

Abstract
Introduction
Residual pluripotent stem cells (PSC) within differentiated populations are problematic because of their potential to form tumors. Simple methods to reduce their occurrence are needed.
Methods
Here, we demonstrate that control of the oxygen partial pressure (pO2) to physiological levels typical of the developing embryo, enabled by culture on a highly oxygen permeable substrate, reduces the fraction of PSC within and the tumorigenic potential of differentiated populations.
Results
Differentiation and/or extended culture at low pO2 reduced measured pluripotency markers by up to four orders of magnitude for mouse PSCs (mPSCs). Combination with cell sorting increased the reduction to as much as six orders of magnitude. Upon implantation into immunocompromised mice, mPSCs differentiated at low pO2 either did not form tumors or formed tumors at a slower rate than at high pO2.
Conclusions
Low pO2 culture alone or in combination with other methods is a potentially straightforward method that could be applied to future cell therapy protocols to minimize the possibility of tumor formation.
Keywords: Pluripotent stem cells, Differentiation, Cell therapy, Teratoma, Oxygen, Hypoxia, Transplantation
Introduction
Pluripotent stem cells (PSCs), including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), have immense potential due to their ability to form many clinically-relevant cell types.31 However, because of their pluripotency, ESC and iPSC can form teratomas, tumors composed of derivatives of the three germ layers, and are a s safety risk if implanted into humans in their undifferentiated state. The tumorigenicity of PSC could be a major obstacle for the safe translation of stem cell biology to regenerative medicine. The presence of any number of PSC is a potential tumor formation risk because as few as two mouse ESC (mESC) have been shown to form tumors in immunocompromised mice.17 There have been no reported cases of undifferentiated PSC being implanted into humans, but in one case study, fetal neural stem cells implanted into a human caused the formation of a glioneuronal neoplasm, derived from donor cells.1 Hence, preclinical testing of any stem cell-derived product for tumor formation is essential.9
Differentiation of PSC reduces the risk of tumor formation.4 However, residual PSC can still persist, even after being subjected to a differentiation protocol, and cause tumor formation upon implantation.19 While studies have reported that differentiation of PSC completely avoided tumor appearance,14,16 these experiments were not designed to sensitively detect residual PSC. The location of transplantation can affect tumor incidence and formation rates in animal models, with injections into subcutaneous and intramuscular tissue being the most convenient, sensitive, and reliable.13,17 Because humans will require a large number of cells for treatment, even a small number of residual PSCs pose a safety risk. Although the incidence of tumor formation can be mitigated by efficient differentiation protocols,14 further reduction of residual PSCs is desired for clinical translation.
A variety of approaches have been examined for this purpose. These include culture conditions harmful to PSCs,30 cytotoxic antibodies,7,28 antibodies or other affinity ligands specific to cell surface markers on PSC that enable positive or negative selection with fluorescent-activated (flow cytometry) or magnetic-activated cell sorting (FACS or MACS),12,29 and genetic engineering in order to selectively kill residual PSCs while retaining the differentiated cells.3,20,25,27 Almost all previous methods suffer from limitations of effectiveness, high cost, and/or reproducibility issues. None are currently in clinical use, and there is need for simple, cost-effective, and efficient methods.
The oxygen partial pressure (pO2) under which embryonic cells develop in vivo, and PSC are cultured in vitro, is an influential parameter. During normal embryonic development, cells are exposed to pO2 much lower than atmospheric (160 mmHg), which influences many aspects of embryogenesis, such as tracheal development, cardiovascular and bone morphogenesis, and adipogenesis. Cellular signaling as a consequence of being exposed to differentiation levels of pO2 often occurs via hypoxia inducible factor-1α (HIF-1α).21,26 Cell culture in vitro is commonly performed in a humidified air phase consisting of 95% air and 5% CO2, resulting in a gas phase partial pressure of oxygen of 142 mmHg (pO2gas), which is much higher than the pO2 levels at which HIF-1 would be enzymatically active. While estimates and direct measurements of actual physiological oxygen levels are much lower than 142 mmHg, this value varies depending on multiple parameters but is generally accepted to be between approximately 36 and 7 mmHg.22
Teratomas form only rarely during embryonic development. Differentiation of PSC in vitro at low pO2 affects the generation of several cell types, such as neurons, hematopoietic progenitors, endothelial cells, cardiomyocytes, and chondrocytes. We have previously shown that pO2 also affects many aspects of pluripotency, such as expression of the pluripotency genes Oct4, Nanog, and Sox2, while not causing PSCs to exit pluripotency with culture in a self-renewal media.24 Others have shown that reprogramming efficiency of mouse and human fibroblasts to iPSC is increased under low pO2.32,33 Given the effect of hypoxia on differentiation and the lack of development of teratomas in the embryo, we hypothesized that the risk of teratoma development during differentiation and culture of PSC might be reduced if carried out under low pO2 conditions.
Here, we show that differentiation and extended culture of mESCs and mouse iPSCs (miPSCs), both containing a green fluorescence protein (GFP) transgene driven by an Oct4 promoter (denoted as Oct4-GFP+), at low pO2 drastically reduced the fraction and number of residual PSC, which, upon implantation into nude mice, slowed or eliminated the appearance of teratomas. Whereas in virtually all previous studies of oxygen effects in culture, reported gas-phase pO2gas differed from the value at the cell because of diffusion gradients,21 these studies were enabled by culture on a high oxygen permeability surface that permitted precise control of the pO2 at the cell surface. Low pO2 culture was combined with FACS removal of residual PSCs to reduce the fraction of residual PSCs much more than either method alone. Recovered residual Oct4-GFP+ mESc after being subjected to the differentiation protocol, had in vitro characteristics similar to those of undifferentiated mESCs.
Materials and Methods
pO2 Control During In Vitro Culture
Oxygen was controlled with gas containing 5% CO2 and 20%, 5%, or 1% O2, balance N2 (Airgas), corresponding to gas-phase oxygen partial pressure (pO2gas) of 142, 36, and 7 mmHg when humidified, flowing in chambers at 37 °C. Cells were cultured under differentiating conditions in custom-made silicone rubber membrane dishes (Specialty Manufacturing, Saginaw, MI, USA) with high O2 permeability. Cellular attachment was promoted by 24-h incubation of 2 µg/mL fibronectin (F1141; Sigma-Aldrich).
