

Abstract
Neurofibrillary tangles composed of aberrantly aggregating tau protein are a hallmark of Alzheimer’s disease and related dementia disorders. Recent work has shown that mammalian suppressor of tauopathy 2 (MSUT2), also named ZC3H14 (Zinc Finger CCCH-Type Containing 14), controls accumulation of pathological tau in cultured human cells and mice. Knocking out MSUT2 protects neurons from neurodegenerative tauopathy and preserves learning and memory. MSUT2 protein functions to bind polyadenosine [poly(A)] tails of mRNA through its C-terminal CCCH type zinc finger domains, and loss of CCCH domain function suppresses tauopathy in Caenorhabditis elegans and mice. Thus, we hypothesized that inhibiting the poly(A):MSUT2 RNA–protein interaction would ameliorate pathological tau accumulation. Here we present a high-throughput screening method for the identification of small molecules inhibiting the poly(A):MSUT2 RNA–protein interaction. We employed a fluorescent polarization assay for initial small molecule discovery with the intention to repurpose hits identified from the NIH Clinical Collection (NIHCC). Our drug repurposing development workflow included validation of hits by dose–response analysis, specificity testing, orthogonal assays of activity, and cytotoxicity. Validated compounds passing through this screening funnel will be evaluated for translational effectiveness in future studies. This preclinical drug development pipeline identified diverse FDA approved drugs duloxetine, saquinavir, and clofazimine as potential repurposing candidates for reducing pathological tau accumulation.
Keywords: Alzheimer’s disease, Neurotherapeutics, RNA binding proteins, MSUT2, Tau, Neurodegeneration
Graphical Abstract
INTRODUCTION
Alzheimer’s disease (AD) causes progressive impairment of cognitive function due to neurodegeneration and atrophy in the brain regions responsible for learning and memory; to date, no known effective disease modifying therapies have been discovered.1 As the population of the United States has aged, the prevalence of AD has increased.2 To put the problem in perspective, heart disease-related deaths dropped 9% between 2000 and 2017, while AD-related deaths increased by 145%.3 AD histopathology consists of neurofibrillary tangles (NFTs) composed of aberrantly aggregating tau protein within neurons and senile plaques composed of amyloid-beta (Aβ) in the interneuronal space.4–6 The complex molecular dynamics underlying AD pathology remain incompletely understood, and the precise causes of disease initiation and progression remain unclear for late onset Alzheimer’s disease.7 However, pathological tau burden detected postmortem by conventional brain histology or antemortem using positron emission tomography imaging tools shows that pathological tau burden correlates well with cognitive decline.8,9 Although a hallmark of AD, NFTs also appear in many other distinct dementia disorders. Disorders exhibiting deposits of pathological tau protein are known as tauopathy disorders and include frontotemporal lobar degeneration, Pick’s disease, progressive supranuclear palsy, corticobasal degeneration, and AD.10 It is hoped that therapeutics targeting tau will have broad impact across these tauopathy disorders.
Although extensive drug-discovery initiatives primarily targeting the pathologically aggregating peptide Aβ have been ongoing for over two decades, this approach has failed to yield viable therapeutics to halt neurodegeneration.11 Recent work has suggested that RNA binding proteins (RBPs) may play a critical role in the progression of diverse neurodegenerative diseases by interacting with the messenger RNA (mRNA) of aberrantly aggregating proteins.12 These studies suggest targeting RBPs could have future therapeutic utility.
The RBP family of proteins is diverse and is composed of over 1500 genes in humans.13 The canonical functions of RBPs reflect this diversity and range from RNA transcription, splicing, polyadenylation, RNA export, localization, to translation.14 An example of one of these RBPs, TAR DNA-binding protein 43 kDa (TDP-43) has a well-defined role in neurodegeneration.15 TDP-43, expressed in the nucleus, functions in RNA transport and processing.16 The accumulation and mislocalization of TDP-43 into insoluble cytoplasmic deposits occurs in nearly all cases of Amyotrophic Lateral Sclerosis (ALS).17 Other RBPs implicated in neurodegenerative disorders include Fused in Sarcoma (FUS) with dominant missense mutations causing ALS and Poly(A) Binding Protein Nuclear 1 (PABPN1) with a coding repeat expansion causing oculopharyngeal muscular dystrophy.18–20
Previous work has shown that the poly(A) binding protein MSUT2 (an alias for ZC3H14) potentiates pathological tau accumulation and may serve as a tractable therapeutic target for intervention in tauopathies including Alzheimer’s disease.21,22 We have shown that RNAi knockdown of MSUT2 in a human cell model overexpressing tau decreases pathological tau species including phosphorylated tau, pretangle conformations, and detergent insoluble tau species. Further and most compelling, tau transgenic MSUT2 knockout mice are protected against tau-mediated neurofibrillary degeneration including decreased pathological tau burden, reduced memory deficits, and neuronal preservation.22 It should be noted that while we have previously shown that MSUT2 zinc finger domains are sufficient in driving tau deposition, the molecular mechanisms underpinning MSUT2 effects on pathological tau are not fully understood. It is hoped that hits found through MSUT2 inhibition screens will serve as chemical probes and tool compounds and lead to further mechanistic insights.
A key consideration in drug discovery for inhibitors of the poly(A):MSUT2 RNA–protein interaction is specificity. MSUT2 works in concert with another important regulator of RNA processing, PABPN1.23 PABPN1 is expressed in the nucleus throughout all tissues and binds to RNA to control the size of mRNA transcript poly(A) tails.18,24 It has been shown that MSUT2, PABPN1, and poly(A) RNA colocalize in nuclear speckles.21 Any therapeutic strategy targeting MSUT2 binding to poly(A) RNA must avoid altering the poly(A):PABPN1 interaction because PABPN1 knock down exacerbates pathological tau accumulation in cultured cells22 and PABPN1 serves as an essential protein (knockout lethal).25
In this study, we have developed a drug repurposing pipeline for the identification of potent and specific small-molecule inhibitors of MSUT2 RBP and poly(A) RNA. To this end, a primary fluorescence polarization (FP) high-throughput screen (HTS) identified compounds which were further validated through a PABPN1 counterscreen, an orthogonal Alpha Screen, and for cell toxicity.
RESULTS AND DISCUSSION
A High-Throughput Fluorescence Polarization Assay for Inhibition of Poly(A):MSUT2 RNA Interaction.
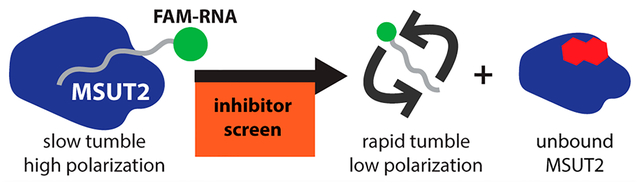
Fluorescence polarization relies on the fact that apparent rotational velocity (tumbling) of molecules is inversely proportional to molecular weight.26 Polarized incident light striking a relatively large, slowly tumbling fluorescently tagged molecule is emitted as polarized light while small, rapidly tumbling molecules emit nonpolarized light.27 In order to screen potential inhibitors of MSUT2 RBP and RNA by FP, fluorescent FAM (fluorescein amidite)-labeled poly(A)15 RNA (FAM-RNA) and recombinant MSUT2 ZF (CCCH zinc finger domain only) protein expressing constructs were generated (Figure 1a). Recombinant MSUT2 ZF protein and FAM-RNA form a complex (FAM-RNA:MSUT2 ZF) in vitro with high affinity. This complex tumbles at a relatively low rate in comparison to free FAM-RNA, resulting in a high emission of polarized light (Figure 1b). Inhibition of the interaction results in unbound FAM-RNA tumbling at a relatively rapid rate and emitting nonpolarized light (Figure 1b).
Figure 1.
