MIBI) | Ionpath | 715601–100 |
| CD8 (MIBI) | Ionpath | 715801–100 |
| CD3 (MIBI) | Ionpath | 715901–100 |
| CD16 (MIBI) | CST | 24326BF |
| TIM3 (MIBI) | CST | 45208S |
| CD14 (MIBI) | Abcam | ab226121 |
| Keratin (MIBI) | Ionpath | 716501–100 |
| S100A12 (MIBI) | Lifespan Biosciences | LS-C785701 |
| NULP1-TCF25 (MIBI) | Invitrogen | PA5–21418 |
| RRBP1 (MIBI) | Millipore Sigma | HPA009026 |
| VIMENTIN (MIBI) | CST | 5741BF |
| ASCL1 (MIBI) | Abcam | ab240385 |
| ASCL1 (MIBI) | Abcam | ab251539 |
| HLA-DR (MIBI) | Ionpath | 717201–100 |
| NeuroD1 (MIBI) | Abcam | ab226489 |
| CD45 (MIBI) | Ionpath | 717501–100 |
| HLA I (176) (MIBI) | Ionpath | 717602–100 |
| ASCL1 (IHC) | BD | #556604 |
| NEUROD1 (IHC) | Abcam | #ab205300 |
| POU2F3 (IHC) | Santa Cruz | #6D1 |
| PLCG2 (IHC) | Sigma-Aldrich | #HPA020100 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Critical Commercial Assays | ||
| Cultrex BME Cell invasion assay kit | R&D Systems | #3455–096-K |
| CellTiter-Glo 2.0 assay | Promega | #G9242 |
| Wnt signaling reporter assay | BPS Biosciences | #60500 |
| Firefly/Renilla luciferase assay kit | Sigma Millipore | #SCT152 |
| Deposited Data | ||
| scRNA-seq and MIBI data | HTAN Data Portal | https://data.humantumoratlas.org/ |
| Experimental Models: Cell Lines | ||
| H82 | ATCC | #HTB-175 |
| SHP-77 | ATCC | #CRL-2195 |
| H526 | ATCC | #CRL-5811 |
| H446 | ATCC | #HTB-171 |
| DMS-114 | ATCC | #CRL-2066 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| PLCG2 overexpression lentiviral plasmid | GeneCopoeia | #EX-A8643-Lv201 |
| PLCG2 CRISPR knock out lentiviral plasmid | Sigma-Aldrich | #HSPD0000031727 |
| Software and Algorithms | ||
| SEQC | Azizi, et al., 2018 | https://github.com/dpeerlab/seqc |
| CB2 | Ni, et al. 2020 | https://github.com/zijianni/scCB2 |
| DoubletDetection | Gayoso, et al. 2018 | https://github.com/dpeerlab/doubletdetection |
| scanpy (suite of single-cell algorithms, including UMAP, tSNE, score_genes, among others) | Wolf, et al., 2018 | https://scanpy.readthedocs.io/en/stable/# |
| PhenoGraph (includes clustering and Markov absorption modeling) | Levine, et al. 2015 | https://github.com/dpeerlab/phenograph |
| fastMNN (through the batchelor package) | Haghverdi, et al. 2018 | https://github.com/LTLA/batchelor/blob/master/R/fastMNN.R |
| MAGIC and knnDREMI | van Dijk, et al. 2018 | https://github.com/dpeerlab/magic |
| MAST | Finak, et al. 2015 | https://github.com/RGLab/MAST |
| limma | Ritchie, et al. 2015 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| fGSEA | Korotkevich, et al. 2019 | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| Ambient RNA detection | Smillie, et al. 2019 | https://github.com/cssmillie/ulcerative_colitis |
| DirichletReg | Maier, et al. 2014 | https://cran.r-project.org/web/packages/DirichletReg/index.html |
| cellphonedb | Efremova, et al. 2020 | https://github.com/Teichlab/cellphonedb |
| survival | Therneau, et al. 2020 | https://cran.r-project.org/web/packages/survival/index.html |
| Non-negative matrix factorization in Scikit-learn v. 20.0 | Pedregosa, et al. 2011 | https://scikit-learn.org/stable/ |
| Vectra Imaging Processing Pipeline | https://github.com/dpeerlab/Vectra_Imaging_pipeline | |
| MaskRCNN_cell (segmentation for Vectra image) | https://github.com/dpeerlab/MaskRCNN_cell | |
| ARK-analysis (MIBI analysis) | https://github.com/angelolab/ark-analysis | |
| Mesmer | Noah F. Greenwald et al. 2021 | https://github.com/vanvalenlab/deepcell-tf |
| squidpy | Giovanni Palla, et al. 2021 | https://github.com/theislab/squidpy/ |
| Other | ||
HIGHLIGHTS.
