

SUMMARY
E-cadherin junctions facilitate assembly and disassembly of cell contacts that drive development and homeostasis of epithelial tissues. In this study, using Xenopus embryonic kidney and Madin-Darby canine kidney (MDCK) cells, we investigate the role of the Wnt/planar cell polarity (PCP) formin Daam1 (Dishevelled-associated activator of morphogenesis 1) in regulating E-cadherin-based intercellular adhesion. Using live imaging, we show that Daam1 localizes to newly formed cell contacts in the developing nephron. Furthermore, analyses of junctional filamentous actin (F-actin) upon Daam1 depletion indicate decreased microfilament localization and slowed turnover. We also show that Daam1 is necessary for efficient and timely localization of junctional E-cadherin, mediated by Daam1’s formin homology domain 2 (FH2). Finally, we establish that Daam1 signaling promotes organized movement of renal cells. This study demonstrates that Daam1 formin junctional activity is critical for epithelial tissue organization.
Graphical abstract
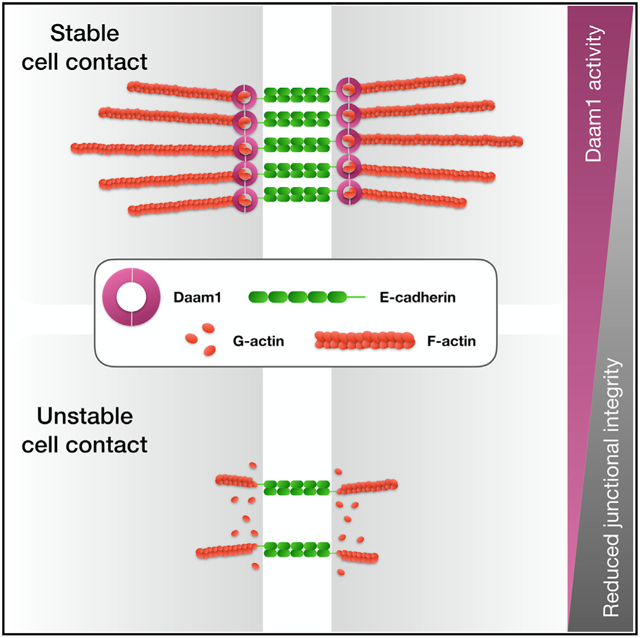
In brief
How cells remodel their adhesions through cell-surface proteins such as E-cadherin is a central question in epithelial tissue biology. Krneta-Stankic et al. show that the Wnt/PCP formin Daam1 regulates cytoskeletal membrane dynamics and E-cadherin localization within developing nephrons. These findings provide a new framework for studying cell-cell adhesion and nephron morphogenesis.
INTRODUCTION
Extensive cellular rearrangements with changes in cell shape drive morphogenesis, including the process of tubulogenesis. To execute these processes, cells must interact with each other and their environment in a timely and coordinated manner. These interactions involve transduction of signals from adhesive contacts with the extracellular matrix (ECM) and neighboring cells. How cells remodel their adhesions is one of the central questions in epithelial biology.
The cadherin family of cell adhesion proteins facilitates intercellular adhesion and formation of cell junctions (Adams et al., 1998; Takeichi, 2014; Yap et al., 2015). Changes in E-cadherin-based adhesion are associated with developmental disorders and disease progression (Friedl and Mayor, 2017; Mendonsa et al., 2018). E-cadherin levels at intercellular contacts depend on actin cytoskeleton organization, but much remains to be learned about regulation of actin assembly at these adhesive sites. Moreover, much of our understanding of in vivo actin regulation and cell adhesion dynamics stems from observations in cell culture, invertebrate embryos, and vertebrate skin. Understanding junction dynamics in intact vertebrate tissues is challenging because of technical limitations and tissue inaccessibility. In this study, we probe the role of the formin protein Dishevelled-associated activator of morphogenesis 1 (Daam1) in intercellular adhesion during kidney development using Xenopus laevis embryonic kidney and Madin-Darby canine kidney (MDCK) cells.
Similar to many organs in our body, the kidney consists of a network of epithelial tubules. The morphology of these tubules, called nephrons, is vital for kidney function. Nephrons arise from the mass of mesenchymal cells that undergo mesenchymal-epithelial transitions (MET) and coordinated cell rearrangements to form nephric tubules consisting of tightly connected epithelial cells (McMahon, 2016; Saxen, 1987). Oriented cell intercalations drive elongation of nephric tubules through a process called convergent extension (CE) (Castelli et al., 2013; Karner et al., 2009; Kunimoto et al., 2017; Lienkamp et al., 2012). Extensive cytoskeletal rearrangements characterized by alterations in cell shape and coordinated cell interactions accompany CE morphogenetic movements. Although E-cadherin-based adhesions are implicated in mediating MET and coordination of cell rearrangements (Campbell and Casanova, 2016), little is known about how they function in nephrogenesis (Combes et al., 2015; Lefevre et al., 2017; Marciano et al., 2011; Vestweber et al., 1985).
Daam1 is a formin protein required for nephric tubulogenesis (Miller et al., 2011). Formin proteins organize the actin cytoskeleton by nucleating and polymerizing unbranched actin filaments. Although Rho GTPases activate most formins, activation of Daam1 depends on its interaction with Dishevelled (Dvl), a key intracellular component of the Wnt signaling pathway (Liu et al., 2008). Wnt signaling plays an essential role in nephron development (McMahon, 2016; Miller and McCrea, 2010). Secreted Wnt ligands bind Frizzled (Fz) receptors and subsequently, via Dvl, regulate canonical (β-catenin-dependent) and non-canonical (β-catenin-independent)/planar cell polarity (PCP) signaling. Although canonical signaling commonly governs inductive events and cell fate, the non-canonical/PCP branch mostly influences cell behaviors and morphology. Nonetheless, the roles of different branches of the Wnt pathway continue to evolve as studies provide evidence of cross-talk (Nagy et al., 2016; O’Brien et al., 2018). Dvl regulates the non-canonical/PCP branch of the Wnt pathway through direct interaction with Daam1 (Liu et al., 2008).
Increasing evidence suggests that formins function as key regulators of actin assembly at cell junctions (Grikscheit and Grosse, 2016). Recent work using mouse mammary gland epithelial cells indicates that Daam1 is important for the stability of epithelial junctions (Nishimura et al., 2016). Here we expand these findings by examining the role of Daam1 in cell junctions in the context of tissue morphogenesis during nephron development. Furthermore, using live imaging, we show that, during establishment of cell junctions, Daam1 localizes to cellular protrusions that initiate cell-cell contact and, subsequently, to newly formed junctions to promote their stability. We find that Daam1 facilitates nephron morphogenesis by regulating junctional filamentous actin (F-actin) assembly and, in turn, promotes E-cadherin-based epithelial adhesion.
RESULTS
Daam1 co-localizes with F-actin and E-cadherin within the nephric primordium
The Xenopus embryonic kidney consists of a single large, fully functional nephron. Pronephric cells are specified from the mesoderm by late gastrula stages, Nieuwkoop and Faber (NF) stage 12.5 (Brennan et al., 1998, 1999; Vize et al., 1995). In the early tail bud (~NF stage 20), the pronephric primordium condenses, becoming visible as a bulge ventral to the anterior somites (Vize et al., 2003). This primordium undergoes MET transition and CE movements to form nephric tubules (Lienkamp et al., 2012; Vize et al., 2003). Although the exact timing of MET is not well understood, the onset of CE begins simultaneously with lumen formation (~NF stage 30). Pronephric function begins around NF stages 37/38 and is fully operative in NF stage 40 tadpoles (Vize et al., 1995).
Knockdown (KD) of Daam1 in Xenopus disrupts nephron morphology without apparent effects on expression of early differentiation genes (Miller et al., 2011). Differentiation signals driving nephric mesoderm development in Xenopus largely function before onset of tubular morphogenesis (Vize et al., 2003). To further probe how Daam1 regulates shaping of nephrons, we analyzed the subcellular localization of Daam1 at the beginning of tubular morphogenesis (~NF stage 30). Through targeted microinjections into embryonic cells fated to contribute to the kidney (DeLay et al., 2016; Moody and Kline, 1990), we expressed fluorescently tagged daam1 mRNA in nephron progenitors and analyzed its localization in fixed and live tissue.
For analyses of fixed tissue, embryos were subjected to whole-mount staining. Samples were stained with an antibody against GFP to visualize Daam1 and an antibody against Lhx1 to label nephron progenitors (DeLay et al., 2018; Venegas-Ferrin et al., 2010). Additional staining with Phalloidin allowed us to visualize the F-actin cytoskeleton; alternatively, another antibody was used to define E-cadherin’s localization (Figure 1). Consistent with its previously reported localization in other cell types (Corkins et al., 2019; Higashi et al., 2019; Jaiswal et al., 2013; Kawabata Galbraith et al., 2018; Kida et al., 2007; Nishimura et al., 2012, 2016), Daam1 co-localizes with patches of F-actin with a diffuse staining pattern in the cytoplasm and strong localization to cell junctions (Figure 1A). Furthermore, co-immunostaining for GFP and E-cadherin demonstrated that E-cadherin is expressed within the nephric primordium and that it co-localizes with Daam1 at cell junctions (Figure 1B).
Figure 1. Daam1 co-localizes with junctional F-actin and E-cadherin during early nephron development.

(A) Confocal maximum image projections of whole-mount immunostaining of Xenopus nephric primordium labeled by Lhx1 (white) and GFP to visualize Daam1 (green) in conjunction with phalloidin staining to visualize F-actin (magenta); scale bar, 50 μm. a’–a”” are close-up images of region a; scale bars, 20 μm. The first panel consists of the entire z stack to show the cell positions within the entire kidney. In contrast, images displayed in a’–a”” contain a subset of the z slices to exclude the intense signal from phalloidin within the Xenopus skin. The white dotted box in a”–a”” marks the junction shown enlarged in the insets (scale bars, 10 mm) and analyzed in (B).
(B) A line plot showing the junctional intensity of F-actin (magenta) and GFP-Daam1 (green) of the junction highlighted in (A), a”–a””, by the white dotted box.
