

Abstract
Powdery mildew is one of the most destructive diseases in the world, causing substantial grain yield losses and quality reduction in cereal crops. At present 23 powdery mildew resistance genes have been identified in rye, of which the majority are in wheat-rye translocation lines developed for wheat improvement. Here, we investigated the genetics underlying powdery mildew resistance in the Gülzow-type elite hybrid rye (Secale cereale L.) breeding germplasm. In total, 180 inbred breeding lines were genotyped using the state-of-the-art 600 K SNP array and phenotyped for infection type against three distinct field populations of B. graminis f. sp. secalis from Northern Germany (2013 and 2018) and Denmark (2020). We observed a moderate level of powdery mildew resistance in the non-restorer germplasm population, and by performing a genome-wide association study using 261,406 informative SNP markers, we identified a powdery mildew resistance locus, provisionally denoted PmNOS1, on the distal tip of chromosome arm 7RL. Using recent advances in rye genomic resources, we investigated whether nucleotide-binding leucine-rich repeat genes residing in the identified 17 Mbp block associated with PmNOS1 on recent reference genomes resembled known Pm genes.
Subject terms: Plant breeding, Plant breeding, Pattern recognition receptors in plants, Genome-wide association studies
Introduction
Powdery mildew (PM) is one of the most devasting diseases globally4. In cereals, the causative agent of PM is the ascomycete fungus Blumeria graminis (DC.) speer (Bg), which is capable of inflicting severe grain yield loss (≥ 20%) and quality reduction in cereals5–8. In periods of conducive conditions, such as frequent precipitation and low to moderate temperatures, Bg can cause severe epidemics by repeated infections and clonal reproduction9. As an obligate biotroph, Bg is highly specialized to its host species, divided into several distinct ‘formae specialis’ (f. sp.)10, and dependent on a living host for survival and reproduction. In the later stages of the growing season, or in periods of unconducive conditions, chasmothecia structures are formed11. The chasmothecia field inoculum facilitates the infection of volunteer plants or the successive autumn sown winter crop, allowing Bg to overwinter as dormant mycelia12. Long-distance dispersal by wind of conidiospores drives the large spatial variability in the Bg population13–15. In barley (Hordeum vulgare L.), the virulence complexity of B. graminis f. sp. hordei has been observed to increase with the prevailing wind direction from west to east in Europe16. Rather than geographical origin, evidence suggests that local use of host resistance and fungicides are the primary factors influencing the European B. graminis f. sp. hordei population17. These findings underline the seriousness of long-distance dispersal of B. graminis spores, which allows novel and aggressive pathotypes to evolve through the recombination of distinct pathotypes and rapidly spread18. In rye, the causative agent of PM, B. graminis f. sp. secalis, has attracted little scientific interest in recent years19,20.
To reduce our dependency on pesticides, host resistance constitutes a sustainable alternative for farmers to ensure crop productivity21. At present, 23 major PM resistance (R) genes have been identified in rye (Table 1). However, it is likely that a subset of colocalized R genes are allelic, as there was no evidence that these genes were distinct from existing R genes prior to denotation.
Table 1.
Location and origin of powdery mildew resistance genes in rye (Secale cereale L.) and wheat- (Triticum aestivum L.) rye translocation lines with rye being the donor parent. After1–3..
| Chromosome | Arm | Gene name | References | |
|---|---|---|---|---|
| Rye | Wheat | |||
| 1R | Short | Pm1 | Pm8 | 22 |
| Pm1 | Pm17 | 23 | ||
| Pm1 | PmCn17 | 24 | ||
| Pm | 2 | |||
| Pm | 25 | |||
| Long | PmSESY | 26 | ||
| ND | Pm7 | – | 27 | |
| 2R | Long | Pm2 | – | 28 |
| – | PmJZHM2RL | 29 | ||
| Pm | – | 30,31 | ||
| ND | – | Pm7 | 32 | |
| Pm8 | Pm3 | 31 | ||
| Pm | 33 | |||
| 3R | Short | Pm3 | – | 34 |
| 4R | Long | Pm | – | 35,36 |
| ND | Pm | – | 37 | |
| Pm6 | – | 34 | ||
| 5R | Long | Pm4 | – | 38 |
| 6R | Short | – | Pm56 | 39 |
| Long | – | Pm20 | 40 | |
| Pm5 | – | 41 | ||
| Pm | – | 31,36 | ||
| 7R | Long | Pm | – | 42 |
ND: Interchromosomal position not determined.
The majority of characterized Pm genes encode intracellular nucleotide-binding leucine-rich repeat (NLR) proteins that recognize pathogen effector molecules, leading to an effector-triggered immunity resistance response43–45. Canonical NLR genes are composed of three domains46. In grasses, the N-terminus is composed of a coiled-coil (CC) domain believed to be involved in signaling and the induction of cell death47. In the center, a nucleotide-binding adaptor shared by APAF-1, R proteins, and the CED-4 (NB-ARC) domain functions as a regulatory domain determining the protein activation state48. Last, in the C-terminus, a leucin-rich repeat (LRR) domain is involved in effector recognition49. In rye, 770 canonical NLR genes have been identified in the ‘Lo7’ reference genome50.
Rye has been an important source for the improvement of PM resistance in wheat (Triticum aestivum L.) by chromosomal translocation of segments housing R genes3,25,42. While PM is effectively controlled by host genetic resistance in cereals, it has historically been rapidly overcome by virulent races of Bg51,52. Currently, several of the top-yielding hybrid rye cultivars examined in official Danish trials are susceptible to PM disease (Supplementary Table S1)53.
Genomic-based breeding techniques have, however, dramatically accelerated the introgression and pyramiding of several R genes for enhanced resistance durability54–56. Recent advances in genomic resources available in rye, including the 600 K SNP array and chromosomal-scale reference genome of the German inbred winter rye line ‘Lo7’ and Chinese population rye variety ‘Weining’, constitute significant milestones in rye genomic breeding50,57,58. Continuous mining for the discovery of novel genetic variability in rye R genes is essential to expand the ‘toolset’ available for resistance breeding. The importance of genetic studies in rye is further supported by the possibility of introgressing novel R genes into the staple cereal wheat by chromosomal translocation lines3.
In this paper, we investigate PM resistance in a less-prevalent elite Gülzow-type hybrid rye breeding germplasm. Our objective was to I) characterize PM resistance in the assayed germplasm, II) identify PM resistance-associated SNP markers to be implemented by marker-assisted selection for breeding novel resistant hybrid rye cultivars, III) investigate whether NLR genes residing in PM resistance-associated blocks on the ‘Lo7’ and ‘Weining’ reference genomes resemble known Pm genes, and IV) develop a marker map for the 600 K high-density SNP array and validate its performance in the assayed germplasm.
Results
600 K SNP genotyping of panel
To investigate the genetics underlying powdery mildew (PM) resistance, the assayed hybrid rye breeding germplasm was genotyped on the rye 600 K SNP array. With only scaffold positional data available for the array, SNP marker sequences were anchored to the recent ‘Lo7’ rye reference genome and stringently filtered to ensure its accuracy. In total, 591,196 markers were successfully mapped to the reference genome, and the developed marker map was made available at https://doi.org/10.5281/ZENODO.5086235. Quality filtration of markers for low minor allele frequency, missing markers, and missing individual scores across the panel led to the identification of 261,406 informative markers (Supplementary material 1). Characterization of fundamental performance-related metrics revealed a homogeneous inter- and intrachromosomal distribution of markers (Table 2). On average, each chromosome housed 32,676 markers with a mean marker-to-marker distance of 25.54 kb. The largest marker-to-marker distance was 9.95 Mbp on chromosome 2R, with a mean of 4.05 Mbp across the chromosomes. As a quality parameter, the polymorphism information content (PIC) was calculated to estimate the ability of markers to detect polymorphisms within the assayed germplasm (Supplementary Table S2). Across the informative marker panel, a mean PIC of 0.234 was identified, with a mean interchromosomal PIC ranging from 0.204 to 0.249 (Table 2). Visualization of these array performance metrics along the rye genome using Circos revealed a drop in marker density and PIC across the pericentromeric region on all chromosomes (Fig. 1).
Table 2.