Most small cell lung cancer (SCLC) tumors share a small PLCG2-high subpopulation
This PLCG2-high SCLC subpopulation is linked to metastasis and poor prognosis
SCLC is enriched in profibrotic and immunosuppressive monocytes/macrophages
The presence of myeloid cells is associated with the PLCG2-high SCLC subpopulation
ACKNOWLEDGEMENTS
This publication is part of the HTAN (Human Tumor Atlas Network) Consortium paper package. A list of HTAN members is available at https://humantumoratlas.org/htan-authors/. This work was supported by NCI U2C CA233284 (DP, CMR), the Robert J. and Helen C. Kleberg Foundation (DP, CMR), NCI U54 CA209975 (DP), NCI R01 CA197936, U24 CA213274, and R35 CA263816 (CMR), the SU2C/VAI Epigenetics Dream Team (CMR), the Alan and Sandra Gerry Metastasis and Tumor Ecosystems Center (DP, JMC, OC, IM, LM), the Druckenmiller Center for Lung Cancer Research (CMR, DRJ, MB, TS, AQV), AACR Lung Cancer Fellowship (JMC), ASCO Young Investigator Award (JMC), Parker Institute for Cancer Immunotherapy grant (TS, DP); International Association for the Study of Lung Cancer grant (TS), NIH K08 CA248723 (AC), NIH K08 CA245206 (MB), NCI R01 CA217169 and R01 CA240472 (DRJ). We gratefully acknowledge use of the Integrated Genomics Operation Core, funded by the NCI Cancer Center Support Grant P30 CA08748, Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. We also acknowledge Kathleen Daniels, David Humphries, Joana Da Silva Leite, Fang Fang, Barbara Oliveira, Magdalena Parys, Mark Kweens and Rui Gardner from the MSKCC Flow Cytometry Core for their invaluable help.
Footnotes
DECLARATION OF INTEREST
JMC reports advisory role in VantAI. AQV reports honoraria from AstraZeneca. MO reports advisory roles for PharMar, Novartis and Targeted Oncology and reports honoraria from Bristol-Myers Squibb and Merck Sharp & Dohme. CMR has consulted regarding oncology drug development with AbbVie, Amgen, Ascentage, Astra Zeneca, Bicycle, Celgene, Daiichi Sankyo, Genentech/Roche, Ipsen, Jazz, Lilly, Pfizer, PharmaMar, Syros, and Vavotek. CMR serves on the scientific advisory boards of Bridge Medicines, Earli, and Harpoon Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES.
- Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M et al. (2020). Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizarani N, Saviano A, Sagar Mailly L, Durand S, Herman JS, Pessaux P, Baumert TF, and Grün D (2019). A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 572, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel Anoop P,*1, 2, 3, 4 Itay Tirosh,*3 Trombetta John J., 3Alex K. Shalek, 3 Gillespie Shawn M., 2, 3, 4 Wakimoto Hiroaki, 1 Cahill Daniel P., 1 Nahed Brian V., 1 Curry William T., 1 Martuza Robert L., 1 Louis David N., 2 Rozenblatt-Rosen Orit, 3 Mari, 4†‡, and Human (2014). R es e a rc h | r e po r ts. Science (80-. ). 344, 1396–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M et al. (2018). Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 174, 1293–1308.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach DH, Park HJ, and Lee SK (2018). The Dual Role of Bone Morphogenetic Proteins in Cancer. Mol. Ther. - Oncolytics 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baine MK, Hsieh M-S, Lai WV, Egger JV, Jungbluth AA, Daneshbod Y, Beras A, Spencer R, Lopardo J, Bodd F et al. (2020). SCLC Subtypes Defined by ASCL1, NEUROD1, POU2F3, and YAP1: A Comprehensive Immunohistochemical and Histopathologic Characterization. J. Thorac. Oncol. 15, 1823–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baras AS, Drake C, Liu JJ, Gandhi N, Kates M, Hoque MO, Meeker A, Hahn N, Taube JM, Schoenberg MP et al. (2016). The ratio of CD8 to Treg tumor-infiltrating lymphocytes is associated with response to cisplatin-based neoadjuvant chemotherapy in patients with muscle invasive urothelial carcinoma of the bladder. Oncoimmunology 5, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basha O, Barshir R, Sharon M, Lerman E, Kirson BF, Hekselman I, and Yeger-Lotem E (2017). The TissueNet v.2 database: A quantitative view of protein-protein interactions across human tissues. Nucleic Acids Res. 45, D427–D431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best SA, Hess JB, Souza-Fonseca-Guimaraes F, Cursons J, Kersbergen A, Dong X, Rautela J, Hyslop SR, Ritchie ME, Davis MJ et al. (2020a). Harnessing Natural Killer Immunity in Metastatic SCLC. J. Thorac. Oncol. 15, 1507–1521. [DOI] [PubMed] [Google Scholar]
