

Abstract
Clonal hematopoiesis of indeterminate potential (CHIP) is the presence of a clonally expanded hematopoietic stem cell caused by a leukemogenic mutation in individuals without evidence of hematologic malignancy, dysplasia, or cytopenia. CHIP is associated with a 0.5-1.0% risk per year of leukemia. Remarkably, it confers a two-fold increase in cardiovascular risk independent of traditional risk factors. Roughly 80% of patients with CHIP have mutations in epigenetic regulators DNMT3A, TET2, ASXL1, DNA damage repair genes PPM1D, TP53, the regulatory tyrosine kinase JAK2, or mRNA spliceosome components SF3B1, and SRSF2. CHIP is associated with a pro-inflammatory state that has been linked to coronary artery disease, myocardial infarction, and venous thromboembolic disease, as well as prognosis among those with aortic stenosis and heart failure. Heritable and acquired risk factors are associated with increased CHIP prevalence, including germline variation, age, unhealthy lifestyle behaviors (i.e. smoking, obesity), inflammatory conditions, premature menopause, HIV and exposure to cancer therapies. This review aims to summarize emerging research on CHIP, the mechanisms underlying its important role in propagating inflammation and accelerating cardiovascular disease, and new studies detailing the role of associated risk factors and co-morbidities that increase CHIP prevalence.
Keywords: CHIP, ASCVD, Inflammation, Heart Failure, Aortic Stenosis, Cardio-Oncology
1. Introduction
Clonal hematopoiesis refers to the presence of clonal populations of hematopoietic stem cells (HSCs). Hematopoiesis is generally a polyclonal process with HSCs of equipotential, giving rise to erythroid, lymphoid, myeloid, or megakaryocytic cells. Mutations may occur in genes that confer selective fitness advantage with aging HSCs less adept to correct for these errors, giving rise to clonally expanded populations of stem cells [1]. Clonal hematopoiesis may occur in the context of selective pressures such as cytotoxic therapies and tobacco smoking, inability to rectify DNA replication errors among aging HSCs or in the context of neutral drift, the random genetic drift of evolutionarily neutral alleles at the molecular level.
While clonal hematopoiesis can result in hematologic malignancy, cooperative mutations in additional genes are required to induce malignant transformation. Consequently, most people with clonal hematopoiesis never develop blood cancer. Therefore, these clonal populations are referred to as having “indeterminate potential.” In 2015, a formal definition of clonal hematopoiesis of indeterminate potential (CHIP) was proposed with the following qualifiers: CHIP must occur in the absence of morphological variation in blood cells; a candidate driver gene mutation should be present at variant allele frequency of at least 2% in peripheral blood; and in the absence of diagnostic criteria for hematologic malignancy. Defining CHIP at a VAF of at least 2% reflects not only technical limits of sequencing technologies, but also a practical cutoff. With the emergence of extremely high-resolution sequencing, nearly all healthy 50-60 year-old patients tested had evidence of clonal hematopoietic populations with VAF of 0.03%, yet studies show that very small clones have minimal clinical consequence [2, 3]. Nevertheless, the trajectory from smaller to larger clones currently remains ill-defined.
2. Early Epidemiologic Evidence Linking CHIP and Cardiovascular Disease
While CHIP carries an increased relative risk of incident hematologic malignancy (HR ~13) [4, 5], the overall increase in absolute risk is comparatively small – roughly 0.5-1% per year [6]. Early epidemiologic studies noted that CHIP increased the risk of death by 40%, a magnitude far greater than could be explained by the risk of hematologic malignancy alone. Indeed, in these initial cohorts only one individual out of 246 carrying mutations associated with clonal hematopoiesis died of hematologic cancer [5]. Subsequent secondary analyses posited increased risk of coronary artery disease (HR 1.8 - 2.0), ischemic stroke (HR 2.6), and premature myocardial infarction (HR 4.0) independent of traditional risk factors for CVD which were later confirmed in independent datasets [5, 7]. In fact, the magnitude of risk elevation from CHIP rivals that of traditional Framingham risk factors (Figure 2) [5]. Subsequent studies have gone on to link CHIP carrier status with poorer prognosis in related cardiovascular conditions such as heart failure [8], and aortic stenosis [9].
Figure 2: Selected hazard ratios (HR) for CHIP and incident coronary heart disease are of similar magnitude to traditional risk factors.
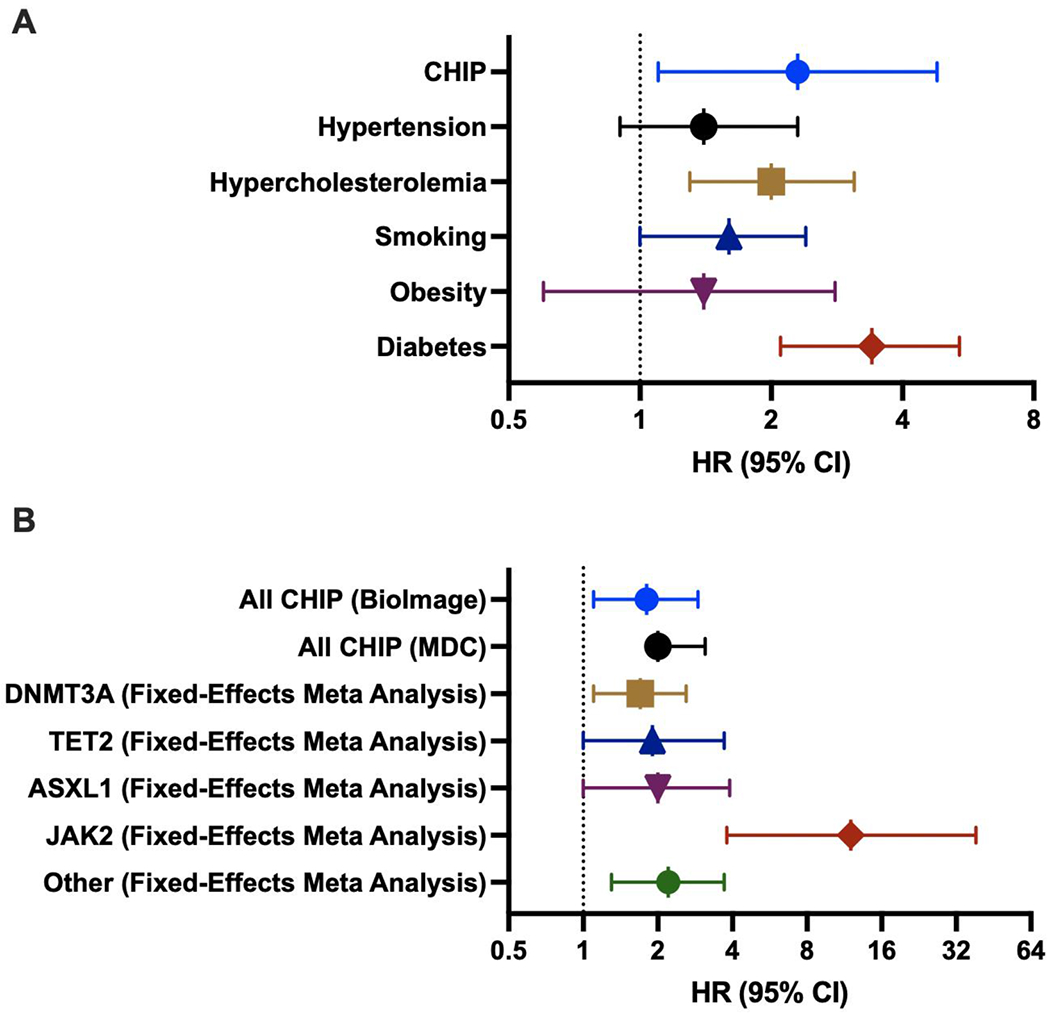
(A) HR with 95% CI for incident coronary heart disease adjusted for age, sex, HTN, HLD, smoking, obesity, diabetes among Jackson Heart Study & FUSION participants, Summarized data from Jaiswal et al 2014 [4]
(B) HR with 95% CI for incident coronary heart disease adjusted for age, sex, HTN, total and HDL cholesterol, triglycerides, smoking, and type 2 diabetes among BioImage, MDC (CHIP) and a fixed-effects meta-analysis for each gene (incorporating BioImage, MDC, JHS/Fusion/FHS). Summarized data from Jaiswal et al 2017 [7]
3. Mechanisms of CHIP Candidate Driver Mutations in Driving Inflammation and CVD
