

Abstract
Traumatic brain injury (TBI) is an established risk factor for the development of psychiatric disorders, especially depression and anxiety. Yet the mechanistic pathways underlying this risk remain unclear, limiting treatment options and hindering the identification of clinically-useful biomarkers. One salient pathophysiological process implicated in both primary psychiatric disorders and TBI is inflammation. An important consequence of inflammation is the increased breakdown of tryptophan to kynurenine and, subsequently, the metabolism of kynurenine into several neuroactive metabolites including the neurotoxic NMDA receptor agonist, quinolinic acid (QuinA), and the neuroprotective NMDA receptor antagonist, kynurenic acid (KynA). Here, we review studies of the kynurenine pathway (KP) in TBI and examine their potential clinical implications. The weight of the literature suggests that there is increased production of neurotoxic kynurenines such as QuinA in TBI of all severities, and that elevated QuinA concentrations in both the cerebrospinal fluid and blood are a negative prognostic indicator, being associated with death, MRI abnormalities, increased depressive and anxiety symptoms, and prolonged recovery. We hypothesize that an imbalance in KP metabolism is also one molecular pathway through which the TBI-induced neurometabolic cascade may predispose to the development of psychiatric sequelae. If this model is correct, KP metabolites could serve to predict who is likely to develop psychiatric illness while drugs that target the KP could help to prevent or treat depression and anxiety arising in the context of TBI.
Keywords: inflammation, kynurenine, TBI, concussion, depression, psychiatric
Introduction
There were approximately 2.87 million traumatic brain injury (TBI)-related emergency department visits, hospitalizations, and deaths in the U.S. in 2014 according to the CDC, an underestimate that does not account for patients who received care in other clinical settings or did not receive care at all (1). TBI is generally classified by severity as being mild, moderate, or severe based on the patient’s level of consciousness (typically assessed using the Glasgow Coma Scale which grades patients based on eye opening, verbal, and motor responses) and the presence and duration of certain injury characteristics such as loss of consciousness and amnesia. Thus, the spectrum of TBI ranges from severe injuries that result in coma or death to mild sport-related concussions (SRC; typically considered a subset of mild TBI) that may only cause transient symptoms without intracranial findings or alterations in mental status.
The pathophysiology of TBI has been reviewed elsewhere (2–5). Briefly, injury to brain tissue and associated vasculature occurs due to external forces, such as blunt trauma, rapid acceleration/deceleration, penetrating injury, or blast. This primary injury can be focal (e.g., contusions) or diffuse (e.g., diffusion axonal injury) and can range from microstructural changes not detectable by conventional neuroimaging to macroscopic lesions. This initial trauma is followed by a range of metabolic and neurochemical changes that can lead to secondary injury. Neurochemical changes include increased glutamate release and ionic changes (i.e., potassium efflux; sodium/calcium influx), an initial increase in metabolism and subsequent depletion of glucose, and neuroinflammation, among others. In most mild cases it is thought these changes cause transient cellular dysfunction, but in more severe cases or instances of repetitive injury they can lead to cell death, abnormal protein accumulation, chronic inflammation, or other complications such as ischemia, hypoxia, or edema (6).
TBI is also associated with increased risk for a variety of negative long-term health outcomes including psychiatric disorders. A meta-analysis of 57 studies demonstrated that prior TBI is associated with an increased risk for developing a subsequent psychiatric disorder (Odds Ratio [OR]=2.00) (7). The same meta-analysis found that the greatest psychiatric risk following TBI was depression (OR=2.14), though TBI was also associated with elevated risk for bipolar disorder (OR=1.85) and mixed anxiety and depression (OR=1.84) (7). Similarly, a meta-analysis of over 90 studies (n>10,000) found that TBI of all severity (average time since injury=33.7 months) was associated with an increased risk of major depressive disorder (MDD) and/or dysthymia (OR=1.66 for formal diagnosis, OR=3.41 for clinically significant scores on self-report metrics), with MDD/dysthymia present in 27% to 38% of TBI patients, depending on the outcome measure (i.e., formal diagnosis versus self-report) (8). Depression is also more common in former athletes with a history of SRC compared to those without (9). Multiple meta-analyses have shown that post-traumatic stress disorder (PTSD) is also more prevalent in both civilian and military TBI patients (10–12). Unfortunately, the mechanistic pathways through which TBI predisposes individuals to psychiatric complications remain unclear, limiting treatment options and hindering the identification of prognostic and monitoring biomarkers.
The etiology of psychiatric symptoms following TBI likely includes a combination of external factors (e.g., psychological trauma), functional disabilities that may result from injury, and neuropathological changes. One physiological process that has been hypothesized to play a mechanistic role in both TBI (13–15) and depression/anxiety (16–20) is inflammation. An under-appreciated consequence of the inflammatory process is activation of the kynurenine pathway (KP) to produce the cellular energy source nicotinamide adenine dinucleotide (NAD+) from tryptophan via several neuroactive intermediates. Preclinical studies demonstrate that KP metabolites are necessary for inflammation-induced depressive-like behaviors, as the depressogenic effects of inflammation can be prevented by blocking activation of the KP (21–23). In humans, elevated concentrations of neurotoxic KP metabolites or reduced concentrations of neuroprotective KP metabolites have been repeatedly associated with MDD and bipolar disorder (24,25). Further, these KP abnormalities are attenuated by effective treatment of depression, including electroconvulsive therapy (26), cognitive behavioral therapy (27), neurofeedback (28), exercise (29,30), escitalopram (31), and ketamine (32–34). In this review, we outline the key metabolites and enzymes of the KP and discuss their neuroactive properties. Subsequently, we review the extant preclinical and clinical literature on the KP in the context of TBI. In doing so, we present the hypothesis that TBI-induced abnormalities in the KP contribute to secondary injury following TBI and may increase the risk for psychiatric disorders following TBI. Finally, we discuss the potential clinical applications of our mechanistic model.
Overview of KP pathway
The KP is the major pathway for tryptophan metabolism in both the periphery and central nervous system (CNS), with 95% of TRP metabolized into kynurenine (KYN) by the enzymes indoleamine 2,3-dioxygenase (IDO) and tryptophan dioxygenase (TDO) (Figure 1). IDO is thought to be predominantly expressed in macrophages and microglia while TDO is primarily expressed in the liver. Because the KP is the major endogenous source of NAD+ and activated immune cells have higher energy requirements (35), several different cytokines increase the activity of IDO, including interferon-gamma (IFNγ), interleukin-1β (IL-1β) and tumor necrosis factor (TNF) (36–39). Conversely, the activity of TDO is primarily increased by cortisol. Although not as extensively documented as inflammation, dysfunction of the hypothalamic-pituitary-adrenal (HPA) axis has been documented following TBI (40–43). KYN is subsequently broken down along one of two separate branches of the KP, in the process producing metabolites that are generally regarded as having opposing properties. The kynurenine aminotransferase (KAT) enzymes, which are thought be expressed in astrocytes in the CNS, convert KYN to kynurenic acid (KynA). KynA is a glycine-site antagonist of N-Methyl-D-aspartate (NMDA) receptors that competitively inhibits other ionotropic glutamate receptors at high concentrations (44). Because of its ability to block NMDA receptor-mediated excitotoxicity, KynA is often considered to be neuroprotective (44–46). KynA has also been hypothesized to act as a negative allosteric modulator of α7-nicotinic receptors (47), an agonist for G protein-coupled receptor 35 (48), and an agonist of the aryl hydrocarbon receptor (49).