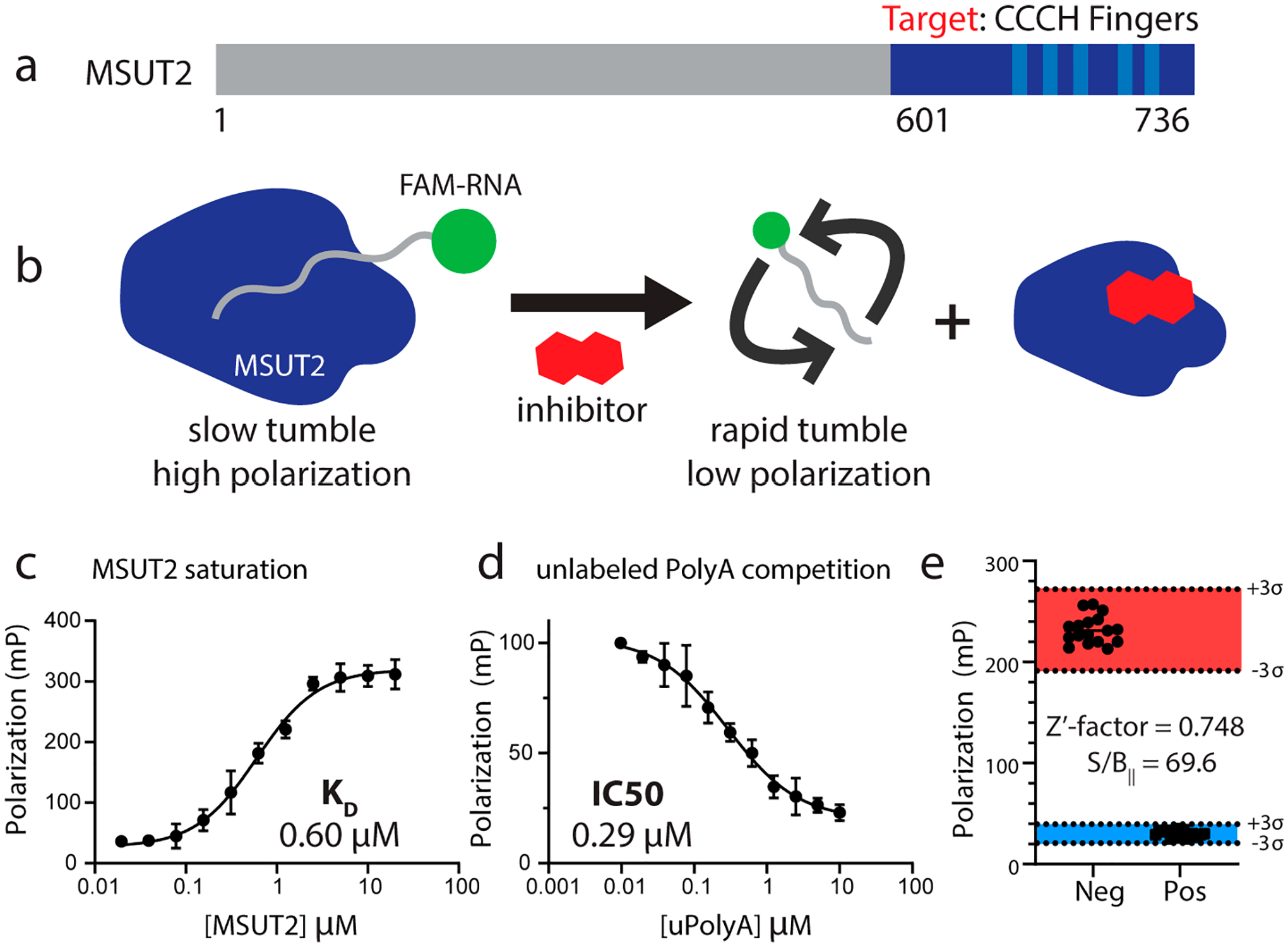
(a) Schematic of MSUT2 (ZC3H14). Targeted construct consisted of only the c-terminal end (dark blue and amino acids 601–736) of MSUT2. Five CCCH finger domains are indicated (light blue). (b) Fluorescence polarization depicting MSUT2 (dark blue) bound to FAM labeled RNA emitting highly polarized light and free FAM-RNA emitting low levels of polarized light after disruption by the inhibitor. (c) Saturation assay holding FAM-RNA concentration constant at 10 nM and increasing concentrations of MSUT2. Curve fitted using a 4-parameter nonlinear regression model (Y = bottom + (top − bottom)/(1 + (IC50/X)HillSlope), with 36 data points (x,y values) analyzed at N = 3 for each point, R2 = 0.9792. (d) Competition assay with MSUT2 concentration at 125 nM and FAM-RNA at 10 nM with increasing concentrations of unlabeled poly(A)15. Curve fitted using a 4-parameter nonlinear regression model (Y = bottom + (top − bottom)/(1 + (IC50/X)HillSlope), with 36 x,y values analyzed at N = 3 for each point, R2 = 0.9630. (e) Z′-factor bar graph showing negative controls on the left and positive controls on the right. Three standard deviations on either side of the means are indicated, and Y-axis values are polarization values in units of mP. Mean of Neg column, 231.8 and mean of Pos column 30.24, N = 17 for each group. Z′-factor determined to be 0.748. Signal to background ratio was determined to be 69.6.
To determine the optimal concentrations of both MSUT2 ZF protein and FAM-RNA probe, we performed a two-dimensional titration of protein and RNA concentrations. This experiment revealed that a ratio of 10 nM FAM-RNA:125 nM MSUT2 ZF resulted in robust and reproducible polarization signal along with a sufficiently high fluorescence intensity to minimize any potential background interference. Next, the binding constant between MSUT2 ZF and FAM-RNA was determined by holding FAM-RNA constant at 10 nM while increasing MSUT2 ZF concentration. An affinity of 0.60 μM for FAM-RNA:MSUT2 ZF interaction was determined (Figure 1c). To develop a positive control for inhibition of FAM-RNA:MSUT2 ZF complex formation, we tested varying concentrations of unlabeled poly(A)15 RNA in an FP competition assay (10 nM FAM-RNA:125 nM MSUT2 ZF) resulting in an IC50 of 0.29 μM (Figure 1d).
The Z′-factor is a well-known statistic for suitability of high-throughput screening (HTS) design28 and is calculated by the equation
where σ = standard deviation, μ = mean, p = positive controls, and n = negative controls. While an ideal assay would have a Z′-factor value of 1, a value of 1 > Z ≥ 0.5 indicates a large separation band between positive and negative controls and shows the assay is robust and suitable for screening and identification of potential inhibitors. As the Z′-factor value drops below 0.5, assays become less reliable with decreasing control separation and increasing signal variation.28 The Z′-factor of our screen, 0.748, indicates an assay capable of reliably detecting potential FAM-RNA:MSUT2 ZF interaction inhibitors and experimental conditions provided for a high signal to background ratio of 69.6 (Figure 1e).
A Drug Repurposing Strategy: Screening of the NIH Clinical Collection Compound Library.
Because of the rapidly increasing costs of drug development from initial screening through clinical trials, repurposing approved drugs for new indications has become an important focus in drug development.29 Our plan to find suitable compounds for drug repurposing began with a screen of the NIH Clinical Collection (NIHCC) library. The NIHCC consists of a diverse array of 700 compounds with historical pharmacological use and well-studied safety profiles. Our workflow for identifying MSUT2 selective inhibitors included a primary FP screen followed by dose validation (Figure 2). Next, an orthogonal poly(A):MSUT2 binding assay Alpha Screen allowed us to rule out assay interference, and selectivity for MSUT2 over PABPN1 was determined by a parallel FP assay with PABPN1 as the RBP. PABPN1 knockdown strongly exacerbates tau accumulation in human cells and Alzheimer’s cases exhibiting depletion of the MSUT2/PABPN1 complex show more severe neurodegenerative changes. Thus, we wished to eliminate compounds interfering with both PABPN1 and MSUT2 and focus on compounds truly specific for poly(A):MSUT2 binding. From the potent, dose responsive, and selective compounds inhibiting MSUT2 RNA binding, hits were validated for low toxicity and considered suitable for further studies in physiologically relevant models.
Figure 2.

