

Abstract
Although only a small fraction will ever develop the active form of tuberculosis (ATB) disease, chemoprophylaxis treatment in latent TB infected (LTBI) individuals is an effective strategy to control pathogen transmission. Characterizing immune responses in LTBI upon chemoprophylactic treatment is important to facilitate treatment monitoring, and thus improve TB control strategies. Here, we studied changes in the blood transcriptome in a cohort of 42 LTBI and 8 ATB participants who received anti-TB therapy. Based on the expression of previously published gene signatures of progression to ATB, we stratified the LTBI cohort in two groups and examined if individuals deemed to be at elevated risk of developing ATB before treatment (LTBI-Risk) differed from others (LTBI-Other). We found that LTBI-Risk and LTBI-Other groups were associated with two distinct transcriptomic treatment signatures, with the LTBI-Risk signature resembling that of treated ATB patients. Notably, overlapping genes between LTBI-Risk and ATB treatment signatures were associated with risk of progression to ATB and interferon (IFN) signaling, and were selectively downregulated upon treatment in the LTBI-Risk but not the LTBI-Other group. Our results suggest that transcriptomic reprogramming following treatment of LTBI is heterogeneous and can be used to distinguish LTBI-Risk individuals from the LTBI cohort at large.
Keywords: latent tuberculosis, blood transcriptomics, prophylaxis treatment
Graphical Abstract
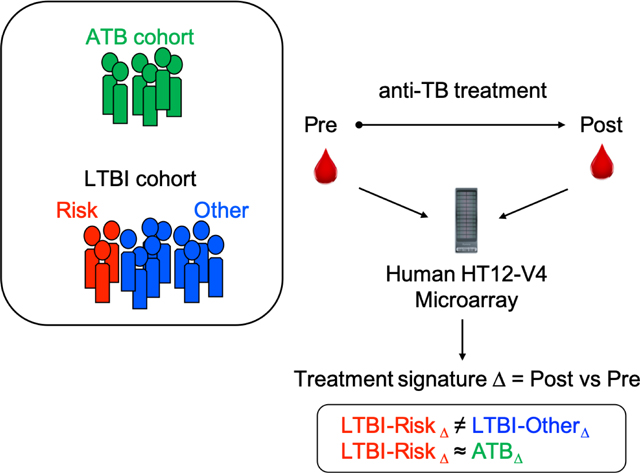
1. Introduction
Tuberculosis (TB), caused by the pathogen Mycobacterium tuberculosis (Mtb), is one of the leading cause of death by a single infectious agent, killing approximately 1.4 million individuals each year 1. The historic binary classification of patients into those with ‘latent TB infection’ (LTBI) and ‘active TB disease’ (ATB), the latter associated with clinical symptoms and morbidity, does not capture the biological heterogeneity and overlap between infection states 2, 3. Indeed, the ‘clinical state’ of LTBI is heterogeneous, and includes individuals who may have eliminated the pathogen, as well as individuals with incipient or subclinical active disease 4.
A key target of TB control strategies is the prevention of Mtb (re-)activation in individuals with LTBI using chemoprophylactic treatment. It is estimated that approximately a quarter of the world’s population had LTBI 5, representing a considerable reservoir for active disease development and therefore transmission. The estimated lifetime risk of individuals with LTBI progressing to ATB is 12% 6, but varies widely depending on several factors (e.g. age and immunosuppression). Thus, although not all LTBI individuals will benefit from therapy, this approach represents an effective way to eliminate Mtb in this population and reduce the possibility of transmission to others. Treatment of LTBI typically consists of either three months of daily rifampicin and isoniazid combination therapy or six months of daily isoniazid monotherapy 7, 8. These treatments can have severe side effects, such as hepatotoxicity which can be fatal. More recently, shorter and lighter regimen such as isoniazid and rifapentine weekly for three months, or daily for one month have proven as efficacious, with higher treatment completion and lower adverse reaction rates, when compared to six or nine months of daily isoniazid 9–12. There is no “gold standard” test for LTBI which is currently diagnosed by either a tuberculin skin test or by measuring IFNγ release in response to Mtb-derived T cell antigens in an Interferon Gamma Release Assay (IGRA). Since these tests only test Mtb immune sensitization, rather than ongoing infection, they correlate poorly with progression to active disease 13 and with chemoprophylactic treatment success 14, 15. Monitoring immune responses to TB preventive therapy might therefore be beneficial to patients, not only to potentially shorten therapy (and reduce the risk of drug-related adverse events) but also to confirm treatment success, and more globally to better understand LTBI heterogeneity and identify the individuals who derive the most benefit from treatment. Even if shorter and simpler regimens are safer, it is logistically and economically not possible to treat the entire LTBI population (especially in high endemic areas where the risk of re-infection is high).
Whole blood transcriptomics is increasingly used in the field of TB research to identify both diagnostic and mechanistic immune signatures of infection and disease, highlighting the heterogeneous spectrum of TB 16. Whole blood gene signatures have recently shown potential to predict ATB progression in LTBI 17–22. With respect to anti-TB therapy, blood transcriptomic changes upon treatment in ATB patients have also been used to monitor treatment efficacy, and identify the individuals at risk of relapse 21, 23–27. More recently, one study has investigated transcriptomic changes in whole blood in response to treatment among individuals with LTBI following Mtb-specific in vitro stimulation 28. Authors identified a dichotomous transcriptomic profile following in vitro stimulation across all participants, and hypothesized it might distinguish between LTBI individuals with viable Mtb bacilli versus those who have controlled or eliminated the bacteria.
In this study, we monitored direct ex vivo blood transcriptomic changes following treatment in a cohort of 42 individuals with LTBI. We hypothesized that individuals with LTBI at risk of developing ATB are immunologically more similar to those with ATB compared to the majority of non-progressing LTBI individuals, and will thus display blood transcriptomic changes following treatment that are similar to those in treated ATB individuals. Establishing such immune heterogeneity in treatment signatures might enable us to better understand the spectrum of LTBI and facilitate chemoprophylactic TB treatment monitoring.
2. Material and methods
2.1. Ethics, consent and permissions
Participants recruited for this study were enrolled under ethical approval granted by the National Research Ethics Service, Heart of England NHS Foundation Trust (2012107RM), Committee South Central - Oxford C (12/SC/0299) and the Ministry of Defence Research Ethics Committee (MoDREC 237/PPE/11), respectively. All samples were obtained for specific use in this study. All clinical investigations were conducted according to the principles of the Declaration of Helsinki, and all participants provided written informed consent prior to participation.
2.2. Study design
This is a longitudinal study of individuals with LTBI or ATB receiving anti-TB treatment, with blood samples collected before and after therapy. Participants were enrolled at one of three clinical sites in Birmingham, Oxford or Catterick, UK. Enrollment of all participant groups was done simultaneously at the three clinical sites. ATB were recruited as symptomatic individuals seeking care at the clinical sites. LTBI were recruited as IGRA+ seeking care at the clinical sites, but were not household contact of ATB participants. For the LTBI group, participants with prior or recent history of ATB were excluded. ATB was defined as either a suspected (based on clinical suspicion and radiological and/or histological evidence) or microbiologically confirmed (by Mtb culture) new diagnosis of pulmonary or extrapulmonary TB disease, in the absence of any other significant co-morbidity. LTBI status was confirmed in participants with a positive IGRA (QuantiFERON-TB Gold In-Tube, Cellestis, or T-SPOT.TB, Oxford Immunotec) and the absence of clinical and radiographic signs of ATB or any other significant co-morbidity. Uninfected control participants (TBneg cohort) were recruited as healthy controls at the same three clinical sites in whom ATB and LTBI were excluded and had no past medical history of ATB or evidence of previous exposure to Mtb, as confirmed by a negative IGRA test. All participants were HIV-negative adults (≥18 years). Anti-TB treatment was based on UK standard regimens of combination drug therapy 29. For the LTBI cohort, treatment was 3 months of daily rifampicin and isoniazid. For the ATB cohort, treatment consisted of 2 months of daily rifampicin, isoniazid, pyrazinamide, and ethambutol, followed by 4 months of daily rifampicin and isoniazid. All ATB participants successfully completed the 6 months treatment course, based on resolution of clinical symptoms and sputum conversion to negativity (where applicable). There was no occurrence of relapse or recurrence. Because all three clinical sites have a low Mtb incidence setting, it was presumed that the risk of re-infection was negligible for both LTBI and ATB participants. For the LTBI cohort, the post-treatment sample was obtained 4 to 10 months after the pre-treatment sample, corresponding to at least one month and up to 7 months after completion of treatment. For the ATB cohort, the post-treatment sample was obtained 7 to 13 months after the pre-treatment sample, corresponding to at least one month and up to 7 months after completion of treatment. For all samples, 2.5mL of blood was collected directly into a PAXgene® tube (BD Biosciences), mixed, and stored at −80°C until processing.
2.3. Full blood count assay
Full blood count (FBC) was performed on whole blood in UKAS-accredited clinical laboratories using standard procedures. Each feature is expressed in its standard reference unit, and values were compared between TB cohorts using the non-parametric Mann-Whitney test.
2.4. Microarray analysis
Total intracellular RNA was extracted from PAXgene® samples using the Blood RNA Kit (Qiagen) according to the manufacturer’s instructions. Globin mRNA was depleted from total RNA extractions using a biotin/streptavidin/magnetic bead-based assay, according to the manufacturer’s instructions (GLOBINclear Kit, Ambion). The depleted RNA was then amplified and biotin-labeled using the TotalPrep RNA Amplification Kit (Illumina), followed by hybridization onto Illumina HumanHT-12 (v4.0) expression beadchips according to the manufacturer’s instructions. Arrays were scanned with an Illumina bead array reader confocal scanner. The idat Illumina HumanHT12-V4 microarray files were converted to raw expression data using the Bioconductor package beadarray v2.38 30 in R. The raw data was quantile normalized and log2 transformed using the Bioconductor package lumi v2.40 31 in R. A comprehensive annotation was performed using annotations provided by biomaRt v2.40.5, the Gene Expression Omnibus (GEO), and platform specific annotation obtained from Illumina. If there was not a consensus among annotations, biomaRt was given preference given its regular updates, followed by GEO, and lastly the platform specific annotation. For non-specific filtering median, coefficient of variation and inter-quartile range for all probes across all samples were calculated. The probes accounting for the bottom 50% in each measurement were overlapped, and those probes that fell in the bottom 50% of all measurements (n=20,147) were removed from further analysis, leaving a final set of 27,960 probes of the initial 48,107 probes. Microarray data has been deposited to NCBI’s Gene Expression Omnibus (GEO) database repository and is available under GEO accession number GSE152532.
