

Abstract
Background:
Ketamine is a highly effective antidepressant for patients with treatment-resistant major depressive disorder (MDD). Resting-state fMRI studies show disruptions of functional connectivity (FC) between limbic regions and resting-state networks (RSNs) including default mode (DMN), central executive (CEN), and salience networks (SN) in MDD. Here, we investigated whether serial ketamine treatments change FC between limbic structures and RSNs.
Methods:
MDD patients (n=44) were scanned at baseline (T1), and 24 hours after the first (T2), and fourth infusion (T3) of ketamine. Healthy controls (n=50) were scanned at baseline with a subgroup (n=17) rescanned at two weeks. Limbic regions included the amygdala and hippocampus and RSNs included the DMN, CEN and SN.
Results:
Ketamine increased right amygdala FC to the right CEN (p=0.05), decreased amygdala FC to the left CEN (p=0.005) at T2 versus T1 (p=0.015), which then increased at T3 versus T2 (p=0.002), and decreased left amygdala FC to the SN (p=0.016). Decreased left amygdala to SN FC at T2 predicted improvements in anxiety at T3 (p=0.006). Ketamine increased right hippocampus FC to left CEN (p=0.001) and this change at T2 predicted decreased anhedonia at T3 (p=0.005).
Conclusions:
Ketamine modulates FC between limbic regions and RSNs implicated in MDD. Increases in FC between limbic regions and the CEN suggest ketamine may be involved in restoring top-down control of emotion processing. FC decreases between the left amygdala and SN suggest ketamine may ameliorate MDD-related dysconnectivity in these circuits. Early FC changes between limbic regions and RSNs may be predictive of clinical improvements.
Keywords: Major depression, mood disorders, antidepressant treatment, ketamine, personalized medicine, functional connectivity
INTRODUCTION
Numerous effective pharmacotherapies are available to treat major depressive disorder (MDD), however, less than half of patients remit within the first three months of treatment (1) and ~30% remain unresponsive to ≥2 pharmacotherapies (2, 3). This may be explained by large heterogeneity of MDD (4). Ketamine, a NMDA receptor antagonist, is shown to induce fast-acting and robust antidepressant effects in 60–70% of treatment-resistant depression (TRD) patients (5). Growing evidence also suggests that multiple ketamine treatments may lead to more durable response (6–8) . Depressive symptoms improve within hours to days post infusion suggesting that ketamine perturbs neural pathways mediating the regulation and expression of mood and emotion, thus playing a downstream role in therapeutic response.
While the cause of MDD still remains elusive, a large body of literature points to systems-level disruptions in cortico-limbic networks mediating mood, emotion, and cognition. Resting-state functional magnetic resonance imaging (rsfMRI) provides a powerful noninvasive means to examine disruptions in functional connectivity (FC) of these networks in MDD, (9–11) implicating several resting state networks (RSNs) (12–14). The default mode network (DMN), including the anterior cingulate cortex (ACC), posterior cingulate cortex (PCC), precuneus, angular gyrus, and medial prefrontal cortex (PFC), is widely implicated in MDD, specifically with features such as rumination, impaired attention and cognitive control (15–17). DMN is hyperactive in patients with MDD (18), and has been reported to normalize with standard antidepressant treatments (19, 20). The salience network (SN) is also hyperactive in MDD (21), and is comprised of the insula, dorsal ACC, and the frontopolar cortex. The SN is involved in detecting and filtering salient stimuli in order to contribute to functions, such as communication, social behavior, and self-awareness through the integration of sensory, emotional, and cognitive information (22, 23). The lateral parietal cortices and dorsolateral PFC (DLPFC) comprise the Central Executive Network (CEN) that is involved in goal-directed functions such as attention, decision-making, working memory, and executive control (24, 25) and has been reported to show decreased connectivity in depression (26, 27). Taken together, rsfMRI studies support the theory that MDD associates with an imbalance between hyperactive ventral and hypoactive dorsal cortico-limbic systems (28–30).
Numerous studies have also implicated the amygdala and hippocampus in the pathophysiology of MDD. In addition to its role in episodic memory, the hippocampus is involved in the regulation of motivation and emotion, responses to emotion, and regulation/susceptibility of stress, and the amygdala is involved in the autonomic responses to emotion, emotional memory, and emotion regulation (31–34). Several neuroimaging studies have reported dysfunction and treatment modulation of the hippocampus in MDD (35–38). Specifically, smaller hippocampal volume is reported in MDD compared to non-depressed individuals (39, 40) and lower nodal centralities of the left hippocampus are related to longer duration of disease (37). MRI investigations have also implicated the amygdala in MDD including disrupted connectivity with the dorsal cingulate (11) and insula (41, 42), increased amygdala activation to faces task that resolves post antidepressant treatment (43), and lower white matter integrity between the amygdala and regions of the CEN, SN, and DMN correlated to symptom severity (44). Notably, both the amygdala and hippocampus are thought to be nodes of the SN (amygdala) and DMN (amygdala and hippocampus); therefore, targeting connectivity between these limbic regions and cortex-dominant RSNs may help illuminate the mechanisms of antidepressant response to interventions like ketamine.
The current neuroimaging literature investigating effects of ketamine in MDD is small; however, a few studies have suggested effects of single infusion of ketamine on the subgenual ACC, posterior parietal cortex, hippocampus, PFC, and DMN (18, 45, 46). In a task-based fMRI study, ketamine treatment associated with a significant increase in activity within the right caudate (47). It is important to note that the studies thus far are limited in finding neural correlates of clinical change with ketamine, and mostly have not addressed the more durable effects of multiple ketamine infusions. However, two recent studies from our group reported changes in cerebral blood flow in precuneus and occipital regions and changes in amygdala activity for emotional face processing after multiple ketamine infusions, though this remains an underrepresented area of research (48, 49).
To address how single and serial ketamine treatment perturbs functional connectivity and clinical correlates of ketamine-related clinical response, using rsfMRI we investigated whether intravenous ketamine leads to similar or distinct changes in connectivity between “limbic” regions (amygdala, hippocampus), and the cortical networks that might be involved in regulating the activity of those structures (CEN, DMN, SN). We hypothesized that FC between amygdala and/or hippocampus and cortical RSNs might be deficient in depression, and could be “restored” following a single and serial infusions of ketamine in patients with treatment-resistant MDD. If a significant change in FC with treatment was observed, post-hoc analyses investigated cross-sectional differences between patients and controls and clinical correlations with longitudinal change in FC to further understand the acute antidepressant effects of ketamine. Previous randomized placebo-controlled trials have clearly established the superiority of ketamine over placebo in improving depression (5, 7, 50); therefore, we chose an open-label design to minimize patient burden and to address our goal of understanding the effects of ketamine on cortico-limbic networks.
MATERIALS AND METHODS
Participants:
Participants included N=44 MDD patients and N=50 non-depressed healthy controls (HC). Handedness was not considered because 94% of participants were right-handed. MDD patients received a clinical evaluation battery at baseline (T1), 24 hours after a single sub-anesthetic dose of 0.5 mg/kg of intravenous ketamine (T2), and 24–72 hours post a fourth infusion of ketamine (Fig. 1). The study lasted 2–2.5 weeks dependent on the day the first infusion started. Data was not collected on weekends. These intervals were predetermined based on scheduling availabilities. Five MDD patients did not complete T3 due to scheduling conflicts. Thirty-three HCs were scanned once only while an additional seventeen HCs were scanned twice 2 weeks apart. HC did not receive ketamine. Patients were recruited from the Southern California region and were consented for participation as approved by the UCLA Institutional Review Board (Table 1). Eligibility criteria for all patients included a diagnosis of MDD by clinical consultation using DSM-V (SCID (51)) criteria, unsuccessful response to ≥2 prior antidepressant trials, 20–65 years of age, pre-treatment 17-item Hamilton Depression Rating Scale (HDRS (52)) of >16 (exhibiting moderate to severe depressive symptoms), and a referral letter from their treating physician. Exclusion criteria included neurological/physical/developmental disorders, substance abuse/dependence history within the preceding 3-months, current or past history of psychosis, schizoaffective disorder or schizophrenia, first episode or late onset of depression (>50 years), depression related to a medical condition, ketamine, ECT or other neuromodulation therapy within the previous 6 months, or suicidal attempt 1 month prior to study start.