Figure 1:
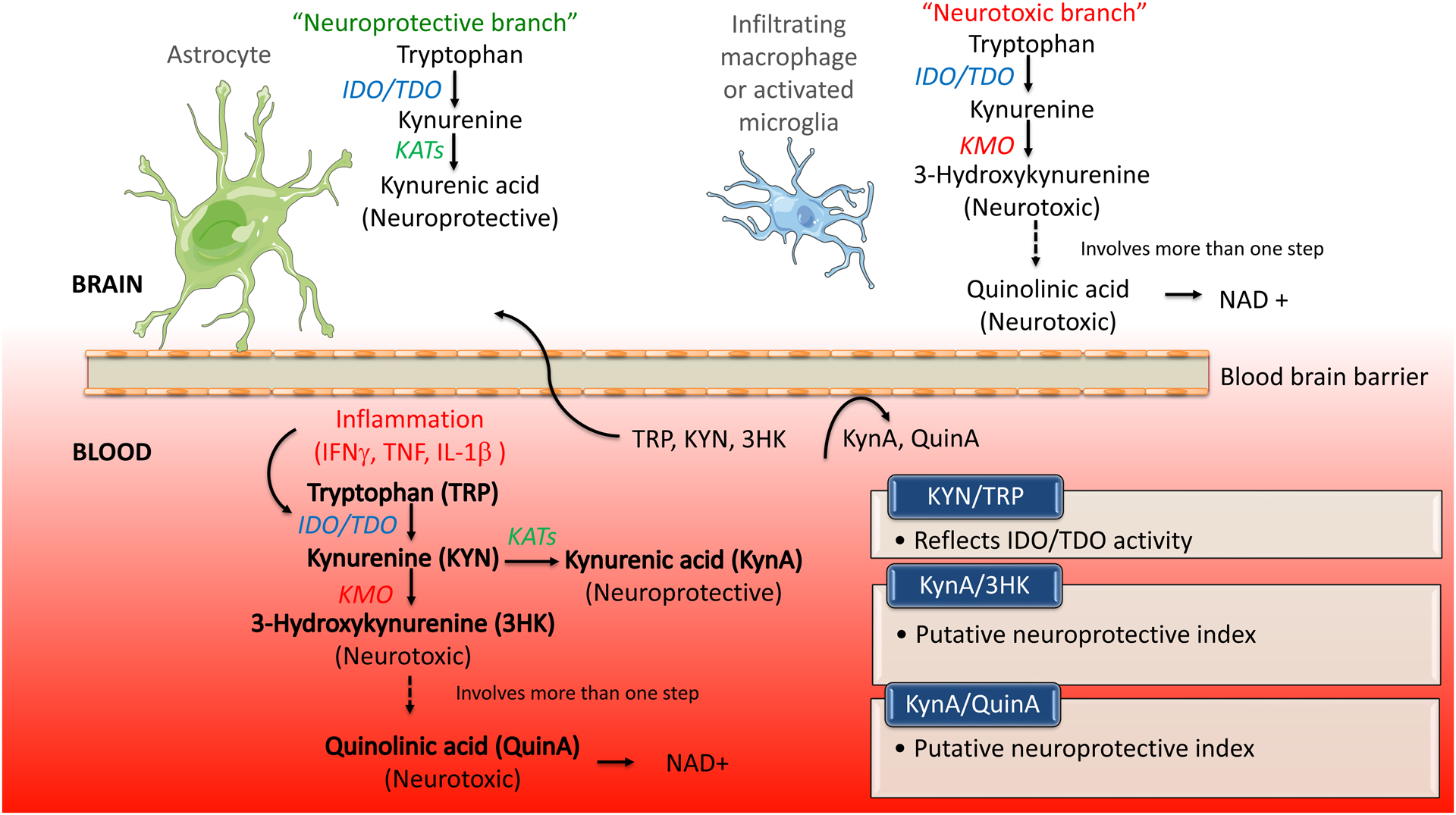
Simplified figure showing the kynurenine pathway (KP) of tryptophan catabolism. Activation of the hypothalamic-pituitary-adrenal axis can lead to the induction of tryptophan 2, 3-dioxygenase (TDO). Pro-inflammatory cytokines such as interferon gamma (IFNγ), tumor necrosis factor (TNF) and interleukin-1β (IL-1β) can activate indoleamine 2, 3-dioxygenase (IDO). TDO and IDO catalyze the conversion of tryptophan (TRP) to kynurenine (KYN). KYN is then converted to either kynurenic acid (KynA) by the kynurenine aminotransferase enzymes (KATs) or 3-hydroxykynurenine (3HK) by kynurenine monooxygenase (KMO). 3HK is further metabolized into several different metabolites, including quinolinic acid (QuinA), a precursor for nicotinamide adenine dinucleotide (NAD+). Within the central nervous system, the KP is thought to be differentially metabolized in astrocytes or microglia. TRP uptake and metabolism in astrocytes leads to the production of the KynA, which is often considered to be neuroprotective. In contrast, TRP uptake and metabolism in microglia leads to the production of so-called neurotoxic metabolites such as 3HK and QuinA. QuinA acts as an agonist at the glutamate N-methyl-D-aspartate (NMDA) receptor and is neurotoxic at elevated concentrations. TRP, KYN, and 3HK can be transported across the blood brain barrier whereas KynA and QuinA are not standardly thought to be able to cross the blood brain barrier. Figure adapted from (28).
Alternatively, inflammatory cytokines also increase the activity of the enzyme kynurenine-3-monooxygenase (KMO) (36), which converts KYN into the free-radical generator 3-hydroxykynurenine (3HK), which is in turn eventually metabolized into the NMDA receptor agonist and potential neurotoxin, quinolinic acid (QuinA). QuinA has potency similar to glutamate at the NMDA receptor but with less-efficient reuptake - therefore it has stronger excitotoxic effects (50). QuinA has multiple other deleterious effects, promoting glutamate release, inhibiting the reuptake of glutamate by astrocytes, generating reactive oxygen species, disrupting the blood brain barrier, promoting tau phosphorylation, destabilizing the cellular cytoskeleton, and disrupting autophagy (51). QuinA is subsequently converted into NAD+ which plays an important role in cellular metabolism. Presumably because inflammation is a metabolically-demanding state, metabolism down the “neurotoxic” QuinA pathway is favored during immune activation (52,53).
TRP, KYN, and 3HK can be transported across the blood brain barrier (BBB). Under physiological conditions 60–80% of KYN in the brain is thought to be from the periphery and this approaches 100% following inflammation (54,55). KynA and QuinA do not readily cross the BBB and thus central KynA and QuinA are thought to be derived from brain KYN (55). However, there is some evidence that this view of blood brain permeability is overly simplistic. First, peripheral administration of KynA has been shown to be neuroprotective following experimental TBI (56–58). Second, there is preclinical evidence that 50%−70% of radiolabeled QuinA infused subcutaneously is eventually detected in the brain and cerebrospinal fluid (CSF) (59). Third, significant correlations between peripheral and CSF levels of QuinA have been documented in unmedicated MDD patients, in patients with hepatitis C after IFNα treatment, and in HIV patients (r-values ~0.5–0.7) (60–62) – though less robust associations have been reported in Alzheimer’s disease patients and controls (63). There is evidence that BBB may be disrupted after TBI (e.g., due to mechanical insult or secondary inflammatory responses) (64–66). Thus, central levels of KP metabolites likely reflect changes in local brain sources as well as peripheral KP concentrations driven by secondary systemic inflammatory processes. Future studies are needed to determine the extent to which peripheral and central levels of KP metabolites are correlated following TBI, as well as the relative role of peripherally versus centrally-derived KP metabolites in TBI.
Evidence of KP involvement in TBI
Preclinical evidence of a role for the KP in TBI
The first reports of KP metabolites in the TBI literature originate from the use of KynA as a broad spectrum excitatory amino acid antagonist in preclinical models of TBI. This fundamental work focused on characterizing the aforementioned neurochemical cascade of TBI, rather than characterizing associations of the KP with TBI. Nevertheless, these findings are illustrative of the potential role of KP metabolites in TBI. Administration of KynA in the hippocampus and cortex reduced potassium increases and decreased glucose utilization and accumulation of lactate typically observed following fluid percussion injury (FPI) (67–69). Administration of KynA into the hippocampus also attenuated both acute astroglia and microglial response in a controlled cortical impact injury model (70). In parallel work, intravenous KynA administration at 15 minutes post-injury reduced cognitive dysfunction and motor deficits following lateral FPI in the rat (58). KynA administration also attenuated edema and calcium increases in multiple brain regions at 48 hours post-injury, and reduced neuronal loss and attenuated immunoreactivity of the cytoskeletal protein microtubule-associated protein-2 in CA3 of the hippocampus at 2 weeks post-injury (56–58).
A limited number of studies have directly investigated the effects of experimental TBI on the KP. These studies have generally shown increased activation of the first step of the KP (i.e., increased KYN or expression of IDO) or alternatively, increased metabolism down the neurotoxic branch of the KP (i.e., increased QuinA or KMO activity). Using a controlled cortical impact model of pediatric TBI in rabbits, Zhang and colleagues reported that IDO expression was upregulated relative to sham and naïve rabbits by 6 hours post-injury at the site of injury and remained elevated for up to 21 days post-injury (71). Similar results were seen for IDO protein and KYN concentrations as well as a corresponding elevation in expression of TNF, IL-1B, and IL-6 at the same time points. Focal cortical injury models in rats showed elevated expression of QuinA in reactive microglial in injured and adjacent sites at 1 day post-injury (72). Similarly, although not an experimental model of TBI, per se, Zakhary and colleagues demonstrated increased expression of QuinA and KynA at 24 and 72 hours, but not 6 or 12 hours, in perilesional cortex following partial frontal lobectomy in rats (73). Post-injury intraperitoneal injection of the KMO inhibitor RO 61–8048 reduced edema, decreased markers of apoptosis, and improved long-term neurological function following the surgically induced injury; these protective effects of the KMO inhibitor were reversed by concurrent administration of a KAT enzyme inhibitor which prevented the formation of KynA. The same KMO inhibitor, administered daily starting prior to injury, was also reported to ameliorate the damage to retinal ganglion cell function and structure observed 5-weeks following a single, high-pressure blast (74).
