

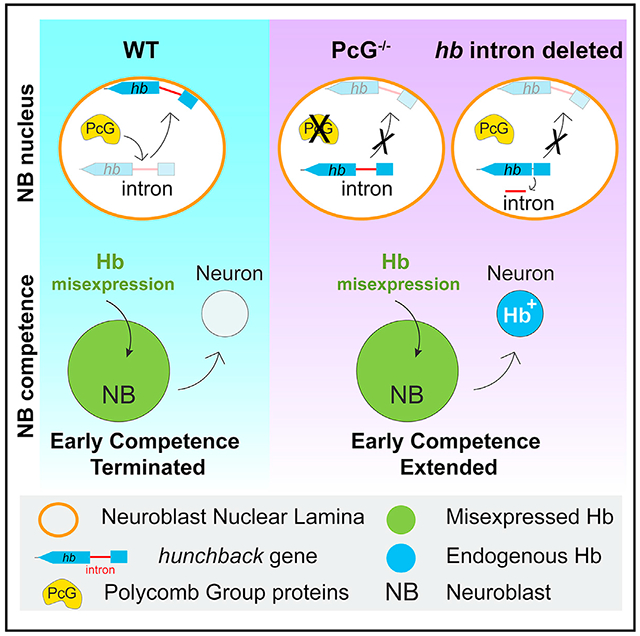
SUMMARY
The nuclear lamina is typically associated with transcriptional silencing, and peripheral relocation of genes highly correlates with repression. However, the DNA sequences and proteins regulating gene-lamina interactions are largely unknown. Exploiting the developmentally timed hunchback gene movement to the lamina in Drosophila neuroblasts, we identified a 250 bp intronic element (IE) both necessary and sufficient for relocation. The IE can target a reporter transgene to the lamina and silence it. Endogenously, however, hunchback is already repressed prior to relocation. Instead, IE-mediated relocation confers a heritably silenced gene state refractory to activation in descendent neurons, which terminates neuroblast competence to specify early-born identity. Surprisingly, we found that the Polycomb group chromatin factors bind the IE and are required for lamina relocation, revealing a nuclear architectural role distinct from their well-known function in transcriptional repression. Together, our results uncover in vivo mechanisms underlying neuroblast competence and lamina association in heritable gene silencing.
Graphical abstract
In brief
In Drosophila neuroblasts, relocation of the hunchback gene locus to the nuclear lamina confers heritable silencing in daughter neurons. Lucas et al. identify a genomic element necessary and sufficient for hunchback gene movement in vivo. Polycomb proteins target this element for lamina relocation, thereby regulating competence, but not hunchback expression.
INTRODUCTION
The metazoan genome is non-randomly distributed in nuclear space, and its three-dimensional organization is considered to play a critical role in gene regulation. High-throughput studies have shown that higher order genome architectures exist across species and are likely important for animal development (Bonev et al., 2017; Lucas and Kohwi, 2019; Van Bortle and Corces, 2013). Though global changes in genome organization have been observed in the context of development, at the smallest scale of individual genes, little is known about how such reorganization translates to gene regulation. This is at least, in part, due to the fact that genome architecture is highly context specific and, thus, requires cell-type and stage-specific analyses in vivo. However, lack of model systems that are amenable to both genetic dissection and genome architecture analyses in intact animals is a major challenge to address this important question in a biologically meaningful manner. To tackle this, we used neural progenitors (neuroblasts) of intact Drosophila embryos and exploited their well-characterized lineages to study mechanisms of gene regulation through genome architecture reorganization in vivo.
The Drosophila embryonic ventral nerve cord (VNC) harbors a bilateral set of 30 neuroblasts in each segment, each of which gives rise to a unique lineage of neural daughters. They undergo multiple rounds of asymmetric divisions that each produce a self-renewed neuroblast and a differentiating ganglion mother cell that divides once again to produce two postmitotic neural cells. As the neuroblast progenitors divide, they sequentially express a series of evolutionarily conserved transcription factors that specify cell fate of the neural progeny descended from each division (Allan and Thor, 2015; Brody and Odenwald, 2000; Doe, 2017; Elliott et al., 2008; Isshiki et al., 2001; Kohwi and Doe, 2013; Mattar et al., 2015; Miyares and Lee, 2019; Rossi et al., 2017; Telley et al., 2019). Hunchback (Hb), the first of such temporal identity factors, specifies the early cohort of neurons, called “early-born” neurons; these postmitotic cells actively maintain hb transcription as a molecular marker of their early temporal birth (Figure 1A). Hb is normally expressed by neuroblasts for one or two divisions and is quickly repressed by a brief, transient pulse of its transcriptional repressor, seven-up (Svp) (Kanai et al., 2005). However, neuroblasts remain competent to specify early-born identity neurons for several additional divisions, indicating that progenitor potential to specify particular cell fates persists longer than the production of that cell type, a phenomenon observed in both insects and mammals (Cleary and Doe, 2006; Desai and McConnell, 2000; Kohwi et al., 2013; Pearson and Doe, 2003). This period of competence, which we refer to as the “early competence window,” is thus a representation of the internal state of the progenitor, reflecting its potential to produce a specific cell type. This internal state can be experimentally observed by transgenically misexpressing Hb in the neuroblast and assessing whether the descendent neuron subsequently activates and sustains hb transcription endogenously, a key molecular characteristic of early-born identity. Note that misexpressed Hb in the neuroblast does not induce hb within the neuroblast itself, nor does misexpression directly in the postmitotic neuron induce hb within the neuron. Only when Hb is supplied to the neuroblast during the early competence window does the postmitotic neuron that descends from that division activate endogenous Hb expression. Thus, the parental neuroblast “primes” the future transcriptional program that will be activated within the neuron, and once the early competence period ends, the neuroblast becomes refractory to Hb and can no longer specify early-born identity (Kohwi et al., 2013; Pearson and Doe, 2003). We previously showed that the early competence window closes when the hb genomic locus relocates to the nuclear lamina, a gene silencing hub (Shevelyov and Nurminsky, 2012; van Steensel and Belmont, 2017), in the neuroblast progenitor (Kohwi et al., 2013). Once this occurs, misexpressed Hb in the neuroblast no longer results in hb expression in the subsequent neuronal progeny. This change in hb gene positioning, which we observe as an increase in the fraction of hb genomic loci peripherally localized in neuroblast nuclei, occurs at mid-embryogenic stage 12, several divisions after hb has been transcriptionally repressed by a transient pulse of Svp (Figure 1B). Lamin loss-of-function mutants result in reduced peripheral localization of the hb gene and a concomitant extension in early competence, indicating a causal relationship between relocation and heritable hb gene silencing (Kohwi et al., 2013). Mechanisms that govern this second type of hb regulation through changes in gene positioning are not known.
Figure 1. Chromatin accessible hb intronic element does not regulate hb transcription.
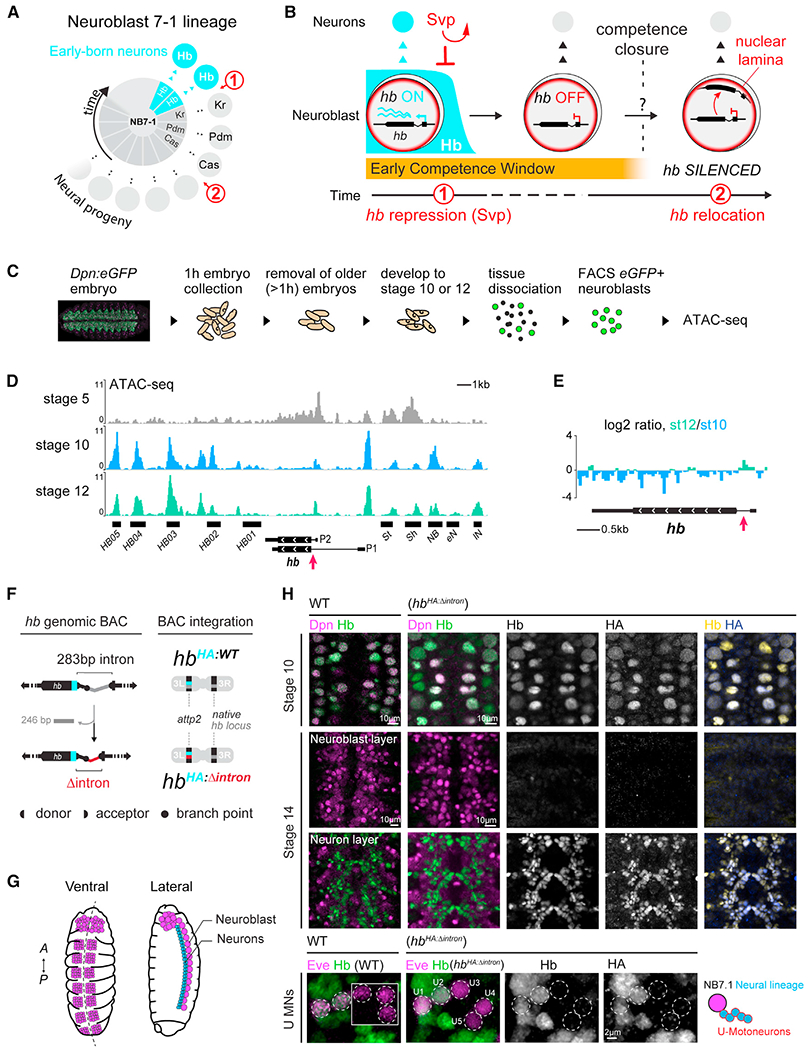
(A) Neuroblast 7–1 (NB7-1, center) and its neural lineage arranged by birth order.
(B) Over time, hunchback (hb) gene in the neuroblast is transcriptionally repressed (1) then relocates to the nuclear lamina (2), closing the early competence window.
(C) Schematic of neuroblast purification.
(D) ATAC-seq profiles of stage 5, 10, and stage 12 neuroblasts (st10 and st12 traces are an average of n = 5 and 4 replicates, respectively) showing accessible chromatin (fragment length < 100 bp). Black horizontal bars indicate known hb transcriptional enhancers/promoters (Hirono et al., 2012; McKay and Lieb, 2013). St, Sh, NB, eN and lN refer to stripe, shadow, neuroblast, early neuron and late neuron enhancer, respectively. P2 and P1 refer to zygotic and maternal promoters. Red arrow, hb intronic element.
(E) Log2 ratio.
(F) Schematic of IE deletion from the 32 kb hb genomic BAC and generation of transgenic animals.
(G) Schematic of ventral and lateral views of the embryonic VNC; neurons (blue), neuroblast (purple).
(H) Ventral views of wild type and HbHA:Δintron embryo VNC immunostained with anti-Hb (all Hb protein), anti-HA (BAC-specific), and anti-Dpn (pan-neuroblast) antibodies. Bottom panels show NB7-1’s U motoneuron (U MNs) Eve+ neural progeny; inset shows neurons from different z planes. Scale bars for st10 and 14 panels, 10 μm; scale bar for U-motoneuron panels, 2 μm. See also Figures S1 and S2.
Here, we used the timed relocation of the endogenous, developmentally regulated hb gene to the neuroblast nuclear lamina in intact Drosophila embryos to investigate mechanisms underlying genome architecture reorganization in vivo. We identify a hitherto undescribed, 250 bp intronic DNA element (IE) necessary and sufficient for gene-lamina relocation. In isolation, the IE is sufficient to target a transcriptionally active reporter gene to the nuclear lamina and silence it. In contrast, removal of the IE from its endogenous locus has no effect on hb’s normal transcriptional dynamics, but instead impairs hb gene relocation and extends progenitor competence to generate hb-expressing neurons. Thus, gene-lamina association in the progenitor can confer a heritably silenced state refractory to activation in the neuronal progeny. Importantly, we identify the evolutionarily conserved Polycomb (PcG) chromatin factors act through this element to control hb gene-lamina interactions. Strikingly, we find that loss of PcG activity impairs hb gene-lamina relocation and extends neuroblast competence without altering transcriptional dynamics, revealing an unexpected role in nuclear architecture. In sum, our results provide mechanistic insights into regulation of gene-lamina associations and progenitor competence transitions in intact animals.
RESULTS
Chromatin profiling identifies a hb intronic DNA element that increases in accessibility upon lamina relocation
The neuroblast early competence window terminates when the key early-born neuron marker gene, hb, relocates to the neuroblasts’ nuclear lamina, which disables it from transcriptional activation in the neural progeny. To generate hypotheses for possible mechanisms underlying this gene repositioning, we examined the chromatin structure at the hb genomic locus in neuroblasts before and after the hb gene relocates to the nuclear lamina. We generated a transgenic fly line (dpn-eGFP) that expresses eGFP driven by an enhancer for the neuroblast-specific gene, deadpan (Dpn) (Awasaki et al., 2014) (Figures 1C and S1A) to purify precisely staged neuroblasts by fluorescence-activated cell sorting (FACS) (Figures S1B and S1C). Neuroblasts were subjected to ATAC-seq to examine their chromatin accessibility profiles (Figures 1D and S1A). We bioinformatically validated the specificity of our data using the available Drosophila embryo single cell ATAC-seq (Cusanovich et al., 2018) and found a strong correlation with the nervous system clusters (Figure S2B). Furthermore, we found that neural genes expressed in neuroblasts, such as worniu and deadpan, were associated with robust chromatin accessibility compared with non-neural genes (Figures S2C and S2D).