- Best SA, Hess JB, Souza-Fonseca-Guimaraes F, Cursons J, Kersbergen A, Dong X, Rautela J, Hyslop SR, Ritchie ME, Davis MJ et al. (2020b). Harnessing Natural Killer Immunity in Metastatic SCLC. J. Thorac. Oncol. 15, 1507–1521. [DOI] [PubMed] [Google Scholar]
- Borromeo MD, Savage TK, Kollipara RK, He M, Augustyn A, Osborne JK, Girard L, Minna JD, Gazdar AF, Cobb MH et al. (2016). ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell Rep. 16, 1259–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botev ZI, Grotowski JF, and Kroese DP (2010). Kernel density estimation via diffusion. Ann. Stat. 38, 2916–2957. [Google Scholar]
- Byers LA, and Rudin CM (2015). Small cell lung cancer: Where do we go from here? Cancer 121, 664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo E, Leon J, Mazzei G, Abolhassani N, Haruyama N, Saito T, Saido T, Hokama M, Iwaki T, Ohara T et al. (2017). Comparative profiling of cortical gene expression in Alzheimer’s disease patients and mouse models demonstrates a link between amyloidosis and neuroinflammation. Sci. Rep. 7, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalishazar MD, Wait SJ, Huang F, Ireland AS, Mukhopadhyay A, Lee Y, Schuman SS, Guthrie MR, Berrett KC, Vahrenkamp JM et al. (2019). MYC-driven small-cell lung cancer is metabolically distinct and vulnerable to arginine depletion. Clin. Cancer Res. 25, 5107–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Wu Q, Zhang Y, Lu T, Yue W, and Zhang D (2016). Tcf4 controls neuronal migration of the cerebral cortex through regulation of Bmp7. Front. Mol. Neurosci. 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN et al. (2015). Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagnostics 17, 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Yu J, Park A, Dubon MJ, Do J, Kim Y, Nam D, Noh J, and Park KS (2019). BMP-4 enhances epithelial mesenchymal transition and cancer stem cell properties of breast cancer cells via Notch signaling. Sci. Rep. 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, Ryu HS, Kim S, Lee JE, Park YH et al. (2017). Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijk D. van, Nainys J, Sharma R, Kathail P, Carr AJ, Moon KR, Mazutis L, Wolf G, Krishnaswamy S, and Pe’er D (2017). MAGIC: A diffusion-based imputation method reveals gene-gene interactions in single-cell RNA-sequencing data. BioRxiv 111591. [Google Scholar]
- Van Dijk D, Sharma R, Nainys J, Wolf G, Krishnaswamy S, Pe D, Dijk D. Van, Sharma R, Nainys J, Yim K et al. (2018). Recovering Gene Interactions from Single-Cell Data Resource Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 174, 716–729.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongre A, and Weinberg RA (2019a). New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 20, 69–84. [DOI] [PubMed] [Google Scholar]
- Dongre A, and Weinberg RA (2019b). New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 20, 69–84. [DOI] [PubMed] [Google Scholar]
- Dora D, Rivard C, Yu H, Bunn P, Suda K, Ren S, Lueke Pickard S, Laszlo V, Harko T, Megyesfalvi Z et al. (2020). Neuroendocrine subtypes of small cell lung cancer differ in terms of immune microenvironment and checkpoint molecule distribution. Mol. Oncol. 14, 1947–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremova M, Vento-Tormo M, Teichmann SA, and Vento-Tormo R (2020). CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 15, 1484–1506. [DOI] [PubMed] [Google Scholar]
- Egeblad M, and Werb Z (2002). New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2, 161–174. [DOI] [PubMed] [Google Scholar]