Workflow schematic for drug selection. The NIH Clinical Collection was first screened by fluorescence polarization followed by a battery of secondary validation measures including dose–response analysis, specificity counterscreening against PABPN1, and replication through alpha assay orthogonal screening. Compounds passing initial filters were then subjected to a cellular toxicity assay to select potentially translationally relevant candidates.
Identification and Validation of MSUT2 Inhibitory Compounds from the NIHCC Library.
The single-point primary screen was conducted in duplicate and utilized the optimal FP assay conditions determined above (125 nM MSUT2 RNABP and 10 nM FAM-RNA probe) with library compounds added to a final concentration of 10 μM in a 96-well format. This screen resulted in 12 initial hits (Figure 3). The hit window corresponded to those compounds with >80% inhibition at 10 μM and corresponded to Z-scores ≤ −4σ (standard deviations) from the mean polarization of all samples (Figure 3a). Hits were further dose–response validated by fluorescence polarization, revealing eight relatively potent compounds showing dose-dependent inhibition with IC50s below 3 μM (Figure 4). Compound specificity was empirically tested by counter screening against PABPN1, a regulator of the length of mRNA poly(A) tails.30,31 The IC50 of each of the previous eight compounds was determined for PABPN1 using fluorescence polarization (Figure 5), and a specificity ratio was determined (Table 1).
Figure 3.
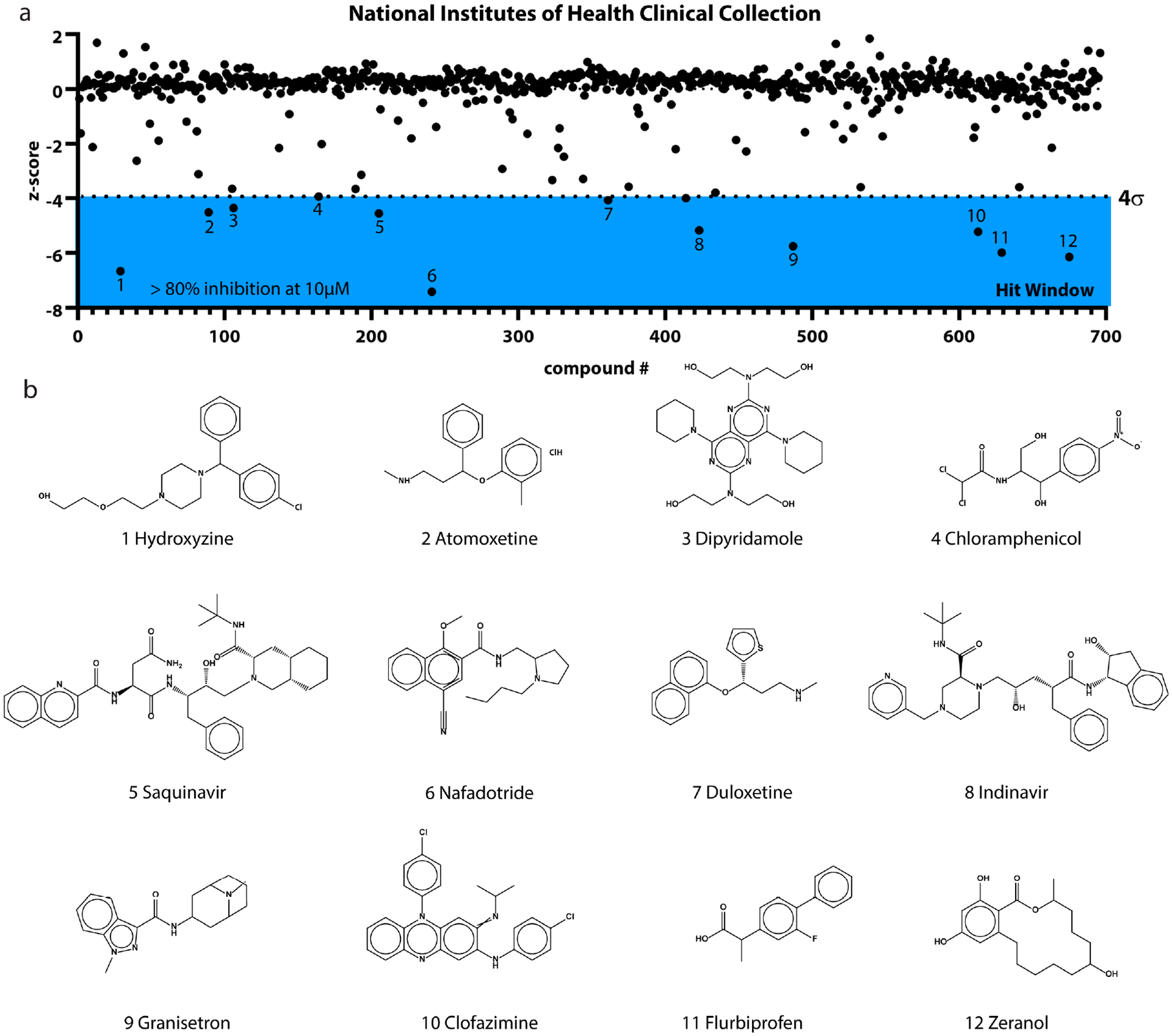
(a) Graph depicting the fluorescence polarization screen of NIH Clinical Collection compounds at 10 μM. Hit window (blue) began below −4σ (standard deviations) from the mean and included 12 primary hits. (b) Structures of compounds identified through the primary screen.
Figure 4.
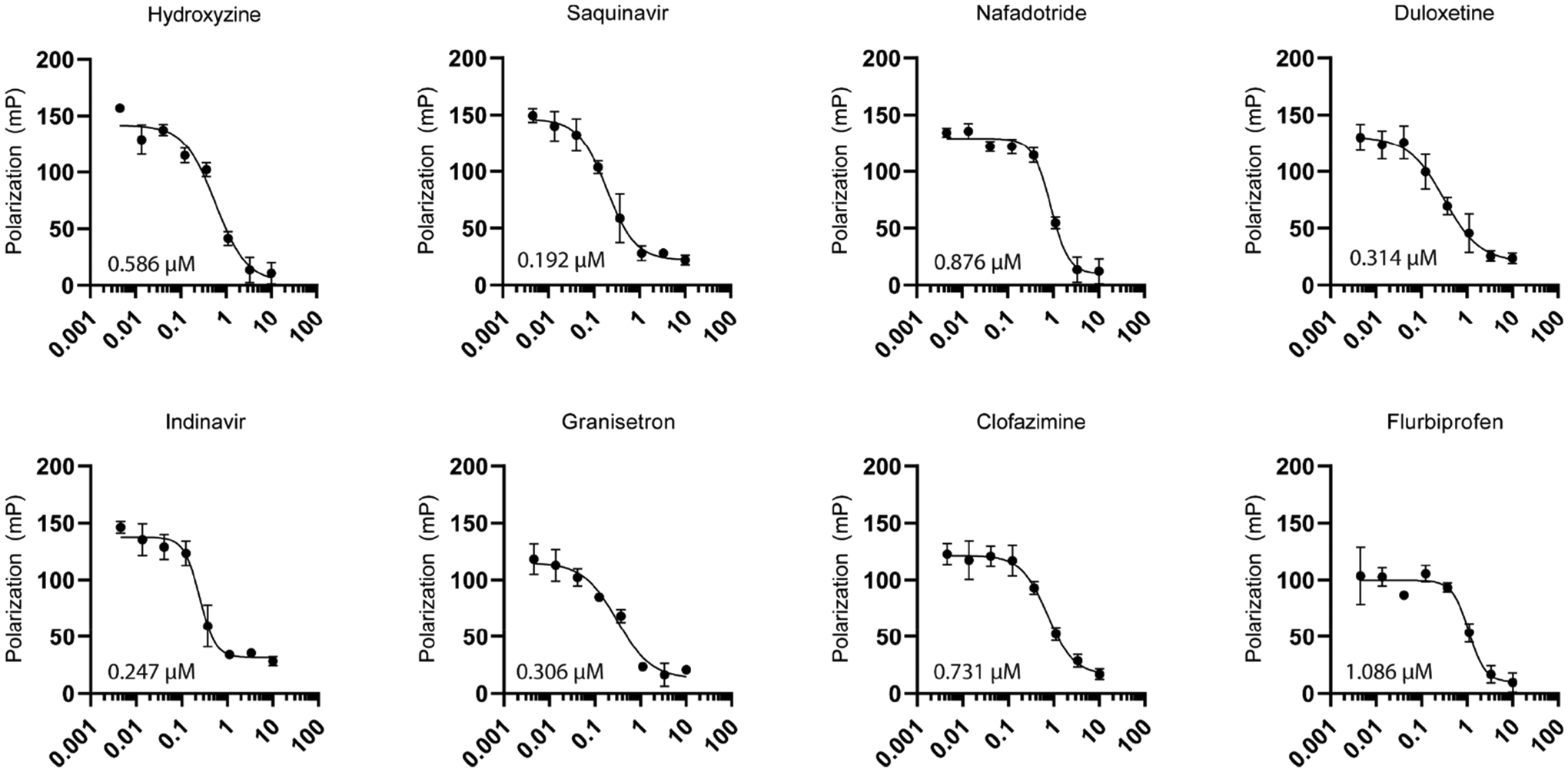
Dose–response by fluorescence polarization of eight positive hits with calculated IC50’s. Relative IC50 determined through a variable slope nonlinear regression model using the equation Y = bottom + (top − bottom)/1 + (IC50/X)HillSlope. Line fitting was performed using GraphPad Prism8 analysis tools. N = 3 biological replicates for each compound.