The hb gene’s spatiotemporal expression pattern throughout embryonic development is well known. At stage 5, half of the embryo actively transcribes hb, and the hb gene body is highly accessible (Figure 1D), consistent with observations by others (Haines and Eisen, 2018). While hb transcriptional enhancers (Hirono et al., 2012; McKay and Lieb, 2013) remained largely accessible at all stages, when the hb gene relocates to the nuclear lamina at stage 12, we found the gene body largely inaccessible; genes associated with the nuclear lamina are typically characterized by repressive features, including reduced chromatin accessibility (Milon et al., 2014; Pickersgill et al., 2006; Shevelyov and Nurminsky, 2012; van Steensel and Belmont, 2017). This decrease in accessibility is already partially apparent at stage 10, the earliest we are able to purify neuroblasts using the GFP reporter, despite active expression of Hb (Figure 1H); this perhaps indicates a change in chromatin state at the hb gene preceding the neuroblast transition to the Kruppel temporal window. Despite this, we found a region within the hb intron just downstream of the transcriptional start site that had increased in accessibility between stage 10 and 12 (p value = 0.0047) (Figure 1E), counter to a general trend that gene body regions tend to decrease in accessibility upon silencing (Milon et al., 2014).
The hb intron element is not required for proper hb transcription in the VNC
To investigate whether the hb intron element (IE) plays any role in hb transcriptional regulation in neuroblasts and/or hb’s potential to be activated in neurons, we took advantage of a previously characterized transgenic line in which we integrated into the genome a bacterial artificial chromosome (BAC) containing the entire hb genomic locus. This 32 kb BAC includes a hemagglutinin (HA) epitope tag fused to Hb (HbHA:WT), and we showed that HbHA:WT faithfully reconstitutes endogenous Hb expression and function (Kohwi et al., 2013). Using this as a base, we used LoxP-mediated recombination to further selectively remove from the BAC ~250 bp of the intron, corresponding to the ATAC peak, while preserving the splice sites (called “hbHA:Δintron” Figure 1F). Both the wild type and the IE-deleted BAC transgenes were integrated into the same genomic locus using phiC31-mediated integration allowing direct comparison of the endogenously expressed HbHA proteins.
Drosophila embryonic development is highly stereotyped, and gross morphological features unique to each developmental stage allow us to precisely compare Hb expression in stage-matched wild-type and transgenic embryos. We found hbHA:Δintron embryos immunostained with anti-HA antibodies, which selectively detects Hb protein derived only from the hbHA:Δintron transgene, and anti-Hb antibodies, which detects Hb protein from both the native hb locus and the hbHA:Δintron transgene, show the same spatiotemporal pattern as wild-type embryos and embryos with the full-length hb BAC (Figures 1G, 1H, and S3A). In neuroblast 7–1 (NB7-1), one of the 30 neuroblasts that has a well-characterized lineage for which the even-skipped (Eve) transcription factor identifies its progeny, we detected HA and Hb in only the first two neurons, marking the early-born neuron fate, as is well-characterized in wild-type embryos (Brody and Odenwald, 2000; Isshiki et al., 2001; Kohwi et al., 2013). Moreover, consistent with normal Hb expression dynamics in neuroblasts, we did not find extra early-born neurons that could have indicated extended neuroblast Hb expression. Thus, the 250 bp hb IE does not play a role in regulating the normal Hb expression dynamics in the embryonic nerve cord.
The hb IE is required for hb gene relocation to the neuroblast nuclear lamina
We next investigated the consequence of the IE deletion on hb gene positioning. We mapped the physical position of the hb gene in neuroblast nuclei by DNA fluorescence in situ hybridization (FISH) and 3D confocal imaging on whole embryos. Embryos were co-stained with Lamin Dm0, the Lamin B intermediate filament of the nuclear lamina, and Worniu, a pan-neuroblast marker. The hb DNA FISH probe targets a ~10-kb region of the hb genomic locus and can detect the native hb gene as well as the 32 kb BAC. We previously showed that the hb BAC, which contains hb’s transcriptional enhancers and regulatory elements, behaves similarly to the native hb gene, including transcriptional regulation as well as the timing and frequency of gene relocation to the nuclear lamina, which reaches a plateau of ~50% (~60% are <0.4 μm from lamin, “near”) at stage 12 when the early competence window closes (Kohwi et al., 2013). The IE-deleted BAC was inserted in the same integration site, thus allowing a side-by-side comparison of the transgene positioning. In the hbHA:Δintron embryos, there are two endogenous hb gene loci and two Δintron hb loci, and thus, failure of the hbHA:Δintron gene locus to relocate to the neuroblast nuclear lamina would result in a concomitant decrease in the percentage of hb DNA FISH signals at the nuclear lamina at stage 12. Consistent with this prediction, at stage 12, 53.8% ± 2.6% hb loci in HbHA:WT neuroblasts compared with only 35.5% ± 3.5% in hbHA:Δintron neuroblasts were at the lamina (p = 0.002), and 62.9% ± 2.7% of hb loci in HbHA:WT neuroblasts compared with only 45.5% ± 2.2% in hbHA:Δintron neuroblasts were within 0.4 μm of the lamina (p = 0.0009) (Figures 2A–2C). The FISH probe can detect all four hb loci (Kohwi et al., 2013), but the images in the figure show a single z-plane of a representative neuroblast nucleus in which the center, brightest point of the DNA FISH signal can be seen. Results from older neuroblasts at stage 14 were similar: 56.6% ± 7.2% of the hb loci were at the lamina in hbHA:WT neuroblasts compared with only 36.7% ± 5.0% in hbHA:Δintron (p = 0.007), and 65.8% ± 4.5% hb loci were near the lamina in hbHA:WT compared with 50.3% ± 5.3% in hbHA:Δintron neuroblasts (p < 0.01). Together, the data show that while deletion of the IE does not impair normal hb transcriptional dynamics, hb IE is required in the neuroblast for gene-lamina association.
Figure 2. The hb intron is required for gene relocation to the nuclear lamina and termination of the early competence window.
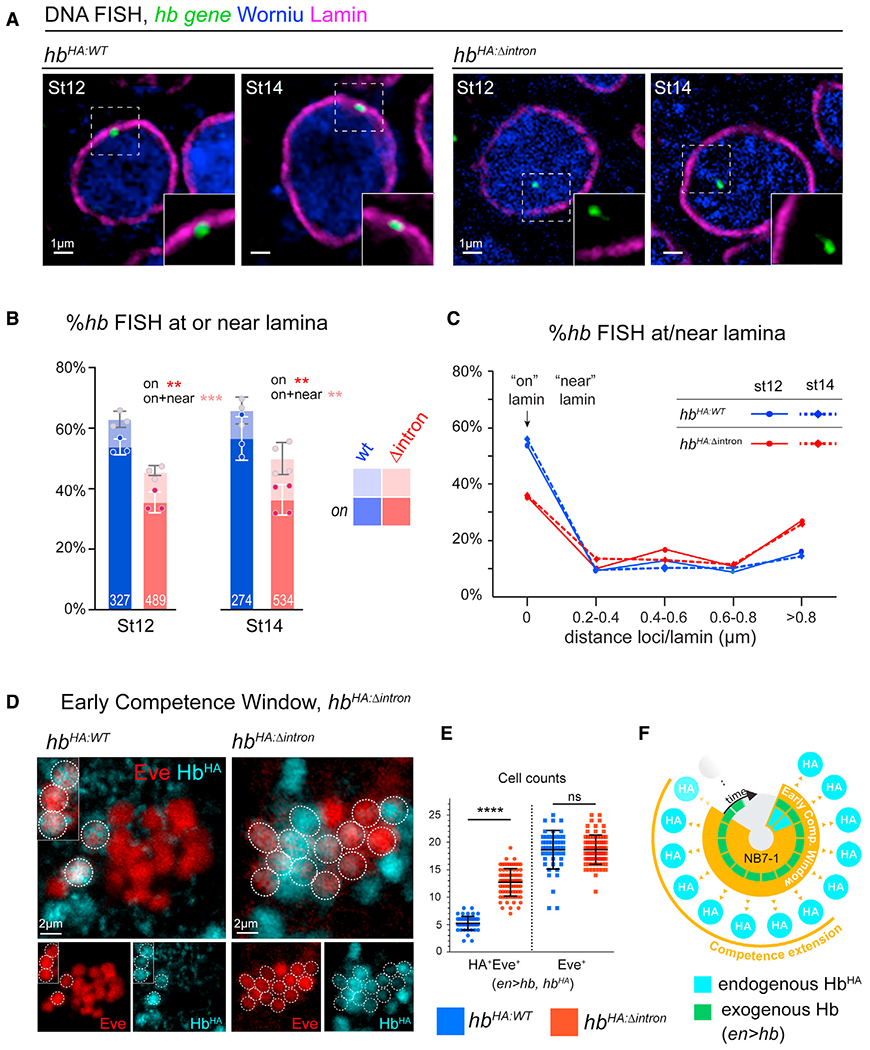
(A) Single z-plane showing DNA FISH signal of representative neuroblast nuclei. Whole stage 12 and 14, HbHA:WT or HbHA:Δintron embryos were hybridized with hb DNA probe and immunostained for Lamin Dm0 and pan-neuroblast marker, Worniu. Scale bars, 1 μm.
(B) Fraction of hb loci at neuroblast nuclear lamina. Each datapoint is from a single embryo; loci quantified shown on each bar.
(C) Spatial distribution of hb loci binned by gene-lamina distances.
(D) NB7-1 lineage early competence assay in HbHA:Δintron vs HbHA:WT (en>GAL4, UAS-hb). Scale bars, 2 μm.
(E) Quantification of early competence window (Eve+HA+) and total lineage length (Eve+). Each data point represents a single NB7-1 lineage quantified from n = 3 wild type embryos and n = 5 HbHA:Δintron embryos.
(F) Schematic summary of early competence window in hbHA:Δintron neuroblasts. All data are represented as mean ± SD. See also Figures S3 and S4. For the above and all subsequent graphs, we applied standard t-tests for statistical analysis. Statistical significance is classified as: * < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001.
The hb IE is required to terminate the neuroblast early competence window
hb gene relocation to the nuclear lamina occurs nearly synchronously across an otherwise heterogeneous group of neuroblasts at mid-embryogenic stage 12, terminating competence to specify early-born molecular identities. To investigate the biological consequence of deleting the IE in competence regulation, we focused on the NB7-1 lineage for which competence has been well-described (Cleary and Doe, 2006; Kohwi et al., 2013; Pearson and Doe, 2003). NB7-1 normally expresses Hb for two divisions and produces two Hb+ U motoneurons (Figure 1A) but can respond to Hb misexpression to produce more (Meng et al., 2019; Seroka and Doe, 2019). However, the period of competence to produce these early-born neurons is limited to the first five divisions on average (Kohwi et al., 2013). For our competence assay, we used the Gal4/UAS system (Brand and Perrimon, 1993) to drive continuous Hb misexpression in the neuroblast via the UAS-hb transgene and quantified early-born neurons molecularly identified by expression of HbHA, a proxy for endogenous hb transcriptional activation (Figures 2D–2F). We used the engrailed (en)-gal4 driver, which drives strong expression in row 6 and 7 neuroblasts, including NB7-1 (Cleary and Doe, 2006; Kohwi et al., 2013; Pearson and Doe, 2003). Compared with hbHA:WT, we found a pronounced increase in the number of HA+ neurons produced in hbHA:Δintron flies (5.2 ± 1.3 neurons in hbHA:WT versus 12.7 ± 2.5 in hbHA:Δintron, p < 0.0001) (Figures 2E and S3B). HbHA:Δintron cannot be induced by misexpression of Hb directly in the postmitotic neurons (Figure S4), just as for HbHA:WT (Kohwi et al., 2013), arguing against the possibility that removing the IE reduces hb repression or lowers the activation threshold in response to Hb misexpression. We conclude that the hb IE functions as a regulatory element for the gene’s relocation to the nuclear lamina, and this topological transition has an important biological consequence on progenitor competence.