Evidence of KP role in severe TBI in humans
The first evidence of KP abnormalities following TBI in humans comes from analysis of QuinA in CSF in severe TBI patients. CSF concentrations of QuinA in adult TBI patients significantly increased across the first 72–83 hours post-injury, reaching concentrations (mean 463 nmol/L) approximately nine times the normal QuinA concentration (75). Moreover, QuinA was significantly elevated in patients that died compared to those that survived, controlling for time since injury. Similar findings were reported in a limited number of children with severe TBI, with CSF concentrations of QuinA increased significantly over approximately 1 week post-injury and higher QuinA concentrations observed in non-survivors versus survivors (76). More recently, increased IDO-positive cells were observed in injured brain tissue post-mortem in TBI patients relative to controls (77). The same study also reported elevated CSF levels of KYN at days 4 and 5 post-injury, elevated KynA at days 2–5 post-injury, and elevated QuinA at days 1–5 post-injury in the CSF of severe TBI patients relative to non-TBI patient controls (77). QuinA showed the greatest elevation, with a tenfold increase by day 4 relative to control levels. Compared to controls, the ratios of QuinA to KYN and QuinA to KynA were also substantially elevated in TBI patients at days 1–5 and days 2–5 post-injury, respectively, but there was no difference in the ratio of KynA to KYN at any visit, indicating a substantial shift of KP metabolism toward neurotoxic QuinA following severe TBI. Moreover, higher QuinA concentrations measured between days 1 and 5 post-injury were associated with worse 6-month outcome in TBI patients. Similar alterations in KP metabolites have also been observed in the plasma of TBI patients. Fifteen chronic severe brain injury patients (primarily TBI with some cases of alternative injury such as hypoxia, cerebral infarcts, or subdural hematoma; 1–35 years post-injury) displayed widespread differences in KP metabolites relative to controls, including higher KYN/TRP, indicating increased IDO activity, as well as lower KynA to KYN ratios and decreased KynA (78). This pattern of elevated IDO activity (KYN/TRP ratio) but decreased KAT activity (KynA/KYN ratio) is suggestive of increased metabolism down the “neurotoxic pathway”, though no differences in QuinA were observed. The observed KP abnormalities in this study were observed several years following injury, which could explain the subtle differences relative to the aforementioned studies (e.g., lack of QuinA differences in the chronic brain injury cohort). Further work is needed to determine the time course of potential KP abnormalities following TBI, though chronic inflammation has been reported years following TBI (79–83).
Evidence of a role for the KP in SRC in humans
To our knowledge, initial studies on the effects of TBI on the KP in humans were restricted to severe TBI, with no reports in less severe injuries (e.g., mild or moderate TBI). Our group has recently begun to address this knowledge gap by investigating KP abnormalities in high school and collegiate athletes with SRC, which is typically considered a subset of mild TBI that is rarely associated with intracranial lesions (84). In our first publication, plasma concentrations of KP metabolites were measured in collegiate football athletes at 1 day, 1 week, and 1 month following SRC (85). Compared to uninjured football athletes, those with SRC had higher QuinA and lower KynA/QuinA at all time points. Furthermore, in athletes with SRC, lower KynA/QuinA and higher QuinA were associated with worse depression and anxiety symptoms and a greater number of days out before return-to-play. One caveat of this study, however, was that the SRC group reported more prior concussions than the control group; thus, we were unable to determine whether the elevations in QuinA were due to the acute or prior injuries (85). Following up on this work, we demonstrated long-term associations between KP metabolites and neuroimaging markers of brain structure and function in the context of SRC. Plasma concentrations of KP metabolites were compared amongst football players with prior concussion (on average 10 months since last concussion), football players without prior concussions, and healthy controls with no prior concussion or football experience (86). Both football players with and without prior concussion had either a trend or significantly lower KYN compared to controls, while plasma QuinA concentrations were elevated in football players with prior concussion compared to those without. We also reported an inverse association between the “neurotoxic” metabolite, 3HK, and bilateral hippocampal volume in football players with prior concussion and a positive association between the “neuroprotective index”, KynA/QuinA, and left hippocampal volume in football players without prior concussion, echoing KP-hippocampal volume relationships observed in psychiatric cohorts (87,88). In a second study in the same sample, we additionally found that circulating KynA/QuinA was associated with resting state functional connectivity (RSFC) of the anterior cingulate cortex and hippocampus. However, the relationship was less straightforward than the structural MRI, differing based on the athletes’ concussion history and football experience (89).
We have recently followed-up our earlier reports in an independent, prospectively-collected cohort of high school and collegiate athletes. Serum was collected in football players prior to injury and then at 6 hours and 1, 8, 15, and 45 days post-injury (90). Uninjured football players and uninjured non-contact sport athletes participated in similar visits as two independent control groups. Somewhat counter-intuitively, the “neuroprotective index”, KynA/3HK was elevated in football players with SRC at the 6-hour visit relative to pre-injury baseline (although it was decreased over the subsequent visits). Nevertheless, we found that higher KynA/3HK at the 6-hour visit was associated with less severe depressive symptoms 1-day post-concussion, suggesting that acute increases in KynA are protective. In contrast to our prior work (85), no differences in KynA/QuinA or QuinA were observed across concussed athletes. However, in additional analyses comparing acutely concussed athletes with and without prior concussion, we found that KynA/QuinA was lower across all visits in the acutely concussed athletes reporting a prior concussion, an effect driven by an increase in QuinA (90). We also reported in the same cohort that serum levels of QuinA were positively associated with RSFC hyperconnectivity at 1, 8, 15, and 45 days post-injury in acutely injured athletes with a prior concussion only (91). No association was observed in uninjured athletes or acutely injured athletes with no prior concussion. RSFC hyperconnectivity has been proposed to be a common functional response to acute neurological injury and is associated with worse symptoms and outcome following SRC (92–94).
The fact that elevated QuinA and the association between QuinA and abnormal RSFC was only observed in acutely injured athletes with prior concussion and not in acutely injured athletes without prior concussion or controls may reflect a form of immune system priming (90,91). In this model, prior concussion may sensitize microglia and macrophages, which are the primary sources of QuinA (95,96), to respond more robustly to a subsequent inflammatory trigger (i.e., a repeat concussion). This is similar to reports of microglial priming in animal models (97–99). Additional studies are needed to directly test this hypothesis.
Summary of TBI-related KP abnormalities and their relevance for psychiatric sequelae
Although small, the existing literature supports the hypothesis that following TBI, tryptophan is preferentially metabolized into KYN and KYN is in turn preferentially metabolized down the “neurotoxic” pathway into QuinA. Elevated QuinA is associated with a worse outcome (i.e. death, MRI abnormalities, psychiatric symptoms, and longer return to play) in human studies of both mild and severe TBI. These data raise the possibility that certain neuroactive KP metabolites contribute to secondary injury following TBI due to their well-established neurotoxic properties. Moreover, given the association of increased QuinA with depressive symptoms and functional abnormalities following SRC, and parallel data in psychiatric patients (100–106), we propose that TBI-mediated abnormalities in the KP constitutes a key molecular pathway mediating the increased risk of psychiatric disorders following TBI (Figure 2).
Figure 2.