(C) Confocal maximum image projections of whole-mount immunostaining of Xenopus nephric primordium labeled by Lhx1 (white) and GFP to visualize Daam1 (green) in conjunction with E-cadherin (magenta); scale bar, 50 μm. c’–c”” are close-up images of region c; scale bars, 20 μm. The first panel consists of the entire z stack to show the cell positions within the entire kidney. In contrast, images displayed in c’–c”” contain a subset of the z slices to exclude the intense signal from E-cadherin within the Xenopus skin. The white dotted box in c”–c”” marks the junction shown enlarged in the insets (scale bars, 10 μm) and analyzed in (D).
(D) A line plot showing the junctional intensity of E-cadherin (magenta) and GFP-Daam1 (green) of the junction highlighted in c”–c”” by the white dotted box.
During early development, the Xenopus epithelium’s opaqueness hinders imaging of fluorescence in internal tissues, including the pronephric primordium. In fixed tissues, this is overcome by using clearing agents such as benzyl alcohol (BA):-benzyl benzoate (BB). However, this process requires embryo dehydration in methanol or ethanol, which is incompatible with use of fluorescent phalloidins (Becker and Gard, 2006). To overcome this obstacle, we briefly washed embryos (<20 s) in isopropanol before clearing with BA:BA (Nworu et al., 2014; Strickland et al., 2004), allowing visualization of Daam1 in conjunction with F-actin in the intact pronephric primordium (Figure 1A).
To overcome an analogous challenge in live embryos, we developed a way of imaging the nephric primordium in vivo (Figure 2A). Previous studies have used a “windowed” embryo approach to microsurgically remove the ectoderm to expose and image underlying tissue (Kim and Davidson, 2013). Adapting this approach, we created kidney-windowed embryos by removing the surface epithelium and exposing the underlying nephric primordium for high-resolution in vivo imaging. In vivo time-lapse imaging of kidney-windowed embryos showed GFP-Daam1 localizing to cell junctions. However, we observed that GFP-Daam1 is also localized to cytoplasmic vesicles and cellular protrusions (Figure 2B; Video S1). These observations are in line with previous reports regarding Daam1’s localization and likely hindered because of unfavorable fixation conditions for observation of these dynamic processes (Corkins et al., 2019; Jaiswal et al., 2013; Kawabata Galbraith et al., 2018; Kida et al., 2007; Nishimura et al., 2012, 2016). Moreover, to better understand the dynamics of Daam1 in cell junctions, we imaged de novo formation of cell junctions in dissociated GFP-Daam1-expressing nephric primordium cells (Figure 2A). In vivo time-lapse analyses showed that Daam1 localizes to filopodium-like protrusions and, subsequently, to newly formed junctions (Figure 2C; Video S2).
Figure 2. Daam1 localizes to newly formed cell-cell contacts.

(A) Schematic showing the steps involved in preparing “windowed kidney” embryos and primary cultures expressing GFP-Daam1. For clarity, the 8-cell GFP-Daam1-injected Xenopus blastomere is fate-mapped strictly to the nephric primordium, and blastomere also contributes to the epidermis, ventral and dorsal somites, hindgut, proctodeum, and trunk neural crest cells (DeLay et al., 2016; Moody and Kline, 1990).
(B) Time-lapse imaging montage of the nephric primordium expressing GFP-Daam1 in “windowed kidney” embryos. Elapsed time is indicated at the top; scale bars, 10 μm. The white arrows point to cellular protrusions shown enlarged in the insets (scale bars, 5 μm); see Video S1.
(C) Time-lapse imaging montage showing cells isolated from a developing nephron expressing GFP-Daam1 mRNA adhering to each other. Elapsed time is indicated at the top; scale bars, 5 μm. The orange dotted line delineates the border arising between two cells, and the white arrows point to filopodium-like cellular protrusions; see Video S2.
Finally, we assessed Daam1’s localization in the mature epithelium of fully developed nephrons (Figure S1). Interestingly, junctional localization of Daam1 was not detected in the mature epithelium. These data suggest a role of Daam1 in regulating intercellular adhesion of renal progenitors, specifically at onset of tubulogenesis and CE.
Daam1 controls organization and assembly of junctional F-actin within the nephric primordium
Daam1’s co-localization with junctional F-actin in developing nephrons led us to ask whether Daam1 regulates F-actin. To address this, we depleted Daam1 in nephron progenitors using Morpholino (MO) oligos in kidney-targeted microinjections (DeLay et al., 2016; Moody and Kline, 1990). A proven Daam1 MO (Habas et al., 2001; Liu et al., 2008; Miller et al., 2011) was co-injected with a membrane GFP (mGFP) mRNA linage tracer. Morphants fixed at onset of tubular morphogenesis (~NF stage 30) were subjected to whole-mount immunostaining and confocal imaging. To verify KD success, we carried out western blot analyses of protein lysates from NF stages 28–32 MO-injected embryos (Figure 3B). Phalloidin staining revealed that, upon Daam1 depletion, F-actin in renal progenitors becomes disorganized and reduced at cell junctions (Figures 3A and 3C; Videos S3 and S4). These results suggest that Daam1 helps organize the F-actin cytoskeleton at cell junctions during nephron development.
Figure 3. Effects of Daam1 depletion on the nephrogenic primordium.

Kidney-targeted microinjections using control or Daam1 MO combined with the membrane-GFP mRNA (used as a lineage tracer) were carried out to analyze the effect of Daam1 depletion on Xenopus developing kidney.
(A) Maximum-projection confocal images of control or Daam1 KD embryos stained whole mount with antibodies against GFP (to detect the membrane-GFP tracer, green) and Lhx1 (to label nephric progenitors, white), in conjunction with phalloidin (to visualize F-actin, magenta). The top panels consist of the entire z stacks to show the cell positions within the entire kidney; scale bars, 50 μm. a’–a” and b’–b” are close-up images of the corresponding regions in white boxes consisting of a subset of the z slices to exclude the intense F-actin signal within the Xenopus skin; scale bars, 20 μm. The “seam” of enriched actin in the nephron center corresponds to the region where the lumen will form; see Videos S3 and S4.
(B) Western blot and graph of densitometry measures showing Daam1 and GAPDH protein levels for uninjected wild-type (WT), control (standard MO)-injected, and Daam1 KD (Daam1 MO)-injected embryos. Embryo lysates were pooled from 10–20 V2-cell injected embryos at stages NF 28–32, and approximately 1/2 embryo was loaded per lane. Individual band intensities are normalized to the uninjected band and plotted in arbitrary units (a.u.) for uninjected (black), control (orange), and Daam1 KD (blue). The results are expressed as means ± SEM from four independent experiments. nsp > 0.05, ***p ≤ 0.001, analyzed by one-way ANOVA.
(C) The graph showing the mean relative fluorescence intensity levels in a.u. of junctional F-actin in the nephric primordia of control (orange) and Daam1 KD (blue) embryos. ncontrol = 40 junctions and nDaam1 KD = 40 junctions on 2 embryos, denoted by open and closed circles, respectively. ****p ≤ 0.0001, analyzed by unpaired t test.
(D–G) Morphometric analyses of control and Daam1-depleted nephric primordia. The thick bars represent the mean for control (orange) and Daam1-depleted (blue) embryos. ****p ≤ 0.0001, analyzed by unpaired t test. Graphs show comparison of control and Daam1-depleted nephric primordia of (D) the average number of Lhx1-positive nephron progenitors, where ncontrol = 5 embryos and nDaam1KD = 5 embryos; (E) the relative distance between nearest neighbors of Lhx1-positive nuclei; (F) the relative cell area; and (G) the relative circularity, where 1 represents a perfect circle.
(E–G) ncontrol = 40 junctions on 2 embryos and nDaam1 KD = 40 junctions on 2 embryos, denoted by open and closed circles, respectively.
Alterations in the positioning and organization of nephron progenitors were observed upon Daam1 depletion. Nephric primordia in these morphants consist of fewer progenitor cells (Figure 3D) spaced farther apart (Figure 3E) compared with controls. In addition to changes in the number and position of progenitors, we also noted cell morphology changes. Nephric progenitors with diminished Daam1 activity display an increase in cell area (Figure 3F) and circularity (Figure 3G). The resultant reduction in Lhx1-positive cells is most likely the outcome of reducing the number of progenitors. Preliminary data show that, upon Daam1 KD, some cells are excluded from the nephric primordium, likely because of competitive elimination (data not shown). Furthermore, the actin cytoskeleton promotes intercellular adhesion, and the reduced number, increased distance, size, and circularity of Daam1-depleted Lhx1 cells may result from reduced cell adhesion.
Because F-actin polymerization is a dynamic process, to better understand the mechanisms underlying the observed morphological perturbations, we assessed F-actin behavior in vivo (Figures 4 and S2). In kidney-windowed NF stage 30 embryos, F-actin dynamics were assessed in intact animals. We labeled F-actin in developing nephrons by co-injecting mCherry-tagged utrophin mRNA (Burkel et al., 2007) with control or Daam1 MO (Figure S2). Daam1 KD cells display cell borders that are less straight than those of control cells. Furthermore, the cortex underlying cell junctions appears less dense and more fragmented in Daam1-depleted cells, implying a mechanically weaker cortex. To further assess the actin remodeling capability of Daam1-depleted junctions, we carried out fluorescence recovery after photobleaching (FRAP) assays (Figure 4). Overall, FRAP experiments suggest slower turnover of junctional F-actin in Daam1-depleted nephrons. In control nephrons, 100% of bleached junctions successfully recover fluorescence. In Daam1-depleted nephrons, only 59% of bleached junctions recover (Figure 4A). We wanted to find out whether F-actin pre-bleach fluorescence or the bleaching depth explain the differences in Daam1 KD cell recovery. Upon quantification (Figure S3), we find that the pre-bleach intensities and bleaching depth are not significantly different between Daam1 KD junctions with and without recovery and unlikely to explain differences in Daam1 KD cell recovery. Moreover, Daam1 KD junctions within the same embryo and with similar pre-bleach intensity and bleaching depth values can have different recovery outcomes (Figure S3, bar graph, embryo 2, blue). Cell junctions comprise dynamically mosaic E-cadherin clusters coupled to different actin dynamics (Cavey et al., 2008; Indra et al., 2018). Therefore, the differences in F-actin dynamics in Daam1 KD cells indicate the potential existence of two actin pools regulated differentially by Daam1 and suggest that other actin regulators are involved in modulating polymerization. However, it possible that the differences stem from cell heterogeneity (e.g., Daam1 expression levels or cell type representation) within the nephric primordium. Because we could not assess F-actin dynamics in junctions that fail to recover quantitatively, only junctions with detectable recovery signals were used to determine the mean half-time to recovery (Figure 4D) and the mean mobile fraction (Figure 4E). For each junction, values were taken from individually fitted curves. These data indicate that the mean recovery half-time for junctional F-actin in Daam1 KD nephrons is significantly slower (5.57 s ± 0.99 SEM) in comparison with control nephrons (3.70 s ± 0.35 SEM) (Figure 4D). In contrast, the mean mobile fractions are relatively similar (56.0%± 4.2 SEM for control and 54.3% ± 4.5 SEM for Daam1 KD) (Figure 4E). From these data, we conclude that Daam1 drives the rate of F-actin turnover to promote polymerization at junctions during nephron development. Moreover, the decrease in F-actin fluorescence levels observed upon Daam1 depletion may be a consequence of impaired actin assembly.