Characteristics of informative 600 K SNP array on the Nordic Seed hybrid rye (Secale cereale L.) elite breeding germplasm (n = 180). Markers were positioned on the ‘Lo7’ reference genome.
| Chromosome | Chromosome length (Mbp) | Markers | Informative SNP markers | |||
|---|---|---|---|---|---|---|
| Markers | Mean inter- marker distance ± SD (kb) | Largest inter- marker distance (Mbp) | PIC ± SD | |||
| 1R | 727.33 | 72,089 | 33,854 | 21.47 ± 54.7 | 2.43 | 0.204 ± 0.134 |
| 2R | 945.85 | 77,774 | 33,698 | 28.07 ± 96.5 | 9.95 | 0.240 ± 0.120 |
| 3R | 965.54 | 69,428 | 31,493 | 30.65 ± 75.4 | 3.60 | 0.238 ± 0.111 |
| 4R | 906.54 | 81,652 | 32,555 | 27.84 ± 75.5 | 2.98 | 0.230 ± 0.125 |
| 5R | 876.06 | 81,842 | 37,073 | 23.63 ± 66.9 | 3.78 | 0.238 ± 0.123 |
| 6R | 885.15 | 84,283 | 36,872 | 24.00 ± 62.2 | 3.31 | 0.249 ± 0.105 |
| 7R | 889.76 | 86,994 | 38,918 | 23.12 ± 56.6 | 2.31 | 0.231 ± 0.120 |
| Unmapped | – | 35,245 | 16,943 | – | – | 0.239 ± 0.123 |
| Mean | 886.59 | 74,869 | 32,676 | 25,54 ± 69.7 | 4.05 | 0.234 ± 0.121 |
SD: Standard deviation, PIC: Polymorphism information content.
Figure 1.

Distribution of 244,463 informative 600 K SNP array markers in 180 Nordic Seed hybrid rye (Secale cereale L.) breeding lines along the ‘Lo7’ reference genome in 10 mb bins. The outer track depicts the marker density per bin. The inner track depicts the polymorphism-information content (PIC) per bin.
Phenotyping of hybrid rye breeding germplasm
To provide a comprehensive phenotypic dataset of PM resistance in the assayed Gülzow-type hybrid rye breeding germplasm, the lines were scored for their infection type (IT) against three distinct Blumeria graminis f. sp. secalis (Bgs) populations (Table 3, Supplementary Table S3). The lines scoring an IT below 1 were considered ‘resistant’ (Supplementary Fig. S1). Across the assayed germplasm, the N13 (Nienstädt, 2013) Bgs population yielded a mean IT of 2.40 ± 1.28 standard deviations (SD), with 47 resistant lines out of which 3 were restorers. The N18 (Nienstädt, 2018) Bgs population yielded a mean IT of 2.71 ± 1.11 SD with 29 resistant lines, out of which 1 was a restorer. D20 (Dyngby, 2020) Bgs population yielded a mean IT of 2.74 ± 1.10 SD with 20 resistant lines out of which 1 was a restorer. Across the assayed germplasms, 20 out of 88 non-restorer germplasm and 1 out of 92 restorer lines were consistently resistant to all three Bgs populations. Both controls, hybrid cv. KWS Binntto (‘susceptible’) and KWS Serafino (‘resistant’) were susceptible to all three Bgs populations. KWS Binntto had a mean IT of 3.56 ± 0.54 SD and KWS Serafino 3.01 ± 1.01 SD across Bgs populations.
Table 3.
Nine-step 0–4 scale for scoring infection types in cereal powdery mildew, after Torp et al.88.
| Phenotype | Infection-type | Mycelium growth | Sporulation | Development of chlorosis/necrosis |
|---|---|---|---|---|
| Resistant | 0 | None | None | No |
| 0–1 | None | None | Yes | |
| 1 | Weak | None | Yes | |
| Partially resistant | 1–2 | Weak | Weak | Yes |
| 2 | Moderate | Weak | Yes | |
| Partially susceptible | 2–3 | Moderate | Moderate | Yes |
| 3 | Strong | Moderate | Yes | |
| Susceptible | 3–4 | Strong | Strong | Yes |
| 4 | Strong | Strong | No |
To visualize the resistance spectrum of breeding lines, a circular neighbor-joining dendrogram was constructed, and concentric circles were added to integrate the scored IT (Fig. 2).
Figure 2.

Circular neighbor-joining dendrogram of 180 hybrid rye (Secale cereale L.) breeding lines. Infection type (0–4) reaction against three powdery mildew populations displayed by concentric circles around the dendrogram.
Statistical analysis of the line infection-type distribution across Bgs population by ANOVA showed that the N13 Bgs population differed significantly from the N18 and D20 Bgs populations (pval < 0.00016) (Fig. 3). Nine non-restorer germplasm lines were found to exhibit a differential resistance response to the N13 Bgs population. These lines were categorized as ‘partially resistant’, with a mean infection type of 1.52 ± 0.58 SD; they exhibited a mean infection type of 2.74 ± 0.70 and 2.80 ± 0.77 against the N18 and D20 populations, respectively.
Figure 3.
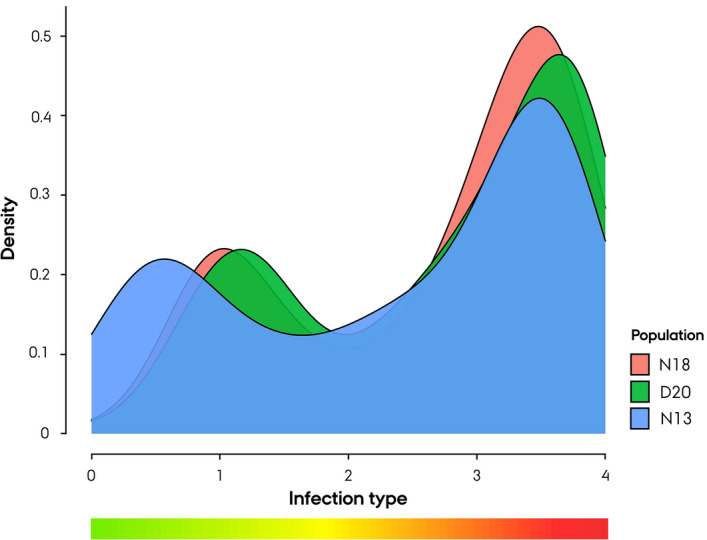
Density plot of the infection type distribution across 180 Nordic Seed hybrid rye (Secale cereale L.) breeding lines against three powdery mildew populations.
Genome-wide association study
For the identification of SNP markers associated with PM resistance, GWAS using MLM and the BLINK method was used for each of the three Bgs population trials and across trials for the entire germplasm and individual parental populations (Supplementary Figs. S2, S3). Isolated single SNPs in GWAS-MLM results were removed from the analysis even if they were significantly associated, as these were interpreted as having spurious associations or incorrect mapping positions. Instead, dense peaks comprising a large number of significant associated markers within a confined region in GWAS-MLM were selected for further analysis. GWAS-MLM on the entire panel led to the identification of a haplotype block on chromosome arm 7RL that was significantly associated (− log10 = 14.6) with powdery mildew resistance spanning from 882 to 898 Mbp (Fig. 4B, C). The haplotype block harbored 244 markers exhibiting an association above the Bonferroni adjusted significance threshold (− log10 ≥ 6.7) (Supplementary Table S4. Successive GWAS-BLINK led to the identification of the top-most resistance-associated (− log10 = 37.9) marker within the haplotype block on chromosome arm 7RL at 892.09 Mbp, which explained 16.8% of the phenotypic variance (Fig. 4A).
Figure 4.

Manhattan plot for the genome-wide association study (GWAS) of powdery mildew disease resistance in Nordic Seed hybrid rye (Secale cereale L.) elite breeding germplasm (n = 180) using 261,406 informative SNP markers. (A) GWAS using BLINK including a Q–Q plot. (B) GWAS using the MLM method including a Q–Q plot. (C) GWAS using the MLM method for the PM resistance-associated region on chromosome arm 7RL. Asterisks denote the position of the top-associated SNP marker identified in BLINK GWAS. NLR genes on the ‘Lo7’ reference genome are represented by green bars. The purple line represents the Bonferroni adjusted significance threshold based on informative markers.
In the non-restorer germplasm population, lines were found to carry a highly conserved haplotype across the significantly associated markers on chromosome arm 7RL (Supplementary Table S6). A resistant haplotype was conserved in 42 out of 48 resistant lines and a susceptible haplotype was found in 28 out of 31 susceptible lines, and the remaining 9 lines showed a differential resistance response (Supplementary material S3).
GWAS using BLINK led to the disappearance of the marker position on the ‘Unmapped’ chromosome that was in linkage disequilibrium with the genomic region on chromosome arm 7RL (Fig. 4A, B). In the non-restorer germplasm, an additional nonsignificant (− log10 = 3.62) peak was identified in GWAS-MLM spanning from 743.9 to 754.4 Mbp on chromosome arm 5RL (Supplementary Fig. S2, Supplementary Table S4). Gene mining by GWAS in the restorer population alone was not performed due to the low number of resistant lines (n = 5).