Figure 1. Study design.
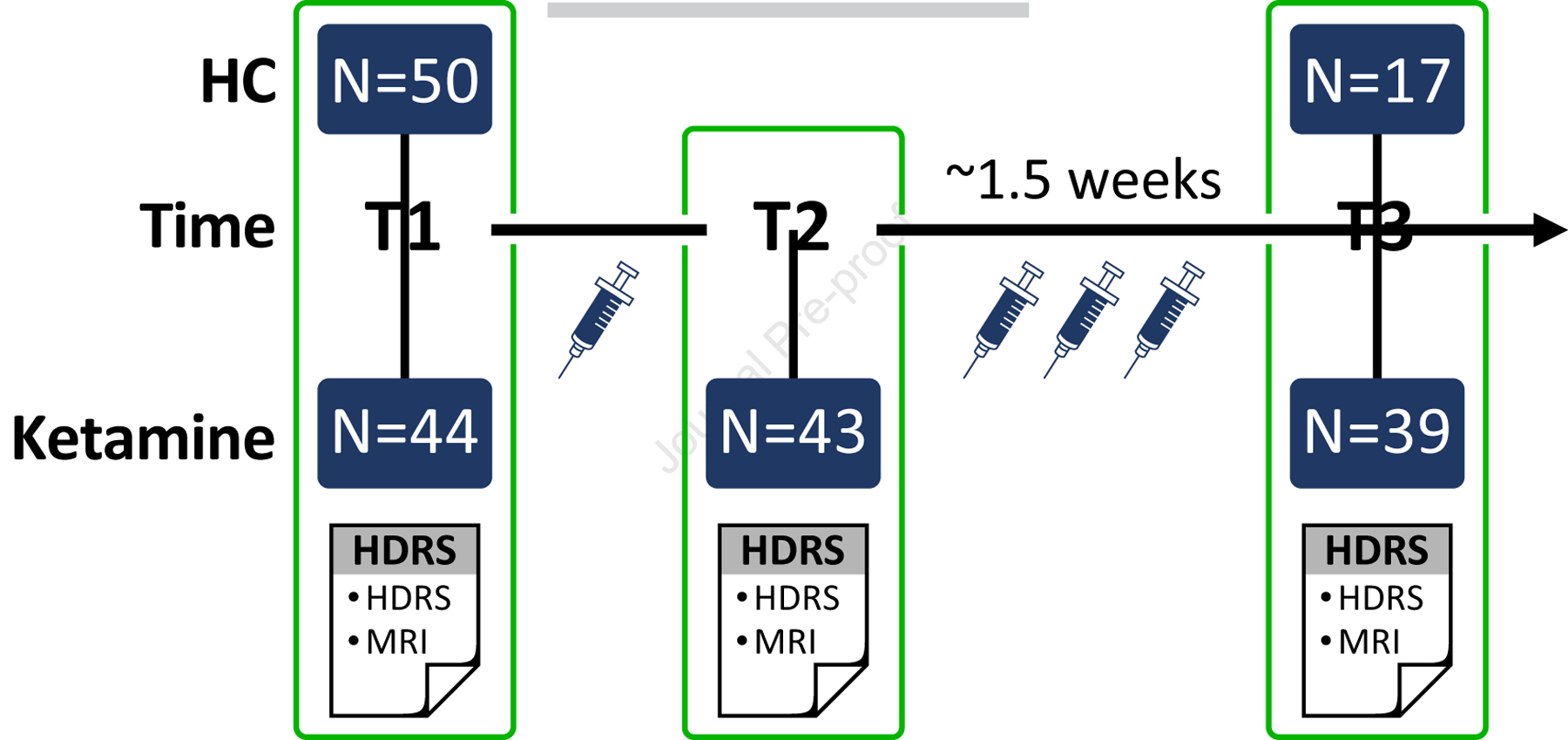
Ketamine group was administered clinical scales and received an MRI at three time points (T1: pre-treatment/baseline, T2: 24 hours post-first infusion of ketamine, and T3: 24–72 hours post fourth infusion of ketamine). The study length varied from 2–2.5 weeks depending on which day the first infusion started on. Fifty non-depressed healthy controls (HC) received an MRI at baseline (T1) and a subset of the fifty (n=17) HCs received a repeat assessment 2 weeks after T1.
Table 1.
Demographics and clinical characteristics
| Ketamine (n= 44) | HC (n=50) | HC with repeat (n=17)b | |
|---|---|---|---|
| Gender (F/M) | 18/26 | 27/23 | 9/8 |
| Age (years) | 38.2±10.9 | 32.3±11.9 | 28.2±6.9 |
| Education | 10.1 ±2.4 | 10.8±1.9 | 11.5±1.5 |
| Lifetime Illness (years) | 20.2±12.1 | -- | |
| Current episode (years) |
5.3±6.6 | -- | |
| Responsea | 23/39 (59%) | -- |
Response was defined as ≥50% improvement in Hamilton Depression Rating Scales (HDRS) 24–72 hours after the fourth ketamine infusion (T3).
HC with repeat (n=17) are a subset of the total cohort of HC (n=50)
Ketamine:
Patients were permitted to remain on stable antidepressant medications (unchanged for at least six weeks prior to treatment). Benzodiazepines that influence cortical excitability and other medications considered a contraindication to ketamine were discontinued 72 hours prior to the first infusion and throughout the treatment trial. Treatment included 40-minute IV infusions of a sub-anesthetic dose (0.5 mg/kg) of ketamine diluted in 60cc of saline with continuous clinical and hemodynamic monitoring (5). Psychotomimetic effects, blood pressure, blood oxygen saturation, heart rate, and respiratory rate were monitored during the infusion by a psychiatrist followed by additional monitoring for 3 hours by a trained nurse.
Clinical outcome measures:
The HDRS- 17 item questionnaire was used to track overall response with ketamine after the fourth infusion (T3). Patients were identified as responders if HDRS scores decreased by ≥50% at T3 from baseline (53). Snaith-Hamilton Pleasure Scale (SHAPs) (54), Depression Anxiety Stress Scale (DASS) (55, 56), and behavioral inhibition (BIS) scale (57), and a combined Rumination scale (58, 59) were administered at all three time points in order to evaluate the effect of ketamine on specific symptoms of MDD. These scales were chosen based on wide use in depression literature.
Imaging Protocol and Processing:
All imaging of participants was performed on a Siemens 3T Prisma MRI system at UCLA’s Brain Mapping Center using a 32 channel head coil. Image acquisition sequences from the Human Connectome Project (HCP) Lifespan studies were utilized in this study. For resting-state scans, two runs of a multiband EPI sequence with inverse phase encoding were acquired: repetition time (TR)=800 ms, echo time (TE)=37 ms, flip angle=52°, 72 axial slices, 2×2×2 mm3 spatial resolution, multiband factor = 8, phase encoding direction=AP/PA, acquisition time (TA)=6:41 per run. The structural scans consisted of one T1 weighed (voxel size (VS)=0.8mm isotropic; TR=2500ms; TE=1.81:3.6:5.39:7.18ms; inversion time (TI)=1000ms; flip angle (FA)=8.0°; TA=8:22min) and one T2 weighted acquisition (VS=0.8mm isotropic; TR=3200ms; TE=564ms; TA=6:35min). All data was preprocessed using the Human Connectome Project minimal preprocessing pipeline (60). Independent components (ICs) representing artifacts were identified using ICA for each run and removed from voxel timecourses using FSL regfilt. To identify resting state networks (RSNs), group ICA was run using FSL MELODIC (50 components) on all MDD and HC volunteers, and dual regression extracted time courses for each IC for each volunteer. Three RSNs associated with MDD (DMN, CEN, and SN) were chosen to investigate changes in functional connectivity (FC) with anatomical ROIs selected a priori, including the hippocampus (right and left) and amygdala (right and left) (13). Group ICA identified five ICs that encompassed the three RSNs of interest: one DMN, two CENs (left and right CEN), and one salience network (SN). Time courses for the amygdala (right and left) and hippocampus (right and left) were extracted using ROI masks derived from the Harvard-Oxford subcortical structural atlases (61). Correlations were calculated between time courses of the networks and seeds (Fischer’s z-scores).
