

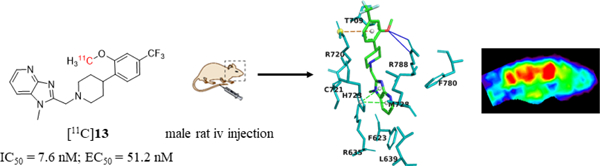
Abstract
Three benzimidazole derivatives (13-15) have been synthetized as potential PET imaging ligands for mGluR2 in the brain. Of these compounds, 13 exhibits potent binding affinity (IC50 = 7.6 ± 0.9 nM), PAM activity (EC50 = 51.2 nM), and excellent selectivity against other mGluR subtypes (> 100-fold). [11C]13 was synthesized via O-[11C]methylation of its phenol precursor 25 with [11C]methyl iodide. The achieved radiochemical yield was 20 ± 2% (n = 10, decay-corrected) based on [11C]CO2 with radiochemical purity > 98% and molar activity 98±30 GBq/µmol EOS. Ex vivo biodistribution studies revealed reversible accumulation of [11C]13 and hepatobiliary and urinary excretions. PET imaging studies in rats demonstrated that [11C]13 accumulated in the mGluR2-rich brain regions. Pre-administration of mGluR2-selective PAM, 17 reduced the brain uptake of [11C]13, indicating a selective binding. However, pre-administration of 13 significantly enhanced [11C]13 uptake in the brain. Therefore, [11C]13 is both a potential PET imaging ligand for mGluR2 and a drug candidate for the treatment of CNS disorders.
Graphical Abstract
INTRODUCTION
The metabotropic glutamate receptor 2 (mGluR2) is widely expressed in the nervous systems.1,2 mGluR2 expression is abundant in brain areas such as prefrontal cortex, hippocampus, amygdala, striatum, thalamus, cerebellum, and nucleus accumbens.3,4 It predominantly localizes on presynaptic nerve terminals and modulates synaptic transmission and neuroplasticity.3 Structurally, mGluR2 has a characteristic extracellular Venus flytrap domain (VFTD), a seven transmembrane (7-TM) domain and a cysteine rich domain (CRD) that connects the mGluR dimers.5 The therapeutic benefits of mGluR2 modulators has been suggested for Alzheimer’s disease6–9, schizophrenia10–13, depression14, anxiety15 and pain16–18.
Initial research and drug discovery efforts had focused on pharmacological ligands for mGluR2/3, which have been published in literature and developed for the treatment of anxiety and schizophrenia in preclinical and clinical studies.10–13,15 Until recently, mGluR2 and mGluR3 have been thought to have similar functions: they share high sequence homology, generally couple to Gi/o signaling, and provide negative feedback to reduce glutamate signaling. However, despite a successful phase 2 study conducted entirely in Russia for patients with schizophrenia, the clinical development of LY2140023, a mGluR2/3 receptor agonist prodrug, was halted due to lack of antipsychotic efficacy compared to placebo in three phase 2 or phase 3 trials.12,19–21 The studies also revealed that the antipsychotic effect of mGluR2/3 agonists was absent in mGluR2 knockout mice but not mGluR3 knockout mice, suggesting the antipsychotic effects might be mediated via the mGluR2 but not mGluR3 receptor and even the effect of mGluR2 and mGluR3 might be different/opposite.22,23
Previously, we have reported two orthosteric antagonists as PET tracers, namely, [11C]MMMHC (1) in 200324 and [11C]CMGDE (2) in 201225, for Group II mGluRs (mGluR2 & mGluR3) (Figure 1). Since then, several PET radiotracers for mGluR2 have been derived from allosteric modulators that target the 7-TM instead of the VFTD region of mGluR2. It is believed that the allosteric modulators would bear higher lipophilicity and mGluR2 selectivity than orthosteric ligands due to the hydrophobicity and heterogeneity of the 7-TM binding pocket across mGlu receptors.26–28 So far, two radioligands in this category have been advanced for human clinical trials, including mGluR2 PAM [11C]JNJ42491293 (3) and a radioligand from Merck. However, [11C]JNJ42491293 (3) was not found useful for the visualization and quantification of mGluR2 in vivo because of its apparent off-target binding.29,30 The Merck radiotracer was only reported in an abstract without information on its chemical structure and detailed imaging results.31,32 The fluorine-18 labeled derivative of 3, [18F]FE-JNJ-42491293 (4), was disclosed in an abstract but it is not clear if this tracer has the similar off-target binding as 3.33 Recently, a mGluR2 PAM tracer [11C]CMDC (5) and its three derivatives 6-8 were reported; however, 5 exhibited an insufficient affinity and low BBB-penetration. PET imaging with 5 did not enable in vivo visualization of the living rat brain.34,35 On the other hand, three different types of mGluR2 NAM-based tracers were also disclosed. The mGluR2 NAM tracers [11C]QCA (9)36 and its analogue [11C]1037 showed off-target binding and limited brain uptake with intensive interaction with brain efflux pumps on the murine BBB. Two other types of NAM-based radiotracers have been disclosed in the patent literature. The compound 11 and its derivatives were patented as PET tracers for mGluR2/3.38 The compound 12 and its derivatives were developed as mGluR2 PET ligands, but no in vivo PET imaging result has been described.39
Figure 1.
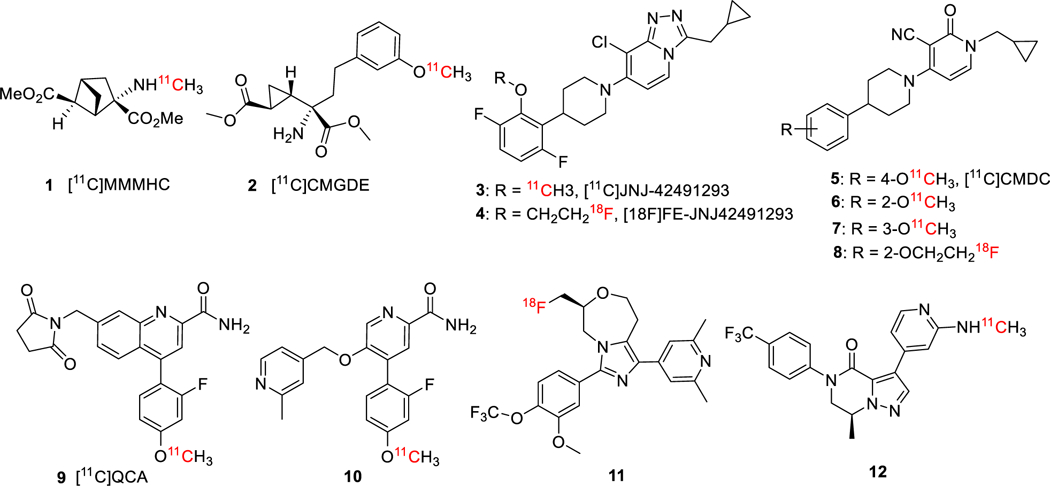
Typical PET radiotracers for mGluR2.
The lack of efficient and efficacious mGluR2 PET tracers prompted us to further extend our previous effort toward exploration of mGluR2 PAMs as suitable PET imaging candidates. The benzimidazole derivatives have been the most widely examined series of mGluR2 PAMs in literature27,40,41 with examples of highly potent mGluR2 ligands of compounds 13 (EC50 = 13 nM)42 and 14 (EC50 = 5 nM, Figure 2)41. The presence of 2-methoxy-4-trifluoromethyl-phenyl group in compounds 13 and 14 allows rapid radiolabeling of their phenol precursors via O-[11C]methylation with [11C]CH3I. We further designed compound 15 as a PET imaging candidate based on a potent mGluR2 PAM [2-(((1R,5S,6r)-6-((4-chloro-2-fluorophenoxy)methyl)-3-azabicyclo[3.1.0]hexan-3-yl)methyl)-1-methyl-1H-benzo[d]imidazole] (16, EC50 = 8 nM)43 by replacing the distal 4-chloro-2-fluorophenoxy group with a 2-methoxy-4-(trifluoromethyl)phenoxy moiety. The structurally distinct compound 17, a potent and selective mGluR2 PAM (EC50 = 78 nM),44 was used as a selective blocking reagent during the investigation of [11C]JNJ42491293 (3) and therefore we used it as a blocking reagent in the present studies.30 Here, we report the design, synthesis and characterization of compounds 13-15 using in silico modeling, in vitro assays and in vivo PET imaging methods to evaluate their potential as mGluR2-selective PET imaging ligands.
Figure 2.
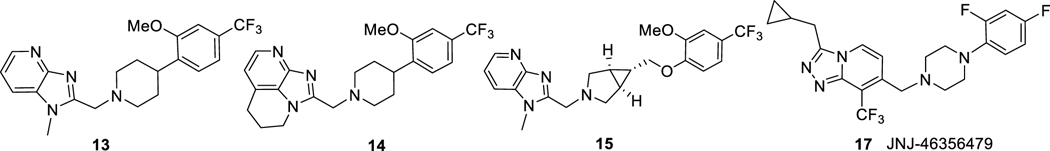
Chemical structures of compounds 13-15 and 17.
RESULTS AND DISCUSSION
Chemistry.
Structurally, compounds 13-15 feature a merged heterocyclic core, a central cyclic amine core and a substituted distal arene. These compounds were synthesized according to reaction sequences delineated in Scheme 1. The intermediates 18-20 were prepared according to the reported procedures.41,42 The intermediate 24 was synthesized in two steps: Mitsunobu reaction of 21 and 22; followed by removal of N-Boc-group. Finally, compounds 13-15 were synthesized by the corresponding reductive amination reactions. Compound 17 was prepared according to the published procedure.44
Scheme 1.
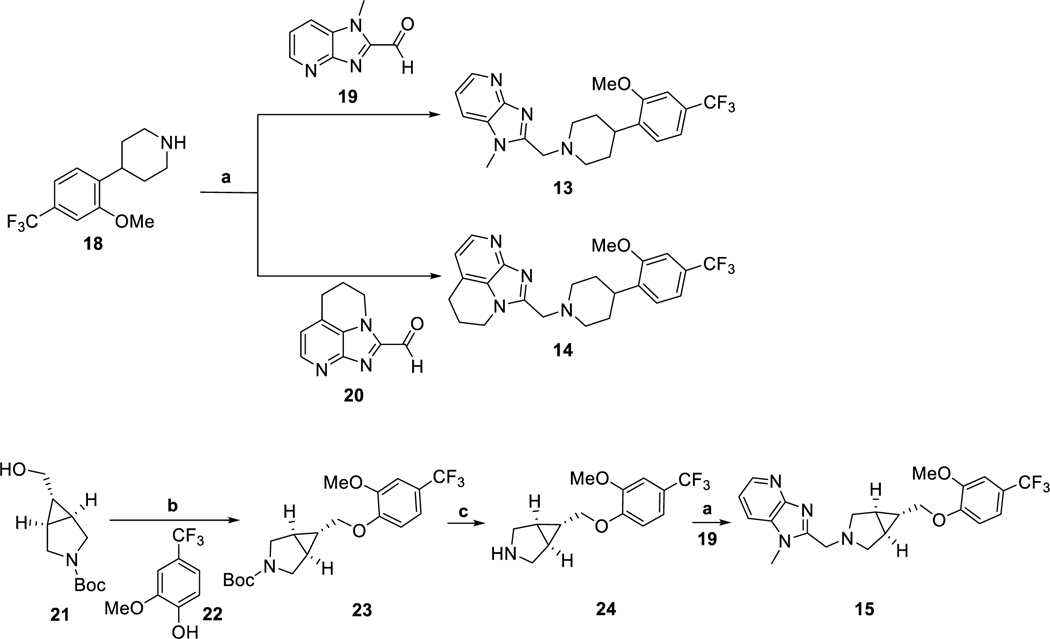
Syntheses of 13–15. Reagents and conditions: (a) Et3N, MgSO4, then Na(OAc)3BH, DCE, rt, overnight; (b) PPh3, diethyl azodicarboxylate solution (40 wt.% in toluene), THF, rt, 16 h; (c) TFA, DCM, rt, 2 h.
Structural insights of compounds 13–15.