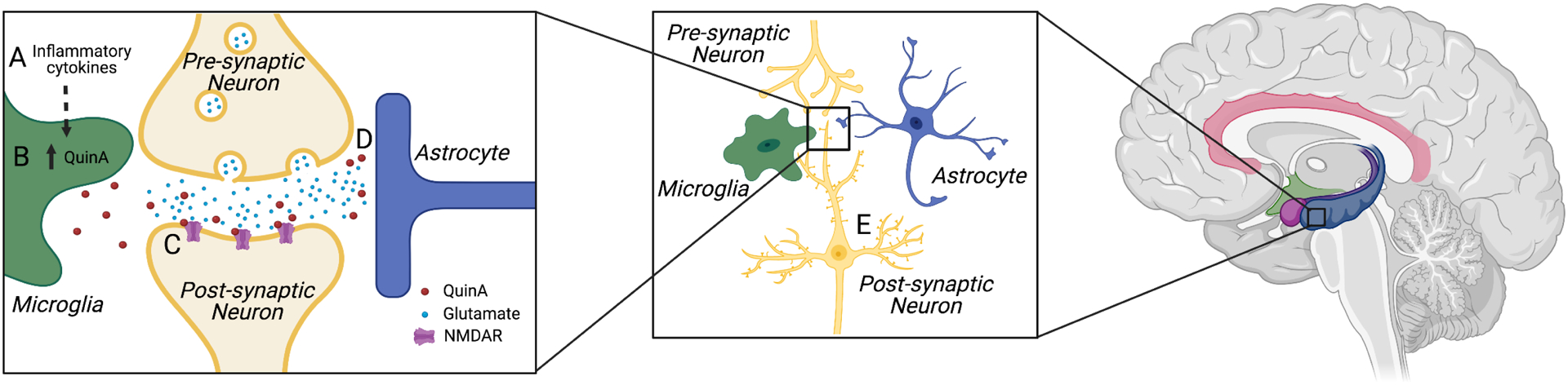
Heuristic mechanistic model of the neuropsychiatric sequelae of TBI.
(A) The inflammatory response to TBI results in an increase in the IDO and KMO enzymes, elevating central KYN concentrations and subsequently increasing the production of “neurotoxic” pathway metabolites such as QuinA (B). These metabolites contribute to secondary injury through a variety of mechanisms. For instance, QuinA acts as an agonist at NMDA receptors (C) and compromises glutamate recycling by astrocytes (D) contributing to excitotoxicity and a cascade of neuronal damage or in the case of milder TBI, dendritic remodeling, loss of synapses and more subtle disturbances in synaptic plasticity (E). When this disturbance of glutamatergic neurotransmission and decrement in synaptic plasticity compromises the function of neural circuitry involved in regulating affect, interoception, or learning and memory, psychiatric symptoms may occur. Limbic structures are highlighted in the brain image on the right. The focus (i.e., zoomed in image) of the current figure is on the hippocampus, though this heuristic model would apply to other brain regions associated with emotional processing or reward. Created with BioRender.com.
It has been known since the 1980’s that QuinA is a potent neurotoxin at supraphysiological doses, levels that may be approached after severe TBI (75). At physiological or nanomolar doses, administration of QuinA in vitro inhibits neuronal synaptic protein expression (107) and decreases the number and length of neuritic branches from primary cortical neurons (108). Similarly, 30 nmol QuinA injections into the medial prefrontal cortex of mice altered hippocampal synaptic transmission, impaired long-term potentiation, disrupted RSFC between various cortical regions, and induced reversal learning defects on a spatial navigation task (109). Thus, the more subtle increases in QuinA observed after mild TBI or SRC may impact neuroplasticity through the NMDA receptor resulting in the synaptic remodeling and loss of dendritic spines which appear as reductions in volume on MRI (110–114).
When disruptions in microstructure or plasticity occur in the neural circuits involved in the processing of emotions, the individual may be at increased risk for developing symptoms of depression and anxiety. Although damage from TBI can occur across the brain, and chronic inflammation (and therefore elevated QuinA) can occur throughout the brain regardless of the initial injury location (79–83), it is noteworthy that the brain regions commonly implicated in mood and anxiety disorders (115–117) are particularly vulnerable to TBI because of the shape of the skull (e.g., the basal forebrain and medial temporal lobe (118,119)). Furthermore, the hippocampus is thought to have the highest density of NMDA receptors in the brain (120) and thus may be particularly vulnerable to QuinA-mediated secondary injury regardless of the anatomical location of the primary injury. This may explain why many of the studies reviewed above reported associations between KP metabolites and hippocampal structure or function. However, damage to other neural circuits may also give rise to symptoms of depression and anxiety. For instance, there is a well-established relationship between anhedonia, disrupted dopaminergic signaling, and inflammation (121). Dopaminergic neurons in the ventral tegmental area are regulated by glutamatergic and GABAergic afferents (122) and thus KP-mediated changes in NMDA receptor signaling could alter basal ganglia function and disrupt motivated behavior, leading to depression.
Clinical implications
According to our mechanistic model, the KP might prove to be a plausible therapeutic target for preventing or ameliorating the neuropathological and neuropsychiatric sequalae of TBI. One approach would be to target inflammation, thus preventing activation of the KP. However, preclinical evidence demonstrates that the depressogenic effects of inflammation can be prevented by blocking activation of the KP even in the presence of high concentrations of inflammatory mediators (21–23). Thus, more direct targeting of the KP might be appropriate, particularly given reports that anti-inflammatory treatments can be counterproductive in primary mood disorder patients (123–126). In theory, augmenting activity of the KAT enzymes or inhibiting KMO could be neuroprotective after injury. KMO inhibitors have shown initial promise in preclinical models of injury and TBI (73,74). Another approach is to augment the neuroprotective effects of KynA. Several clinical trials for depression are currently underway with the KynA analogue, 4-chlorokynurenine (AV-101) which acts as a selective antagonist at the glycine-binding site of the NMDA receptor. Nevertheless, AV-101 failed to beat placebo in a small clinical trial in treatment-resistant depression (127), though similar compounds (e.g., ketamine, a competitive NMDA receptor antagonist) have recently received FDA approval for treatment resistant depression. A third approach is to prevent the transport of KYN into brain by blocking the large amino acid transporter (LAT1) with a competing amino acid (e.g., leucine). This treatment approach successfully prevented LPS-induced depression-like behavior in mice without affecting inflammation or sickness behavior (128) and clinical trials are underway or planned in both primary MDD (NCT03079297) and for sleep disturbances in mild TBI patients (NCT04603443).
KP abnormalities are reported across a wide range of injury mechanisms in the context of TBI, as well as other forms of CNS injury (e.g., stroke; (129,130)), non-head trauma (131) and several psychiatric and neurological diseases (24,132–135). Thus, KP abnormalities are not specific to TBI, and their potential as TBI-specific (i.e., diagnostic) biomarkers is limited. However, KP metabolites might have value in monitoring recovery in TBI patients (monitoring biomarker) or predicting outcome following TBI - for instance, identifying TBI patients at increased risk for developing negative long-term outcomes such as psychiatric or neurodegenerative disease (prognostic biomarker).
Summary and future directions
The extant literature suggests that increased production of neurotoxic KP metabolites after TBI may contribute to secondary injury and increase the risk of psychiatric morbidity. Nevertheless, most of the clinical data are cross-sectional in nature or derived from limited sample sizes and should therefore be treated with caution. Furthermore, studies in humans have to date focused on patients at opposite ends of the TBI severity spectrum (i.e., severe TBI patients and athletes with concussions). Additional work is needed to identify the effects of TBI across the range of severity on the KP. Experimental medicine studies in TBI populations are needed to disambiguate the mechanistic effects of KP activation from general inflammatory processes and to evaluate the therapeutic efficacy of treatments targeting the KP. Longitudinal cohort studies explicitly designed to test whether KP abnormalities predict the development of de novo psychiatric disease following TBI and determine the utility of KP metabolites as monitoring or prognostic biomarkers are also needed to move the field forward.
Acknowledgements
Support for this work was provided by the National Institute of Neurological Disorders And Stroke of the National Institutes of Health under Award Number R01NS102225 (TM) and the National Institute of Mental Health under award number R21MH113871 (JS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
Dr. Meier reports no biomedical financial interests or potential conflicts of interest.
Dr. Savitz reports no biomedical financial interests or potential conflicts of interest.