Figure 4. Daam1 regulates assembly of junctional F-actin in developing nephrons.

F-actin dynamics at cell junctions of control and Daam1 KD developing nephrons expressing mCherry-Utrophin were assessed using FRAP. (A–E) Control (black, ntotal = 27 junctions, 1–5 junctions/embryo) and Daam1 KD (purple, 16 junctions; orange, 11 junctions; ntotal = 27 junctions, 1–5 junctions/embryo).
(A) Percentage of junctions showing recovery of fluorescence after bleaching in control and Daam1 KD nephrons.
(B) Typical time-lapse images of control and Daam1 KD cell junctions before and after photobleaching. In each image, the bleached region is highlighted with a circle (black, control junction showing recovery; purple, Daam1 KD junction showing recovery; orange, Daam1 KD junction showing no recovery of fluorescence after photobleaching). Scale bars, 10 μm.
(C) Graph showing average recovery curves in a.u. obtained from individual best-fit plots for control (black) and Daam1 KD junctions with (purple) and without (orange) recovery of fluorescence after photobleaching.
(D and E) Bar graphs comparing control and Daam1 KD profiles calculated from individual best-fit curves for control (black) and Daam1 KD junctions with recovery of fluorescence after photobleaching (purple). The dots represent analyzed junctions. Data represent the mean ± SEM from three independent experiments. The p values were analyzed by unpaired t test.
(D) Bar graph of the relative half-times for F-actin.
(E) Bar graph of the relative mobile fraction for F-actin.
Daam1 promotes localization of E-cadherin at cell contacts
The interplay between E-cadherin and the actin cytoskeleton promotes intercellular adhesion and assembly of junctions. Therefore, we wanted to find out whether changes observed in junctional F-actin dynamics upon Daam1 KD alter the adhesion between nephron progenitors. We analyzed the effect of Daam1 depletion on E-cadherin localization in developing nephrons (Figures 5 and S4). Daam1 morphants displayed reduced E-cadherin levels at interfaces between neighboring cells during these early stages of pronephric morphogenesis (Figure 5A), which was quantified by measuring E-cadherin’s fluorescence intensity profiles along the length of individual junctions (Figure 5B). In contrast, there was no difference in overall E-cadherin levels between Daam1 KD and control embryos, as determined by western blotting (Figure 5C), suggesting that Daam1 is more critical for localization of E-cadherin at cell-cell contacts than for its expression levels. Moreover, the results support prior studies in which depletion of Daam1 led to diffusion of lateral E-cadherin without apparent changes in its protein levels (Nishimura et al., 2016). We next examined the effect of Daam1-depletion on E-cadherin localization in developing nephrons in vivo (Figure S4). Overexpression of GFP-tagged E-cadherin led to severe phenotypes in control and Daam1 morphants. Nonetheless, E-cadherin localization between neighboring Daam1 KD cells appeared to be more diffuse than in controls.
Figure 5. Daam1 promotes localization of junctional E-cadherin.

(A) Maximum-projection confocal images of whole-mount immunostaining of Xenopus nephric primordium labeled by Lhx1 (magenta) and E-cadherin (cyan) in control and Daam1 KD embryos; scale bars, 50 μm. a’–a” and b’–b” are close-up images of the corresponding regions in the white boxes; scale bars, 20 μm. The top panels consist of the entire z stacks to show nephric progenitors’ cell positions within the developing kidney. In contrast, images displayed in a’–a” and b’–b” contain a subset of the z slices to exclude the intense signal from E-cadherin expressed in the epithelium of the Xenopus skin. a’–b’, nephric cell progenitors labeled by Lhx1 (magenta) and E-cadherin (cyan); a”–b”, E-cadherin (cyan).
(B) Violin plots depicting the relative fluorescence intensity in a.u. of junctional E-cadherin in the nephric primordia of control (orange) and Daam1 KD (blue). ncontrol = 88 junctions on 4 embryos and nDaam1 KD = 84 junctions on 4 embryos. Centerlines represents the median; limits show the first and third quartiles. ****p ≤ 0.0001, analyzed by unpaired t test.
(C) Western blot and graph of densitometry measures in a.u. showing Daam1, E-cadherin, and GAPDH protein levels in uninjected WT, control (standard MO)-injected, and Daam1 KD (Daam1 MO)-injected embryos. Embryo lysates were pooled from 10–20 1-cell injected embryos at stages NF 11–12, and approximately 1/2 embryo was loaded per lane. Individual band intensities are normalized to the uninjected band and plotted in a.u. for uninjected (black), control (orange), and Daam1 KD (blue). The results are expressed as means ± SEM from four independent experiments. nsp > 0.05, ****p ≤ 0.0001, analyzed by one-way ANOVA.
Interestingly, Daam1 KD cells remain capable of forming nephrons. Studies in MDCK cells using the pan-formin inhibitor SMIFH2 suggest that early but not later stages of cell adhesion require formins (Collins et al., 2017). Moreover, Daam1 KD mammary epithelia form a monolayer characterized by irregular tilting of lateral cell membranes and distorted cell morphology (Nishimura et al., 2016). To further understand the function of Daam1 in nephron assembly, we analyzed the epithelium of mature nephrons in NF stages 39/40 embryos (Figure S5). Indeed, we did not observe any apparent changes in the local concentration of junctional E-cadherin in Daam1 KD and control embryos (Figure S5A). We considered the possibility that Daam1 protein levels have recovered because of the MO pools’ consumption over time. However, western blot analyses show significantly reduced Daam1 levels at this late stage (Figure S5B). Nonetheless, the tubular lumens in Daam1 KD nephrons displayed defects in size and variability in diameter compared with controls. Similar to observations in mammary epithelia (Nishimura et al., 2016), tubular cells in Daam1-depleted nephrons are less uniform in shape and have irregular tilting of lateral membranes (Figure S5A; Videos S5 and S6). When visualized by transmission electron microscopy (TEM), Daam1-depleted cells appear less columnar with indistinct and wavy cell borders (Figure S5C). Our results suggest that Daam1 regulates adhesion between nephron progenitor cells and, subsequently, the mature nephric epithelium’s morphology.
The Daam1 FH2 domain mediates E-cadherin localization
Daam1 is known to act upstream of small Rho GTPases, which regulate the actin cytoskeleton (Habas et al., 2001; Liu et al., 2008); therefore, we tested the importance of the actin polymerization activity of Daam1 for intercellular adhesion. Formins are defined by a conserved formin homology 2 (FH2) domain. Daam1 forms a dimer via its FH2 domain, which nucleates and elongates actin filaments (Lu et al., 2007; Yamashita et al., 2007). A isoleucine-to-alanine (Ile698Ala) mutation in this domain abolishes the actin polymerizing activity of Daam1 in vitro (Lu et al., 2007) and in vivo (Liu et al., 2008; Nishimura et al., 2016). We expressed full-length GFP-Daam1 or GFP-Daam1 FH2 mutant (Ile698Ala) mRNA and analyzed their effect on E-cadherin localization in the nephric progenitors (Figure 6). Expression of GFP-Daam1 FH2 mutant mRNA showed reduced E-cadherin levels at cell interfaces of nephric progenitors compared with those expressing GFP-Daam1 (Figures 6A and 6B). However, the E-cadherin phenotype was less prominent than in Daam1 KD. Functional studies showed that, although this FH2 mutation abolishes Daam1’s ability to polymerize actin, it does not prevent its activation of Rho (Liu et al., 2008). Moreover, expression of dominant-negative Rho within the nephric primordium produces a similar phenotype as depletion of Daam1 (Miller et al., 2011). These results suggest that Rho activity in the FH2 mutant likely diminishes its effect on E-cadherin localization in nephric progenitors compared with Daam1 KD. Furthermore, we assessed Daam1 protein levels to determine whether observed differences in E-cadherin localization were due to disparities in translation efficiency or stability of the GFP-Daam1 and FH2 proteins (Figure 6C). Overall, Daam1 protein was present at comparable levels, making these possibilities unlikely. Ultimately, the FH2-mutant-expressing progenitors mature into nephrons characteristic of Daam1 KD (Figure S6). These results show that the Daam1 FH2 domain is necessary to localize E-cadherin to cell-cell contacts in nephron progenitors.
Figure 6. The FH2 domain of Daam1 mediates E-cadherin localization to cell junctions.

(A) Maximum-projection confocal images of whole-mount immunostaining showing E-cadherin (cyan) in Xenopus nephric primordium labeled by Lhx1 (magenta) expressing GFP-Daam1 or GFP-Daam1 FH2 mutant mRNA (labeled by anti-GFP, green); scale bars, 50 μm. a’–a” and b’–b” are close-up images of the corresponding regions in the white boxes; scale bars, 20 μm. The top panels consist of the entire z stacks to show nephric progenitors’ cell positions within the developing kidney. In contrast, images displayed in a–a’ and b–b’ contain a subset of the z slices to exclude the intense signal from E-cadherin expressed in the epithelium of the Xenopus skin. a’–b’, nephric cell progenitors labeled by Lhx1 (magenta), anti-GFP (green), and E-cadherin (cyan); a”–b”, E-cadherin (cyan).