Undifferentiated mESC and miPSC Culture
Undifferentiated R1 mESC and miPSC with an inserted GFP coding sequence driven by an Oct4 promoter (Oct4-GFP mESC)15,34 and R1 mESC with a homozygous HIF-1α gene knockout (HIF-1α−/− mESC)5 were propagated without mouse embryonic fibroblasts (MEFs) in Dulbecco’s modified Eagles medium (DMEM; SCRR-2010; American Type Culture Collection (ATCC), Manassas, VA, USA) supplemented with 10% (v/v) fetal bovine serum (FBS; SCRR 30-2020; ATCC), 2 mM L-alanyl-L-glutamine (SCRR 30-2115; ATCC), 50 U/mL penicillin, and 50 µg/mL streptomycin (15070; Invitrogen, Carlsbad, CA, USA), 1x MEM non-essential amino acids (SCRR 30-2116; ATCC), 100 µM 2-mercaptoethanol (M7522; Sigma-Aldrich; St. Louis, MO, USA), and 103 units/mL leukemia inhibitory factor (LIF; LIF2005; Millipore, Billerica, MA, USA).
Differentiation of mESC and miPSC
mESC, miPSC, and HIF-1α-/- mESC were differentiated using a protocol that enhanced differentiation to cardiomyocytes at reduced pO2.23 Embryoid bodies (EBs) were formed in propagation medium without LIF using 500 cells per 20-µL hanging drop. After 2 days, 30 EBs per well were transferred to fibronectin-coated (F1141; Sigma) silicone rubber membrane-based 24-well plates filled with 2 mL of propagation medium without LIF. After an additional 3 d, medium was replaced with 2.5 mL of serum-free medium, consisting of 50% (v/v) DMEM (90-133-PB; Mediatech) and 50% (v/v) Ham’s F-12 (10-080-CV; Mediatech) supplemented with 0.75% (w/v) sodium bicarbonate (25-035-CI; Mediatech), 9 mM glucose (G8769; Sigma-Aldrich), 5 µg/mL human insulin (I9278; Sigma-Aldrich), 50 µg/mL holo transferrin (T1283; Sigma-Aldrich), 31.2 nM sodium selenite (S9133; Sigma-Aldrich), 50 units/ml penicillin, and 50 µg/ml streptomycin. Cells were differentiated for 10 d. As noted, pO2gas was kept constant throughout differentiation in some experiments and was varied in others, and 0.2 mM ascorbic acid (A4034; Sigma-Aldrich) was included in the medium.
Extended culture of differentiated PSC
After the 10-d (mESC, miPSC, or HIF-1α-/- mESC) differentiation protocol, cells were subjected to additional extended culture for up to 80 d. This medium consisted of 50% (v/v) DMEM (90-133-PB; Mediatech) and 50% (v/v) Ham’s F-12 (10-080-CV; Mediatech) supplemented with 0.75% (w/v) sodium bicarbonate (25-035-CI; Mediatech), 9 mM glucose (G8769; Sigma-Aldrich), 5 µg/mL human insulin (I9278; Sigma-Aldrich), 50 µg/mL holo transferrin (T1283; Sigma-Aldrich), 31.2 nM sodium selenite (S9133; Sigma-Aldrich), 50 units/ml penicillin, and 50 µg/ml streptomycin. In some experiments, pO2gas was identical to pO2gas during differentiation, while in others, pO2gas was changed. Cell samples were acquired for analysis by detachment with a 5-min incubation of 0.25% trypsin at 37 °C.
Flow cytometric analysis of MF-20 immunostained cells
The fraction of MF-20+ cells in dispersed cells was acquired with flow cytometry. Samples were fixed by 20 min incubation in 1% (w/v) paraformaldehyde in PBS. Fixed cells numbering 3x105 were permeabilized in 0.5% saponin (S-4521; Sigma-Aldrich) for 10 min, incubated for 30 min in 2% (v/v) FBS in PBS to block non-specific binding, and incubated at room temperature with primary antibody sarcomeric myosin heavy chain (MF-20; MF-20 supernatant; Developmental Studies Hybridoma Bank, Iowa City, IA, USA) diluted 1:10 in 2% FBS solution for 1 h. Samples were then incubated with goat anti-mouse phycoerythrin-conjugated secondary antibody (115-116-146; Jackson ImmunoResearch; West Grove, PA) diluted 1:250 in 2% FBS solution for 30 min at room temperature in the dark, washed 3x in 2% FBS solution, and staining data were measured using a Guava PCA flow cytometer (Millipore).
Flow Cytometric Analysis of Oct4-GFP Expression
Oct4-GFP expression was measured with a flow cytometer (FACScan; Becton Dickinson). A negative control of R1 mESC without the Oct4-GFP reporter was differentiated under the same conditions as the Oct4-GFP cells, and a gating threshold was set so that all of these cells were counted as negative for Oct4-GFP. Between 105 and 106 cells total were analyzed. The fraction of cells expressing Oct4-GFP (Oct4-GFP+) was calculated as the number of cells above the threshold divided by the total number (GFP+ plus GFP-) of cells in the sample. This was used to estimate the number of detected cells in some subsequent experiments by multiplying this measured fraction of Oct4-GFP+ cells by the total cell number for the experiment. In cases where no cells were detected in 106 cells, ‘none detected’ was reported, but that finding does not mean that an Oct4-GFP cell would not be detected with a higher cell count.
Quantitative Real-Time PCR
Total RNA was isolated (74104, Qiagen, Valencia, CA), cDNA synthesized (4368814, Applied Biosystems, Foster City, CA), and qPCR performed on a Fast qPCR System (7900HT, Applied Biosystems) using Power SYBR Green PCR Master Mix (4367659, Applied Biosystems). 28S ribosomal RNA (rRNA) was used as an endogenous control. Primer sequences used for 28S rRNA, Oct4, Nanog, Nestin, and Nkx2.5 have been previously reported.24 Additional primers sequences used (in order of forward and reverse primer and 5′ to 3′) were Brachyury T TCCCGGTGCTGAAGGTAAAT and CCGTCACGAAGTCCAGCAA, Foxa2 TCAAGGCCTACGAACAGGTCAT and GCCCGCTTTGTTCGTGACT, cTnT GCTTCCGCTGTCCAGAGACT and CACCAAGTTGGGCATGAAGA, cardiac-α-Actin GCTTCCGCTGTCCAGAGACT and GCTTCCGCTGTCCAGAGACT, Mef2c TGCTGGTCTCACCTGGTAAC and ATCCTTTGATTCACTGATGGCAT.