The most commonly mutated candidate driver genes in CHIP are DNMT3A, TET2, and ASXL1, which increase the risk of atherosclerotic cardiovascular disease with HR of 1.7, 1.9 and 2.0 respectively (Table 1). These three mutations, referred to as “DTA mutations” in the myeloid leukemia literature, account for ~80% of all CHIP cases [7]. Additional mutations are seen in JAK2, which is particularly associated with increased rates of thrombosis, as well as the DNA damage response pathway genes PPM1D and TP53, and mRNA splicing factors SRSF2 and SF3B1. Mutations in these sets of genes are commonly observed in myelodysplastic syndrome (MDS), myeloproliferative neoplasms (MPNs) and acute myeloid leukemia [10–13].
Table 1:
Common CHIP Driver Mutations
| Candidate Driver | % of CHIP1 | Mechanism | References |
|---|---|---|---|
| DNMT3A | ~58.5% | Methyltransferase enzyme that catalyzes DNA methylation at CpG sites and alters epigenetic signature; tumor suppressor gene | [9, 14, 15, 17, 18] |
| TET2 | ~20% | DNA demethylase TET2 (ten-eleven translocation-2) augments DNA methylation and affects transcription by recruiting histone deacetylases toward promoters; tumor suppressor gene | [9, 20, 21, 23, 25] |
| ASXL1 | ~8.0% | Epigenetic modulator and chromatin-binding protein, function relatively unknown | [27, 29] |
| JAK2 | ~3.2% | Transmits intracellular signals downstream of cytokine receptors. JAK2 tyrosine phosphorylates and activates TET2 in response to cytokines, linking extracellular signals with epigenetic changes in hematopoiesis | [24, 31, 33, 34] |
| PPM1D, TP53 | ~3.8%, 1.9% | DNA damage response pathway in regulatory feedback loop with the tumor suppressor p53. | [35, 36] |
| SF3B1, SRSF2 | 2%, 2% | mRNA spliceosome complex components | [24] |
| No candidate driver mutation | Limits of detection methods, epigenetic changes not detectable, neutral drift, or mosaic chromosomal alterations |
[37, 40–42] |
Approximate percentages among CHIP with candidate driver mutations as detected in “Inherited causes of clonal haematopoiesis in 97,691 whole genomes, Bick et al Nature 2020” [24]
DNMT3A
DNA methyltransferase 3a (DNMT3A) is the most commonly mutated gene in CHIP. DNMT3A encodes a methyltransferase enzyme that catalyzes DNA methylation at CpG sites and is a critical epigenetic regulator of gene expression. The majority of pathogenic mutations are loss-of-function including disruptive missense mutations in regulatory and catalytic domains, nonsense mutations, insertions-deletions, and splice site mutations. DNMT3A pathogenic mutations enhance HSC self-renewal [14] and promote the expression of multipotency genes while suppressing differentiation factor expression [15]. This enables DNMT3A mutations to affect all hematopoietic lineages, inducing pro-inflammatory T-cell polarization and activating the inflammasome complex.
Mouse model experiments utilizing CRISPR gene editing establish that DNMT3A CHIP causes aberrant inflammation but may also be fostered by inflammation itself. Murine macrophage cell lines bearing DNMT3A mutation show increased inflammatory gene induction in response to lipopolysaccharide challenge, with increased expression of Cxcl1, Cxcl2, IL-6 and Ccl5. Studies also show that a pro-inflammatory environment can reciprocally drive DNMT3A CH expansion. In chronic infection, DNMT3A-mutant HSCs outcompete wild-type HSCs and lead to DNMT3A-CH in peripheral blood, via increased resistance to stress-induced apoptosis and differentiation defects. Inflammatory interferon-gamma was sufficient to drive this clonal expansion of DNMT3A-mutant HSCs [16]. This pro-inflammatory environment has cardiac consequences: in a mouse model, DNMT3A deletion in HSCs leads to increased angiotensin II-mediated cardiac hypertrophy, reduced cardiac function, and greater cardiac and renal fibrosis [17].
Studies of DNMT3A CHIP in humans demonstrate similar downstream effects. Transcriptomic profiling of peripheral blood mononuclear cells derived from heart failure patients who were DNMT3A-mutant CHIP carriers demonstrated significantly increased expression of inflammatory interleukins IL-1β, IL-5, IL-8, activation of the NLRP3 inflammasome, macrophage inflammatory proteins CCL3 and CCL35, and resistin. Studies of this population also reveal a pro-inflammatory circulating monocyte signature and marked induction of T-cell stimulating genes like CD58, increased expression of T-cell alpha receptor and changes in T-cell subtype signature [18]. This pro-inflammatory T-cell polarization signature is observed in DNMT3A CHIP carriers in a cohort of aortic stenosis patients undergoing transcatheter aortic valve replacement, with a significantly increased Th17/Treg ratio in such patients [9].
TET2
The second most commonly mutated CHIP gene is DNA demethylase TET2 (ten-eleven translocation-2). Notably while DNMT3A adds methyl groups at CpG sites, TET2 oxidizes the methyl group which is the first step in removing the mark. Remarkably these two biochemically opposing functions have a convergent stem cell phenotype. In mouse models, TET2 loss of function enhances HSC self-renewal and preferentially leads to differentiation toward myeloid lineages [19]. TET2 has an important role in restraining the expression of inflammatory genes in macrophages. TET2-deficient macrophages show increased inflammation, both spontaneous and in response to lipopolysaccharide, further potentiating an activated pro-inflammatory state [20]. TET2 deficiency is associated with higher circulating levels of IL-1β through induction of the NLRP3 inflammasome [21], IL-6 [22], and IL-8 [7].