To provide structural insights on ligand-protein binding, compounds 13–15 were docked into a mGluR2 homology model, which was built in YASARA45 (Supporting Information Figure S1 and Table S1) and validated by a series of structural analysis tools of ModFOLD46, ERRAT and VERIFY 3D47–49 (see Supporting Information Figures S2-S7). The key binding residues were predicted by Partial Order Optimum Likelihood (POOL)50, DEPTH51 and MetaPocket52 (Supporting Information Figure S8). The docking experiments were performed at the 7-TM region with AutoDock embedded in YASARA (Supporting Information Table S2).53 As shown in Figure 3, compounds 13–15 localize similarly at the entrance of the 7-TM region with their heterocyclic cores projecting to the bottom hydrophobic pocket and the distal substituted arenes interacting with residues at the extracellular loop 2 (EL2). Compound 13 has the best docking score of 8.7 kcal/mol compared to the values of 8.6 kcal/mol and 7.3 kcal/mol for compounds 14 and 15, respectively. Compound 13 shows a hydrogen bonding interaction with Arg788, a π-cation interaction with Arg720, and a π- π stacking interaction with His723 (Figure 3). His723 has been previously reported as a key hydrophobic residue that interacts with several mGluR2 PAMs.54,55 Compound 14 has similar key binding interactions as that of compound 13, whereas, compound 15 exhibited fewer contacts in the binding pocket than compounds 13 and 14, consistent with the decreased docking score. Overall, the in silico simulations suggest compounds 13-15 as potent mGluR2 binding ligands.
Figure 3.
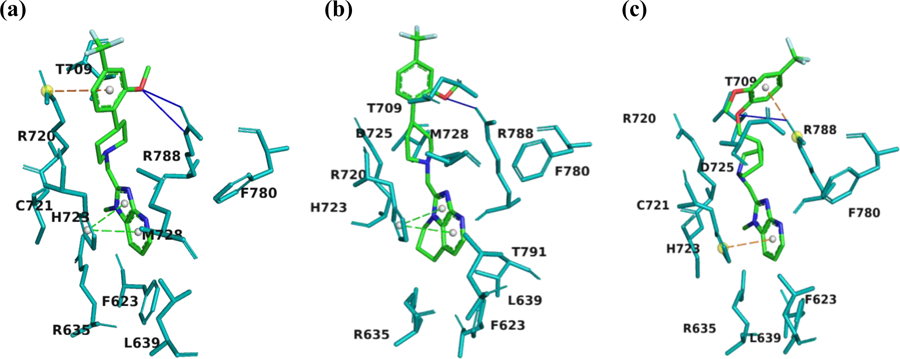
Snapshots of the docking results for compounds 13 (a), 14 (b) and 15 (c). Pictures were rendered in PyMol 2.3.3. The interacting residues are shown in teal. The ligand atoms are rendered as carbon in green, nitrogen in blue, oxygen in red, and fluorine in cyan. Blue lines represent H-bonds, green dotted lines show π-π stacking, and orange dotted lines indicate π-cation interaction.
In vitro pharmacochemical properties.
To evaluate compounds 13-15, the pharmacochemical properties including affinity to mGluR2, mGluR2 PAM activity, selectivity toward other mGluRs, lipophilicity, plasma protein binding, metabolic and solution stabilities as well as blood brain barrier (BBB) penetration capability were determined. In these studies, compound 13 was compared to the other two mGluR2 ligands 14 and 15.
The binding affinity of compounds 13-15 was measured by the competitive binding assay in mGluR2 transfected CHO cells at the presence of 10 nM tritium-labeled radioligand [3H]JNJ-46281222 (see Supporting Information).56–58 The concentration of compounds 13-15 was increased from 0.01 nM to 10 μM to generate a competitive binding curve, with which the IC50 values were determined. As displayed in Table 1, compound 13 has potent binding toward mGluR2 (IC50 = 7.6 ± 0.9 nM), which is slightly stronger than those of compounds 14 (IC50 = 10.5 ± 0.5 nM) and 15 (IC50 = 10.5 ± 7.9 nM). The results also indicated that compounds 13-15 shared the same allosteric binding site of mGluR2.
Table 1.
Binding affinity of compounds 13-15 to mGluR2.
| Compound | 13 | 14 | 15 |
|---|---|---|---|
| IC50 ± SEM (nM) | 7.6 ± 0.9 | 10.5 ± 0.5 | 10.5 ± 7.9 |
| log (IC50 ± SEM) | −8.12 ± 0.79 | −7.98 ± 0.21 | −7.98 ± 1.39 |
Previously reported EC50 values for compounds 13 and 14 (EC50 = 13 nM and 5 nM, respectively)36, 37 were determined by forced-coupling of mGluR2 to Gα15 or Gα16 followed by fluorescence detection of calcium flux upon activation. However, this assay is sub-optimal as it does not signal through the biorelevant cAMP pathway. Here, the mGluR2 PAM activity of compounds 13–15 was determined using Promega’s split luciferase based GloSensor cAMP biosensor assay,59,60 where, with this live cell assay, the mGluR2 PAM activity was evaluated in the presence of EC20 amount of L-glutamate by measuring changes in intracellular cAMP concentration, the relevant second messenger mechanism. An mGluR2 PAM, [3’-(((2-cyclopentyl-6,7-dimethyl-1-oxo-2,3-dihydro-1H-inden-5-yl)oxy)methyl)-[1,1’-biphenyl]-4-carboxylic acid] (BINA),61 was used as the reference compound for the assay. Figure 4 shows, the EC50 values of 13, 14 and 15 are 51.2 nM, 101 nM and 7.8 μM, respectively, suggesting that 13 is a very potent mGluR2 PAM. The selectivity of 13–15 was also analyzed among the various mGluR subtypes, in which the Gq coupled receptors (mGluR1 and mGluR5) were tested using Ca2+ mobilization assay and the Gi/o coupled receptors (mGluR2, mGluR3, mGluR4, mGluR6 and mGluR8) using cAMP assay. Results demonstrate that 13 has good selectivity against other mGlu receptors (> 100-fold, Supporting Information Table S3).
Figure 4.
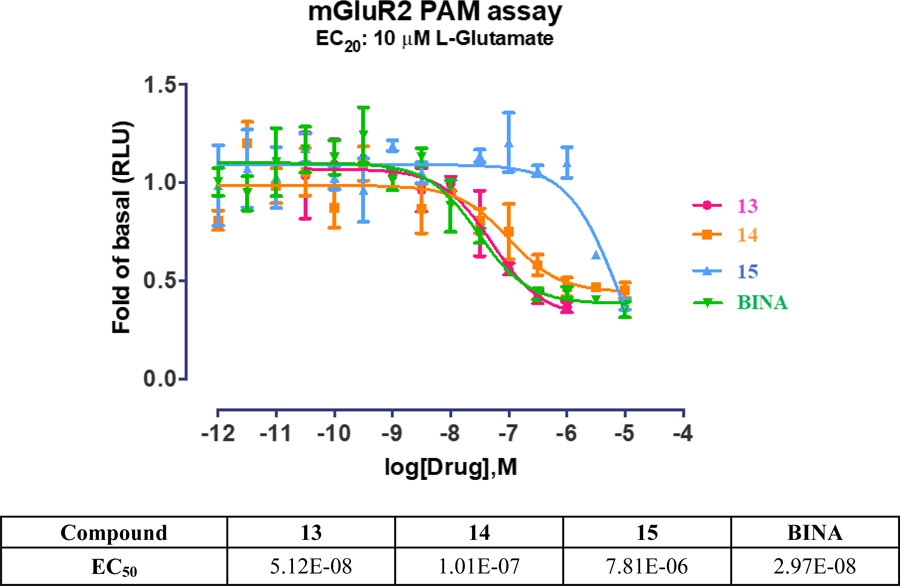
The mGluR2 PAM activity.
The physicochemical properties of compounds 13–15 were determined via ChemBiodraw (version 16.0) based on the molecular weight (MW), topological polar surface area (tPSA), and cLogP (Table 2). The experimental lipophilicity was measured by using liquid-liquid partition between n-octanol and water (“shake-flask method”).62 The LogP values obtained for compounds 13–15 were 3.65, 3.86 and 3.30 respectively, indicating their satisfactory CNS penetrating potentials (Supporting Information Table S5).63 The plasma protein binding comprises compounds’ binding to albumin, α1-acid glycoprotein and lipoproteins once delivered to the bloodstream. This property was evaluated for compounds 13 and 14 by equilibrium dialysis,64 where two chambers were separated by a dialysis membrane (MWCO 8 kD). The plasma protein bindings of 13 and 14 are 87.2% and 88.7%, respectively (Supporting Information Tables 1 and S6). Therefore, the high plasma free fraction of compounds 13 and 14 (> 10%) would allow enough free drug concentration in blood stream to reach the brain targets.
Table 2.
Physicochemical properties of compounds 13-15
| Compound | MW (g/mol) | tPSA | HBD | cLogP | Log P | PPB ± SEM (%) |
|---|---|---|---|---|---|---|
| 13 | 404.44 | 40.43 | 1 | 3.57 | 3.65 | 87.2 ± 0.1 |
| 14 | 430.48 | 40.43 | 1 | 4.08 | 3.86 | 88.7 ± 0.1 |
| 15 | 432.45 | 49.66 | 1 | 2.89 | 3.3 | --- |
The in vitro plasma and liver microsomal stability of 13 and 14 were studied by incubating the test compounds in rat serum and rat liver microsomes as well as NADPH cofactor, respectively, using previously published methods.65,66 Diltiazem and ML128 (a mGluR4 PAM)67,68 were used as co-assay QC controls for plasma and microsomal stability assays, respectively, to ensure that the assays were operating properly, and that the activity of the plasma and microsomes were consistent with established criteria. Compounds 13 and 14 are much more stable than diltiazem in rat plasma (Supporting Information Tables 3 and S7). The results also show that 13 and 14 exhibit reasonable microsomal stability and are much more stable than ML128, in which the suitable hepatic clearance of 13 and 14 is predicted (Supporting Information Tables 2 and S8-S9). The solution stability of 13 was evaluated with buffer solutions at pH 5.0, 7.4 and 9.4, respectively (Supporting Information Tables 3 and S10).69 The results indicate that 13 is relatively stable in pH ranging from 5.0 to 9.4.
BBB penetration was a major barrier for some recently reported mGluR2 PET tracers that otherwise could have efficacy for imaging the brain target as shown by radiotracers [11C]9 and [11C]10.36,37 We have studied BBB penetration potential of compounds 13–15 with two in vitro assays, namely, parallel artificial membrane permeability assay (PAMPA) and Pgp-Glo™ assay. The PAMPA assay was carried out to predict passive BBB permeability.70 Quality control standards were run with each sample set to monitor the consistency of the analysis. Verapamil was used as a high permeability standard (Pe = 16 × 10−6 cm/s) and theophylline was used as a low permeability standard (Pe = 0.12 × 10−6 cm/s). As Figure 5a shows, compound 13 has the best membrane permeability with an average effective permeability (Pe) value of 9.3 × 10−6 cm/s.
Figure 5.
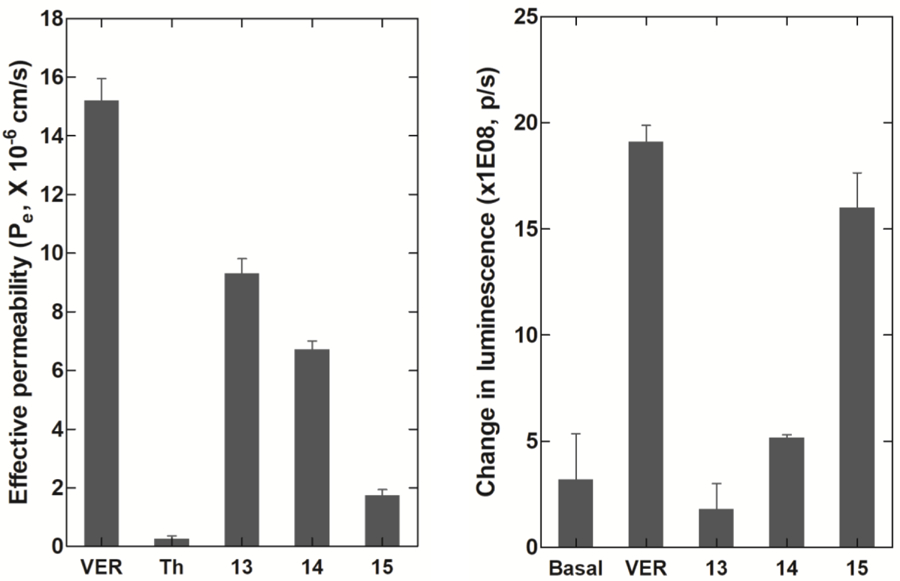
Assessment of BBB permeability for compounds 13–15 via a) PAMPA assay and b) Pgp-Glo™ assay. Pictures were rendered from Prism 5.0.