Cell Enumeration
Cells were lysed by vigorous vortexing in 1% (v/v) Triton X-100 (T-9284; Sigma-Aldrich) and 0.1 M citric acid (0627; Mallinckrodt Specialty Chemicals Co., Paris, KY, USA), stained with Guava ViaCount (4000-0041; Millipore), and counted using a Guava PCA flow cytometer (Millipore).
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde and then incubated for 45 min in 3% (v/v) donkey serum (DS; D9663; Sigma-Aldrich) and 0.1% Triton X-100. Cells were incubated overnight at 4 °C with primary antibodies against Oct4 (sc-9081; Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted 1:100, Nanog (IHC-00205; Bethyl Laboratories, Montogomery, TX, USA) diluted 1:100, SSEA-1 (MC-480; DSHB) diluted 1:10, Foxa2 (sc-6554; Santa Cruz Biotechnology) diluted 1:200, Nestin diluted 1:50, cardiac troponin T (cTnT; MS-295-P1; NeoMarkers, Fremont, CA, USA) diluted 1:50, or Nkx2.5 (sc-8697; Santa Cruz Biotechnology) in 3% DS, 0.1% Triton X-100 solution, washed once with PBS, and incubated with secondary donkey AlexaFluor 488 (green) or 594 (red) antibodies (Invitrogen) against the appropriate species diluted 1:250 in 3% DS, 0.1% Triton X-100 solution for 2 h in the dark. Images were acquired with an Axiovert 200 fluorescence microscope).
Teratoma Appearance in Mice
Samples containing 105 (mESC, miPSC, or HIF-1α−/− mESC) in 200 μL of 50% (v/v) Matrigel (BD Biosciences) and 50% (v/v) serum-free differentiation medium were injected subcutaneously into the lower flank of female nude mice, 43–56 days old (Charles River Laboratories International), using a 23G needle in a laminar-flow hood. Mice were visually observed every 3–4 days for the appearance of masses under the skin. The protocol was similar to one designed for biosafety testing of stem cell products, for which implantation of only two ES cells was sufficient to form detectable tumors.17 All procedures were approved by the MIT Committee on Animal Care.
Histology
Tumors were excised, fixed overnight in 4% (w/v) paraformaldehyde (36606; Alfa Aesar, Ward Hill, MA, USA), paraffin embedded, sectioned, and stained with hematoxylin and eosin (H&E) at the Division of Comparative Medicine (DCM) at MIT. Stained histological sections were analyzed by an American College of Veterinary Pathologist board-certified pathologist.
Fluorescence-Activated Cell Sorting (FACS)
mESC that underwent 10 days differentiation and an additional 20 days extended culture were detached with trypsin and then sorted (BD FACSAria II, Becton Dickinson). mESC were sorted into Oct4-GFP+ and Oct4-GFP- populations or on the basis of expression of the mESC surface marker stage-specific embryonic antigen-1 (SSEA-1). Cells were treated with a PE-conjugated anti-SSEA-1 antibody (12-8813; eBioscience, San Diego, CA, USA) before sorting into SSEA-1+ and SSEA-1- populations. We adjusted parameters to be extremely conservative to minimize the chances of false negatives.
Recovery of Undifferentiated Cells from Differentiated Population
After 10 days of differentiation and 20 days of extended culture of the Oct4-GFP mESCs, FACS was used to collect residual Oct4-GFP+ cells, which were then plated onto polystyrene cell culture flasks and cultured as undifferentiated mESCs for 2 weeks, as described earlier. These cells were then subjected to the 10-day differentiation protocol described earlier.
Phase Contract Microscopy
Images were acquired with an Axiovert 200 fluorescence microscope (Carl Zeiss, Oberkochen, Germany).
Statistical Analysis
Unpaired Student’s t-test and ANOVA followed by Tukey’s post-hoc tests were used where indicated with p < 0.05 considered statistically significant. Data is presented as mean ± SD unless otherwise noted.
Results
Differentiated and Culture at low pO2 Increases Differentiation of PSCs to Cardiomyocytes and Reduces Residual PSCs
We differentiated mESC at atmospheric and physiological pO2 to examine cardiomyocyte differentiation (Fig. 1). Differentiating mESC at various, constant pO2gas for 11 days resulted in a large increase in the fraction of cells expressing the cardiomyocyte marker MF-20 with decreasing values of pO2gas that was reproducibly observed in 7 additional independent experimental runs (Fig. 1a). The highest number fraction of MF-20+ cells was 31% with a pO2gas of 7 mmHg, compared to 23% and 9% at 36 and 142 mmHg, respectively. Culture at 7 mmHg resulted in a substantial decrease in total cell number relative to 36 and 142 mmHg, respectively, both of which had comparable cell numbers (Fig. 1b). As a result, the number of MF-20+ cells was higher at 36 mmHg by a factor of 2.3 and 2.9 compared to 142 and 7 mmHg, respectively, resulting in 55 MF-20+ cells being generated per initial undifferentiated cell. The expression of the cardiac marker genes cTnT, Mef2c, and Nkx2.5 were also higher at low compared to high pO2gas (Figs. 1d–1f). Low pO2-differentiated mESC immunostained positive for cTnT, which were striated and demonstrated sarcomeric organized (Fig. 1g) and for Nkx2.5 (Fig. 1h). While this protocol was developed for generating cardiomyocytes, other differentiated cells are also present, and therefore throughout this study we generally refer to these cells as differentiated cells.
Figure 1.

Low pO2 increases mESC differentiation to cardiomyocytes. mESC were differentiated 11 days culture at 142, 36, or 7 mmHg pO2gas with ascorbic acid. (a) Fraction of MF-20+ cells from mESC acquired with flow cytometry (n = 7). (b) The total cell number per well from mESC acquired with nuclei counts (n = 7). (c) The number of MF-20+ cells from mESC calculated by multiplying the fraction of MF-20+ cells (a) and the total cell number (b) (n = 7). (d)–(f) Gene expression of cTnT, Mef2c, and Nkx2.5 acquired with real-time PCR for (n = 4). (g) and (h) En face fluorescence images of mESC differentiated at 7 mmHg pO2gas and stained against cTnT (green) and with DAPI (blue) and against Nkx2.5 (green). * indicates statistical difference (p < 0.05) compared to 142 mmHg (black bar), and # indicates statistical difference compared to 36 mmHg (grey bar).