(B) Violin plots depicting the relative fluorescence intensity of junctional E-cadherin in nephric primordia expressing GFP-Daam1 (orange) and GFP-Daam1 FH2 mutant (blue) mRNA. nDaam1 = 60 junctions on 3 embryos and nDaam1FH2mutant = 55 junctions on 3 embryos. Centerlines show the median; limits show the first and third quartiles. ***p ≤ 0.001, analyzed by unpaired t test.
(C) Western blot and the graph of densitometry measures showing the exogenous and endogenous protein levels of Daam1 in uninjected WT embryos and embryos injected with 1 ng Daam1 or Daam1 FH2 mutant mRNA. The non-specific band confirms equal loading. Embryo lysates were pooled from 10–20 1-cell injected embryos at stages NF 11–12, and approximately 1/2 embryo per lane. The results are expressed as means ± SEM from three independent experiments. Individual band intensities are normalized to the uninjected band and plotted in a.u. for uninjected (black), Daam1 (orange), and Daam1 FH2 mutant (blue). nsp > 0.05, ***p ≤ 0.001, ****p ≤ 0.0001, analyzed by one-way ANOVA.
Daam1 mediates cohesion of MDCK cells
Nephron morphogenesis is achieved through CE (Lienkamp et al., 2012). CE is collective cell movement characterized by coordinated and directed cell rearrangements (Huebner and Wallingford, 2018; Tada and Heisenberg, 2012). In recent years, E-cadherin has emerged as a key mediator of coordinating cohesion and directional persistence of collective movements in epithelial and mesenchymal clusters (Cai et al., 2014; Campbell and Casanova, 2015; Cohen et al., 2016). Our results demonstrate that Daam1 is necessary for organization of the nephrogenic primordium (Figure 3) and proper localization of E-cadherin (Figures 5, S4, and 6); therefore, we wanted to determine whether Daam1 is necessary for coordination of direction between renal cells. Because Xenopus nephron progenitors’ opaqueness prevents in vivo tracking of their movements in 3D, we generated MDCK cells constitutively expressing an shRNA against Daam1 and analyzed whether E-cadherin localization is affected in these cells (Figures 7A and S7). The efficiency of Daam1 KD was confirmed by western blot (Figure 7B). Similar to what we saw in Xenopus nephrons, we observed E-cadherin’s impaired localization in nascent (Figure 7A) but not mature adhesions (Figure S7). We next examined Daam1 KD cells’ migratory behavior in time-lapse imaging of wound healing assays (Figures 7C-7E; Video S7). We found that these cells exhibit a delay in wound closure compared with control cells (Figure 7C; Video S7). Additionally, we observed their random detachment from migrating epithelial sheets (Video S7). To better understand the behavior of these cells, we tracked their movement within the migrating sheets over time (Figure S7). We obtained the relative distances over which cells traveled from these tracks and extrapolated cell velocities (Figure 7D). These analyses show that Daam1 KD cells move faster than controls, demonstrating that delayed wound closure in Daam1 KD cells is not due to impaired velocity. However, mapping the trajectory paths (Figure 7E) revealed that the movement of Daam1 KD cells is less directed than that of controls. Thus, Daam1 is necessary for cells’ organization during collective movement of epithelial sheets. These results indicate that Daam1 contributes to cohesion by regulating the connectedness of cells through E-cadherin.
Figure 7. Daam1-depleted MDCK cells display compromised localization of E-cadherin at cell-cell contacts and impaired cohesion during collective movement.

(A) Maximum-projection confocal images of E-cadherin (green), F-actin (magenta), and DAPI (cyan) in subconfluent MDCK control and shDaam1 KD cells. The first column shows merged images of individual color channels. Images of individual color channels for F-actin and E-cadherin are shown in the second and third columns, respectively. Scale bars, 50 μm. E-cadherin localization in nascent cell-cell contacts (marked by white brackets and shown enlarged in the corresponding white boxes; scale bars, 10 μm) is impaired in shDaam1 KD cells.
(B) Western blot showing Daam1 and GAPDH protein levels in MDCK control and shDaam1 KD cells. Individual band intensities are normalized to the control band and plotted in a.u. for control (orange) and Daam1 KD (blue). The graph of densitometry measures represents the means ± SEM of two independent experiments. *p ≤ 0.05, analyzed by unpaired t test.
(C–F) Summary of the wound healing experiments for MDCK control and Daam1 KD cells; see Video S7.
(C) Daam1 depletion impairs wound closure. The graph represents the percentage of the wound surface area over time for control (purple) and Daam1 KD (orange) cells. Error bars indicate SE of the mean of 4 assays.
(D–F) Manually tracking single cells’ migration paths during wound closure demonstrates that Daam1 organizes collective movement of MDCK epithelial monolayers by modulating the speed and directionality of individual cells. Daam1 results in increased velocity and random migration. ncontrol = 52 cells from 4 assays and nDaam1 KD = 42 cells tracked from 4 assays. Cells were tracked in 15-min increments for 12 h.
(D) Violin plots represent migration velocity calculated from tracking traveled distances of single cells for control and Daam1 KD cells. Centerlines show the median; limits show the first and third quartiles. ***p ≤ 0.001, analyzed by unpaired t test.
(E) Wind rose plot showing migration tracks of individual control and Daam1 KD cells.
DISCUSSION
Using Xenopus embryonic kidney and MDCK cells as models, we show that Daam1 mediates E-cadherin dependent intercellular adhesion and organization of nephrogenic primordia by regulating polymerization of junctional actin filaments. Ultimately, this affects the morphology of the mature nephric epithelium. These findings have several important implications for regulation of intercellular adhesion and epithelial tubulogenesis.
First we show that Daam1 and E-cadherin localize to cell-cell contacts during nephron development and that Daam1 promotes E-cadherin localization at sites of cell-cell contact. In contrast to early stages of nephron development, junctional Daam1 was not detected in mature nephrons. Likewise, we did not observe any perturbations of E-cadherin localizing to intercellular junctions. These data suggest that Daam1 is necessary for localization of E-cadherin at cell junctions in nephron progenitor cells during the early stages of nephron morphogenesis.
This conclusion builds upon assumptions that overexpression of GFP-Daam1 does not affect our localization analyses. However, multiple lines of evidence suggest that this scenario is improbable because prior studies indicate that overexpression of full-length Daam1 has little to no effect on the actin cytoskeleton or other cellular processes (Liu et al., 2008). Additionally, several studies demonstrate that the distribution of GFP-Daam1 recapitulates its endogenous localization, as discerned via immunostaining (Jaiswal et al., 2013; Li et al., 2011; Nishimura et al., 2012, 2016). Moreover, consistent with our observations in Xenopus nephrons, sub-confluent cultures of MDCK cells stably expressing shDaam1 show transient repression of E-cadherin localization to cell-cell contacts, and it appears to be lost as the cells become confluent.
Our data support recent findings suggesting that actin nucleating proteins and Rho GTPases are required for early stages of E-cadherin-mediated cell adhesion in MDCK epithelia and not in mature junctions (Collins et al., 2017). They also imply that morphological defects we see in mature nephrons are consequences of earlier events. It is interesting that, similarly, silencing of E-cadherin expression in MDCK cells disrupts formation of cell junctions. In contrast, E-cadherin signaling seems to be mostly dispensable in the established epithelium when cells are not mechanically stressed (Capaldo and Macara, 2007).
Second, we show that Daam1 localizes to actin protrusions and newly formed cell-cell contacts in developing nephrons, suggesting that Daam1 coordinates assembly of cell junctions during CE via E-cadherin. Actin polymerization at the cell membrane mediates polarized movement and intercalation of cells during CE through cadherin engagement (Huebner and Wallingford, 2018; Huebner et al., 2020). Here we show that Daam1 functions to ensure proper organization and size of the nephric primordium at the time of CE and polarized movement of renal epithelial sheets. Our data support previous studies implicating Daam1 in CE and polarized cell movements (Ang et al., 2010; Kida et al., 2007; Liu et al., 2008). Furthermore, we show that Daam1 mediates polarized movement and cohesion of MDCK cells without slowing the motility of individual cells. These data suggest that Daam1 promotes collective cell movements by controlling actin polymerization at cell-cell contacts and strengthening E-cadherin-based adhesion.
Finally, Wnt9b and Wnt11 regulate tubular nephron morphogenesis, and disruptions of cell behaviors traditionally regulated by the PCP pathway define their loss-of-function phenotypes (Karner et al., 2009; O’Brien et al., 2018). We show that inhibiting the signaling activity of Daam1 in the nephron results in phenotypic characteristics comparable with those reported for Wnt11 and Wnt9b. Moreover, earlier studies have demonstrated that Daam1 rescues Wnt11-induced CE gastrulation defects (Liu et al., 2008). These data point to Daam1 as a potential downstream effector of Wnt11 and Wnt9b signaling in nephric tubulogenesis. Interestingly, wnt9b and wnt9a are also induced in injured nephrons, and mutation of fzd9b is associated with reduced regenerative capacity of nephric tubules (Kamei et al., 2019), suggesting a potential role of Daam1 in repair and regeneration of nephric tubules. This hypothesis is further supported by increasing evidence that actin-based protrusions are important for repairing cell junctions (Li et al., 2020) and studies showing that Daam1 promotes assembly of actin-based protrusions (Jaiswal et al., 2013; Nishimura et al., 2016).