Figure 5.
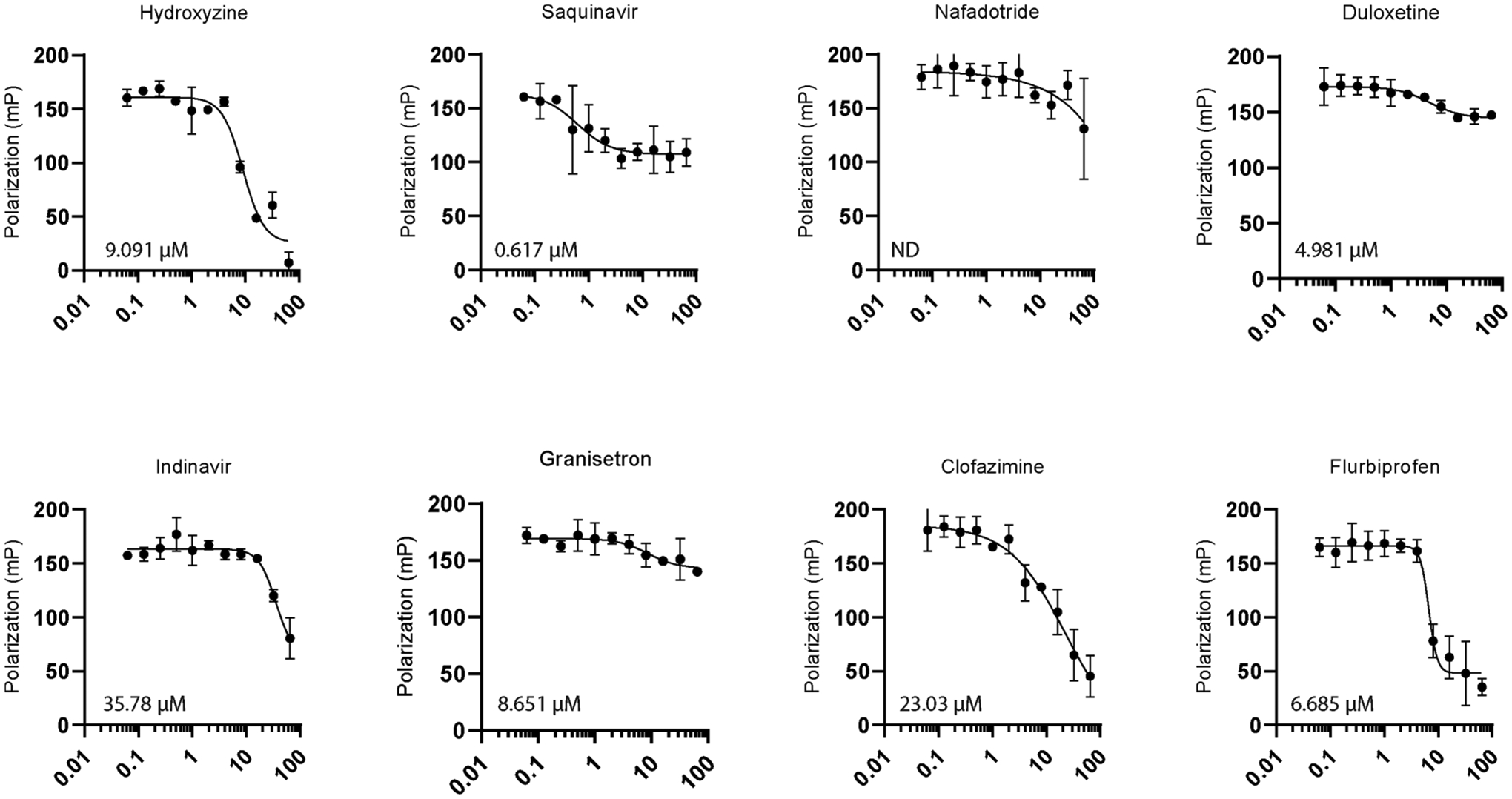
PABPN1 fluorescence polarization counter screen for compounds which passed the dose–response filter. Relative IC50 determined through a variable slope nonlinear regression model using equation Y = bottom + (top − bottom)/1 + (IC50/X)HillSlope. Line fitting was performed using GraphPad Prism8 analysis tools. N = 2 biological replicates for each compound. ND denotes IC50 not determined as a graph could not be fitted to a regression model (ambiguous fit).
Table 1.
Specificity Ratio of Hits
| IC50,MSUT2 | IC50,PABPN1 | PABPN1/MSUT2 | |
|---|---|---|---|
| hydroxyzine | 0.586 | 9.091 | 15.51 |
| saquinavir | 0.192 | 0.617 | 3.21 |
| nafadotride | 0.876 | ND | ND |
| duloxetine | 0.314 | 4.981 | 15.86 |
| indinavir | 0.247 | 35.78 | 144.9 |
| granisetron | 0.306 | 8.651 | 28.27 |
| clofazimine | 0.731 | 2303 | 31.50 |
| flurbiprofen | 1.086 | 6.685 | 6.156 |
An AlphaScreen (AS) assay was developed as an orthogonal screen. Briefly, in this screen, biotinylated RNA binds streptavidin-coated donor beads, while GST-MSUT2 ZF binds glutathione-coated acceptor beads. When donor beads and acceptor beads are brought within close proximity of one another, laser excitation leads to fluorophore emission of the acceptor bead via an excited singlet oxygen activating chemiluminescence in the acceptor beads. Active compounds prevent beads from being arranged in close proximity and decrease the alpha signal. Compounds were only considered validated if they showed activity both by FP and AS. Duloxetine, saquinavir, and clofazimine displayed dose-dependent inhibition activity by AS (Figure 6) and were further tested for toxicity. Variation between IC50s determined by the primary FP and orthogonal AS is expected due to the underlying differences in detection methods, fluorescence polarization versus singlet oxygen transfer, as well as variation in reagent concentrations. Further, the AS screen is more complex and consists of four components (donor beads, acceptor beads, biotinylated RNA, and GST-MSUT2), while the FP assay is only composed of FAM-RNA and GST-MSUT2. Another discrepancy between assays is that while the inhibitory response for duloxetine acted over 4 log (0.01–10 μM) units in the FP assay (indicative of multiple ligand binding sites), duloxetine inhibition as measured by AS occurred over a single log unit ~1.0–10 μM, indicating a single binding site. Toxicity of these compounds in a HEK cell model was determined using a standard Promega Cell Titer Glo assay. Saquinavir showed some toxicity at the highest tested dose of 40 μM (65% viability), while duloxetine and clofazimine viability was above 90% for all tested doses when normalized to the 2% DMSO vehicle control (Figure 7).