Next, we examined the effects of physiological pO2 on expression of residual PSC markers in vitro by differentiating and performing extended culture on Oct4-GFP mESCs, Oct4-GFP miPSCs, and HIF-1α-/- mESCs at various pO2gas. During the 10-d differentiation, the fraction of mESC and miPSC expressing Oct4-GFP decreased rapidly to a range of < 10−1 to nearly 10−4. This reduction was larger at 36 and 7 than at 142 mmHg, and the difference was more pronounced for miPSC (Figs. 2a and 2b). We hypothesized that these cells were residual PSC that could develop into tumors upon implantation into animals.
Figure 2.

Expression of pluripotency markers decrease substantially with time during differentiation and extended culture under reduced oxygen. Cells were differentiated for 10 days followed by up to 80 days of extended culture (90 days total). (a) Flow cytometric quantification of the fraction and number of Oct4-GFP+ for differentiating mESCs and miPSCs at 142, 36, and 7 mmHg pO2gas (n = 3). (b) Representative en face bright field and Oct4-GFP images of cell aggregates after extended culture at 142 or 7 mmHg pO2gas. (c) Relative expression of Oct4 and Nanog mRNA with time in mESCs, miPSCs, and HIF-1α−/− mESCs at 142, 36, and 7 mmHg pO2gas (n = 3) measured with qPCR.
During extended culture for an additional 80 days, the fraction of Oct4-GFP+ cells changed little at 142 mmHg, continued to decrease at reduced pO2 to levels nearly as low as 10−6, and was not statistically different between 36 and 7 mmHg (Figs. 2a and 2b). The fraction of Oct4-GFP+ cells at 142 mmHg was greater than that at 7 mmHg by factors of 1.3 × 103 and 2.3 × 104 for mESC and miPSC, respectively. Culture at reduced pO2 similarly lowered the number of residual PSCs. For mESC populations, the number of Oct4-GFP+ cells dropped from 2 × 104 at d 10 to 4 × 103 at day 90 for culture at 142 mmHg and from 4 × 103 to 4 at 7 mmHg. Thus, culture of mESC at 7 mmHg led to a 1000-fold greater reduction from day 10 to day 90 in the number of Oct4-GFP+ cells compared to culture at 142 mmHg and to a 5000-fold reduction compared to the numbers of Oct4-GFP+ cells present immediately after differentiation at 142 mmHg. Trends for miPSC were similar, with the corresponding fold reduction values being 1200 and 14,000, respectively. The expression of Oct4 and Nanog mRNA was also lowered with culture at reduced pO2 (Fig. 2c). This trend was consistent with reduced Oct4-GFP measurements (Figs. 2a and 2b). To test if the reduction in these pluripotency genes required HIF-1α, we differentiated and performed extended culture of the HIF-1α−/− mESC line. We observed that Oct4 and Nanog relative gene expression were not different at 142 and 7 mmHg pO2gas (Fig. 2c), indicating a role of HIF-1α in the benefits of reduced pO2gas.
The use of different pO2gas values during differentiation and extended culture was investigated by differentiating mESC for 10 days at either 142 or 7 mmHg, followed by 20 days extended culture at the same or the other pO2gas (Fig. 3). After 10 days of differentiation at 142 mmHg, the fraction of Oct4-GFP+ cells was 1.4 × 10−2. Extended culture for 20 days at 142 mmHg did not change the fraction of Oct4-GFP+ cells. However, extended culture at 7 mmHg decreased the fraction to 5 × 10−4. Differentiation for 10 days at 7 mmHg resulted in a fraction of Oct4-GFP+ cells of 6 × 10−4, a factor of 22 lower than at 142 mmHg. Extended culture for 20 days at 7 mmHg further decreased the fraction to 3 × 10−5. However, extended culture at 142 mmHg increased the mean fraction to 3 × 10−3. This data demonstrates that differentiation can be performed at a pO2gas higher than 7 mmHg and still get the benefit of reduced residual PSCs by extended culture at low pO2.
Figure 3.
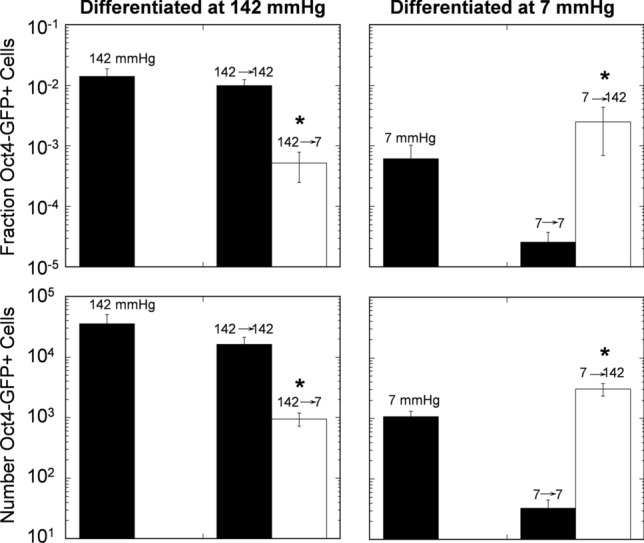
Expression of Oct4-GFP in mESC decreased with extended culture at 7 mmHg after differentiation at 142 mmHg pO2gas. Shown are flow cytometric quantification of the fraction and number of Oct4-GFP of mESCs differentiated at 142 or 7 mmHg pO2gas for 10 days and then subjected to 20 days extra culture at the same (black bar) or the other (white bar) pO2gas (n = 3). *indicates statistical difference compared to the black bar of the same time as determined by t-test.
Differentiated and Culture at Low pO2 delays tumor Appearance After Implantation
We next examined the effects of reducing the fraction and number of PSCs by in vitro differentiation for 10 days and extended culture for 20 days (30 days in total) at 142, 36, or 7 mmHg pO2gas on the timing of teratoma formation in vivo (Fig. 4). Undifferentiated PSCs were used as a positive control and formed tumors 14 days (mESC, miESC) or 20 days (HIF-1α-/- mESC) after implantation (Fig. 4a). Differentiation followed by extended culture at the same pO2gas increased the time for the first appearance to 17 days at 142 mmHg (mESC and miESC) and to 31 and 35 days for 36 mmHg and 7 mmHg, respectively (mESC) (Fig. 4a). Differentiated HIF-1α−/− mESCs received no benefit from reduced pO2 (Fig. 4a). Tumors were excised from euthanized mice, and tissue sections were routinely examined histopathologically (Fig. 4b). The tumors contained derivatives of all three germ layers and were identified as teratomas. Together, these transplantation data are consistent with the in vitro data from Fig. 2 and demonstrate that extended hypoxic culture reduces the rate of teratoma formation for mPSCs.