Cadherin localization is a complex process, and current studies propose at least three different ways of achieving localization of E-cadherin: (1) clustering enforced by cortical F-actin, (2) clustering regulated by exposure of neighboring cells through actin-based protrusions, and (3) clustering promoted by condensation of F-actin networks via myosin (Yap et al., 2015). However, further research into E-cadherin’s timely regulation is required to characterize the precise molecular details and specify relationships between different E-cadherin clustering modes. Ultimately, investigations of how the regulation of junctional E-cadherin by Daam1 fits in with different E-cadherin clustering modes will also be essential.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rachel K. Miller (Rachel.K.Miller@uth.tmc.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Xenopus laevis
Xenopus laevis adult male and female frogs were obtained from Nasco (LM00531MX and LM00713M) (Fort Atkins, WI, USA) and maintained according to standard procedures. Xenopus embryos were obtained by in vitro fertilization (Sive et al., 2010) and staged as previously described by Nieuwkoop and Faber (1994). Studies were conducted on a range of embryonic stages spanning NF stages 1 - 40. All work was carried out following the University of Texas Health Science Center at Houston, Institutional Animal Care and Use Committee (IACUC) protocol #AWC-19-0081.
MDCK cell lines
Madin-Darby Canine Kidney (MDCK) II cell lines were purchased from the American Type Culture Collection (ATCC). MDCK cells were cultured at 37°C with 5% CO2 in Dulbecco’s Modified Eagle’s Medium (DMEM) (Sigma, D6429) supplemented with 10% fetal bovine serum (FBS) (Sigma, F0926) and 1% Antibiotic-antimycotic solution (Sigma, A5955).
METHOD DETAILS
Embryo microinjections
Xenopus embryos were microinjected at one-cell or into the V2 blastomere at the eight-cell stage, targeting embryonic kidney (DeLay et al., 2016; Moody and Kline, 1990; Nieuwkoop and Faber, 1994). Embryos were injected with synthetic mRNAs alone or in combination with antisense morpholino oligonucleotides (MOs). For mRNA injections, capped mRNA transcripts were synthesized from DNA-plasmids using SP6 mMessage mMachine transcription kit (ThermoFisher, AM1340M) and purified. pCS2-GFP-Daam1 and pCS2-GFP-Daam1 (Ile698Ala) plasmids were gifts from Dr. Raymond Habas’s and Dr. Bruce Goode’s labs, respectively (Lu et al., 2007). A mutation A2822G discovered in these plasmids was corrected by site-directed mutagenesis as previously reported (Corkins et al., 2019) before mRNA synthesis. pCS2-membrane-tagged-RFP (mRFP) (Davidson et al., 2006), pCS2-membrane-tagged-EGFP (mEGFP) (Shindo and Wallingford, 2014), and pCS2-mCherry-Utrophin (mCherry-UtrCH) (Burkel et al., 2007) constructs were gifts from Dr. Raymond Keller’s lab, Dr. John Wallingford’s lab, and Dr. William Bement’s lab, respectively. Formerly developed translation-blocking Daam1 (5′GCCGCAGGTCTGTCAGTTGCTTCTA 3′) (Corkins et al., 2018; Habas et al., 2001; Miller et al., 2011) and standard control (5′CCTCTTACCTCAGTTACAATTTATA 3′) MOs were purchased from GeneTools, LLC (Philomath, OR, USA). MOs were injected at 20ng per embryo while the amount of injected mRNA per embryo were as follows: GFP-Daam1 [1ng], mCherry-Daam1 [1ng], GFP-Daam1(I698A) [1ng], mCherry-Daam1(I698A) [1ng], mRFP [0.5ng], mGFP [0.5ng] and mCherry-UtrCH [1ng].
Generation of stable MDCK shDaam1 cell lines
shDaam1 KD cell lines were generated by a retrovirus-based transduction method as previously described (Corkins et al., 2019). Briefly, the pLKO.1 lentiviral shDaam1 constructs (TTTCAGGAGATAGTATTGTGC, AAACAGGTCTTTAGCTTCTGC) were purchased from GE-Dharmacon (Clone ID: TRCN0000122999, Clone ID: TRCN0000123000). HEK293T cells were co-transfected with shDaam1 and virus packaging plasmids (psPAX2 and pMD2.G) using Polyethylenimine (PEI). The viral titers were collected starting 24 hours post-transfection over the course of two days and purified using 0.22 μm Polyethersulfone (PES) syringe filters. Infections were carried out in the presence of polybrene. MDCK II cells remained in infection media for 24 hours, followed by puromycin selection with the final concentration of 0.70 μg/ml. Lastly, MDCK II shDaam1 KD stable cell lines were validated by western blotting.
Western blotting
Western blotting was carried out using published protocols (Kim et al., 2002; Williams et al., 2017). In short, Xenopus embryos were cultured to the desired stage and collected. The whole-embryo lysates were prepared by resuspending 10-20 embryos in a prechilled TX100-lysis buffer (10 mM HEPES, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.5% Triton X-100, pH 7.4) and centrifuged at 18,407 RCF at 4°C for 5 minutes. The resulting protein lysates were resuspended in an equal volume 2X Laemmli (BioRad,161-0737) + dithiothreitol (Fisher BioReagents, BP17225) solution and incubated at 95°C for 2 minutes. For making protein lysates using MDCK cells, cells were washed with PBS and pelleted by centrifugation at 18,407 RCF at 4°C for 5 minutes. Cell pellets were resuspended in in a prechilled Triton-lysis buffer (50mM Tris pH 7.4, 1% Triton X-100, 150mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, 1 mM Na3VO4, 10 mM sodium fluoride, 10 mM β-glycerophosphate, 1 mg/ml aprotinin, and 1 mg/ml leupeptin). The cell lysates were incubated on ice for 20 minutes, followed by sonication and centrifugation at 18,407 RCF for 10 minutes at 4°C. Bradford assay was used to determine the total amount of protein in the lysates. The protein samples were run on an 8% SDS-PAGE gel and transferred to nitrocellulose or polyvinylidene difluoride (PVDF) membranes. The blots were blocked for at least 1.5 hours at room temperature using the KPL Detector Block Kit (Sera Care, 5920-0004, 71-83-00) and probed with primary antibodies overnight at 4°C. The next day, after a series of washes with Tris-buffered saline containing 0.1% Tween 20 (TBST), blots were incubated with secondary antibodies for at least 1 hour at room temperature. Protein expression levels were detected with enhanced chemiluminescence (SuperSignal West Pico PLUS Chemiluminescent Substrate, Thermo Fisher, 34580) using Li-Cor and BioRad ChemiDoc XRS imagers. The following antibodies were used: rabbit anti-Daam1 (1:1000, Proteintech, 14876-1-AP), rabbit anti-Daam1 (1:1000, gift from Dr. Raymond Habas), rabbit anti-GAPDH (1:1000, Santa Cruz, sc-25778), mouse anti-E-cadherin (1:1000, BD Transduction Laboratories, 610182), rabbit anti-GFP (1:250, ICL Lab, RGFP-45A), anti-rabbit IgG (H + L)-HRP (1:5000, BioRad,1706515) and anti-mouse IgG (H + L)-HRP (1:5000, BioRad, 1706516).
Immunostaining and staining
Xenopus embryos were fixed in MEMFA (3.7% formaldehyde, 4 mM MOPS, 2 mM EGTA, and 1 mM MgSO4, pH 7.4) for 1 hour at room temperature or overnight at 4°C. Embryos to be stained with Phalloidin were fixed using methanol-free formaldehyde (Thermo Scientific, 28908). Immunostaining was carried out according to previously published methods (Hemmati-Brivanlou and Melton, 1994; Krneta-Stankic et al., 2010). In short, fixed-embryos were washed 3 times for 15 minutes at room temperature with phosphate-buffered saline (PBS) containing 0.1% Triton X-100 and 0.2% bovine serum albumin (BSA) and blocked using 10% goat serum diluted in PBST for 1 hour at room temperature. Embryos were incubated with the primary antibodies overnight at 4°C. The next day, embryos were washed 5 times for 1 hour with PBS-T at room temperature and then incubated with the secondary antibodies overnight at 4°C. The following day, embryos were washed 3 times for 1 hour at room temperature and dehydrated in methanol before clearing. To preserve Phalloidin-labeling, isopropanol was used to dehydrate Phalloidin-stained embryos (Nworu et al., 2014). Embryos were cleared using BA:BB (1-part benzyl alcohol: 2-parts benzyl benzoate) clearing solution and imaged. MDCK cells were fixed in 4% paraformaldehyde (PFA) for 10 minutes at room temperature. Cultures were washed 3 times with PBS and incubated with 50mM ammonium chloride for 10 minutes at room temperature to neutralize the PFA. Next, samples were washed 3 times with PBS and blocked using 10% goat serum/ PBST for 1 hour and incubated with primary antibodies overnight at 4°C. The following day, MDCK cells were washed 3 times with PBS and incubated with secondary antibodies for 1 hour at room temperature. Lastly, stained samples were rewashed 3 times with PBS before mounting in Fluoromount-G medium (Southern Biotech, 0100-01) for imaging. The following primary antibodies were used: chicken anti-GFP (1:250, Abcam, ab13970), rabbit anti-RFP (1:250, MBL International, PM005), rabbit anti-GFP (1:250, ICL Lab, RGFP-45A), rabbit anti-Lhx1 (1:250, a gift from Dr. Masanori Taira) and mouse anti-E-cadherin (1:100, BD Transduction Laboratories, 610182). For detection of primary antibodies the following secondary antibodies were used: anti-rabbit IgG Alexa 488 (1:500, Invitrogen, A-11008), anti-rabbit IgG Alexa 555 (1:500, Invitrogen, A-21428), anti-rabbit IgG Alexa 647 (1:500, Invitrogen, A-21244), anti-mouse IgG Alexa 488 (1:500, Invitrogen, A-11001), anti-mouse IgG Alexa 555 (1:500, Invitrogen, A-21422), anti-mouse IgG Alexa 647 (1:500, Invitrogen, A-21235), anti-mouse IgG Alexa 488 (1:500, Jackson ImmunoResearch, 715-545-150) and anti-chicken IgY Alexa 488 (1:500, Invitrogen, A-11039). Fluorescent probes used for staining were as follows: FITC-conjugated lectin from Erythrina cristagalli (1:500, Vector labs, FL-1141), Phalloidin-Alexa 568 (embryos- 1:40 and cells-1:200, Invitrogen, A12380), and diamidino-2-phenylindole (DAPI) (1:500, Thermo Scientific, 62247).