The ~250 bp hb IE is sufficient for nuclear lamina relocation and silencing of reporter gene driven by a constitutively active neuroblast enhancer
To analyze how the hb IE functions when taken out of context of the endogenous genomic locus, we generated a neuroblast GFP reporter construct, in which its synthetic intervening sequence (IVS intron) was swapped with the hb IE sequence. We implemented the ParS-ParB system (Saad et al., 2014) to compare the subnuclear positioning of the control ParS-dpn-IVS-mCD4-GFP construct versus one in which the IVS intron was replaced with either one or three tandem copies of the ~250 bp hb IE, ParS-dpn-1x(3x)hbintron-mCD4-GFP. Briefly, a ParS sequence element inserted into the reporter transgene recruits mNeon-fused ParB proteins (Figure 3A), allowing fluorescent visualization of the transgene locus within the nucleus (Figure 3B). Both the control and the IE-swapped constructs were inserted into the identical genomic locus for direct comparison in situ. In the ParS-dpn-IVS-mCD4-GFP control fly line, we found 21.6% ± 2.6% loci at the lamina and 30.7% ± 2.3% loci near (<0.4 μm) the lamina in stage 14 neuroblasts (297 loci quantified from n = 3 embryos), consistent with observations from others that the genome is non-static (Kind et al., 2015), and even non-LADs (lamina associated domains) can be peripherally localized ~30% of the time (Guelen et al., 2008; Harr et al., 2015; Zullo et al., 2012). In contrast to the control, in the ParS-dpn-1xhbintron-mCD4-GFP fly line, we found 46.7% ± 1.5% loci at or 59.4% ± 5.3% near the lamina (304 loci quantified from 3 embryos; p values < 0.0001 and = 0.001, respectively), and in the ParS-dpn-3xhbintron-mCD4-GFP fly line, we found 53.5% ± 2.8% loci at or 62.7% ± 3.2% near the lamina (494 loci quantified from n = 4 embryos; p values < 0.0001 and < 0.0001, respectively). These are significant increases in localization at the nuclear lamina (Figure 3C) compared with the control transgene, despite being driven by a constitutively active neuroblast enhancer. Thus, the hb IE is both necessary and sufficient for gene-lamina relocation in vivo.
Figure 3. The hb intronic element is sufficient to relocate a GFP reporter gene to the neuroblast nuclear lamina.
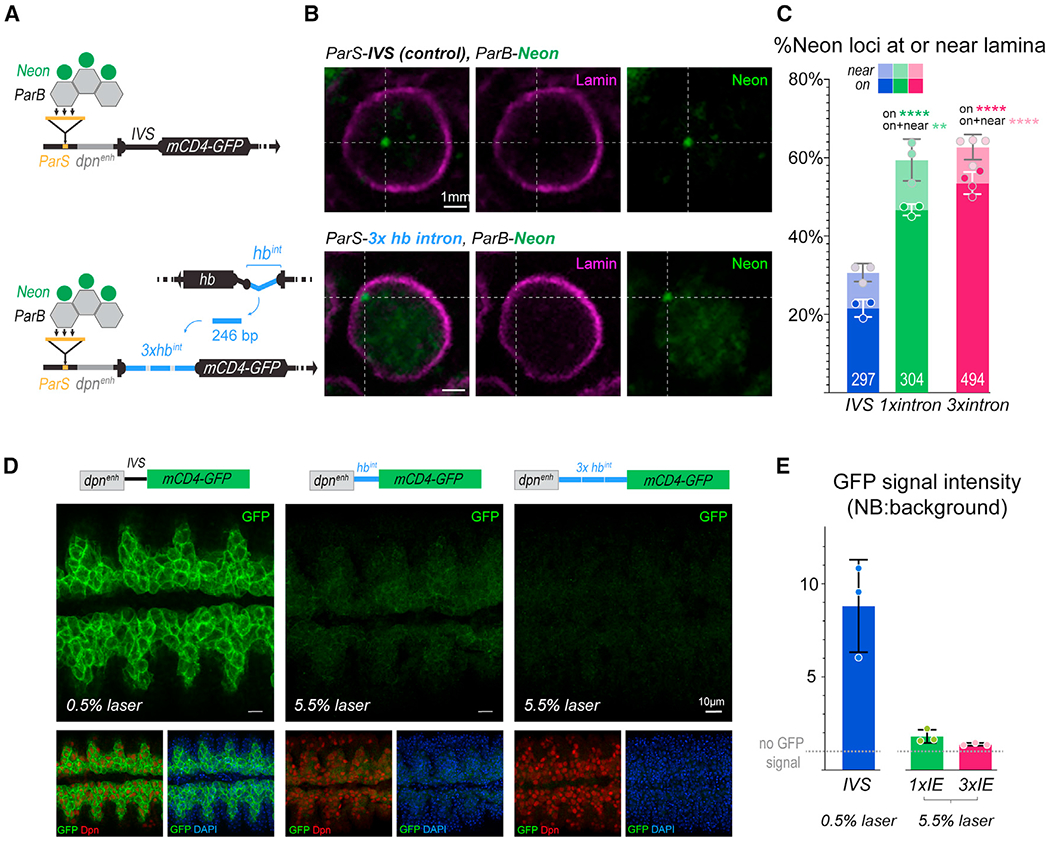
(A) Schematic of genotypes.
(B) In situ detection of reporter transgene in whole, stage 14 embryo neuroblasts. Representative image of single z-plane of neuroblast showing ParB-neon (transgene locus) and lamin from either IVS control (top panels) or hb intron-swapped reporter constructs. Scale bars, 1μm.
(C) Quantification of transgene loci localized at or near the lamina. Each datapoint is from a single embryo; loci quantified shown on each bar.
(D) Ventral views of 50-μm z projections through the neuroblast layer of control, 1× intron-swapped, 3× intron-swapped GFP reporter embryos showing dose-dependent effects of the hb intron element insertion. Embryos are stained with GFP, Dpn (pan-neuroblast), and DAPI. Scale bars, 10μm.
(E) Neuroblast GFP signal quantified as a ratio of neuroblasts to background. Each datapoint represents the average of 50 neuroblasts from one embryo. All data are represented as mean ± SD.
The ParS-dpn-IVS-mCD4-GFP control reporter line expresses GFP in the neuroblasts of the VNC and is detectable from stage 10. However, in the ParS-dpn-3xhbintron-mCD4-GFP embryos, GFP is silenced and never detectable in the neuroblast at any stage, even after allowing time for GFP accumulation in the neuroblast and imaging with ten times the laser power as used for the control reporter line (5.5% versus 0.5%, respectively) (Figures 3D and 3E). In the ParS-dpn-1xhbintron-mCD4-GFP embryo, the GFP signal was also severely reduced, but became visually detectable by stage 14, indicating a dosage effect of the hb intron element replacement and the sufficiency for even one copy of the hb intron element to nearly fully silence an otherwise transcriptionally active GFP transgene (Figure 3E). The silencing of the transgene with only ~60% localization at the lamina likely reflects the mobility of the genome. Indeed, live-imaging studies have shown confined diffusion of LADs (Bronshtein et al., 2016; Qin et al., 2017). Such dynamics cannot be fully captured in fixed embryo preparations, in which we observe only a snapshot of the genome at a time and likely underestimate the fraction of gene loci functionally associated with the lamina. As the peripheral localization and transcriptional silencing are coincident, it is difficult to determine whether the reporter transgene silencing was the cause or consequence of relocation to the lamina. However, our observations are consistent with others’ that relocation to the lamina can silence transcriptionally active reporter genes (Finlan et al., 2008; Reddy et al., 2008). Importantly, in the biological context of the endogenous hb gene, in which transcriptional repression occurs significantly prior to lamina relocation (several divisions in NB7-1), removal of the IE blocks lamina relocation without disrupting transcriptional dynamics. Thus, the data show that lamina association can serve a function different from transcriptional repression, to confer a heritable, refractory gene state that blocks activation in descendent neurons born after mitotic divisions.
The Polycomb (PcG) chromatin factors target the hb IE
Though subnuclear repositioning of genes is an important component of gene regulation and has been observed across multiple animal models (Kohwi et al., 2013; Meister et al., 2010; Peric-Hupkes et al., 2010; Williams et al., 2006), proteins that recognize specific gene elements to facilitate relocation are not known. Given the increase in chromatin accessibility of the hb intronic element at the stage when the gene relocates to the neuroblast nuclear periphery for silencing, we hypothesized that the IE may be a binding target for a protein(s) mediating gene relocation. We identified two juxtaposing putative binding motifs for Pho, the PcG subunit with a sequence-specific DNA-binding domain (Brown et al., 2018; Oktaba et al., 2008) (Figure 4A) coinciding with the dip in reads at the ATAC-seq peak, a typical profile for a chromatin accessible site in which bound proteins interfere with Tn5 transposition (Bentsen et al., 2020; Buenrostro et al., 2013; Hesselberth et al., 2009). PcG proteins are chromatin factors that are well known for their important role in the epigenetic regulation of developmental genes through the cooperative activity of two multimeric protein complexes, Polycomb repressive complex 1 (PRC1) and PRC2, which are recruited to chromatin through different paths (Beuchle et al., 2001; Grossniklaus and Paro, 2014; Nègre et al., 2006; Papp and Müller, 2006; Schuettengruber and Cavalli, 2009; Schwartz et al., 2006; Struhl, 1981; Tolhuis et al., 2006). By immunostaining for Posterior sex combs (Psc) and Enhancer of Zeste [E(z)], the core subunits of PRC1 and PRC2, respectively, we found that both are enriched in embryonic neuroblasts (Figure 4B). Taking advantage of available Drosophila ChIP-seq data sets from larval brain and whole embryo (Bonnet et al., 2019; Brown et al., 2018), we identified strong ChIP enrichment of both PRC1 and PRC2 subunits across the hb genomic region of ~25 kb, with robust binding of multiple subunits directly aligning at the hb intron site (Figure 4C). Interestingly, putative Hb target genes in the CNS (Sen et al., 2019) are significantly enriched for chromatin accessible Pho sites like the hb intron, compared with non-Hb targets across the genome, (Figure S5). Although hb is the only known universal marker of early-born neurons, which consist of diverse cell types, perhaps genes associated with early-born identity similarly become refractory to activation in neurons at the end of the neuroblast’s early competence window.
Figure 4. Chromatin profiling reveals highly accessible PcG-binding site in hb intron.
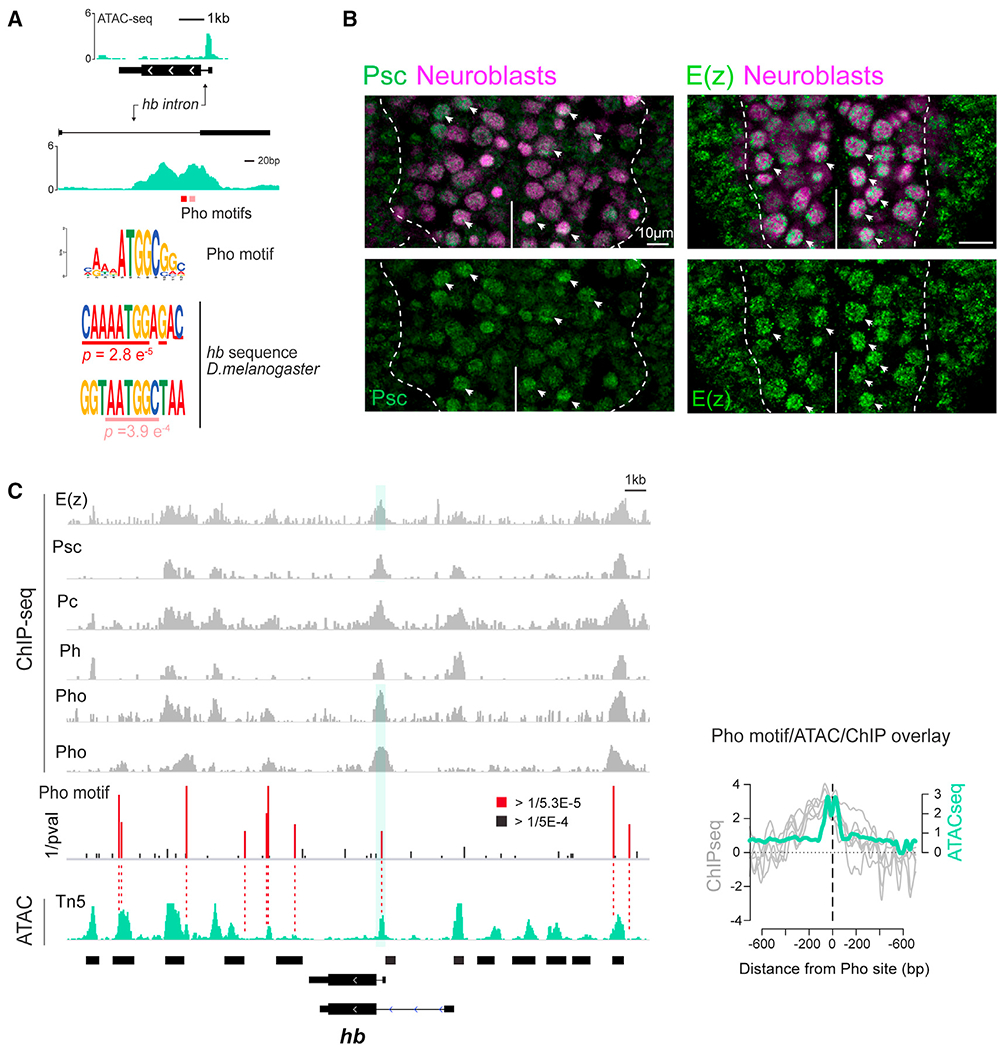
(A) ATAC-seq signal (green) at the hb locus shown at two scales. Two putative Pho binding motifs indicated in red; respective position weight matrix within hb intron ATAC peak are shown below.