The Pgp-Glo™ assay was carried out on recombinant human P-gp in a cell membrane fraction to investigate whether the brain penetration will be affected by P-glycoprotein (P-gp) efflux transporter.71 The effect of compounds 13–15 on P-gp ATPase activity was examined by comparing the untreated samples and the samples treated with 13–15 to sodium orthovanadate (Na3VO4)-treated control. The difference in luminescent signal between Na3VO4-treated samples and samples treated with the test compounds implied P-gp ATPase activity in the presence of the test compound. Verapamil, a P-gp substrate, was used as a positive control in the assay. By comparing basal and verapamil activities to that of 13–15, it is clearly indicated that 13 is not a P-gp substrate and 15 is a potential P-gp substrate, while 13 displays a moderate P-gp ATPase activity (Figure 5b).
The in vitro pharmacological studies reveal that compound 13 has many CNS drug-like properties, including the potent mGluR2 PAM activity and good selectivity against other mGluRs, suitable lipophilicity and PPB, adequate metabolic stability, favorable passive permeability as measured by PAMPA, and no P-gp liability. Based on these results, compound 13 was selected for the radiolabeling and for in vivo evaluation as potential mGluR2 PET radioligand.
Radiochemistry.
The radiosynthesis of [11C]13 was achieved via the one-step O-methylation of its phenol precursor 23. Compound 23 was synthesized by demethylation of 13 using boron tribromide (Scheme 2). The radiosynthesis of [11C]13 was performed by the reaction of 23 (0.5 ± 0.1 mg) with [11C]CH3I in the presence of aqueous NaOH (5N, 3 μL) in dry DMF (250 μL). The reaction was carried out at 80 °C for 2 min, followed by purification using a semi-preparative HPLC (Figure S9). The identity of [11C]13 was confirmed by co-injection with the unlabeled 13 on an analytical HPLC (Figure S10). The radiochemical yield was 20 ± 2% decay-corrected (n = 10), calculated from starting [11C]CO2. The [11C]13 was then formulated into 10% ethanolic saline solution (pH = 5–6) before injection. The radiochemical and chemical purity were greater than 98%, and the molar activity was 98 ± 30 GBq/µmol at the end of synthesis (EOS). The overall synthesis time was ca. 50 min, and no radiolysis was observed up to 90 min.
Scheme 2.
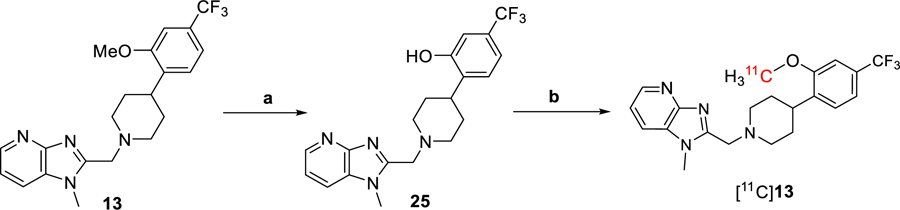
Synthesis of [11C]13. Reagents and conditions: (a) BBr3, DCM, 0 ℃ then rt, 2 h. (b) [11C]CH3I, 5N NaOH, DMF, rt, then 80 ℃, 2 min.
Ex vivo biodistribution studies.
The ex vivo whole body biodistribution of [11C]13 was performed in 16 normal male Sprague Dawley rats after intravenous injection of [11C]13 at several time points (5, 20, 30 and 40 min). The uptake value is expressed in the unit of % ID/g. These studies support reversible accumulation of [11C]13 with the highest accumulation 5 min after administration of radioactivity in other investigated tissue areas but the lungs where the maximum accumulation was at 20 min and the muscle where the radioactivity steadily increased up to 40 min (Figure 6). The highest accumulation was measured in the liver (2.73 ± 0.02% ID/g) followed by kidney (1.05 ± 0.07% ID/g), spleen (0.67 ± 0.05% ID/g), lung (0.59 ± 0.04% ID/g), and heart (0.58 ± 0.05% ID/g). The high radioactivity uptake in liver and kidney suggest that hepatobiliary elimination and renal excretion contribute to the whole body distribution of [11C]13. The average accumulation of [11C]13 in the rat brain at 5 min was 0.49 ± 0.07% ID/g. This result indicates a rapid BBB penetration of [11C]13, which was consistent with the following in vivo brain imaging studies.
Figure 6.
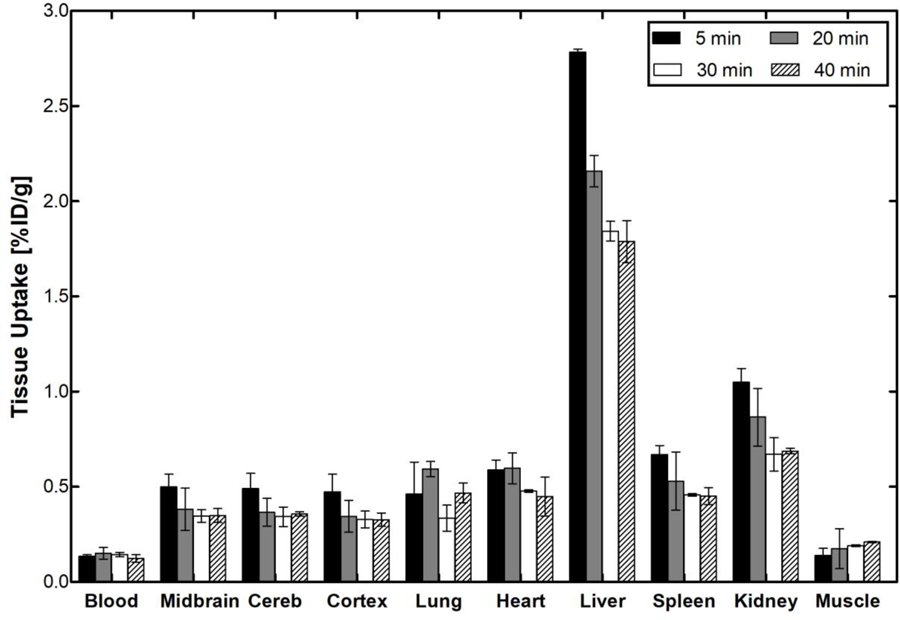
The ex vivo biodistribution in rat at four different time points post-[11C]13 injection. Picture was rendered from Prism 5.0.
PET imaging.
In vivo characterization of [11C]13 was conducted with PET imaging using rat (male Sprague-Dawley) models. Dynamic PET scans were performed for 60 min after tail vein injection of [11C]13. Representative PET images of cumulative volumetric distribution of [11C]13 at time interval of 10–15 min are shown on five coronal, axial and sagittal levels (Figure 7). The accumulation of [11C]13 clearly delineates the mGluR2-rich regions in the rat brain. Time-activity curves (TACs) showed fast radioactivity uptake (SUVmax = 1.8 ± 0.2, n = 9) and time-dependent accumulation of radioactivity in different brain regions. The highest accumulation of [11C]13 was in the thalamus, followed by striatum, cerebellum, and cortex. (Figure 8a). Blocking studies were conducted to investigate mGluR2-selective binding of [11C]13. Pretreatment with the structurally distinct in vivo active mGluR2 PAM ligand 17 (4 mg/kg i.v.) 10 min before [11C]13 injection resulted in a 28–37% decrease of [11C]13 uptake in different brain areas at the 10–30 min time window (Figure 8b). On the other hand, administration of unlabeled compound 13, using a dose of 4 mg/kg iv. 10 min before [11C]13 injection, resulted in a 33–49% enhancement of radioactivity uptake in the different brain areas at the same time window as mentioned above. These results confirm that [11C]13 has in vivo mGluR2-selective binding in the rat brain. The significant increase of radioactivity uptake after self-blocking indicates that the compound 13, as a mGluR2 PAM, is capable of potentiating strong pharmacological effects, making 13 a potential candidate for therapeutic approaches.
Figure 7.
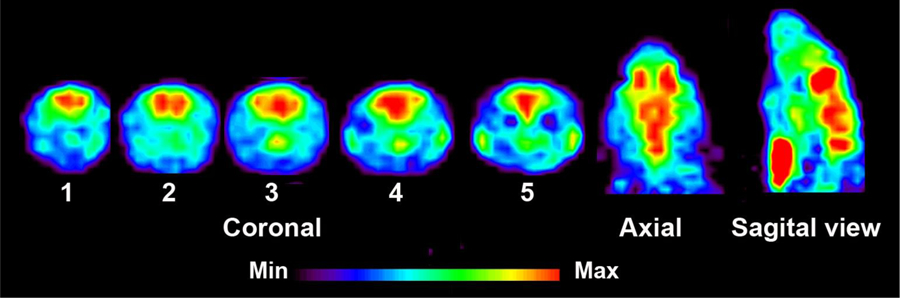
PET images of [11C]13 uptake in the rat brain at the time interval 10–15 min. Coronal level 1 shows uptake in the cingulate and motor cortex; level 2 in the striatum, level 3 in the thalamus and striatum, level 4 in the thalamus and hippocampus and level 5 in the cerebellum. Axial and sagittal views show activity distribution in the midbain level. Slice thickness is 1.25 mm.
Figure 8.
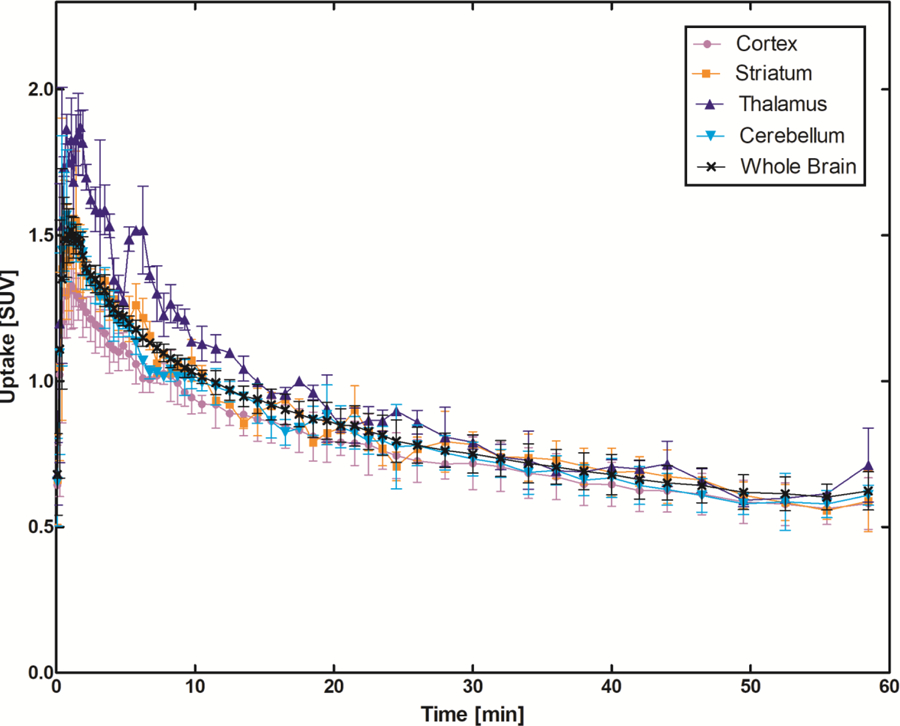
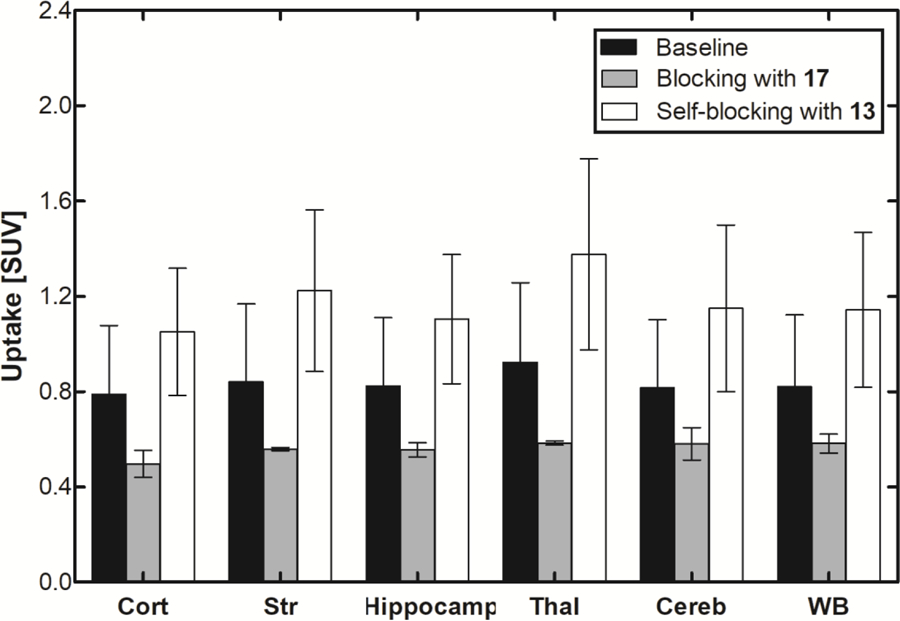
In vivo binding profile of [11C]13 in the rat brain. a) Time–activity distribution of [11C]13 in different brain areas show fast accumulation and reversible binding. The data is averaged of six normal Sprague Dawley rats. b) The blocking effect was calculated in the time interval 10–30 min after administration of [11C]13. Cort = cortex, Str = striatum, Hippocamp = hippocampus, Thal = thalamus, Cereb = cerebellum and WB = whole brain. Pictures were rendered from Prism 5.0.