Transmission electron microscopy (TEM)
Xenopus embryos were fixed in 2%formaldehyde+0.5% glutaraldehyde (Ted Pella Inc., 18505 and 18426). Fixed embryos were washed with 0.1 M sodium cacodylate buffer and treated with 0.1% Millipore-filtered cacodylate buffered tannic acid. Embryos were post-fixed using 1% buffered osmium, followed by staining using 1% Millipore-filtered uranyl acetate. Stained embryos were dehydrated by washing in increasing ethanol concentrations, permeated, and embedded in LX-112 medium. After embedding, embryos were placed in a 60°C oven for approximately 3 days to polymerize. Polymerized samples were sectioned using Leica Ultracut microtome (Leica, Deerfield, IL). Collected ultrathin sections were stained with uranyl acetate and lead citrate in a Leica EM Stainer and subjected to imaging.
Isolation and live imaging of Xenopus kidney cells
Around embryonic NF stage 30, developing nephrons were surgically dissected under a fluorescent stereomicroscope using a pair of sharpened forceps (Fisher, NC9404145). Microsurgical dissections were performed in plastic Petri dishes coated with 2% agar containing Danilchik’s for Amy (DFA) solution (53 mM NaCl, 5 mM Na2CO3, 4.5 mM Potassium Gluconate, 32 mM Sodium Gluconate, 1 mM CaCl2, 1 mM MgSO4, buffered to pH 8.3 with 1 M bicine) supplemented with 1 g/l of and Antibiotic antimycotic solution (1:100, Sigma, A5955). To dissociate into single cells, the isolated nephrons were transferred to fibronectin-coated (1 μg/ml, Roche, 10-838-039-001) glass-bottom imaging chambers (Thermo, A7816) prefilled with Calcium Magnesium Free Media (CMFM) (88 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 7.5 mM Tris pH 7.6). After ~30 minutes at room temperature, the CMFM media was aspirated from the chamber’s top without disturbing the cells. Fresh DFA media was added and again removed by careful aspiration. To ensure the complete removal of the CMFM media, this process was repeated at least 5 times. The cells were left undisturbed at room temperature for 15-30 minutes before imaging.
Wound-healing assay
MDCK cells were seeded in 6-well culture plates at a density of 100,000 cells/well and allowed to reach confluency. Before inducing a “wound” or scratch or in a confluent cell monolayer, the cells were treated with Mitomycin C (10 μg/ml) for 3 hours at 37°C to prevent future proliferation. A linear scratch was made by gliding the 200 μL sterile tip across the bottom of each well. After making a scratch, cells were washed with 1X PBS and refed with 5 mL of 10%FBS/DEMEM supplemented with a 1%Antibiotic-antimycotic solution. Samples were placed in a 37°C heated imaging chamber with 5% CO2 and subjected to time-lapse imaging.
Fluorescence recovery after photobleaching (FRAP)
To visualize actin dynamics, embryos were injected into V2 blastomere at the 8-cell stage with 1 ng mCherry-UtrCH mRNA along with 20 ng of Daam1 or Standard (Control) morpholino (MO). Injected embryos were cultured to around NF stage 30. Since Xenopus epithelium’s opaqueness prevents direct in vivo imaging of developing nephrons at this early embryonic stage, “windowed” embryos were generated. To make windowed embryos, embryos were anesthetized in 0.04% (0.15%) Ethyl 3-aminobenzoate methanesulfonate (Sigma, E10521) diluted in DFA. Next, the epithelium covering the developing nephron was surgically removed under a fluorescence-dissecting microscope, exposing the developing nephron for in vivo imaging. Windowed embryos were mounted under glass-coverslips and subjected to fluorescence recovery after photobleaching (FRAP). FRAP assays were performed on an inverted 3i spinning disk microscope-integrated to a NIKON TiE with perfect focus and equipped with a Vector FRAP scanning module and a Hamamatsu Flash 4.0 camera. Images were acquired with a NIKON Plan-Apo 60X water 1.2 NA objective. A small fragment at the midpoint of a cell junction was bleached with the 561nm laser line at 100% power. A series of twenty pre-bleach images were captured, and post-bleach recovery was recorded continuously until the fluorescent signal reached a steady state. Movies were analyzed using SlideBook 6.0, and curve fitting was done with SigmaPlot and GraphPad Prism 8.0 software. For curve fitting, a single exponential function (f(t) = α(1-e−kT), where T1/2 (half-time of recovery) is ln 0.5/(-k), and α is the mobile fraction was used. Raw recovery curves were corrected for background and photofading. The lowest fluorescence signal and the time-point after bleaching were scaled to 0, and curves were normalized to 1 based on the reference signal before bleaching. Per embryo, between 1 and 5 single junctions were photobleached. To avoid bleaching-induced variation in fluorescence, junctions selected for photobleaching were spaced far apart. All FRAP experiments were carried out using multiple embryos and repeated at least three times.
Image acquisition and processing
Olympus SZX16 fluorescent stereomicroscope equipped with Olympus DP71 camera was used for carrying out Xenopus microsurgical manipulations, embryo mounts, and scoring of kidney phenotypes in NF stage 40 embryos. Zeiss LSM800 with Airyscan detector, LeicaSP5, and NikonA1 microscopes were used for confocal imaging of fixed and live samples. Captured images and time-lapse movies were exported as original files and processed using ImageJ (Fiji plugin). Image panels were built using the FigureJ plugin. Final figures were assembled in Adobe Photoshop CC. TEM imaging was carried out in a JEM 1010 transmission electron microscope (JEOL, USA, Inc., Peabody, MA) with the AMT Imaging System (Advanced Microscopy Techniques Corp, Danvers, MA).
QUANTIFICATION AND STATISTICAL ANALYSIS
ImageJ (Fiji plugin) software was used for quantitative image analyses. For quantification of E-cadherin and F-actin staining (Figures 3, 5, and 6), images were captured using the same settings. The mean fluorescence intensity along the length of the selected junction was measured. To analyze tissue organization during nephron development (Figure 3), cells were manually counted using the Cell Counter tool. The shortest distance between two neighboring nuclei was measured using the straight-line selection tool. The area and circularity were measured using the Analyze Particles tool. Circularity is defined as 4π x [Area]/[Perimeter]2, ranging from 0 to 1, where 0 represents infinitely elongated polygon, and 1 represents the perfect circle. Wound area (Figure 7) was also measured using the Analyze Particles tool. To obtain traveled distance, trajectories of randomly selected cells were traced manually in each frame of the time-lapse using Plugin for Motion Tracking and Analysis (MTrackJ) (The Biomedical Imaging Group of Erasmus University Medical Centre, Rotterdam, the Netherlands). Additionally, the intensity measurements of the western blot bands were performed in ImageJ. The mean gray values of individual protein bands were corrected for the background and loading. The target proteins’ final values were normalized to the protein amount measured in uninjected wild-type embryos (Figures 3, S5, 5, and 6) or wild-type cells (Figure 7). All experiments were repeated at least two times, except for TEM studies, representing single-trial analyses due to the experiment’s prohibitive costs. The exact sample size and statistical analysis for each experiment are presented in the corresponding figure legend. Statistical analyses were carried out using GraphPad Prism 8.0 software.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| chicken anti-GFP | Abcam | Cat# ab13970; RRID:AB_300798 |
| rabbit anti-RFP | MBL International | Cat# PM005; RRID:AB_591279 |
| rabbit anti-GFP | ICL Lab | Cat# RGFP-45A |
| rabbit anti-Lhx1 | gift from Dr. Masanori Taira, Chou University | N/A |
| mouse anti-E-cadherin | BD Transduction Laboratories | Cat# 610182; RRID:AB_397581 |
| anti-rabbit IgG Alexa 488 | Invitrogen | Cat# A-11008; RRID:AB_143165 |
| anti-rabbit IgG Alexa 555 | Invitrogen | Cat# A-21428; RRID:AB_2535849 |
| anti-rabbit IgG Alexa 647 | Invitrogen | Cat# A-21244; RRID:AB_2535812 |
| anti-mouse IgG Alexa 488 | Invitrogen | Cat# A-11001; RRID:AB_2534069 |
| anti-mouse IgG Alexa 555 | Invitrogen | Cat# A-21422; RRID:AB_2535844 |
| anti-mouse IgG Alexa 647 | Invitrogen | Cat# A-21235; RRID:AB_2535804 |
| anti-mouse IgG Alexa 488 | Jackson ImmunoResearch | Cat# 715-545-150; RRID:AB_2340846 |
| anti-chicken IgY Alexa 488 | Invitrogen | Cat# A-11039; RRID:AB_2534096 |
| rabbit anti-Daam1 | Proteintech | Cat# 14876-1-AP; RRID:AB_2089444 |
| rabbit anti-Daam1 | a gift from Dr. Raymond Habas’s lab, Temple University | N/A |
| rabbit anti-GAPDH | Santa Cruz | Cat# sc-25778; RRID:AB_10167668 |
| anti-rabbit IgG (H + L)-HRP | BioRad | Cat# 1706515; RRID:AB_11125142 |
| anti-mouse IgG (H + L)-HRP | BioRad | Cat# 1706516; RRID:AB_11125547 |
| Chemicals, peptides, and recombinant proteins | ||
| FITC-conjugated lectin from Erythrina cristagalli | Vector labs | Cat# FL-1141; RRID:AB_2336437 |
| Phalloidin-Alexa 568 | Invitrogen | Cat# A12380 |
| diamidino-2-phenylindole (DAPI) | Tdermo Scientific | Cat# 62247 |
| Dulbecco’s Modified Eagle’s Medium (DMEM) | Sigma | Cat# A5955 |
| fetal bovine serum (FBS) | Sigma | Cat# F0926 |
| Antibiotic-antimycotic solution | Sigma | Cat# A5955 |
| 2x Laemmli Sample Buffer | BioRad | Cat# 610737 |
| Dithiothreitol | Fisher BioReagents | Cat# BP172-25 |
| 16% Formaldehyde (w/v), Methanol-free | Thermo Scientific | Cat# 28908 |