(B) VNC of stage 12 embryos showing Psc or E(z) (green), pan-neuroblast markers, Dpn, and Wor (magenta). Scale bars, 10 μm.
(C) ChIP-seq profiles of PRC1 and PRC2 subunits across ~25 kb hb genomic region (gray) aligned with stage 12 neuroblast-specific ATAC-seq data (green). ChIP-seq profiles (Brown et al., 2018); last Pho ChIP-seq profile (Bonnet et al., 2019). Red/black bars, statistically significant putative Pho binding motifs (red bars, inverse of p value < Q1 ± 1.5 × IQR). Normalized ChIP (log2 ChIP/input ratio, gray lines) peaks directly align with the ATAC signal (RPM normalized, green line) peak at the Pho motif site (center dashed line). See also Figure S5.
PRC1 is required for hb gene relocation to the neuroblast nuclear lamina, but not for hb repression
To test whether PcG factors are required for hb gene-lamina relocation, we examined neuroblast hb gene positioning in Psc loss-of-function mutants. Psc is central to PRC1 repressive activity and plays a primary role in gene silencing (King et al., 2005). We found that Psch27 null neuroblasts showed a significant reduction in hb loci at the nuclear lamina (stage 12: 53.4% ± 4.3% in wild type versus 26.7% ± 5.7% in Psc−/−, p = 0.001; stage 14: 57.1% ± 7.9% in wild type versus 32.2% ± 5.5% in Psc−/−, p = 0.01) (Figure 5A). Thus, PRC1 is required for hb gene-lamina association.
Figure 5. PRC1 is required for hb relocation to nuclear lamina in neuroblasts, but not in hb repression.
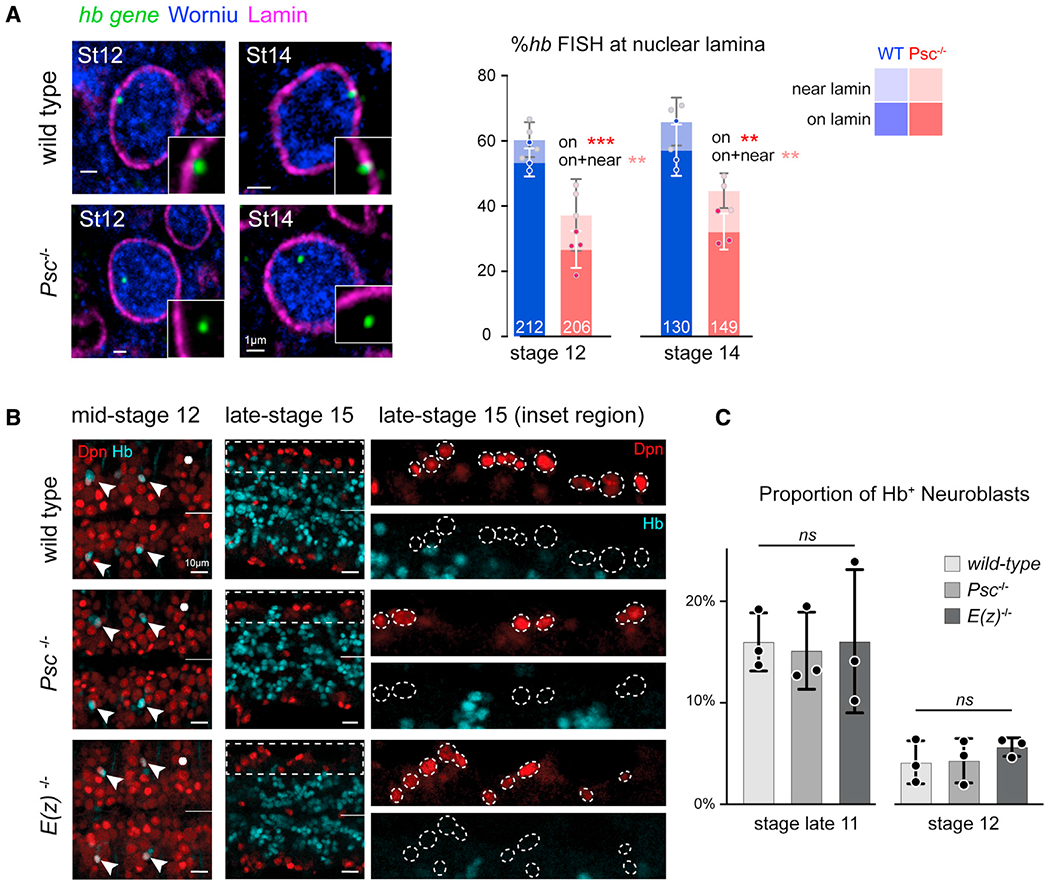
(A) Left: single z-plane of DNA FISH image of representative neuroblast nuclei from intact stage 12 and 14, WT, or Psc−/− embryos. Right: fraction of hb loci at or near neuroblast nuclear lamina. Each datapoint is from a single embryo; loci quantified shown on each bar. Scale bars, 1 μm.
(B) Ventral view of stage 12 and 15 VNC from WT, Psc−/−, and E(z)−/− embryos immunostained for Hb and Dpn. At late stage 12, Hb+ neuroblasts detected only in thoracic segments (arrows); no Hb+ neuroblasts detected in abdominal segments (*) (midline, solid lines). Scale bars, 10 μm.
(C) Proportion of Hb+ neuroblasts in Psc−/− or E(z)−/− embryos. Each datapoint is from a single embryo. All data are represented as mean ± SD; ns, not significant. See also Figure S6.
PcG proteins are critical players in stabilizing cell type-specific gene expression programs by maintaining gene repression. Given that actively transcribed genes are often localized away from the repressive nuclear periphery (Meister et al., 2010; Peric-Hupkes and van Steensel, 2010), one possible explanation for reduced hb gene-lamina association could be that hb becomes de-repressed in Psc mutant neuroblasts. Surprisingly, we found no differences in Hb spatiotemporal expression in Psce24 or E(z)731 loss-of-function mutant neuroblasts; Hb was expressed in newly formed neuroblasts and rapidly downregulated with the same timing as in wild type (Figures 5B and 5C). Importantly, we did not observe any Hb de-repression in neuroblasts of older mutant embryos at stage 15.
To address the possibility of maternal effects that could mask zygotic mutant phenotypes, we examined a maternal and zygotic loss-of-function mutants for extra sex combs (escmat-zyg-) (Struhl, 1981; Tie et al., 1998), a component of PRC2 essential for E(z) methyltransferase activity (Bonnet et al., 2019). Though escmat-zyg- mutants show strong de-repression of the PcG Hox target, Abdominal B, we did not find Hb de-repression even in late-stage neuroblasts (Figures S6A and S6B). We also examined a zygotic double mutant for Psc and Su(z)2 (PscP3C), which has a severe PRC1 loss-of-function phenotype (Classen et al., 2009). Similar to escmat-zyg- mutants, PscP3C also showed Abdominal B de-repression across the animal by mid-embryogenesis but no de-repression of Hb in late-stage mutant neuroblasts. Further supporting the lack of PcG-mediated repression of hb in neuroblasts, in wild-type embryos, we find markedly low levels of histone H3 Lysine 27 trimethylation (H3K27me3), the primary histone modification associated with PcG- mediated gene silencing, in neuroblasts compared with neighboring cells (Figure S6C). Histone H3 as well as both PRC1 and PRC2 components, however, are readily detectable in neuroblasts (Figure S6C and 4B), and loss of PcG results in a reduction in hb relocation to the nuclear lamina and an extension in early competence. Together, the data show that in neuroblasts, PcG activity is required for hb gene-lamina relocation, but not hb repression.
PcG chromatin factors are required to terminate the early competence window
Given the effects of PcG loss on hb gene positioning, we next tested PcG proteins in neuroblast competence using our competence assay on NB7-1 (Figures 6, S7A, and S7B). We found an extension in the early competence window in Psch27 null mutants (4.5 ± 2.3 HbHA+ neurons in wild type compared with 7.2 ± 2.0 in Psch27, p < 0.0001), and phenotypes were consistent with a different Psc null allele (Psce24, 8.6 ± 2.5 HbHA+ neurons, p < 0.0001). Similarly, we found that the early competence window was extended in E(z) mutants. Compared with wild type (5.3 ± 1.2 HbHA+ neurons), E(z)731 mutants had an extended early competence window (8.7 ± 2.7 HbHA+ neurons in E(z)−/−, p < 0.0001). E(z) heterozygous embryos had a small, but statistically significant increase in HbHA+ neurons as well (6.3 ± 1.6 in E(z)+/−, p = 0.0006). Together, we show that either loss of PcG or deletion of the 250 bp IE containing the PcG-binding site disrupts hb gene relocation to the neuroblast nuclear lamina and a failure in the timely closure of the early competence window. However, in both cases, normal hb transcriptional dynamics are not compromised, indicating that gene-lamina interactions and transcriptional regulation can be controlled separately, and that in neuroblasts, PcG can play a unique role in nuclear architecture by regulating gene-lamina association (Figure 7).
Figure 6. PcG chromatin factors are required to terminate the early competence window.
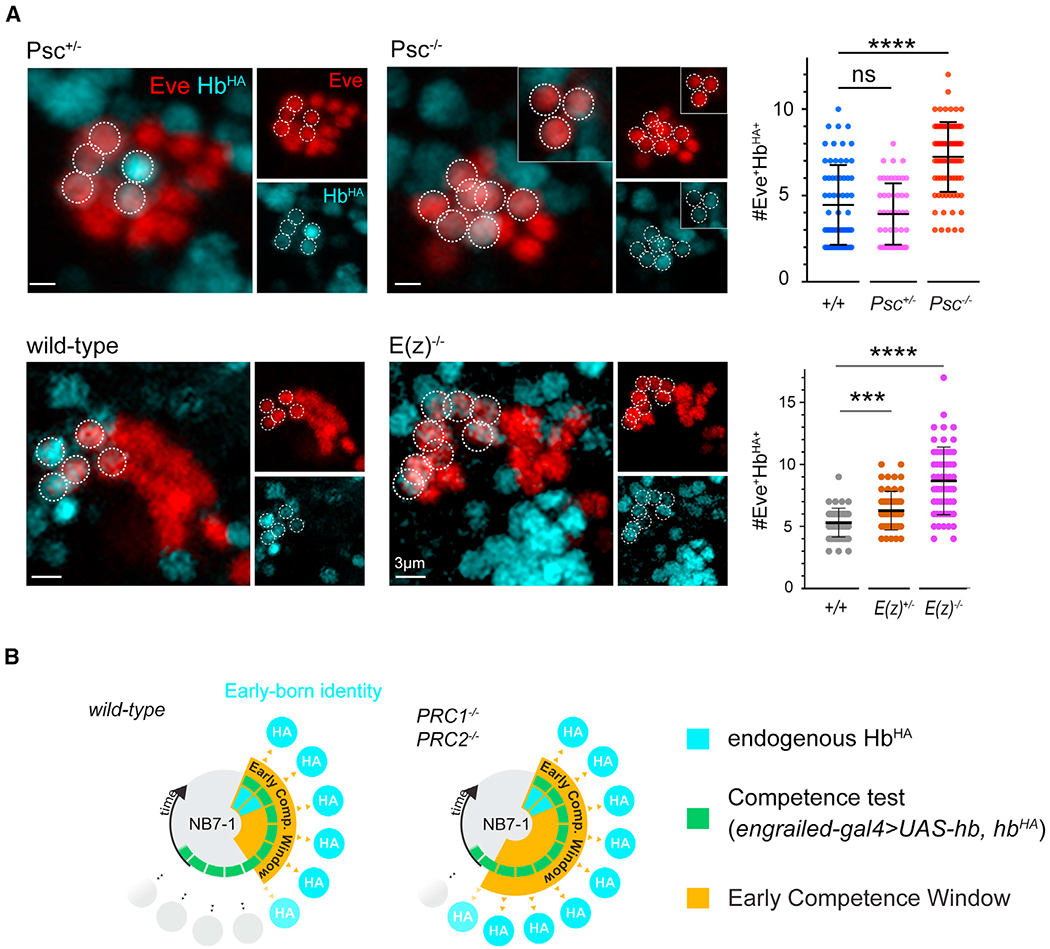
(A) Early competence assay of Psch27 and E(z)731 embryos; representative images from control and mutant genotypes shown. Dotted circles indicate early-born neurons of NB7-1 lineage. Insets show neural progeny from overlapping z planes. Quantification shown at the right. Each datapoint represents a single NB7-1 lineage from n = 3–4 embryos; bars show mean ± SD.