2.5. Bioinformatic analysis
Batch effect was removed using the Combat function from the Bioconductor package sva v3.36 32 in R (Figure S1). Differential expression analysis was performed on combat normalized data using the Bioconductor package limma v3.44.1 33 in R and probes as entities, and the Benjamini and Hochberg’s method for p-value adjustment. Differentially expressed probes were then converted to corresponding genes for biological pathways enrichment and protein-protein interaction analyses, and for defining signature overlaps between cohorts. When differentially expressed probes mapped to the same gene, it was ensured that directionality was similar. If no known gene was associated to the probe, then the entity was ignored. Gene signature scores were calculated using a standard z-score formula, by summing the normalized expression values for all upregulated genes (or probes if a gene was mapped to several probes) and subtracting the sum of the normalized expression of all downregulated genes (or probes if a gene was mapped to several probes) present in the signature, and correcting for the average and standard deviation of the sums across all samples, as well as the number of samples. Heatmaps and PCA components were extracted from normalized expression values using the bioinformatic software analysis Qlucore omics explorer. Expression of gene signatures in immune blood cell types were calculated with the CellTypeScore tool in DICE database (http://dice-database.org/). The DICE database contains gene expression data generated from 13 immune cell types (and 2 activation conditions) isolated from peripheral blood mononuclear cells of 91 healthy individuals 34. For a given gene list, the CellTypeScore tool sum the normalized expression values of each individual gene constituting the list for each of the cell types/stimulation conditions. Pathway analysis was performed with the online platform enrichR 35, 36 and the BioPlanet resource database 37. Functional protein-protein association networks were defined by querying the online STRING database (version 11.0) using a minimum required interaction score of 0.4 (medium confidence). Statistical significance of overlap between treatment signatures was calculated using the hypergeometric distribution test and considering all 27,960 probes that passed the initial filtering as the total number of probes. All R scripts used to analyze the data have been deposited to GitHub and are publicly available (https://github.com/akulsinghania/burel_et_al_LTBI_heterogeneity).
3. Results
We enrolled a total of 69 LTBI and 17 ATB participants into the study. Blood samples were obtained before treatment for all participants (LTBIpre and ATBpre cohorts), and post anti-TB therapy for 42/69 LTBI and 8/17 ATB participants (LTBIpost and ATBpost cohorts). A blood sample was also obtained from 11 Mtb uninfected individuals (TBneg) that were recruited at the same sites. Participant demographics metadata are available in Table S1 and summarized for each cohort in Table S2. The TBneg cohort was age-matched to the LTBI cohort (mean age 27), whereas the ATB cohort was older (mean age 34) (Table S2). In terms of sex, the TBneg cohort was about equally split between female and male (55% female), whereas both LTBI and ATB cohorts had a lower percent of female (38% and 18% female, respectively) (Table S2). The TBneg cohort was predominantly composed of individuals of European ethnicity (55%) compared to LTBI (10%) or ATB (12%) (Table S2). For the ATB cohort, about a third (29%) of participants had microbiologically confirmed presence of Mtb (Table S2). For each blood sample, RNA content was assessed for gene expression using microarrays. Full blood counts were also obtained from blood samples collected prior to treatment for 7/11 TBneg, 15/17 ATB and 64/69 LTBI participants.
3.1. Stratification of the LTBI cohort based on previously published gene signatures of risk of progression to ATB
To identify individuals within the LTBI cohort that were potentially at increased risk of progression to active disease, we stratified the cohort based on their transcriptomic status pre-treatment using ‘risk of progression to ATB’ gene signatures previously identified 17–21. The seminal study by Zak et al. reported a 16-gene signature (termed Zak16 henceforth) that predicted progression from LTBI to ATB up to one year before reactivation 17. We assessed the expression of the Zak16 signature pre-treatment in the three cohorts: LTBIpre, ATBpre and TBneg. As expected, the Zak16 signature could discriminate between ATBpre and LTBIpre (AUC of 0.86, p < 0.0001), as well as ATBpre and TBneg (AUC of 0.87, p = 0.001), but not between LTBIpre and TBneg (AUC of 0.56, p = 0.51) (Figure 1A). For each sample, we calculated a Zak16 gene signature z-score by summing the normalized expression values of all probes mapped to each gene present in the Zak16 signature using a z-score standard formula. The ATBpre cohort had an increased Zak16 z-score compared to LTBIpre or TBneg participants, and the LTBIpre cohort showed the highest inter-individual variability (Figure 1B). Based on the maximum Zak16 z-score value in the TBneg cohort, the LTBIpre cohort was sub-divided into 2 groups: a LTBI-Risk group (n=10, 14%), comprising LTBI participants with a score expression higher than the maximum value of the TBneg cohort; and a LTBI-Other group (n=59, 86%), encompassing the remaining LTBI participants (Figure 1B). The level of expression of all genes included in the Zak16 signature was highly similar within a given LTBI individual (Figure 1C), indicating that all genes contributed to the calculated z-scores. Since Zak16 is not the only published TB progression signature, we also looked for expression of Sweeney3 18, 19, BATF2 20 and RISK6 21 signatures to ensure reproducibility. All three signatures included at least one gene overlapping with the Zak16 signature (Figure S2A), and performed equally well at discriminating ATBpre from LTBIpre (AUC of 0.92, p < 0.0001 for Sweeney3; AUC of 0.83, p < 0.0001 for BATF2; AUC of 0.80, p = 0.0002 for RISK6) or ATBpre from TBneg (AUC of 0.90, p = 0.0003 for Sweeney3; AUC of 0.84, p = 0.003 for BATF2; AUC of 0.87, p = 0.001 for RISK6) (Figure S2B). Expression of the Zak16 signature was also highly correlated with all three signatures (p < 0.0001 and spearman correlation coefficient r of 0.85, 0.91 and 0.68 for Sweeney3, BATF2, and RISK6, respectively) (Figure S2C). All 10 individuals identified as LTBI-Risk according to the Zak16 signature showed an increased z-score compared to the LTBI-Other group (Figure S2D) and were also ranked in the top 10 highest z-score for at least two of the three other signatures. Six of the 10 genes making up the three other signatures showed a high level of correlation of expression within each individual (BATF2; DUSP3 and GBP5 in Sweeney3; FCGR1B, GBP2 and SERPING1 in RISK6, Figure S2E) including all overlapping genes with Zak16 (Figure S2A). We applied the Zak16 signature classification for subsequent analyses as it is the only signature presenting an overlap with all three others, and thus the most representative of the literature.
Figure 1: Identification of LTBI participants at higher risk progression based on the expression of a previously published ‘risk of progression to ATB’ signature prior the start of treatment.
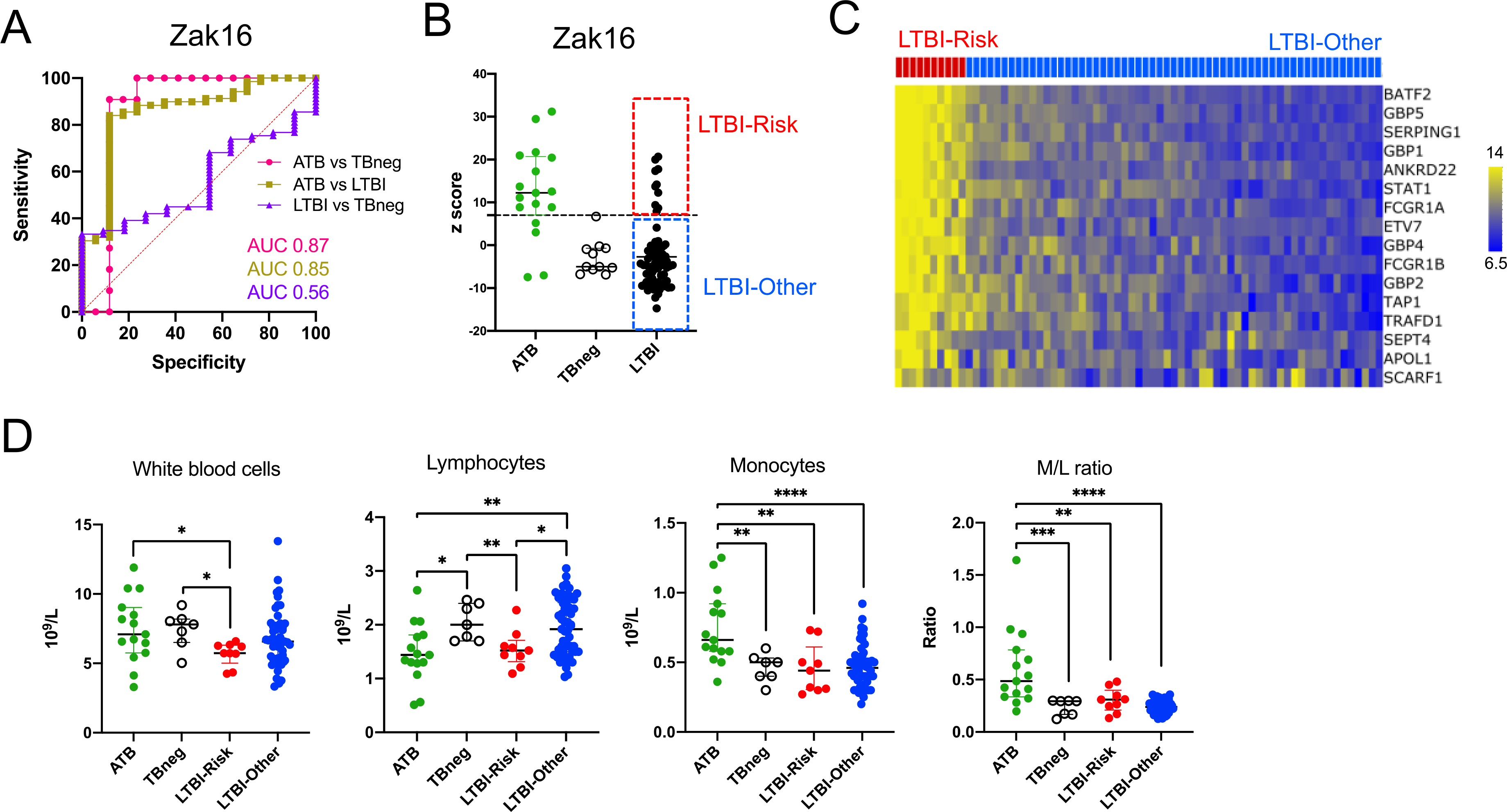
A) Receiver operating characteristic (ROC) curves and B) individual z-scores for the Zak16 gene signature 17 in blood samples collected prior to treatment in ATB (n=17), LTBI (n=69) and TBneg (n=11) individuals. The upper value in the TBneg cohort was used to stratify the LTBI cohort into LTBI-Risk (n=10), and LTBI-Other (n=59) groups. C) Heatmap representing the normalized expression of individuals genes of the Zak16 gene signature in blood samples collected prior to treatment in LTBI-Risk and LTBI-Other individuals. If a gene was mapped to several probes, average expression of all probes was shown. D) Differences in full blood counts parameters in blood samples collected prior to treatment in ATB (n=15), TBneg (n=7), LTBI-Risk (n=9) and LTBI-Other (n=55). Statistical differences between cohorts were defined with the non-parametric unpaired Mann-Whitney test (* p < 0.05; ** p < 0.01; *** p < 0.001).