- Elakad O, Lois AM, Schmitz K, Yao S, Hugo S, Lukat L, Hinterthaner M, Danner BC, von Hammerstein-Equord A, Reuter-Jessen K et al. (2020). Fibroblast growth factor receptor 1 gene amplification and protein expression in human lung cancer. Cancer Med. 9, 3574–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farabaugh SM, Micalizzi DS, Jedlicka P, Zhao R, and Ford HL (2012). Eya2 is required to mediate the pro-metastatic functions of Six1 via the induction of TGF-Β signaling, epithelial-mesenchymal transition, and cancer stem cell properties. Oncogene 31, 552–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferone G, Lee MC, Sage J, and Berns A (2020). Cells of origin of lung cancers: Lessons from mouse studies. Genes Dev. 34, 1017–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo CR, Azevedo RA, Mousdell S, Resende-Lara PT, Ireland L, Santos A, Girola N, Cunha RLOR, Schmid MC, Polonelli L et al. (2018). Blockade of MIF-CD74 signalling on macrophages and dendritic cells restores the antitumour immune response against metastatic melanoma. Front. Immunol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M et al. (2015). MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, Sadik H, Argani P, Wagner P, Vahdat LT et al. (2012). Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res. 72, 1384–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay CM, Stewart CA, Park EM, Diao L, Groves SM, Heeke S, Nabet BY, Fujimoto J, Solis LM, Lu W et al. (2021a). Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 39, 346–360.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay CM, Stewart CA, Park EM, Diao L, Groves SM, Heeke S, Nabet BY, Fujimoto J, Solis LM, Lu W et al. (2021b). Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 39, 346–360.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar AF, Carney DN, Nau MM, and Minna JD (1985). Characterization of Variant Subclasses of Cell Lines Derived from Small Cell Lung Cancer Having Distinctive Biochemical, Morphological, and Growth Properties. Cancer Res. 45, 2924–2930. [PubMed] [Google Scholar]
- George J, Lim JS, Jang SJ, Cun Y, Ozretia L, Kong G, Leenders F, Lu X, Fernández-Cuesta L, Bosco G et al. (2015). Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giopanou I, Lilis I, Papaleonidopoulos V, Agalioti T, Kanellakis NI, Spiropoulou N, Spella M, and Stathopoulos GT (2017). Tumor-derived osteopontin isoforms cooperate with TRP53 and CCL2 to promote lung metastasis. Oncoimmunology 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald NF, Miller G, Moen E, Kong A, Kagel A, Camacho C, Mcintosh BJ, Leow K, Schwartz MS, Dougherty T et al. (2021). Whole-cell segmentation of tissue images with human-level performance using large-scale data annotation and deep learning. BioRxiv 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R, Rowe PSN, Liu S, Simpson LG, Xiao ZS, and Darryl Quarles L (2002). Inhibition of MEPE cleavage by Phex. Biochem. Biophys. Res. Commun. 297, 38–45. [DOI] [PubMed] [Google Scholar]
- Hamilton NE, and Ferry M (2018). Ggtern: Ternary diagrams using ggplot2. J. Stat. Softw. 87. [Google Scholar]
- Horn L, Mansfield AS, Szczęsna A, Havel L, Krzakowski M, Hochmair MJ, Huemer F, Losonczy G, Johnson ML, Nishio M et al. (2018a). First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 379, 2220–2229. [DOI] [PubMed] [Google Scholar]
- Horn L, Mansfield AS, Szczȩsna A, Havel L, Krzakowski M, Hochmair MJ, Huemer F, Losonczy G, Johnson ML, Nishio M et al. (2018b). First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N. Engl. J. Med. 379, 2220–2229. [DOI] [PubMed] [Google Scholar]
- Hrckulak D, Janeckova L, Lanikova L, Kriz V, Horazna M, Babosova O, Vojtechova M, Galuskova K, Sloncova E, and Korinek V (2018). Wnt effector TCF4 is dispensable for Wnt signaling in human cancer cells. Genes (Basel). 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubaux R, Thu KL, Coe BP, Macaulay C, Lam S, and Lam WL (2013). EZH2 promotes E2F-driven SCLC tumorigenesis through modulation of apoptosis and cell-cycle regulation. J. Thorac. Oncol. 8, 1102–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland AS, Micinski AM, Kastner DW, Guo B, Wait SJ, Spainhower KB, Conley CC, Chen OS, Guthrie MR, Soltero D et al. (2020). MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate. Cancer Cell 60–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Cui L, Yie TA, Rom WN, Cheng H, and Tchou-Wong KM (2001). Inhibition of anchorage-independent growth and lung metastasis of A549 lung carcinoma cells IκBβ. Oncogene 20, 2254–2263. [DOI] [PubMed] [Google Scholar]