Statistical Analyses:
Baseline demographic and clinical measures were evaluated using chi-square tests for categorical variables and independent 2 sample t-tests for continuous variables. Main effects of ketamine treatment on FC were investigated using linear mixed models (compound symmetry covariance) on Fischer’s z-scores with time, run and hemisphere as repeated factors in MDD patients. These omnibus analyses were Bonferroni corrected for the two main hypotheses tested (α=0.05/2=0.025): (1) FC of the amygdala to RSNs and (2) FC of the hippocampus to RSNs change with ketamine treatment in patients with MDD.
A number of follow-up analyses were considered. If a time by hemisphere effect was present (pcorr<0.05), follow-up analyses investigated the main effect of treatment (time) separately for each hemisphere with time and run as repeated measures. If an effect of time was present, cross-sectional post-hoc analyses (independent samples t-test) were performed to study differences between healthy controls and MDD patients at baseline. In addition, to study clinical correlations 1) change in FC after a single infusion of ketamine and percent change in clinical scores (BIS, SHAPs, Rumination, and DASS) after full treatment and 2) change in FC after serial infusions of ketamine and percent change in clinical scores (BIS, SHAPs, Rumination, and DASS) after full treatment were investigated. These clinical correlations were only investigated if an effect of time was present in MDD patients. These analyses were Bonferroni corrected for 8 tests α=0.05/8=0.006; 2 hemispheres and 4 clinical scales) within each metric (limbic structure to 3 RSNs). The approach for post-hoc cross-sectional and clinical correlation investigation was chosen to facilitate interpretation of the significant effects of ketamine and reduce multiple comparisons. In order to further investigate significant treatment effects, paired t-tests were performed to examine effects of time in HC (n=17).
RESULTS
Subject characteristics.
The MDD and HC groups did not differ significantly in gender (χ2=1.6, p=0.205) or education (t(1,91)=1.58, p=0.12). A significant difference in age (t(1,92)=−2.52, p=0.013) was observed; therefore, cross-sectional analysis included age as a covariate (Table 1). Overall there was a 54.9% decrease in HDRS for all patients at T3. A significant decrease during treatment was observed for the SHAPS, DASS, and rumination scales, while the BIS scale did not significantly change (Table 2).
Table 2:
Clinical Measures for patients at baseline (TP1); 24 hours after the first infusion (TP2) and 24–72 hours after fourth infusion (TP3).
| TP1 | TP2 | TP3 | ANOVA | ||
|---|---|---|---|---|---|
| Mean (SD) |
Mean (SD) | Mean (SD) | F | p | |
| HDRS | 19.3 (5.2) | 13.0 (4.6) | 8.3 (4.2) | 57.8 | <0.0001 |
| Rumination | 13.0 (3.1) | 11.7 (3.2) | 10.3 (2.2) | 9.4 | <0.0001 |
| BIS | 23.8 (3.2) | 23.0 (4.0) | 22.5 (3.4) | 1.3 | 0.27 |
| SHAPS | 7.7 (4.0) | 6.4 (4.4) | 3.2 (3.7) | 13.4 | <0.0001 |
| DASS | 5.1 (4.7) | 3.7 (3.7) | 1.6 (2.0) | 9.2 | <0.0001 |
HDRS: Hamilton Depression Rating Scale- 17 item, BIS: behavioral inhibition scale, SHAPS: Snaith-Hamilton Pleasure Scale, DASS: Depression Anxiety Stress Scale
Effects of ketamine on amygdala to RSN connectivity.
The omnibus mixed model used to investigate effects of time showed significant effects for FC between the amygdala and two RSNs of interest. Amygdala FC to the left CEN showed a significant effect of time (F(2, 461.1)=5.33, p=0.005), which was consistent across hemispheres (i.e., no significant time-by-hemisphere interaction; Fig. 2). Pairwise comparisons showed a drop in FC after a single infusion (T1 vs T2, p=0.015) and a subsequent increase after serial infusion (T2 vs T3, p=0.002). In healthy controls, FC between amygdala and left CEN did not differ from patients at baseline or change over time (p>0.05 for both).
Figure 2. Amygdala connectivity to resting-state networks (RSNs).
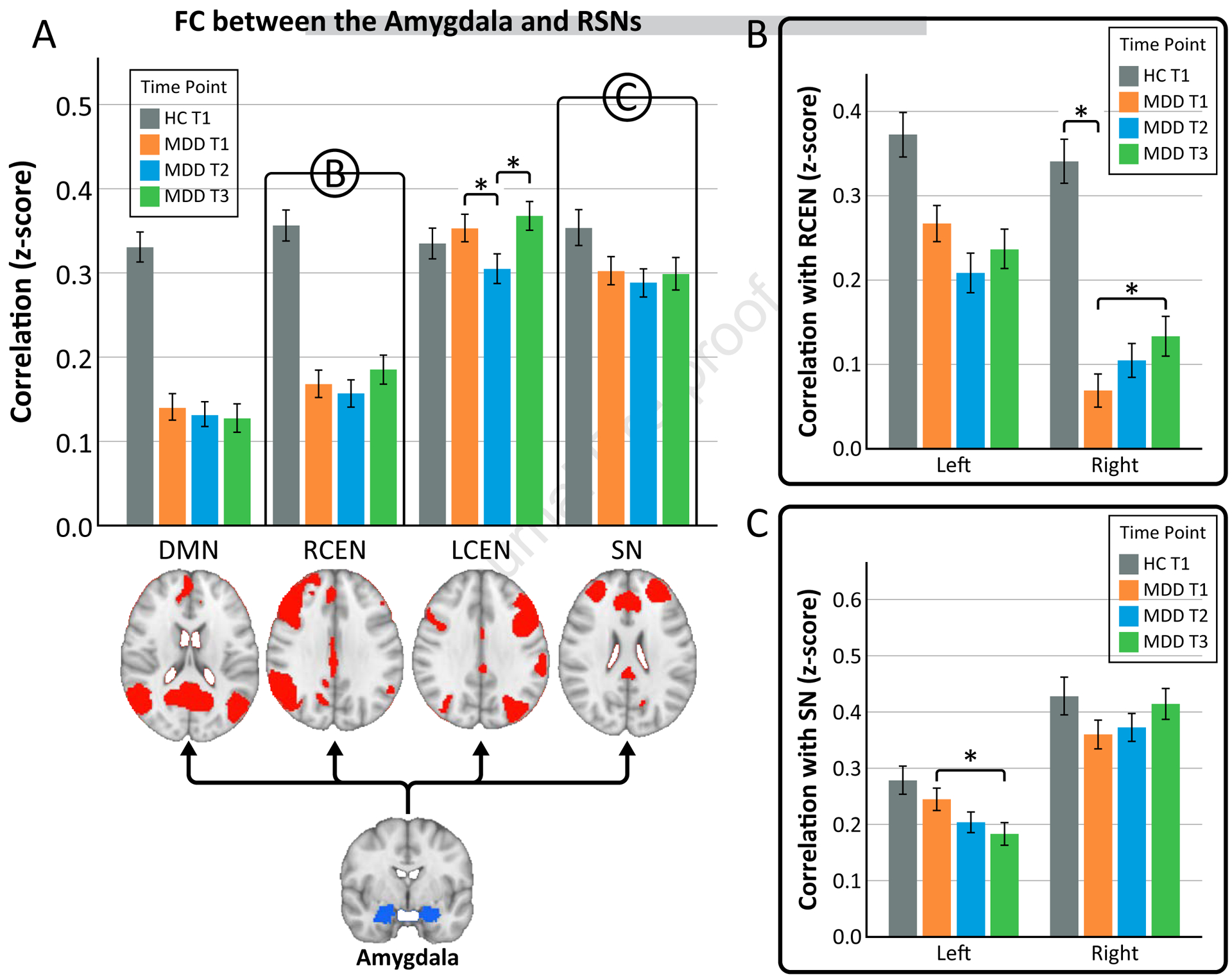
A) Functional connectivity between the bilateral amygdala and RSNs (default mode network (DMN), right central executive network (RCEN), left central executive network (LCEN), and salience network (SN)). Connectivity between the amygdala and the RCEN and SN showed a time by hemisphere effect, therefore, the right and left amygdala were looked at separately in B and C. B) Functional connectivity between the left and right amygdala and RCEN. C) Functional connectivity between the left and right amygdala and SN.