This pro-inflammatory state potentiated by TET2 CHIP leads to accelerated atherosclerosis. Initial studies of Ldlr−/− atherosclerogenic mouse models marrow reconstituted with Tet2−/− HSCs via irradiation and bone marrow transplantation demonstrated greater atherosclerotic plaque burden [7]. In a similar mouse model, Fuster et al mimicked the effect of variant allele frequencies observed in humans by studying mice with ~10% Tet2 −/− bone marrow with 90% Tet2 WT, followed by ~10% Tet2+/− bone marrow with 90% Tet2 WT. They observed a similar acceleration of atherosclerotic plaque burden at this clinically relevant VAF although speed of Tet2 haploinsufficient clonal dominance is likely faster in the model than in humans [21]. Subsequent murine studies showed Tet2 loss of function accelerates myocardial fibrosis and heart failure in pressure-overload- and ischemia-induced murine models of heart failure, modulated through the induction of the IL-1β/NLRP3 inflammasome [23].
Many findings extend to human populations to-date, with in silico analysis of the TOPMed cohort revealing significantly increased serum IL-1β levels among TET2 CHIP carriers [24]. Cardiovascular risk conferred in DNMT3A- and TET2-mutant CHIP carriers could be abrogated by an inhibitory IL-6 receptor gene variant (IL6R p.Asp358Ala) [25], using a stratified Mendelian randomization approach to demonstrate the central role of pro-inflammatory mediators NLRP3, IL-1β and IL6 in mediating the development of CHIP-associated atherosclerosis. A cohort of patients with severe degenerative aortic stenosis undergoing transcatheter aortic valve implantation (TAVI) noted an association between TET2 CHIP carrier status and higher circulating levels of non-classical monocytes (CD14dimCD16++), which secrete higher concentrations of pro-inflammatory cytokines including TNF-α, IL-1β, and IL-8. Compared to those without CHIP, these patients had an increased medium-term all-cause mortality following TAVI [9]. Deep-targeted amplicon sequencing of bone-marrow derived mononuclear cells among a cohort of chronic heart failure patients also showed an association between TET2 CHIP carrier status and HF progression and poorer clinical outcomes. This included a dose-response relationship with increasing TET2 variant allele frequency [8].
ASXL1
ASXL1 (additional sex combs-like 1) is the third most commonly mutated gene in CHIP, with its gene product regulating polycomb-mediated transcriptional repression. ASXL1 mutations are typically frameshift or nonsense mutations occurring near the 5′ end of the gene and likely lead to gain of function and aberrant histone modifications [26]. Observational studies detail a link between ASXL1 mutations in blood cells with smoking [27], and among patients with HIV [28]. ASXL1 deletion facilitates aberrant gene expression and results in myeloid transformation [29]. However, ASXL1 deletion in mice also impairs HSC functioning, and so the mechanisms by which ASXL1 mutations lead to clonal hematopoiesis are not clear.
Similarly, the mechanism by which ASXL1 enhances inflammation and atherosclerosis is also poorly understood. No in vivo data has demonstrated the downstream inflammatory effects of ASXL1 CHIP carriers, but one can postulate that the shift toward myeloid transformation in ASXL1 CHIP carriers may carry similar downstream effects to TET2 loss of function.
JAK2
JAK2 is a non-receptor tyrosine kinase that transmits intracellular signals downstream of cytokine receptors. JAK2 tyrosine phosphorylates and activates TET2 in response to cytokines, linking extracellular signals with epigenetic changes in hematopoiesis [30]. JAK2 p.V617F gain-of-function mutations in hematopoietic cells are associated with myeloproliferative neoplasm (MPNs) like polycythemia vera, essential thrombocytopenia and myelofibrosis, which are in turn associated with myocardial infarction, deep vein thrombosis and stroke. JAK2 p.V617F mutations enhance formation of neutrophil extracellular traps (NET) that promote thrombosis. Indeed, in population studies, JAK2 p.V617F CHIP carriers demonstrate increased incidence of deep vein thrombosis and pulmonary embolus [31]. JAK2 CHIP carrier status is associated with higher levels of IL-18, and downstream increases in IL-6 production and inflammation [24].
JAK2 p.V617F mutations in CHIP tend to occur at a younger age and carry an up to a 10-fold increased risk of coronary artery disease – the strongest risk of premature cardiac disease among CHIP variants [7, 24]. Interestingly, the induction of atherosclerosis in JAK2 CHIP carriers occurs in the presence of reduced serum cholesterol: a negative correlation is observed between JAK2 CHIP-carrier status and both total and LDL cholesterol among studies in large WGS databases [24]. This is in contrast to the lack of association between CHIP overall and lipid profile [7].
Studies in Ldlr-deficient mice bearing JAK2 p.V617F mutations demonstrate accelerated atherosclerosis [32]. JAK2 mutant CH leads to increased proliferation of macrophages, and necrotic core formation in atherosclerotic lesions in mouse models. These necrotic core lesions demonstrate increased AIM2 inflammasome expression (as opposed to NLRP3), oxidative DNA damage, and DNA replication stress, effects blunted by IL-1β inhibition [33]. Additionally, murine heart failure models transplanted with JAK2 p.V617F myeloid clones show evidence of accelerated pathologic remodeling, revealing a role for JAK2 CHIP in potentiating fibrosis in heart failure [34].
CHIP with less common driver mutations: TP53, PPM1D, SF3B1, SRSF2
The next most frequent CHIP mutations are in DNA damage repair genes TP53 and PPM1D. PPM1D (protein phosphatase Mn2+/Mg2+-dependent 1D) is part of the DNA damage response pathway and in regulatory feedback loop with the tumor suppressor p53. Activated p53 induces PPM1D expression, leading to downstream dephosphorylation of p53 and downregulation of apoptosis. PPM1D loss-of-function mutations are in particular associated with CH in the context of prior exposure to cytotoxic chemotherapies such as cisplatin, etoposide and doxorubicin [35]. p53 mutations promote HSC expansion in response to radiation-induced stress, and mutant p53 interacts with EZH2 through epigenetic mechanisms to enhance its association with chromatin, increasing H3K27 tri-methylation of genes regulating differentiation and self-renewal of HSCs [36].
CHIP driver mutations in SF3B1 and SRSF2 are key components of the mRNA spliceosome. Mutations in these genes lead to defects in splicing and export of mRNAs encoding genes involved in translation. While these CHIP mutations are not well studied with respect to cardiovascular disease, studies have shown that patients with SF3B1 mutant-CHIP have higher circulating levels of IL-18 [24].