Figure 6.
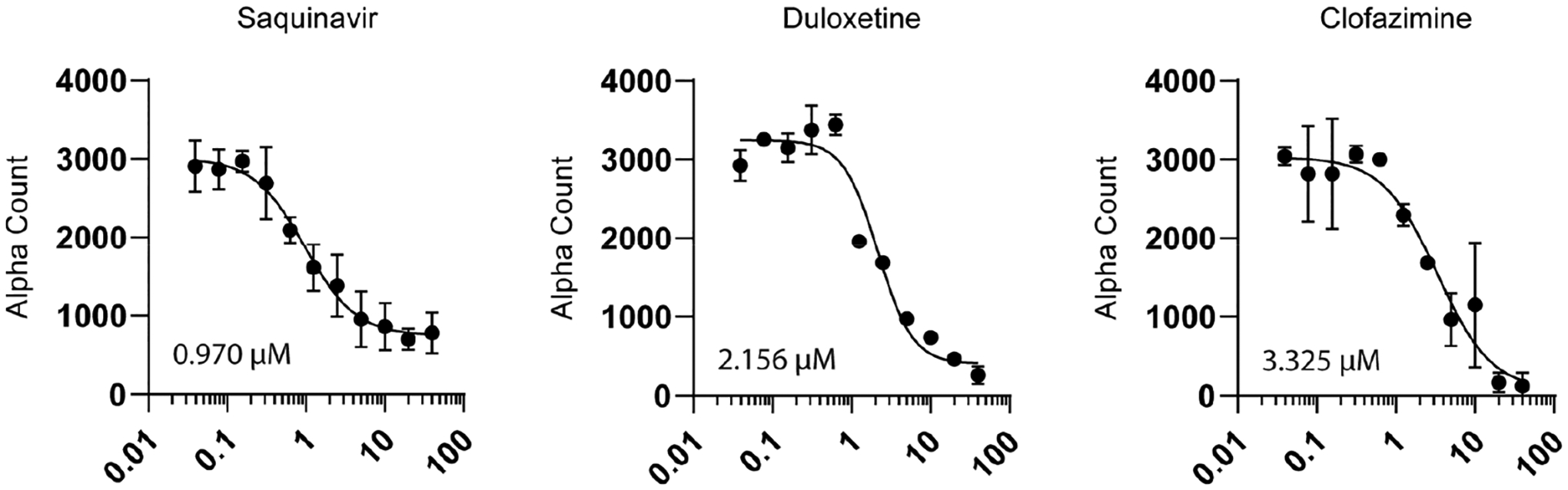
Alpha assay orthogonal dose–response validation where Y-axis is alpha count for compounds which showed dose–response activity. Relative IC50 was determined through a variable slope nonlinear regression model using the equation Y = bottom + (top − bottom)/1 + (IC50/X)HillSlope. Line fitting was performed using GraphPad Prism8 analysis tools. Twenty-two x,y values were analyzed for each compound, N = 2 biological replicates at each point.
Figure 7.
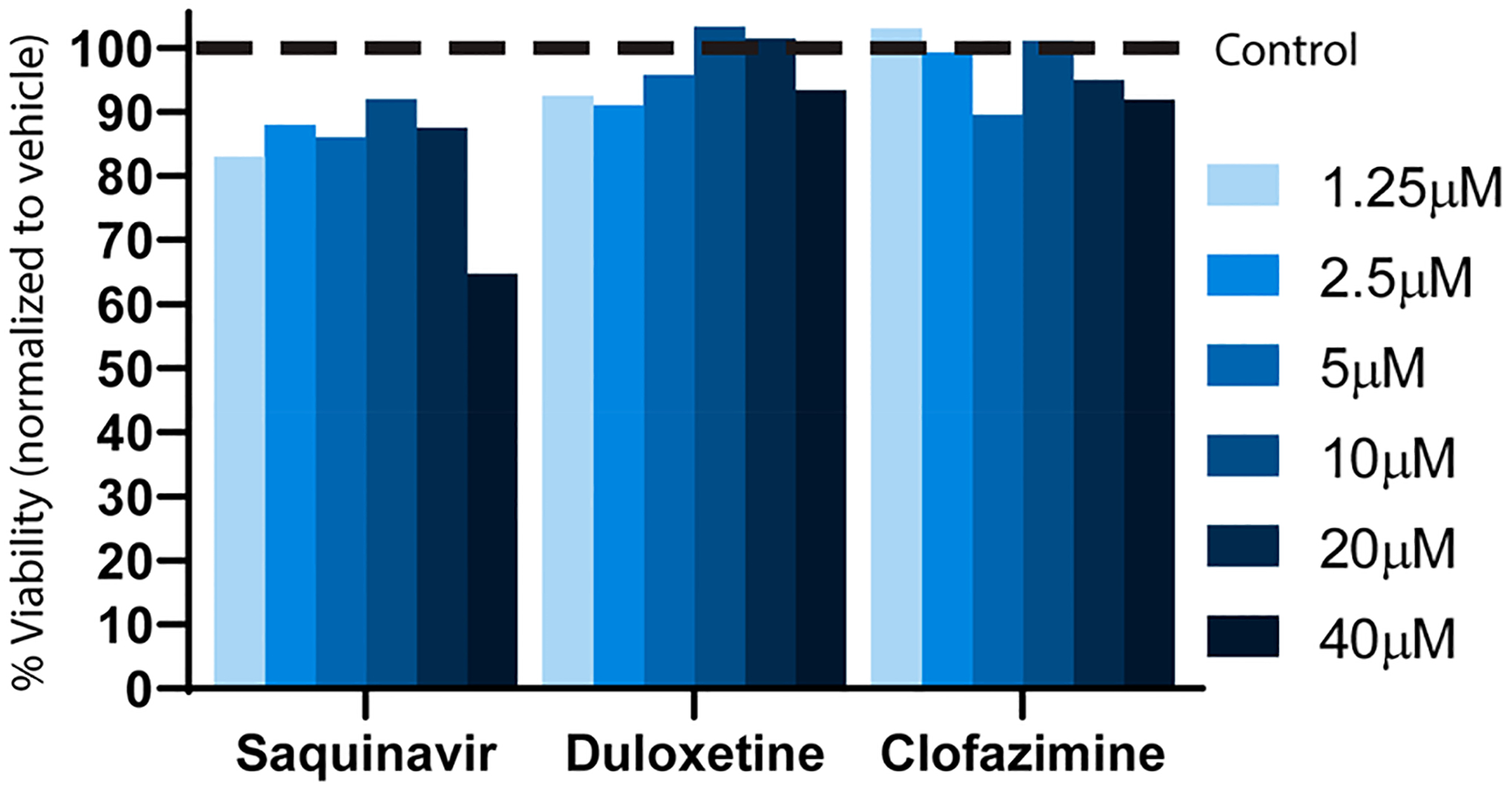
Cell viability assay (Promega Cell Titer Glo) for three validated compounds at indicated concentrations. Dashed line represents 100% normalized viability for 2% DMSO vehicle.
The Utility of Drug Repurposing.
We screened the NIH Clinical Collection (NIHCC) library for small molecules that inhibit the poly(A):MSUT2 RNA-protein interaction as a first effort at MSUT2 drug development. The NIHCC is ideally suited for drug repurposing, a drug development approach that seeks to discover new indications for previously approved drugs.29,32 The advantages realized by drug repurposing include the elimination of over a decade’s worth of preclinical drug discovery work and streamlined clinical trials. This approach has become particularly important when one considers that drug development spending has doubled while very few therapeutics targeting neurodegeneration have proven efficacious.33 Drug collections suitable for repurposing include safe drugs which may have failed to show efficacy in clinical trials, off-patent generics, or those abandoned because of commercial reasons.32 Efforts to find novel indications for existing drugs is largely driven by the extensive costs in time and money to bring drugs to market, estimated at 13 years and $1.8 billion on average.34 Highlighting the merit to this approach, 90% of blockbuster drugs from 1993 now have secondary indications.35 Because safety and efficacy have already been well-established, drug candidates from repurposing collections such as the NIHCC have a much higher likelihood of reaching Phase II trials and beyond.