Full blood counts from samples collected prior to treatment indicated significant differences between all four cohorts (Figure 1D). ATB has been previously associated with increased monocyte counts and M/L ratio compared to LTBI 38, 39. We found that monocyte counts and monocyte to lymphocyte ratio (M/L ratio) were increased in ATB compared to TBneg and LTBI cohorts and that lymphocyte counts were reduced in ATB compared to TBneg or LTBI-Other cohorts (Figure 1D). Interestingly, the LTBI-Risk group also showed a reduction in lymphocyte counts, as well as total white blood cells counts compared to TBneg or LTBI-Other cohorts (Figure 1D). Thus, in terms of white blood cell composition, it appears that LTBI-Risk participants share some similarities with the ATB cohort. No quantitative differences were observed for polymorphonuclear cells or platelets between any of the cohorts (Figure S3).
3.2. Identification of an ATB treatment signature
In order to identify transcriptomic changes in the LTBI groups upon treatment and define its overlap with ATB, we first identified the treatment signature in our ATB cohort. Several studies have reported whole blood gene signatures of treatment in ATB 23, 24, 26. To ensure that our experimental data and analysis strategy were consistent with previous findings, we set out to identify the treatment signature of the ATB cohort by comparing ATBpost and ATBpre samples (ATBΔ) and define its overlap to previously published signatures. Despite the low number of participants (n=8, including 3 microbiologically confirmed cases), we found significant changes upon treatment with a total of 262 differentially expressed probes (which mapped to 238 unique genes) (adjusted p < 0.1, Figure 2A and 2B, Table S3). The expression pattern of the ATB treatment gene signature was similar in ATB cases that were either confirmed (n=3) or suspected (n=5) for both upregulated and downregulated genes (Figure 2A and 2C). No differences were observed for participants with pulmonary (n=6) vs extra-pulmonary (n=2) ATB (data not shown). Genes that were upregulated after treatment in ATB were associated with the highest cumulative expression level in NK cells (Figure 2D) and were significantly enriched for one pathway (tyrosine kinase activity upon TCR activation, Figure 2E). In contrast, genes that were downregulated after treatment in ATB were strongly associated with classical and non-classical monocytes (Figure 2F) and significantly enriched for several cytokine signaling pathways including IFNγ, IL-5, and IL-12 (Figure 2G). We compared our ATBΔ treatment signature (designated Burel238) to previously published host transcriptomic signatures following anti-TB therapy in ATB patients (Bloom267 23, Cliff62 24, and Thompson9 26) and found a significant overlap between our ATBΔ treatment signature and these signatures (Figure 2H, Table S4). This confirmed that our approach for defining an ATB-treatment signature identifies sets of genes that are comparable to those identified in previous studies.
Figure 2: Blood transcriptomic signature of treatment in the ATB cohort is associated with NK cells, monocytes and IFN signaling.
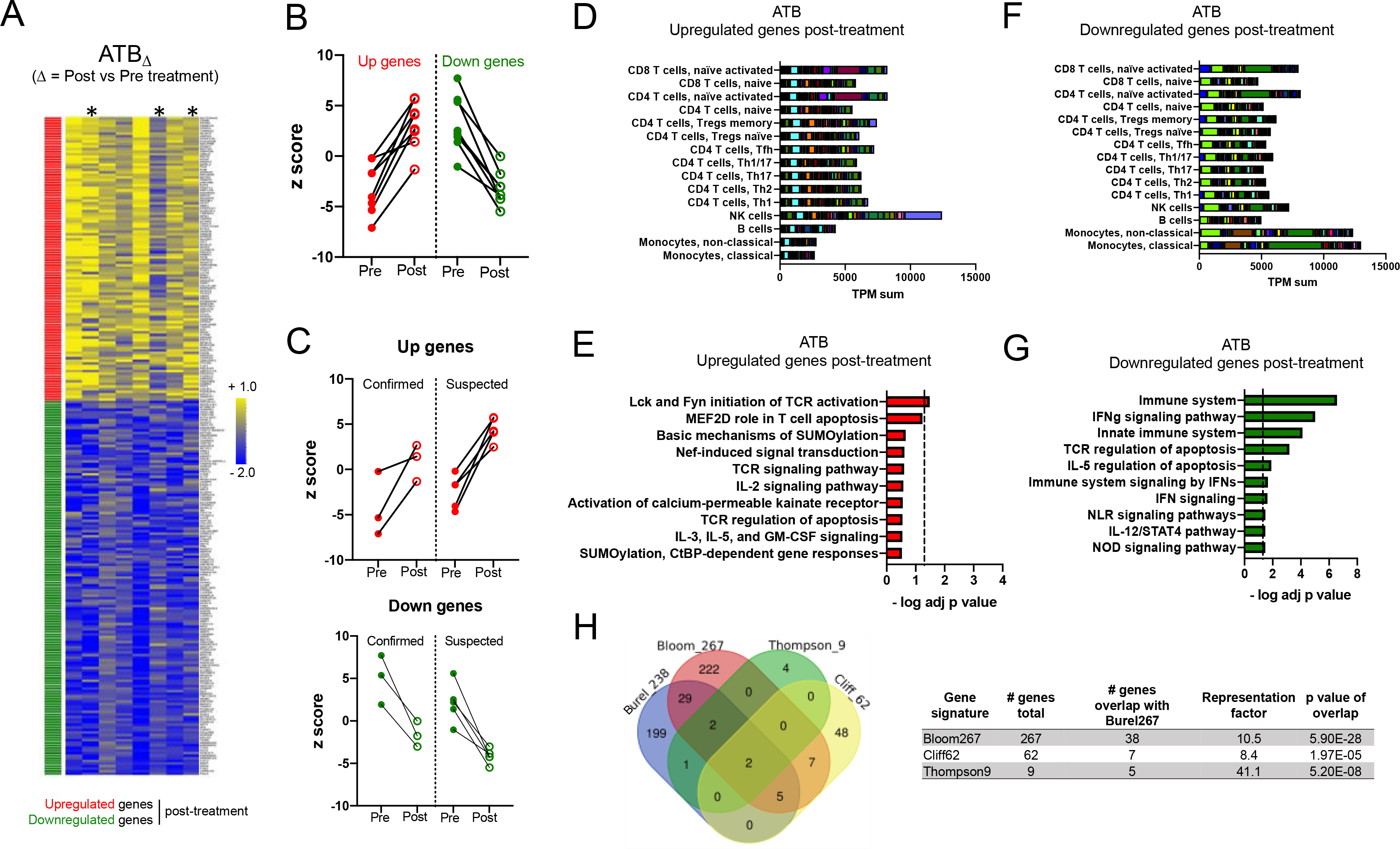
A) Heatmap representing the log fold change expression upon treatment of the upregulated (red) and downregulated (green) gene-mapped probes in the ATBΔ signature derived from differential expression analysis (adjusted p value < 0.1) between post-and pre-treatment paired blood samples in the ATB cohort (n=8) (Table S3). Asterisks show microbiologically confirmed cases. B) Individual z-scores pre and post treatment for the upregulated and downregulated genes in the ATBΔ signature in B) all ATB participants (n=8) and C) microbiologically confirmed (n=3) vs suspected (n=5) ATB participants. D) Immune cell type-specific expression and E) top-10 biological pathways enriched in the upregulated genes of the ATBΔ signature. F) Immune cell type-specific expression and G) top-10 biological pathways enriched in the downregulated genes of the ATBΔ signature. (D,F) For immune cell type-specific expression, each bar consists of stacked sub-bars showing the TPM normalized expression of every gene in corresponding cell type, extracted from the DICE database 34 (http://dice-database.org/). (E,G) Biological pathways were ranked with increasing adjusted p value and dotted line represents significance threshold (adjusted p value < 0.05). H) Overlap between our newly identified ATBΔ treatment signature (Burel238) and previously reported signatures associated with anti-TB therapy 23, 24, 26.
3.3. Identification of LTBI-Risk and LTBI-Other treatment signatures
Next, we investigated the transcriptional changes associated with anti-TB therapy in the LTBI cohort by identifying differentially expressed genes between LTBIpost and LTBIpre samples in both LTBI-Risk and LTBI-Other cohorts (LTBI-RiskΔ and LTBI-OtherΔ treatment signatures) for which a paired blood sample was available (6/10 for LTBI-Risk and 39/59 LTBI-Other participants). There was only one differentially expressed probe when comparing post and pre-treatment samples in the LTBI-Risk group using the same cutoff that was used for the ATB cohort (adjusted p < 0.1, Table S5). The probe was mapped to the gene GBP6, which has been repeatedly associated with whole blood transcriptomic signatures of ATB 23, 40, 41. In the LTBI-Other group, the same cutoff identified seven probes (which mapped to seven genes) as differentially expressed, all upregulated (Table S6). These results indicate that anti-TB therapy in LTBI leads to minor changes in the blood transcriptome compared to the ATB cohort.
Since our study aimed to identify global similarities in blood transcriptome changes upon treatment in LTBI and ATB, it sought to identify a higher number of genes that were dysregulated upon treatment in the LTBI groups. We therefore modified the statistical threshold to a non-adjusted p value lower than 0.01 and an absolute log fold change higher than 0.2. This fold change threshold was selected to compensate for the lowest stringency of the non-adjusted p value in comparison to the adjusted p value, and eliminate half of the differentially expressed genes meeting the new criteria of non-adjusted p < 0.01. With this modified threshold, we identified 140 differentially expressed probes (which mapped to 125 genes) following treatment in the LTBI-Risk group (Figure 3A, Table S5), and 195 differentially expressed probes (which mapped to 185 genes) after treatment in the LTBI-Other group (Figure 4A, Table S6).
Figure 3: Blood transcriptomic signature of treatment in the LTBI-Risk cohort is associated with activated T cells and IFN signaling.