Figure 4.

Reduced pO2 culture slowed the appearance of teratoma formation in mice. (a) Survival curve of the emergence of teratomas in immunocompromised mice transplanted with mESCs, miPSCs, or HIF-1α−/− mESCs either undifferentiated (n = 8) or differentiated for 10 days followed by extended culture for 20 days at 142, 36, 7 mmHg (n = 8). Matrigel alone (n = 6) served as a negative control. (b) Representative H&E-stained tissue section of a tumor derived from mESC differentiated at 7 mmHg pO2gas displaying (i) small glandular structure, (ii) hyaline cartilage, and (iii) neuroepithelial structures.
Differentiated and Culture at Low pO2 Combined with FACS Reduces Residual PSCs and Delays Tumor Appearance After Implantation
In another set of experiments, we combined low pO2gas differentiation with FACS based on Oct4-GFP and SSEA-1 expression with mESC (Fig. 5). Sorting based on Oct4-GFP expression decreased the fraction and number of residual Oct4-GFP+ cells in the Oct4-GFP- population by a factor of approximately 50 for extended culture at 142 mmHg and to zero at 7 mmHg, as determined by subsequent flow cytometric analysis. However, because sorting based on GFP expression required genetic modifications of the cells, which might be problematic for clinical application, we also investigated low pO2 culture combined with FACS based on mESC cell surface markers stained with a phycoerythrin (PE)-conjugated anti-SSEA-1 antibody. Sorting based on SSEA-1 expression decreased the fraction and number of residual Oct4-GFP+ cells in the Oct4-GFP- population by factors of 16 and 34 for extended culture at 142 and 7 mmHg, respectively, as determined by subsequent flow cytometric analysis. mESC exposed to both differentiation and extended culture at 7 mmHg followed by FACS removal of SSEA-1+ cells had a fraction of Oct4-GFP+ cells that were lower by a factor of 1000 (p < 0.001) compared to unsorted cells cultured at 142 mmHg.
Figure 5.
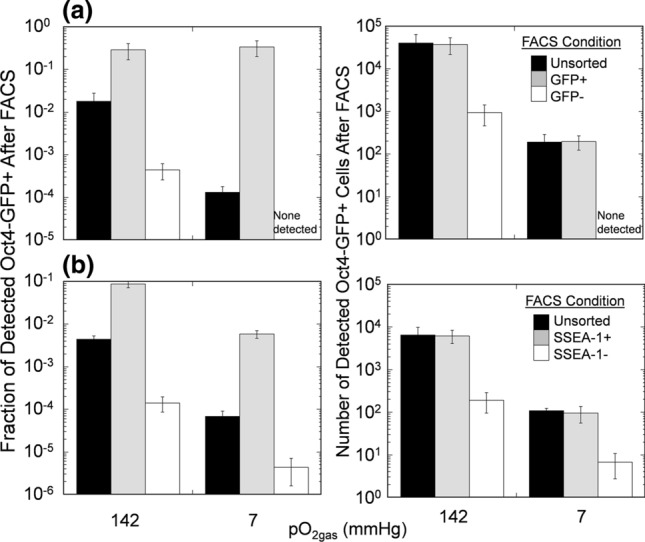
Differentiation at reduced pO2gas combined with subsequent sorting further reduces residual undifferentiated mPSCs. mESC cells underwent 10-day differentiation at 7 mmHg followed by 20-day extended culture at the indicated pO2gas, then were sorted based on (a) Oct4-GFP expression or (b) SSEA-1 expression (n = 3). Data shown are measurements of the fraction (left) and number (right) of cells that were Oct4-GFP+ within the resulting population after FACS.
After FACS, unsorted cells and sorted populations (Oct4-GFP+ and Oct4-GFP- or SSEA-1+ and SSEA-1-), each differentiated and then cultured at either 142 or 7 mmHg, were implanted into immunocompromised mice (Fig. 6). Each injectate of an unsorted cell population contained, on average, 1770 and 13 Oct4-GFP+ cells at 142 and 7 mmHg, respectively; each injectate of a sorted Oct4-GFP+ population contained 1729 and 13 Oct4-GFP+ cells at 142 and 7 mmHg, respectively; and each injectate of a sorted Oct4-GFP- population contained 41 and 0 Oct4-GFP+ cells at 142 and 7 mmHg, respectively, out of 105 cells total in each preparation. For cells cultured at 142 mmHg, tumors appeared at approximately the same time, 18 days after implantation, in both unsorted and sorted GFP+ populations; The sorted Oct4-GFP- population had an additional 11-day delay in tumor appearance (total delay 29 days). For 7 mmHg, initial tumor appearance occurred 34 days after implantation for the unsorted cells and 8 days later (total delay 42 days) with the sorted GFP+ population; 50% of the sorted GFP+ population never formed tumors. With the sorted Oct4-GFP- population, no tumors formed during 210 days of observation.
Figure 6.
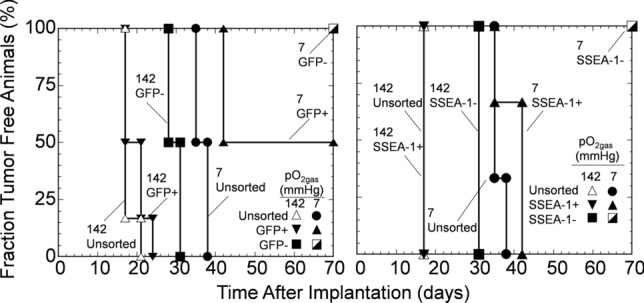
Transplantation of mPSCs differentiated and cultured at low pO2, followed by FACS to remove residual undifferentiated mPSCs, eliminated appearance of teratoma formation. Data shown is survival curve of the emergence of teratomas in immunocompromised mice transplanted with mPSCs that underwent both differentiation and extended culture at either 142 or 7 mmHg pO2gas and were then sorted based on Oct4-GFP expression (left) or SSEA-1 expression (right) (n = 6). Data points placed at the upper right corner denote no tumors formed with the studied population over 210 days, after which observation stopped.