Therapeutic Potential of MSUT2 as a Target.
Due to its recent identification as a driver of mammalian tauopathy, targeting MSUT2 for therapeutic development remains in the very early preclinical discovery stages.22 Targeting MSUT2 is challenging as MSUT2 is nonenzymatic and has no known targetable binding pocket. Additionally, while the structure of Nab2, the yeast homologue of MSUT2, has been determined by crystallography and NMR,36,37 the MSUT2 protein structure remains unsolved and is required for extensive in silico drug discovery efforts. Although a homologue of Nab2, MSUT2 is structurally distinct. MSUT2 contains five CCCH zinc finger domains instead of seven found in NAB2. Further, NAB2 contains RNA-binding domains not found in MSUT2. Finally, the key MSUT2 binding partner PABPN1 is not found in yeast. Furthermore, until development of methods presented here, there have been no adequate high-throughput screening assays for MSUT2 function and inhibition. Moreover, bulk poly(A) RNA maintenance is a ubiquitous function, and off-target effects are an important consideration as it is suspected that MSUT2 may interact with most mRNA poly(A) tails given its high affinity for poly(A) RNA. Further, there could be other binding partners for MSUT2 beyond PABPN1, and blocking these interactions could have off-target consequences and must be carefully evaluated in future in vivo studies. Despite these concerns, MSUT2 does present an intriguing novel target for drug discovery given its strong effects on pathological tau and that MSUT2 function is dispensable for mouse development as MSUT2 knockout mice appear normal. Further, compounds identified in this and future drug screening efforts will provide tool compounds for probing the underlying pathological mechanism of MSUT2 in models of tauopathy.
Overexpression of MSUT2 in tau-transgenic mouse hippocampi leads to increases in pathological tau deposition, neuroinflammation, and neurodegeneration. Conversely, knocking out MSUT2 in mouse models of tauopathy has the reciprocal effect and reduces hyperphosphorylated, pretangle, and tau tangle burden while being anti-inflammatory and neuroprotective.22 Further, MSUT2 knockout protects against memory deficits as shown by the Barnes maze paradigm. MSUT2 knockout mice (in a non-tau background) are healthy, lack cognitive deficits, and display normal neurological function.22 As mentioned previously, MSUT2 interacts with PABPN1 and forms a complex within neurons. Evidence suggests that while some MSUT2 positive neurons have tau tangles, depletion of the MSUT2:PABPN1 complex exacerbates Alzheimer’s disease pathology with higher pathological tau burden, increased neuronal loss, and an early onset of AD.22 Thus, PABPN1 would appear to be a critical antitarget for poly(A):MSUT2 inhibitors.
Perspective on Hit Compounds and Future Screening.
The screen methodology presented here identifies three potential repurposing candidates. Duloxetine, a serotonin-norepinephrine reuptake inhibitor known as Cymbalta, has indications for depression, anxiety, and pain caused by neuropathy as well as fibromyalgia. Notably, while first approved for major depressive disorder in 2004, additional indications were not approved until years later: fibromyalgia in 200838,39 and musculoskeletal pain in 2010.40 Duloxetine became available as a generic in 2013. Notably, a 10 year study of 20,215 elderly patients prescribed various serotonin reuptake inhibitors showed that duloxetine use was associated with a reduced risk of dementia.41 Further, in a rat model of chronic cerebral hypoperfusion, duloxetine attenuated neuronal loss in the hippocampus.42 Despite the promising activity of duloxetine, we predict it will most likely have limited therapeutic potential as an inhibitor of MSUT2-mediated tauopathy. Potential therapeutics must be very potent as our previous work has indicated at least 75% knockdown of MSUT2 must be achieved. Because duloxetine is heavily bound (~90%) to albumin and α1-acid glycoprotein in the plasma, very high and toxic doses would be required to achieve required MSUT2 knockdown.43 Finally, although known to cross the blood-brain barrier to exert therapeutic effects, it has been shown that duloxetine crosses into cerebral spinal fluid at a much lower rate than other antidepressants44 and this low penetration may prevent duloxetine from having a substantial effect on MSUT2 activity. Despite these limitations, duloxetine could prove to have an important role as a tool compound in probing the molecular mechanism of MSUT2.
Saquinavir, developed by Roche under the brand name Invirase, is a protease inhibitor prescribed as an antiretroviral to treat HIV infection. Saquinavir’s mechanism of action is to bind and inhibit viral proteases HIV-1 and HIV-2, leading to the prevention of viral maturation.45 There are currently no secondary indications for saquinavir, although there have been studies highlighting its activity against other targets. For example, a recent repurposing effort identified saquinavir (as well as clofazimine) as a potential therapeutic for Chagas disease.46 It should be noted that saquinavir and many other HIV targeting drugs have low brain penetrance. Saquinavir has been shown as a substrate of efflux transporters which may further reduce its ability to accumulate in the brain.47
Clofazimine, though primarily used as a treatment for leprosy caused by Mycobacerium leprae, has been studied for other indications. Clofazimine is currently being looked at for its effectiveness in treating tuberculosis, caused by the related bacteria Mycobacterium tuberculosis.48 Its canonical mode of action for these indications is to preferentially bind guanine-rich areas of Mycobacterium DNA and prevent bacterial development. Additional research is ongoing into clofazimine treatment as an anticancer agent.49 Although it has been shown that there is little interaction of clofazimine with poly(A) in a previous study,50 it is a reasonable assumption that clofazimine could be binding directly to poly(A) and inhibiting poly(A):MSUT2 interaction in our in vitro studies. Further, because it is known that clofazimine is unable to cross the blood-brain barrier, any further translational studies will require generation of brain-penetrant derivatives.49
Future drug discovery efforts against MSUT2 and other potential targets exacerbating neurodegeneration will require much larger repositories of compounds to find lead candidates as well as discovery of new screening methods and alternative therapeutic strategies. Further, advances are being made using in silico or virtual drug screening in regard to algorithms predicting protein:inhibitor conformations as well as in scoring potential therapeutics, highlighting the need for a molecular structure of MSUT2. This so-called virtual docking of ligands to MSUT2 will be a key avenue in increasing throughput for lead candidate discovery. Our work here demonstrates MSUT2 activity is clearly targetable and a strong candidate for further small molecule screening campaigns.
METHODS
RNA.
5′ Fluorescein labeled and unlabeled poly(A) RNA were purchased from IDT (sequences 5′-AAAAAAAAAAAAAAA-3′ and 5′FAM-AAAAAAAAAAAAAAA-3′). Both FAM-labeled and unlabeled RNA were diluted to 100 μM in RNase/DNase free Qiagen water and stored at −80 °C, away from light.
Recombinant Protein.
MSUT2 ZF (CCCH domains only) and PABNP1 cDNA were cloned into the pGEX-6P-1 vector (Pharmacia). MSUT2 ZF and PABPN1 encoding plasmids were transformed into BL21 (DE3) bacteria. Terrific broth (TB, 10 mL) starter cultures were grown overnight at 37 °C in a shaking incubator. The following morning, 1 L TB cultures were inoculated and grown at 37 °C with shaking to the log phase and induced with 1 mM final concentration of IPTG for 4 h at 37 °C. Following induction, DNA and RNA were degraded using benzonase nuclease or a cocktail of DNase I and RNase A. Affinity based gravity column purification was performed by binding GST-tagged MSUT2 or PABPN1 to sepharose-glutathione resin and subsequently eluting with 20 mM glutathione. The resulting eluate was buffer exchanged into PBS and stored at −80 °C. Protein purity and yield were analyzed via Bradford assay and Coomassie-stained SDS-PAGE.
Chemical Library.