- Kassis J, Moellinger J, Lo H, Greenberg NM, Kim HG, and Wells A (1999). A role for phospholipase C-γ-mediated signaling in tumor cell invasion. Clin. Cancer Res. 5, 2251–2260. [PubMed] [Google Scholar]
- Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, Yang SR, Kurian A, Van Valen D, West R et al. (2018a). A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging. Cell 174, 1373–1387.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, Yang SR, Kurian A, Van Valen D, West R et al. (2018b). A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging. Cell 174, 1373–1387.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G, Sukhov V, and Sergushichev A (2016). Fast gene set enrichment analysis. 1–29. [Google Scholar]
- Kusakari S, Nawa M, Sudo K, and Matsuoka M (2018). Calmodulin-like skin protein protects against spatial learning impairment in a mouse model of Alzheimer disease. J. Neurochem. 144, 218–233. [DOI] [PubMed] [Google Scholar]
- Laughney AM, Hu J, Campbell NR, Bakhoum SF, Setty M, Lavallée VP, Xie Y, Masilionis I, Carr AJ, Kottapalli S et al. (2020). Regenerative lineages and immune-mediated pruning in lung cancer metastasis (Springer US; ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lee SJ, Conway OJ, Jansen I, Carrasquillo MM, Kleineidam L, van den Akker E, Hernández I, van Eijk KR, Stringa N, Chen JA et al. (2019). A nonsynonymous mutation in PLCG2 reduces the risk of Alzheimer’s disease, dementia with Lewy bodies and frontotemporal dementia, and increases the likelihood of longevity. Acta Neuropathol. 138, 237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Simonds EF, Bendall SC, Davis KL, Amir EAD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER et al. (2015). Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 162, 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitin HM, Yuan J, Cheng YL, Ruiz FJ, Bush EC, Bruce JN, Canoll P, Iavarone A, Lasorella A, Blei DM et al. (2019). De novo gene signature identification from single- cell RNA - seq with hierarchical Poisson factorization. Mol. Syst. Biol. 15, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Diao B, Guo S, Huang X, Yang C, Feng Z, Yan W, Ning Q, Zheng L, Chen Y et al. (2017). VSIG4 inhibits proinflammatory macrophage activation by reprogramming mitochondrial pyruvate metabolism. Nat. Commun. 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Guo L, Lin G, Chen Z, Chen T, Lin J, Zhang B, and Gu X (2015). Clinical and prognostic significance of OPN and VEGF expression in patients with non-small-cell lung cancer. Cancer Epidemiol. 39, 539–544. [DOI] [PubMed] [Google Scholar]
- McInnes L, Healy J, and Melville J (2018). UMAP: Uniform manifold approximation and projection for dimension reduction. ArXiv. [Google Scholar]
- Mollaoglu G, Guthrie MR, Böhm S, Brägelmann J, Can I, Ballieu PM, Marx A, George J, Heinen C, Chalishazar MD et al. (2017). MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 31, 270–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, Yuan F, Chen S, Leung HM, Villoria J et al. (2018). A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno Ayala MA, Gottardo MF, Zuccato CF, Pidre ML, Nicola Candia AJ, Asad AS, Imsen M, Romanowski V, Creton A, Isla Larrain M et al. (2020). Humanin Promotes Tumor Progression in Experimental Triple Negative Breast Cancer. Sci. Rep. 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris DL, Johnson S, Bleck CKE, Lee D-Y, and Tjandra N (2020). Humanin selectively prevents the activation of pro-apoptotic protein BID by sequestering it into fibers. J. Biol. Chem. jbc.RA120.013023. [DOI] [PubMed] [Google Scholar]