References
- 1.Centers for Disease Control and Prevention (2019): Surveillance Report of Traumatic Brain Injury-related Emergency Department Visits, Hospitalizations, and Deaths-United States, 2014. Centers for Disease Control and Prevention, U.S. Department of Health and Human Services. Retrieved from www.cdc.gov/TraumaticBrainInjury [Google Scholar]
- 2.Ladak AA, Enam SA, Ibrahim MT (2019): A Review of the Molecular Mechanisms of Traumatic Brain Injury. World Neurosurgery. 10.1016/j.wneu.2019.07.039 [DOI] [PubMed] [Google Scholar]
- 3.Prins M, Greco T, Alexander D, Giza CC (2013): The pathophysiology of traumatic brain injury at a glance. DMM Dis Model Mech. 10.1242/dmm.011585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barkhoudarian G, Hovda DA, Giza CC (2016): The Molecular Pathophysiology of Concussive Brain Injury - an Update. Physical Medicine and Rehabilitation Clinics of North America. 10.1016/j.pmr.2016.01.003 [DOI] [PubMed] [Google Scholar]
- 5.McGinn MJ, Povlishock JT (2016): Pathophysiology of Traumatic Brain Injury. Neurosurgery Clinics of North America. 10.1016/j.nec.2016.06.002 [DOI] [PubMed] [Google Scholar]
- 6.Giza CC, Hovda DA (2014): The new neurometabolic cascade of concussion. Neurosurgery 75: S24–S33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perry DC, Sturm VE, Peterson MJ, Pieper CF, Bullock T, Boeve BF, et al. (2016): Association of traumatic brain injury with subsequent neurological and psychiatric disease: A meta-analysis. Journal of Neurosurgery, vol. 124. pp 511–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osborn AJ, Mathias JL, Fairweather-Schmidt AK (2014): Depression following adult, non-penetrating traumatic brain injury: A meta-analysis examining methodological variables and sample characteristics. Neuroscience and Biobehavioral Reviews. 10.1016/j.neubiorev.2014.07.007 [DOI] [PubMed] [Google Scholar]
- 9.Manley G, Gardner AJ, Schneider KJ, Guskiewicz KM, Bailes J, Cantu RC, et al. (2017): A systematic review of potential long-term effects of sport-related concussion. British Journal of Sports Medicine, vol. 51. pp 969–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loignon A, Ouellet MC, Belleville G (2020): A systematic review and meta-analysis on PTSD following TBI among military/veteran and civilian populations. Journal of Head Trauma Rehabilitation. 10.1097/HTR.0000000000000514 [DOI] [PubMed] [Google Scholar]
- 11.Van Praag DLG, Cnossen MC, Polinder S, Wilson L, Maas AIR (2019): Post-Traumatic Stress Disorder after Civilian Traumatic Brain Injury: A Systematic Review and Meta-Analysis of Prevalence Rates. Journal of Neurotrauma. 10.1089/neu.2018.5759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iljazi A, Ashina H, Al-Khazali HM, Lipton RB, Ashina M, Schytz HW, Ashina S (2020): Post-Traumatic Stress Disorder After Traumatic Brain Injury—A Systematic Review and Meta-Analysis. Neurological Sciences. 10.1007/s10072-020-04458-7 [DOI] [PubMed] [Google Scholar]
- 13.Simon DW, McGeachy MJ, Baylr H, Clark RSB, Loane DJ, Kochanek PM (2017): The far-reaching scope of neuroinflammation after traumatic brain injury. Nature Reviews Neurology, vol. 13. pp 171–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J (2017): Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron, vol. 95. pp 1246–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morganti-Kossmann MC, Semple BD, Hellewell SC, Bye N, Ziebell JM (2019): The complexity of neuroinflammation consequent to traumatic brain injury: from research evidence to potential treatments. Acta Neuropathologica, vol. 137. pp 731–755. [DOI] [PubMed] [Google Scholar]
- 16.Miller AH, Raison CL (2016): The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nature Reviews Immunology. 10.1038/nri.2015.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008): From inflammation to sickness and depression: When the immune system subjugates the brain. Nature Reviews Neuroscience. 10.1038/nrn2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irwin MR, Cole SW (2011): Reciprocal regulation of the neural and innate immune systems. Nature Reviews Immunology. 10.1038/nri3042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Michopoulos V, Powers A, Gillespie CF, Ressler KJ, Jovanovic T (2017): Inflammation in Fear-and Anxiety-Based Disorders: PTSD, GAD, and beyond. Neuropsychopharmacology. 10.1038/npp.2016.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beurel E, Toups M, Nemeroff CB (2020): The Bidirectional Relationship of Depression and Inflammation: Double Trouble. Neuron. 10.1016/j.neuron.2020.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim H, Chen L, Lim G, Sung B, Wang S, McCabe MF, et al. (2012): Brain indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depression. J Clin Invest. 10.1172/JCI61884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lawson MA, Parrott JM, McCusker RH, Dantzer R, Kelley KW, O’Connor JC (2013): Intracerebroventricular administration of lipopolysaccharide induces indoleamine-2,3-dioxygenase-dependent depression-like behaviors. J Neuroinflammation. 10.1186/1742-2094-10-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Connor JC, Lawson MA, André C, Moreau M, Lestage J, Castanon N, et al. (2009): Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry. 10.1038/sj.mp.4002148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Savitz J (2020): The kynurenine pathway: a finger in every pie. Molecular Psychiatry, vol. 25. pp 131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bartoli F, Misiak B, Callovini T, Cavaleri D, Cioni RM, Crocamo C, et al. (2020): The kynurenine pathway in bipolar disorder: a meta-analysis on the peripheral blood levels of tryptophan and related metabolites. Mol Psychiatry. 10.1038/s41380-020-00913-1 [DOI] [PubMed] [Google Scholar]
- 26.Schwieler L, Samuelsson M, Frye MA, Bhat M, Schuppe-Koistinen I, Jungholm O, et al. (2016): Electroconvulsive therapy suppresses the neurotoxic branch of the kynurenine pathway in treatment-resistant depressed patients. J Neuroinflammation. 10.1186/s12974-016-0517-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savitz J, Ford BN, Yeh HW, Akeman E, Cosgrove K, Clausen AN, et al. (2020): Behavioral activation therapy for depression is associated with a reduction in the concentration of circulating quinolinic acid. Psychol Med. 10.1017/S0033291720004389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsuchiyagaito A, Smith JL, El-Sabbagh N, Zotev V, Misaki M, Al Zoubi O, et al. (2021): Real-time fMRI neurofeedback amygdala training may influence kynurenine pathway metabolism in major depressive disorder. NeuroImage Clin. 10.1016/j.nicl.2021.102559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schlittler M, Goiny M, Agudelo LZ, Venckunas T, Brazaitis M, Skurvydas A, et al. (2016): Endurance exercise increases skeletal muscle kynurenine aminotransferases and plasma kynurenic acid in humans. Am J Physiol - Cell Physiol. 10.1152/ajpcell.00053.2016 [DOI] [PubMed] [Google Scholar]
- 30.Javelle F, Bloch W, Knoop A, Guillemin GJ, Zimmer P (2021): Toward a neuroprotective shift: eight weeks of high intensity interval training reduces the neurotoxic kynurenine activity concurrently to impulsivity in emotionally impulsive humans - A randomized controlled trial. Brain Behav Immun S0889-1591: 00178–1. [DOI] [PubMed] [Google Scholar]
- 31.Halaris A, Myint AM, Savant V, Meresh E, Lim E, Guillemin G, et al. (2015): Does escitalopram reduce neurotoxicity in major depression? J Psychiatr Res. 10.1016/j.jpsychires.2015.04.026 [DOI] [PubMed] [Google Scholar]
- 32.Zhou Y, Zheng W, Liu W, Wang C, Zhan Y, Li H, et al. (2018): Antidepressant effect of repeated ketamine administration on kynurenine pathway metabolites in patients with unipolar and bipolar depression. Brain Behav Immun. 10.1016/j.bbi.2018.09.007 [DOI] [PubMed] [Google Scholar]
- 33.Verdonk F, Petit AC, Abdel-Ahad P, Vinckier F, Jouvion G, de Maricourt P, et al. (2019): Microglial production of quinolinic acid as a target and a biomarker of the antidepressant effect of ketamine. Brain Behav Immun. 10.1016/j.bbi.2019.06.033 [DOI] [PubMed] [Google Scholar]
- 34.Kadriu B, Farmer CA, Yuan P, Park LT, Deng Z De, Moaddel R, et al. (2019): The kynurenine pathway and bipolar disorder: intersection of the monoaminergic and glutamatergic systems and immune response. Mol Psychiatry. 10.1038/s41380-019-0589-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Straub RH (2017): The brain and immune system prompt energy shortage in chronic inflammation and ageing. Nature Reviews Rheumatology. 10.1038/nrrheum.2017.172 [DOI] [PubMed] [Google Scholar]