Nucleotide-binding leucine-rich repeat proteins in powdery mildew resistance-associated blocks on chromosome arm 7RL
Annotation of NLR genes in the reference genomes ‘Lo7’ and ‘Weining’ led to the identification of 770 and 1027 full-length (‘complete’) NLR genes, respectively. Positional information, annotation, and NB-ARC and NLR sequences of all ‘complete’ and ‘partial’ NLR genes for both reference genomes have been made available at https://doi.org/10.5281/zenodo.5085854. The PM resistance-associated block residing in the subtelomeric region of chromosome arm 7RL spanned 17 Mbp on the ‘Lo7’ reference genome and harbored 25 NLR genes (Fig. 4C, Supplementary Table S8). Out of the 244 markers residing in the block significantly associated (− log10(p) > 6.72) with PM resistance, 155 were accurately positioned on the ‘Weining’ reference genome (Supplementary Table S7). The markers mapped to a site spanning 14 Mbp from 994.6 to 1,008.4 Mbp on chromosome arm 7RL and housed 16 NLR genes (Supplementary Table S8). A search in the protein database using the NCBI Blastx function showed that the majority of NLRs shared similarities with resistance gene analogs (RGAs) and Pik-2-like and Pik6-NP-like disease resistance proteins in the diploid parental species of wheat. In both reference genomes, two NLRs shared similarities with Rpp13-like disease resistance proteins in Triticum urartu and barley.
Phylogenetic analysis using the NB-ARC domain of NLRs residing in the PM resistance-associated haplotype block on chromosome arm 7RL led to the finding that the majority of the NLRs were represented in both reference genomes (Fig. 5). The ‘Weining’ reference genome exhibited one unique NLR not present in ‘Lo7’, while the latter exhibited eight unique NLRs, of which five formed a distinct clade. Eight homogeneously represented NLRs from each reference genome clustered in a large clade (‘clade 1’).
Figure 5.

Phylogenetic tree of nucleotide-binding leucine-rich repeat (NLR) protein NB-ARC domains in the ‘Weining’ and ‘Lo7’ rye (Secale cereale L.) reference genomes. NLR genes are located at a powdery mildew resistance-associated site in the subtelomeric region of chromosome arm 7RL.
An additional phylogenetic analysis was performed using the entire reference genome NLR repertoire including NLR genes of characterized R genes as references (Fig. 6, Supplementary Fig. S4). The NLR genes residing within the haplotype block were found to span much of the reference NLR repertoire diversity. Clade 1 remained intact in both reference genome NLR repertoire trees positioned in a section harboring four out of five reference Pm genes included, with the closest being Pm60. In both reference genome NLR repertoire trees, a single NLR (Lo7_chr7R_nlr_94, Wei_chr7R_nlr_139) showed an evolutionary relationship with an Rpp13-like disease resistance gene and four Mla alleles.
Figure 6.

Phylogenetic tree of 770 nucleotide-binding leucine-rich repeat (NLR) protein NB-ARC domains in the ‘Lo7’ rye (Secale cereale L.) reference genome. The NB-ARC domains of known NLR genes have been included as references. NLR genes residing in a powdery mildew resistance block in the subtelomeric region of chromosome arm 7RL (Lo7–7RL PM) are colored teal.
Characterization of an Rpp13-like NLR gene residing in the resistance-associated block on chromosome arm 7RL
While several of the NLR genes residing within the PM resistance-associated haplotype block on chromosome arm 7RL chromosome showed evidence of an evolutionary relationship with known Pm genes, we selected the Rpp13-like NLR gene for further investigation on the basis of its close proximity. The Rpp13-like NLR gene (Lo7_chr7R_nlr_94, Wei_chr7R_nlr_139) was the closest NLR gene, residing approximately 2 Mbp from the top-most PM resistance-associated marker identified by BLINK (Fig. 4C, Supplementary Table S8).
The homologous Rpp13-like NLR genes in the ‘Lo7’ and ‘Weining’ reference genomes encoded canonical NLR proteins of 922 to 947 aa, showing 97% sequence similarity, with sequences differing by three single amino acid variants and an indel of 25 aa in the NB-ARC domain (Supplementary material 2). Protein BLAST of the NLR gene led to 88% sequence similarity with disease resistance Rpp13-like protein 4 in T. urartu, which encodes a 924 aa NLR protein sequence.
Discussion
In Denmark, the top-yielding hybrid rye cultivars and population varieties were evaluated in Danish official trials as an advisory service for farmers53. In the last decade, a high level of powdery mildew infection in rye was recorded only once in the trials in 2017 at a site in southern Denmark (Supplementary Fig. S1). At the trial site, several hybrid rye cultivars displayed up to 18% leaf area covered by PM. While no recent studies have investigated the effect of powdery mildew in top-yielding hybrid rye cultivars, Matzen et al. (2019) reported a 16% yield reduction in triticale at a disease severity of ≤ 10% leaf area covered by powdery mildew under field conditions in 2017. It is, therefore, reasonable to presume that under certain conducive conditions, PM is capable of causing substantial grain yield and quality losses in rye. Furthermore, on the basis of the trial records, we selected a hybrid cv. KWS Serafino as a resistant control in our study, as it showed less than 1% leaf area covered by PM at the site in 2017. However, under a high disease pressure, cv. KWS Serafino was found to be susceptible in the current study, suggesting a potential lower level of resistance against PM in the evaluated top-yielding hybrid cultivars than evident from the official trials.
In this study, we investigated PM resistance in the less-prevalent Gülzow-type elite hybrid rye breeding germplasm against three distinct Blumeria graminis f. sp. secalis (Bgs) populations from Denmark and Northern Germany. We observed a moderate level of powdery mildew resistance in the non-restorer germplasm population, and by performing a genome-wide association study (GWAS) using 261,406 informative SNP markers, we identified a strong PM resistance-associated site on the distal region of chromosome arm 7RL.
Hybrid rye breeding germplasms are highly secluded with little exchange of material. The existing exchange is subsequently influenced by the deployed fertility control system, which determines the compatibility of foreign material for introgression into heterotic parental populations59. Additionally, the 600 K SNP array was developed using lines from a German hybrid rye breeding germplasm deploying the predominant Pampa-type cytoplasmic male sterility system (CMS)57. In contrast, the germplasm assayed in this study deploys the less-prevalent Gülzow-type CMS system60. Due to the distinct nature of the different hybrid rye breeding germplasms, we investigated the performance of the 600 K SNP array on the Gülzow-type germplasm. With no physical position data available for the 600 K SNP markers, we developed a marker map by anchoring the marker sequences to the ‘Lo7’ reference genome. In brief, we found that the 600 K SNP array yielded a dense panel of informative SNP markers in the Gülzow-type germplasm with markers homogeneously distributed across the rye genome. The level of marker informativeness measured by the polymorphism information content (PIC) was largely comparable with observations made in maize (Zea mays L.). Here, high-density SNP genotyping of 544 diverse CIMMYT inbred lines yielded 362 K informative SNP markers with a mean PIC of 0.2561. In this study, however, we did observe a drop in marker informativeness and density across the pericentromeric region. Similar observations were made in the study by Rabanus-Wallace, et al.50, who reported a considerable reduction in both genetic diversity and gene density within the pericentromeric region of the inbred rye line ‘Lo7’. In barley, this region has furthermore been observed to display a 20-fold lower recombination rate62. Thus, we concluded that the 600 K SNP array performed satisfactorily on the Gülzow-type germplasm in relation to the fundamental characteristics investigated. The high-density array constitutes a milestone in rye genomic resources transitioning SNP-based genomic studies and replaces the previous rye 5 K SNP array by Haseneyer, et al.57,63.
GWAS on SNP genotype data has become an effective tool in genome-based plant breeding for the study of oligogenic traits often governed by a few genes with large effects, such as the major monogenic inherited resistance (R) genes63. The use of high-density SNP typing has allowed whole-genome scans for the identification of often small haplotype blocks significantly associated with resistance64. The creation of the 600 K SNP array and the chromosomal-scale reference genomes ‘Lo7’ and ‘Weining’ in rye has significantly changed the genomic toolbox available for mining novel resistance genes50,57. Using these recent advances in rye genomic resources, we successfully managed to identify a site on chromosome arm 7RL that was significantly associated with PM resistance. The high level of resolution provided by the dense marker panel revealed that the PM resistance-associated haplotype block spanned 17 Mbp on the distal tip of the chromosome arm 7RL subtelomeric region.
Until recently, no PM resistance gene had been identified on the rye 7R chromosome. However, during the development of translocation lines for wheat improvement using a local Chinese variety of rye ‘Baili’, Ren et al.42 discovered a PM-resistant 7BS:7RL translocation line. With the recipient wheat parent being susceptible, the PM resistance gene traced back to the rye donor chromosome arm 7RL. Resistance phenotyping of the translocation line demonstrated that it displayed a high level of PM resistance against prevalent Bg. tritici pathotypes in China, making it very promising for the development of novel PM-resistant wheat cultivars. Although a novel finding in rye, several PM resistance genes have been identified in wheat chromosomal segments syntenic to rye chromosome arm 7RL. During Triticeae speciation, a series of recurrent translocation events gave rise to major patterns of chromosomal rearrangements65. In rye, the distal region of chromosome arm 7RL, therefore, shows high homology with the wheat 2A/B/D chromosomes58,66. Currently, more than five Pm genes have been identified in wheat on the 2A/B/D chromosomes67,68.