Similar tumor formation trends observed with Oct4-GFP sorting were seen in experiments based on sorting for SSEA-1 expression after differentiation and extended culture at 142 or 7 mmHg being (Fig. 6). Animals that received unsorted or SSEA-1+ sorted populations from 142 mmHg formed tumors sooner than animals that received cells from 7 mmHg or SSEA-1-. Cells cultured at 7 mmHg then sorted into SSEA-1- populations formed no tumors during 210 days of observation. On average, 0 detected Oct4-GFP+ cells from the 7 mmHg/SSEA-1- cell culture condition were transplanted into mice, as the frequency of residual Oct4-GFP+ cells was less than 1 in 105 (Fig. 5) and only 105 cells total were transplanted into the mice. Taken together, combining low pO2 culture with a cell sorting strategy to further remove residual undifferentiated PSCs resulted in no observed teratomas.
Residual PSCs After Undergoing Differentiation and Extended Culture Resemble Undifferentiated PSCs
Lastly, we further investigated residual Oct4-GFP+ mESC by removing Oct4-GFP+ cells from differentiated mixtures and culturing them in self-renewal media as undifferentiated mESCs (Fig. 7). These cells continued to express Oct4-GFP in a manner similar to undifferentiated mESC before differentiation and morphologically resembled undifferentiated mESC (Figs. 7a and 7b). They expressed the protein pluripotency markers Oct4, Nanog, and SSEA-1 (Fig. 7c) and could be induced to differentiate in vitro and express markers of all three germ layers (Fig. 8). Taken together, the data indicate that residual Oct4-GFP+ cells sorted from differentiated populations have the same characteristics as mESC that have never been exposed to differentiation conditions.
Figure 7.

Residual undifferentiated mPSCs were recovered from differentiation cell culture and reestablished as an undifferentiated mPSC population in self-renewal media. (a) Histogram of the distribution of Oct4-GFP fluorescence intensity per cell as determined with flow cytometry. Cell populations characterized were (I) Oct4-GFP+ mESC that have never undergone differentiation and extended culture, (II) Oct4-GFP+ cells sorted with FACS from mESC that have undergone 10-day differentiation and 20-day extended culture at 142 mmHg, and (III) Oct4-GFP+ cells cultured similar to case II, but at 7 mmHg. (b) Representative en face bright field image of mESC colony from cell population II. (c) Representative en face images from cell population II immunostained for pluripotency markers.
Figure 8.

Residual undifferentiated mPSCs recovered and reestablished as an undifferentiated mPSC population were capable of differentiation. (a) Relative gene expression of differentiation markers of the three cell populations identified in Fig. 6 after 10 days of differentiation (n = 3). (b) Representative en face immunostaining images of cells of cell population III after differentiation.
Discussion
We performed a quantitative study on the effects of low pO2 on the risk of tumor development by residual PSCs with accurate control of cellular pO2 by culture on highly oxygen-permeable silicone rubber membrane dishes. Differentiation and extended culture of both mESCs and miPSCs at physiological pO2 reduced residual PSCs in vitro. The order of magnitude of this reduction ranged from 102 to 104, depending on the duration of low pO2 culture and the cell type. This benefit could be enhanced when combined with FACS removal of residual PSCs. Upon transplantation into nude mice, the rate of tumor formation was reduced with prior extended culture of cells at low pO2, and no tumors were observed when low pO2 was combined with FACS (Fig. 9).
Figure 9.

Schematic diagram of steps in process. PSCs are subjected to 10 days of differentiation that results in a mixture of differentiated and residual PSCs. Extended culture at low pO2 reduces this residual PSC population. These cells are then transplanted into mice, with an optional FACS step to further reduce residual PSCs and thereby reduce the incidence of teratoma formation.
Differentiation could be performed at any pO2 followed by an extended culture period at low pO2 to reduce the fraction of Oct4-GFP+ cells. Modulation of pO2 during differentiation of PSCs benefits the generation of different cell types, with the extent of benefit dependent on O2 concentration.10,11,23 Separation of the steps allows differentiation at the optimal pO2 for that cell type and extended culture at a different pO2 so as to optimally reduce the fraction of residual PSCs. However, a limitation of this study is that we did not test differentiation and extended culture across a range of differentiated cell types and differentiation protocols, as it is possible that some cell types or protocols may not be compatible with the residual PSC reduction methods demonstrated in this study.
The mechanism of PSC reduction at low pO2 is not understood. Low pO2 could cause residual PSC to differentiate or die. Female hESC derived under low pO2 maintain two active X chromosomes and upon culture at atmospheric pO2, irreversibly silence one X chromosome.18 However, in those studies, cells were cultured under self-renewal conditions, which are very different than the culture conditions used in this study during differentiation and extended culture. Elucidating the mechanism by which low pO2 reduces residual PSCs could allow for improved methodologies and provide further insights into the role of oxygen in stem cell identity. In addition, the aggregate-nature of the differentiation procedure from some stages of the process created differential cellular pO2 exposure. This likely creates cellular heterogeneity in cellular pO2 exposure, which could affect translation of these findings that need to be taken into account.
Low pO2 culture can be used on its own or in conjunction with any other method to reduce or eliminate residual PSCs. In this study, combination of low pO2 culture with FACS removal of SSEA-1-expressing residual PSCs with a commercially available antibody was more effective than either method alone. Low pO2 culture alone or in combination with other methods is a potentially straightforward method that could be applied to future cell therapy protocols to minimize the possibility of tumor formation, particularly as manufacturing processes continue to increase in complexity.2 As technologies using PSC differentiation increase in efficacy in animal models and immune modulatory strategies are developed,6,8,14 translational to the clinic will need to continue to ensure safety of the final cell product and also manage the costs associated with necessary technologies, such as cell sorting. Extension of this approach to human PSCs and use of current directed differentiation protocols would be the immediate next steps towards translating extended low oxygen culture. In addition, accurate quantification of the very scarce frequency of PSCs within a differentiation run will become increasingly important with clinical translation. Specific to this study, a limitation is that it was only feasible to quantify up to 106 cells with flow cytometry. This led to only single-digital number of Oct4-GFP+ cells detected under the conditions where this population was the most infrequent, which by multiplying with the total cell number allowed us to estimate the total number of Oct4-GFP+ cells used in our studies. However, while supporting the fundamental findings of study, this likely led to increased variation in our estimations of residual PSC populations under the conditions where this population was rare.