CH without candidate driver mutations, neutral evolution, and mosaic chromosomal alterations
Despite the identification of multiple driver mutations associated with CHIP, in a significant proportion of cases of clonal hematopoiesis no clear candidate driver mutation is identified. As CHIP is defined as somatic mutation with VAF >2%, CH without known candidate driver mutations is technically excluded from this classification. Despite this, clonal hematopoiesis without driver mutations carries increased risk of hematologic cancers and all-cause mortality, although its links to cardiovascular disease are poorly understood [37].
Whole-exome sequencing of peripheral blood DNA from an unselected cohort of 12,380 Swedish patients was among the first to identify that a large fraction of clonal hematopoiesis carriers (defined as at least 3 somatic mutations at detectable allele frequency) do not carry obvious driver mutations (none of the previously identified candidate driver mutations identified) [4]. Similarly, in whole-genome sequencing of 11,262 Icelandic patients (deCODE Genetics), the majority of subjects with clonal hematopoiesis (defined as >20 single somatic mutations with VAF 0.10-0.20) did not carry a clear driver mutation [37].
CH without driver mutations may be the result of limitations in detection methods (i.e. if driver mutations exist in non-exonic areas) or may be driven primarily by epigenetic encoding to enhance HSCs self-renewal and proliferation. CH without driver mutations might also be the consequence of neutral drift of small populations of active HSCs [37]. This theory focusing on genetic drift and neutral evolution has important potential implications – if the primary downstream effects of CHIP are mediated more by enhanced leukocytosis and less by the effects of individual driver mutations, then a focus on driver mutations as detailed above may not inform a therapeutic approach [38].
Mosaic chromosomal alteration (mCA) represents an additional mechanism of CH without driver mutations. mCAs include larger structural somatic alterations such as deletions, duplications, or copy number neutral loss of heterozygosity (CN-LOH). Similar to candidate driver mutations in CHIP, mCA accumulate with age, rising to a prevalence as high as 35% in the older than 90 population [39]. mCA-driven CH predisposes primarily to lymphoid malignancies like CLL and carries a roughly two-fold increase in all-cause mortality [40, 41]. However, mCA-driven CH (even when associated with DNMT3A or TET2 loss) did not appear to be associated with a significant increase in cardiovascular risk, with the notable exception of JAK2-related CN-LOH events [41]. In addition to an association with incident blood cancer, mCA-driven CH may lead to impaired immunity and predispose to infection [42].
4. Risk Factors Associated with Clonal Hematopoiesis of Indeterminate Potential
Epidemiologic and genetic studies have focused on identifying the interplay between germline variation, co-morbid conditions and lifestyle factors and their association with CHIP. These associations are important foundations for hypothesis generation regarding mechanisms, and for informing the clinical care of CHIP patients (Table 3).
Genotypic Associations with CHIP
While CHIP driver mutations are acquired, somatic mutations, germline variation has an important role in predisposing the development of CHIP. Hinds et al performed initial genome-wide association studies of 726 individuals with myeloproliferative neoplasms, 497 individuals with JAK2 p.V617F clonal hematopoiesis, and 252,140 controls. This identified associations with germline genetic variants of TERT, SH2B3, TET2, ATM, CHEK2, PINT and GF11B [43]. Using whole genome sequencing data from 11,262 participants, Zink et al detected a strong association with CHIP development and an 8-bp deletion in intron 3 of TERT, which correlated with shorter telomere length [37]. Extending these observations to a larger cohort unselected for candidate driver mutation, whole genome sequencing studies of 97,691 individuals identified three gene risk loci associated with a predilection to TET2 CHIP. One set of loci were associated with genes facilitating genomic integrity and telomere length (TERT and CHEK2) that also raised the risk of neoplasm in multiple organ systems; another, with HSC self-renewal (TET2) was only associated with hematologic malignancies; and a final locus at the TCL1A promoter specifically associated with increased risk of DNMT3A CHIP alone [24].
Phenotypic Associations with CHIP
There are numerous phenotypic associations with CHIP, ranging from aging to lifestyle factors. A selection of emerging research is summarized below highlighting the links between these conditions and CHIP risk. The directionality of causality is an area of active research, in part due to the paucity of longitudinal samples limiting the study design and methodologies used to establish these associations.
Aging: Cumulative Genomic Damage and Leukocyte Telomere Length
Aging is a potent risk factor for atherosclerotic cardiovascular disease [44], and aging is marked by the acquisition of somatic mutations in hematopoietic stem cells due to cumulative genomic DNA damage [45]. Multiple studies have demonstrated that the proportion of CHIP carriers increases exponentially with age [4, 5, 46].
Leukocyte telomere length and CHIP have a complex relationship. Patients with inherited telomeropathies show increased CH prevalence [47]. Common variants at TERT predisposing to prolonged leukocyte telomere length (LTL) are associated with increased CHIP odds [24], and CHIP carriers possess significantly shorter leukocyte telomere length (LTL) compared to those without [37]. Mendelian randomization studies in particular support an inverse relationship between LTL and coronary artery disease [48–50], and Nakao et al now systematically show with bidirectional Mendelian randomization that processes promoting LTL lengthening at first increase the propensity for CHIP development, with CHIP then promoting accelerated LTL shortening [51]. LTL length additionally mediated a modest association between CHIP and CAD but this may be limited by the bidirectional opposing associations between LTL and CHIP [51].
Based on these observations, some have proposed a “telomere brink” hypothesis of CHIP. This posits that as individuals age, age-dependent telomere shortening particularly affects the highly proliferative hematopoietic system, and that candidate driver mutations enrich in hematopoietic cells as a means of delaying reaching their “telomere brink” when the replicative potential of HSCs is reached [52].
Complex Interactions with Hyperlipidemia and the “Atherosclerosis Trait Complex”
Chronic elevation of blood lipid levels promotes the formation of atherosclerotic plaques, potentiated by immune cell recruitment and local inflammation. Increased cholesterol levels stimulate proliferation and mobilization of HSCs as well as myeloid cell expansion [53, 54], and high levels of HDL suppress HSC proliferation [55]. In mouse models, Apoe −/− and Ldlr −/− mice fed high-lipid diets demonstrate elevated HSC proliferation [56].
Despite these associations, in large-scale association studies CHIP is not consistently associated with lipid levels (triglycerides, total cholesterol, LDL-C or HDL-C) apart from the association with JAK2 CHIP, which is in fact correlated with a decrease in total cholesterol and LDL-C despite elevated CAD risk [24, 57]. Seeking to explain this discrepancy, Heyde et al demonstrated that atherosclerosis induces HSC proliferation in both mice and humans, and that this induction of HSC proliferation in atherosclerosis is sufficient to drive clonal expansion of mutant Tet2 −/− HSCs in Ldlr−/− mice. Notably, the induction of mild hypercholesterolemia in non-atherosclerotic wild-type did not induce TET2−/− clonal expansion [38], suggesting that in the absence of the inflammatory milieu of atherosclerosis, elevated cholesterol alone is not sufficient to drive clonal hematopoiesis. The extent to which blood cholesterol concentrations differentially promote atherogenesis in the context of CHIP versus no CHIP requires further study.