With the Pm gene discovered by Ren, et al.42 originating from a forage-type population of rye in a gene pool distinct from the germplasm investigated in this study, it is reasonable to presume that the two Pm genes could be either distinct or allelic69. We provisionally denote the novel Pm locus residing in the subtelomeric region of chromosome arm 7RL as PmNOS1.
Comparative analysis of the three Bgs populations used in the study revealed that the N13 population significantly differed from the two more recent populations, showing a less virulent composition of pathotypes. In addition, nine non-restorer germplasm breeding lines displaying a differential resistance profile were identified, showing ‘partial resistance’ only to the N13 population. With none of these lines carrying the resistance haplotype associated with the resistance locus on chromosome arm 7RL, our findings suggest that these do not carry the PmNOS1 locus. While this observation can be explained by the occurrence of recombination events between the PM resistance-associated marker and the causative gene, this result seems less likely due to the consistent divergence in the haplotype70. Instead, it seems more likely that the differentially resistant non-restorer germplasm lines carry a distinct Pm gene that lost its effect during the period 2013–2018 in northern Germany. As a result of host genetic uniformity and the high evolutionary capacity of Bg to acquire virulence and migrate rapidly over long distances, the effectiveness of Pm genes is often rapidly lost. An example of this is the Mla13 Pm gene introduced in a former Czechoslovakian barley cultivar, ‘Koral’, in 198017. After years of providing effective resistance against PM disease, virulence was observed in England in 1988 in a pathotype believed to originate from Czechoslovakia, having migrated by wind across the European continent and North Sea71. Ongoing monitoring of the virulence structure of Bg was conducted to survey the effectiveness of deployed Pm genes in elite cultivars72,73. While the Pm gene present in the differentially resistant non-restorer germplasm lines has seemingly been overcome, our findings suggest that PmNOS1 remains effective.
Enabled by recent advances in rye genomic resources, we investigated whether any of the NLR genes residing in the region harboring the PmNOS1 locus resembled known Pm genes. In several crop species, including rye, NLR genes have been observed to accumulate at recombination hotspots in subtelomeric chromosomal regions50,74,75. Additionally, we identified several large clusters of NLR genes residing in the PM resistance-associated block harboring PmNOS1 in both of the reference genomes. In addition to the PM resistance gene identified in our study, Fusarium head blight and leaf rust resistance genes have been mapped to the subtelomeric region of chromosome arm 7RL76,77. Stem and stripe rust resistance genes reported in the 7BS:7RL translocation line developed by Ren et al.42 are furthermore likely to reside in the subtelomeric region. To investigate the likely diversity of R genes residing in the PM resistance-associated block, we conducted a phylogenetic analysis using the NB-ARC domain sequence of NLR genes residing in the block78,79. In contrast to the rapidly evolving LRR domain often exhibiting intraspecific polymorphism, the NB-ARC domain is largely conserved and suited for the study of evolutionary relationships among NLR genes80,81. As expected, the NLR genes residing in the block represented a large proportion of the NLR repertoire diversity in rye, accentuating the evolutionary plasticity of the NLRs residing in the subtelomeric region75. Intriguingly, guided by a panel of isolated NLR genes as a reference, we observed a predisposition of NLRs within the block to be in close proximity to known Pm genes in wheat and barley. This evolutionary relationship could hint at a common attribute among the NLRs79.
Phylogenetic analysis led to the identification of an NLR gene with an evolutionary relationship and protein sequence similarity to Rpp13-like protein 4 in T. urartu. Based on its close proximity to the marker displaying the strongest association with the PmNOS1 locus, the Rpp13-like NLR gene was selected for further characterization. In A. thaliana, Rpp13 confers resistance against Peronospora parasitica, the causative agent of downy mildew disease82. Recent studies have, however, identified Rpp13-like NLR genes associated with powdery mildew resistance in cereals. In wheat, Liu et al.83 showed that silencing of the Rpp13 homologous gene TaRPP13-3 in resistant wheat cv. ‘Brock’ induced susceptibility to powdery mildew. In barley, Cheng et al.84 found that the expression of an Rpp13-like NLR gene was highly upregulated after inoculation with powdery mildew.
In rye, while the rate of decay has only been determined in a few genes related to frost response, which showed a rapid linkage decay85, these genes have been demonstrated to decay after 3.76 kb, on average, across the genome in a similar hybrid breeding germplasm in maize61. Due to the heterogenic nature of outcrossing species, their rate of decay is often rapid86. However, in a recent population study on assayed germplasm, non-restorer germplasm was found to exhibit relatively low genetic diversity, low effective population size and high linkage disequilibrium59. Consistent with their observations, we found a large conserved haplotype on chromosome arm 7RL harboring the PmNOS1 locus59. The linkage decay in the non-restorer germplasm is likely considerably reduced by the low genetic diversity and effective population size, resulting in a similarly low frequency of effective recombination events. The large amount of linkage, while beneficial for trait discovery in GWAS at lower marker density, impedes the identification of a narrow and precise genomic region that may harbor the gene of interest, even at high marker resolution. In the case of the PmNOS1 locus, the haplotype block on chromosome arm 7RL was found to span 17 Mbp, harboring between 17 and 25 potential candidate NLR genes in the ‘Lo7’ and ‘Weining’ reference genomes. For more accurate mapping of the PmNOS1 locus, the development of multiparent mapping populations could be conducted, allowing several generations of potential effective recombination events in the region87,88. Identification of the causative gene could be performed by resistance gene enrichment sequencing (RenSeq) analysis followed by transformation of a susceptible non-restorer germplasm line to validate the gene89. This would in turn equally show whether the gene is present in the reference genomes and whether PmNOS1 encodes an Rpp13-like NLR protein.
In conclusion, our study demonstrates the immediate value of recent advances in rye genomic resources for the mining of novel resistance genes. These resources now permit accurate identification of delimited resistance-associated haplotype blocks and scanning for trait-associated genes residing within. With pathogens such as Bg displaying a large evolutionary plasticity, shortening the process from identification of resistance-associated sites to isolation of the underlying R gene is important for the development of novel resistant cultivars. The relevance of studies in rye is accentuated by the possibility of introgressing novel R genes into the staple cereal wheat by chromosomal translocation lines. To aid further studies in the field, we have provided both a rye600K SNP array marker map anchored using the ‘Lo7’ reference genome and NLR repertoire information of the ‘Lo7’ and ‘Weining’ reference genomes in open-access data repositories.
Materials and methods
Plant material and DNA extraction
A panel of 180 inbred rye (Secale cereale L.) lines, 92 restorer and 88 non-restorer germplasm, belonging to the elite Gülzow-type hybrid rye breeding germplasm at Nordic Seed A/S (Dyngby, Denmark) were investigated. Population structure and information on the genetic characteristics of the germplasm were presented in a recent study by Vendelbo et al.59. The parental populations represent genetically secluded gene pools with restorer (paternal) lines carrying a dominant allele for the restoration of male fertility and non-restorer germplasm lines carrying a recessive allele and fertile cytoplasm, which is used to maintain cytoplasmic male sterile (maternal) lines. DNA extraction was performed using an adapted SDS-based method according to the USDA90 after Pallotta et al.91 on an equivalent of 75 mg of plant material collected from the coleoptiles and primary leaves of two seven-day-old seedlings per line. The DNA concentration and 260/280 nm ratio of the samples were measured using an Epoch™ microplate spectrophotometer (Biotek®), and evidence of fragmentation was obtained by size visualization on a 1.2% agarose gel.
Molecular marker resource and SNP genotyping
Samples of each line containing 200 ng of high molecular weight gDNA with a ≥ 1.8 260/280 nm ratio were sent for single nucleotide polymorphism (SNP) genotyping at Eurofins Genomics Europe Genotyping (Aarhus, Denmark). Genotyping was performed using a 600 K SNP array with 600,843 SNP markers on an Affymetrix GeneTitan™ Scanner platform57.
Collection and multiplication of Blumeria graminis f. sp. secalis populations
As there was no unified information on germplasm resistance to powdery mildew, three field populations of Bgs were sampled in Northern Germany and Denmark in the period from 2013 to 2020 to screen the lines against a broad range of Bgs pathotypes prevalent in Northern Europe. Two Bgs populations were collected at the Nordic Seed rye multiplication site in Germany in 2013 (N13) and 2018 (N18) (52.29254°N, E9.14896°E). Ten leaves exhibiting PM disease were carefully collected and rinsed in 0.5 L of water to release Bgs spores. Six pots containing 15–20 susceptible 12-day-old seedlings of the restorer line R277 were then inoculated by spraying a fine mist of the spore solution using an atomizer bottle. Pots were transferred to a separate climate chamber for each population and incubated at 18 °C with 12 h of light using 400 W high-pressure Phillips SON-T Agro lamps. Populations were continuously multiplied in an overlapping two-week cycle. Each week, the tray of 2-week-old seedlings was substituted with a tray of fresh 12-day-old seedlings. Inoculation was achieved by passive dissemination of spores from the ‘older’ tray to the tray with new plants through a steel-grid shelf.