The NIH Chemical Collection (NIHCC) was purchased from Evotec and contains a total of nine 96-well plates (700 compounds at 10 mM concentration at 10 μL volumes). For screening, compound was diluted to 250 μM in a separate working drug dilution plate. A volume of 2 μL of compound was transferred to 50 μL final assay volume via an Integra Viaflo pipet, for a final compound concentration of 10 μM.
Fluorescence Polarization Assay.
Fluorescence polarization assay was performed in 1/2 area black plates (Corning 3686). Reaction mixtures were a total volume of 50 μL and contained final concentrations of 125 nM MSUT2 RNABP and 10 nM FAM-RNA in PBS, transferred using an Integra Viaflo pipet with a 96/50 μL head. Then 2 μL of stock compound was transferred yielding a 10 μM final concentration. Plates were then incubated at room temperature, without shaking, for 20 min in a BioSpa8 automated incubator and transferred by robotic arm to a Biotek Cytation 5 with a preconfigured green polarization filter cube (8040561) at excitation 485/20 and emission 528/20, a dichroic mirror at 510 nm, and a read height of 10 mm. Fluorescence polarization was calculated by first subtracting background from a buffer-only control well and then using the equation to determine the polarization (P).
Alpha Screen Assay.
Samples were set up in 96-well PerkinElmer 1/2 area white opaque-bottom plates (PE06). A total reaction volume of 50 μL was used by first adding 20 μL of donor beads (10 μg/mL final concentration), then 10 μL of biotinylated RNA (50 nM final concentration), and next 10 μL of GST-MSUT2 protein (50 nM final concentration). This mixture was incubated at room temperature for 30 min away from light after which 10 μL of acceptor beads (1.25 μg/mL final concentration) was added. The plate was then incubated again at room temperature away from light for 60 min and subsequently read on a PerkinElmer EnSpire multimode microplate reader using a standard 96-well alpha assay protocol.
Cell Culture and Assays.
HEK293 cells were cultured under standard tissue culture conditions [DMEM, 10% defined fetal bovine serum, penicillin (1000 IU/mL), and streptomycin (1000 μg/mL)] as previously described.22 Cell viability was assessed as previously described. Briefly, HEK-293 cells grown to 70% confluence in a 96-well plate were treated with varying concentrations of compound (final concentration of 2% DMSO) and incubated for 72 h. Next, Promega Cell Titer Glo assay was used to assess viability per the manufacturer’s instructions (Promega G7570).
Statistical Analyses and Figures.
Graphs were generated using GraphPad Prism 8. IC50 calculations were performed using GraphPad Prism 8 curve fitting using 4-parameter nonlinear regression. Data points consisted of at least two biological replicates. Error bars represent ± standard error of the mean. Polarization is reported as mP (millipolarization) = 1000 × (I∥ − I⊥)/(I∥ + I⊥), where I∥ is equal to the intensity of the parallel component and I⊥ is the intensity of the perpendicular component. Background correction was done for each plate.
ACKNOWLEDGMENTS
We thank Timothy Martins, Elaine Loomis, Dale Whittington, Grace Scuderi, Riley Weaver, and Jeanna Wheeler for sharing their technical expertise.
Funding
This work was funded by the National Institutes of Health (R56AG057642 and RF1AG055474 to B.C.K.), the Department of Veterans Affairs (Merit Review Grant #I01BX002619 to B.C.K.) and a Postdoctoral Fellowship from the Washington Research Foundation (WRF to J.D.B.)
ABBREVIATIONS
- Aβ
amyloid-beta
- AS
alpha screen
- AD
Alzheimer’s disease
- FAM
fluorescein amidite
- FAM-RNA
fluorescein amidite labeled RNA
- FUS
fused in sarcoma
- FP
fluorescence polarization
- GST
glutathione S-transferase
- HTS
high-throughput screen
- MSUT2
mammalian suppressor of tauopathy 2
- PABPN1
poly(A) Binding Protein Nuclear 1
- poly(A)
polyadenosine
- NFT
neurofibrillary tangles
- NIHCC
NIH Clinical Collection
- RBP
RNA binding protein
- TB
terrific broth
- TDP-43
TAR DNA-binding protein 43 kDa
- ZF
zinc fingers
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acschemneuro.0c00214
The authors declare the following competing financial interest(s): T.J.S., J.D.B., and B.C.K. have a patent application pending (assigned U.S. Patent Application No. 16/383,178) “COMPOSITIONS AND METHODS FOR SUPPRESSING MSUT2”. The remaining authors declare no conflicts of interest.
Contributor Information
Jeremy D. Baker, Division of Gerontology and Geriatric Medicine, Department of Medicine, University of Washington, Seattle, Washington 98104, United States; Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, Washington 98108, United States
Rikki L. Uhrich, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, Washington 98108, United States
Timothy J. Strovas, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, Washington 98108, United States
Aleen D. Saxton, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, Washington 98108, United States
Brian C. Kraemer, Division of Gerontology and Geriatric Medicine, Department of Medicine, Department of Psychiatry and Behavioral Sciences, and Department of Pathology, University of Washington, Seattle, Washington 98104, United States; Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, Washington 98108, United States.
REFERENCES
- (1).Congdon EE, and Sigurdsson EM (2018) Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol 14, 399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Corrada MM, Brookmeyer R, Paganini-Hill A, Berlau D, and Kawas CH (2010) Dementia incidence continues to increase with age in the oldest old: the 90+ study. Ann. Neurol 67, 114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Alzheimer’s Association (2019) 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dementia 15, 321–387. [Google Scholar]
- (4).Wood JG, Mirra SS, Pollock NJ, and Binder LI (1986) Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc. Natl. Acad. Sci. U. S. A 83, 4040–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Glenner GG, and Wong CW (2012) Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. 1984. Biochem. Biophys. Res. Commun 425, 534–539. [DOI] [PubMed] [Google Scholar]
- (6).Bloom GS (2014) Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71, 505–508. [DOI] [PubMed] [Google Scholar]
- (7).Hanseeuw BJ, Betensky RA, Jacobs HIL, Schultz AP, Sepulcre J, Becker JA, Cosio DMO, Farrell M, Quiroz YT, Mormino EC, Buckley RF, Papp KV, Amariglio RA, Dewachter I, Ivanoiu A, Huijbers W, Hedden T, Marshall GA, Chhatwal JP, Rentz DM, Sperling RA, and Johnson K (2019) Association of Amyloid and Tau With Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol 76, 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Arriagada PV, Growdon JH, Hedley-Whyte ET, and Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631. [DOI] [PubMed] [Google Scholar]
- (9).Xia C, Makaretz SJ, Caso C, McGinnis S, Gomperts SN, Sepulcre J, Gomez-Isla T, Hyman BT, Schultz A, Vasdev N, Johnson KA, and Dickerson BC (2017) Association of In Vivo [18F]AV-1451 Tau PET Imaging Results With Cortical Atrophy and Symptoms in Typical and Atypical Alzheimer Disease. JAMA Neurol 74, 427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Rojas JC, and Boxer AL (2016) Targeting tauopathies for therapeutic translation. Nat. Rev. Neurol 12, 74–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Huang Y, and Mucke L (2012) Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wolozin B, and Ivanov P (2019) Stress granules and neurodegeneration. Nat. Rev. Neurosci 20, 649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Gerstberger S, Hafner M, and Tuschl T (2014) A census of human RNA-binding proteins. Nat. Rev. Genet 15, 829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hentze MW, Castello A, Schwarzl T, and Preiss T (2018) A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol 19, 327–341. [DOI] [PubMed] [Google Scholar]