CONCLUSION
We have synthesized and characterized three benzimidazole derivatives (13–15) as mGluR2 PAMs. Compound 13 demonstrated nanomolar binding potency toward mGluR2 and excellent selectivity over other mGluRs. Further in vitro pharmacological and brain permeability evaluations confirmed the potential of compound 13 as PET imaging ligand. A robust and reliable one-step radiosynthetic procedure was established for radiolabeling compound 13 with carbon-11. The desired product [11C]13 was obtained with a radiochemical yield of 20 ± 2 % (n = 10, decay-corrected) based on [11C]CO2 and a molar activity of 98 ± 30 GBq/µmol at the end of synthesis (50 min). The ex vivo pharmacokinetic results of [11C]13 suggested its reversible accumulation in most tissue areas and hepatobiliary & urinary excretions. PET imaging studies indicated that [11C]13 crossed the BBB rapidly and was mainly accumulated in the mGluR2-rich regions of the rat brain such as the thalamus, cerebellum, striatum and cortex. The blocking studies using mGluR2-selective PAM (17) significantly reduced the [11C]13 uptake in these brain regions, indicating the highly selective uptake of [11C]13 in rat brain. Distinct from previous observations of mGluR2 PET radioligands, self-blocking of [11C]13 resulted in an apparent uptake increase in the accumulation by almost 50%. This result indicates a significant modulation effect of compound 13 in vivo as mGluR2 PAM, which bears promising therapeutic applications for translational studies in neurological conditions and/or disorders.
Altogether, these results suggest that compound 13 might have two different application areas. When radiolabeled with carbon-11 it will be a potential PET imaging ligand for mGluR2 in the brain and when used as a drug (cold compound) it might be a therapeutic drug for different neurological conditions and diseases which are affected by mGluR2 malfunction.
EXPERIMNETAL SECTION
Animal Procedures.
The animal studies were approved and done under the guidelines of the Subcommittee on Research Animals of the Massachusetts General Hospital and Harvard Medical School in accordance with the Guide of NIH for the Care and Use of Laboratory Animals.
Materials and Methods.
All reagents and starting materials were obtained from the commercial sources including Sigma-Aldrich (St. Louis, MO), Thermo Fisher Scientific, Oakwood Products, Inc., Matrix Scientific, Acros Organics and used as received. The reactions were monitored by TLC using a UV lamp monitored at 254 nm. If necessary, the reactions were also checked by LC−MS using the Agilent 1200 series HPLC system coupled with a multi-wavelength UV detector and a model 6310 ion trap mass spectrometer (Santa Clara, CA) equipped with an Agilent Eclipse C8 analytical column (150 mm × 4.6 mm, 5 μm). Elution was with a 0.1% formic acid solution of water (A) and acetonitrile (B). The silica gel used in flash column chromatography was from Aldrich (Cat. 60737, pore size 60 Å, 230–400 mesh). Flash chromatography was also performed with a CombiFlash Rf Purification System (Teledyne Isco) using a Silica ReadySep Rf column. The products were identified by LC−MS as well as 1H NMR, 13C NMR and 19F NMR using a Varian 500 MHz spectrometer. All NMR samples were dissolved in chloroform-d (CDCl3), methanol-d4 (CD3OD) or DMSO-d6 [(CD3)2SO] containing tetramethylsilane as a reference standard. Chemical shifts were expressed as ppm and calculated downfield or upfield from the NMR signal of reference standard. J was expressed as Hz, and its splitting patterns were reported as s, d, t, q, or m. HRMS was obtained from the High-Resolution Mass Spectrometry Facility at the University California, Riverside, using electrospray ionization (ESI)/atmospheric pressure chemical ionization (APCI) technique (Agilent Time of Flight (TOF) LC−MS). Unless otherwise specified, the purities of all new compounds were over 95% determined by HPLC.
Chemistry. tert-Butyl(1R,5S,6R)-6-((2-methoxy-4-(trifluoromethyl)phenoxy)methyl)-3-azabicyclo [3.1.0] hexane-3-carboxylate (23).
2-methoxy-4-(trifluoromethyl)phenol (22, 0.45 g, 2.3 mmol) and triphenyl phosphine (0.9 g, 3.5 mmol) were added to a solution of (1R,5S,6R)-tert-butyl 6-(hydroxymethyl)-3-azabicyclo[3.1.0]hexane-3-carboxylate (21, 0.5 g, 2.3 mmol) in THF under nitrogen. Diethyl azodicarboxylate solution (40 wt. % in toluene, 1.5 g, 3.5 mmol) was added and the reaction was stirred for 16 h. The reaction mixture was stripped in vacuum to give orange oil. The crude product was purified via flash chromatography to give 23 as a white solid (0.44 g, 1.14 mmol, 48% yield). 1H NMR (500 MHz, CDCl3) δ ppm 7.18 (dd, J = 1.0, 8.5 Hz, 1H), 7.08 (d, J = 1.8 Hz, 1H), 6.89 (d, J = 8.4 Hz, 1H), 4.04–4.08 (m, 1H), 3.92 (s, 3H), 3.82–3.92 (m, 1H), 3.58–3.72 (m, 2H), 3.33–3.42 (m, 2H), 1.59 (d, J = 2.7 Hz, 2H), 1.45 (s, 9H), 1.18–1.25 (m, 1H). 13C NMR (125 MHz, CDCl3) δ ppm 154.8, 150.8, 149.5, 124.3 (q, J = 271.3 Hz), 123.3 (q, J = 32.7 Hz), 118.2 (q, J = 4.2 Hz), 112.7, 108.5 (q, J = 3.6 Hz), 79.4, 70.7, 56.1, 28.5, 21.4. LC-MS calculated for C19H24F3NO4: 387.17; observed: m/z 410.0 [M+Na]+.
(1R,5S,6R)-6-((2-methoxy-4-(trifluoromethyl)phenoxy)methyl)-3-azabicyclo[3.1.0] hexane (24).
Trifluoroacetic acid (1 mL) was added to a solution of 23 (0.44 g, 1.14 mmol) in dichloromethane (5 mL). The mixture was stirred at room temperature for 2 h. The solvent was removed under reduced pressure to give 24 as a yellow oil (0.3 g, 1.05 mmol, 92% yield). 1H NMR (300 MHz, methanol-d4) δ ppm 7.16–7.24 (m, 2H), 7.05 (d, J = 8.3 Hz, 1H), 4.0 (d, J = 6.7 Hz, 2H), 3.88 (s, 3H), 3.42–3.53 (m, 4H), 1.94–1.98 (m, 2H), 1.39–1.47 (m, 1H). 13C NMR (75 MHz, methanol-d4) δ ppm 152.4, 151.1, 125.8 (q, J = 270.5 Hz), 124.4 (q, J = 32.6 Hz), 119.4 (q, J = 4.2 Hz), 114.4, 109.7 (q, J = 3.6 Hz), 70.7, 56.7, 22.2, 21.0. LC-MS calculated for C14H16F3NO2: 287.11; observed: m/z 288.1 [M+H]+.
2-((4-(2-Methoxy-4-(trifluoromethyl)phenyl)piperidin-1-yl)methyl)-1-methyl-1H-imidazo[4,5-b]pyridine (13).
Trimethylamine (0.22 g, 2.16 mmol), magnesium sulfate (0.65 g, 5.41 mmol) and 1-methyl-1H-imidazo[4,5-b]pyridine-2-carbaldehyde (19, 0.81 mmol, 0.16 g) were added to a solution of 4-(2-methoxy-4-(trifluoromethyl)phenyl)piperidine (18, 0.54 mmol, 0.13 g) in 1,2-dichloroethane (5 mL) under nitrogen. The mixture was stirred at room temperature for 30 min before sodium triacetoxyborohydride (0.17 g, 0.81 mmol) was added. The reaction mixture was then stirred overnight at room temperature and then quenched with dichloromethane. The organic phase was washed with water and brine. The aqueous phase was extracted with dichloromethane. Combined organic layer was dried over sodium sulfate. The solvent was removed at reduced pressure, and the residue was purified by flash column chromatography to give the product as a white solid (0.35 mmol, 0.14 g, 65% yield). 1H NMR (500 MHz, CD3OD): δ 8.41 (d, J = 5.0 Hz, 1H), 8.02 (d, J = 8.5 Hz, 1H), 7.34–7.37 (m, 2H), 7.20 (d, J = 7.5 Hz, 1H), 7.14 (s, 1H), 4.01 (s, 3H), 3.94 (s, 2H), 3.89 (s, 3H), 3.31–3.32 (m, 3H), 2.33–2.37(m, 2H), 1.72–1.83 (m, 4H). LC-MS calculated for C21H23F3N4O: 404.18; observed: m/z 405.15 [M+H]+.
2-((4-(2-Methoxy-4-(trifluoromethyl)phenyl)piperidin-1-yl)methyl)-5,6-dihydro-4H-imidazo [4,5,1-ij][1,7]naphthyridine (14).
In a similar procedure as described for synthesizing 13, compound 14 was prepared by using 18 (100 mg, 0.34 mmol) and 5,6-Dihydro-4H-imidazo[4,5,1-ij][1,7]naphthyridine-2-carbaldehyde (20, 110 mg, 0.51 mmol) to give the product as a white solid (76 mg, 0.18 mmol, 52% yield). 1H NMR (500 MHz, (CD3)2SO): δ 8.50 (d, J = 5.0 Hz, 1H), 7.40 (d, J = 8.5 Hz, 1H), 7.25 (d, J = 8.5 Hz, 1H), 7.20 (s, 1H), 7.01 (d, J = 4.5 Hz, 1H,), 4.33 (t, J = 6.0 Hz, 2H), 4.02 (s, 1H), 3.29 (s, 1H), 3.86 (s, 3H), 2.93–2.97 (m, 5H), 2.15–2.48 (m, 5H), 1.60–1.71 (m, 4H). LC-MS calculated for C23H25F3N4O: 430.20; observed: m/z 431.20 [M+H]+.
2-(((1R,5S,6R)-6-((2-methoxy-4-(trifluoromethyl)phenoxy)methyl)-3-azabicyclo[3.1.0] hexan-3-yl)methyl)-1-methyl-1H-imidazo[4,5-b]pyridine (15).