Acknowledgments
Oct4-GFP mESC and miPSC lines were generously donated by Professor Douglas Melton (Harvard University). HIF-1α−/− mESC were generously donated by Professor Peter Carmeliet (Katholieke Universiteit Leuven). Helpful discussion and technical assistance with aspects of this work were provided by Professor Susan Bonner-Weir (Joslin Diabetes Center). Helpful discussion and technical assistance with aspects of the cardiomyocyte aspects of this work were provided by Dr. Daryl Powers (MIT). Histopathological assessment was provided by Dr. Nicola M. A. Parry (MIT). The FACS was operated by Michael Jennings and Michele Griffin (MIT). Rose E. Yu, Giulia M. Dula, Julie Paul, and Bhargavi Chevva (MIT) provided technical assistance.
Author Contributions
JRM and CKC conceived the experimental design. JRM and JT conducted the in vitro experiments. JRM conducted the in vivo experiments. JRM and CKC wrote the manuscript. All authors edited and reviewed the manuscript.
Funding
This study was supported by a grant from the Juvenile Diabetes Research Foundation (41-2009-803).
Data Availability
All data associated with this study is presented in the paper. All cell lines used were acquired from third parties.
Conflict of interest
J.R.M. and C.K.C. are inventors on issued patent number 10,179,901 entitled “Methods and compositions for increased safety of stem cell-derived populations”. J.R.M. is a consultant for Sana Biotechnology. J.R.M. is co-founder of Salentra Biosciences. J.T. has no declarations.
Ethical Approval
All animal work was approved by the MIT Committee on Animal Care.
Informed Consent
There were no research subject participants. All authors consent to publication.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jeffrey R. Millman, Email: jmillman@wustl.edu
Clark K. Colton, Email: ckcolton@mit.edu
References
- 1.Amariglio N, Hirshberg A, Scheithauer BW, Cohen Y, Loewenthal R, Trakhtenbrot L, Paz N, Koren-Michowitz M, Waldman D, Leider-Trejo L, Toren A, Constantini S, Rechavi G. Donor-derived brain tumor following neural stem cell transplantation in an ataxia telangiectasia patient. PLoS Med. 2009;6:e1000029. doi: 10.1371/journal.pmed.1000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Augsornworawat P, Velazco-Cruz L, Song J, Millman JR. A hydrogel platform for in vitro three dimensional assembly of human stem cell-derived islet cells and endothelial cells. Acta Biomater. 2019;97:272–280. doi: 10.1016/j.actbio.2019.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blum B, Bar-Nur O, Golan-Lev T, Benvenisty N. The anti-apoptotic gene survivin contributes to teratoma formation by human embryonic stem cells. Nat. Biotechnol. 2009;27:281–287. doi: 10.1038/nbt.1527. [DOI] [PubMed] [Google Scholar]
- 4.Brederlau A, Correia AS, Anisimov SV, Elmi M, Paul G, Roybon L, Morizane A, Bergquist F, Riebe I, Nannmark U, Carta M, Hanse E, Takahashi J, Sasai Y, Funa K, Brundin P, Eriksson PS, Li JY. Transplantation of human embryonic stem cell-derived cells to a rat model of Parkinson’s disease: effect of in vitro differentiation on graft survival and teratoma formation. Stem Cells. 2006;24:1433–1440. doi: 10.1634/stemcells.2005-0393. [DOI] [PubMed] [Google Scholar]
- 5.Brusselmans K, Bono F, Collen D, Herbert JM, Carmeliet P, Dewerchin M. A novel role for vascular endothelial growth factor as an autocrine survival factor for embryonic stem cells during hypoxia. J. Biol. Chem. 2005;280:3493–3499. doi: 10.1074/jbc.M406613200. [DOI] [PubMed] [Google Scholar]
- 6.Chong JJ, Yang X, Don CW, Minami E, Liu YW, Weyers JJ, Mahoney WM, Van Biber B, Cook SM, Palpant NJ, Gantz JA, Fugate JA, Muskheli V, Gough GM, Vogel KW, Astley CA, Hotchkiss CE, Baldessari A, Pabon L, Reinecke H, Gill EA, Nelson V, Kiem HP, Laflamme MA, Murry CE. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature. 2014;510:273–277. doi: 10.1038/nature13233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choo AB, Tan HL, Ang SN, Fong WJ, Chin A, Lo J, Zheng L, Hentze H, Philp RJ, Oh SK, Yap M. Selection against undifferentiated human embryonic stem cells by a cytotoxic antibody recognizing podocalyxin-like protein-1. Stem Cells. 2008;26:1454–1463. doi: 10.1634/stemcells.2007-0576. [DOI] [PubMed] [Google Scholar]
- 8.Deuse T, Hu X, Gravina A, Wang D, Tediashvili G, De C, Thayer WO, Wahl A, Garcia JV, Reichenspurner H, Davis MM, Lanier LL, Schrepfer S. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019;3:252. doi: 10.1038/s41587-019-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fink DW., Jr FDA regulation of stem cell-based products. Science. 2009;324:1662–1663. doi: 10.1126/science.1173712. [DOI] [PubMed] [Google Scholar]
- 10.Garreta E, Melo E, Navajas D, Farré R. Low oxygen tension enhances the generation of lung progenitor cells from mouse embryonic and induced pluripotent stem cells. Physiol. Rep. 2014;2:e12075. doi: 10.14814/phy2.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hakim F, Kaitsuka T, Raeed JM, Wei FY, Shiraki N, Akagi T, Yokota T, Kume S, Tomizawa K. High oxygen condition facilitates the differentiation of mouse and human pluripotent stem cells into pancreatic progenitors and insulin-producing cells. J. Biol. Chem. 2014;289:9623–9638. doi: 10.1074/jbc.M113.524363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haramoto Y, Onuma Y, Mawaribuchi S, Nakajima Y, Aiki Y, Higuchi K, Shimizu M, Tateno H, Hirabayashi J, Ito Y. A technique for removing tumourigenic pluripotent stem cells using rBC2LCN lectin. Regen. Ther. 2020;14:306–314. doi: 10.1016/j.reth.2020.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hentze H, Soong PL, Wang ST, Phillips BW, Putti TC, Dunn NR. Teratoma formation by human embryonic stem cells: evaluation of essential parameters for future safety studies. Stem Cell Res. 2009;2:198. doi: 10.1016/j.scr.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Hogrebe NJ, Augsornworawat P, Maxwell KG, Velazco-Cruz L, Millman JR. Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat. Biotechnol. 2020;38:460–470. doi: 10.1038/s41587-020-0430-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huangfu D, Maehr R, Guo W, Eijkelenboom A, Snitow M, Chen AE, Melton DA. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 2008;26:795–797. doi: 10.1038/nbt1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim JH, Auerbach JM, Rodriguez-Gomez JA, Velasco I, Gavin D, Lumelsky N, Lee SH, Nguyen J, Sanchez-Pernaute R, Bankiewicz K, McKay R. Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson’s disease. Nature. 2002;418:50–56. doi: 10.1038/nature00900. [DOI] [PubMed] [Google Scholar]