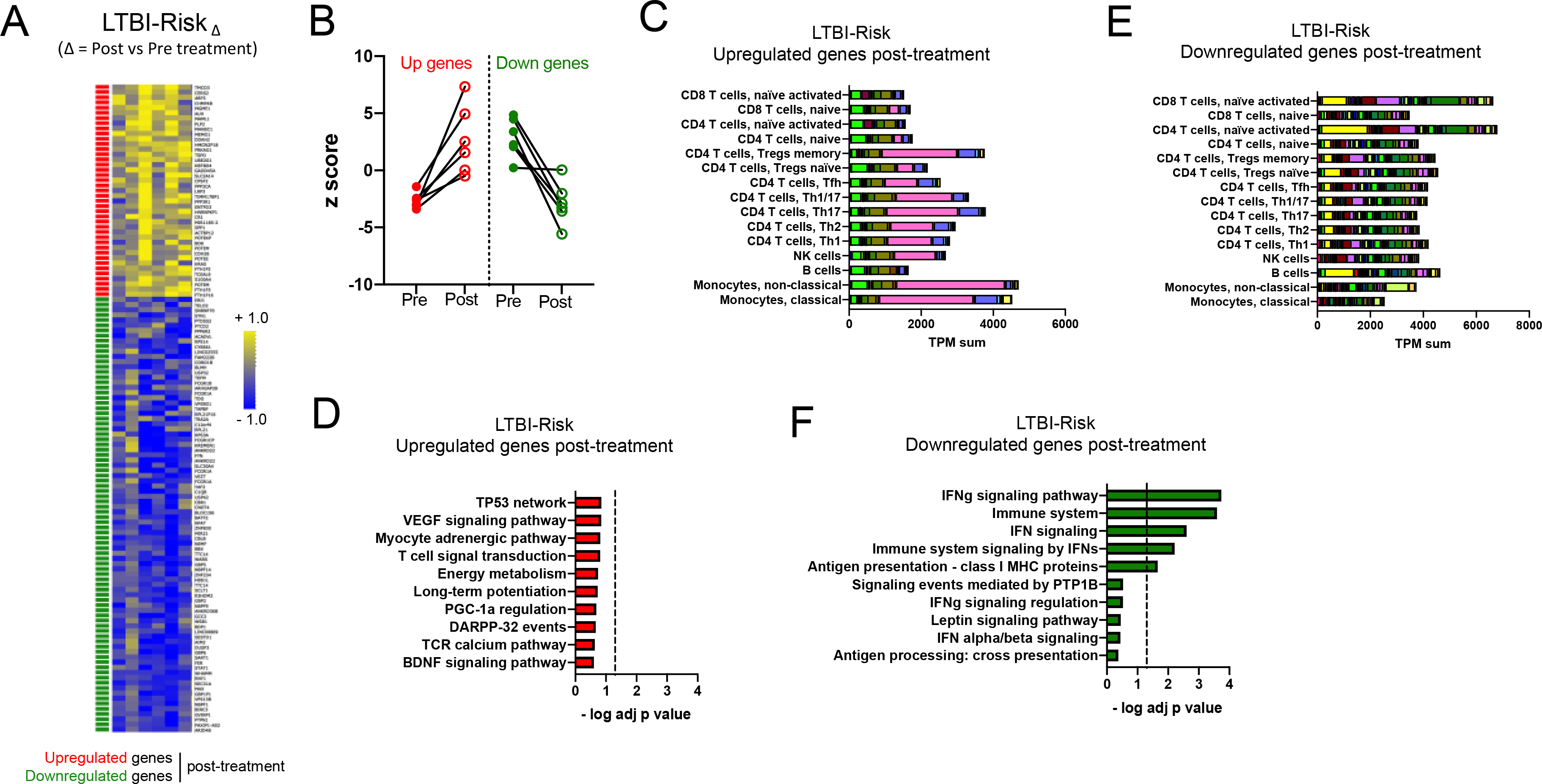
A) Heatmap representing the log fold change expression upon treatment and B) individual z-scores pre and post treatment for the upregulated (red) and downregulated (green) gene-mapped probes in the LTBI-RiskΔ signature derived from differential expression analysis (non-adjusted p value < 0.01, absolute log fold change > 0.2) between post-and pre-treatment paired blood samples in the LTBI-Risk cohort (n=6) (Table S5). C) Immune cell type-specific expression and D) top-10 biological pathways enriched in the upregulated genes of the LTBI-RiskΔ signature. E) Immune cell type-specific expression and F) top-10 biological pathways enriched in the downregulated genes of the LTBI-RiskΔ signature. (C,E) For immune cell type-specific expression, each bar consists of stacked sub-bars showing the TPM normalized expression of every gene in corresponding cell type, extracted from the DICE database 34 (http://dice-database.org/). (D,F) Biological pathways were ranked with increasing adjusted p value and dotted line represents significance threshold (adjusted p value < 0.05).
Figure 4: Blood transcriptomic signature of treatment in the LTBI-Other cohort is associated with NK cells, B cells and platelet-related pathways.
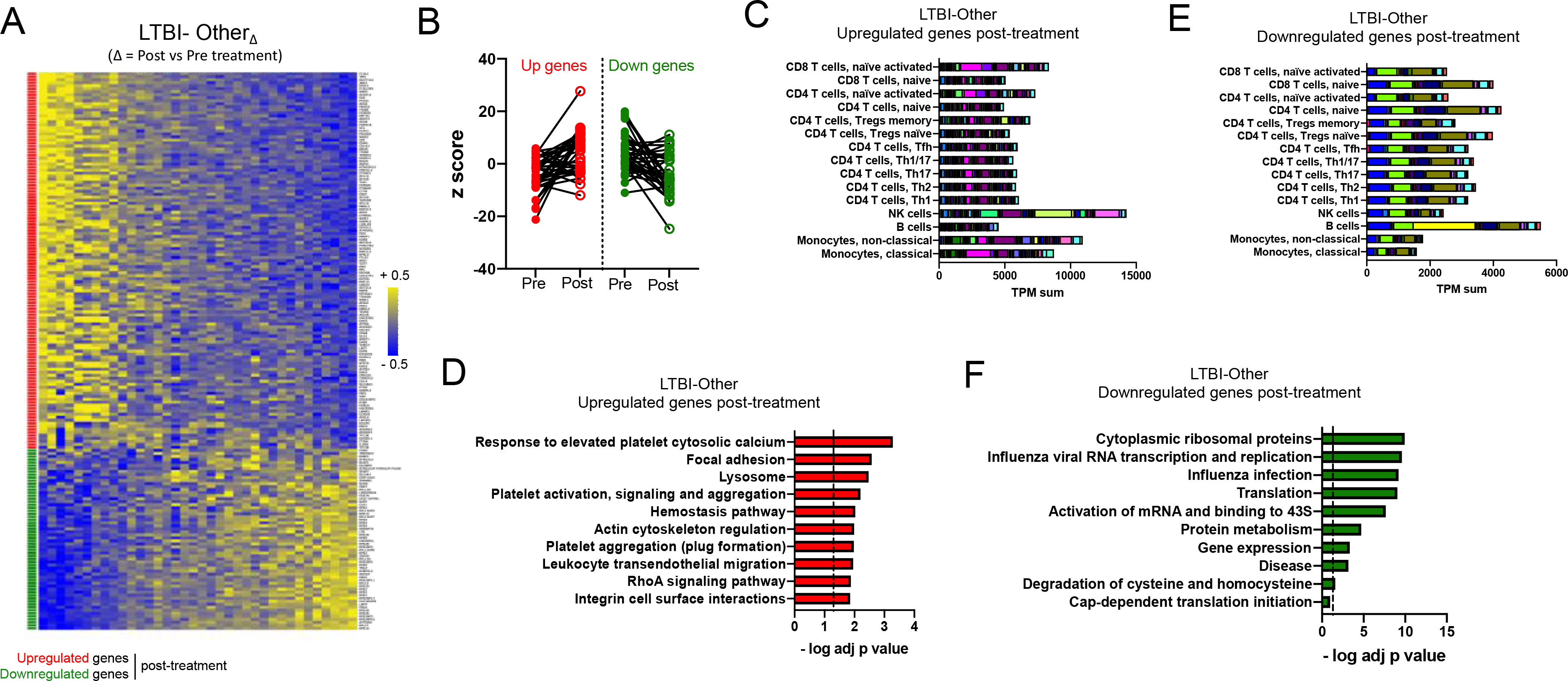
A) Heatmap representing the log fold change expression upon treatment and B) individual z-scores pre and post treatment for the upregulated (red) and downregulated (green) gene-mapped probes in the LTBI-OtherΔ signature derived from differential expression analysis (non-adjusted p value < 0.01, absolute log fold change > 0.2) between post-and pre-treatment paired blood samples in the LTBI-Other cohort (n=36) (Table S6). C) Immune cell type-specific expression and D) top-10 biological pathways enriched in the upregulated genes of the LTBI-OtherΔ signature. E) Immune cell type-specific expression and F) top-10 biological pathways enriched in the downregulated genes of the LTBI-OtherΔ signature. (C,E) For immune cell type-specific expression, each bar consists of stacked sub-bars showing the TPM normalized expression of every gene in corresponding cell type, extracted from the DICE database 34 (http://dice-database.org/). (D,F) Biological pathways were ranked with increasing adjusted p value and dotted line represents significance threshold (adjusted p value < 0.05).
The LTBI-RiskΔ signature contained mostly genes that were downregulated upon treatment (Figure 3A). Despite a variation in magnitude, all participants showed a relatively homogeneous expression profile except for one participant who did not show any changes in expression upon treatment for the downregulated genes (Figure 3B). Upregulated genes were associated with the highest cumulative expression level in classical and non-classical monocytes (Figure 3C) and no statistically significant enrichment for biological pathways (Figure 3D). Downregulated genes were associated with the highest cumulative expression level in activated T cells (Figure 3E) and significant enrichment for IFN signaling pathway (Figure 3F).
In contrast, the LTBI-OtherΔ signature showed much higher heterogeneity across participants and the majority of genes were upregulated upon treatment (Figure 4A and 4B). Interestingly, some participants showed an antagonistic pattern with downregulation of the expected upregulated genes and vice-versa (Figure 4A and 4B). Further analysis identified that age, but not sex, ethnicity, or time since completion of treatment was significantly positively correlated with the LTBI-OtherΔ signature score (Figure S4). This observation highlights the significant heterogeneity within the LTBI cohort and suggests the LTBI-Other group could be further divided into several subgroups, in particular age groups. Upregulated genes in the LTBI-OtherΔ signature had the highest cumulative expression level in NK cells, followed by non-classical monocytes (Figure 4C). Biological pathways significantly enriched in the upregulated genes upon treatment included platelet-associated pathways (response to elevated platelet cytosolic calcium; platelet activation, signaling and aggregation; platelet aggregation (plug formation)) and adhesion/migration pathways (focal adhesion; leukocyte transendothelial migration; integrin cell surface interactions) (Figure 4D). Downregulated genes had the highest cumulative expression level in B cells (Figure 4E) and significant enrichment for ribosomal gene associated pathways (top 8 pathways, Figure 4F).