- 36.Zunszain PA, Anacker C, Cattaneo A, Choudhury S, Musaelyan K, Myint AM, et al. (2012): Interleukin-1β: A new regulator of the kynurenine pathway affecting human hippocampal neurogenesis. Neuropsychopharmacology 37: 939–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujigaki H, Saito K, Fujigaki S, Takemura M, Sudo K, Ishiguro H, Seishima M (2006): The signal transducer and activator of transcription 1α and interferon regulatory factor 1 are not essential for the induction of indoleamine 2,3-dioxygenase by lipopolysaccharide: Involvement of p38 mitogen-activated protein kinase and nuclear factor-κB. J Biochem 139: 655–662. [DOI] [PubMed] [Google Scholar]
- 38.O’Connor JC, André C, Wang Y, Lawson MA, Szegedi SS, Lestage J, et al. (2009): Interferon-γ and tumor necrosis factor-α mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus calmette-guérin. J Neurosci 29: 4200–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lestage J, Verrier D, Palin K, Dantzer R (2002): The enzyme indoleamine 2,3-dioxygenase is induced in the mouse brain in response to peripheral administration of lipopolysaccharide and superantigen. Brain, Behavior, and Immunity, vol. 16 16: 596–601. [DOI] [PubMed] [Google Scholar]
- 40.La PL, Kline GA, Black A, Wynne-Edwards K, Schneider K, Emery CA, Debert CT (2019): The relationship between saliva cortisol measures, symptom burden, length of recovery and concussion history following pediatric sport-related concussion. Clin J Sport Med 29: e69. [Google Scholar]
- 41.Yang WH, Chen PC, Wang TC, Kuo TY, Cheng CY, Yang YH (2016): Endocrine dysfunction following traumatic brain injury: A 5-year follow-up nationwide-based study. Sci Rep 6. 10.1038/srep32987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lieberman SA, Oberoi AL, Gilkison CR, Masel BE, Urban RJ (2001): Prevalence of neuroendocrine dysfunction in patients recovering from traumatic brain injury. J Clin Endocrinol Metab 86: 2752–2756. [DOI] [PubMed] [Google Scholar]
- 43.Schneider HJ, Kreitschmann-Andermahr I, Ghigo E, Stalla GK, Agha A (2007): Hypothalamopituitary dysfunction following traumatic brain injury and aneurysmal subarachnoid hemorrhage: A systematic review. Journal of the American Medical Association, vol. 298. pp 1429–1438. [DOI] [PubMed] [Google Scholar]
- 44.Kessler M, Terramani T, Lynch G, Baudry M (1989): A Glycine Site Associated with N-Methyl-d-Aspartic Acid Receptors: Characterization and Identification of a New Class of Antagonists. J Neurochem 52: 1319–1328. [DOI] [PubMed] [Google Scholar]
- 45.Foster AC, Vezzani A, French ED, Schwarcz R (1984): Kynurenic acid blocks neurotoxicity and seizures induced in rats by the related brain metabolite quinolinic acid. Neurosci Lett 48: 273–278. [DOI] [PubMed] [Google Scholar]
- 46.Harris CA, Miranda AF, Tanguay JJ, Boegman RJ, Beninger RJ, Jhamandas K (1998): Modulation of striatal quinolinate neurotoxicity by elevation of endogenous brain kynurenic acid. Br J Pharmacol. 10.1038/sj.bjp.0701834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hilmas C, Pereira EFR, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX (2001): The brain metabolite kynurenic acid inhibits α7 nicotinic receptor activity and increases non-α7 nicotinic receptor expression: Physiopathological implications. J Neurosci 21: 7463–7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang J, Simonavicius N, Wu X, Swaminath G, Reagan J, Tian H, Ling L (2006): Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J Biol Chem 281: 22021–22028. [DOI] [PubMed] [Google Scholar]
- 49.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. (2011): An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478: 197–203. [DOI] [PubMed] [Google Scholar]
- 50.Foster AC, Miller LP, Oldendorf WH, Schwarcz R (1984): Studies on the disposition of quinolinic acid after intracerebral or systemic administration in the rat. Exp Neurol. 10.1016/0014-4886(84)90239-5 [DOI] [PubMed] [Google Scholar]
- 51.Guillemin GJ (2012): Quinolinic acid, the inescapable neurotoxin. FEBS Journal, vol. 279. pp 1356–1365. [DOI] [PubMed] [Google Scholar]
- 52.Saito K, Markey SP, Heyes MP (1992): Effects of immune activation on quinolinic acid and neuroactive kynurenines in the mouse. Neuroscience. 10.1016/0306-4522(92)90467-G [DOI] [PubMed] [Google Scholar]
- 53.Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B, et al. (2013): NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology. 10.1038/npp.2013.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kita T, Morrison PF, Heyes MP, Markey SP (2002): Effects of systemic and central nervous system localized inflammation on the contributions of metabolic precursors to the L-kynurenine and quinolinic acid pools in brain. J Neurochem 82: 258–268. [DOI] [PubMed] [Google Scholar]
- 55.Fukui S, Schwarcz R, Rapoport SI, Takada Y, Smith QR (1991): Blood–Brain Barrier Transport of Kynurenines: Implications for Brain Synthesis and Metabolism. J Neurochem 56: 2007–2017. [DOI] [PubMed] [Google Scholar]
- 56.Hicks RR, Smith DH, Gennarelli TA, McIntosh T (1994): Kynurenate is neuroprotective following experimental brain injury in the rat. Brain Res 655: 91–96. [DOI] [PubMed] [Google Scholar]
- 57.Hicks RR, Smith DH, McIntosh TK (1995): Temporal response and effects of excitatory amino acid antagonism on microtubule-associated protein 2 immunoreactivity following experimental brain injury in rats. Brain Res 678: 151–160. [DOI] [PubMed] [Google Scholar]
- 58.Smith DH, Okiyama K, Thomas MJ, McIntosh TK (1993): Effects of the excitatory amino acid receptor antagonists kynurenate and indole-2-carboxylic acid on behavioral and neurochemical outcome following experimental brain injury. J Neurosci 13: 5383–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heyes MP, Morrison PF (1997): Quantification of local de novo synthesis versus blood contributions to quinolinic acid concentrations in brain and systemic tissues. J Neurochem 68: 280–288. [DOI] [PubMed] [Google Scholar]
- 60.Raison CL, Dantzer R, Kelley KW, Lawson MA, Woolwine BJ, Vogt G, et al. (2010): CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-α: Relationship to CNS immune responses and depression. Mol Psychiatry 15: 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haroon E, Welle JR, Woolwine BJ, Goldsmith DR, Baer W, Patel T, et al. (2020): Associations among peripheral and central kynurenine pathway metabolites and inflammation in depression. Neuropsychopharmacology 45: 998–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heyes MP, Brew BJ, Saito K, Quearry BJ, Price RW, Lee K, et al. (1992): Inter-relationships between quinolinic acid, neuroactive kynurenines, neopterin and β2-microglobulin in cerebrospinal fluid and serum of HIV-1-infected patients. J Neuroimmunol 40: 71–80. [DOI] [PubMed] [Google Scholar]
- 63.Jacobs KR, Lim CK, Blennow K, Zetterberg H, Chatterjee P, Martins RN, et al. (2019): Correlation between plasma and CSF concentrations of kynurenine pathway metabolites in Alzheimer’s disease and relationship to amyloid-β and tau. Neurobiol Aging 80: 11–20. [DOI] [PubMed] [Google Scholar]
- 64.Hay JR, Johnson VE, Young AMH, Smith DH, Stewart W (2015): Blood-brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J Neuropathol Exp Neurol 74: 1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnson VE, Weber MT, Xiao R, Cullen DK, Meaney DF, Stewart W, Smith DH (2018): Mechanical disruption of the blood–brain barrier following experimental concussion. Acta Neuropathol 135: 711–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cash A, Theus MH (2020): Mechanisms of blood–brain barrier dysfunction in traumatic brain injury. International Journal of Molecular Sciences. 10.3390/ijms21093344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Katayama Y, Becker DP, Tamura T, Hovda DA (1990): Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J Neurosurg 73: 889–900. [DOI] [PubMed] [Google Scholar]
- 68.Kawamata T, Katayama Y, Hovda DA, Yoshino A, Becker DP (1992): Administration of excitatory amino acid antagonists via microdialysis attenuates the increase in glucose utilization seen following concussive brain injury. J Cereb Blood Flow Metab 12: 12–24. [DOI] [PubMed] [Google Scholar]
- 69.Kawamata T, Katayama Y, Hovda DA, Yoshino A, Becker DP (1995): Lactate accumulation following concussive brain injury: the role of ionic fluxes induced by excitatory amino acids. Brain Res 674: 196–204. [DOI] [PubMed] [Google Scholar]