- Ni Z, Chen S, Brown J, and Kendziorski C (2020). CB2 improves power of cell detection in droplet-based single-cell RNA sequencing data. Genome Biol. 21, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owonikoko TK, Dwivedi B, Chen Z, Zhang C, Barwick B, Ernani V, Zhang G, Gilbert-Ross M, Carlisle J, Khuri FR et al. (2021). YAP1 Expression in SCLC Defines a Distinct Subtype With T-cell–Inflamed Phenotype. J. Thorac. Oncol. 16, 464–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palla G, Spitzer H, Klein M, Fischer D, Christina Schaar A, Benedikt Kuemmerle L, Rybakov S, Ibarra IL, Holmberg O, Virshup I et al. (2021). Squidpy: a scalable framework for spatial single cell analysis. BioRxiv 2021.02.19.431994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang X, Xie R, Zhang Z, Liu Q, Wu S, and Cui Y (2019). Identification of SPP1 as an Extracellular Matrix Signature for Metastatic Castration-Resistant Prostate Cancer. Front. Oncol. 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HJ, and Helfman DM (2019). Up-regulated fibronectin in 3D culture facilitates spreading of triple negative breast cancer cells on 2D through integrin β−5 and Src. Sci. Rep. 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, Statsenko G, Hochmair MJ, Özgüroğlu M, Ji JH et al. (2019). Durvalumab plus platinum–etoposide versus platinum–etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet 394, 1929–1939. [DOI] [PubMed] [Google Scholar]
- Pearsall SM, Humphrey S, Revill M, Morgan D, Frese KK, Galvin M, Kerr A, Carter M, Priest L, Blackhall F et al. (2020). The Rare YAP1 Subtype of SCLC Revisited in a Biobank of 39 Circulating Tumor Cell Patient Derived Explant Models: A Brief Report. J. Thorac. Oncol. 15, 1836–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitulescu ME, and Adams RH (2010). Eph/ephrin molecules - A hub for signaling and endocytosis. Genes Dev. 24, 2480–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier JT, Gardner EE, Connis N, Moreira AL, De Stanchina E, Hann CL, and Rudin CM (2015). DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene 34, 5869–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston CC, Maurer MJ, Oberg AL, Visscher DW, Kalli KR, Hartmann LC, Goode EL, and Knutson KL (2013). The ratios of CD8+ T cells to CD4+CD25+ FOXP3+ and FOXP3− T cells correlate with poor clinical outcome in human serous ovarian cancer. PLoS One 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS et al. (2017). Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 171, 1611–1624.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanal-Villalonga A, Molina-Pinelo S, Cirauqui C, Ojeda-Márquez L, Marrugal Á, Suarez R, Conde E, Ponce-Aix S, Enguita AB, Carnero A et al. (2019). FGFR1 Cooperates with EGFR in Lung Cancer Oncogenesis, and Their Combined Inhibition Shows Improved Efficacy. J. Thorac. Oncol. 14, 641–655. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J et al. (2012). Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet. 44, 1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, Heymach JV, Johnson JE, Lehman JM, MacPherson D et al. (2019). Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat. Rev. Cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Brambilla E, Faivre-Finn C, and Sage J (2021). Small-cell lung cancer. Nat. Rev. Dis. Prim. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala G, Dituri F, Raimondi C, Previdi S, Maffucci T, Mazzoletti M, Rossi C, Iezzi M, Lattanzio R, Piantelli M et al. (2008). Phospholipase Cγ1 is required for metastasis development and progression. Cancer Res. 68, 10187–10196. [DOI] [PubMed] [Google Scholar]
- Saunders LR, Bankovich AJ, Anderson WC, Aujay MA, Bheddah S, Black KA, Desai R, Escarpe PA, Hampl J, Laysang A et al. (2015). A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci. Transl. Med. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Edelkraut U, Daniel G, Hoffmann A, and Spengler D (2014). Zac1 Regulates Cell Cycle Arrest in Neuronal Progenitors via Tcf4. Mol. Cell. Biol. 34, 1020–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seabold S, and Perktold J (2010). Statsmodels: Econometric and Statistical Modeling with Python. Proc. 9th Python Sci. Conf. 92–96. [Google Scholar]
- Shang B, Liu Y, Jiang SJ, and Liu Y (2015). Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 5, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shurin MR (2018). Osteopontin controls immunosuppression in the tumor microenvironment. J. Clin. Invest. 128, 5209–5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2020). Cancer statistics, 2020. CA. Cancer J. Clin. 70, 7–30. [DOI] [PubMed] [Google Scholar]
- Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, Herbst RH, Rogel N, Slyper M, Waldman J et al. (2019). Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 178, 714–730.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkin D, Thomas A, and Teicher BA (2019). Are neuroendocrine negative small cell lung cancer and large cell neuroendocrine carcinoma with WT RB1 two faces of the same entity? Lung Cancer Manag. 8, LMT13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sricharan K, Raich R, and Hero AO (2012). Estimation of nonlinear functionals of densities with confidence. IEEE Trans. Inf. Theory 58, 4135–4159. [Google Scholar]
- Stein-O’Brien GL, Arora R, Culhane AC, Favorov AV, Garmire LX, Greene CS, Goff LA, Li Y, Ngom A, Ochs MF et al. (2018). Enter the Matrix: Factorization Uncovers Knowledge from Omics. Trends Genet. 34, 790–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al. (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M, Talavera-López C, Maatz H, Reichart D, Sampaziotis F et al. (2020). SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 26, 681–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Koga K, Linge HM, Zhang Y, Lin X, Metz CN, Al-Abed Y, Ojamaa K, and Miller EJ (2009). Macrophage CD74 contributes to MIF-induced pulmonary inflammation. Respir. Res. 10, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therneau TM, and Grambsch PM (2000). Modeling survival analysis: Extending cox models.
- Traag VA, Waltman L, and van Eck NJ (2019). From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, Chang S, Conley SD, Mori Y, Seita J et al. (2019). A molecular cell atlas of the human lung from single cell RNA sequencing. BioRxiv 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallières E, Shepherd FA, Crowley J, Van Houtte P, Postmus PE, Carney D, Chansky K, Shaikh Z, and Goldstraw P (2009). The IASLC lung cancer staging project: Proposals regarding the relevance of TNM in the pathologic staging of small cell lung cancer in the forthcoming (seventh) edition of the TNM classification for lung cancer. J. Thorac. Oncol. 4, 1049–1059. [DOI] [PubMed] [Google Scholar]
- Wang JP, and Hielscher A (2017). Fibronectin: How its aberrant expression in tumors may improve therapeutic targeting. J. Cancer 8, 674–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, and Werb Z (2020). Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 11, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooten DJ, Groves SM, Tyson DR, Liu Q, Lim JS, Albert R, Lopez CF, Sage J, and Quaranta V (2019). Systems-level network modeling of Small Cell Lung Cancer subtypes identifies master regulators and destabilizers. PLoS Comput. Biol. 15, e1007343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, and Zhou BP (2010). TNF-α/NFκ-B/Snail pathway in cancer cell migration and invasion. Br. J. Cancer 102, 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Qu F, Cai H, Chuang C-H, Lim JS, Jahchan N, Grüner BM, S Kuo C, Kong C, Oudin MJ et al. (2019). Axon-like protrusions promote small cell lung cancer migration and metastasis. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarchoan M, Albacker LA, Hopkins AC, Montesion M, Murugesan K, Vithayathil TT, Zaidi N, Azad NS, Laheru DA, Frampton GM et al. (2019). PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y (2012). Semaphorin Signaling in Vertebrate Neural Circuit Assembly. Front. Mol. Neurosci. 5, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Ergin V, Lin L, Stork C, Chen L, and Zheng S (2019a). Axonogenesis Is Coordinated by Neuron-Specific Alternative Splicing Programming and Splicing Regulator PTBP2. Neuron 101, 690–706.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, Modak M, Carotta S, Haslinger C, Kind D et al. (2019b). Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 179, 829–845.e20. [DOI] [PubMed] [Google Scholar]
- Zhao DH, Hong JJ, Guo SY, Yang RL, Yuan J, Wen CJ, Zhou KY, and Li CJ (2004). Aberrant expression and function of TCF4 in the proliferation of hepatocellular carcinoma cell line BEL-7402. Cell Res. 14, 74–80. [DOI] [PubMed] [Google Scholar]
- Zimmerman KD, Espeland MA, and Langefeld CD (2020). Pseudoreplication bias in single-cell studies; A practical solution. BioRxiv 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical characteristics of samples analyzed by single-cell RNA-seq, Related to Figures 1 and S1