An additional Bgs population was collected in autumn 2020 (D20) at Nordic Seed (Dyngby, Denmark) (55.94944°N, E10.25414°E) using a mixture of hybrid cvs. KWS Binntto, KWS Bono and KWS Florano. Pots with 15 to 20 seedlings of the susceptible mixture were placed outdoors 12 days after sowing (DAS) in August-October 2020. Plants were controlled regularly, and all leaves showing PM disease within the period were collected. Prior to inoculation, leaves were placed in Petri dishes with moist filter paper and set to sporulate at room temperature in light for four to eight hours. Then, a pot containing 15–20 seedlings of the susceptible mixture was inoculated at 12 DAS by horizontally stroking the collected leaves across the seedlings. Inoculated pots were sprayed with a fine mist of water, placed in a container with transparent lids to ensure 100% RH and incubated in the dark at 10–15 °C for 24 h. After incubation in the dark, the pots were transferred to an isolated greenhouse cabin and incubated under 16 h of daylight at 18–24 °C and 8 h of dark at 14–16 °C.
Multiplication of trial inoculum for the three Bgs populations was performed using the same procedure as described above. For each population, two trays containing 35 pots of the susceptible mixture were inoculated by brushing 3–4 highly infected pots inoculated 20 days earlier across the tray.
Infection and scoring
All lines were phenotyped for the infection-type response to the Bgs populations. In each greenhouse trial, eight seeds per line were sown in a 28-hole tray using a completely randomized design with two repetitions for each of the two trial replicates. For each tray, a positive (‘susceptible’) control consisting of hybrid cv. KWS Binntto and negative (‘resistant’) control cv. KWS Serafino was included. At 14 DAS, trays were inoculated as described above by brushing 3–4 highly infected pots inoculated 20 days earlier across the tray. After 14 days of incubation, the lines were phenotyped by scoring the infection response on the first and second leaves for each of the eight seedlings per repetition in accordance with a 9-step 0–4 scale by Torp et al.88 (Table 3).
Data analysis
Bioinformatic analysis of SNP marker data was performed with the R studio (v. 1.3.959) interface in R statistical software (v. 4.0.1) by applying various predesigned packages92,93.
Mapping of 600 K SNP array markers to the ‘Lo7’ reference genome
Positional data of the 600 K SNP markers were obtained by mapping each of the 600,843 SNP marker sequences to the rye reference genome ‘Lo7’ using the NCBI blastn (v. 2.9.0 +) function50,94. The mapping positions of SNPs were hereafter stringently filtered for I) complete SNP sequence alignment and II) a maximum of 1 mismatch to ensure accurate positioning.
Molecular markers and characterization of 600 K SNP array performance
Prior to analysis, markers were filtered for a marker allele frequency ≥ 0.005, missing individual score ≤ 0.2 and missing marker score ≤ 0.1. Fundamental characteristics of SNP marker informativeness, including polymorphism information content (PIC), were calculated using the SnpReady (v. 0.9.6) R package95. The interchromosomal distribution of the informative marker PIC, marker-to-marker distance and marker density in 10 mb bins on the ‘Lo7’ rye genome were visualized using Circos (v. 0.69.8) in the Galaxy online interface96,97. A Circos plot was constructed using the pipeline developed by Hiltemann, et al.98.
Analysis of phenotypic data
The distribution of infection types against the three respective PM populations was visualized by density plots using ggplot2 (v. 3.3.3) R package99. To determine whether the population infection-type distribution differed significantly, ANOVA was conducted using R.
To correct the resistance phenotype for the effects of replication and population, we fitted the data to a linear mixed model using the lme4 (v. 1.1.26) package in R:
where µ is the general mean, P is the population, R represents the replications, l is the line id, and ɛ is the residuals. P and R were set as fixed effects, and l was set as a random effect. The random effect and residuals were assumed to be independent normally distributed variables described as follows: l ~ N (0, I ơ2l), and ɛ ~ N (0, I ơ2ɛ). The BLUP solutions for the line effect were used in GWAS. Data were also fitted to a linear mixed model for each of the populations to correct for the effect of replication. In this model ‘P’ was removed.
Genome-wide association study
Discovery of PM resistance-associated SNP markers was performed by a genome-wide association study (GWAS) using the genomic association and prediction integration tool (GAPIT) (v.3) package in R100. The Manhattan plot was colorized using the RColorBrewer (v.1.1–2) R package color palette99. GWAS using a mixed linear model (MLM) was performed to identify discrete haplotype blocks associated with powdery mildew resistance. Additionally, the Bayesian-information and linkage-disequilibrium iteratively nested keyway (BLINK) method was performed to identify the top-most powdery mildew resistance-associated marker within each haplotype block101. BLINK uses a multiple loci test for MLM by combining a fixed effects model, Bayesian information content and linkage disequilibrium information to collectively improve the statistical power while simultaneously reducing the computational run time. Markers that are in linkage disequilibrium with the top-most significant marker at a site are excluded in BLINK. A standard Bonferroni-corrected threshold of α = 0.05 was used as the significance threshold.
Annotation of nucleotide-binding leucine-rich repeat proteins in the reference genome
To investigate whether PM-associated sites in the ‘Lo7’ and ‘Weining’ reference genomes housed nucleotide-binding leucine-rich repeat (NLR) genes, annotation was performed using NLR-parser (v.3) and NLR-annotator (https://github.com/steuernb/NLR-Annotator)79,102.
Characterization of nucleotide-binding leucine-rich repeat proteins
The gene structure, coding sequence and NLR protein sequence of candidate genes were extracted from RNA-seq and de novo protein data provided in ‘Weining’ and ‘Lo7’ reference genome data repositories50,58. To investigate whether NLR genes residing in PM resistance-associated sites resembled known genes, the NCBI blastx function was used for protein–protein searches in the online database94. For functional analysis and the prediction of protein domains, InterPro Scan was used103. Identification of sequence divergence between reference genome homologs was performed by multiple sequence alignment using the multiple sequence comparison by log-expectation (MUSCLE) method for coding sequences and the ‘Clustal Omega’ method for NLR protein sequences in Geneious Prime (v. 2020.2.3).
Phylogenetic analysis
Neighbor-joining clustering analysis of breeding lines was performed with Euclidean genetic distance measurement using the ape (v. 5.3) R package104. The tree was constructed after 10,000 bootstrapping iterations with weak nodes (≤ 80% recurrence) collapsed into multifurcations. A circular neighbor-joining tree was generated using the iTOL (v. 5) online tool (http://itol.embl.de/), enabling a colorful visualization of each line infection type spectrum against the three Bgs populations105.
To investigate the sequence similarity between nucleotide-binding leucine-rich repeat (NLR) genes at PM resistance-associated sites in the ‘Lo7’ and ‘Weining’ reference genomes, neighbor-joining clustering analysis using NB-ARC (nucleotide-binding adaptor shared by APAF-1, R proteins, and CED-4) domain sequences was performed. Identification of PM resistance-associated sites on ‘Weining’ was performed by mapping PM resistance-associated markers using the same procedure as described previously for ‘Lo7’. As references, NB-ARC domain sequences of available leaf rust (Lr1, Lr10, Lr21, Lr22a), stem rust (Sr13, Sr22), yellow rust (Yr5, Yr10, Yr28), powdery mildew (Pm2, Pm3, Pm8, Pm12, Pm17, Pm60, Mla1, Mla6, Mla7, Mla10, RPP13, RPP-like-T. urartu) and resistance gene analogs (RGA1, RGA2, RGA3, RGA4, RGA5) were included. NB-ARC sequences were obtained from the UniProt online database106. Phylogenetic analysis was conducted using a pipeline developed by Toparslan, et al.107 in R. Multiple sequence alignment of NB-ARC domain sequences was performed via msa (v. 1.20.1) using the ‘Clustal Omeage’ method and pairwise genetic distance based on identity calculated with the seqinr (4.2–8) package in R108,109. A tree was constructed for the ‘Lo7’ and ‘Weining’ NLR repertoires separately and visualized using ggtree (v. 2.2.4) R package110.
Graphical editing
Graphs and figures were outputted from R in .svg format and manually curated using Inkscape (v. 1.1) (https://inkscape.org/).
Ethical statement
Experimental research and field studies on plants (either cultivated or wild), including the collection of plant material,complies with relevant institutional, national, and international guidelines and legislation.
Supplementary Information
Acknowledgements
We would like to thank laboratory technician Hanne Svenstrup at Nordic Seed A/S for contributing to the genotypic data collection and Anette Deterding and Marlene Walbrodt at Nordic Seed Germany GmbH (Nienstädt, Germany) for multiplying the N13 and N18 Bgs populations. We thank Professor Mogens Hovmøller and senior researchers Dr. Annemarie Fejer Justesen and Dr. Chris Khadgi Sørensen at the Institute of Agroecology Aarhus University (Flakkebjerg, Denmark) for their intellectual input for the study. The plant material included in this study was provided by Nordic Seed Germany GmbH (Nienstädt, Germany). 600K SNP genotyping was provided by Eurofind Genomics (Skejby, Denmark).