- (15).Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, and Oda T (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun 351, 602–611. [DOI] [PubMed] [Google Scholar]
- (16).Ito D, Hatano M, and Suzuki N (2017) RNA binding proteins and the pathological cascade in ALS/FTD neurodegeneration. Sci. Transl. Med 9, eaah5436. [DOI] [PubMed] [Google Scholar]
- (17).Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, and Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. [DOI] [PubMed] [Google Scholar]
- (18).Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N, Tome FM, Lafreniere RG, Rommens JM, Uyama E, Nohira O, Blumen S, Korczyn AD, Heutink P, Mathieu J, Duranceau A, Codere F, Fardeau M, and Rouleau GA (1998) Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet 18, 164–167. [DOI] [PubMed] [Google Scholar]
- (19).Kwiatkowski TJ Jr., Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, and Brown RH Jr. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. [DOI] [PubMed] [Google Scholar]
- (20).Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, and Shaw CE (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Guthrie CR, Greenup L, Leverenz JB, and Kraemer BC (2011) MSUT2 is a determinant of susceptibility to tau neurotoxicity. Hum. Mol. Genet 20, 1989–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wheeler JM, McMillan P, Strovas TJ, Liachko NF, Amlie-Wolf A, Kow RL, Klein RL, Robinson PS, Guthrie C, Saxton A, Kanaan NM, Raskind M, Peskind E, Trojanowski JQ, Lee VMY, Wang L-S, Keene CD, Bird T, Schellenberg GD, and Kraemer B (2019) The poly(A) binding protein MSUT2 controls resistance to both pathological tau and gliosis. Sci. Transl. Med 11, eaao6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wigington CP, Williams KR, Meers MP, Bassell GJ, and Corbett AH (2014) Poly(A) RNA-binding proteins and polyadenosine RNA: new members and novel functions. Wiley Interdiscip. Rev. RNA 5, 601–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wahle E (1991) A novel poly(A)-binding protein acts as a specificity factor in the second phase of messenger RNA poly-adenylation. Cell 66, 759–768. [DOI] [PubMed] [Google Scholar]
- (25).Malerba A, Klein P, Bachtarzi H, Jarmin SA, Cordova G, Ferry A, Strings V, Espinoza MP, Mamchaoui K, Blumen SC, St Guily JL, Mouly V, Graham M, Butler-Browne G, Suhy DA, Trollet C, and Dickson G (2017) PABPN1 gene therapy for oculopharyngeal muscular dystrophy. Nat. Commun 8, 14848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jameson DM, and Ross JA (2010) Fluorescence polarization/anisotropy in diagnostics and imaging. Chem. Rev 110, 2685–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Weber G (1952) Polarization of the fluorescence of macromolecules. I. Theory and experimental method. Biochem. J 51, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhang JH, Chung TD, and Oldenburg KR (1999) A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screening 4, 67–73. [DOI] [PubMed] [Google Scholar]
- (29).Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, Doig A, Guilliams T, Latimer J, McNamee C, Norris A, Sanseau P, Cavalla D, and Pirmohamed M (2019) Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discovery 18, 41. [DOI] [PubMed] [Google Scholar]
- (30).Apponi LH, Leung SW, Williams KR, Valentini SR, Corbett AH, and Pavlath GK (2010) Loss of nuclear poly(A)-binding protein 1 causes defects in myogenesis and mRNA biogenesis. Hum. Mol. Genet 19, 1058–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Benoit B, Mitou G, Chartier A, Temme C, Zaessinger S, Wahle E, Busseau I, and Simonelig M (2005) An essential cytoplasmic function for the nuclear poly(A) binding protein, PABP2, in poly(A) tail length control and early development in Drosophila. Dev. Cell 9, 511–522. [DOI] [PubMed] [Google Scholar]
- (32).Sleigh S, and Barton C (2010) Repurposing Strategies for Therapeutics. Pharm. Med 24, 151–159. [Google Scholar]
- (33).Cummings JL, Morstorf T, and Zhong K (2014) Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimer’s Res. Ther 6, 37–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Gupta SC, Sung B, Prasad S, Webb LJ, and Aggarwal BB (2013) Cancer drug discovery by repurposing: teaching new tricks to old dogs. Trends Pharmacol. Sci 34, 508–517. [DOI] [PubMed] [Google Scholar]
- (35).Gelijns AC, Rosenberg N, and Moskowitz AJ (1998) Capturing the Unexpected Benefits of Medical Research. N. Engl. J. Med 339, 693–698. [DOI] [PubMed] [Google Scholar]
- (36).Brockmann C, Soucek S, Kuhlmann SI, Mills-Lujan K, Kelly SM, Yang J-C, Iglesias N, Stutz F, Corbett AH, Neuhaus D, and Stewart M (2012) Structural Basis for Polyadenosine-RNA Binding by Nab2 Zn Fingers and Its Function in mRNA Nuclear Export. Structure 20, 1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kuhlmann SI, Valkov E, and Stewart M (2014) Structural basis for the molecular recognition of polyadenosine RNA by Nab2 Zn fingers. Nucleic Acids Res 42, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wright CL, Mist SD, Ross RL, and Jones KD (2010) Duloxetine for the treatment of fibromyalgia. Expert Rev. Clin. Immunol 6, 745–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lunn MP, Hughes RA, and Wiffen PJ (2014) Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst. Rev, Cd007115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Smith HS, Smith EJ, and Smith BR (2012) Duloxetine in the management of chronic musculoskeletal pain. Ther. Clin. Risk Manage 8, 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kostev K, Bohlken J, and Jacob L (2019) Analysis of the Effects of Selective Serotonin (and Noradrenaline) Reuptake Inhibitors on the Risk of Dementia in Patients with Depression. J. Alzheimer’s Dis 69, 577–583. [DOI] [PubMed] [Google Scholar]
- (42).Park JA, and Lee CH (2018) Neuroprotective Effect of Duloxetine on Chronic Cerebral Hypoperfusion-Induced Hippocampal Neuronal Damage. Biomol. Ther 26, 115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Knadler MP, Lobo E, Chappell J, and Bergstrom R (2011) Duloxetine: clinical pharmacokinetics and drug interactions. Clin. Pharmacokinet 50, 281–294. [DOI] [PubMed] [Google Scholar]
- (44).Paulzen M, Gründer G, Veselinovic T, Wolf B, Hiemke C, and Lammertz SE (2016) Duloxetine enters the brain – But why is it not found in the cerebrospinal fluid. J. Affective Disord 189, 159–163. [DOI] [PubMed] [Google Scholar]
- (45).Pribis JP, Al-Abed Y, Yang H, Gero D, Xu H, Montenegro MF, Bauer EM, Kim S, Chavan SS, Cai C, Li T, Szoleczky P, Szabo C, Tracey KJ, and Billiar TR (2015) The HIV Protease Inhibitor Saquinavir Inhibits HMGB1-Driven Inflammation by Targeting the Interaction of Cathepsin V with TLR4/MyD88. Mol. Med 21, 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Bellera CL, Balcazar DE, Vanrell MC, Casassa AF, Palestro PH, Gavernet L, Labriola CA, Gálvez J, Bruno-Blanch LE, Romano PS, Carrillo C, and Talevi A (2015) Computer-guided drug repurposing: Identification of trypanocidal activity of clofazimine, benidipine and saquinavir. Eur. J. Med. Chem 93, 338–348. [DOI] [PubMed] [Google Scholar]
- (47).Park S, and Sinko PJ (2005) P-Glycoprotein and Mutlidrug Resistance-Associated Proteins Limit the Brain Uptake of Saquinavir in Mice. J. Pharmacol. Exp. Ther 312, 1249. [DOI] [PubMed] [Google Scholar]
- (48).Bahuguna A, and Rawat DS (2020) An overview of new antitubercular drugs, drug candidates, and their targets. Med. Res. Rev 40, 263–292. [DOI] [PubMed] [Google Scholar]
- (49).Mulkearns-Hubert EE, Torre-Healy LA, Silver DJ, Eurich JT, Bayik D, Serbinowski E, Hitomi M, Zhou J, Przychodzen B, Zhang R, Sprowls SA, Hale JS, Alban TJ, Berezovsky A, Bell BA, Lockman PR, Jha BK, and Lathia JD (2019) Development of a Cx46 Targeting Strategy for Cancer Stem Cells. Cell Rep 27, 1062–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Morrison NE, and Marley GM (1976) Clofazimine binding studies with deoxyribonucleic acid. Int. J. Lepr. Other Mycobact. Dis 44, 475–481. [PubMed] [Google Scholar]