(B) Schematic summary of results. See also Figure S7.
Figure 7. Summary model.
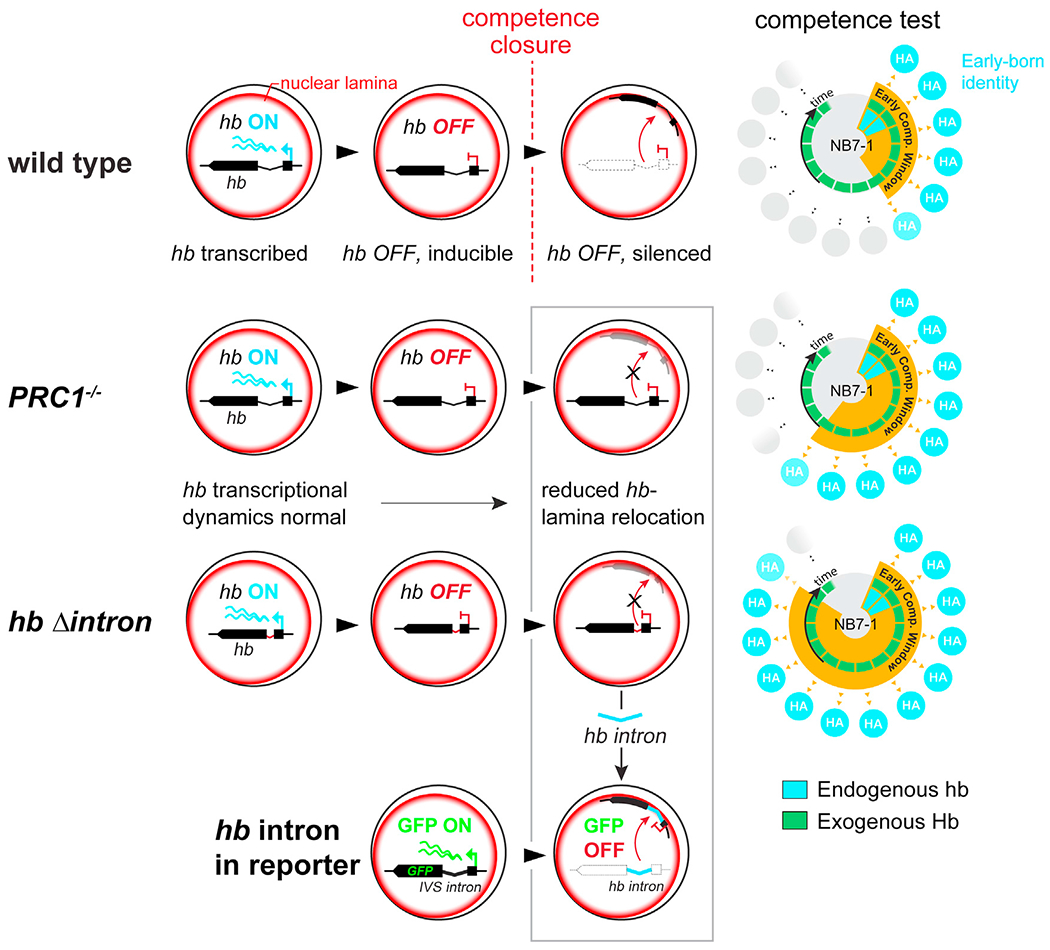
cis-acting, 250 bp intronic element regulates hb gene relocation to the neuroblast nuclear lamina in mid-embryonic neuroblasts. The intronic element is bound by PcG chromatin factors which are required for hb gene relocation. Schematic to the right shows the early competence window for each genotype. Bottom, the hb intron element is sufficient to relocate a GFP reporter construct to the neuroblast nuclear lamina and silence it. Gray box highlights the gene positioning phenotypes as a result of various genetic manipulations.
DISCUSSION
While changes in nuclear architecture has increasingly become a major focus of understanding gene regulation during development, how genome reorganization is controlled and how those changes influence subsequent cell fate decisions in vivo are not known. To address this challenging question, we took advantage of the Drosophila embryo neuroblast model system in which we had previously documented the developmentally timed relocation of the hb gene to the nuclear lamina in vivo. This relocation, a critical step in terminating neuroblast competence to generate neurons of early-born identity, indicates that the underlying genome architecture of the progenitor determines the transcriptional identity of the descendent neuron. Here, we identified a hitherto uncharacterized core 250 bp genomic element within the hb intronic DNA sequence that is required for this topological relocation, without affecting the gene’s normal transcriptional dynamics within the neuroblast. Upon deletion of this element, the hb gene fails to relocate to the lamina, extending neuroblast competence to produce hb-expressing neurons. Thus, the IE does not directly regulate hb transcription per se. Rather, the potential for hb expression in neurons, and consequently neuronal identity, is established by the subnuclear hb gene positioning within the progenitor, and this topological organization is regulated by the IE.
The hb IE in isolation, however, is sufficient to reposition a reporter gene driven by a constitutively active enhancer to the nuclear lamina and can repress it in vivo. Thus, by examining its function in both an artificial reporter context as well as in its endogenous context of a developmentally regulated gene in intact animals, we demonstrate that this genomic element is responsible for gene-lamina association. However, the biological consequence of the presence of this element, whether it leads to transcriptional repression or a heritably refractory gene state, depends on the specific genomic context. The fact that hb gene-lamina association in the progenitor affects the hb gene state in the neuron indicates that changes in nuclear architecture can have functional consequences in cells born after multiple mitotic divisions.
Identification of the hb intron as a core regulatory element led to our findings that PcG chromatin factors are key regulators of gene-lamina interactions. In both Drosophila and mammalian cells, PcG proteins have been shown to engage in long-range interactions to facilitate repression (Bantignies et al., 2011; Bonev et al., 2017; Eagen et al., 2017; Gentile et al., 2019; Gozalo et al., 2020; Kundu et al., 2017; Tolhuis et al., 2011), and loss of PRC1 was shown to lead to Hox gene decompaction prior to de-repression (Cheutin and Cavalli, 2018). Thus, PcG function in higher order genome architecture has been intimately tied to its role as a developmental repressor. Whether PcG proteins have a repression-independent function in regulating genome architecture is an important and emerging question with very few in vivo studies. In murine embryonic stem cells (ESCs), loss of PRC2- or PRC1-mediated chromatin interactions is not necessarily accompanied by gene activation (Boyle et al., 2020; Cruz-Molina et al., 2017), suggesting that PcG factors might regulate a subset of target genes at the level of higher order chromatin architecture without affecting their repressive state. However, determining whether such regulation is biologically meaningful requires careful validation in tissue context. We find that in neuroblasts neither loss of PRC2 or PRC1 results in de-repression of Hb in neuroblasts once its normal expression window ends, even upon loss of both maternal and zygotic esc, the most severe genetic loss of PRC2 function that causes genome-wide loss of H3K27me3 deposition (Bonnet et al., 2019). This is further supported by our observations that neuroblasts have markedly lower H3K27me3 signals compared with their neighboring cells. Thus, while maintenance of hb repression once it is silenced in neuroblasts does not appear to rely on PcG proteins or H3K27me3 chromatin modifications, PcG activity is essential for hb gene-lamina association.
Furthermore, in the context of the developing animal, we found that the PcG-mediated gene-lamina association of the hb gene plays an essential role in regulating neuroblast competence to specify early-born fate neurons. Investigating mechanisms controlling stem/progenitor competence is crucial to understanding how specific cell fates are established in an organized, coordinated manner within the developing brain. PcG function has been implicated in stem cell competence in many animal systems, but mechanisms remain largely unknown. Upon removal of PRC2, germ cells in C. elegans become competent to respond to terminal selector transcription factors and directly convert to specific neuronal cell types (Patel et al., 2012). Loss of PcG activity has been shown to extend Drosophila neuroblast competence to respond to the temporal identity factor Kruppel (Touma et al., 2012). During the neurogenesis-to-gliogenesis transition in mouse, loss of a PRC1 subunit, Ring1B, leads to an increase in progenitor competence to respond to Wnt signaling to induce neurogenesis (Hirabayashi et al., 2009). In murine ESCs, loss of PRC2 reduces competence to activate anterior neural genes in response to differentiation (Cruz-Molina et al., 2017). Thus, PcG protein function in neural competence is likely highly conserved, underscoring the importance of understanding the mechanisms of their function in vivo.
In summary, our results provide important mechanistic insights into nuclear architecture reorganization in vivo by identifying a small genomic element responsible for gene-lamina association of a developmentally regulated gene. We identified PcG chromatin factors as key regulators that associate with this gene element for lamina relocation without affecting the gene’s transcriptional dynamics, demonstrating a non-canonical role in nuclear architecture. Importantly, we provide functional evidence that topology-specific gene regulation plays a critical role in vivo, to control neuroblast early competence. Competence transitions are an important developmental feature of neural progenitors that not only determine the timeframe when each neural cell type can be generated during development but also provide the heritable blueprint upon which the transcriptional program of the descendent neurons is executed. Future work will elucidate whether similar processes are employed during mammalian neurogenesis and how such mechanisms have contributed to the evolution of neural diversity.
Limitations of the study
While our results show that the IE and PcG proteins are required for hb gene relocation to the neuroblast nuclear lamina to confer a heritably silenced gene state, there may be additional factors that play a role in establishing and/or maintaining hb association with the nuclear periphery. Deletion of the IE results in a greater extension in early competence compared with loss of Psc or E(z), which could be due to compensatory function between PcG subunits or yet unknown factors that target the IE. Furthermore, we do not have the full picture of PcG function within embryonic neuroblasts. For the hb gene, we have shown that PcG function is required for its relocation to the nuclear lamina and not for its repression. Further studies will be required to determine whether PcG in neuroblasts regulates gene-lamina interactions across the genome and the extent to which such interactions are also decoupled from the repressive functions for which PcG proteins are well known. PcG has been associated with maintenance of gene repression through catalyzing H3K27me3 histone marks, and these marks have also been proposed to prevent spreading of repression beyond LAD borders (Harr et al., 2015; Guelen et al., 2008). However, given the dearth in H3K27me3 signals in neuroblasts compared with other cell types and the lack of repressive effects that loss of E(z) and Esc has on hb, it would be fascinating and important to map H3K27me3 deposition in neuroblasts and determine the functional relationship between PcG proteins and H3K27me3 in the context of nuclear architecture.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Minoree Kohwi (mk3632@columbia.edu).