Author contributions
All authors were involved in the study design. N.M.V. performed the phenotyping, bioinformatic analysis, and visual output and wrote the manuscript. K.M. assisted in the DNA extraction of the germplasm. P.S.K oversaw the multiplication of Bgs populations. J.O. was responsible for all communication with Trait Genetics and Eurofins Genomics conducting the SNP genotyping. K.M., P.S., J.O. and A.J. were involved in the intellectual input for the study, including the interpretation of the results. All authors were involved in the conceptualization of the study and revision of the manuscript.
Funding
The research was funded by Innovation Fund Denmark (Grant No. 8053-00085B) and the Pajbjerg Foundation.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-02488-5.
References
- 1.Schlegel RHJ. Rye genetics, breeding and cultivation. CRC Press Taylor & Francis Group; 2014. [Google Scholar]
- 2.Schlegel, R. H. J. & Korzun, V. Genes, markers and linkage data of rye (Secale cereale L.), 11th updated inventory, V. 0.1.21. 1–115. http://www.rye-gene-map.de (2021).
- 3.Crespo-Herrera LA, Garkava-Gustavsson L, Åhman I. A systematic review of rye (Secale cereale L.) as a source of resistance to pathogens and pests in wheat (Triticum aestivum L.) Hereditas. 2017;154:1–9. doi: 10.1186/s41065-017-0033-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Savary S, et al. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evolut. 2019;3:430–439. doi: 10.1038/s41559-018-0793-y. [DOI] [PubMed] [Google Scholar]
- 5.Savary S, Ficke A, Aubertot J-N, Hollier C. Crop losses due to diseases and their implications for global food production losses and food security. Food Secur. 2012;4:519–537. doi: 10.1007/s12571-012-0200-5. [DOI] [Google Scholar]
- 6.Griffey CA, Das MK, Stromberg EL. Effectiveness of adult-plant resistance in reducing grain yield loss to powdery mildew in winter wheat. Plant Dis. 1993;77:618–622. doi: 10.1094/PD-77-0618. [DOI] [Google Scholar]
- 7.Conner RL, Kuzyk AD, Su H. Impact of powdery mildew on the yield of soft white spring wheat cultivars. Can. J. Plant Sci. 2003;83:725–728. doi: 10.4141/P03-043. [DOI] [Google Scholar]
- 8.Matzen N, Heick TM, Jørgensen LN. Control of powdery mildew (Blumeria graminis spp.) in cereals by Serenade® ASO (Bacillus amyloliquefaciens (former subtilis) strain QST 713) Biol. Control. 2019;139:104067. doi: 10.1016/j.biocontrol.2019.104067. [DOI] [Google Scholar]
- 9.Te Beest DE, Paveley ND, Shaw MW, Van Den Bosch F. Disease-weather relationships for powdery mildew and yellow rust on winter wheat. Phytopathology. 2008;98:609–617. doi: 10.1094/PHYTO-98-5-0609. [DOI] [PubMed] [Google Scholar]
- 10.Troch V, et al. Formae speciales of cereal powdery mildew: Close or distant relatives? Mol. Plant Pathol. 2014;15:304–314. doi: 10.1111/mpp.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jorgensen JH. Erysiphe graminis, powdery mildew of cereals and grasses. Genet. Plant Pathogenic Fungi Adv. Plant Pathol. 1988;6:137–157. doi: 10.1016/B978-0-12-033706-4.50012-8. [DOI] [Google Scholar]
- 12.Jankovics T, et al. New insights into the life cycle of the wheat powdery mildew: direct observation of ascosporic Infection in Blumeria graminis f. sp. tritici. Phytopathology. 2015;105:797–804. doi: 10.1094/PHYTO-10-14-0268-R. [DOI] [PubMed] [Google Scholar]
- 13.Cowger C, Parks R, Kosman E. Structure and migration in U.S. Blumeria graminis f. sp. tritici populations. Phytopathology. 2016;106:295–304. doi: 10.1094/PHYTO-03-15-0066-R. [DOI] [PubMed] [Google Scholar]
- 14.Dreiseitl A. Great pathotype diversity and reduced virulence complexity in a Central European population of Blumeria graminis f. sp. hordei in 2015–2017. Europ. J. Plant Pathol. 2018;153:801–811. doi: 10.1007/s10658-018-1593-6. [DOI] [Google Scholar]
- 15.Zhu J, et al. Genetic evidence of local adaption and long distance migration in Blumeria graminis f. sp. hordei populations from China. J. General Plant Pathol. 2016;82:69–81. doi: 10.1007/s10327-016-0643-1. [DOI] [Google Scholar]
- 16.Limpert E, Godet F, Müller K. Dispersal of cereal mildews across Europe. Agric. Forest Meterol. 1999;97:293–308. doi: 10.1016/S0168-1923(99)00073-8. [DOI] [Google Scholar]
- 17.Wolfe MS, Brändle U, Koller B, Limpert E, McDermott J. Barley mildew in Europe: population biology and host resistance. Euphytica. 1992;63:125–139. doi: 10.1007/BF00023918. [DOI] [Google Scholar]
- 18.Brown, J. K. & Hovmoller, M. S. Aerial dispersal of pathogens on the global and continental scales and its impact on plant disease. Science297 (2002). [DOI] [PubMed]
- 19.Kast, W. K. & Geiger, H. H. Studies on the inheritance of mildew resistance in rye. I. Results from inbred lines and F1 crosses. (1982).
- 20.Miedaner T, Schmidt H, Geiger H. Components of variation for quantitative adult-plant resistance to powdery mildew in winter rye. Phytopathology. 1993;83:1071–1075. doi: 10.1094/Phyto-83-1071. [DOI] [Google Scholar]
- 21.Nelson R, Wiesner-Hanks T, Wisser R, Balint-Kurti P. Navigating complexity to breed disease-resistant crops. Nat. Rev. Genet. 2018;19:21–33. doi: 10.1038/nrg.2017.82. [DOI] [PubMed] [Google Scholar]
- 22.Frieb, B., Heun, M. & Bushuk, W. Cytological characterization, powdery mildew resistance and storage protein composition of tetraploid and hexaploid 1BL/1RS wheat-rye translocation lines. Theor. Appl. Genet.78 (1989). [DOI] [PubMed]
- 23.Hsam SLK, Zeller FJ. Evidence of allelism between genes pm8 and pm17 and chromosomal location of powdery mildew and leaf rust resistance genes in the common wheat cultivar 'Amigo'. Plant Breed. 1997;116:119–122. doi: 10.1111/j.1439-0523.1997.tb02164.x. [DOI] [Google Scholar]
- 24.Ren T-H, et al. Development and characterization of a new 1BL.1RS translocation line with resistance to stripe rust and powdery mildew of wheat. Euphytica. 2009;169:207–213. doi: 10.1007/s10681-009-9924-5. [DOI] [Google Scholar]
- 25.Han G, et al. Identification of an elite wheat-Rye T1RS.1BL translocation line conferring high resistance to powdery mildew and stripe rust. Plant Dis. 2020;104:2940–2948. doi: 10.1094/PDIS-02-20-0323-RE. [DOI] [PubMed] [Google Scholar]
- 26.He H, et al. Characterization of a new gene for resistance to wheat powdery mildew on chromosome 1RL of wild rye Secale sylvestre. Theor. Appl. Genet. 2021;134:887–896. doi: 10.1007/s00122-020-03739-1. [DOI] [PubMed] [Google Scholar]
- 27.Koebner, R. M. D. Introduction into wheat of disease resistance from related species. Annual report of the Plant Breeding Institute, 69–70 (1986).
- 28.Driscoll CJ, Jensen NF. A genetic method for detecting introduced intergenetic translocations. Genetics. 1963;48:459–468. doi: 10.1093/genetics/48.4.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhuang LF, Sun L, Li AX, Chen TT, Qi ZJ. Identification and development of diagnostic markers for a powdery mildew resistance gene on chromosome 2R of Chinese rye cultivar Jingzhouheimai. Mol. Breed. 2010;27:455–465. doi: 10.1007/s11032-010-9443-z. [DOI] [Google Scholar]
- 30.Hysing S-C, et al. Agronomic performance and multiple disease resistance in T2BS.2RL wheat-rye translocation lines. Crop Sci. 2007;47:254–260. doi: 10.2135/cropsci2006.04.0269. [DOI] [Google Scholar]
- 31.Heun M, Friebe B. Introgression of powdery mildew resistance from rye into wheat. Phytopathology. 1990;80:242–245. doi: 10.1094/Phyto-80-242. [DOI] [Google Scholar]
- 32.Driscoll CJ, Jensen NF. Release of a wheat-rye translocation stock involving leaf rust and powdery mildew resistances 1. Crop Sci. 1965;5:279–280. doi: 10.2135/cropsci1965.0011183X000500030031x. [DOI] [Google Scholar]
- 33.An DG, Li LH, Li JM, Li HJ, Zhu YG. Introgression of resistance to powdery mildew conferred by chromosome 2R by crossing wheat nullisomic 2D with rye. J. Integr. Plant Biol. 2006;48:838–847. doi: 10.1111/j.1744-7909.2006.00275.x. [DOI] [Google Scholar]
- 34.Lind, V. Analysis of the resistance of wheat-rye addition lines to powdery mildew of wheat Tagungsbericht Akademie der Landwirtschaftswissenschaften der DDR (1982).