Table S2. Shared mutations detected in SCLC-A and SCLC-N subpopulations of the biphenotypic sample Ru1215, Related to Figure S2
Table S3. Differentially expressed genes comparing SCLC-A versus SCLC-N in single-cell RNA-seq of SCLC cells using MAST, Related to Figures 2 and S2
Table S4. Differentially expressed genes comparing SCLC-A versus rest in single-cell RNA-seq of SCLC cells using MAST, Related to Figures 2 and S2
Table S5. Differentially expressed genes comparing SCLC-N versus rest in single-cell RNA-seq of SCLC cells using MAST, Related to Figures 2 and S22
Table S6. Differentially expressed genes comparing SCLC-P versus rest in single-cell RNA-seq of SCLC cells using MAST, Related to Figures 2 and S2
Table S7. Pathway enrichment of SCLC-A versus SCLC-N in SCLC cells using GSEA, Related to Figures 2 and S2
Table S8. Pathway enrichment of SCLC-P versus rest in SCLC cells using GSEA, Related to Figures 2 and S2
Table S9. Pathway enrichment in the recurrent, PLCG2-high SCLC cluster versus rest of SCLC cells using GSEA, Related to Figures 3 and S3
Table S10. Differentially expressed genes of SCLC recurrent subclone (cluster 22) versus rest in single-cell RNA-seq using MAST, Related to Figures 3 and S3
Table S11. Differentially expressed genes that are recurrently overexpressed in the recurrent, PLCG2-high SCLC cluster across samples, ranked by the Bonferroni-adjusted Edgington’s combined p-value, Related to Figures 3 and S3
Table S12. Gene modules with high knnDREMI conditioned on PLCG2, divided by low, medium, and high PLCG2 expression, Related to Figure S3
Table S13. Pathways with average z-scores of gene expression correlated with the average z-score of gene expression in the high-PLCG2 gene module, Related to Figure S3
Table S14. Clinical characteristics and ASCL1/NEUROD1 positivity on immunohistochemistry for an independent SCLC cohort analyzed by Vectra and MIBI, Related to Figures 4, 5, 7, S4, S7
Table S15. A summary of antibodies, MIBItag, and concentrations used in MIBI-TOF, Related to Figures 4, 5, 7, S4, S7
Table S16. Survival data and clinical covariates of samples analyzed by single-cell RNA-seq and stratified by fraction of the recurrent, PLCG2-high SCLC cluster, Related to Figures 4 and S4
Table S17. Clinical characteristics, CD45+ percentage, and ASCL1/NEUROD1 positivity on immunohistochemistry for an independent SCLC cohort analyzed by flow cytometry, Related to Figure S4
Table S18. Number of immune cell types per sample in scRNA-seq, Related to Figure S5
Table S19. Differentially expressed genes of Mono/Mφ cluster 1 vs other Mono/Mφ subsets in single-cell RNA-seq using MAST, Related to Figure S7
Table S20. Differentially expressed genes of SCLC-A versus rest in bulk RNA-seq data from (George et al., 2015) and (Rudin et al., 2012) using limma, Related to STAR Methods
Table S21. Differentially expressed genes of SCLC-N versus rest in bulk RNA-seq data from (George et al., 2015) and (Rudin et al., 2012) using limma, Related to STAR Methods
Table S22. Differentially expressed genes of SCLC-P versus rest in bulk RNA-seq data from (George et al., 2015) and (Rudin et al., 2012) using limma, Related to STAR Methods
Table S23. Differentially expressed genes of SCLC-Y versus rest in bulk RNA-seq data from (George et al., 2015) and (Rudin et al., 2012) using limma, Related to STAR Methods
Table S24. Pathway enrichment in Mono/Mφ cluster 1 vs other Mono/ Mφ subsets using GSEA, Related to STAR Methods
Data S1. Gene sets curated from MSigDB and literature, used for pathway enrichment analysis in SCLC subtypes and clusters, Related to STAR Methods
Data S2. Markers used for cell type annotation, curated from literature, Related to STAR Methods
Data Availability Statement
Software and tools used for the enclosed data analysis will be provided open source at http://github.com/dpeerlab. In collaboration with the NIH-funded HTAN Data Coordinating Center (U24), single-cell analysis at time of publication will be made available as an interactive, online platform for independent visualization and analysis. MIBI-TOF data will be made available at https://mskcc.ionpath.com/tracker.