- 70.Suma T, Koshinaga M, Fukushima M, Kano T, Katayama Y (2008): Effects of in situ administration of excitatory amino acid antagonists on rapid microglial and astroglial reactions in rat hippocampus following traumatic brain injury. Neurol Res 30: 420–429. [DOI] [PubMed] [Google Scholar]
- 71.Zhang Z, Rasmussen L, Saraswati M, Koehler RC, Robertson C, Kannan S (2019): Traumatic Injury Leads to Inflammation and Altered Tryptophan Metabolism in the Juvenile Rabbit Brain. J Neurotrauma 36: 74–86. [DOI] [PubMed] [Google Scholar]
- 72.Chung RS, Leung YK, Butler CW, Chen Y, Eaton ED, Pankhurst MW, et al. (2009): Metallothionein Treatment Attenuates Microglial Activation and Expression of Neurotoxic Quinolinic Acid Following Traumatic Brain Injury. Neurotox Res 15: 381–389. [DOI] [PubMed] [Google Scholar]
- 73.Zakhary G, Sherchan P, Li Q, Tang J, Zhang JH (2020): Modification of kynurenine pathway via inhibition of kynurenine hydroxylase attenuates surgical brain injury complications in a male rat model. J Neurosci Res 98: 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harper MM, Woll AW, Evans LP, Delcau M, Akurathi A, Hedberg-Buenz A, et al. (2019): Blast preconditioning protects retinal ganglion cells and reveals targets for prevention of neurodegeneration following blast-mediated traumatic Brian injury. Investig Ophthalmol Vis Sci 60: 4159–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sinz EH, Kochanek PM, Heyes MP, Wisniewski SR, Bell MJ, Clark RSB, et al. (1998): Quinolinic acid is increased in CSF and associated with mortality after traumatic brain injury in humans. J Cereb Blood Flow Metab 18: 610–615. [DOI] [PubMed] [Google Scholar]
- 76.Bell MJ, Kochanek PM, Heyes MP, Wisniewski SR, Sinz EH, Clark RSB, et al. (1999): Quinolinic acid in the cerebrospinal fluid of children after traumatic brain injury. Critical Care Medicine, vol. 27. pp 493–497. [DOI] [PubMed] [Google Scholar]
- 77.Yan EB, Frugier T, Lim CK, Heng B, Sundaram G, Tan M, et al. (2015): Activation of the kynurenine pathway and increased production of the excitotoxin quinolinic acid following traumatic brain injury in humans. J Neuroinflammation 12. 10.1186/s12974-015-0328-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mackay GM, Forrest CM, Stoy N, Christofides J, Egerton M, Stone TW, Darlington LG (2006): Tryptophan metabolism and oxidative stress in patients with chronic brain injury. Eur J Neurol 13: 30–42. [DOI] [PubMed] [Google Scholar]
- 79.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, et al. (2011): Inflammation after trauma: Microglial activation and traumatic brain injury. Ann Neurol. 10.1002/ana.22455 [DOI] [PubMed] [Google Scholar]
- 80.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W (2013): Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 10.1093/brain/aws322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Folkersma H, Boellaard R, Yaqub M, Kloet RW, Windhorst AD, Lammertsma AA, et al. (2011): Widespread and prolonged increase in (R)-11C-PK11195 binding after traumatic brain injury. J Nucl Med. 10.2967/jnumed.110.084061 [DOI] [PubMed] [Google Scholar]
- 82.Coughlin JM, Yuchuanwang Y, Minn I, Bienko N, Ambinder EB, Xu X, et al. (2017): Imaging of glial cell activation and white matter integrity in brains of active and recently retired national football league players. JAMA Neurol. 10.1001/jamaneurol.2016.3764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Coughlin JM, Wang Y, Munro CA, Ma S, Yue C, Chen S, et al. (2015): Neuroinflammation and brain atrophy in former NFL players: An in vivo multimodal imaging pilot study. Neurobiol Dis 74: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Klein AP, Tetzlaff JE, Bonis JM, Nelson LD, Mayer AR, Huber DL, et al. (2019): Prevalence of Potentially Clinically Significant Magnetic Resonance Imaging Findings in Athletes with and without Sport-Related Concussion. J Neurotrauma. 10.1089/neu.2018.6055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Singh R, Savitz J, Teague TK, Polanski DW, Mayer AR, Bellgowan PSF, Meier TB (2016): Mood symptoms correlate with kynurenine pathway metabolites following sports-related concussion. J Neurol Neurosurg Psychiatry. 10.1136/jnnp-2015-311369 [DOI] [PubMed] [Google Scholar]
- 86.Meier TB, Savitz J, Singh R, Teague TK, Bellgowan PSF (2016): Smaller Dentate Gyrus and CA2 and CA3 Volumes Are Associated with Kynurenine Metabolites in Collegiate Football Athletes. J Neurotrauma 33: 1349–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Savitz J, Dantzer R, Wurfel BE, Victor TA, Ford BN, Bodurka J, et al. (2015): Neuroprotective kynurenine metabolite indices are abnormally reduced and positively associated with hippocampal and amygdalar volume in bipolar disorder. Psychoneuroendocrinology 52: 200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Savitz J, Drevets WC, Smith CM, Victor TA, Wurfel BE, Bellgowan PSF, et al. (2015): Putative neuroprotective and neurotoxic kynurenine pathway metabolites are associated with hippocampal and amygdalar volumes in subjects with major depressive disorder. Neuropsychopharmacology 40: 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meier TB, Lancaster MA, Mayer AR, Teague TK, Savitz J (2017): Abnormalities in Functional Connectivity in Collegiate Football Athletes with and without a Concussion History: Implications and Role of Neuroactive Kynurenine Pathway Metabolites. J Neurotrauma 34: 824–837. [DOI] [PubMed] [Google Scholar]
- 90.Meier TB, Nitta ME, Teague TK, Nelson LD, McCrea MA, Savitz J (2020): Prospective study of the effects of sport-related concussion on serum kynurenine pathway metabolites. Brain Behav Immun 87: 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Meier TB, España L, Nitta ME, Kent Teague T, Brett BL, Nelson LD, et al. (2021): Positive association between serum quinolinic acid and functional connectivity following concussion. Brain Behav Immun 91: 531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hillary FG, Roman CA, Venkatesan U, Rajtmajer SM, Bajo R, Castellanos ND (2015): Hyperconnectivity is a fundamental response to neurological disruption. Neuropsychology 29: 59–75. [DOI] [PubMed] [Google Scholar]
- 93.Kaushal M, España LY, Nencka AS, Wang Y, Nelson LD, McCrea MA, Meier TB (2019): Resting-state functional connectivity after concussion is associated with clinical recovery. Hum Brain Mapp. 10.1002/hbm.24440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Van Der Horn HJ, Scheenen ME, De Koning ME, Liemburg EJ, Spikman JM, Van Der Naalt J (2017): The Default Mode Network as a Biomarker of Persistent Complaints after Mild Traumatic Brain Injury: A Longitudinal Functional Magnetic Resonance Imaging Study. J Neurotrauma. 10.1089/neu.2017.5185 [DOI] [PubMed] [Google Scholar]
- 95.Guillemin GJ, Smythe G, Takikawa O, Brew BJ (2005): Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 49: 15–23. [DOI] [PubMed] [Google Scholar]
- 96.Espey MG, Chernyshev ON, Reinhard JF, Namboodiri MAA, Colton CA (1997): Activated human microglia produce the excitotoxin quinolinic acid. Neuroreport. 10.1097/00001756-199701200-00011 [DOI] [PubMed] [Google Scholar]
- 97.Perry VH, Holmes C (2014): Microglial priming in neurodegenerative disease. Nature Reviews Neurology, vol. 10. pp 217–224. [DOI] [PubMed] [Google Scholar]
- 98.Witcher KG, Eiferman DS, Godbout JP (2015): Priming the Inflammatory Pump of the CNS after Traumatic Brain Injury. Trends in Neurosciences, vol. 38. pp 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dilger RN, Johnson RW (2008): Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. J Leukoc Biol 84: 932–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wichers MC, Koek GH, Robaeys G, Verkerk R, Scharpé S, Maes M (2005): IDO and interferon-α-induced depressive symptoms: A shift in hypothesis from tryptophan depletion to neurotoxicity. Mol Psychiatry. 10.1038/sj.mp.4001600 [DOI] [PubMed] [Google Scholar]
- 101.Myint AM, Kim YK, Verkerk R, Scharpé S, Steinbusch H, Leonard B (2007): Kynurenine pathway in major depression: Evidence of impaired neuroprotection. J Affect Disord. 10.1016/j.jad.2006.07.013 [DOI] [PubMed] [Google Scholar]
- 102.Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z, et al. (2011): Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: Evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflammation. 10.1186/1742-2094-8-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Savitz J, Drevets WC, Wurfel BE, Ford BN, Bellgowan PSF, Victor TA, et al. (2015): Reduction of kynurenic acid to quinolinic acid ratio in both the depressed and remitted phases of major depressive disorder. Brain Behav Immun. 10.1016/j.bbi.2015.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bay-Richter C, Linderholm KR, Lim CK, Samuelsson M, Träskman-Bendz L, Guillemin GJ, et al. (2015): A role for inflammatory metabolites as modulators of the glutamate N-methyl-d-aspartate receptor in depression and suicidality. Brain Behav Immun. 10.1016/j.bbi.2014.07.012 [DOI] [PubMed] [Google Scholar]