*p<0.05
FC between the amygdala to both the right CEN and the SN showed time by hemisphere effects (F(2, 455.6)=3.98, p=0.019; F(2, 456.0)=4.24, p=0.015 respectively), and therefore follow-up analysis were completed for the left and right amygdala separately. FC between the right amygdala and the right CEN showed significant changes with ketamine treatment (F(2,207.2)=2.99, p=0.05). Specifically, FC increased after serial ketamine infusion (T1 vs T3, p=0.016). Cross-sectional analysis showed HC had greater connectivity between the right amygdala and right CEN than MDD at baseline (F=9.9, df=1, p=0.002); therefore, serial ketamine could be considered to have a “normalizing” effect towards controls. No effect of time was observed between the right amygdala and right CEN for HC (p>0.05). FC between the left amygdala and the right CEN showed a decreasing trend but did not reach significance (F(2, 206.2)=2.61, p=0.076).
FC between the left amygdala and SN showed significant decreases with ketamine treatment (F(2,206.8)=4.25, p=0.016) (Fig. 2). After a single infusion, a trend towards lower FC was apparent (T1 vs. T2, p=0.065). After serial infusions, FC decreased significantly (T1 vs T3, p=0.005). In healthy controls, FC between left amygdala and SN did not differ from MDD patients at baseline or change over time (p>0.05 for both). FC between the right amygdala and SN did not change significantly with ketamine treatment (F(2, 206.7)=2.07, p=0.13) (Fig. 2).
Effects of ketamine on hippocampus to RSN connectivity.
The omnibus mixed model used to investigate effects of time showed significant effects for the hippocampus FC to one RSN of interest. Hippocampal FC to left CEN showed a time by hemisphere effect (F(2, 455.9)=3.9, p=0.022) and therefore follow-up analysis were completed for the left and right hippocampus separately. FC of the right hippocampus to the left CEN went from no connectivity to negative connectivity (“anticorrelated”) after ketamine treatment (F(2,206.6)=7.75, p=0.001) (Fig. 3). Pairwise comparisons for left CEN showed increased negative connectivity after both single (T1 vs T2, p=0.001) and serial ketamine (T1 vs T3, p=0.001). In healthy controls, FC between the right hippocampus and left CEN did not differ from MDD patients or change over time (p>0.05 for both). FC between the left hippocampus and left CEN showed no significant change with ketamine treatment (F(2, 206.6)=0.77, p=0.46) (Fig. 3).
Figure 3. Hippocampal connectivity to resting-state networks (RSNs).
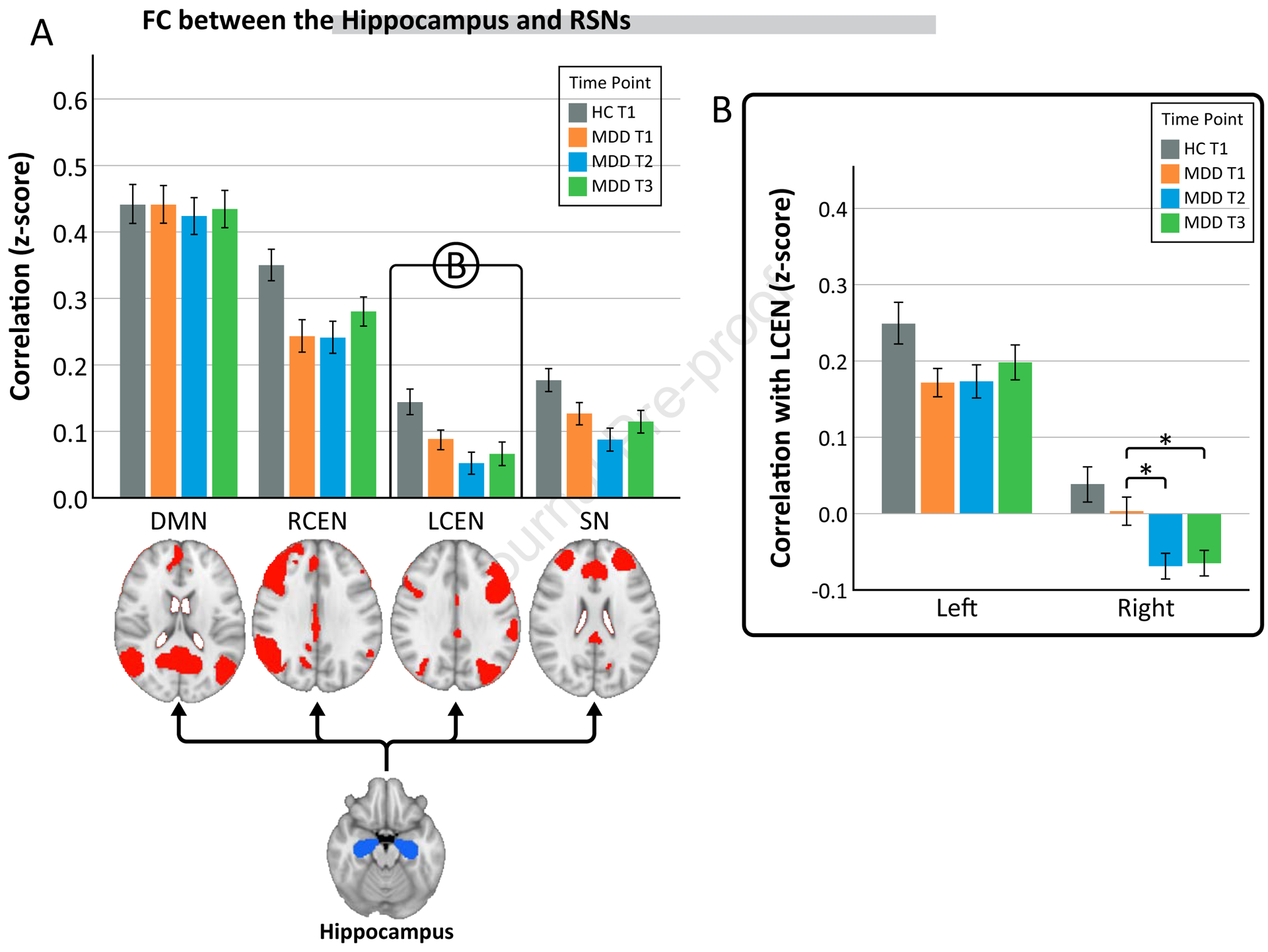
A) Functional connectivity between the bilateral hippocampus and RSNs (default mode network (DMN), right central executive network (RCEN), left central executive network (LCEN), and salience network (SN)). Connectivity between the hippocampus and the LCEN showed a time by hemisphere effect therefore the right and left hippocampus were looked at separately in B. B) Functional connectivity between the left and right hippocampus and LCEN.