| Fibronectin | Roche | Cat# 10 838 039 001 |
| Ethyl 3-aminobenzoate methanesulfonate | Sigma | Cat# E10521 |
| Critical commercial assays | ||
| SP6 mMessage mMachine transcription kit | TdermoFisher | Cat# AM1340M |
| KPL Detector Block Kit | Sera Care | Cat# 5920-0004, 71-83-00 |
| SuperSignal West Pico PLUS Chemiluminescent Substrate | Thermo Fisher | Cat# 34580 |
| Experimental models: Cell lines | ||
| Madin-Darby Canine Kidney (MDCK) II | American Type Culture Collection (ATCC) | Cat# ATCC® CRL-2936 |
| HEK293T cells | a gift from Dr. Andrew Gladden’s lab, North Carolina University | N/A |
| Experimental models: Organisms/strains | ||
| Xenopus laevis adult male frogs | Nasco | Cat# LM00713M |
| Xenopus laevis adult female frogs | Nasco | Cat# LM00531MX |
| Oligonucleotides | ||
| Daam1 morpholino-5′GCCGCAGGTCTGTCAGTTGCTTCTA 3′ | GeneTools, LLC | Corkins et al., 2018; Habas et al., 2001; Miller et al., 2011 |
| Control/Standard morpholino-5′CCTCTTACCTCAGTTACAATTTATA 3′ | GeneTools, LLC | Corkins et al., 2018; Miller et al., 2011 |
| pLKO.1 lentiviral shDaam1 construct-TTTCAGGAGATAGTATTGTGC | GE-Dharmacon | Clone ID: TRCN0000122999 |
| pLKO.1 lentiviral shDaam1 construct-AAACAGGTCTTTAGCTTCTGC | GE-Dharmacon | Clone ID: TRCN0000123000 |
| Recombinant DNA | ||
| pCS2-GFP-Daam1 | a gift from Dr. Raymond Habas’s lab, Temple University | Liu et al., 2008 |
| pCS2-GFP-Daam1 (Ile698Ala) | a gift from Dr. Bruce Goode’s lab, Brandeis University | Lu et al., 2007 |
| pCS2-membrane-tagged-RFP | a gift from Dr. Raymond Keller’s lab, University of Virginia | Davidson et al., 2006 |
| pCS2- mCherry -Daam1 | Dr. Rachel Miller’s lab, McGovern Medical School | Corkins et al., 2019 |
| pCS2-mCherry-Daam1 (Ile698Ala) | Dr. Rachel Miller’s lab, McGovern Medical School | Corkins et al., 2019 |
| pCS2-membrane-tagged-EGFP | a gift from Dr. John Wallingford’ lab, The University of Texas at Austin | Shindo and Wallingford, 2014 |
| pCS2-mCherry-Utrophin | a gift from Dr. William Bement’s lab, University of Wisconsin-Madison | Burkel et al., 2007 |
| virus packaging plasmids psPAX2 and pMD2.G | a gift from Dr. Andrew Gladden’s lab, University of North Carolina | Williams et al., 2017 |
| Software and algorithms | ||
| Slidebook 6.0 | 3i-Intelligent Imaging Innovations Software | https://www.intelligent-imaging.com/slidebook |
| SigmaPlot | Systat Software | https://systatsoftware.com/products/sigmaplot/ |
| Prism 8.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ (Fiji plugin) | Open-source program | https://imagej.nih.gov/ij/ |
| Adobe Photoshop CC | Adobe Inc. Software | https://www.adobe.com/products/photoshop.html |
| Other | ||
| Tweezer - Dumont no.5 | Fisher | Cat# NC9404145 |
| Glass-bottom imaging chambers | Tdermo | Cat# A7816 |
Highlights.
Daam1 localizes to cell-cell contacts in developing nephron
Daam1 regulates the cytoskeletal membrane dynamics
Daam1 promotes E-cadherin localization
Formin homology domain 2 (FH2) of Daam1 mediates E-cadherin localization
ACKNOWLEDGMENTS
We thank the Miller lab, Pierre McCrea, and Jae-II Park lab members for lively discussions and suggestions regarding the manuscript. We also thank Richard Behringer, Yoshihiro Komatsu, Oleh Pochynyuk, and Anna Marie Sokac for help with the project. We thank Kenneth Dunner Jr. at the UT MD Anderson High-Resolution Electron Microscopy Facility and CCSG grant NIH P30CA016672 for supporting the TEM data. We are also thankful to Raymond Habas, Bruce Goode, John Wallingford, Norihiro Sudou, Masanori Taira, Raymond Keller, and William Bement for providing antibodies and constructs. We thank the instructors and teaching assistants of the 2015 National Xenopus Resource Advanced Imaging Course. We are grateful to J.C. Whitney and T.H. Gomez for taking care of the animals. We are grateful to the UTHealth Office of the Executive Vice President and Chief Academic Officer and the Department of Pediatrics Microscopy Core for funding the Zeiss LSM800 confocal microscope. We also thank the BSRB Microscopy Facility in the Department of Genetics, UT-MD Anderson Cancer Center. This work was funded by NIH NIDDK grants (K01DK092320, R03DK118771, and R01DK115655 to R.K.M.); startup funding from the Department of Pediatrics, Pediatric Research Center at the McGovern Medical School (to R.K.M.); The Antje Wuelfrath Gee and Harry Gee, Jr. Family Legacy Scholarship (to V.K.-S.), and The Gigli Family Endowed Scholarships (to V.K.-S.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109340.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams CL, Chen YT, Smith SJ, and Nelson WJ (1998). Mechanisms of epithelial cell-cell adhesion and cell compaction revealed by high-resolution tracking of E-cadherin-green fluorescent protein. J. Cell Biol 142, 1105–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang SF, Zhao ZS, Lim L, and Manser E (2010). DAAM1 is a formin required for centrosome re-orientation during cell migration. PLoS ONE 5, e13064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker BE, and Gard DL (2006). Visualization of the Cytoskeleton In Xenopus Oocytes and Eggs by Confocal Immunofluorescence Microscopy. Methods Mol. Biol 322, 69–86. [DOI] [PubMed] [Google Scholar]
- Brennan HC, Nijjar S, and Jones EA (1998). The specification of the pronephric tubules and duct in Xenopus laevis. Mech. Dev 75, 127–137. [DOI] [PubMed] [Google Scholar]
- Brennan HC, Nijjar S, and Jones EA (1999). Glomus specification and induction in Xenopus. Development 126, 5847–5856. [DOI] [PubMed] [Google Scholar]
- Burkel BM, von Dassow G, and Bement WM (2007). Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil. Cytoskeleton 64, 822–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Chen S-C, Prasad M, He L, Wang X, Choesmel-Cadamuro V, Sawyer JK, Danuser G, and Montell DJ (2014). Mechanical feedback through E-cadherin promotes direction sensing during collective cell migration. Cell 157, 1146–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K, and Casanova J (2015). A role for E-cadherin in ensuring cohesive migration of a heterogeneous population of non-epithelial cells. Nat. Commun 6, 7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K, and Casanova J (2016).A common framework for EMT and collective cell migration. Development 143, 4291–4300. [DOI] [PubMed] [Google Scholar]
- Capaldo CT, and Macara IG (2007). Depletion of E-cadherin disrupts establishment but not maintenance of cell junctions in Madin-Darby canine kidney epithelial cells. Mol. Biol. Cell 18, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli M, Boca M, Chiaravalli M, Ramalingam H, Rowe I, Distefano G, Carroll T, and Boletta A (2013). Polycystin-1 binds Par3/aPKC and controls convergent extension during renal tubular morphogenesis. Nat. Commun 4, 2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavey M, Rauzi M, Lenne PF, and Lecuit T (2008). A two-tiered mechanism for stabilization and immobilization of E-cadherin. Nature 453, 751–756. [DOI] [PubMed] [Google Scholar]
- Cohen DJ, Gloerich M, and Nelson WJ (2016). Epithelial self-healing is recapitulated by a 3D biomimetic E-cadherin junction. Proc. Natl. Acad. Sci. USA 113, 14698–14703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C, Denisin AK, Pruitt BL, and Nelson WJ (2017). Changes in E-cadherin rigidity sensing regulate cell adhesion. Proc. Natl. Acad. Sci. USA 114, E5835–E5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combes AN, Davies JA, and Little MH (2015). Cell–Cell Interactions Driving Kidney Morphogenesis. Curr. Top. Dev. Biol 112, 467–508. [DOI] [PubMed] [Google Scholar]
- Corkins ME, Hanania HL, Krneta-Stankic V, DeLay BD, Pearl EJ, Lee M, Ji H, Davidson AJ, Horb ME, and Miller RK (2018). Transgenic Xenopus laevis Line for In Vivo Labeling of Nephrons within the Kidney. Genes (Basel) 9, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corkins ME, Krneta-Stankic V, Kloc M, McCrea PD, Gladden AB, and Miller RK(2019). Divergent roles of the Wnt/PCP Formin Daam1 in renal ciliogenesis. PLoS ONE 14, e0221698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson LA, Marsden M, Keller R, and Desimone DW (2006). Integrin α5β1 and fibronectin regulate polarized cell protrusions required for Xenopus convergence and extension. Curr. Biol 16, 833–844. [DOI] [PubMed] [Google Scholar]
- DeLay BD, Krneta-Stankic V, and Miller RK (2016). Technique to Target Microinjection to the Developing Xenopus Kidney. J. Vis. Exp, 53799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLay BD, Corkins ME, Hanania HL, Salanga M, Deng JM, Sudou N, Taira M, Horb ME, and Miller RK (2018). Tissue-Specific Gene Inactivation in Xenopus laevis: Knockout of lhx1 in the Kidney with CRISPR/Cas9. Genetics 208, 673–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, and Mayor R (2017). Tuning Collective Cell Migration by Cell-Cell Junction Regulation. Cold Spring Harb. Perspect. Biol 9, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grikscheit K, and Grosse R (2016). Formins at the Junction. Trends Biochem. Sci. 41, 148–159. [DOI] [PubMed] [Google Scholar]
- Habas R, Kato Y, and He X (2001). Wnt/Frizzled activation of Rho regulates vertebrate gastrulation and requires a novel Formin homology protein Daam1. Cell 107, 843–854. [DOI] [PubMed] [Google Scholar]
- Hemmati-Brivanlou A, and Melton DA (1994). Inhibition of activin receptor signaling promotes neuralization in Xenopus. Cell 77, 273–281. [DOI] [PubMed] [Google Scholar]