3.4. Significant overlap between LTBI-Risk and ATB treatment signatures
To investigate if treatment-induced transcriptional changes in LTBI-Risk or LTBI-Other cohort resembled those seen in ATB, we defined the overlap between the treatment signatures in all three cohorts: ATBΔ (Figure 2A, Table S3), LTBI-RiskΔ (Figure 3A, Table S5) and LTBI-OtherΔ (Figure 4A, Table S6). There were four genes shared between LTBI-RiskΔ and LTBI-OtherΔ (R3HDM2, SEC31A, SNRNP70, WARS) and five genes shared between LTBI-OtherΔ and ATBΔ (DISC1, IL2RB, SP100, TTC38, TUT7B) (Figure 5A). Neither of these two overlaps was significant. In contrast, a significant overlap was found between LTB-RiskΔ and ATBΔ, with 21 genes shared (adjusted p < 5e-17, Figure 5A). All 21 genes were downregulated upon treatment in both LTB-Risk and ATB cohorts (Figure 5B and 5C). Interestingly, the LTBI-Other cohort presented an opposite pattern with prevailing upregulation of all these genes upon treatment (Figure 5B and 5C). The 21 genes were associated with the highest cumulative expression level in T cells (Figure 5D), particularly activated T cells, and were significantly enriched for IFN signaling pathway (Figure 5E). Thus, transcriptomic changes upon anti-TB therapy in the LTBI-Risk group resemble those occurring in ATB, with shared downregulated genes associated with IFN signaling.
Figure 5: Blood transcriptomic signatures of treatment in the LTBI-Risk cohort significantly overlaps with the ATB cohort.
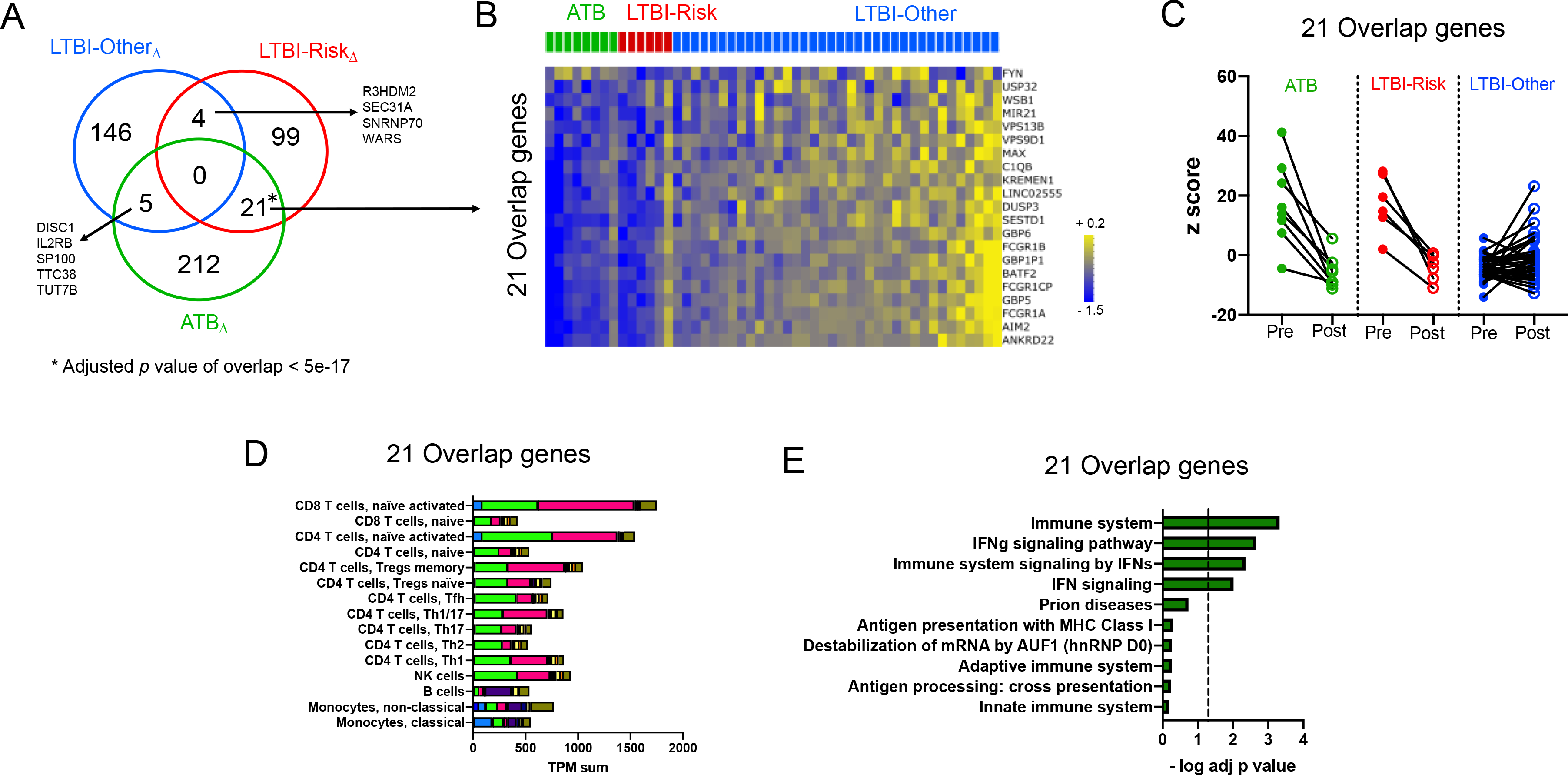
A) Venn-diagram representing overlaps between individual genes from the ATBΔ, LTBI-RiskΔ and LTBIΔ-Other treatment signatures as defined in Figures 2, 3 and 4. B) Heatmap representing the log fold change expression upon treatment and C) individual z-scores pre and post treatment for the 21 overlapping genes between the LTBI-RiskΔ and ATBΔ treatment signatures across LTBI-Risk, LTBI-Other and ATB cohorts. For B), if a gene was mapped to several probes, average expression of all probes was shown. D) Immune cell type-specific expression in the 21 overlapping genes between the LTBI-RiskΔ and ATBΔ treatment signatures. Each bar consists of stacked sub-bars showing the TPM normalized expression of every gene in corresponding cell type, extracted from the DICE database 34 (http://dice-database.org/). E) top-10 biological pathways enriched in the 21 overlapping genes between the LTBI-RiskΔ and ATBΔ treatment signatures. Biological pathways were ranked with increasing adjusted p value and dotted line represents significance threshold (adjusted p value < 0.05).
3.5. Expression changes of ‘risk of progression to active TB’ signatures during treatment in LTBI
We identified that the 21 overlapping genes shared between LTB-RiskΔ and ATBΔ contained several genes from ‘risk of progression to ATB’ signatures (ANKRD22, BATF2, DUSP3, FCGR1A/B and GBP5, Figure 5B), which we used to stratify the LTBI cohort into LTBI-Risk and LTBI-Other groups prior the start of treatment (Figure 1A and Figure S2). This is not unexpected, since the Zak16 signature expression has been shown to be reduced upon treatment in ATB 17. This prompted us to further analyze expression changes of individual genes from risk progression signatures upon anti-TB therapy in LTBI.
We classified genes present in any of the four progression signatures (Zak16 17, Sweeney3 18, 19, BATF2 20 and RISK6 21) as either downregulated, upregulated or not significantly changed upon anti-TB therapy in the LTBI-Risk or LTBI-Other cohorts. Out of the total number of genes present in any of the four progression signatures, 8 were downregulated upon treatment in the LTBI-Risk group but not the LTBI-Other group (Figure 6A, Figure S5). The remaining genes were not differentially expressed upon treatment in either the LTBI-Risk or the LTBI-Other group, according to our criteria of significance (p < 0.01 and absolute log fold change > 0.2; Figure 6A, Figure S6). Genes from the progression signatures that were selectively downregulated in the LTBI-Risk group showed significant enrichment for IFN signaling (Figure 6B) and for functional protein-protein associations (p value of enrichment = 1.1E-16, Figure 6C) in particular between IFN signaling associated genes. In contrast, no statistically significant enrichment for biological pathways or functional protein-protein associations was found in the ‘not significant’ genes upon treatment (Figure 6D, Figure 6E). Overall, this shows that several genes from previously described progression to ATB signatures, in particular those that are significantly associated with IFN signaling, have a reduced expression upon TB-preventive treatment in LTBI-Risk but not LTBI-Other participants.
Figure 6: Genes from ‘risk of progression to ATB’ signatures that are associated with IFN signaling are downregulated upon anti-TB treatment in LTBI-Risk but not LTBI-Other participants.