- 35.Ma P, et al. Development of novel wheat-rye chromosome 4R translocations and assignment of their powdery mildew resistance. Plant Dis. 2020;104:260–268. doi: 10.1094/PDIS-01-19-0160-RE. [DOI] [PubMed] [Google Scholar]
- 36.Fu S, et al. New wheat-rye 5DS-4RS.4RL and 4RS-5DS.5DL translocation lines with powdery mildew resistance. J. Plant Res. 2014;127:743–753. doi: 10.1007/s10265-014-0659-6. [DOI] [PubMed] [Google Scholar]
- 37.An D, et al. Molecular cytogenetic characterization of a new wheat-rye 4R chromosome translocation line resistant to powdery mildew. Chromosome Res. 2013;21:419–432. doi: 10.1007/s10577-013-9366-8. [DOI] [PubMed] [Google Scholar]
- 38.Riley, R. & Macer, R. C. F. The chromosomal distribution of genetic resistance of rye to wheat pathogens. Canad. J. Genet. Cytol.8 (1966).
- 39.Hao M, et al. Introgression of powdery mildew resistance gene Pm56 on rye chromosome arm 6RS into wheat. Front. Plant Sci. 2018;9:1–8. doi: 10.3389/fpls.2018.01040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Friebe B, Heun M, Tuleen N, Zeller FJ, Gill BS. Cytogenetically monitored transfer of powdery mildew resistance from rye into wheat. Crop Sci. 1994;34:621–625. doi: 10.2135/cropsci1994.0011183X003400030003x. [DOI] [Google Scholar]
- 41.Miller TE. The homoeologous relationship between the chromosomes of rye and wheat. Current status. Canad. J. Genet. Cytol. 1984;26:578–589. doi: 10.1139/g84-091. [DOI] [Google Scholar]
- 42.Ren T, et al. Molecular and cytogenetic characterization of a wheat-rye 7BS7RL translocation line with resistance to stripe rust, powdery mildew, and fusarium head blight. Phytopathology. 2020;110:1713–1720. doi: 10.1094/PHYTO-02-20-0061-R. [DOI] [PubMed] [Google Scholar]
- 43.Elmore JM, Lin ZJ, Coaker G. Plant NB-LRR signaling: Upstreams and downstreams. Curr. Opin. Plant Biol. 2011;14:365–371. doi: 10.1016/j.pbi.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hurni S, et al. Rye Pm8 and wheat Pm3 are orthologous genes and show evolutionary conservation of resistance function against powdery mildew. Plant J. 2013;76:957–969. doi: 10.1111/tpj.12345. [DOI] [PubMed] [Google Scholar]
- 45.Xie J, et al. A rare single nucleotide variant in Pm5e confers powdery mildew resistance in common wheat. New Phytol. 2020;228:1011–1026. doi: 10.1111/nph.16762. [DOI] [PubMed] [Google Scholar]
- 46.Marone D, Russo MA, Laido G, De Leonardis AM, Mastrangelo AM. Plant nucleotide binding site-leucine-rich repeat (NBS-LRR) genes: Active guardians in host defense responses. Int. J. Mol. Sci. 2013;14:7302–7326. doi: 10.3390/ijms14047302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang J, Han M, Liu Y. Diversity, structure and function of the coiled-coil domains of plant NLR immune receptors. J. Integr. Plant Biol. 2021;63:283–296. doi: 10.1111/jipb.13032. [DOI] [PubMed] [Google Scholar]
- 48.Takken FL, Goverse A. How to build a pathogen detector: Structural basis of NB-LRR function. Curr. Opin. Plant Biol. 2012;15:375–384. doi: 10.1016/j.pbi.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 49.Krasileva KV, Dahlbeck D, Staskawicz BJ. Activation of an Arabidopsis resistance protein is specified by the in planta association of its leucine-rich repeat domain with the cognate oomycete effector. Plant Cell. 2010;22:2444–2458. doi: 10.1105/tpc.110.075358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rabanus-Wallace MT, et al. Chromosome-scale genome assembly provides insights into rye biology, evolution and agronomic potential. Nat. Genet. 2021;53:564–573. doi: 10.1101/2019.12.11.869693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ryan P, Carbone L, Marshall D, Cowger C. Virulence structure of eastern US wheat powdery mildew population. Plant Dis. 2008;92:1074–1082. doi: 10.1094/PDIS-92-7-1074. [DOI] [PubMed] [Google Scholar]
- 52.Cowger C, Mehra L, Arellano C, Meyers E, Murphy JP. Virulence differences in Blumeria graminis f. sp. tritici from the Central and Eastern United States. Phytopathology. 2018;108:402–411. doi: 10.1094/PHYTO-06-17-0211-R. [DOI] [PubMed] [Google Scholar]
- 53.Sortsinfo. The Danish official trial records, <https://sortinfo.dk/> (2021).
- 54.Miedaner T, Korzun V. Marker-assisted selection for disease resistance in wheat and barley breeding. Phytopathology. 2012;102:560–566. doi: 10.1094/PHYTO-05-11-0157. [DOI] [PubMed] [Google Scholar]
- 55.Mundt CC. Pyramiding for resistance durability: Theory and practice. Phytopathology. 2018;108:792–802. doi: 10.1094/PHYTO-12-17-0426-RVW. [DOI] [PubMed] [Google Scholar]
- 56.Koller T, Brunner S, Herren G, Hurni S, Keller B. Pyramiding of transgenic Pm3 alleles in wheat results in improved powdery mildew resistance in the field. Theor. Appl. Genet. 2018;131:861–871. doi: 10.1007/s00122-017-3043-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bauer E, et al. Towards a whole-genome sequence for rye (Secale cereale L.) Plant J. 2017;89:853–869. doi: 10.1111/tpj.13436. [DOI] [PubMed] [Google Scholar]
- 58.Li G, et al. A high-quality genome assembly highlights rye genomic characteristics and agronomically important genes. Nat. Genet. 2021;53:574–584. doi: 10.1038/s41588-021-00808-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vendelbo NM, Sarup P, Orabi J, Kristensen PS, Jahoor A. Genetic structure of a germplasm for hybrid breeding in rye (Secale cereale L.) PLoS ONE. 2020;15:e0239541. doi: 10.1371/journal.pone.0239541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vendelbo, N. M. et al. Genomic scan of male fertility restoration genes in a ‘Gülzow’ type hybrid 1 breeding system of rye (Secale cereale L.). Int. J. Mol. Sci., Submitted. [DOI] [PMC free article] [PubMed]
- 61.Wu Y, et al. Molecular characterization of CIMMYT maize inbred lines with genotyping-by-sequencing SNPs. Theor. Appl. Genet. 2016;129:753–765. doi: 10.1007/s00122-016-2664-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baker K, et al. The low-recombining pericentromeric region of barley restricts gene diversity and evolution but not gene expression. Plant J. 2014;79:981–992. doi: 10.1111/tpj.12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu HJ, Yan J. Crop genome-wide association study: A harvest of biological relevance. Plant J. 2019;97:8–18. doi: 10.1111/tpj.14139. [DOI] [PubMed] [Google Scholar]
- 64.Brachi B, Moris GP, Borevitz JO. Genome-wide association studies in plants: The missing heritability is in the field. Genome Biol. 2011;12:1–8. doi: 10.1186/gb-2011-12-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Genes G, Li W, Challa GS, Zhu H, Wei W. Recurrence of chromosome rearrangements and reuse of dna breakpoints in the evolution of the triticeae genomes. G3 Genetics. 2016;6:3837–3847. doi: 10.1534/g3.116.035089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martis MM, et al. Reticulate evolution of the rye genome. Plant Cell. 2013;25:3685–3698. doi: 10.1105/tpc.113.114553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang XQ, Röder MS. Molecular mapping of powdery mildew resistance genes in wheat: A review. Euphytica. 2004;137:203–223. doi: 10.1023/B:EUPH.0000041576.74566.d7. [DOI] [Google Scholar]
- 68.Zhang R, et al. Pm62, an adult-plant powdery mildew resistance gene introgressed from Dasypyrum villosum chromosome arm 2VL into wheat. Theor. Appl. Genet. 2018;131:2613–2620. doi: 10.1007/s00122-018-3176-5. [DOI] [PubMed] [Google Scholar]
- 69.Ma R, Yli-Mattila A, Pulli S. Phylogenetic relationships among genotypes of worldwide collection of spring and winter ryes (Secale cereale L.) determined by RAPD-PCR markers. Hereditas. 2004;140:210–221. doi: 10.1111/j.1601-5223.2004.01844.x. [DOI] [PubMed] [Google Scholar]
- 70.Andersen JR, Lubberstedt T. Functional markers in plants. Trends Plant Sci. 2003;8:554–560. doi: 10.1016/j.tplants.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 71.Brown JK, Jessop AC, Rezanoor NH. Genetic uniformity in barley and its powdery mildew pathogen. Proc. Royal Soc. Lond. Ser. B Biol. Sci. 1991;246:83–90. doi: 10.1098/rspb.1991.0128. [DOI] [Google Scholar]