Smoking and Obstructive Airway Disease
Smoking has been consistently observed to associate with CH [4, 37], and never smoked status compared to current smoking status is associated with a reduced risk of developing CHIP [58]. Research suggests that ASXL1 mutations in particular are enriched in current and past smokers [27, 59]. While two groups have reported associations with CHIP and COPD, the association of CHIP and smoking likely confound these associations [37, 60].
Obesity and Type 2 Diabetes
In a study of 8709 post-menopausal women, having a normal body mass index compared to being obese was associated with lower frequency of CHIP [58], and obesity-related insulin resistance appears to be associated with increased TET2 CHIP status. Jaiswal et al reported a 1.3 fold increased odds of CHIP in diabetes patients [5]. Subsequently, studies in mouse models showed that TET2-driven clonal hematopoiesis led to increased expression of IL-1β in white adipose tissue, aggravating age and obesity-related insulin resistance and worsening hyperglycemia [61]. Whether CHIP is associated with the risk for incident diabetes mellitus requires further investigation.
Sleep Fragmentation
Sleep disruption increases the risk of cardiovascular disease, diabetes, obesity, and cancer, and studies in mouse models demonstrate increased atherosclerotic lesions in sleep-deprived mice, with systemic monocytosis and neutrophilia [62]. The emergence of CH of Tet2−/− clones in Ldlr−/− mouse models is accelerated by a factor of 1.6 with sleep fragmentation [38], suggesting that sleep deprivation may accelerate CHIP development. The association of addiction and psychiatric diseases with CHIP might be driven by the fact that sleep fragmentation is a critical component these conditions [37].
Premature Menopause
Premature menopause, both natural and surgical, is associated with an increased risk of cardiovascular events in women (including coronary artery disease, heart failure, ischemic stroke, PAD, VTE, MR and aortic stenosis) [63]. A follow-up cohort study of women from the UK Biobank and Women’s Health Initiative databases established premature menopause was associated with increased CHIP prevalence (OR 1.40) and incident CAD (HR 1.36, rising to 1.48 for VAF > 0.1), with a stronger association observed with natural premature menopause (HR 1.73) versus surgical premature menopause [64]. DNMT3A was the only candidate driver mutation found to be associated with premature menopause.
Chronic Inflammatory Conditions
A broad range of pro-inflammatory and rheumatologic conditions associate with CHIP, including chronic pulmonary disease [37]. Systemic sclerosis (SSc) is characterized by immune dysregulation, aberrant fibrosis and microangiopathy. A study of 90 SSc patients and 44 healthy donors found the prevalence of CHIP was elevated in SSc patients (25%) compared to healthy donors (4%), with DNTM3A mutations most common. However, no clinical differences were apparent between CHIP and non-CHIP carriers in this small cohort [65]. Increased CH prevalence was also found in rheumatoid arthritis (RA) patients. Savola et al studied a cohort of 59 RA patients and found a 17% prevalence of CHIP, increasing to up to 25% among 70-79 year old patients studied. DNMT3A and TET2 mutations were most common [66]. Whether CHIP associates with other closely related rheumatologic conditions such as systemic lupus erythematosus (SLE) – which itself carries a significantly increased risk of early MI and CVD [67] - is unknown. Limited data suggest an increased risk of myeloid neoplasms in both RA and SLE populations [68].
Ulcerative colitis (UC), an inflammatory bowel disease (IBD) marked by T-cell infiltration in the colon and overproduction of TNF-alpha and interferon-gamma, has also been associated with CHIP. While cardiovascular manifestations in IBD tend to cluster around immune-related phenomena (pericarditis, myocarditis, thromboembolism), there is also an increased risk of ischemic heart disease in UC patients [69–71]. UC patients with CHIP exhibited a distinctive mutational spectrum, with DNMT3A and PPM1D mutations most commonly seen in this patient cohort, and evidence of a correlation found between elevated levels of serum interferon-gamma and DNMT3A mutation [72]. Elevated levels of Th17 cells and Th17 related cytokines are important in the pathogenesis of mucosal damage in IBD [73], similar to observations of pro-inflammatory T cell polarization (increased Th17 ratio) in DNMT3A-CHIP carriers with severe degenerative aortic stenosis [9].
Chronic Infection and HIV
Chronic infections may also increase the risk of developing CHIP. In mouse models, chronic mycobacterial infection led to DNMT3A-CHIP clone expansion via an interferon-gamma dependent mechanism [16], and mCA-driven CH was associated with an increased risk of sepsis, pneumonia, and coronavirus disease 2019 hospitalization [42].
Among HIV patients, coronary artery disease is a major source of morbidity, the consequence of accelerated biological aging, chronic inflammation and immune dysregulation. HIV is associated with a greater risk of myelodysplastic syndromes [74]. In a multi-ethnic sample of 600 people living with HIV (PLWH) derived from the Swiss HIV Cohort Study, a two-fold increased prevalence of CHIP was observed when compared to 8,111 participants from the ARIC (Atherosclerotic Risk in the Community) cohort. In particular, there was a greater prevalence of ASLX1 mutations among PLWH [28]. Similar findings were observed in the ARCHIVE study, which included 220 HIV positive and 226 HIV-negative Australian participants, and demonstrated a two-fold increased risk of CHIP with DNMT3A (48.5%), TET2 (20.5%) and ASXL1 (11.4%) mutations [75].
Cancer Treatment
Clonal hematopoiesis has been observed in the context of a wide variety of solid and liquid malignancies, often considered to be a consequence of cytotoxic chemotherapies. Among patients treated with stem-cell transplantation for non-Hodgkin’s lymphoma, the incidence of CH was 30% [76]. Cancer therapies shape the fitness landscape of clonal hematopoiesis, with ionizing radiation, topoisomerase II inhibitors and cisplatin selecting for mutations in DNA damage response genes TP53, PPM1D and CHEK2 [35, 59]. Importantly, immune checkpoint blockade did not appear to be associated with expansion of clonal hematopoiesis [77]. A cohort of 135 invasive glioma patients undergoing next-generation sequencing of cfDNA treated with temozolomide treatment showed increased CH-type mutations, most commonly TP53, followed by ATM, GNAS and JAK2, that correlated with risk of shorter survival [78]. Understanding the degree of cardiovascular risk conveyed by higher CHIP prevalence in these populations will be important in informing oncologic survivorship care.
Conclusions and Future Directions for CHIP Prevention and Therapy
As studies detail new genotypic and phenotypic associations with CHIP, researchers and clinicians are confronted with the question of what preventative and therapeutic interventions could be taken to mitigate disease risk. Understanding which cohorts of patients could benefit from targeted CHIP testing could enable a precision-medicine approach to risk reduction. Additionally, many CHIP patients may be identified incidentally. Cell-free DNA analysis, intended to detect circulating tumor DNA to aid in early-cancer detection, is invariably confounded by the presence of CHIP, as the vast majority of cell-free DNA arises from hematopoietic cells [79–81].