- Higashi T, Stephenson RE, and Miller AL (2019). Comprehensive analysis of formin localization in Xenopus epithelial cells. Mol. Biol. Cell 30, 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner RJ, and Wallingford JB (2018). Coming to Consensus: A Unifying Model Emerges for Convergent Extension. Dev. Cell 46, 389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner RJ, Malmi-kakkada AN, Sarikaya S, Weng S, and Wallingford JB (2020). Cadherin clustering controls heterogeneous, asymmetric junction dynamics during vertebrate axis elongation. bioRxiv. 10.1101/2020.02.11.944033. [DOI] [Google Scholar]
- Indra I, Choi J, Chen CS, Troyanovsky RB, Shapiro L, Honig B, and Troyanovsky SM (2018). Spatial and temporal organization of cadherin in punctate adherens junctions. Proc. Natl. Acad. Sci. USA 115, E4406–E4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal R, Breitsprecher D, Collins A, Corrêa IR Jr., Xu MQ, and Goode BL (2013). The formin Daam1 and fascin directly collaborate to promote filopodia formation. Curr. Biol 23, 1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei CN, Gallegos TF, Liu Y, Hukriede N, and Drummond IA (2019). Wnt signaling mediates new nephron formation during zebrafish kidney regeneration. Development 146, dev168294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karner CM, Chirumamilla R, Aoki S, Igarashi P, Wallingford JB, and Carroll TJ (2009). Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nat. Genet 41, 793–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata Galbraith K, Fujishima K, Mizuno H, Lee SJ, Uemura T, Sakimura K, Mishina M, Watanabe N, and Kengaku M (2018). MTSS1 Regulation of Actin-Nucleating Formin DAAM1 in Dendritic Filopodia Determines Final Dendritic Configuration of Purkinje Cells. Cell Rep. 24, 95–106.e9. [DOI] [PubMed] [Google Scholar]
- Kida YS, Sato T, Miyasaka KY, Suto A, and Ogura T (2007). Daam1 regulates the endocytosis of EphB during the convergent extension of the zebrafish notochord. Proc. Natl. Acad. Sci. USA 104, 6708–6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, and Davidson LA (2013). Microsurgical approaches to isolate tissues from Xenopus embryos for imaging morphogenesis. Cold Spring Harb. Protoc 2013, 362–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SW, Fang X, Ji H, Paulson AF, Daniel JM, Ciesiolka M, van Roy F, and McCrea PD (2002). Isolation and characterization of XKaiso, a transcriptional repressor that associates with the catenin Xp120(ctn) in Xenopus laevis. J. Biol. Chem 277, 8202–8208. [DOI] [PubMed] [Google Scholar]
- Krneta-Stankic V, Sabillo A, and Domingo CR (2010). Temporal and spatial patterning of axial myotome fibers in Xenopus laevis. Dev. Dyn 239, 1162–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunimoto K, Bayly RD, Vladar EK, Vonderfecht T, Gallagher AR, and Axelrod JD (2017). Disruption of Core Planar Cell Polarity Signaling Regulates Renal Tubule Morphogenesis but Is Not Cystogenic. Curr. Biol 27, 3120–3131.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefevre JG, Chiu HS, Combes AN, Vanslambrouck JM, Ju A, Hamilton NA, and Little MH (2017). Self-organisation after embryonic kidney dissociation is driven via selective adhesion of ureteric epithelial cells. Development 144, 1087–1096. [DOI] [PubMed] [Google Scholar]
- Li D, Hallett MA, Zhu W, Rubart M, Liu Y, Yang Z, Chen H, Haneline LS, Chan RJ, Schwartz RJ, et al. (2011). Dishevelled-associated activator of morphogenesis 1 (Daam1) is required for heart morphogenesis. Development 138, 303–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JXH, Tang VW, and Brieher WM (2020). Actin protrusions push at apical junctions to maintain E-cadherin adhesion. Proc. Natl. Acad. Sci. USA 117, 432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienkamp SS, Liu K, Karner CM, Carroll TJ, Ronneberger O, Wallingford JB, and Walz G (2012). Vertebrate kidney tubules elongate using a planar cell polarity-dependent, rosette-based mechanism of convergent extension. Nat. Genet 44, 1382–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Sato A, Khadka D, Bharti R, Diaz H, Runnels LW, and Habas R (2008). Mechanism of activation of the Formin protein Daam1. Proc. Natl. Acad. Sci. USA 105, 210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Meng W, Poy F, Maiti S, Goode BL, and Eck MJ (2007). Structure of the FH2 domain of Daam1: implications for formin regulation of actin assembly. J. Mol. Biol 369, 1258–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciano DK, Brakeman PR, Lee CZ, Spivak N, Eastburn DJ, Bryant DM, Beaudoin GM 3rd, Hofmann I, Mostov KE, and Reichardt LF (2011). p120 catenin is required for normal renal tubulogenesis and glomerulogenesis. Development 138, 2099–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon AP (2016). Development of the Mammalian Kidney. Curr. Top. Dev. Biol 117, 31–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendonsa AM, Na TY, and Gumbiner BM (2018). E-cadherin in contact inhibition and cancer. Oncogene 37, 4769–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RK, and McCrea PD (2010). Wnt to build a tube: Contributions of Wnt signaling to epithelial tubulogenesis. Dev. Dyn 239, 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RK, Canny SG, Jang C-W, Cho K, Ji H, Wagner DS, Jones EA, Habas R, and McCrea PD (2011). Pronephric tubulogenesis requires Daam1-mediated planar cell polarity signaling. J. Am. Soc. Nephrol 22, 1654–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody SA, and Kline MJ (1990). Segregation of fate during cleavage of frog (Xenopus laevis) blastomeres. Anat. Embryol. (Berl.) 182, 347–362. [DOI] [PubMed] [Google Scholar]
- Nagy II, Xu Q, Naillat F, Ali N, Miinalainen I, Samoylenko A, and Vainio SJ (2016). Impairment of Wnt11 function leads to kidney tubular abnormalities and secondary glomerular cystogenesis. BMC Dev. Biol 16, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwkoop PD, and Faber J (1994). Normal table of Xenopus laevis (Daudin) : a systematical and chronological survey of the development from the fertilized egg till the end of metamorphosis (Garland Publishing; ). [Google Scholar]
- Nishimura T, Honda H, and Takeichi M (2012). Planar cell polarity links axes of spatial dynamics in neural-tube closure. Cell 149, 1084–1097. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Ito S, Saito H, Hiver S, Shigetomi K, Ikenouchi J, and Takeichi M (2016). DAAM1 stabilizes epithelial junctions by restraining WAVE complex-dependent lateral membrane motility. J. Cell Biol 215, 559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nworu CU, Krieg PA, and Gregorio CC (2014). Preparation of developing Xenopus muscle for sarcomeric protein localization by high-resolution imaging. Methods 66, 370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien LL, Combes AN, Short KM, Lindström NO, Whitney PH, Cullen-McEwen LA, Ju A, Abdelhalim A, Michos O, Bertram JF, et al. (2018). Wnt11 directs nephron progenitor polarity and motile behavior ultimately determining nephron endowment. eLife 7, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxen L (1987). Organogenesis of the Kidney (Cambridge University Press; ). [Google Scholar]
- Shindo A, and Wallingford JB (2014). PCP and septins compartmentalize cortical actomyosin to direct collective cell movement. Science 343, 649–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sive HL, Grainger RM, and Harland RM (2010). Microinjection of Xenopus Oocytes. Cold Spring Harb. Protoc 2010, pdb.prot5536. [DOI] [PubMed] [Google Scholar]
- Strickland L, von Dassow G, Ellenberg J, Foe V, Lenart P, and Burgess D (2004). Light microscopy of echinoderm embryos. Methods Cell Biol. 74, 371–409. [DOI] [PubMed] [Google Scholar]
- Tada M, and Heisenberg CP (2012). Convergent extension: using collective cell migration and cell intercalation to shape embryos. Development 139, 3897–3904. [DOI] [PubMed] [Google Scholar]
- Takeichi M (2014). Dynamic contacts: rearranging adherens junctions to drive epithelial remodelling. Nat. Rev. Mol. Cell Biol 15, 397–410. [DOI] [PubMed] [Google Scholar]
- Venegas-Ferrín M, Sudou N, Taira M, and del Pino EM (2010). Comparison of Lim1 expression in embryos of frogs with different modes of reproduction. Int. J. Dev. Biol 54, 195–202. [DOI] [PubMed] [Google Scholar]
- Vestweber D, Kemler R, and Ekblom P (1985). Cell-adhesion molecule uvomorulin during kidney development. Dev. Biol 112, 213–221. [DOI] [PubMed] [Google Scholar]
- Vize PD, Jones EA, and Pfister R (1995). Development of the Xenopus pronephric system. Dev. Biol 171, 531–540. [DOI] [PubMed] [Google Scholar]
- Vize PD, Woolf AS, and Bard JBL (2003). The kidney: from normal development to congenital disease (Academic Press; ). [Google Scholar]
- Williams E, Villar-Prados A, Bowser J, Broaddus R, and Gladden AB (2017). Loss of polarity alters proliferation and differentiation in low-grade endometrial cancers by disrupting Notch signaling. PLoS ONE 12, e0189081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Higashi T, Suetsugu S, Sato Y, Ikeda T, Shirakawa R, Kita T, Takenawa T, Horiuchi H, Fukai S, and Nureki O (2007). Crystal structure of human DAAM1 formin homology 2 domain. Genes Cells 12, 1255–1265. [DOI] [PubMed] [Google Scholar]
- Yap AS, Gomez GA, and Parton RG (2015). Adherens Junctions Revisualized: Organizing Cadherins as Nanoassemblies. Dev. Cell 35, 12–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.