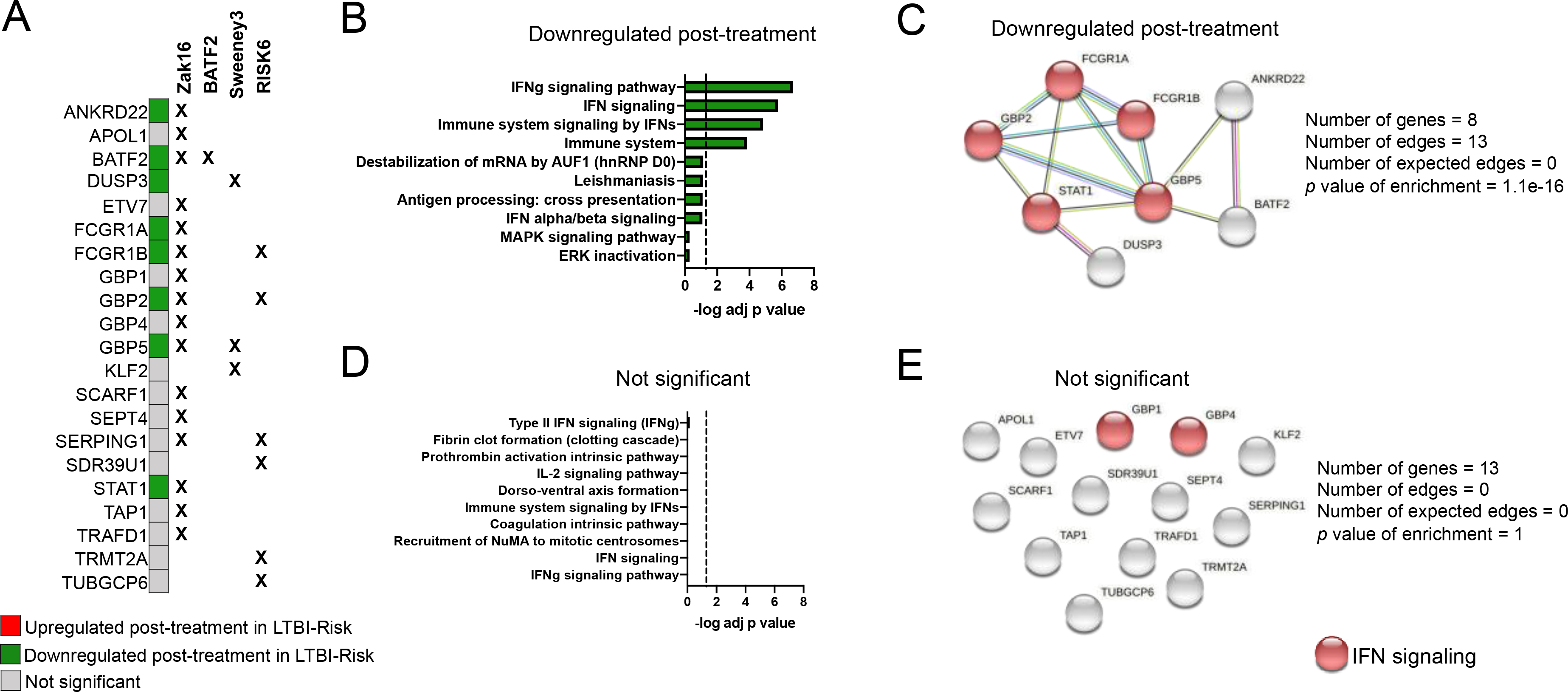
A) Individual classification of genes within the ‘risk of progression to ATB’ signatures (reported in references 17, 18, 20, 21) based on whether they are differentially expressed in the LTBI-Risk or the LTBI-Other group upon treatment (LTBI-RiskΔ and LTBI-OtherΔ treatment signatures defined in Figures 3 and 4). None of the genes were differentially expressed in the LTBI-Other group upon treatment. B) top-10 biological pathways enriched and C) functional protein-protein association network in the downregulated genes upon treatment in the LTBI-Risk group. D) top-10 biological pathways enriched and E) functional protein-protein association network in genes that are not significantly dysregulated upon treatment in either the LTBI-Risk or LTBI-Other groups. (B,D) Biological pathways were ranked with increasing adjusted p value and dotted line represents significance threshold (adjusted p value < 0.05). (C,E) Functional protein-protein association networks were defined by querying the online STRING database (version 11.0).
4. Discussion
We studied changes in the host blood transcriptome following anti-TB therapy in LTBI to investigate immune heterogeneity in response to treatment in this cohort. We stratified LTBI participants into two groups based on Zak16, the seminal ‘risk of progression to ATB’ gene signature reported in LTBI17 (and which correlated well with subsequent signatures 18–21): LTBI-Risk and LTBI-Other, displaying high and low expression of the Zak16 gene signature prior to treatment, respectively. We identified 10 out of 69 LTBI participants (14%) as LTBI-Risk. This prevalence is much higher than the expected rate of progression from LTBI to ATB 22, 42, therefore it is unlikely that all of the LTBI-Risk individuals would develop active disease in their lifetime. A distinct classification might have been obtained with another method to calculate the gene signature score, and set the threshold for positivity. We used a standard z-score approach (see methods) without gene pairing to calculate gene signature scores in our study. This differs from the methods that were used for the calculation of Zak16 17 and RISK6 21 signature scores, which used customized gene pairing approaches, but allowed us to use a consistent method across all gene signatures analyzed in this study (i.e. previously defined and newly discovered). As for the threshold, we used the highest value within the TBneg cohort to divide the LTBI cohort into LTBI-Risk and LTBI-Other, which is subjective to our study. This is based on the hypothesis that the TBneg cohort contain exclusively Mtb uninfected individuals. Thus, the maximum value of the TBneg cohort is the maximum value that would be expected for individuals with no evidence of current or past Mtb infection, and any higher value potentially reflects incipient disease (LTBI-Risk). We elected this approach as the Zak16 signature was originally measured with RNAseq and qRT-PCR, and using a different method (as mentioned above)17, thus cut-offs used in that study could not be directly translated to our microarray expression data.
Using our classification, we identified that LTBI-Risk individuals displayed a whole blood transcriptomic signature following preventative therapy that overlapped significantly with that observed among the ATB cohort in response to treatment, but differed from the remaining LTBI cohort. We contend that such overlap is due to immunological similarities in response to treatment and corroborates the hypothesis that the LTBI-Risk cohort includes individuals with incipient disease. Our findings strike a chord with those of a very recent study in a small LTBI population which reported stratification of the cohort into two distinct populations based on their whole blood transcriptome upon treatment 28. The authors suggested that this dichotomy might differentiate between individuals with viable bacilli from immunological sensitization without viable infection 28. There was no overlap between their and our treatment signatures, likely due to the fact that their gene expression data was generated after Mtb-specific in vitro stimulation rather than measured directly ex vivo in whole blood. In addition, we also found full blood count differences between LTBI-Risk and LTBI-Other cohorts. Similar to ATB, the LTBI-Risk group showed reduced lymphocyte counts compared to TBneg or LTBI-Other groups. Taken together, our findings support our hypothesis that the LTBI-Risk cohort is at higher risk for progression to active disease compared to the LTBI cohort at large, and thus resembles more closely ATB participants. More importantly, they suggest that monitoring blood transcriptomic changes upon treatment might help to identify which individuals have incipient disease and viable bacilli from those who do not, and thus identify which LTBI individuals might more benefit from chemoprophylaxis treatment.
The LTBI-RiskΔ treatment signature contained a significant overlap with ‘risk of progression to ATB’ signatures. Looking at four predictive gene signatures (Zak16 17, Sweeney3 18, 19, BATF2 20 and RISK6 21), we have shown that in the subset of LTBI-Risk individuals (who do express high level of these genes prior to treatment), several of the ‘risk of progression to ATB’ genes are downregulated upon anti-TB therapy. These genes were significantly enriched for IFN signaling and functional protein-protein associations, in comparison to those that are not significantly affected by treatment. The IFN signaling pathway has been repeatedly associated with host RNA signatures in TB 23, 24, 40, 43, and in our study, it was also significantly enriched in downregulated genes in both LTBI-RiskΔ and ATBΔ treatment signatures. This likely reflects the higher activated state of both innate and adaptive immune cells present in the blood of clinically infected individuals and incipient cases, which is then switched off upon treatment and highlights the importance of this pathway in LTBI progression to ATB. The presence of an IFN signature in the blood has been repeatedly associated with other respiratory infections including Influenza 44, SARS-CoV-2 45, but also auto-immune diseases 46. Thus, an alternative explanation for the elevated expression of the IFN pathway in the blood of LTBI-Risk participants could be the presence of another underlying infection or illness at the time of enrollment.
In addition to IFN associated genes, downregulated genes in the ATBΔ treatment signature showed the highest cumulative expression in monocytes, which is consistent with previous findings that monocyte absolute counts and monocyte to lymphocyte ratio are prospective markers of active TB risk 47, 48, and that these parameters are reduced following TB therapy in ATB 39. Conversely, upregulated genes in the ATB and the LTBI-Other treatment signature showed highest expression in NK cells. NK cells have been previously associated with protective immune responses to TB 49, and their frequency and cytolytic potential is increased in LTBI individuals compared to ATB or TBneg cohorts 50. Upon TB treatment, NK cells are increased in frequency 50 and display increased activation and cytokine production 51. Taken together, these results suggest monocytes and NK cells might play a key role in Mtb immune responses. Alternatively, this may reflect changes in frequency of white blood cell populations over time in response to TB treatment, rather than qualitative changes within these subsets.
Regardless of whether dysregulated gene expression profiles in the blood upon treatment have a quantitative or qualitative origin, our study shows its potential for monitoring immune responses associated with chemoprophylactic treatment of TB. There are currently no clinical biomarkers for monitoring the success of anti-TB therapy in LTBI. Previous studies have found no evidence that IGRA tests are a reliable metric for monitoring LTBI treatment 14, 15. A recent study has reported the presence of Mtb DNA in CD34+ PBMC, which was reduced upon TB chemoprophylaxis therapy in HIV infected individuals 52. Importantly, their marker correlated poorly with IGRA status, suggesting it might reflect “true” LTBI rather than Mtb sensitization. The main limitation of this test is the requirement of a large volume of blood (100mL) which would be challenging to implement in low resource countries with high Mtb prevalence. Nevertheless, it highlights the promise of blood-based biomarkers other than IGRA for monitoring TB therapy in LTBI. One gene expression study has found a handful of genes that are universally downregulated in LTBI upon treatment, but did not consider LTBI as a heterologous population, and limited their analysis to 10 genes 53. Only one study has previously investigated blood transcriptomic changes upon LTBI treatment on a genome-wide scale in 18 participants, but relied on Mtb in vitro stimulation to identify gene signatures 28. Thus, our genome-wide longitudinal investigation of the ex vivo blood transcriptome upon treatment in 42 LTBI participants constitutes a significant novel addition to the literature.
By monitoring blood transcriptomic changes directly ex vivo upon treatment, we observed a selective downregulation of several of the IFN associated progression signatures genes upon anti-TB therapy in LTBI-Risk but not LTBI-Other participants, highlighting the potential value of these signatures and the IFN pathway to monitor LTBI treatment in individuals with increased risk of progression to ATB. This observation requires further validation in larger studies as we identified that preventive TB treatment induces subtle longitudinal gene expression changes ex vivo in blood in LTBI. In particular, our small cohort of paired blood samples collected pre and post treatment in LTBI-Risk (n=6) and ATB (n=8) participants precluded us to use “strict” statistical cutoffs, that could be reused in future studies. Additionally, since our study is the first of its kind, we have no previous datasets to include as validation cohorts, compare our findings, or run meta-analyses as was elegantly done in the context of ATB in Chowdhury et al 50. Such studies would increase the robustness of the statistical thresholds required to select genes used as biomarkers. Specifically, it would be of interest to compare our results to LTBI samples collected in areas with higher endemicity where it is logistically more challenging to treat the LTBI population, and where increased exposure to Mtb might shape differently the immune responses to treatment. Finally, blood transcriptomic changes upon treatment need to be investigated in association with clinical parameters such as occurrence of progression to ATB. A recent prospective longitudinal study in South Africa found no evidence that preventative treatment reduced the rate of incident tuberculosis over 15 months in LTBI participants classified as higher risk of progression to ATB based on the expression of a subset of the Zak16 signature prior to initiation of therapy 42. This important result suggests that the LTBI-Risk group might be further heterogeneous, and that only assessing blood RNA signatures prior to initiation of treatment is not informative enough to predict progression to ATB upon treatment completion. Additionally, Gupta et al. in their multicohort meta-analysis found that whole blood gene signatures of incipient TB, including Zak16, have a relatively short window of prognostic utility 22. Close prospective monitoring of blood transcriptomic changes occurring early after initiation of preventive therapy in such cohorts could shed light on the immune events specifically occurring among individuals who may re-activate, and might ultimately allow the prediction of treatment failure or success in LTBI.