An understanding of CHIP biology and its reciprocal relationship to a pro-inflammatory state presents numerous potential targets for therapy. As a genetic proxy of IL-6 inhibition, the presence of the inhibitory IL-6 receptor gene variant (IL6R p.Asp358Ala) reduced the CVD risk in DNMT3A and TET2 CHIP carriers by ~50%, highlighting that inhibiting IL-6 signaling can decrease the risk of cardiovascular disease to a much greater degree among individuals with CHIP versus without [25]. This is in keeping with studies which have found that IL-1β blockade with canakinumab after MI reduced risk of death form cardiovascular disease, rates of nonfatal AMI and nonfatal stroke (CANTOS [Canakinumab Anti-inflammatory Thrombosis Outcomes Study]). Non-prespecified post hoc exploratory analyses demonstrated that a greater reduction was seen in those with TET2 CHIP [82]. In a JAK2 CHIP model, both non-selective IL-1 receptor blockade with anakinra and targeted IL-1β blockade led to reduced atherosclerotic plaque instability, with normalized macrophage proliferation and density in early atherosclerotic plaques, and reduced core necrosis and increased cap thickness in advanced atherosclerotic plaques [33]. Targeted inhibition of the inflammasome itself may also have protective benefits against atherogenesis. In mouse models of TET2 deficient mice, pharmacologic inhibition of the NLRP3 inflammasome with MCC950 led to a 50% decrease in aortic atherosclerotic plaque size, greater than the non-significant reduction observed in wild-type mice [21]. Targeted inhibition of the AIM2 inflammasome, which is characteristic of JAK2 mutant CHIP, might also have similar benefit [33].
Targeting candidate driver mutations in CHIP could represent an alternative therapeutic strategy. Vitamin C metabolites activate TET2 and can mimic restoration of TET2 via enhancing 5-hydroxymethycytosine formation in TET2-deficient mice to reverse aberrant HSC self-renewal, presenting a potential preventive therapy for TET2-CHIP carriers [83]. In JAK2 mutant CHIP, treatment with the approved JAK2 inhibitor ruxolitinib reduced abnormal neutrophil extracellular trap formation and deep vein thrombosis [31] as well as circulating IL-18 levels [33], and JAK2 inhibition with fedratinib in Apoe −/− mice suppressed myelopoiesis and the development of atherosclerosis [84].
Additionally, understanding the associations of race, ethnicity and ancestry with CHIP prevalence is a crucial area for future research. The majority of early CHIP studies have been conducted in European Caucasian cohorts. However, a modestly lower prevalence of CHIP has been observed in individuals of Hispanic and East Asian ancestry compared to those of other ethnicities [7, 25]. Additionally, the germline variant rs144418061, which conveys an increased risk of CHIP, is a variant present only in individuals of African ancestry [25], recognized in part due to the expanded diversity of the TOPMED cohort (40% European, 32% African, 16% Hispanic and 10% Asian). Further studies in racially and ethnically diverse populations will be important in understanding the implications of a CHIP diagnosis in broader populations and may help to identify further associations between certain ethnic groups and germline variants or specific candidate driver mutations. Many questions remain in understanding the potent cardiovascular disease risk conveyed by CHIP. Ongoing research focused on elucidating the underlying genetic and biological mechanisms driving CHIP, and the environmental risk factors modulating CHIP risk will be critical in addressing this newly recognized risk factor. CHIP represents a unique shift in the study of cardiovascular genetics and atherosclerosis biology from inherited germline mutations to an understanding of the critical role of acquired somatic mutations. As identification of CHIP patients becomes more common, this represents a unique and emerging opportunity for multidisciplinary collaboration between hematologists, oncologists, and cardiologists.
Figure 1: CHIP associates with an altered inflammatory state, elevating cardiovascular risk.
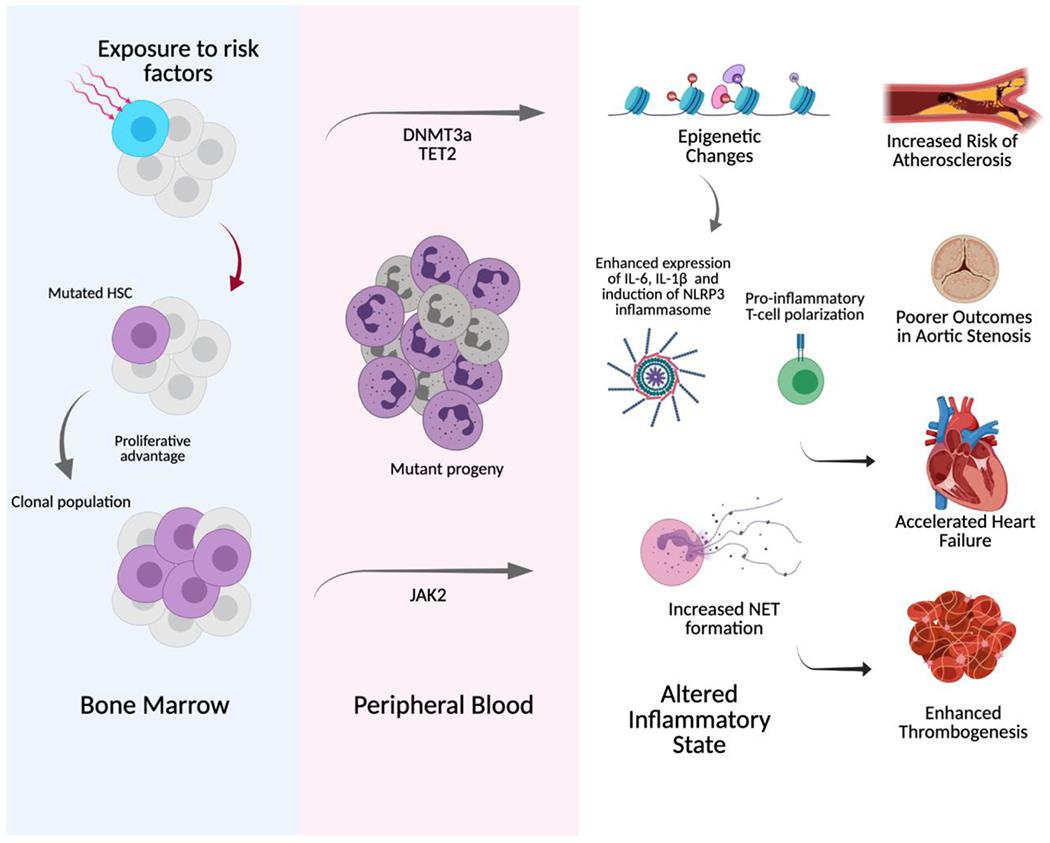
The acquisition of driver mutations (in DNMT3A, TET2, and JAK2 shown as a selection here) leads to clonal hematopoiesis of indeterminate potential (CHIP), which induces an altered inflammatory state that is associated with an increased risk of atherosclerosis, poorer outcomes in aortic stenosis and heart failure, and enhanced thrombogenesis. HSC= hematopoietic stem cell, NET = neutrophil extracellular traps (created with Biorender.com)
Table 2:
Emerging Evidence of Genotypic and Phenotypic Associations with CHIP
| Risk Factor | Relevant Studies |
|---|---|
| Germline Mutation | [24, 37, 43] |
| Aging and leukocyte telomere length | [24, 37, 48, 50, 51] |
| Smoking | [4, 27, 37, 58, 59] |
| Obesity, Insulin resistance & Type 2 Diabetes | [5, 58, 85] |
| Hyperlipidemia & Atherosclerosis | [53, 54, 58] |
| Sleep Deprivation | [38, 62] |
| Premature Menopause | [64] |
| Chronic Inflammatory Conditions | [37] Systemic Sclerosis [65] Rheumatoid Arthritis [66] Ulcerative colitis [72] |
| Chronic Infection, including HIV | Chronic Infection [16] HIV [28, 75] |
| Cancer therapy | [35, 59, 78] |
Highlights.