In a similar procedure as described for synthesizing 13, compound 15 was prepared by using 19 (HCl salt, 50 mg, 0.25 mmol) and 24 (HCl salt, 98 mg, 0.304 mmol) to give product as a white solid (31 mg, 0.072 mmol, 28% yield). 1H NMR (500 MHz, CDCl3): δ 8.47 (d, J = 4.5 Hz, 1H), 7.59 (d, J = 7.5 Hz, 1H), 7.29 (m, 2H), 7.02 (s, 1H), 6.83 (d, J = 8.5 Hz, 1H), 3.93 (s, 2H), 3.85 (s, 3H), 3.83 (d, J = 7.5 Hz, 2H), 3.78 (s, 3H), 2.96 (d, J = 9.0 Hz, 2H), 2.57 (d, J = 8.0 Hz, 2H), 1.67(m, 1H), 1.50 (s, 2H). 13C NMR (125 MHz, CDCl3): δ 155.0, 154.4, 151.0, 149.3, 144.5, 128.4, 123.1, 122.8, 118.2, 117.6, 116.9, 112.4, 108.4, 71.3, 56.0, 54.5, 51.5, 30.1, 21.5, 18.6. 19F NMR (470 MHz, CDCl3): δ −57.6. LC-MS calculated for C22H23F3N4O2: 432.18; observed: m/z 433.15 [M+H]+. HRMS m/z calculated for C22H24F3N4O2 [M+H]+, 433.1851, found m/z 433.1863.
3-(Cyclopropylmethyl)-7-((4-(2,4-difluorophenyl)piperazin-1-yl)methyl)-8-(trifluoromethyl)[1,2,4]triazolo[4,3-a]pyridine (17, JNJ-46356479).
In a similar procedure as described for synthesizing 13, compound 17 was prepared by using 3-(cyclopropylmethyl)-8-(trifluoromethyl)-[1,2,4]triazolo[4,3-a]pyridine-7-carbaldehyde (50 mg, 0.19 mmol) and 1-(2,4-difluorophenyl)piperazine (41.6 mg, 0.21 mmol), TEA (0.11 mL, 0.76 mmol), MgSO4 (0.229 g, 1.9 mmol) and NaBH(OAc)3 (60.4 mg, 0.285 mmol) in DCM (3 mL) to give product as a pale-yellow solid (44.2 mg, 0.098 mmol, 51.6% yield). 1H NMR (500 MHz, CDCl3) δ 8.23 (d, J = 7.5 Hz, 1H), 7.45 (d, J = 6.5 Hz, 1H), 6.90−6.91 (m, 1H), 6.78–6.81 (m, 2H), 3.81 (s, 2H), 3.10 (d, J = 6.5 Hz, 2H), 3.01–3.09 (m, 4H), 2.63–2.75 (m, 4H), 1.21–1.26 (m, 1H), 0.60–0.62 (m, 2H), 0.33–0.34 (m, 2H). LC-MS calculated for C22H22F5N5: 451.18; observed: m/z 452.05 [M+H]+.
2-(1-((1-Methyl-1H-imidazo[4,5-b]pyridin-2-yl)methyl)piperidin-4-yl)-5-(trifluoromethyl)phenol (25).
The boron tribromide solution (1 mL, 1 M in DCM, 1 mmol) was added dropwise to a solution of compound 13 (70 mg, 0.173 mmol) in 3 mL dichloromethane at 0 ℃. The mixture was slowly warmed to room temperature and stirred for another 2 h. After the reaction was completed, 5 mL of saturated sodium bicarbonate was added, and the mixture was extracted with dichloromethane. The crude product was purified by flash column chromatography to give product as a white solid (60 mg, 0.153 mmol, 88% yield). 1H NMR (500 MHz, CD3OD): δ 8.42 (d, J = 5.0 Hz, 1H), 8.03 (d, J = 8.5 Hz, 1H), 7.34–7.37 (m, 1H), 7.28 (d, J = 7.5 Hz, 1H), 7.05 (d, 1H, J = 7.5 Hz), 7.00 (s, 1H), 4.01 (m, 3H), 3.94, (m, 2H), 2.99–3.06 (m, 3H), 2.34–2.38 (m, 2H), 1.73–1.86 (m, 4H). 13C NMR (125 MHz, CD3OD): δ 155.4, 155.0, 153.7, 143.6, 136.6, 128.7, 126.8, 125.4, 123.3, 118.7, 118.0, 115.3, 115.3, 111.0, 54.6, 54.2, 35.1, 31.3, 29.5. LC-MS calculated for C20H21F3N4O: 390.17; observed: m/z 391.10 [M+H]+. HRMS m/z calculated for C20H22F3N4O [M+H]+, 391.1746, found m/z 391.1765.
Radiochemistry.
11CO2 was obtained via the 14N(p,α)11C reaction on nitrogen with 2.5% oxygen, 16 MeV protons (GE Healthcare, PETtrace), and trapped on molecular sieves in a TRACERlab FX-MeI synthesizer (GE Healthcare). 11CH4 was obtained by the reduction of 11CO2 in the presence of hydrogen at 350 °C and passed through an oven containing I2 to produce 11CH3I via a radical reaction. 11CH3I was trapped in a 5 mL V-vial containing a solution of excess 25 (0.5 ± 0.2 mg) and an aqueous 5N NaOH (3 uL) in dry dimethylformamide (250 μL) at room temperature and then heated at 80 °C for 2 min. The reaction mixture was diluted with 1.0 mL of water and purified using a HPLC system equipped with a semi-preparative column (Waters XBridge, C18, 250 × 10 mm, 5 μ), a UV detector monitored at 254 nm, and a radioactivity detector. The product was eluted with acetonitrile/water/TFA (30/70/0.7) at a flow rate of 5 mL/min. The fractions corresponding to [11C]13 (tR = 11 min) were collected into a large dilution vessel, which was pre-loaded with 2 mL of 8.4% sodium bicarbonate for injection, USP (Hospira) and 23 mL of sterile water for injection, USP. The product was loaded onto a C18 light cartridge, (Waters; pre-activated with 4 mL of EtOH followed by 10 mL of SWFI). The C18 light cartridge was washed with 10 mL of SWFI to remove traces of salts, residual acetonitrile and TFA. The C18 light cartridge was then eluted with 1 mL of dehydrated ethyl alcohol (USP) and followed by 10 mL of 0.9% sodium chloride solution (USP) into a product collection vessel. The formulated solution was filtered through a vented Millipore-GV 0.22μ sterilizing filter (EMD Millipore) into a 10 mL vented sterile vial.
Radiochemical purity and chemical quality were measured by an analytical HPLC equipped with an analytical column (Waters, XBridge, C18, 3.5 μ, 4.6 × 150 mm), a UV detector monitored at 254 nm, and a radioactivity detector, which was eluted with a solution (acetonitrile/0.1%TFA water = 30/70) at a flow rate of 1 mL/min. [11C]13 was eluted ~6 min (chemical and radiochemical purities > 98%, n = 10). The radiosynthesis time was 50 min from the end of bombardment (EOB). The molar activity was 98 ± 30 GBq/µmol at the end of synthesis (EOS).
Molecular modeling.
The mGluR2 receptor model structure was built in YASARA45 using a series of structures from the Protein Data Bank (PDB). These structures were obtained after a BLAST72 search of the mGluR2 sequence against the PDB. The model was built by manually selecting from these template structures with sequence homology to mGluR2. These templates are mGluR1 complexed with glutamate (PDB ID:1EWK)73, mGluR5 complexed with glutamate (PDB ID: 3LMK)74 and Metabotropic Glutamate Receptor 5 Apo Form (PDB ID 6N52)75. Using these three structures as templates, a hybrid model for mGluR2 was built in YASARA. Results of model evaluations are given in the Supporting Information.
To prepare the ligands for docking, the ligands were drawn on ChemDraw Professional 16.0 by PerkinElmer and were converted into PDB format in Avogadro 1.276. These ligands were further optimized in Avogadro before docking. Docking was performed into the model structure with AutoDock53 embedded in YASARA45.
GloSensor cAMP functional assay.
HEK-293 cells were maintained with complete Dulbecco’s modified Eagle’s medium (DMEM), which was composed of 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 units/mL penicillin G, 100 μg/mL streptomycin at 37 °C in the presence of 5% CO2. HEK-293 stable cell lines with tetracycline inducible expression of mGluR1, mGluR2, mGluR4, mGluR6 or mGluR8 were maintained with complete DMEM with Hygromycin B (100 μg/mL), Blasticidin (15 μg/mL) at 37°C in the presence of 5% CO2.
The Gq coupled receptors (mGluR1 and mGluR5) were tested using Ca2+ mobilization assay. mGluR1 stable cell lines were plated into poly-L-lysine (PLL) coated 384-well black clear bottom cell culture plates with complete Basal Medium Eagle (BME) buffer, which was composed of 10% dialyzed FBS, penicillin G (100 units/mL), streptomycin (100 μg/mL) with Tetracycline (1 μg/mL) at density of 20,000 cells in 40 µl per well for overnight. On the other hand, HEK-293 Cells transiently transfected using the calcium phosphate method with cDNA encoding mGluR5 for 40 h were plated into the plate with complete BME at density of 20,000 cells in 40 µL per well for 8 h. mGluR1 stable cells or cells transiently expressing mGluR5 were incubated with 20 μL of the calcium dye (FLIPR Calcium 4 Assay Kit; Molecular Devices) diluted in the assay buffer (1× HBSS, 2.5 mM probenecid, and 20 mM HEPES, pH 7.4) for 45 min at 37 °C and 15 min at room temperature. To measure agonist activity of receptors, the drug plates were prepared with different concentrations of test or reference compound at 3 times the desired final concentration. When measuring antagonist activity, another drug plate which contained EC80 concentration of the reference drug was prepared. Once loaded in FLIPR (Molecular Devices), basal fluorescence was measured for 10 s, then 10 μL of test or reference compounds were added, followed by continued fluorescence measurement for an additional 120 s. Raw data were plotted as a function of molar concentration of the compound with Prism 5.0 (GraphPad Software).
The Gi/o coupled receptors (mGluR2, mGluR3, mGluR4, mGluR6 and mGluR8) were tested using cAMP assay. Promega’s split luciferase based GloSensor cAMP biosensor technology was used in determining Gi-GPCR mediated cAMP production in live cells. On the cells stably expressing mGluR2, mGluR3, mGluR4, mGluR6 or mGluR8, GloSensor cAMP DNA construct was transfected overnight. Cells were seeded into PLL coated 384-well white clear bottom cell culture plates with complete BME Buffer with Tetracycline (1 μg/mL) at a density of 20,000 cells for another 24 h. The cell medium was removed and then 20 μl of buffer was loaded. To measure the agonist activity, 10 μL of 3x test compound solution was added 15 min before addition of 10 μl of luciferin/isoproterenol mixture at a final concentration of 4 mM and 200 nM, respectively, followed by counting of the plate. To measure the PAM or antagonist activity, cells were pre-incubated with test compound for 15 min before addition of EC20 or EC80 concentration of a reference agonist for another 15 min. Then 10 μl of luciferin/isoproterenol mixture at a final concentration of 4 mM and 200 nM, respectively, was added for 15 min followed by counting of the plate. In these experiments, isoproterenol was used to activate endogenous β2 adrenergic receptors expressed in HEK293 T cells to activate the endogenous Gs protein. Luminescence was counted in a TriLux luminescence counter. Data were analyzed with Prism 5.0 (GraphPad software).
Compounds were tested for their potency in dose-response experiments. Eight-point dose response curves were performed in duplicate twice on two separate lots of cells (sometimes a third curve might be needed if in the first experiment the range of concentrations used was outside of the active range). For antagonists, these curves were performed in the presence of the EC80 concentration of the agonist. For each compound, the results from four replicates were averaged and then either EC50 or IC50 values were calculated by non-linear regression using the 4-parameter logistic equation. Results were reported as EC50 or IC50 values for each tested compound (and receptor) and include the EC50 or IC50 values of a known agonist or antagonist for comparison purposes.
In vitro characterization.