*p<0.05
Clinical correlations
Amygdala.
Correlations between acute change in FC between the left amygdala and SN after a single infusion was correlated with post-treatment improvement in BIS (Pearson’s r=0.44, p=0.006) after all infusions (Fig. 4a). BIS improvement was also correlated with change in FC between left amygdala and SN at the end of treatment (Pearson’s r=0.46, p=0.004) (Fig. 4c). No other correlations were significant for amygdala FC.
Figure 4. Correlations between measures of clinical improvement and reductions in FC between limbic regions and resting-state networks.
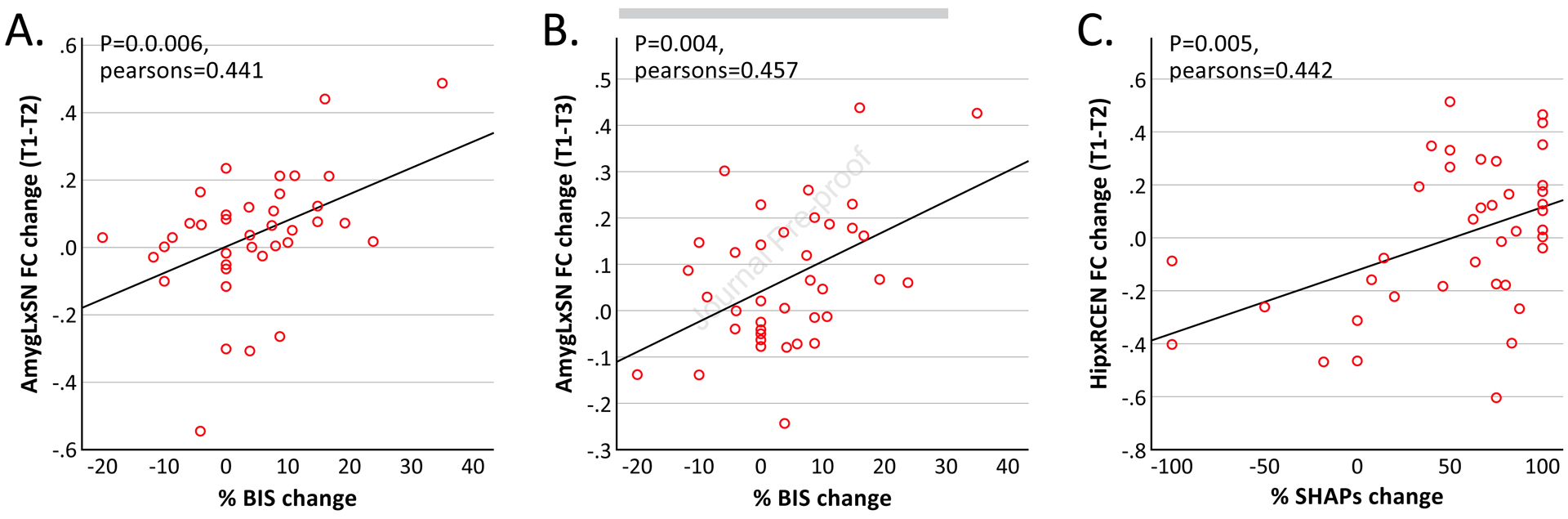
(A) Change in FC between the left amygdala (AmygL) and salience network (SN) after a single infusion of ketamine correlated with change in BIS at the end of treatment. (B) Change in FC between the AmygL and SN at the end of treatment correlated with change in BIS at the end of treatment. (C) Change in FC between the hippocampus (Hip) and right central executive network (RCEN) after a single infusion of ketamine correlated with change in SHAPs at the end of treatment. BIS, behavioral inhibition system scale; SHAPs, Snaith-Hamilton Pleasure Scale; T1, time 1 (baseline); T2, time 2 (24 hours after first infusion of ketamine); T3, time 3 (24 hours after fourth infusion of ketamine).
Hippocampus.
Acute change in FC between the hippocampus and right CEN after a single infusion of ketamine correlated with SHAPs (Pearson’s r=0.45, p=0.004) (Fig. 4b). No other correlations with hippocampal FC reached our criterion for significance.
DISCUSSION
Experimental models informed by current evidence suggest MDD is a brain-network disorder affecting several regions and networks within the brain (13) that can be mediated with antidepressant treatments (14, 62). Here, we studied the acute effects of single and serial ketamine infusions in patients with treatment-resistant MDD on the modulation of functional connectivity between two limbic regions (amygdala and hippocampus) and three target RSNs (SN, CEN, and DMN). The regions and RSNs were selected a-priori based on their implications in MDD (12–14, 37, 49). Investigations included longitudinal analysis, cross-sectional analysis, and clinical correlations of longitudinal changes in FC. Our results showed that connectivity of the amygdala and hippocampus to RSNs changed with treatment and these changes related to improvements in features of depression such as anxiety and anhedonia. FC also did not change over time in controls, providing further evidence that these effects in MDD are related to ketamine and not due to poor test-retest reliability.
Longitudinal changes with ketamine treatment
Effects of treatment on FC between amygdala and RSNs.
FC of the amygdala to the left CEN showed a significant decrease after a single infusion of ketamine which then stabilized after serial ketamine infusions, increasing towards FC observed in healthy controls. While this change in FC between the amygdala and left CEN was higher at T3 than T1 it did not reach significance. Similarly, amygdala connectivity to the right CEN increased with treatment in the direction of healthy controls implying normalization, though this effect did not reach significance for the left amygdala. This is in line with previous literature implicating CEN and prefrontal cortex hypoconnectivity in MDD (26, 63). An exploratory investigation reported decreased amygdala connectivity with regions involved in cognition and executive control such as the dorsolateral prefrontal cortex and the inferior frontal gyrus (both regions are a part of the CEN) in women with MDD (64). Cognitive behavioral therapy is also shown increases in CEN connectivity (65) and increases in FC between amygdala and the cognitive control network (66). Jenkins et al. also reported recently that retention of FC between the amygdala and CEN is important in the cognitive control of emotion, which may improve performance during emotional faces recognition tasks (67). Therefore, ketamine may be involved in increasing connectivity between the amygdala and CEN leading to increased top-down control of emotion processing frequently observed as disturbed in MDD patients (10, 27, 68, 69).
FC of the left amygdala to the SN was decreased in MDD at baseline, and further decreased with ketamine treatment in the MDD group. This is consistent with a previous study reporting that a single infusion of ketamine reduced FC between the insula (part of the SN) and the DMN, which was also decreased in MDD compared to healthy controls (45). However, prior findings have also reported increased SN FC in MDD compared to healthy controls. An analysis of causal connectivity showed significantly higher effective connectivity of amygdala to the anterior insula (a node of the SN) in MDD (41), a rsfMRI analysis demonstrated increased insula FC with the amygdala (42), and another study showed increased SN connectivity in MDD (21). With the role of the amygdala and SN in emotion processing and perception, disruptions in this network may be the cause of decreased ability to process emotions (70). Our results show ketamine may resolve disrupted connectivity and lead to more normalized FC potentially underlying emotion regulation.
Effects of treatment on FC between hippocampus and RSNs.