- 105.Wurfel BE, Drevets WC, Bliss SA, McMillin JR, Suzuki H, Ford BN, et al. (2017): Serum kynurenic acid is reduced in affective psychosis. Transl Psychiatry. 10.1038/tp.2017.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Young KD, Drevets WC, Dantzer R, Teague TK, Bodurka J, Savitz J (2016): Kynurenine pathway metabolites are associated with hippocampal activity during autobiographical memory recall in patients with depression. Brain Behav Immun. 10.1016/j.bbi.2016.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bhat A, Tan V, Heng B, Lovejoy DB, Sakharkar MK, Essa MM, et al. (2020): Roflumilast, a cAMP-Specific Phosphodiesterase-4 Inhibitor, Reduces Oxidative Stress and Improves Synapse Functions in Human Cortical Neurons Exposed to the Excitotoxin Quinolinic Acid. ACS Chem Neurosci. 10.1021/acschemneuro.0c00636 [DOI] [PubMed] [Google Scholar]
- 108.O’Reilly K, O’Farrell K, Midttun O, Rakovets Y, David -Bercholz J, Harkin A (2021): Kynurenic Acid Protects Against Reactive Glial-associated Reductions in the Complexity of Primary Cortical Neurons. J Neuroimmune Pharmacol. 10.1007/s11481-020-09976-x [DOI] [PubMed] [Google Scholar]
- 109.Latif-Hernandez A, Shah D, Ahmed T, Lo AC, Callaerts-Vegh Z, Van Der Linden A, et al. (2016): Quinolinic acid injection in mouse medial prefrontal cortex affects reversal learning abilities, cortical connectivity and hippocampal synaptic plasticity. Sci Rep. 10.1038/srep36489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cobb JA, Simpson J, Mahajan GJ, Overholser JC, Jurjus GJ, Dieter L, et al. (2013): Hippocampal volume and total cell numbers in major depressive disorder. J Psychiatr Res. 10.1016/j.jpsychires.2012.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gosche KM, Mortimer JA, Smith CD, Markesbery WR, Snowdon DA (2002): Hippocampal volume as an index of Alzheimer neuropathology: Findings from the Nun study. Neurology. 10.1212/WNL.58.10.1476 [DOI] [PubMed] [Google Scholar]
- 112.Keifer OP, Hurt RC, Gutman DA, Keilholz SD, Gourley SL, Ressler KJ (2015): Voxel-based morphometry predicts shifts in dendritic spine density and morphology with auditory fear conditioning. Nat Commun. 10.1038/ncomms8582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.D. P, D.C. L, A.J. E (2012): The neurogenesis hypothesis of affective and anxiety disorders: Are we mistaking the scaffolding for the building? Neuropharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stockmeier CA, Mahajan GJ, Konick LC, Overholser JC, Jurjus GJ, Meltzer HY, et al. (2004): Cellular changes in the postmortem hippocampus in major depression. Biol Psychiatry. 10.1016/j.biopsych.2004.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Logue MW, van Rooij SJH, Dennis EL, Davis SL, Hayes JP, Stevens JS, et al. (2018): Smaller Hippocampal Volume in Posttraumatic Stress Disorder: A Multisite ENIGMA-PGC Study: Subcortical Volumetry Results From Posttraumatic Stress Disorder Consortia. Biol Psychiatry. 10.1016/j.biopsych.2017.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schmaal L, Veltman DJ, Van Erp TGM, Smann PG, Frodl T, Jahanshad N, et al. (2016): Subcortical brain alterations in major depressive disorder: Findings from the ENIGMA Major Depressive Disorder working group. Mol Psychiatry. 10.1038/mp.2015.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wise T, Radua J, Via E, Cardoner N, Abe O, Adams TM, et al. (2017): Common and distinct patterns of grey-matter volume alteration in major depression and bipolar disorder: Evidence from voxel-based meta-analysis. Mol Psychiatry. 10.1038/mp.2016.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McAllister TW (2011): Neurobiological consequences of traumatic brain injury. Dialogues Clin Neurosci. 10.31887/dcns.2011.13.2/tmcallister [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bigler ED (2007): Anterior and Middle Cranial Fossa in Traumatic Brain Injury: Relevant Neuroanatomy and Neuropathology in the Study of Neuropsychological Outcome. Neuropsychology. 10.1037/0894-4105.21.5.515 [DOI] [PubMed] [Google Scholar]
- 120.Monaghan DT, Cotman CW (1985): Distribution of N-methyl-D-aspartate-sensitive L-[3H]glutamate-binding sites in rat brain. J Neurosci. 10.1523/jneurosci.05-11-02909.1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Felger JC, Miller AH (2012): Cytokine effects on the basal ganglia and dopamine function: The subcortical source of inflammatory malaise. Frontiers in Neuroendocrinology. 10.1016/j.yfrne.2012.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Morales M, Margolis EB (2017): Ventral tegmental area: Cellular heterogeneity, connectivity and behaviour. Nature Reviews Neuroscience. 10.1038/nrn.2016.165 [DOI] [PubMed] [Google Scholar]
- 123.Warner-Schmidt JL, Vanover KE, Chen EY, Marshall JJ, Greengard P (2011): Antidepressant effects of selective serotonin reuptake inhibitors (SSRIs) are attenuated by antiinflammatory drugs in mice and humans. Proc Natl Acad Sci U S A. 10.1073/pnas.1104836108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Stolk P, Souverein PC, Wilting I, Leufkens HGM, Klein DF, Rapoport SI, Heerdink ER (2010): Is aspirin useful in patients on lithium? A pharmacoepidemiological study related to bipolar disorder. Prostaglandins Leukot Essent Fat Acids. 10.1016/j.plefa.2009.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kessing LV, Rytgaard HC, Gerds TA, Berk M, Ekstrøm CT, Andersen PK (2019): New drug candidates for depression – a nationwide population-based study. Acta Psychiatr Scand. 10.1111/acps.12957 [DOI] [PubMed] [Google Scholar]
- 126.Berk M, Agustini B, Woods RL, Nelson MR, Shah RC, Reid CM, et al. (2021): Effects of aspirin on the long-term management of depression in older people: a double-blind randomised placebo-controlled trial. Mol Psychiatry. 10.1038/s41380-021-01020-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Park LT, Kadriu B, Gould TD, Zanos P, Greenstein D, Evans JW, et al. (2020): A Randomized Trial of the N-Methyl-d-Aspartate Receptor Glycine Site Antagonist Prodrug 4-Chlorokynurenine in Treatment-Resistant Depression. Int J Neuropsychopharmacol. 10.1093/ijnp/pyaa025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Walker AK, Wing EE, Banks WA, Dantzer R (2019): Leucine competes with kynurenine for blood-to-brain transport and prevents lipopolysaccharide-induced depression-like behavior in mice. Mol Psychiatry. 10.1038/s41380-018-0076-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Colpo GD, Venna VR, McCullough LD, Teixeira AL (2019): Systematic review on the involvement of the kynurenine pathway in stroke: Pre-clinical and Clinical Evidence. Front Neurol 10. 10.3389/fneur.2019.00778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Isabel Cuartero M, de la Parra J, García-Culebras A, Ballesteros I, Lizasoain I, Ángeles Moro M (2016): The Kynurenine Pathway in the Acute and Chronic Phases of Cerebral Ischemia. Curr Pharm Des 22: 1060–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ploder M, Spittler A, Schroecksnadel K, Neurauter G, Pelinka LE, Roth E, Fuchs D (2009): Tryptophan degradation in multiple trauma patients: Survivors compared with non-survivors. Clin Sci 116: 593–598. [DOI] [PubMed] [Google Scholar]
- 132.Lim CK, Fernández-Gomez FJ, Braidy N, Estrada C, Costa C, Costa S, et al. (2017): Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Progress in Neurobiology, vol. 155. pp 76–95. [DOI] [PubMed] [Google Scholar]
- 133.Lovelace MD, Varney B, Sundaram G, Lennon MJ, Lim CK, Jacobs K, et al. (2017): Recent evidence for an expanded role of the kynurenine pathway of tryptophan metabolism in neurological diseases. Neuropharmacology, vol. 112. pp 373–388. [DOI] [PubMed] [Google Scholar]
- 134.Cervenka I, Agudelo LZ, Ruas JL (2017): Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science. 10.1126/science.aaf9794 [DOI] [PubMed] [Google Scholar]
- 135.Heilman PL, Wang EW, Lewis MM, Krzyzanowski S, Capan CD, Burmeister AR, et al. (2020): Tryptophan Metabolites Are Associated With Symptoms and Nigral Pathology in Parkinson’s Disease. Mov Disord. 10.1002/mds.28202 [DOI] [PMC free article] [PubMed] [Google Scholar]