Clonal hematopoiesis of indeterminate potential (CHIP) is a new, potentially causal cardiovascular risk factor.
CHIP associates with an altered, chronic inflammatory state marked by enhanced cytokine expression (e.g., NLPR3 inflammasome activation) and T-cell polarization.
Emerging research has identified inherited and acquired risk factors associated with increased CHIP prevalence, which may provide important insights about the genesis of CHIP as well as CHIP-associated cardiovascular disease.
Conflict of Interest
P.N. reports grant support from Amgen, Apple, and Boston Scientific, consulting income from Apple, Blackstone Life Sciences, Genentech, and Novartis, and spousal employment at Vertex, all unrelated to the present work. The other authors do not report any disclosures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Watson CJ, et al. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science, 2020. 367(6485): p. 1449–1454. [DOI] [PubMed] [Google Scholar]
- 2.Bowman RL, Busque L, and Levine RL, Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell Stem Cell, 2018. 22(2): p. 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Young AL, et al. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun, 2016. 7: p. 12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Genovese G, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med, 2014. 371(26): p. 2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jaiswal S, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med, 2014. 371(26): p. 2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steensma DP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood, 2015. 126(1): p. 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaiswal S, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med, 2017. 377(2): p. 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dorsheimer L, et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol, 2019. 4(1): p. 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mas-Peiro S, et al. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur Heart J, 2020. 41(8): p. 933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas Research, N., et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med, 2013. 368(22): p. 2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindsley RC, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood, 2015. 125(9): p. 1367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bejar R, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med, 2011. 364(26): p. 2496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papaemmanuil E, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood, 2013. 122(22): p. 3616–27; quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeong M, et al. Loss of Dnmt3a Immortalizes Hematopoietic Stem Cells In Vivo. Cell Rep, 2018. 23(1): p. 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Challen GA, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet, 2011. 44(1): p. 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hormaechea-Agulla D, et al. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNgamma signaling. Cell Stem Cell, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sano S, et al. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ Res, 2018. 123(3): p. 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abplanalp WT, et al. Clonal Hematopoiesis-Driver DNMT3A Mutations Alter Immune Cells in Heart Failure. Circ Res, 2021. 128(2): p. 216–228. [DOI] [PubMed] [Google Scholar]
- 19.Buscarlet M, et al. Lineage restriction analyses in CHIP indicate myeloid bias for TET2 and multipotent stem cell origin for DNMT3A. Blood, 2018. 132(3): p. 277–280. [DOI] [PubMed] [Google Scholar]
- 20.Cull AH, et al. Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol, 2017. 55: p. 56–70 e13. [DOI] [PubMed] [Google Scholar]
- 21.Fuster JJ, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science, 2017. 355(6327): p. 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Q, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature, 2015. 525(7569): p. 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sano S, et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1beta/NLRP3 Inflammasome. J Am Coll Cardiol, 2018. 71(8): p. 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bick AG, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature, 2020. 586(7831): p. 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bick AG, et al. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation, 2020. 141(2): p. 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asada S and Kitamura T, Aberrant histone modifications induced by mutant ASXL1 in myeloid neoplasms. Int J Hematol, 2019. 110(2): p. 179–186. [DOI] [PubMed] [Google Scholar]
- 27.Dawoud AAZ, Tapper WJ, and Cross NCP, Clonal myelopoiesis in the UK Biobank cohort: ASXL1 mutations are strongly associated with smoking. Leukemia, 2020. 34(10): p. 2660–2672. [DOI] [PubMed] [Google Scholar]
- 28.Bick AG, et al. Increased CHIP Prevalence Amongst People Living with HIV. medRxiv, 2020. [Google Scholar]
- 29.Fujino T and Kitamura T, ASXL1 mutation in clonal hematopoiesis. Exp Hematol, 2020. 83: p. 74–84. [DOI] [PubMed] [Google Scholar]
- 30.Jeong JJ, et al. Cytokine-Regulated Phosphorylation and Activation of TET2 by JAK2 in Hematopoiesis. Cancer Discov, 2019. 9(6): p. 778–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolach O, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med, 2018. 10(436). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang W, et al. Macrophage Inflammation, Erythrophagocytosis, and Accelerated Atherosclerosis in Jak2 (V617F) Mice. Circ Res, 2018. 123(11): p. e35–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fidler TP, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sano S, et al. JAK2 (V617F)-Mediated Clonal Hematopoiesis Accelerates Pathological Remodeling in Murine Heart Failure. JACC Basic Transl Sci, 2019. 4(6): p. 684–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsu JI, et al. PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell, 2018. 23(5): p. 700–713 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen S, et al. Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nat Commun, 2019. 10(1): p. 5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zink F, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood, 2017. 130(6): p. 742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heyde A, et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell, 2021. 184(5): p. 1348–1361 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Terao C, et al. Chromosomal alterations among age-related haematopoietic clones in Japan. Nature, 2020. 584(7819): p. 130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loh PR, et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature, 2018. 559(7714): p. 350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loh PR, Genovese G, and McCarroll SA, Monogenic and polygenic inheritance become instruments for clonal selection. Nature, 2020. 584(7819): p. 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zekavat SM, et al. Hematopoietic mosaic chromosomal alterations and risk for infection among 767,891 individuals without blood cancer. medRxiv, 2020. [Google Scholar]
- 43.Hinds DA, et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood, 2016. 128(8): p. 1121–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang JC and Bennett M, Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res, 2012. 111(2): p. 245–59. [DOI] [PubMed] [Google Scholar]