- 17.Lawrenz B, Schiller H, Willbold E, Ruediger M, Muhs A, Esser S. Highly sensitive biosafety model for stem-cell-derived grafts. Cytotherapy. 2004;6:212–222. doi: 10.1080/14653240410006031. [DOI] [PubMed] [Google Scholar]
- 18.Lengner CJ, Gimelbrant AA, Erwin JA, Cheng AW, Guenther MG, Welstead GG, Alagappan R, Frampton GM, Xu P, Muffat J, Santagata S, Powers D, Barrett CB, Young RA, Lee JT, Jaenisch R, Mitalipova M. Derivation of pre-X inactivation human embryonic stem cells under physiological oxygen concentrations. Cell. 2010;141:872–883. doi: 10.1016/j.cell.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 19.Ma S, Viola R, Sui L, Cherubini V, Barbetti F, Egli D. Beta cell replacement after gene editing of a neonatal diabetes-causing mutation at the insulin locus. Stem Cell Rep. 2018;11:1407–1415. doi: 10.1016/j.stemcr.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menendez S, Camus S, Herreria A, Paramonov I, Morera LB, Collado M, Pekarik V, Maceda I, Edel M, Consiglio A, Sanchez A, Li H, Serrano M, Belmonte JC. Increased dosage of tumor suppressors limits the tumorigenicity of iPS cells without affecting their pluripotency. Aging Cell. 2012;11:41–50. doi: 10.1111/j.1474-9726.2011.00754.x. [DOI] [PubMed] [Google Scholar]
- 21.Millman JR, Tan JH, Colton CK. The effects of low oxygen on self-renewal and differentiation of embryonic stem cells. Curr. Opin. Organ Transplant. 2009;14:694–700. doi: 10.1097/MOT.0b013e3283329d53. [DOI] [PubMed] [Google Scholar]
- 22.Pabon JE, Jr, Findley WE, Gibbons WE. The toxic effect of short exposures to the atmospheric oxygen concentration on early mouse embryonic development. Fertil. Steril. 1989;51:896–900. doi: 10.1016/S0015-0282(16)60688-X. [DOI] [PubMed] [Google Scholar]
- 23.Powers DE, Millman JR, Bonner-Weir S, Rappel MJ, Colton CK. Accurate control of oxygen level in cells during culture on silicone rubber membranes with application to stem cell differentiation. Biotechnol. Prog. 2010;26:805–818. doi: 10.1002/btpr.359. [DOI] [PubMed] [Google Scholar]
- 24.Powers DE, Millman JR, Huang RB, Colton CK. Effects of oxygen on mouse embryonic stem cell growth, phenotype retention, and cellular energetics. Biotechnol. Bioeng. 2008;101:241–254. doi: 10.1002/bit.21986. [DOI] [PubMed] [Google Scholar]
- 25.Qadir MMF, Alvarez-Cubela S, Belle K, Sapir T, Messaggio F, Johnson KB, Umland O, Hardin D, Klein D, Perez-Alvarez I, Sadiq F, Alcazar O, Inverardi LA, Ricordi C, Buchwald P, Fraker CA, Pastori RL, Dominguez-Bendala J. A double fail-safe approach to prevent tumorigenesis and select pancreatic beta cells from human embryonic stem cells. Stem Cell Rep. 2019;12:611–623. doi: 10.1016/j.stemcr.2019.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat. Rev. Mol. Cell Biol. 2008;9:285–296. doi: 10.1038/nrm2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sułkowski M, Konieczny P, Chlebanowska P, Majka M. Introduction of exogenous HSV-TK suicide gene increases safety of keratinocyte-derived induced pluripotent stem cells by providing genetic “emergency exit” switch. Int. J. Mol. Sci. 2018;19:197. doi: 10.3390/ijms19010197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan HL, Tan BZ, Goh WXT, Cua S, Choo A. In vivo surveillance and elimination of teratoma-forming human embryonic stem cells with monoclonal antibody 2448 targeting annexin A2. Biotechnol. Bioeng. 2019;116:2996–3005. doi: 10.1002/bit.27135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang C, Lee AS, Volkmer JP, Sahoo D, Nag D, Mosley AR, Inlay MA, Ardehali R, Chavez SL, Pera RR, Behr B, Wu JC, Weissman IL, Drukker M. An antibody against SSEA-5 glycan on human pluripotent stem cells enables removal of teratoma-forming cells. Nat. Biotechnol. 2011;29:829–834. doi: 10.1038/nbt.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, Hashimoto H, Suzuki T, Yamashita H, Satoh Y, Egashira T, Seki T, Muraoka N, Yamakawa H, Ohgino Y, Tanaka T, Yoichi M, Yuasa S, Murata M, Suematsu M, Fukuda K. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell. 2013;12:127–137. doi: 10.1016/j.stem.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 31.Velazco-Cruz L, Goedegebuure MM, Millman JR. Advances toward engineering functionally mature human pluripotent stem cell-derived beta cells. Front. Bioeng. Biotechnol. 2020;8:786. doi: 10.3389/fbioe.2020.00786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, Ebina W, Mandal PK, Smith ZD, Meissner A, Daley GQ, Brack AS, Collins JJ, Cowan C, Schlaeger TM, Rossi DJ. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshida Y, Takahashi K, Okita K, Ichisaka T, Yamanaka S. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell. 2009;5:237–241. doi: 10.1016/j.stem.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 34.Zhou Q, Chipperfield H, Melton DA, Wong WH. A gene regulatory network in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA. 2007;104:16438–16443. doi: 10.1073/pnas.0701014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data associated with this study is presented in the paper. All cell lines used were acquired from third parties.