Materials availability
Drosophila stocks and other reagents from in this study are available upon request from the Lead Contact, Minoree Kohwi, or from public/commercial sources that are listed in Key Resources.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken polyclonal anti-HA | Bethyl Laboratories | Cat#A190-106A; RRID: AB_66692 |
| Chicken polyclonal anti-GFP | Aves Labs | Cat#GFP-1010; RRID: AB_2307313 |
| Rat monoclonal anti-HA (3F10) | Sigma-Aldrich | Cat#11867423001; RRID: AB_390918 |
| Rat monoclonal anti-Deadpan (11D1CH11) | Abcam | Cat#ab195172 |
| Rat monoclonal anti-Worniu (5A3AD2) | Abcam | Cat#ab196362 |
| Rabbit polyclonal anti-Hunchback | Kind gift from Dr. Chris Doe, University of Oregon | N/A |
| Mouse monoclonal anti-Eve (3C10) | Developmental Studies Hybridoma Bank (DSHB) | Cat#3C10-c; RRID: AB_528229 |
| Mouse monoclonal anti-Elav (9F8A9) | DSHB | Cat#9F8A9-s; RRID: AB_2314364 |
| Rabbit polyclonal anti-Lamin R-836 | Kind gift from Dr. Paul Fischer,Stony Brook University | N/A |
| Mouse monoclonal anti-Psc (6E8) | DSHB | Cat#6E8-s; RRID: AB_528437 |
| Rabbit polyclonal anti-E(z) | Kind gift from Dr. Jürg Müller, Max-Planck Institute of Biochemistry | N/A |
| Mouse monoclonal anti-Abd-B (1A2E9) | DSHB | Cat#1A2E9; RRID: AB_528061 |
| Rabbit polyclonal anti-H3K27me3 | Sigma-Aldrich | Cat#07-449; RRID: AB_310624 |
| Mouse anti-mNeonGreen | Antibodies-online | Cat#ABIN5608502 |
| Rabbit polyclonal anti-Histone H3 | Abcam | Cat#ab5176; RRID: AB_304763 |
| Bacterial and Virus Strains | ||
| SW102 E. coli containing 32kb-HA-hb BAC | Kohwi et al. 2013 | N/A |
| TransforMax EPI300 Electrocompetent E. coli | Lucigen | EC300110 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Halocarbon oil 27 | Sigma-Aldrich | H8773; Cas: 9002-83-9 |
| Fetal Bovine Serum | Sigma-Aldrich | 12306C |
| Calcein Violet 450AM Viability Dye | Thermo Fisher Scientific | 65-0854-39 |
| 7-AAD Viability Staining Solution | Thermo Fisher Scientific | 00-6993-50 |
| SYBR® Green I Nucleic Acid Gel Stain | Thermo Fisher Scientific | S7563 |
| DAPI | Thermo Fisher Scientific | D3571 |
| AMPure XP beads | Beckman Coulter | A63880 |
| TDE1 (Tagment DNA Enzyme, Tn5) | Illumina | 15027865 |
| NEBNext High-Fidelity 2x PCR MM | NEB | m0541s |
| Normal Goat Serum | Jackson ImmunoResearch | 005-000-121 |
| Aqua-Poly/Mount | Polysciences | 18606 |
| Chan and Gehring’s Balanced Saline (C&G Medium) | Chan and Gehring, 1971 | N/A |
| Fisherbrand Sterile Cell Strainers (40μm) | Fisher Scientific | 22363547 |
| Bel-Art SP Scienceware Flowmi Cell Strainers (40μm) | Fisher Scientific | 14100150 |
| DWK Life Sciences Wheaton Dounce Tissue Grinders | Fisher Scientific | 357542 |
| Critical Commercial Assays | ||
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | 20024906 |
| PhiX CONTROL V3 KIT | Illumina | FC-110-3001 |
| High Sensitivity DNA Kit | Agilent | 5067-4626 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Q32851 |
| DNA Clean & Concentrator-5 | Zymo Research | D4013 |
| FISH Tag DNA Orange Kit, with Alexa Fluor 555 dye | Thermo Fisher Scientific | F32948 |
| HiSpeed Plasmid Midi Kit | Qiagen | 12643 |
| Deposited Data | ||
| Stages 5, 10, and 12_sortedNB_ATACseq | This study | NCBI SRA: PRJNA754600 |
| Experimental Models: Organisms/Strains | ||
| D. melanogaster: Psch27 : Psc[h27]/CyO | Bloomington Drosophila Stock Center (BDSC) | Cat#5547; RRID:BDSC_5547 |
| D. melanogaster: Psc[e24]: y[1] w[*]; P{ry [+t7.2]=neoFRT}42D Psc[e24]/SM6b, P{ry [+t7.2]=eve-lacZ8.0}SB1 | BDSC | Cat#24155; RRID:BDSC_24155 |
| D. melanogaster: E(z)[731]: w[*]; E(z)[731] P{1xFRT.G}2A/TM6C, Sb[1] Tb[1] | BDSC | Cat#24470; RRID:BDSC_24470 |
| D. melanogaster: inscutable-gal4: w[*];P{w [+mW.hs]=GawB}insc[Mz1407] | BDSC | Cat#8751; RRID:BDSC_8751 |
| D. melanogaster:CQ2-GAL4: y[1] w[*]; P{w [+mC]=CQ2-GAL4}O | BDSC | Cat#7466; RRID:BDSC_7466 |
| D. melanogaster: en-GAL4: w[1118]; P{y [+t7.7] w[+mC]=GMR60F10-GAL4}attP2 | BDSC | Cat#46438; RRID:BDSC_46438 |
| D. melanogaster: UAS-hb | Wimmer et al., 2000 | N/A |
| D. melanogaster: Psc-Su(z)2P3C (P3C) | Classen et al., 2009 | N/A |
| D. melanogaster: esc[6]/CyO | Struhl, 1981 | N/A |
| D. melanogaster: esc[5]/CyO | Struhl, 1981 | N/A |
| D. melanogaster: HbHA:WT | Kohwi et al., 2013 | N/A |
| D. melanogaster: Dpn-eGFP | This paper | N/A |
| D. melanogaster: Hb[HA:Δintron] | This paper | N/A |
| D. melanogaster: w[1118] | BDSC | Cat#3605; RRID:BDSC_3605 |
| D. melanogaster: PBac{UAS-CD4-tdGFP} | BDSC | Cat#35836; RRID: BDSC_35836 |
| Oligonucleotides | ||
| Primers for the Hb DNA FISH probe, see Table S1 | Kohwi et al., 2013 | N/A |
| Recombinant DNA | ||
| NLS pEGFP-N3(1299) | Addgene | Cat#62043; RRID: Addgene_62043 |
| Dpn-Gal80 | Kind gift from Dr. Chris Doe, University of Oregon | N/A |
| Dpn-NLSpEGFP-N3 | This paper | N/A |
| Hb[HA:WT] BAC | Kohwi et al., (2013) | N/A |
| Hb[HA:Δintron] BAC | This paper | N/A |
| PL-452 N-EGFP | AddGene | Cat#19173; RRID: Addgene_19173 |
| pJFRC81 | AddGene | Cat#36432; RRID: Addgene_36432 |
| pBPGUw-attB | AddGene | Cat#17575; RRID: Addgene_17575 |
| Software and Algorithms | ||
| bcl2fastq conversion software | Illumina | N/A |
| FASTQC | S. Andrews | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Cutadapt | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| bowtie2 | Langmead and Salzberg, 2012 | https://github.com/BenLangmead/bowtie2 |
| Picard Toolkit | Broad Institute | http://broadinstitute.github.io/picard |
| samtools | Li et al., 2009 | http://www.htslib.org |
| BEDTools | Quinlan and Hall, 2010 | https://github.com/arq5x/bedtools2 |
| BedGraphToBigWig | Kent et al., 2010 | http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/bedGraphToBigWig |
| MACS2 | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| deepTools2 | Ramírez et al., 2016 | https://github.com/deeptools/deepTools |
| DiffBind | Ross-Innes et al., 2012 | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| Fiji | Schindelin et al., 2012 | https://fiji.sc |
Data and code availability
The ATAC sequence data reported in this paper have been deposited to the NCBI Sequence Read Archive with the accession number SRA:PRJNA754600 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA754600). Any additional information required to reanalyze the data reported in this manuscript will available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Fly lines
Wild-type (w1118), Psch27 (Bloomington, BDSC#5547), Psce24 (BDSC#24155), E(z)731 (BDSC#24470), 1407-GAL4 (BDSC#8751), CQ2-GAL4 (BDSC#7466), en-GAL4 (chrom III, BDSC#46438), en-GAL4 (chrom II, (Harrison et al., 1995; Isshiki et al., 2001), UAS-hb (Wimmer et al., 2000), esc6 and esc5 (Struhl, 1981; Tie et al., 1998), Psc-Su(z)2P3C (P3C) (Classen et al., 2009), hbHA:WT (Kohwi et al., 2013). Flies were raised on a standard cornmeal/molasses medium at 25C.
METHOD DETAILS
Plasmids and BAC
dpn-eGFP was generated by modifying a dpn-GAL80 construct (kind gift from Chris Doe) to replace GAL80 with eGFP (Addgene #1299) and was inserted into attP2 and attP40 integration sites. hbHA:Δintron was generated by using recombineering (Venken et al., 2009) to remove a portion of the hb intron from a 32kb BAC containing the hb gene locus with an N-terminal HA epitope tag (Kohwi et al., 2013). ParS-dpn-1xhbintron-mCD4-tdGFP and ParS-dpn-3xhbintron-mCD4-tdGFP were generated by replacing the 10XUAS and GFP of the pJFRC81-10XUAS-IVS-Syn21-GFP-p10 (pJFRC81) (Addgene #36432), with dpn (from the dpn-gal80 construct, above) and mCD4 (BL #35836), and then subsequently replacing the IVS intron with either a single copy, or three tandem copies of the 250bp element of the hb intron. The ParS sequence was cloned from pFG4-INT2. The dpn-ParB-Neon2 construct was generated from the pBPGUw-attB (Addgene #17575). ParB-Neon sequence from an hsp70-ParB-Neon plasmid was cloned into an intermediate plasmid containing the Dpn enhancer followed by the Drosophila Synthetic Core Promoter (DSCP) and terminated by the 3’UTR of hsp70. Both pFG4-INT2 and hsp70-ParB-Neon plasmids were a kind gift from Dr. Richard Mann. All transgenes above were generated using Sequence Ligation Independent Cloning (SLIC)(Jeong et al., 2012). To generate the Δintron:HA:hb construct, we synthesized primers (below) with 80nt homology to sequences flanking the hb intron to amplify a lox flanked Kanamycin resistance (KanR) cassette within the PL-452 N-eGFP plasmid. This fragment was used to recombineer the 32kb HA:hb BACampR, which replaced 246bp of the intron with 86bp sequence from the PL-452N-eGFP plasmid. The final construct was inserted at attp2 (3L 68A4) using site specific integration.
5’HR Forward: tcatgatctttataatcaccgtcatggtctttgtagtccatcttggcggctctagacggctgtaatggataagaaaacag agagagtggggttagtctcggcatggacgagctgtacaag; 3’HR Reverse: ataatatacttgcaaatccttacgaaaatcccgacaa atttggaatatacttcgatacaatcgcaatcatacgcactgagcggccacgaaacggtctagtggatcccctcgagggaccta.
Cell dissociation and FACS
dpn-eGFP embryos were collected for one hour, dechorionated, and immersed in halocarbon oil (Sigma-Aldrich) to identify stage. For stage 5 ATAC-seq, five embryos were collected after aging for one hour at 25°C, dissociated and directly processed for ATAC-seq. For stage 10 and 12 neuroblasts, embryos older than stage 4 were removed by hand to ensure developmental stage uniformity. Embryos were then aged at 18°C for 16 hours to reach stage 12, or at 29°C for 3 hours to reach stage 10 before dissociating in Chan and Gehring’s Balanced Saline (C&G) buffer (Chan and Gehring, 1971) with 2% fetal bovine serum. Cells were incubated with Calcein Violet 450 AM and 7-AAD (Thermo Fisher Scientific) to label live and dead cells, respectively, and sorted on a Beckman Coulter MoFlo Astrios EQ (Figure S3B). FACS based on their viability, size, granularity and GFP fluorescence intensity (Figure S3C). Neuroblasts were collected into cold ATAC-seq lysis buffer and incubated for 10 min on ice prior to transposition. A fraction of sorted cells were plated on glass coverslips and fixed for 15min at room temperature in 4% formaldehyde for immunostaining (Figure S3D). FACS gates were adjusted to include single round neuroblasts and exclude Elav+ neurons that formed neuroblast-sized clusters. During each FACS, cells were examined under brightfield to confirm these criteria.
Low input ATAC-seq
ATAC-seq was performed as described in (Buenrostro et al., 2015) with minor modifications for low-input. After sorting, the transposition reaction mix (13.5μl 2x TD Buffer + 1.5μl Tn5 Transposase, Tn5 Illumina) was added to neuroblasts and incubated for 30min at 37°C. ATAC-seq libraries were generated using NEB Next High Fidelity 2xPCR Master Mix to amplify transposed DNA using barcoded primers (Buenrostro et al., 2013). Concentrations and fragment size distributions were checked on a 2100 Bioanalyzer. Libraries were pooled and sequenced paired-end on an Illumina NextSeq500 sequencer. Four biological replicates of neuroblasts were sequenced. Paired-end sequencing data were aligned to the Dm6 Drosophila genome using bowtie2. PCR duplicates were removed and read positions relative to Tn5 cut site were adjusted as in (Buenrostro et al., 2013). For visualization purposes, all samples were normalized by Reads per million (RPM). Peaks were called using MACS2 from <100bp fragment pairs representing open regions and ranked by fold change.
ATAC-seq data processing
Raw sequencing data (basecall files (BCLs)) were converted to FASTQ format using bcl2fastq conversion software (Illumina). Quality assurance was performed using FASTQC (Andrews, 2010). Adaptor sequences were removed using cutadapt in paired-end mode (-m5 -e 0.2 -o {R1} -p {R2}) (Martin, 2011). Paired-end reads were aligned against dm6 (dmel_r6, Flybase) using bowtie2 (-k 4 -X2000 –local –mm) (Langmead and Salzberg, 2012). PCR duplicate were remove using Picard MarkDuplicatesReads (Picard Toolkit, Broad Institute) and further filtering was done using samtools (-F 1804 -f 2 -q 30 chrX chrY chr4 chr2L chr2R chr3L chr3R) (Li et al., 2009). Read positions were adjusted as in (Buenrostro et al., 2013) on bedgraph file using +5/−4 bp shift for first in pair and second in pair respectively and read per million (RPM) normalization was applied to all samples using BEDTools genomecov (-scale {RPM} -d) (Quinlan and Hall, 2010) before generating bigwig file for visualization using bedGraphToBigWig (Kent et al., 2010).