The right hippocampus showed greater negative FC (“anti-correlation”) to the left CEN with treatment. Our results showing an anticorrelation effect after a single infusion and then sustained anticorrelation after serial infusion shows that ketamine may be restoring negative connectivity. Negative FC between the hippocampus and regions of the CEN including bilateral PFC, bilateral parietal lobe have been reported in healthy controls (71). These results are consistent with the limbic-cortical dysregulation model of MDD as proposed by Mayberg (27, 69, 72). The cortical compartment, inferior parietal cortex and DLPFC, is associated with depressive symptoms including apathy, anhedonia, and cognitive performance while the limbic compartment, including the hippocampus, mediate vegetative and somatic aspects of MDD. Depressive symptoms can be linked to decreases in activity in cortical regions and increases in limbic areas (27, 33, 69, 72, 73) implicating different and interacting roles of cortical and limbic areas in regulation of emotion and cognition as well as in the pathology of MDD. Therefore, we propose, weaker or absence of this negative FC in MDD may relate to dysfunction of the functional segregation, perhaps leading to cognitive and emotional symptoms in MDD. Notably, our findings show ketamine restored the functional segregation between the right hippocampus and the left CEN, which is compatible with the idea that ketamine restores top-down regulation of ventral limbic structures and may be related to symptom remission. This interpretation is consistent with previous neuroimaging studies indicating that single infusions of ketamine can induce treatment effects in similar regions, such as subgenual ACC, posterior parietal cortex, hippocampus, PFC, caudate, and DMN (18, 45–47). Our results provide further evidence of single ketamine induced BOLD changes and provide the first evidence of changes due to serial ketamine treatment.
Neural correlates of clinical change
Previous investigations have reported relationships between changes in global depression score and fMRI changes after a single infusion of ketamine. For example, a recent study demonstrated increased global connectivity in the PFC, insula and caudate in responders to a single ketamine infusion, suggesting baseline prefrontal and striatal circuitry may be relevant to successful clinical outcomes (46). Another recent study reported that FC increases between the lateral PFC and subgenual ACC were associated with symptom reduction and that lower baseline FC predicted response (74). A pilot study also showed differences in diffusion metrics in fronto-limbic pathways between responders and nonresponders after a single infusion of ketamine (75). As a follow-up analyses to further understand the changes in FC observed, we targeted several different symptom dimensions (inhibition/avoidance, anxiety, rumination, anhedonia). We investigated potential associations of these clinical scales with FC changes after a single and multiple ketamine infusions.
The BIS scale, a measure of inhibition and avoidance that is typically elevated in depression (76,77), did not change after ketamine treatment on average. However, posttreatment decreases (improvements) in the BIS scale were significantly correlated with acute and posttreatment decreases in FC between the left amygdala and SN. Critically, this initial decrease in FC may show plasticity of this network that relates to improvements in anxiety symptoms elevated in MDD (78). We also report that improvement in the SHAPs, a measure of anhedonia that is increased in depression (79), was correlated with acute decreases in FC between the hippocampus and right CEN. Taken together, these results indicate that acute FC change after a single infusion of ketamine predicts improved anxious avoidance and anhedonia after serial ketamine infusions.
Limitations
Several limitations with this investigation need to be considered. First, the results of this study should be validated in a larger sample, particularly with respect to less powered cross-sectional effects. Second, we designed this mechanistic trial as open-label, both because the primary goal targeted longitudinal effects of ketamine on brain connectivity (i.e., not efficacy of ketamine to treat depression) and because of the ethical implications of including a placebo group in TRD volunteers. We felt it unnecessarily burdensome for suffering patients to receive a placebo treatment in this multi-visit study. Given that previous randomized placebo-controlled trials have clearly established the superiority of ketamine to improve the symptoms of depression compared to placebo, it is very unlikely that the changes in connectivity we report here can be explained by the placebo effect. Nevertheless, the neurobiological basis of the placebo effect in depression in an underrepresented area of research, and could be addressed by larger multi-site mechanistic studies. Our MDD participants had a limited range of symptoms with a mean of 19.3 on the 17-item HDRS. This may affect their baseline FC, however, we show no correlation between baseline FC and HDRS scores and ketamine is also most likely to be used in a population exhibiting moderate to severe depressive symptoms (5, 50, 80).
Conclusion
This is the first paper investigating imaging effects of serial ketamine infusions on functional connectivity. Findings from the current analysis support previous findings and demonstrate that ketamine therapy leads to neuroplasticity between limbic regions (amygdala and hippocampus) and RSNs that are essential for emotion regulation, executive function, goal-orientated behavior, self-awareness, and social behavior. A restoration of FC is observed between the amygdala and SN, amygdala and the right CEN, and between the hippocampus and the left CEN with ketamine treatment. Neuroplasticity of these networks also related to clinical improvements in anxiety and anhedonia. Further, results suggest early neuroplasticity may serve as a biomarker for clinical outcomes. Although ketamine did not appear to influence DMN FC in our study, future studies targeting other aspects of DMN connectivity beyond the amygdala and hippocampus may be more informative given the importance of the DMN to the neurobiology of depression. Overall, findings support repeated ketamine therapy leads to regulation of limbic regions by large scale RSNs and this reestablished regulation may be a neural correlate of symptom reduction.
Acknowledgements
This study was supported by Award Numbers F32MH111193 (to MMV), K24MH102743 (to KLN), and U01MH110008 (to KLN and RE) from the National Institute of Mental Health and the Muriel Harris Chair in Geriatric Psychiatry (to RE). This research was additionally supported by the UCLA Depression Grand Challenge, support for which is provided by the UCLA Office of the Chancellor and philanthropy. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Mental Health or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ClinicalTrials.gov: Biomarkers of Fast Acting Therapies in Major Depression, https://clinicaltrials.gov/ct2/show/NCT02165449, NCT02165449
Disclosures
The authors report no biomedical financial interests or potential conflicts of interest.
References:
- 1.Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, et al. (2006): Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 163:28–40. [DOI] [PubMed] [Google Scholar]
- 2.McGrath PJ, Stewart JW, Fava M, Trivedi MH, Wisniewski SR, Nierenberg AA, et al. (2006): Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry. 163:1531–1541; quiz 1666. [DOI] [PubMed] [Google Scholar]
- 3.Mrazek DA, Hornberger JC, Altar CA, Degtiar I (2014): A review of the clinical, economic, and societal burden of treatment-resistant depression: 1996–2013. Psychiatr Serv. 65:977–987. [DOI] [PubMed] [Google Scholar]
- 4.Zimmerman M, Ellison W, Young D, Chelminski I, Dalrymple K (2015): How many different ways do patients meet the diagnostic criteria for major depressive disorder? Compr Psychiatry. 56:29–34. [DOI] [PubMed] [Google Scholar]
- 5.Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. (2006): A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 63:856–864. [DOI] [PubMed] [Google Scholar]
- 6.Murrough JW, Perez AM, Pillemer S, Stern J, Parides MK, aan het Rot M, et al. (2013): Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biol Psychiatry. 74:250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh JB, Fedgchin M, Daly EJ, De Boer P, Cooper K, Lim P, et al. (2016): A Double-Blind, Randomized, Placebo-Controlled, Dose-Frequency Study of Intravenous Ketamine in Patients With Treatment-Resistant Depression. Am J Psychiatry. 173:816–826. [DOI] [PubMed] [Google Scholar]
- 8.aan het Rot M, Collins KA, Murrough JW, Perez AM, Reich DL, Charney DS, et al. (2010): Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry. 67:139–145. [DOI] [PubMed] [Google Scholar]