- 45.Welch JS, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell, 2012. 150(2): p. 264–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie M, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med, 2014. 20(12): p. 1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perdigones N, et al. Clonal hematopoiesis in patients with dyskeratosis congenita. Am J Hematol, 2016. 91(12): p. 1227–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ojha J, et al. Genetic Variation Associated with Longer Telomere Length Increases Risk of Chronic Lymphocytic Leukemia. Cancer Epidemiol Biomarkers Prev, 2016. 25(7): p. 1043–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Telomeres Mendelian Randomization, C., et al. Association Between Telomere Length and Risk of Cancer and Non-Neoplastic Diseases: A Mendelian Randomization Study. JAMA Oncol, 2017. 3(5): p. 636–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li C, et al. Genome-wide Association Analysis in Humans Links Nucleotide Metabolism to Leukocyte Telomere Length. Am J Hum Genet, 2020. 106(3): p. 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakao T, et al. Bidirectional Mendelian randomization supports bidirectional causality between telomere length and clonal hematopoiesis of intermediate potential. medRxiv, 2021: p. 2021.02.26.21252199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aviv A and Levy D, Hemothelium, Clonal Hematopoiesis of Indeterminate Potential, and Atherosclerosis. Circulation, 2019. 139(1): p. 7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oguro H, The Roles of Cholesterol and Its Metabolites in Normal and Malignant Hematopoiesis. Front Endocrinol (Lausanne), 2019. 10: p. 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morgan PK, et al. Hematopoiesis is regulated by cholesterol efflux pathways and lipid rafts: connections with cardiovascular diseases. J Lipid Res, 2020. 61(5): p. 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yvan-Charvet L, et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science, 2010. 328(5986): p. 1689–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murphy AJ, et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest, 2011. 121(10): p. 4138–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu DJ, et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat Genet, 2017. 49(12): p. 1758–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haring B, et al. Healthy Lifestyle and Clonal Hematopoiesis of Indeterminate Potential: Results From the Women’s Health Initiative. J Am Heart Assoc, 2021. 10(5): p. e018789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bolton KL, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet, 2020. 52(11): p. 1219–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Buscarlet M, et al. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood, 2017. 130(6): p. 753–762. [DOI] [PubMed] [Google Scholar]
- 61.Fuster JJ, et al. TET2-Loss-of-Function-Driven Clonal Hematopoiesis Exacerbates Experimental Insulin Resistance in Aging and Obesity. Cell Rep, 2020. 33(4): p. 108326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McAlpine CS, et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature, 2019. 566(7744): p. 383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Honigberg MC, et al. Association of Premature Natural and Surgical Menopause With Incident Cardiovascular Disease. JAMA, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Honigberg MC, et al. Premature Menopause, Clonal Hematopoiesis, and Coronary Artery Disease in Postmenopausal Women. Circulation, 2021. 143(5): p. 410–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ricard L, et al. Clonal haematopoiesis is increased in early onset in systemic sclerosis. Rheumatology (Oxford), 2020. 59(11): p. 3499–3504. [DOI] [PubMed] [Google Scholar]
- 66.Savola P, et al. Clonal hematopoiesis in patients with rheumatoid arthritis. Blood Cancer J, 2018. 8(8): p. 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu Y and Kaplan MJ, Cardiovascular disease in systemic lupus erythematosus: an update. Curr Opin Rheumatol, 2018. 30(5): p. 441–448. [DOI] [PubMed] [Google Scholar]
- 68.Bekele DI and Patnaik MM, Autoimmunity, Clonal Hematopoiesis, and Myeloid Neoplasms. Rheum Dis Clin North Am, 2020. 46(3): p. 429–444. [DOI] [PubMed] [Google Scholar]
- 69.Card TR, Langan SM, and Chu TP, Extra-Gastrointestinal Manifestations of Inflammatory Bowel Disease May Be Less Common Than Previously Reported. Dig Dis Sci, 2016. 61(9): p. 2619–26. [DOI] [PubMed] [Google Scholar]
- 70.Kristensen SL, et al. Disease activity in inflammatory bowel disease is associated with increased risk of myocardial infarction, stroke and cardiovascular death--a Danish nationwide cohort study. PLoS One, 2013. 8(2): p. e56944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Singh S, et al. Inflammatory bowel disease is associated with an increased risk of melanoma: a systematic review and meta-analysis. Clin Gastroenterol Hepatol, 2014. 12(2): p. 210–8. [DOI] [PubMed] [Google Scholar]
- 72.Zhang CRC, et al. Inflammatory cytokines promote clonal hematopoiesis with specific mutations in ulcerative colitis patients. Exp Hematol, 2019. 80: p. 36–41 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jiang W, et al. Elevated levels of Th17 cells and Th17-related cytokines are associated with disease activity in patients with inflammatory bowel disease. Inflamm Res, 2014. 63(11): p. 943–50. [DOI] [PubMed] [Google Scholar]
- 74.Kaner JD, et al. HIV portends a poor prognosis in myelodysplastic syndromes. Leuk Lymphoma, 2019. 60(14): p. 3529–3535. [DOI] [PubMed] [Google Scholar]
- 75.Dharan NJ, et al. Age-related clonal haematopoiesis is more prevalent in older adults with HIV: the ARCHIVE study. medRxiv, 2020: p. 2020.11.19.20235069. [Google Scholar]
- 76.Gibson CJ, et al. Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J Clin Oncol, 2017. 35(14): p. 1598–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miller PG, et al. Fitness Landscape of Clonal Hematopoiesis Under Selective Pressure of Immune Checkpoint Blockade. JCO Precision Oncology, 2020(4): p. 1027–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Okamura R, et al. High prevalence of clonal hematopoiesis-type genomic abnormalities in cell-free DNA in invasive gliomas after treatment. Int J Cancer, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Snyder MW, et al. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell, 2016. 164(1-2): p. 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moss J, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun, 2018. 9(1): p. 5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lui YY, Chik KW, and Lo YM, Does centrifugation cause the ex vivo release of DNA from blood cells? Clin Chem, 2002. 48(11): p. 2074–6. [PubMed] [Google Scholar]
- 82.Svensson EC, et al. Abstract 15111: TET2-Driven Clonal Hematopoiesis Predicts Enhanced Response to Canakinumab in the CANTOS Trial: An Exploratory Analysis. Circulation, 2018. 138(Suppl_1): p. A15111–A15111. [Google Scholar]
- 83.Cimmino L, et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell, 2017. 170(6): p. 1079–1095 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tang Y, et al. Inhibition of JAK2 Suppresses Myelopoiesis and Atherosclerosis in Apoe(−/−) Mice. Cardiovasc Drugs Ther, 2020. 34(2): p. 145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fuster JJ and Walsh K, Somatic Mutations and Clonal Hematopoiesis: Unexpected Potential New Drivers of Age-Related Cardiovascular Disease. Circ Res, 2018. 122(3): p. 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]