Peak detection for ATAC-seq data and differential analysis
Peaks were called from fragment-size filtered bam files (< 100bp, representing open region only) using MACS2 (callpeak -f BAMPE -g dm -q 1.10−6 –slocal 10000 –nomodel –shift - 100 –extsize 200 –nolambda) (Zhang et al., 2008). Peaks were ranked using their MACS2 fold change and the top 20% were analyzed for association with neural genes. Replicates were correlated with single cell ATAC data (Cusanovich et al., 2018)by extracting bigwig tracks from their scATAC-seq browser and running plotCorrelation from deepTools (Ramírez et al., 2016). Differential analysis for ATACseq peaks at Stage 10 vs. Stage 12 were performed using DiffBind (Ross-Innes et al., 2012) (all aligned file replicates were used along with their specific peak list). We used 0.05 as threshold of significance for differential analysis results.
Immunohistochemistry
Embryos were immunostained following standard protocols (Rothwell and Sullivan, 2000). Briefly, embryos were fixed in a 1:1 mixture of 4% formaldehyde in PEM buffer (0.1M Pipes, 1mM MgS04, 2mM EGTA) and N-heptane and rocked for 22 min at room temperature. Embryos were devitellinized by vigorous shaking in a 1:1 mixture of methanol:heptane and washed with PBS before staining. Embryos were incubated in primary antibodies diluted in PBS-0.1% Tween 20 (PBT) overnight at 4°C, secondary antibodies at room temperature for 1.5hrs, and streptavidin for 20min at room temperature.
Immuno-DNA FISH
We performed DNA in situ hybridization (DNA-FISH) as previously described (Bantignies et al., 2011; Kohwi et al., 2013). Briefly, embryos were fixed in 4% formaldehyde in PIPES buffer (60mM KCl, 15mM NaCl, 0.5mM spermidine, 0.15mM spermine, 2mM EDTA, 0.5mM EGTA, 15mM PIPES pH 7.4) and rocked for 25 min at room temperature with an equal volume of heptane, followed by vigorous shaking in a 1:1 mixture of methanol:heptane to release the embryos (methanol cracking). We made the hb DNA FISH probe by amplifying genomic DNA spanning a ~10kb region at the hb locus using PCR (Table S1) and generated fluorescent probes using the DNA FISH Tag kit (Thermo Fisher Scientific). Rehydrated embryos were treated with RNaseA (150mg/ml) for 2hr at room temperature and washed 1hr in PB-0.3%Triton-X (PBTx). Embryos were gradually stepped into 100% pre-hybridization mixture (pHM: 50% formamide; 4XSSC; 100 mM NaH2PO4, pH 7.0; 0.1% Tween 20) by a series of PBTx:pHM washes for 20min each. Embryos were pre-hybridized 1h at 37°C, denatured 15min at 80°C and rocked 16h at 37°C in FISH DNA hybridization buffer (10% Dextran sulfate, 50% deionized formamide, 2XSSC, 0.5 mg/ml Salmon Sperm DNA) containing denatured probe. Embryos were washed in a series of decreasing formamide/0.3% CHAPS solutions at 37°C with final washes at room temperature. Embryos were subsequently immunostained according to standard immunochemistry protocol, mounted in vectashield (Vectorlabs) and imaged on a confocal microscope, Zeiss LSM 700.
Confocal imaging
3D imaging was done on a Zeiss 700 Axio Imager 2 laser scanning confocal. DNA-FISH and ParB-ParS images were taken at a 0.4μm step size, and pinholes were adjusted to have equal optical section thickness for all channels.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image analysis
All image analyses were done on Fiji (Schindelin et al., 2012). For DNA-FISH, we identified the z-plane in which the FISH signal was strongest and measured the shortest distance to the nuclear lamina. FISH signals for which pixels overlapped with those of lamin signal was scored to be “on” lamin, and FISH foci not touching lamin but still within 0.4μm of the lamina was scored as “near.” Embryos were scored twice by two independent observers.
ChIP-seq analysis
Raw ChIP-seq datasets from Brown et al. (2018) (GSE102339) (Brown et al., 2018) for E(z), Pc, Ph, Psc, Pho and input and Bonnet et al. (2019) (GSE114832) (Bonnet et al., 2019) for Pho and input were processed as follows: adaptor removal using Cutadapt (-m 5, -e 0.2), alignment to dm6 using bowtie2 (-local, –very-sensitive-local), removal of PCR duplicates using PICARD Mark duplicates, removal of mitochondrial DNA and read quality filter at 30 using Samtools (-q 30). Resulting bam files were merged by replicate, and log2 (RPM-normalized ChIP) / (RPM-normalized Input) bigwig files were generated using Bedtools (bamcompare). We identified putative Pho motifs occurrence within the hb genomic region using PWMScan (PWMTools, ccg.epfl.ch) (Ambrosini et al., 2018) by selecting Pho_DROME_B1H OTF0277.1 (OnTheFly 2014 motif library) with a p-value cutoff at 5.10−4.
Hb vs Pho target analysis
Putative Hb binding target genes were extracted from pan-neuroblast Hb DamID sequencing data (Sen et al., 2019). Dam-only and Hb-Dam raw reads were re-analyzed for peak detection using damidseq_pipeline (Marshall and Brand, 2015). The resulting list was further filtered to include only those with genome-wide hb PWM occurrences (PWMscan (Ambrosini et al., 2018) for HUNB_DROME_SELEX_OTF0231.1 from OnTheFly2014 library using 1e−4 p-value) resulting in 884 genes, annotated by PAVIS (Huang et al., 2013) (dm6.05, 5kb upstream and 1kb downstream), that contain Hb peaks overlapping with a hb binding motif. Chromatin accessible (“open”) Pho motifs in stage 12 neuroblasts were extracted by intersecting the genome-wide Pho-PWM/ChIP list with Stage 12 ATAC-seq peak summit data, as described below: The Pho PWM/ChIP list contains genomic Pho motif occurrences (see ChIP-seq analysis section) that also have Pho ChIP peaks in L3 larval brain (Brown et al., 2018) or late stage embryo Pho ChIP peaks (Bonnet et al., 2019). Stage 12 ATAC-seq peak summit list was generated by running MACS2 (Zhang et al., 2008) on the ATAC Stage 12 neuroblast replicate-merged bam file (Huang et al., 2013) (callpeak with following option: -f BAMPE, -q 0.05, -slocal 10000, -nomodel, -shift −100, -extsize 200, -nolambda). To score for the presence of Pho-motifs within the chromatin accessible regions, each analyzed ATAC peak was restricted to a region that contained the summit and surrounding accessible sequences corresponding to 20% of the peak width. The resulting list was annotated to the closest gene using the PAVIS webtool (manticore.niehs.nih.gov/pavis2). Finally, the Hb putative target genes list and the Stage-12-Open-Pho-PWM/ChIP target genes list were compared (210 genes intersect) and the result was tested against a random model. The random model consists of randomly picking 210 non-Hb target 10,000 times and quantify their intersection with the Stage-12-Open-Pho-PWM/ChIP target list. All data intersections were done using Bedtools (Quinlan and Hall, 2010) (intersect -wa).
Statistical analysis
We applied standard t-tests using Prism v8. Statistical significance was classified as follows: * < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001. For competence window tests, n’s represent each neuroblast lineage quantified by a minimum of three embryos per genotype. For DNA FISH tests, n’s represent each embryo, and number of FISH signals quantified are shown on the graph. Graphs show each data point used for statistical analysis, and bars indicate mean and standard deviation. To estimate the P-value of bootstrap simulation we use the following formula: pval = (1 + sum(s >= s0)) / (N+1), in which s is random values and s0 is the observed value.
Supplementary Material
Highlights.
A 250 bp intronic element (IE) is necessary and sufficient for gene-lamina relocation
IE-mediated gene relocation in neuroblasts confers heritable silencing in progeny
PcG proteins target IE for gene-lamina association, but not gene repression
PcG-mediated gene mobility regulates competence to specify neural identity in vivo
ACKNOWLEDGMENTS
We greatly thank Drs. Gary Struhl and Stavros Lomvardas for critical scientific discussion and feedback on the manuscript. We thank Dr. Jürg Müller on discussion on PcG biology. We additionally thank Drs. Hynek Wichterle, Steve Siegelbaum, Wes Grueber, Carol Mason, Chris Q. Doe, Rui Galvão, and Raphaelle Laureau for comments on the manuscript. We thank members of the Kohwi lab, especially Daiki Tagami and Sofiya Patra, for technical assistance with experiments. M.K. received support from NIH grants R00HD072035 and R01HD092381, Rita Allen Foundation, and Whitehall Foundation. T.L. received support from the NYSTEM training grant C030291. H.Z. received support from Columbia University’s Science Research Fellows Program.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.devcel.2021.08.020.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Allan DW, and Thor S (2015). Transcriptional selectors, masters, and combinatorial codes: regulatory principles of neural subtype specification. Wiley Interdiscip. Rev. Dev. Biol 4, 505–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosini G, Groux R, and Bucher P (2018). PWMScan: a fast tool for scanning entire genomes with a position-specific weight matrix. Bioinformatics 34, 2483–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S (2010). FastQC: a quality control tool for high throughput sequence data https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Awasaki T, Kao CF, Lee YJ, Yang CP, Huang Y, Pfeiffer BD, Luan H, Jing X, Huang YF, He Y, et al. (2014). Making Drosophila lineage-restricted drivers via patterned recombination in neuroblasts. Nat. Neurosci 17, 631–637. [DOI] [PubMed] [Google Scholar]
- Bantignies F, Roure V, Comet I, Leblanc B, Schuettengruber B, Bonnet J, Tixier V, Mas A, and Cavalli G (2011). Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell 144, 214–226. [DOI] [PubMed] [Google Scholar]
- Bentsen M, Goymann P, Schultheis H, Klee K, Petrova A, Wiegandt R, Fust A, Preussner J, Kuenne C, Braun T, et al. (2020). ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat. Commun 11, 4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuchle D, Struhl G, and Müller J (2001). Polycomb group proteins and heritable silencing of Drosophila Hox genes. Development 128, 993–1004. [DOI] [PubMed] [Google Scholar]
- Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot JP, Tanay A, and Cavalli G (2017). Multiscale 3D genome rewiring during mouse neural development. Cell 171, 557–572.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet J, Lindeboom RGH, Pokrovsky D, Stricker G, Çelik MH, Rupp RAW, Gagneur J, Vermeulen M, Imhof A, and Müller J (2019). Quantification of proteins and histone marks in Drosophila embryos reveals stoichiometric relationships impacting chromatin regulation. Dev. Cell 51, 632–644.e6. [DOI] [PubMed] [Google Scholar]
- Boyle S, Flyamer IM, Williamson I, SenGupta D, Bickmore WA, and Illingworth RS (2020). A central role for canonical PRC1 in shaping the 3D nuclear landscape. Genes Dev. 34, 931–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, and Perrimon N (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415. [DOI] [PubMed] [Google Scholar]
- Brody T, and Odenwald WF (2000). Programmed transformations in neuroblast gene expression during Drosophila CNS lineage development. Dev. Biol 226, 34–44. [DOI] [PubMed] [Google Scholar]
- Bronshtein I, Kanter I, Kepten E, Lindner M, Berezin S, Shav-Tal Y, and Garini Y (2016). Exploring chromatin organization mechanisms through its dynamic properties. Nucleus 7, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JL, Sun MA, and Kassis JA (2018). Global changes of H3K27me3 domains and Polycomb group protein distribution in the absence of recruiters spps or Pho. Proc. Natl. Acad. Sci. USA 115, E1839–E1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 109, 21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan LN, and Gehring W (1971). Determination of blastoderm cells in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 68, 2217–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheutin T, and Cavalli G (2018). Loss of PRC1 induces higher-order opening of Hox loci independently of transcription during Drosophila embryogenesis. Nat. Commun 9, 3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Classen AK, Bunker BD, Harvey KF, Vaccari T, and Bilder D (2009). A tumor suppressor activity of Drosophila Polycomb genes mediated by JAK-STAT signaling. Nat. Genet 41, 1150–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary MD, and Doe CQ (2006). Regulation of neuroblast competence: multiple temporal identity factors specify distinct neuronal fates within a single early competence window. Genes Dev 20, 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Molina S, Respuela P, Tebartz C, Kolovos P, Nikolic M, Fueyo R, van Ijcken WFJ, Grosveld F, Frommolt P, Bazzi H, and Rada-Iglesias A (2017). PRC2 facilitates the regulatory topology required for poised enhancer function during pluripotent stem cell differentiation. Cell Stem Cell 20, 689–705.e9. [DOI] [PubMed] [Google Scholar]
- Cusanovich DA, Reddington JP, Garfield DA, Daza RM, Aghamirzaie D, Marco-Ferreres R, Pliner HA, Christiansen L, Qiu X, Steemers FJ, et al. (2018). The cis-regulatory dynamics of embryonic development at single-cell resolution. Nature 555, 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai AR, and McConnell SK (2000). Progressive restriction in fate potential by neural progenitors during cerebral cortical development. Development 127, 2863–2872. [DOI] [PubMed] [Google Scholar]