The Log P was determined using a reversed-phase HPLC method. First, seven reference compounds were examined to obtain the linear regression of the log P against the log of capacity factors by the expression: logPow = a + b * logk. The Log P of these reference compounds was already been determined. The capacity factor k was calculated by the expression: k = (tR – t0)/t0. The retention time tR of test compound was determined on the HPLC (Agilent 1260 infinity II LC System, XTerraTM MS C18 5μ 2.1 × 250 mm, methanol/water=75/25, 0.25 mL/min). The dead-time t0 was measured by using thiourea. All measurements were done with triplicate three parallels and results are given in Table S4. The linear regression equation of the Log P against the log of capacity factors was generated in Excel: logPow = 3.049 + 2.429 * logk , where R2 was 0.9964 (Figure S9). The retention time of compound 13–15 was also determined on the HPLC under the same condition and each test was repeated three times (Table S5).
In the plasma protein binding assay, disposable RED device inserts (product 90006) were from Thermos Scientific (Waltham, MA). Each insert was made of two side-by-side chambers separated by a vertical cylinder of dialysis membrane (MWCO ~8,000) validated for minimal non-specific binding. A stock solution of the test compound in DMSO was spiked into the rat plasma to reach a concentration of 10 μM. 400 μL of sample solution was placed into the sample chamber of the RED device, and 600 μL of phosphate-buffered saline (PBS) was added to the buffer chamber of the RED device. Samples were prepared in triplicates. The plate was covered with aluminum sealing cover and incubated at 37 °C on an orbital shaker at approximately 200 rpm for 5 h. After incubation, 300 μL of post-dialysis samples from the buffer and sample chambers were transferred to different microcentrifuge tubes. To the buffer sample was added 300 μL of plasma, and an equal volume of buffer was added to the collected plasma sample. 600 μL of cold acetonitrile was added to the samples, and the samples were vortexed and incubated for 30 min on ice and then were centrifuged at 14000 rpm for 10 min. Supernatant was transferred to vial for HPLC analysis (XTerraTM MS C18 5μ, 2.1 × 250 mm column; Gradient elution from 5% to 90% B in 30 min; 0.1 M ammonium formate in water (A) and acetonitrile (B); UV 254 nm; 100 uL of injection volume). The percentage of the test bound compound was calculated as % Free = (Concentration in buffer chamber/Concentration in plasma chamber) ×100%; % Bound = 100% - % Free (Table S6).
Compound stability in rat serum was examined using a published method.65 Rat serum (100 μL, Abcam, Inc, No. ab7488) and test compound or control compound (2.5 μL, 1 mM in DMSO) was added to the individual tube. The tube was vortexed and incubated at 37 °C. During the incubation, aliquots of 50 uL samples were quenched with ice-cold acetonitrile at 0, 15, 30, 60, and 120 min time points, respectively. After mixing, the quenched samples were centrifuged, and the supernatant was withdrawn for analysis by HPLC (Agilent 1260 infinity II LC System, XTerraTM MS C18, 5 μ, 2.1 × 250 mm, 20 mM ammonium formate (A)/acetonitrile (B), 0.25 mL/min, gradient of 5% to 100% B). The samples were assayed at least three times. Compound 25 was used as internal standard while diltiazem was used as a positive control. The percentage remaining was calculated by (peak area at the specific time point)/(peak area at 0 min) × 100% (Table S7).
Compound stability in rat liver microsomal was tested using a published method.65,66 In a vial 1.5 μL of test compound (1 mM in DMSO stock solution) was mixed with 432 μL of PBS (50 mM, pH 7.4). The mixture was kept at 37 °C for 10 min before adding 13 μL of Sprague−Dawley rat liver microsomes (Sigma-Aldrich, No. M9066). The vial was vortexed and shaken at 37 °C for 5 min, followed by addition of 50 μL of NADPH (10 mM in PBS stock solution) to start the reaction. The mixture was incubated at 37 °C for 0, 5, 15, 30, 45 min, respectively, and quenched by addition of 250 μL of ice-cold acetonitrile and 3 μL of the internal standard (0.5 mM in DMSO). The quenched solutions were centrifuged at 10,000g for 15 min. The supernatant was collected and quantitated by RP-HPLC (Phenomenex Luna® column 5 µ C18, 100 Å, 250 × 4.6 mm; 0.7 mL/min, 15 min, Acetonitrile/water/0.1% FA). The procedure was repeated three times for each compound. Compound 25 was used as internal standard and compound ML128 served as positive control. The percentage of remaining intact test-compound was calculated by (peak area at the specific time point)/(peak area at 0 min)×100%. Each procedure was repeated three times (Tables S8 & S9).
The solution stability of 13 was examined in the aqueous buffers at different pH values. 50 μL of compound in DMSO (0.25 mM) was added to the sodium acetate-KCl-HCl buffer (950 μL, 20 mM, pH 5.0), phosphate buffer (950 μL, 20 mM, pH 7.4), and boric acid-KCl-NaOH buffer (950 μL, 20 mM, pH 9.4), respectively. The mixtures were incubated for 2 h at 37 °C and analyzed by HPLC (Phenomenex Luna® column, 5 µm C18, 100 Å, 250 × 4.6 mm, eluents: CH3CN/H2O in 0.1% formic acid). The area under curve (AUC) values of 13 was monitored at 0, 15, 30, 60, and 120 min time points (n = 2, Table S10).
In the PAMPA assay, polar brain lipid (PBL) was purchased from Avanti Polar Lipids (Alabaster, AL). Theophylline, caffeine, and dodecane were purchased from Sigma-Aldrich. The 96-well acceptor filter plate (polyvinylidene difluoride membrane, pore size 0.45 μm) and the donor microplate were obtained from Merck Millipore Bioscience (Bedford, MA). Test compound was dissolved in DMSO at 5 mg/mL, and further diluted in phosphate buffer (pH 7.4) to obtain the sample solution at a final concentration of 25 μg/mL. The acceptor wells were coated with 4 μL of porcine polar brain lipid (PBL) in dodecane (20 mg/mL) before 200 μL of phosphate buffer was added. To the corresponding donor well, 300 μL of the sample solution (n = 5) was added. The acceptor well was carefully put on the donor plate and kept for 18 h. After incubation, the acceptor plate was separated from the donor plate and the concentration of the test compounds in both acceptor and donor wells was determined using a UV plate reader (SpectraMax M Series Multi-Mode Microplate Readers). Verapamil (Pe = 16 ×10−6 cm/s) and theophylline (Pe = 0.12 ×10−6 cm/s) were used as positive and negative control compounds, respectively.
The P-gp ATPase activity was measured with the Pgp-Glo™ assay system with human P-gp membrane by following the manufacturer’s instructions (Promega, Co. USA). The assay relies on the ATP dependence of the light-generating reaction of firefly luciferase. Briefly, 25 μg of P-gp membrane was incubated at 37 °C with one of these samples including Na3VO4 (100 μM), solvent control (0.1% DMSO), quercetin (100 μM), the test compound (200 μM), verapamil (100 μM), verapamil (100 μM) plus the test compound (100 μM). The ATPase reaction was initiated by addition of MgATP (5 mM) and followed by incubation for 40 min at 37 °C. The reaction was stopped, and the remaining unmetabolized ATP was detected as a luciferase-generated luminescence signal by addition of ATP detection reagent. Following a room-temperature signal-stabilization period (20 min), luminescence was read on a Veritas microplate luminometer (Tuner Designs, San Francisco, CA). P-gp ATPase activity was presented as a drop-in luminescence of samples compared to that treated with Na3VO4.
Whole body biodistribution study.
The quantitative biodistribution of [11C]13 was done using 16 healthy Sprague Dawley rats (weight 330–370 g). After anesthetization (2% isoflurane with oxygen flow of 1.5 L/min) the rats were administrated with the [11C]13 (30–42 MBq (0.81–1.14 mCi) using tail vein injection and sacrificed by decapitation at the time points 5, 20, 30 or 40 min after administration of the radioactivity. The tissue samples including blood, midbrain, cerebellum, cortex, lung, heart, liver, spleen, kidney and muscle were rapidly collected into pre-weighted gamma-counting tubes and measured with standards (samples of [11C]13) using PerkinElmer Wizard2 2480 gamma-counter. Tubes were weighted, and the net mass of the tissue samples was determined and the percent of the injected radioactivity (% ID/g) in the samples was calculated.
In vivo characterization.
Altogether twelve normal Sprague Dawley rats (male, 275–500 g) were used in sixteen studies to investigate in vivo imaging characteristics of [11C]13. Four rats had control studies followed by the “blocking” studies while three rats had only “blocking” studies and 5 rats had only control studies to investigate binding characteristics of [11C]13. For the imaging studies rats were anesthetized with isoflurane/nitrous oxide (1.0–1.5% isoflurane, with oxygen flow of 1–1.5 L/min) and the tail vein was catheterized for administration of the imaging ligand ([11C]13). The rats were adjusted into the scanner for imaging position (Triumph II Preclinical Imaging System, Trifoil Imaging,LLC, Northridge, CA). The vital signs such as heart rate and/or breathing were monitored throughout the imaging. Data acquisition of 60 min was started from the injection of radioligand [11C]13 (20–41 MBq (0.54–1.11mCi) i.v.).
The “cold” compounds 13 and 17 were used to investigate selectivityand sensitivity of [11C]13 for the mGluR2. For injection 13 was dissolved into a saline solution with 10% DMSO, 5% Tween-20 and 85% PBS with a pH of 7.4 and 17 was dissolved into saline with 20% HP-B-CD with pH under 5.5. The “cold” compounds were administered (i.v., 4 mg/kg) 10 min before the radioactivity.
CT scan was performed after every PET imaging study to obtain anatomical information and correction for attenuation. The PET imaging data were corrected for uniformity, scatter, and attenuation and processed by using maximum-likelihood expectation-maximization (MLEM) algorithm with 30 iterations to dynamic volumetric images (18×10”, 14×30”, 20×60”, 10×180”). CT data were reconstructed by the modified Feldkamp algorithm using matrix volumes of 512×512×512 and pixel size of 170 μm. The ROIs, i.e., whole brain, thalamus, hippocampus, cortex, striatum, and cerebellum, were drawn onto coronal PET slices according to the brain outlines as derived from the rat brain atlas and corresponding TACs (time-activity curves) were created by PMOD 3.2 (PMOD Technologies Ltd., Zurich, Switzerland). Percent changes between the control and blocking studies were calculated in the selected brain areas at the 10–30 min time window after injection of [11C]13.
Supplementary Material
Table 3.
The in vitro stability of compounds 13-15
| Compound | The plasma stability | The microsome stability | The solution stability | |||
|---|---|---|---|---|---|---|
| The intact ± SEM at 120 min (%) | t1/2 (min) | CLint (mL/min/mg protein) | The intact ± SEM at 120 min (%) | |||
| pH = 5.0 | pH = 7.4 | pH = 9.4 | ||||
| 13 | 96.6 ± 2.1 | 74.5 | 17.9 | 95.3 ± 0.3 | 94.5 ± 0.4 | 96.1 ± 0.7 |
| 14 | 89.2 ± 0.6 | 73.7 | 18.1 | --- | --- | --- |
| Diltiazem | 43.2 ± 0.8 | --- | --- | --- | --- | --- |
| ML128 | --- | 4.2 | 317.1 | --- | --- | --- |
ACKNOWLEDGEMENTS
This project was financially supported by NIH grants [1R01EB021708 and 1R01NS100164] and the grants 1S10RR023452–01 and 1S10OD025234–01 for the imaging instrumentation and characterization of the organic compounds. mGluR1–6 and mGluR8 agonist and antagonist functional data as well as mGluR2 PAM activity were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271–2013-00017-C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth (mailto:bryan_roth@med.unc.edu) at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll (mailto:jdrisco1@mail.nih.gov) at NIMH, Bethesda MD, USA. For experimental details please refer to the PDSP web site https://pdspdb.unc.edu/pdspWeb/ (https://pdspdb.unc.edu/pdspWeb/).