- 72.Xue AG, et al. Virulence structure of Blumeria graminis f sp tritici, the causal agent of wheat powdery mildew, in Ontario, Canada, in 2018 and 2019. Canad. J. Plant Pathol. 2021 doi: 10.1080/07060661.2021.1915876. [DOI] [Google Scholar]
- 73.Liu N, et al. Virulence structure of blumeria graminis f sp tritici and Its genetic diversity by ISSR and SRAP profiling analyses. PLoS ONE. 2015;10:e0130881. doi: 10.1371/journal.pone.0130881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nieri D, Di Donato A, Ercolano MR. Analysis of tomato meiotic recombination profile reveals preferential chromosome positions for NB-LRR genes. Euphytica. 2017 doi: 10.1007/s10681-017-1982-5. [DOI] [Google Scholar]
- 75.Chen NWG, et al. Common bean subtelomeres are hot spots of recombination and favor resistance gene evolution. Front. Plant Sci. 2018 doi: 10.3389/fpls.2018.01185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gruner P, Schmitt AK, Flath K, Piepho HP, Miedaner T. Mapping and validating stem rust resistance genes directly in self-incompatible genetic resources of winter rye. Theor. Appl. Genet. 2021 doi: 10.1007/s00122-021-03800-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wehling P, et al. Leaf-rust resistance in rye (Secale cereale L.) 1 Genetic analysis and mapping of resistance genes Pr1 and Pr2. Theor. Appl. Genet. 2003;107:432–438. doi: 10.1007/s00122-003-1263-7. [DOI] [PubMed] [Google Scholar]
- 78.Van de Weyer AL, et al. A species-wide inventory of NLR genes and alleles in Arabidopsis thaliana. Cell. 2019;178:1260–1272. doi: 10.1016/j.cell.2019.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Steuernagel B, et al. The NLR-annotator tool enables annotation of the intracellular immune receptor repertoire. Plant Physiol. 2020;183:468–482. doi: 10.1104/pp.19.01273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mondragon-Palomino M, Meyers BC, Michelmore RW, Gaut BS. Patterns of positive selection in the complete NBS-LRR gene family of Arabidopsis thaliana. Genome Res. 2002;12:1305–1315. doi: 10.1101/gr.159402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Florence J, Vernaldi S, Maekawa T. Evolution and conservation of plant NLR functions. Front. Immunol. 2013;4:297. doi: 10.3389/fimmu.2013.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bittner-Eddy PD, Crute IR, Holub EB, Beynon JL. RPP13 is a simple locus in Arabidopsis thaliana for alleles that specify downy mildew resistance to different avirulence determinants in Peronospora parasitica. Plant J. 2000;21:177–188. doi: 10.1046/j.1365-313x.2000.00664.x. [DOI] [PubMed] [Google Scholar]
- 83.Liu X, et al. TaRPP13-3, a CC-NBS-LRR-like gene located on chr 7D, promotes disease resistance to wheat powdery mildew in Brock. J. Phytopathol. 2020;168:688–699. doi: 10.1111/jph.12949. [DOI] [Google Scholar]
- 84.Cheng J, et al. Genome-wide Identification and expression analyses of RPP13-like genes in barley. BioChip J. 2018;12:102–113. doi: 10.1007/s13206-017-2203-y. [DOI] [Google Scholar]
- 85.Li Y, et al. High levels of nucleotide diversity and fast decline of linkage disequilibrium in rye (secale cereale L.) genes involved in frost response. BMC Plant Biol. 2011;11:6. doi: 10.1186/1471-2229-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gupta PK, Rustgi S, Kulwal PL. Linkage disequilibrium and association studies in higher plants: present status and future prospects. Plant Mol. Biol. 2005;57:461–485. doi: 10.1007/s11103-005-0257-z. [DOI] [PubMed] [Google Scholar]
- 87.Ren, Y. et al. QTL analysis and nested association mapping for adult plant resistance to powdery mildew in two bread wheat populations. Front. Plant Sci.8 (2017). [DOI] [PMC free article] [PubMed]
- 88.Novakazi F, et al. You had me at “MAGIC”!: Four barley MAGIC populations reveal novel resistance QTL for powdery mildew. Genes. 2020;11:1512. doi: 10.3390/genes11121512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Arora S, et al. Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nat. Biotechnol. 2019;37:139–143. doi: 10.1038/s41587-018-0007-9. [DOI] [PubMed] [Google Scholar]
- 90.USDA. United States Department of Agriculture: wheat and barley DNA extraction protocol (96-well plate format), <https://www.ars.usda.gov/ARSUserFiles/60701500/SmallGrainsGenotypingLaboratory/Protocols/wheat%20and%20barleyDNA%20extraction_original.pdf> (2006).
- 91.Pallotta, M. A. et al. Marker assisted wheat breeding in the southern region of Australia. Proceedings of the Tenth International Wheat Genetics Symposium, 789–791 (2003).
- 92.RStudio Team. Rstudio: integrated development for R. RStudio, Inc., Boston, <http://www.rstudio.com > (2015).
- 93.R Core Team. R a language and environment for statistical computing. R foundation for statistical Computing, Vienna, Austria, <https://www.R-project.org/> (2021).
- 94.NCBI. National Center for Biotechnology Information, <https://www.ncbi.nlm.nih.gov> (2021).
- 95.Granato ISC, et al. snpReady: A tool to assist breeders in genomic analysis. Mol. Breed. 2018;38:102. doi: 10.1007/s11032-018-0844-8. [DOI] [Google Scholar]
- 96.Afghan E, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analysis. Nucleic Acids Res. 2018;46:537–544. doi: 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rasche H, Hiltemann S. Galactic circos: User-friendly circos plots within the galaxy platform. GigaScience. 2020;9:gaa06565. doi: 10.1093/gigascience/giaa065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hiltemann, S., Rasche, H. & Gallardi, C. Visualization with circos (galaxy training materials) <https://training.galaxyproject.org/training-material/topics/visualisation/tutorials/circos/tutorial.html> (2021).
- 99.Wickham H. ggplot2. Wiley Interdisciplin. Rev. Comput. Stat. 2011;3:180–185. doi: 10.1002/wics.147. [DOI] [Google Scholar]
- 100.Lipka AE, et al. GAPIT: Genome association and prediction integrated tool. Bioinformatics. 2012;28:2397–2399. doi: 10.1093/bioinformatics/bts444. [DOI] [PubMed] [Google Scholar]
- 101.Huang M, Liu X, Zhou Y, Summers RM, Zhang Z. BLINK: A package for the next level of genome-wide association studies with both individuals and markers in the millions. Gigascience. 2019 doi: 10.1093/gigascience/giy154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Steuernagel B, Jupe F, Witek K, Jones JD, Wulff BB. NLR-parser: Rapid annotation of plant NLR complements. Bioinformatics. 2015;31:1665–1667. doi: 10.1093/bioinformatics/btv005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jones P, et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Paradis E, Schliep K. ape 50: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2019;35:526–528. doi: 10.1093/bioinformatics/bty633. [DOI] [PubMed] [Google Scholar]
- 105.Letunic I, Bork P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019;47:256–259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.UniProt C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:480–489. doi: 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Toparslan E, Karabag K, Bilge U. A workflow with R: Phylogenetic analyses and visualizations using mitochondrial cytochrome b gene sequences. PLoS ONE. 2020;15:e0243927. doi: 10.1371/journal.pone.0243927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bodenhofer U, Bonatesta E, Horejs-Kainrath C, Hochreiter S. msa: An R package for multiple sequence alignment. Bioinformatics. 2015;31:3997–3999. doi: 10.1093/bioinformatics/btv494. [DOI] [PubMed] [Google Scholar]
- 109.Charif, D. & Lobry, J. R. SeqinR 1.0–2: A contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis. Struct. Approach. Sequence Evolut. 207–232 (2007).
- 110.Yu G. Using ggtree to visualize data on tree-like structures. Current Protocols Bioinform. 2020;69:e96. doi: 10.1002/cpbi.96. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.