5. Conclusions
In summary, our study clearly demonstrates that individuals with LTBI display a heterogenous blood transcriptome profile upon anti-TB therapy, which might reflect distinct “Mtb infection states” within LTBI. Upon chemoprophylactic treatment, individuals at higher risk of developing ATB (LTBI-Risk) showed similar blood transcriptomic changes to ATB infected individuals undergoing anti-TB therapy in comparison to the remaining LTBI cohort. In particular, both ATB and LTBI-Risk groups showed selective downregulation of genes that are associated with IFN signaling and that were previously identified in progression signatures. This corroborates the hypothesis that LTBI-Risk individuals have incipient disease and that treatment elicits immune responses that lead to elimination of Mtb bacilli similar to those occurring upon ATB treatment. It also suggests that IFN associated genes from progression signatures might be a promising target to monitor the course of treatment in these individuals. Overall, this study confirms the value of using blood transcriptomics to monitor immune changes occurring upon preventive therapy in LTBI. Future larger studies will be necessary to further dissect LTBI heterogeneity in immune responses to treatment, in particular in the context of post-treatment clinical outcomes and identify genes that could be used as biomarkers to monitor the course of treatment within each group.
Supplementary Material
Acknowledgments
We thank the staff at the clinical sites for assistance with patient recruitment and obtaining samples, particularly Donna Tupper (Catterick).
Funding
This work was funded by the Wellcome Trust (grant number 103420/Z/13/Z), the British Lung Foundation (grant number BHPG13–2), and the National Institute of Allergy and Infectious Diseases of the National Institute of Health (grant numbers R01 AI137681 and U19 AI118626). The funders were not involved in study design, data collection and analysis, decision to publish, nor in preparation of the manuscript. C. Broderick et al.
Footnotes
Declaration of competing interest
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.World Health Organization. Global tuberculosis report 2020. 2020.
- 2.Barry CE 3rd, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, Schnappinger D, Wilkinson RJ, Young D. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol 2009;7:845–855. doi: 10.1038/nrmicro2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Shea MK, Tanner R, Muller J, Harris SA, Wright D, Stockdale L, Stylianou E, Satti I, Smith SG, Dunbar J, Fletcher TE, Dedicoat M, Cunningham AF, McShane H. Immunological correlates of mycobacterial growth inhibition describe a spectrum of tuberculosis infection. Sci Rep 2018;8:14480. doi: 10.1038/s41598-018-32755-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drain PK, Bajema KL, Dowdy D, Dheda K, Naidoo K, Schumacher SG, Ma S, Meermeier E, Lewinsohn DM, Sherman DR. Incipient and Subclinical Tuberculosis: a Clinical Review of Early Stages and Progression of Infection. Clin Microbiol Rev 2018;31 doi: 10.1128/CMR.00021-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Houben RM, Dodd PJ. The Global Burden of Latent Tuberculosis Infection: A Re-estimation Using Mathematical Modelling. PLoS Med 2016;13:e1002152. doi: 10.1371/journal.pmed.1002152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vynnycky E, Fine PE. The natural history of tuberculosis: the implications of age-dependent risks of disease and the role of reinfection. Epidemiol Infect 1997;119:183–201. doi: 10.1017/s0950268897007917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.England PH. Latent TB infection testing and treatment programme: technical guidance and specification. 2019 [Google Scholar]
- 8.World Health Organization. Latent TB Infection: Updated and consolidated guidelines for programmatic management. accessed at https://www.who.int/tb/publications/2018/latent-tuberculosis-infection/en/ 2018 [PubMed]
- 9.Sterling TR, Villarino ME, Borisov AS, Shang N, Gordin F, Bliven-Sizemore E, Hackman J, Hamilton CD, Menzies D, Kerrigan A, Weis SE, Weiner M, Wing D, Conde MB, Bozeman L, Horsburgh CR Jr., Chaisson RE, Team TBTCPTS. Three months of rifapentine and isoniazid for latent tuberculosis infection. N Engl J Med 2011;365:2155–2166. doi: 10.1056/NEJMoa1104875 [DOI] [PubMed] [Google Scholar]
- 10.Sharma SK, Sharma A, Kadhiravan T, Tharyan P. Rifamycins (rifampicin, rifabutin and rifapentine) compared to isoniazid for preventing tuberculosis in HIV-negative people at risk of active TB. Cochrane Database Syst Rev 2013:CD007545. doi: 10.1002/14651858.CD007545.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sterling TR, Scott NA, Miro JM, Calvet G, La Rosa A, Infante R, Chen MP, Benator DA, Gordin F, Benson CA, Chaisson RE, Villarino ME, Tuberculosis Trials Consortium tACTGftPTBTTiotTBTC, the Aids Clinical Trials Group for the Prevent Tb Trial are listed in the Supplement i. Three months of weekly rifapentine and isoniazid for treatment of Mycobacterium tuberculosis infection in HIV-coinfected persons. AIDS 2016;30:1607–1615. doi: 10.1097/QAD.0000000000001098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaisson RE, Ramchandani R, Swindells S. One Month of Rifapentine plus Isoniazid to Prevent HIV-Related Tuberculosis. Reply. N Engl J Med 2019;381:e23. doi: 10.1056/NEJMc1908492 [DOI] [PubMed] [Google Scholar]
- 13.Pai M, Denkinger CM, Kik SV, Rangaka MX, Zwerling A, Oxlade O, Metcalfe JZ, Cattamanchi A, Dowdy DW, Dheda K, Banaei N. Gamma interferon release assays for detection of Mycobacterium tuberculosis infection. Clin Microbiol Rev 2014;27:3–20. doi: 10.1128/CMR.00034-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson JL, Geldenhuys H, Thiel BA, Toefy A, Suliman S, Pienaar B, Chheng P, Scriba T, Boom WH, Hanekom W, Hatherill M. Effect of isoniazid therapy for latent TB infection on QuantiFERON-TB gold in-tube responses in adults with positive tuberculin skin test results in a high TB incidence area: a controlled study. Chest 2014;145:612–617. doi: 10.1378/chest.13-1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dyrhol-Riise AM, Gran G, Wentzel-Larsen T, Blomberg B, Haanshuus CG, Morkve O. Diagnosis and follow-up of treatment of latent tuberculosis; the utility of the QuantiFERON-TB Gold In-tube assay in outpatients from a tuberculosis low-endemic country. BMC Infect Dis 2010;10:57. doi: 10.1186/1471-2334-10-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burel JG, Babor M, Pomaznoy M, Lindestam Arlehamn CS, Khan N, Sette A, Peters B. Host Transcriptomics as a Tool to Identify Diagnostic and Mechanistic Immune Signatures of Tuberculosis. Front Immunol 2019;10:221. doi: 10.3389/fimmu.2019.00221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zak DE, Penn-Nicholson A, Scriba TJ, Thompson E, Suliman S, Amon LM, Mahomed H, Erasmus M, Whatney W, Hussey GD, Abrahams D, Kafaar F, Hawkridge T, Verver S, Hughes EJ, Ota M, Sutherland J, Howe R, Dockrell HM, Boom WH, Thiel B, Ottenhoff THM, Mayanja-Kizza H, Crampin AC, Downing K, Hatherill M, Valvo J, Shankar S, Parida SK, Kaufmann SHE, Walzl G, Aderem A, Hanekom WA, Acs, groups GCcs. A blood RNA signature for tuberculosis disease risk: a prospective cohort study. Lancet 2016;387:2312–2322. doi: 10.1016/S0140-6736(15)01316-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sweeney TE, Braviak L, Tato CM, Khatri P. Genome-wide expression for diagnosis of pulmonary tuberculosis: a multicohort analysis. Lancet Respir Med 2016;4:213–224. doi: 10.1016/S2213-2600(16)00048-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warsinske HC, Rao AM, Moreira FMF, Santos PCP, Liu AB, Scott M, Malherbe ST, Ronacher K, Walzl G, Winter J, Sweeney TE, Croda J, Andrews JR, Khatri P. Assessment of Validity of a Blood-Based 3-Gene Signature Score for Progression and Diagnosis of Tuberculosis, Disease Severity, and Treatment Response. JAMA Netw Open 2018;1:e183779. doi: 10.1001/jamanetworkopen.2018.3779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roe J, Venturini C, Gupta RK, Gurry C, Chain BM, Sun Y, Southern J, Jackson C, Lipman MC, Miller RF, Martineau AR, Abubakar I, Noursadeghi M. Blood Transcriptomic Stratification of Short-term Risk in Contacts of Tuberculosis. Clin Infect Dis 2020;70:731–737. doi: 10.1093/cid/ciz252 [DOI] [PubMed] [Google Scholar]
- 21.Penn-Nicholson A, Mbandi SK, Thompson E, Mendelsohn SC, Suliman S, Chegou NN, Malherbe ST, Darboe F, Erasmus M, Hanekom WA, Bilek N, Fisher M, Kaufmann SHE, Winter J, Murphy M, Wood R, Morrow C, Van Rhijn I, Moody B, Murray M, Andrade BB, Sterling TR, Sutherland J, Naidoo K, Padayatchi N, Walzl G, Hatherill M, Zak D, Scriba TJ, Adolescent Cohort Study t, Consortium GC, Clinical S, Laboratory T, Screen TBC, Consortium A-T, Re PBT, Peruvian Household Contacts Cohort T, team CI. RISK6, a 6-gene transcriptomic signature of TB disease risk, diagnosis and treatment response. Sci Rep 2020;10:8629. doi: 10.1038/s41598-020-65043-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gupta RK, Turner CT, Venturini C, Esmail H, Rangaka MX, Copas A, Lipman M, Abubakar I, Noursadeghi M. Concise whole blood transcriptional signatures for incipient tuberculosis: a systematic review and patient-level pooled meta-analysis. Lancet Respir Med 2020;8:395–406. doi: 10.1016/S2213-2600(19)30282-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bloom CI, Graham CM, Berry MP, Wilkinson KA, Oni T, Rozakeas F, Xu Z, Rossello-Urgell J, Chaussabel D, Banchereau J, Pascual V, Lipman M, Wilkinson RJ, O’Garra A. Detectable changes in the blood transcriptome are present after two weeks of antituberculosis therapy. PLoS One 2012;7:e46191. doi: 10.1371/journal.pone.0046191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cliff JM, Lee JS, Constantinou N, Cho JE, Clark TG, Ronacher K, King EC, Lukey PT, Duncan K, Van Helden PD, Walzl G, Dockrell HM. Distinct phases of blood gene expression pattern through tuberculosis treatment reflect modulation of the humoral immune response. J Infect Dis 2013;207:18–29. doi: 10.1093/infdis/jis499 [DOI] [PubMed] [Google Scholar]