ABBREVIATIONS USED
- BBB
blood-brain barrier
- VFTD
Venus flytrap domain
- 7-TM
seven transmembrane
- CRD
cysteine rich domain
- POOL
Partial Order Optimum Likelihood
- EL2
extracellular loop 2
- PDB
Protein Data Bank
- PAM
positive allosteric modulator
- NAM
negative allosteric modulator
- MW
molecular weight
- tPSA
topological polar surface area
- Clint
the intrinsic clearance
- Gi
adenylate cyclase inhibitory G-protein
- MWCO
molecular weight cut off
- ND
not determined
- PBL
Polar brain lipid
- PBL
porcine polar brain lipid
- P-gp
P-glycoprotein
- PAMPA
parallel artificial membrane permeability assay
- Pe
effective permeability
- EOS
end of synthesis
- SWFI
sterile water for injection
- TAC
time-activity curve
- SUV
standardized uptake value
- USP
United States Pharmacopeia
- % ID/g
percentage of injected dose per gram of wet tissue
Footnotes
ASSOCIATED CONTENT
Supporting Information
The detailed in vitro assays and molecular modeling work are described in the Supporting Information.
The authors declare no conflict interest.
REFERENCES
- (1).Tanabe Y; Masu M; Ishii T; Shigemoto R; Nakanishi S A family of metabotropic glutamate receptors. Neuron 1992, 8, 169–179. [DOI] [PubMed] [Google Scholar]
- (2).Cartmell J; Schoepp DD Regulation of neurotransmitter release by metabotropic glutamate receptors. J. Neurochem 2000, 75, 889–907. [DOI] [PubMed] [Google Scholar]
- (3).Gu G; Lorrain DS; Wei H; Cole RL; Zhang X; Daggett LP; Schaffhauser HJ; Bristow LJ; Lechner SM Distribution of metabotropic glutamate 2 and 3 receptors in the rat forebrain: Implication in emotional responses and central disinhibition. Brain Res 2008, 1197, 47–62. [DOI] [PubMed] [Google Scholar]
- (4).Wright RA; Johnson BG; Zhang C; Salhoff C; Kingston AE; Calligaro DO; Monn JA; Schoepp DD; Marek GJ CNS distribution of metabotropic glutamate 2 and 3 receptors: transgenic mice and [3H]LY459477 autoradiography. Neuropharmacology 2013, 66, 89–98. [DOI] [PubMed] [Google Scholar]
- (5).Yin S; Noetzel MJ; Johnson KA; Zamorano R; Jalan-Sakrikar N; Gregory KJ; Conn PJ; Niswender CM Selective actions of novel allosteric modulators reveal functional heteromers of metabotropic glutamate receptors in the CNS. J. Neurosci 2014, 34, 79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kim SH; Fraser PE; Westaway D; George-Hyslop PHS; Ehrlich ME; Gandy S Group II metabotropic glutamate receptor stimulation triggers production and release of Alzheimer’s amyloid β42 from isolated intact nerve terminals. J. Neurosci 2010, 30, 3870–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Richards G; Messer J; Faull RLM; Stadler H; Wichmann J; Huguenin P; Bohrmann B; Mutel V Altered distribution of mGlu2 receptors in β-amyloid-affected brain regions of Alzheimer cases and aged PS2APP mice. Brain Res 2010, 1363, 180–190. [DOI] [PubMed] [Google Scholar]
- (8).Lee H.-g.; Ogawa O; Zhu X; O’Neill MJ; Petersen RB; Castellani RJ; Ghanbari H; Perry G; Smith MA Aberrant expression of metabotropic glutamate receptor 2 in the vulnerable neurons of Alzheimer’s disease. Acta Neuropathol 2004, 107, 365–371. [DOI] [PubMed] [Google Scholar]
- (9).Caraci F; Molinaro G; Battaglia G; Giuffrida ML; Riozzi B; Traficante A; Bruno V; Cannella M; Merlo S; Wang X; Heinz BA; Nisenbaum ES; Britton TC; Drago F; Sortino MA; Copani A; Nicoletti F Targeting group II metabotropic glutamate (mGlu) receptors for the treatment of psychosis associated with Alzheimer’s disease: selective activation of mGlu2 receptors amplifies β-amyloid toxicity in cultured neurons, whereas dual activation of mGlu2 and mGlu3 receptors is neuroprotective. Mol. Pharmacol 2011, 79, 618–626. [DOI] [PubMed] [Google Scholar]
- (10).Moreno JL; Sealfon SC; Gonzalez-Maeso J Group II metabotropic glutamate receptors and schizophrenia. Cell. Mol. Life Sci 2009, 66, 3777–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chaki S Group II metabotropic glutamate receptor agonists as a potential drug for schizophrenia. Eur. J. Pharmacol 2010, 639, 59–66. [DOI] [PubMed] [Google Scholar]
- (12).Downing AM; Kinon BJ; Millen BA; Zhang L; Liu L; Morozova MA; Brenner R; Rayle TJ; Nisenbaum L; Zhao F; Gomez JC A double-bind, placebo-controlled comparator study of LY2140023 monohydrate in patients with schizophrenia. BMC Psychiatry 2014, 14, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Patil S; Zhang L; Martenyi F; Lowe SL; Jackson KA; Andreev BV; Avedisova AS; Bardenstein LM; Gurovich IY; Morozova MA; Mosolov SN; Neznanov NG; Reznik AM; Smulevich AB; Tochilov VA; Johnson BG; Monn JA; Schoepp DD Activation of mGluR2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat. Med 2007, 13, 1102–1107. [DOI] [PubMed] [Google Scholar]
- (14).Feyissa AM; Woolverton WL; Miguel-Hidalgo JJ; Wang Z; Kyle PB; Hasler G; Stockmeier CA; Iyo AH; Karolewicz B Elevated level of metabotropic glutamate receptor 2/3 in the prefrontal cortex in major depression. Prog. Neuro-psychoph 2010, 34, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Johnson MP; Barda D; Britton TC; Emkey R; Hornback WJ; Jagdmann GE; McKinzie DL; Nisenbaum ES; Tizzano JP; Schoepp DD Metabotropic glutamate 2 receptor potentiators: receptor modulation, frequency-dependent synaptic activity, and efficacy in preclinical anxiety and psychosis model(s). Psychopharmacology (Berl) 2005, 179, 271–83. [DOI] [PubMed] [Google Scholar]
- (16).Mazzitelli M; Palazzo E; Maione S; Neugebauer V Group II metabotropic glutamate receptors: role in pain mechanisms and pain modulation. Front Mol. Neurosci 2018, 11, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chiechio S; Copani A; Zammataro M; Battaglia G; Gereau RWIV; Nicoletti F Transcriptional regulation of type-2 metabotropic glutamate receptors: an epigenetic path to novel treatments for chronic pain. Trends Pharmacol. Sci 2010, 31, 153–160. [DOI] [PubMed] [Google Scholar]
- (18).Niswender CM; Conn PJ Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol 2010, 50, 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Marek GJ When is a Proof-of-Concept (POC) not a POC? Pomaglumetad (LY2140023) as a Case Study for Antipsychotic Efficacy. Curr. Pharm. Des 2015, 21, 3788–3796. [DOI] [PubMed] [Google Scholar]
- (20).Adams DH; Kinon BJ; Baygani S; Millen BA; Velona I; Kollack-Walker S; Walling DP A long-term, phase 2, multicenter, randomized, open-label, comparative safety study of pomaglumetad methionil (LY2140023 monohydrate) versus atypical antipsychotic standard of care in patients with schizophrenia. BMC Psychiatry 2013, 13, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Stauffer VL; Millen BA; Andersen S; Kinon BJ; Lagrandeur L; Lindenmayer JP; Gomez JC Pomaglumetad methionil: no significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophr Res 2013, 150, 434–441. [DOI] [PubMed] [Google Scholar]
- (22).Fell MJ; Svensson KA; Johnson BG; Schoepp DD Evidence for the role of metabotropic glutamate (mGlu)2 not mGlu3 receptors in the preclinical antipsychotic pharmacology of the mGlu2/3 receptor agonist (−)-(1R,4S,5S,6S)-4-amino-2- sulfonylbicyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY404039). J. Pharmacol. Exp. Ther 2008, 326, 209–217. [DOI] [PubMed] [Google Scholar]
- (23).Woolley ML; Pemberton DJ; Bate S; Corti C; Jones DNC The mGlu2 but not the mGlu3 receptor mediates the actions of the mGluR2/3 agonist, LY379268, in mouse models predictive of antipsychotic activity. Psychopharmacology (Berlin, Germany) 2008, 196, 431–440. [DOI] [PubMed] [Google Scholar]
- (24).Yu M; Nagren K; Chen YI; Livni E; Elmaleh D; Kozikowski A; Wang X; Jokivarsi K; Brownell A-L Radiolabeling and biodistribution of methyl 2-(methoxycarbonyl)-2-(methylamino) bicyclo [2.1.1]-hexane-5-carboxylate, a potent neuroprotective drug. Life Sci 2003, 73, 1577–1585. [DOI] [PubMed] [Google Scholar]
- (25).Wang J-Q; Zhang Z; Kuruppu D; Brownell A-L Radiosynthesis of PET radiotracer as a prodrug for imaging group II metabotropic glutamate receptors in vivo. Bioorg. Med. Chem. Lett 2012, 22, 1958–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhang Z; Brownell A-L Imaging of metabotropic glutamate receptors (mGluRs). In Neuroimaging-clinical applications, Bright P, Ed. In Tech-Open Access Publisher: Rijeka, Croatia, 2012; pp 499–532. [Google Scholar]
- (27).Trabanco AA; Cid JM; Lavreysen H; MacDonald GJ; Tresadern G Progress in the development of positive allosteric modulators of the metabotropic glutamate receptor 2. Curr. Med. Chem 2011, 18, 47–68. [DOI] [PubMed] [Google Scholar]
- (28).Sheffler DJ; Pinkerton AB; Dahl R; Markou A; Cosford NDP Recent progress in the synthesis and characterization of group II metabotropic glutamate receptor allosteric modulators. ACS Chem. Neurosci 2011, 2, 382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Andres J-I; Alcazar J; Cid JM; De Angelis M; Iturrino L; Langlois X; Lavreysen H; Trabanco AA; Celen S; Bormans G Synthesis, evaluation, and radiolabeling of new potent positive allosteric modulators of the metabotropic glutamate receptor 2 as potential tracers for positron emission tomography imaging. J. Med. Chem 2012, 55, 8685–8699. [DOI] [PubMed] [Google Scholar]
- (30).Leurquin-Sterk G; Van Laere K; Koole M; Celen S; Bormans G; Langlois X; Te Riele P; Schmidt Mark E; Van Hecken A; Verbruggen A; de Hoon J; Alcazar J; Andres J-I What we observe in vivo is not always what we see in vitro: development and validation of 11C-JNJ-42491293, a novel radioligand for mGluR2. J. Nucl. Med 2017, 58, 110–116. [DOI] [PubMed] [Google Scholar]
- (31).Lohith T; McQuade P; Salinas C; Anderson M; Reynders T; Bautmans A; Bormans G; Serdons K; Laere KV; Hostetler E First-in-human PET imaging of mGluR2 receptors. J. Nucl. Med 2016, 57, 213. [Google Scholar]
- (32).McQuade P; Joshi A; Miller P; Zeng Z; Purcell M; Gantert L; Holahan M; Meissner R; Uslaner J; Hostetler E Discovery and preclinical evaluation of an mGluR2-NAM PET radioligand. J. Nucl. Med 2016, 57, 290. [Google Scholar]
- (33).Majo V; Prabhakaran J; Simpson N; Arango V; Mann JJ; Kumar DJ Development of a [18F]-labeled positive allosteric modulator of the metabotropic glutamate receptor 2 (mGluR2) as a potential PET tracer. J. Nucl. Med 2016, 54, 1072. [Google Scholar]
- (34).Ma Y; Kumata K; Yui J; Zhang Y; Yamasaki T; Hatori A; Fujinaga M; Nengaki N; Xie L; Wang H; Zhang M-R Synthesis and evaluation of 1-(cyclopropylmethyl)-4-(4-[11C]methoxyphenyl)-piperidin-1-yl-2-oxo-1,2-dihydropyridine-3-carbonitrile ([11C]CMDC) for PET imaging of metabotropic glutamate receptor 2 in the rat brain. Bioorg. Med. Chem 2017, 25, 1014–1021. [DOI] [PubMed] [Google Scholar]