- Doe CQ (2017). Temporal patterning in the Drosophila CNS. Annu. Rev. Cell Dev. Biol 33, 219–240. [DOI] [PubMed] [Google Scholar]
- Eagen KP, Aiden EL, and Kornberg RD (2017). Polycomb-mediated chromatin loops revealed by a subkilobase-resolution chromatin interaction map. Proc. Natl. Acad. Sci. USA 114, 8764–8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott J, Jolicoeur C, Ramamurthy V, and Cayouette M (2008). Ikaros confers early temporal competence to mouse retinal progenitor cells. Neuron 60, 26–39. [DOI] [PubMed] [Google Scholar]
- Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, and Bickmore WA (2008). Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet. 4, e1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile C, Berlivet S, Mayran A, Paquette D, Guerard-Millet F, Bajon E, Dostie J, and Kmita M (2019). PRC2-associated chromatin contacts in the developing limb reveal a possible mechanism for the atypical role of PRC2 in HoxA gene expression. Dev. Cell 50, 184–196.e4. [DOI] [PubMed] [Google Scholar]
- Gozalo A, Duke A, Lan Y, Pascual-Garcia P, Talamas JA, Nguyen SC, Shah PP, Jain R, Joyce EF, and Capelson M (2020). Core components of the nuclear pore bind distinct states of chromatin and contribute to polycomb repression. Mol. Cell 77, 67–81.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossniklaus U, and Paro R (2014). Transcriptional silencing by polycomb-group proteins. Cold Spring Harb. Perspect. Biol 6, a019331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, and van Steensel B (2008). Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453, 948–951. [DOI] [PubMed] [Google Scholar]
- Haines JE, and Eisen MB (2018). Patterns of chromatin accessibility along the anterior-posterior axis in the early Drosophila embryo. PLoS Genet. 14, e1007367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr JC, Luperchio TR, Wong X, Cohen E, Wheelan SJ, and Reddy KL (2015). Directed targeting of chromatin to the nuclear lamina is mediated by chromatin state and A-type lamins. J. Cell Biol 208, 33–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DA, Binari R, Nahreini TS, Gilman M, and Perrimon N (1995). Activation of a Drosophila janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. EMBO J. 14, 2857–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, et al. (2009). Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat. Methods 6, 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi Y, Suzki N, Tsuboi M, Endo TA, Toyoda T, Shinga J, Koseki H, Vidal M, and Gotoh Y (2009). Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 63, 600–613. [DOI] [PubMed] [Google Scholar]
- Hirono K, Margolis JS, Posakony JW, and Doe CQ (2012). Identification of hunchback cis-regulatory DNA conferring temporal expression in neuroblasts and neurons. Gene Expr. Patterns 12, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Loganantharaj R, Schroeder B, Fargo D, and Li L (2013). PAVIS: a tool for peak annotation and visualization. Bioinformatics 29, 3097–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isshiki T, Pearson B, Holbrook S, and Doe CQ (2001). Drosophila neuroblasts sequentially express transcription factors which specify the temporal identity of their neuronal progeny. Cell 106, 511–521. [DOI] [PubMed] [Google Scholar]
- Jeong JY, Yim HS, Ryu JY, Lee HS, Lee JH, Seen DS, and Kang SG (2012). One-step sequence- and ligation-independent cloning as a rapid and versatile cloning method for functional genomics studies. Appl. Environ. Microbiol 78, 5440–5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai MI, Okabe M, and Hiromi Y (2005). Seven-up Controls switching of transcription factors that specify temporal identities of Drosophila neuroblasts. Dev. Cell 8, 203–213. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J, Pagie L, de Vries SS, Nahidiazar L, Dey SS, Bienko M, Zhan Y, Lajoie B, de Graaf CA, Amendola M, et al. (2015). Genome-wide maps of nuclear lamina interactions in single human cells. Cell 163, 134–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King IF, Emmons RB, Francis NJ, Wild B, Müller J, Kingston RE, and Wu CT (2005). Analysis of a polycomb group protein defines regions that link repressive activity on nucleosomal templates to in vivo function. Mol. Cell. Biol 25, 6578–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohwi M, and Doe CQ (2013). Temporal fate specification and neural progenitor competence during development. Nat. Rev. Neurosci 14, 823–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohwi M, Lupton JR, Lai SL, Miller MR, and Doe CQ (2013). Developmentally regulated subnuclear genome reorganization restricts neural progenitor competence in Drosophila. Cell 152, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu S, Ji F, Sunwoo H, Jain G, Lee JT, Sadreyev RI, Dekker J, and Kingston RE (2017). Polycomb repressive complex 1 generates discrete compacted domains that change during differentiation. Mol. Cell 65, 432–446.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas T, and Kohwi M (2019). From insects to mammals: regulation of genome architecture in neural development. Curr. Opin. Neurobiol 59, 146–156. [DOI] [PubMed] [Google Scholar]
- Marshall OJ, and Brand AH (2015). damidseq_pipeline: an automated pipeline for processing DamID sequencing datasets. Bioinformatics 31, 3371–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J 17, 3. [Google Scholar]
- Mattar P, Ericson J, Blackshaw S, and Cayouette M (2015). A conserved regulatory logic controls temporal identity in mouse neural progenitors. Neuron 85, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay DJ, and Lieb JD (2013).A common set of DNA regulatory elements shapes Drosophila appendages. Dev. Cell 27, 306–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister P, Towbin BD, Pike BL, Ponti A, and Gasser SM (2010). The spatial dynamics of tissue-specific promoters during C. elegans development. Genes Dev 24, 766–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng JL, Marshall ZD, Lobb-Rabe M, and Heckscher ES (2019). How prolonged expression of Hunchback, a temporal transcription factor, re-wires locomotor circuits. eLife 8, e46089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milon B, Sun Y, Chang W, Creasy T, Mahurkar A, Shetty A, Nurminsky D, and Nurminskaya M (2014). Map of open and closed chromatin domains in Drosophila genome. BMC Genomics 15, 988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyares RL, and Lee T (2019). Temporal control of Drosophila central nervous system development. Curr. Opin. Neurobiol 56, 24–32. [DOI] [PubMed] [Google Scholar]
- Nègre N, Hennetin J, Sun LV, Lavrov S, Bellis M, White KP, and Cavalli G (2006). Chromosomal distribution of PcG proteins during Drosophila development. PLoS Biol. 4, e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oktaba K, Gutiérrez L, Gagneur J, Girardot C, SenGupta AK, Furlong EE, and Müller J (2008). Dynamic regulation by polycomb group protein complexes controls pattern formation and the cell cycle in Drosophila. Dev. Cell 15, 877–889. [DOI] [PubMed] [Google Scholar]
- Papp B, and Müller J (2006). Histone trimethylation and the maintenance of transcriptional ON and OFF states by trxG and PcG proteins. Genes Dev. 20, 2041–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel T, Tursun B, Rahe DP, and Hobert O (2012). Removal of Polycomb repressive complex 2 makes C. elegans germ cells susceptible to direct conversion into specific somatic cell types. Cell Rep. 2, 1178–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson BJ, and Doe CQ (2003). Regulation of neuroblast competence in Drosophila. Nature 425, 624–628. [DOI] [PubMed] [Google Scholar]
- Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SW, Solovei I, Brugman W, Gräf S, Flicek P, Kerkhoven RM, van Lohuizen M, et al. (2010). Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell 38, 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peric-Hupkes D, and van Steensel B (2010). Role of the nuclear lamina in genome organization and gene expression. Cold Spring Harb. Symp. Quant. Biol 75, 517–524. [DOI] [PubMed] [Google Scholar]
- Pickersgill H, Kalverda B, de Wit E, Talhout W, Fornerod M, and van Steensel B (2006). Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat. Genet 38, 1005–1014. [DOI] [PubMed] [Google Scholar]
- Qin P, Parlak M, Kuscu C, Bandaria J, Mir M, Szlachta K, Singh R, Darzacq X, Yildiz A, and Adli M (2017). Live cell imaging of low- and non-repetitive chromosome loci using CRISPR-Cas9. Nat. Commun 8, 14725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy KL, Zullo JM, Bertolino E, and Singh H (2008). Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 452, 243–247. [DOI] [PubMed] [Google Scholar]
- Rossi AM, Fernandes VM, and Desplan C (2017). Timing temporal transitions during brain development. Curr. Opin. Neurobiol 42, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell WF, and Sullivan W (2000). Fluorescent Analysis of Drosophila Embryos (Cold Spring Harbor Press; ). [Google Scholar]
- Saad H, Gallardo F, Dalvai M, Tanguy-le-Gac N, Lane D, and Bystricky K (2014). DNA dynamics during early double-strand break processing revealed by non-intrusive imaging of living cells. PLoS Genet 10, e1004187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B, and Cavalli G (2009). Recruitment of polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development 136, 3531–3542. [DOI] [PubMed] [Google Scholar]
- Schwartz YB, Kahn TG, Nix DA, Li XY, Bourgon R, Biggin M, and Pirrotta V (2006). Genome-wide analysis of Polycomb targets in Drosophila melanogaster. Nat. Genet 38, 700–705. [DOI] [PubMed] [Google Scholar]
- Sen SQ, Chanchani S, Southall TD, and Doe CQ (2019). Neuroblast-specific open chromatin allows the temporal transcription factor, Hunchback, to bind neuroblast-specific loci. eLife 8, e44036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seroka AQ, and Doe CQ (2019). The Hunchback temporal transcription factor determines motor neuron axon and dendrite targeting in Drosophila. Development 146, dev175570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevelyov YY, and Nurminsky DI (2012). The nuclear lamina as a gene-silencing hub. Curr. Issues Mol. Biol 14, 27–38. [PubMed] [Google Scholar]
- Struhl G (1981). A gene product required for correct initiation of segmental determination in Drosophila. Nature 293, 36–41. [DOI] [PubMed] [Google Scholar]
- Telley L, Agirman G, Prados J, Amberg N, Fievre S, Oberst P, Bartolini G, Vitali I, Cadilhac C, Hippenmeyer S, et al. (2019). Temporal patterning of apical progenitors and their daughter neurons in the developing neocortex. Science 364, eaav2522. [DOI] [PubMed] [Google Scholar]
- Tie F, Furuyama T, and Harte PJ (1998). The Drosophila Polycomb group proteins ESC and E(Z) bind directly to each other and co-localize at multiple chromosomal sites. Development 125, 3483–3496. [DOI] [PubMed] [Google Scholar]
- Tolhuis B, Blom M, Kerkhoven RM, Pagie L, Teunissen H, Nieuwland M, Simonis M, de Laat W, van Lohuizen M, and van Steensel B (2011). Interactions among Polycomb domains are guided by chromosome architecture. PLoS Genet 7, e1001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolhuis B, de Wit E, Muijrers I, Teunissen H, Talhout W, van Steensel B, and van Lohuizen M (2006). Genome-wide profiling of PRC1 and PRC2 Polycomb chromatin binding in Drosophila melanogaster. Nat. Genet 38, 694–699. [DOI] [PubMed] [Google Scholar]
- Touma JJ, Weckerle FF, and Cleary MD (2012). Drosophila Polycomb complexes restrict neuroblast competence to generate motoneurons. Development 139, 657–666. [DOI] [PubMed] [Google Scholar]
- Van Bortle K, and Corces VG (2013). Spinning the web of cell fate. Cell 152, 1213–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, and Belmont AS (2017). Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJ, Carlson JW, Schulze KL, Pan H, He Y, Spokony R, Wan KH, Koriabine M, de Jong PJ, White KP, et al. (2009). Versatile P[acman] BAC libraries for transgenesis studies in Drosophila melanogaster. Nat. Methods 6, 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RR, Azuara V, Perry P, Sauer S, Dvorkina M, Jørgensen H, Roix J, McQueen P, Misteli T, Merkenschlager M, and Fisher AG (2006). Neural induction promotes large-scale chromatin reorganisation of the Mash1 locus. J. Cell Sci 119, 132–140. [DOI] [PubMed] [Google Scholar]
- Wimmer EA, Carleton A, Harjes P, Turner T, and Desplan C (2000). Bicoid-independent formation of thoracic segments in Drosophila. Science 287, 2476–2479. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zullo JM, Demarco IA, Piqué-Regi R, Gaffney DJ, Epstein CB, Spooner CJ, Luperchio TR, Bernstein BE, Pritchard JK, Reddy KL, and Singh H (2012). DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell 149, 1474–1487. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ATAC sequence data reported in this paper have been deposited to the NCBI Sequence Read Archive with the accession number SRA:PRJNA754600 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA754600). Any additional information required to reanalyze the data reported in this manuscript will available from the Lead Contact upon request.