- 25.Cliff JM, Cho JE, Lee JS, Ronacher K, King EC, van Helden P, Walzl G, Dockrell HM. Excessive Cytolytic Responses Predict Tuberculosis Relapse After Apparently Successful Treatment. J Infect Dis 2016;213:485–495. doi: 10.1093/infdis/jiv447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson EG, Du Y, Malherbe ST, Shankar S, Braun J, Valvo J, Ronacher K, Tromp G, Tabb DL, Alland D, Shenai S, Via LE, Warwick J, Aderem A, Scriba TJ, Winter J, Walzl G, Zak DE, Catalysis TBBC. Host blood RNA signatures predict the outcome of tuberculosis treatment. Tuberculosis (Edinb) 2017;107:48–58. doi: 10.1016/j.tube.2017.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tornheim JA, Madugundu AK, Paradkar M, Fukutani KF, Queiroz ATL, Gupte N, Gupte AN, Kinikar A, Kulkarni V, Balasubramanian U, Sreenivasamurthy S, Raja R, Pradhan N, Shivakumar S, Valvi C, Hanna LE, Andrade BB, Mave V, Pandey A, Gupta A, Team CTRIS. Transcriptomic Profiles of Confirmed Pediatric Tuberculosis Patients and Household Contacts Identifies Active Tuberculosis, Infection, and Treatment Response among Indian Children. J Infect Dis 2019. doi: 10.1093/infdis/jiz639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broderick C, Cliff JM, Lee JS, Kaforou M, Moore DA. Host transcriptional response to TB preventive therapy differentiates two sub-groups of IGRA-positive individuals. Tuberculosis (Edinb) 2021;127:102033. doi: 10.1016/j.tube.2020.102033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.National Institute for Health and Care Excellence. Tuberculosis: clinical diagnosis and management of tuberculosis, and measures for its prevention and contro. accessed at https://www.nice.org.uk/guidance/cg117 2011 [PubMed]
- 30.Dunning MJ, Smith ML, Ritchie ME, Tavare S. beadarray: R classes and methods for Illumina bead-based data. Bioinformatics 2007;23:2183–2184. doi: 10.1093/bioinformatics/btm311 [DOI] [PubMed] [Google Scholar]
- 31.Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224 [DOI] [PubMed] [Google Scholar]
- 32.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012;28:882–883. doi: 10.1093/bioinformatics/bts034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. doi: 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmiedel BJ, Singh D, Madrigal A, Valdovino-Gonzalez AG, White BM, Zapardiel-Gonzalo J, Ha B, Altay G, Greenbaum JA, McVicker G, Seumois G, Rao A, Kronenberg M, Peters B, Vijayanand P. Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell 2018;175:1701–1715 e1716. doi: 10.1016/j.cell.2018.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 2013;14:128. doi: 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma’ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44:W90–97. doi: 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang R, Grishagin I, Wang Y, Zhao T, Greene J, Obenauer JC, Ngan D, Nguyen DT, Guha R, Jadhav A, Southall N, Simeonov A, Austin CP. The NCATS BioPlanet - An Integrated Platform for Exploring the Universe of Cellular Signaling Pathways for Toxicology, Systems Biology, and Chemical Genomics. Front Pharmacol 2019;10:445. doi: 10.3389/fphar.2019.00445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.La Manna MP, Orlando V, Dieli F, Di Carlo P, Cascio A, Cuzzi G, Palmieri F, Goletti D, Caccamo N. Quantitative and qualitative profiles of circulating monocytes may help identifying tuberculosis infection and disease stages. PLoS One 2017;12:e0171358. doi: 10.1371/journal.pone.0171358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang W, Wang LF, Liu YY, Yang F, Zhu L, Zhang XH. Value of the Ratio of Monocytes to Lymphocytes for Monitoring Tuberculosis Therapy. Can J Infect Dis Med Microbiol 2019;2019:3270393. doi: 10.1155/2019/3270393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O’Garra A. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010;466:973–977. doi: 10.1038/nature09247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaforou M, Wright VJ, Oni T, French N, Anderson ST, Bangani N, Banwell CM, Brent AJ, Crampin AC, Dockrell HM, Eley B, Heyderman RS, Hibberd ML, Kern F, Langford PR, Ling L, Mendelson M, Ottenhoff TH, Zgambo F, Wilkinson RJ, Coin LJ, Levin M. Detection of tuberculosis in HIV-infected and -uninfected African adults using whole blood RNA expression signatures: a case-control study. PLoS Med 2013;10:e1001538. doi: 10.1371/journal.pmed.1001538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scriba TJ, Fiore-Gartland A, Penn-Nicholson A, Mulenga H, Kimbung Mbandi S, Borate B, Mendelsohn SC, Hadley K, Hikuam C, Kaskar M, Musvosvi M, Bilek N, Self S, Sumner T, White RG, Erasmus M, Jaxa L, Raphela R, Innes C, Brumskine W, Hiemstra A, Malherbe ST, Hassan-Moosa R, Tameris M, Walzl G, Naidoo K, Churchyard G, Hatherill M, Team C-S. Biomarker-guided tuberculosis preventive therapy (CORTIS): a randomised controlled trial. Lancet Infect Dis 2021;21:354–365. doi: 10.1016/S1473-3099(20)30914-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maertzdorf J, Repsilber D, Parida SK, Stanley K, Roberts T, Black G, Walzl G, Kaufmann SH. Human gene expression profiles of susceptibility and resistance in tuberculosis. Genes Immun 2011;12:15–22. doi: 10.1038/gene.2010.51 [DOI] [PubMed] [Google Scholar]
- 44.Dunning J, Blankley S, Hoang LT, Cox M, Graham CM, James PL, Bloom CI, Chaussabel D, Banchereau J, Brett SJ, Moffatt MF, O’Garra A, Openshaw PJM, Investigators M. Progression of whole-blood transcriptional signatures from interferon-induced to neutrophil-associated patterns in severe influenza. Nat Immunol 2018;19:625–635. doi: 10.1038/s41590-018-0111-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, Choi B, Nam SK, Sa M, Kwon JS, Jeong SJ, Lee HK, Park SH, Park SH, Choi JY, Kim SH, Jung I, Shin EC. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol 2020;5 doi: 10.1126/sciimmunol.abd1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Higgs BW, Liu Z, White B, Zhu W, White WI, Morehouse C, Brohawn P, Kiener PA, Richman L, Fiorentino D, Greenberg SA, Jallal B, Yao Y. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann Rheum Dis 2011;70:2029–2036. doi: 10.1136/ard.2011.150326 [DOI] [PubMed] [Google Scholar]
- 47.Naranbhai V, Kim S, Fletcher H, Cotton MF, Violari A, Mitchell C, Nachman S, McSherry G, McShane H, Hill AV, Madhi SA. The association between the ratio of monocytes:lymphocytes at age 3 months and risk of tuberculosis (TB) in the first two years of life. BMC Med 2014;12:120. doi: 10.1186/s12916-014-0120-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rakotosamimanana N, Richard V, Raharimanga V, Gicquel B, Doherty TM, Zumla A, Rasolofo Razanamparany V. Biomarkers for risk of developing active tuberculosis in contacts of TB patients: a prospective cohort study. Eur Respir J 2015;46:1095–1103. doi: 10.1183/13993003.00263-2015 [DOI] [PubMed] [Google Scholar]
- 49.Venkatasubramanian S, Cheekatla S, Paidipally P, Tripathi D, Welch E, Tvinnereim AR, Nurieva R, Vankayalapati R. IL-21-dependent expansion of memory-like NK cells enhances protective immune responses against Mycobacterium tuberculosis. Mucosal Immunol 2017;10:1031–1042. doi: 10.1038/mi.2016.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roy Chowdhury R, Vallania F, Yang Q, Lopez Angel CJ, Darboe F, Penn-Nicholson A, Rozot V, Nemes E, Malherbe ST, Ronacher K, Walzl G, Hanekom W, Davis MM, Winter J, Chen X, Scriba TJ, Khatri P, Chien YH. A multi-cohort study of the immune factors associated with M. tuberculosis infection outcomes. Nature 2018;560:644–648. doi: 10.1038/s41586-018-0439-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garand M, Goodier M, Owolabi O, Donkor S, Kampmann B, Sutherland JS. Functional and Phenotypic Changes of Natural Killer Cells in Whole Blood during Mycobacterium tuberculosis Infection and Disease. Front Immunol 2018;9:257. doi: 10.3389/fimmu.2018.00257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Belay M, Tulu B, Younis S, Jolliffe DA, Tayachew D, Manwandu H, Abozen T, Tirfie EA, Tegegn M, Zewude A, Forrest S, Mayito J, Huggett JF, Jones GM, O’Sullivan DM, Martineau HM, Noursadeghi M, Chandran A, Harris KA, Nikolayevskyy V, Demaret J, Berg S, Vordermeier M, Balcha TT, Aseffa A, Ameni G, Abebe M, Reece ST, Martineau AR. Detection of Mycobacterium tuberculosis complex DNA in CD34-positive peripheral blood mononuclear cells of asymptomatic tuberculosis contacts: an observational study. Lancet Microbe 2021;2:e267–e275. doi: 10.1016/S2666-5247(21)00043-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Oyarzabal E, Garcia-Garcia L, Rangel-Escareno C, Ferreyra-Reyes L, Orozco L, Herrera MT, Carranza C, Sada E, Juarez E, Ponce-de-Leon A, Sifuentes-Osornio J, Wilkinson RJ, Torres M. Expression of USP18 and IL2RA Is Increased in Individuals Receiving Latent Tuberculosis Treatment with Isoniazid. J Immunol Res 2019;2019:1297131. doi: 10.1155/2019/1297131 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.