- 9.Dichter GS, Gibbs D, Smoski MJ (2015): A systematic review of relations between resting-state functional-MRI and treatment response in major depressive disorder. J Affect Disord. 172:8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnstone T, van Reekum CM, Urry HL, Kalin NH, Davidson RJ (2007): Failure to regulate: counterproductive recruitment of top-down prefrontal-subcortical circuitry in major depression. J Neurosci. 27:8877–8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matthews SC, Strigo IA, Simmons AN, Yang TT, Paulus MP (2008): Decreased functional coupling of the amygdala and supragenual cingulate is related to increased depression in unmedicated individuals with current major depressive disorder. J Affect Disord. 111:13–20. [DOI] [PubMed] [Google Scholar]
- 12.Menon V (2011): Large-scale brain networks and psychopathology: a unifying triple network model. Trends Cogn Sci. 15:483–506. [DOI] [PubMed] [Google Scholar]
- 13.Price JL, Drevets WC (2012): Neural circuits underlying the pathophysiology of mood disorders. Trends Cogn Sci. 16:61–71. [DOI] [PubMed] [Google Scholar]
- 14.Leaver AM, Espinoza R, Joshi SH, Vasavada M, Njau S, Woods RP, et al. (2015): Desynchronization and Plasticity of Striato-frontal Connectivity in Major Depressive Disorder. Cerebral cortex. [DOI] [PMC free article] [PubMed]
- 15.Sheline YI, Barch DM, Price JL, Rundle MM, Vaishnavi SN, Snyder AZ, et al. (2009): The default mode network and self-referential processes in depression. Proc Natl Acad Sci U S A. 106:1942–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheline YI, Price JL, Yan Z, Mintun MA (2010): Resting-state functional MRI in depression unmasks increased connectivity between networks via the dorsal nexus. Proc Natl Acad Sci U S A. 107:11020–11025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marchetti I, Koster EH, Sonuga-Barke EJ, De Raedt R (2012): The default mode network and recurrent depression: a neurobiological model of cognitive risk factors. Neuropsychol Rev. 22:229–251. [DOI] [PubMed] [Google Scholar]
- 18.Ionescu DF, Felicione JM, Gosai A, Cusin C, Shin P, Shapero BG, et al. (2018): Ketamine-Associated Brain Changes: A Review of the Neuroimaging Literature. Harv Rev Psychiatry. 26:320–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Posner J, Hellerstein DJ, Gat I, Mechling A, Klahr K, Wang Z, et al. (2013): Antidepressants normalize the default mode network in patients with dysthymia. JAMA Psychiatry. 70:373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nemati S, Akiki TJ, Roscoe J, Ju Y, Averill CL, Fouda S, et al. (2020): A Unique Brain Connectome Fingerprint Predates and Predicts Response to Antidepressants. iScience. 23:100800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uddin LQ (2015): Salience processing and insular cortical function and dysfunction. Nat Rev Neurosci. 16:55–61. [DOI] [PubMed] [Google Scholar]
- 22.Chang LJ, Yarkoni T, Khaw MW, Sanfey AG (2013): Decoding the role of the insula in human cognition: functional parcellation and large-scale reverse inference. Cerebral cortex. 23:739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Avery JA, Drevets WC, Moseman SE, Bodurka J, Barcalow JC, Simmons WK (2014): Major depressive disorder is associated with abnormal interoceptive activity and functional connectivity in the insula. Biol Psychiatry. 76:258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bressler SL, Menon V (2010): Large-scale brain networks in cognition: emerging methods and principles. Trends Cogn Sci. 14:277–290. [DOI] [PubMed] [Google Scholar]
- 25.Cole MW, Repovs G, Anticevic A (2014): The frontoparietal control system: a central role in mental health. Neuroscientist. 20:652–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaiser RH, Andrews-Hanna JR, Wager TD, Pizzagalli DA (2015): Large-Scale Network Dysfunction in Major Depressive Disorder: A Meta-analysis of Resting-State Functional Connectivity. JAMA Psychiatry. 72:603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mayberg HS (2003): Modulating dysfunctional limbic-cortical circuits in depression: towards development of brain-based algorithms for diagnosis and optimised treatment. Br Med Bull. 65:193–207. [DOI] [PubMed] [Google Scholar]
- 28.Koenigs M, Grafman J (2009): The functional neuroanatomy of depression: distinct roles for ventromedial and dorsolateral prefrontal cortex. Behav Brain Res. 201:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C, et al. (2005): Deep brain stimulation for treatment-resistant depression. Neuron. 45:651–660. [DOI] [PubMed] [Google Scholar]
- 30.Akil H, Gordon J, Hen R, Javitch J, Mayberg H, McEwen B, et al. (2018): Treatment resistant depression: A multi-scale, systems biology approach. Neurosci Biobehav Rev. 84:272–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nestler EJ, Carlezon WA Jr. (2006): The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 59:1151–1159. [DOI] [PubMed] [Google Scholar]
- 32.Groenewold NA, Opmeer EM, de Jonge P, Aleman A, Costafreda SG (2013): Emotional valence modulates brain functional abnormalities in depression: evidence from a meta-analysis of fMRI studies. Neurosci Biobehav Rev. 37:152–163. [DOI] [PubMed] [Google Scholar]
- 33.Seminowicz DA, Mayberg HS, McIntosh AR, Goldapple K, Kennedy S, Segal Z, et al. (2004): Limbic-frontal circuitry in major depression: a path modeling metanalysis. Neuroimage. 22:409–418. [DOI] [PubMed] [Google Scholar]
- 34.Phillips ML, Drevets WC, Rauch SL, Lane R (2003): Neurobiology of emotion perception II: Implications for major psychiatric disorders. Biol Psychiatry. 54:515–528. [DOI] [PubMed] [Google Scholar]
- 35.Goldapple K, Segal Z, Garson C, Lau M, Bieling P, Kennedy S, et al. (2004): Modulation of cortical-limbic pathways in major depression: treatment-specific effects of cognitive behavior therapy. Arch Gen Psychiatry. 61:34–41. [DOI] [PubMed] [Google Scholar]
- 36.Gong L, Hou Z, Wang Z, He C, Yin Y, Yuan Y, et al. (2018): Disrupted topology of hippocampal connectivity is associated with short-term antidepressant response in major depressive disorder. J Affect Disord. 225:539–544. [DOI] [PubMed] [Google Scholar]
- 37.Zhang J, Wang J, Wu Q, Kuang W, Huang X, He Y, et al. (2011): Disrupted brain connectivity networks in drug-naive, first-episode major depressive disorder. Biol Psychiatry. 70:334–342. [DOI] [PubMed] [Google Scholar]
- 38.Leaver AM, Vasavada M, Joshi SH, Wade B, Woods RP, Espinoza R, et al. (2019): Mechanisms of Antidepressant Response to Electroconvulsive Therapy Studied With Perfusion Magnetic Resonance Imaging. Biol Psychiatry. 85:466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joshi SH, Espinoza RT, Pirnia T, Shi J, Wang Y, Ayers B, et al. (2015): Structural Plasticity of the Hippocampus and Amygdala Induced by Electroconvulsive Therapy in Major Depression. Biol Psychiatry. [DOI] [PMC free article] [PubMed]
- 40.Campbell S, Marriott M, Nahmias C, MacQueen GM (2004): Lower hippocampal volume in patients suffering from depression: a meta-analysis. Am J Psychiatry. 161:598–607. [DOI] [PubMed] [Google Scholar]
- 41.Kandilarova S, Stoyanov D, Kostianev S, Specht K (2018): Altered Resting State Effective Connectivity of Anterior Insula in Depression. Front Psychiatry. 9:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng X, Lin P, Wu X, Gong R, Yang R, Wang J (2018): Insular subdivisions functional connectivity dysfunction within major depressive disorder. J Affect Disord. 227:280–288. [DOI] [PubMed] [Google Scholar]
- 43.Sheline YI, Barch DM, Donnelly JM, Ollinger JM, Snyder AZ, Mintun MA (2001): Increased amygdala response to masked emotional faces in depressed subjects resolves with antidepressant treatment: an fMRI study. Biol Psychiatry. 50:651–658. [DOI] [PubMed] [Google Scholar]
- 44.De Witte NAJ, Mueller SC (2017): White matter integrity in brain networks relevant to anxiety and depression: evidence from the human connectome project dataset. Brain Imaging Behav. 11:1604–1615. [DOI] [PubMed] [Google Scholar]
- 45.Evans JW, Szczepanik J, Brutsche N, Park LT, Nugent AC, Zarate CA Jr. (2018): Default Mode Connectivity in Major Depressive Disorder Measured Up to 10 Days After Ketamine Administration. Biol Psychiatry. 84:582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abdallah CG, Averill LA, Collins KA, Geha P, Schwartz J, Averill C, et al. (2017): Ketamine Treatment and Global Brain Connectivity in Major Depression. Neuropsychopharmacology. 42:1210–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murrough JW, Collins KA, Fields J, DeWilde KE, Phillips ML, Mathew SJ, et al. (2015): Regulation of neural responses to emotion perception by ketamine in individuals with treatment-resistant major depressive disorder. Transl Psychiatry. 5:e509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sahib AK, Loureiro JRA, Vasavada MM, Kubicki A, Joshi SH, Wang K, et al. (2020): Single and repeated ketamine treatment induces perfusion changes in sensory and limbic networks in major depressive disorder. Eur Neuropsychopharmacol. [DOI] [PMC free article] [PubMed]
- 49.Loureiro JRA, Leaver A, Vasavada M, Sahib AK, Kubicki A, Joshi S, et al. (2020): Modulation of amygdala reactivity following rapidly acting interventions for major depression. Hum Brain Mapp. [DOI] [PMC free article] [PubMed]
- 50.Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, Perez AM, et al. (2013): Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 170:1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.First MB WJ, Karg RS, Spitzer RL (2015): Structured Clinical Interview for DSM-5-Research Version (SCID-5 for DSM-5, Research Version; SCID-5-RV).