- (35).Kumata K; Yamasaki T; Hatori A; Zhang Y; Mori W; Fujinaga M; Xie L; Okubo T; Nengaki N; Zhang M-R Synthesis and in vitro evaluation of three novel radiotracers for imaging of metabotropic glutamate receptor subtype 2 in rat brain. Bioorg. Med. Chem. Lett 2017, 27, 3139–3143. [DOI] [PubMed] [Google Scholar]
- (36).Zhang X; Kumata K; Yamasaki T; Cheng R; Hatori A; Ma L; Zhang Y; Xie L; Wang L; Kang HJ; Sheffler DJ; Cosford NDP; Zhang MR; Liang SH Synthesis and preliminary studies of a novel negative allosteric modulator, 7-((2,5-Dioxopyrrolidin-1-yl)methyl)-4-(2-fluoro-4-[11C]methoxyphenyl) quinoline-2-carboxamide, for imaging of metabotropic glutamate receptor 2. ACS Chem. Neurosci 2017, 8, 1937–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kumata K; Hatori A; Yamasaki T; Zhang Y; Mori W; Fujinaga M; Xie L; Nengaki N; Zhang MR Synthesis and evaluation of 4-(2-fluoro-4-[11C]methoxyphenyl)-5-((2-methylpyridin-4-yl)methoxy)picolinamide for PET imaging of the metabotropic glutamate receptor 2 in the rat brain. Bioorg. Med. Chem 2019, 27, 483–491. [DOI] [PubMed] [Google Scholar]
- (38).Li Z; Krause S; Suzuki M; Sasaki T Radiotracer compounds. WO/2016033190-A1, 2016. [Google Scholar]
- (39).Van Gool MLM; Andres-Gil JI; Alcazar-Vaca MJ; Bormans GMR; Celen SJL; Joost V Radiolabelled mGluR2 PET ligands. WO/2016087489-A1, 2016. [Google Scholar]
- (40).D’Alessandro PL; Corti C; Roth A; Ugolini A; Sava A; Montanari D; Bianchi F; Garland SL; Powney B; Koppe EL; Rocheville M; Osborne G; Perez P; de la Fuente J; De Los Frailes M; Smith PW; Branch C; Nash D; Watson SP The identification of structurally novel, selective, orally bioavailable positive modulators of mGluR2. Bioorg. Med. Chem. Lett 2010, 20, 759–762. [DOI] [PubMed] [Google Scholar]
- (41).Efremov IV; Rogers BN; Duplantier AJ; Zhang L; Zhang Q; Maklad NS Azabenzimidazolyl compounds as potentiators of mGluR2 subtype of glutamate receptor and their preparation, pharmaceutical compositions and use in the treatment of diseases. WO/2008012622, 2008. [Google Scholar]
- (42).Zhang L; Brodney MA; Candler J; Doran AC; Duplantier AJ; Efremov IV; Evrard E; Kraus K; Ganong AH; Haas JA; Hanks AN; Jenza K; Lazzaro JT; Maklad N; McCarthy SA; Qian W; Rogers BN; Rottas MD; Schmidt CJ; Siuciak JA; Tingley FD; Zhang AQ 1-[(1-Methyl-1H-imidazol-2-yl)methyl]-4-phenylpiperidines as mGluR2 Positive Allosteric Modulators for the Treatment of Psychosis. J. Med. Chem 2011, 54, 1724–1739. [DOI] [PubMed] [Google Scholar]
- (43).Zhang L; Rogers BN; Duplantier AJ; McHardy SF; Efremov I; Berke H; Qian W; Zhang AQ; Maklad N; Candler J; Doran AC; Lazzaro JT; Ganong AH 3-(imidazolylmethyl)-3-aza-bicyclo[3.1.0]hexan-6-yl)methyl ethers: a novel series of mGluR2 positive allosteric modulators. Bioorg. Med. Chem. Lett 2008, 18, 5493–5496. [DOI] [PubMed] [Google Scholar]
- (44).Cid JM; Tresadern G; Vega JA; de Lucas AI; del Cerro A; Matesanz E; Linares ML; Garcia A; Iturrino L; Perez-Benito L; Macdonald GJ; Oehlrich D; Lavreysen H; Peeters L; Ceusters M; Ahnaou A; Drinkenburg W; Mackie C; Somers M; Trabanco AA Discovery of 8-trifluoromethyl-3-cyclopropylmethyl-7-[(4-(2,4-difluorophenyl)-1-piperazinyl)methyl]-1,2,4-triazolo[4,3-a]pyridine (JNJ-46356479), a selective and orally bioavailable mGlu2 receptor positive allosteric modulator (PAM). J. Med. Chem 2016, 59, 8495–8507. [DOI] [PubMed] [Google Scholar]
- (45).Krieger E; Joo K; Lee J; Lee J; Raman S; Thompson J; Tyka M; Baker D; Karplus K Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).McGuffin LJ; Shuid AN; Kempster R; Maghrabi AHA; Nealon JO; Salehe BR; Atkins JD; Roche DB Accurate template-based modeling in CASP12 using the IntFOLD4-TS, ModFOLD6, and ReFOLD methods. Proteins 2018, 86, 335–344. [DOI] [PubMed] [Google Scholar]
- (47).Waterhouse A; Bertoni M; Bienert S; Studer G; Tauriello G; Gumienny R; Heer FT; de Beer TAP; Rempfer C; Bordoli L; Lepore R; Schwede T SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 2018, 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Eisenberg D; Luthy R; Bowie JU VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol 1997, 277, 396–404. [DOI] [PubMed] [Google Scholar]
- (49).Colovos C; Yeates TO Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 1993, 2, 1511–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Tong WX; Wei Y; Murga LF; Ondrechen MJ; Williams RJ Partial Order Optimum Likelihood (POOL): Maximum likelihood prediction of protein active site residues using 3D structure and sequence properties. PLoS Comput. Biol 2009, 5, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Tan KP; Varadarajan R; Madhusudhan MS DEPTH: a web server to compute depth and predict small-molecule binding cavities in proteins. Nucleic Acids Res 2011, 39, W242–W248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Huang BD MetaPocket: A meta spproach to improve protein ligand binding site prediction. OMICS 2009, 13, 325–330. [DOI] [PubMed] [Google Scholar]
- (53).Morris GM; Goodsell DS; Halliday RS; Huey R; Hart WE; Belew RK; Olson AJ Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem 1998, 19, 1639–1662. [Google Scholar]
- (54).Perez-Benito L; Doornbos MLJ; Cordomi A; Peeters L; Lavreysen H; Pardo L; Tresadern G Molecular switches of allosteric modulation of the metabotropic glutamate 2 receptor. Structure 2017, 25, 1153–1162. [DOI] [PubMed] [Google Scholar]
- (55).Doornbos MLJ; Cid JM; Haubrich J; Nunes A; van de Sande JW; Vermond SC; Mulder-Krieger T; Trabanco AA; Ahnaou A; Drinkenburg WH; Lavreysen H; Heitman LH; AP IJ; Tresadern G Discovery and kinetic profiling of 7-aryl-1,2,4-triazolo[4,3-a]pyridines: positive allostericmodulators of the metabotropic glutamate receptor 2. J. Med. Chem 2017, 60, 6704–6720. [DOI] [PubMed] [Google Scholar]
- (56).Doornbos ML; Perez-Benito L; Tresadern G; Mulder-Krieger T; Biesmans I; Trabanco AA; Cid JM; Lavreysen H; AP IJ; Heitman LH Molecular mechanism of positive allosteric modulation of the metabotropic glutamate receptor 2 by JNJ-46281222. Br. J. Pharmacol 2016, 173, 588–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Poutiainen P; Kil KE; Zhang Z; Kuruppu D; Tannous B; Brownell A-L Co-operative binding assay for the characterization of mGlu4 allosteric modulators. Neuropharmacology 2015, 97, 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Kil KE; Poutiainen P; Zhang Z; Zhu A; Choi JK; Jokivarsi K; Brownell A-L Radiosynthesis and evaluation of an 18F-labeled positron emission tomography (PET) radioligand for metabotropic glutamate receptor subtype 4 (mGlu4). J. Med. Chem 2014, 57, 9130–9138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Roth BL Assay protocol book. In Version III ed.; PDSP, N., Ed. Department of Pharmacology, University of North Carolina; at Chapel Hill, 2018. [Google Scholar]
- (60).DiRaddo JO; Miller EJ; Hathaway HA; Grajkowska E; Wroblewska B; Wolfe BB; Liotta DC; Wroblewski JT A real-time method for measuring cAMP production modulated by Gαi/o-coupled metabotropic glutamate receptors. J. Pharmacol. Exp. Ther 2014, 349, 373–382. [DOI] [PubMed] [Google Scholar]
- (61).Jin X; Semenova S; Yang L; Ardecky R; Sheffler DJ; Dahl R; Conn PJ; Cosford NDP; Markou A The mGluR2 positive allosteric modulator BINA decreases cocaine self-administration and cue-induced cocaine-seeking and counteracts cocaine-induced enhancement of brain reward function in rats. Neuropsychopharmacol 2010, 35, 2021–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Minick DJ; Frenz JH; Patrick MA; Brent DA A comprehensive method for determining hydrophobicity constants by reversed-phase high-performance liquid chromatography. J. Med. Chem 1988, 31, 1923–1933. [DOI] [PubMed] [Google Scholar]
- (63).Pike VW PET radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol. Sci 2009, 30, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Banker MJ; Clark TH; Williams JA Development and validation of a 96-well equilibrium dialysis apparatus for measuring plasma protein binding. J. Pharm. Sci 2003, 92, 967–974. [DOI] [PubMed] [Google Scholar]
- (65).Di L; Kerns EH; Hong Y; Chen H Development and application of high throughput plasma stability assay for drug discovery. Int. J. of Pharm 2005, 297, 110–119. [DOI] [PubMed] [Google Scholar]
- (66).Houston JB Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharmacol 1994, 47, 1469–79. [DOI] [PubMed] [Google Scholar]
- (67).Engers DW; Niswender CM; Weaver CD; Jadhav S; Menon UN; Zamorano R; Conn PJ; Lindsley CW; Hopkins CR Synthesis and evaluation of a series of heterobiarylamides that are centrally penetrant metabotropic glutamate receptor 4 (mGluR4) positive allosteric modulators (PAMs). J. Med. Chem 2009, 52, 4115–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Kil K-E; Zhang Z; Jokivarsi K; Gong C; Choi J-K; Kura S; Brownell A-L Radiosynthesis of N-(4-chloro-3-[11C]methoxyphenyl)-2-picolinamide ([11C]ML128) as a PET radiotracer for metabotropic glutamate receptor subtype 4 (mGlu4). Bioorg. Med. Chem 2013, 21, 5955–5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Di L; Kerns EH; Chen H; Petusky SL Development and application of an automated solution stability assay for drug discovery. J. Biomol. Screen 2006, 11, 40–47. [DOI] [PubMed] [Google Scholar]
- (70).Di L; Kerns EH; Fan K; McConnell OJ; Carter GT High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem 2003, 38, 223–232. [DOI] [PubMed] [Google Scholar]
- (71).Promega. Pgp-Glo Assay System, Instructions for use of products V3591 and V3601. In Technical bulletin, Promega: 2015. [Google Scholar]
- (72).Altschul SF; Madden TL; Schaffer AA; Zhang JH; Zhang Z; Miller W; Lipman DJ Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997, 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Kunishima N; Shimada Y; Tsuji Y; Sato T; Yamamoto M; Kumasaka T; Nakanishi S; Jingami H; Morikawa K Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 2000, 407, 971–977. [DOI] [PubMed] [Google Scholar]
- (74).Dobrovetsky E; Khutoreskaya G; Seitova A; Cossar D; Edwards AM; Arrowsmith CH; Bountra C; Weigelt J; Bochkarev A Ligand binding domain of metabotropoc glutamate receptor mGluR5 complexed with glutamate RCSB Protein Data Bank, 2017, DOI: 10.2210/pdb3LMK/pdb [DOI] [Google Scholar]
- (75).Koehl A; Hu HL; Feng D; Sun BF; Zhang Y; Robertson MJ; Chu M; Kobilka TS; Laermans T; Steyaert J; Tarrasch J; Dutta S; Fonseca R; Weis WI; Mathiesen JM; Skiniotis G; Kobilka BK Structural insights into the activation of metabotropic glutamate receptors. Nature 2019, 566, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Hanwell MD; Curtis DE; Lonie DC; Vandermeersch T; Zurek E; Hutchison GR Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.