- 52.Hamilton M (1960): A rating scale for depression. J Neurol Neurosurg Psychiatry. 23:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nierenberg AA, DeCecco LM (2001): Definitions of antidepressant treatment response, remission, nonresponse, partial response, and other relevant outcomes: a focus on treatment-resistant depression. J Clin Psychiatry. 62 Suppl 16:5–9. [PubMed] [Google Scholar]
- 54.Nakonezny PA, Carmody TJ, Morris DW, Kurian BT, Trivedi MH (2010): Psychometric evaluation of the Snaith-Hamilton pleasure scale in adult outpatients with major depressive disorder. Int Clin Psychopharmacol. 25:328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lovibond PF, Lovibond SH (1995): The structure of negative emotional states: comparison of the Depression Anxiety Stress Scales (DASS) with the Beck Depression and Anxiety Inventories. Behav Res Ther. 33:335–343. [DOI] [PubMed] [Google Scholar]
- 56.Braund TA, Palmer DM, Williams LM, Harris AWF (2020): Dimensions of anxiety in Major depressive disorder and their use in predicting antidepressant treatment outcome: an iSPOT-D report. Psychol Med. 50:1032–1042. [DOI] [PubMed] [Google Scholar]
- 57.Carver CS, & White TL. (1994): Behavioral inhibition, behavioral activation, and affective responses to impending reward and punishment: The BIS/BAS Scales. Journal of Personality and Social Psychology. 67:319–333. [Google Scholar]
- 58.Wells A, Davies MI (1994): The Thought Control Questionnaire: a measure of individual differences in the control of unwanted thoughts. Behav Res Ther. 32:871–878. [DOI] [PubMed] [Google Scholar]
- 59.Nolen-Hoeksema S (2000): The role of rumination in depressive disorders and mixed anxiety/depressive symptoms. J Abnorm Psychol. 109:504–511. [PubMed] [Google Scholar]
- 60.Glasser MF, Sotiropoulos SN, Wilson JA, Coalson TS, Fischl B, Andersson JL, et al. (2013): The minimal preprocessing pipelines for the Human Connectome Project. Neuroimage. 80:105–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Desikan RS, Segonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, et al. (2006): An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 31:968–980. [DOI] [PubMed] [Google Scholar]
- 62.Leaver AM, Espinoza R, Pirnia T, Joshi SH, Woods RP, Narr KL (2016): Modulation of intrinsic brain activity by electroconvulsive therapy in major depression. Biol Psychiatry Cogn Neurosci Neuroimaging. 1:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murrough JW, Abdallah CG, Anticevic A, Collins KA, Geha P, Averill LA, et al. (2016): Reduced global functional connectivity of the medial prefrontal cortex in major depressive disorder. Hum Brain Mapp. 37:3214–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Satterthwaite TD, Cook PA, Bruce SE, Conway C, Mikkelsen E, Satchell E, et al. (2016): Dimensional depression severity in women with major depression and post-traumatic stress disorder correlates with fronto-amygdalar hypoconnectivty. Mol Psychiatry. 21:894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang Z, Oathes DJ, Linn KA, Bruce SE, Satterthwaite TD, Cook PA, et al. (2018): Cognitive Behavioral Therapy Is Associated With Enhanced Cognitive Control Network Activity in Major Depression and Posttraumatic Stress Disorder. Biol Psychiatry Cogn Neurosci Neuroimaging. 3:311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shou H, Yang Z, Satterthwaite TD, Cook PA, Bruce SE, Shinohara RT, et al. (2017): Cognitive behavioral therapy increases amygdala connectivity with the cognitive control network in both MDD and PTSD. Neuroimage Clin. 14:464–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jenkins LM, Stange JP, Barba A, DelDonno SR, Kling LR, Briceno EM, et al. (2017): Integrated cross-network connectivity of amygdala, insula, and subgenual cingulate associated with facial emotion perception in healthy controls and remitted major depressive disorder. Cogn Affect Behav Neurosci. 17:1242–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Erk S, Mikschl A, Stier S, Ciaramidaro A, Gapp V, Weber B, et al. (2010): Acute and sustained effects of cognitive emotion regulation in major depression. J Neurosci. 30:15726–15734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA, et al. (1999): Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry. 156:675–682. [DOI] [PubMed] [Google Scholar]
- 70.Seeley WW, Menon V, Schatzberg AF, Keller J, Glover GH, Kenna H, et al. (2007): Dissociable intrinsic connectivity networks for salience processing and executive control. J Neurosci. 27:2349–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cao X, Liu Z, Xu C, Li J, Gao Q, Sun N, et al. (2012): Disrupted resting-state functional connectivity of the hippocampus in medication-naive patients with major depressive disorder. J Affect Disord. 141:194–203. [DOI] [PubMed] [Google Scholar]
- 72.Mayberg HS (1997): Limbic-cortical dysregulation: a proposed model of depression. J Neuropsychiatry Clin Neurosci. 9:471–481. [DOI] [PubMed] [Google Scholar]
- 73.Fitzgerald PB, Laird AR, Maller J, Daskalakis ZJ (2008): A meta-analytic study of changes in brain activation in depression. Hum Brain Mapp. 29:683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gartner M, Aust S, Bajbouj M, Fan Y, Wingenfeld K, Otte C, et al. (2019): Functional connectivity between prefrontal cortex and subgenual cingulate predicts antidepressant effects of ketamine. Eur Neuropsychopharmacol. 29:501–508. [DOI] [PubMed] [Google Scholar]
- 75.Vasavada MM, Leaver AM, Espinoza RT, Joshi SH, Njau SN, Woods RP, et al. (2016): Structural connectivity and response to ketamine therapy in major depression: A preliminary study. J Affect Disord. 190:836–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Depue RA, Iacono WG (1989): Neurobehavioral aspects of affective disorders. Annu Rev Psychol. 40:457–492. [DOI] [PubMed] [Google Scholar]
- 77.Kasch KL, Rottenberg J, Arnow BA, Gotlib IH (2002): Behavioral activation and inhibition systems and the severity and course of depression. J Abnorm Psychol. 111:589–597. [DOI] [PubMed] [Google Scholar]
- 78.Koster EH, De Lissnyder E, Derakshan N, De Raedt R (2011): Understanding depressive rumination from a cognitive science perspective: the impaired disengagement hypothesis. Clin Psychol Rev. 31:138–145. [DOI] [PubMed] [Google Scholar]
- 79.Lally N, Nugent AC, Luckenbaugh DA, Niciu MJ, Roiser JP, Zarate CA Jr. (2015): Neural correlates of change in major depressive disorder anhedonia following open-label ketamine. J Psychopharmacol. 29:596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Niciu MJ, Luckenbaugh DA, Ionescu DF, Guevara S, Machado-Vieira R, Richards EM, et al. (2014): Clinical predictors of ketamine response in treatment-resistant major depression. J Clin Psychiatry. 75:e417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]