

SUMMARY
Tonic inhibition mediated by extrasynaptic GABAARs regulates various brain functions. However, the mechanisms that regulate tonic inhibition remain largely unclear. Here, we report distinct actions of GluN2A- and GluN2B-NMDA receptors (NMDARs) on tonic inhibition in hippocampal neurons under basal and high activity conditions. Specifically, overexpression of GluN2B, but not GluN2A, reduces α5-GABAAR surface expression and tonic currents. Additionally, knockout of GluN2A and GluN2B decreases and increases tonic currents, respectively. Mechanistically, GluN2A-NMDARs inhibit and GluN2B-NMDARs promote α5-GABAAR internalization, resulting in increased and decreased surface α5-GABAAR expression, respectively. Furthermore, GluN2A-NMDARs, but not GluN2B-NMDARs, are required for homeostatic potentiation of tonic inhibition induced by prolonged increase of neuronal activity. Last, tonic inhibition decreases during acute seizures, whereas it increases 24 h later, involving GluN2-NMDAR-dependent signaling. Collectively, these data reveal an NMDAR subunit-specific regulation of tonic inhibition in physiological and pathological conditions and provide mechanistic insight into activity-dependent modulation of tonic inhibition.
Graphical Abstract
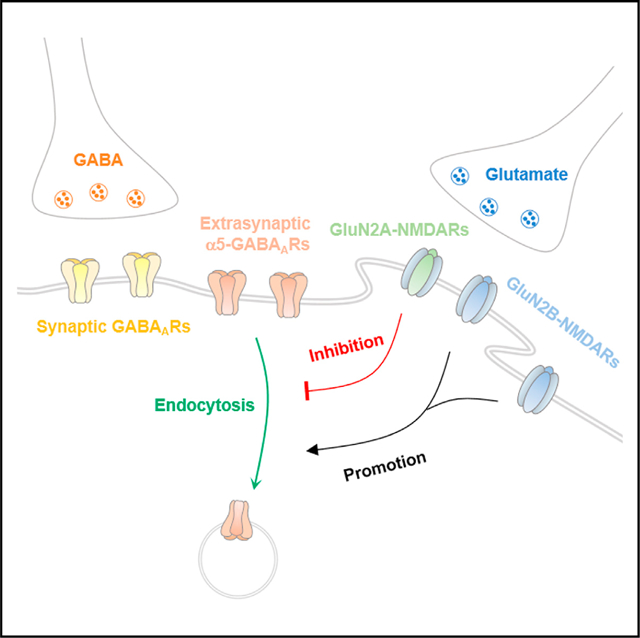
In brief
Wu et al. find differential modulation of tonic inhibition by NMDA receptor subtypes and reveal distinct roles of GluN2A- and GluN2B-NMDA receptors in regulating α5-GABAA receptor trafficking, tonic inhibition, and its homeostatic plasticity. They also demonstrate the regulation of tonic inhibition by NMDA receptors in a kainate-induced seizure model.
INTRODUCTION
γ-Aminobutyric acid (GABA), the major inhibitory neurotransmitter in the adult mammalian brain, exerts fast inhibitory effects on ubiquitously expressed γ-aminobutyric acid type A receptors (GABAARs). Generally, GABAARs can be classified as mediating either phasic or tonic inhibition (Belelli et al., 2009; Farrant and Nusser, 2005). Phasic inhibition is mediated by synaptically localized GABAARs that respond to presynaptic GABA release, whereas tonic inhibition is mediated by GABAARs localized either extrasynaptically or perisynaptically that respond to low ambient levels of GABA. With respect to tonic inhibition, accumulating evidence has revealed its involvement in a variety of neurophysiological processes such as regulating neuronal excitability, synaptic plasticity, and neuronal development (Belelli et al., 2009; Brickley and Mody, 2012; Farrant and Nusser, 2005; Holter et al., 2010; Lee and Maguire, 2014). Although the molecular and cellular mechanisms regulating phasic inhibition have been extensively studied (Han et al., 2021; Jacob et al., 2008; Luscher et al., 2011; Vithlani et al., 2011), those involved in tonic inhibition remain less clear.
NMDA receptors (NMDARs) are heteromeric complexes assembled from the GluN1 and GluN2 or GluN3 subunit (Chatterton et al., 2002; McBain and Mayer, 1994). The GluN2 subunit has four isoforms (GluN2A to GluN2D), which are differently distributed across the brain (Monyer et al., 1992). Specifically, both GluN2A and GluN2B are the predominant GluN2 subunits expressed in the adult hippocampus (Monyer et al., 1994). Importantly, GluN2A- and GluN2B-NMDARs exhibit differences in kinetics, ligand sensitivity, ion permeability, and interactions with intracellular proteins (Vieira et al., 2020). They also have distinct roles in regulating neuronal development (Gambrill and Barria, 2011; Gonda et al., 2020; Sepulveda et al., 2010) and are implicated in pathological conditions such as epilepsy (Chen et al., 2007) and stroke (Chen et al., 2008).
Recent studies have documented a critical role for NMDAR-dependent regulation of GABAergic synapse development and function (Chiu et al., 2018; Gaïarsa, 2004; Gu and Lu, 2018; Gu et al., 2016; Henneberger et al., 2005; Horn and Nicoll, 2018; Marsden et al., 2007; Petrini et al., 2014; Rajgor et al., 2020). However, much less is known about how NMDARs regulate tonic inhibition. It has been shown that genetic deletion of GluN1, the obligatory subunit of the NMDAR, enhances tonic inhibition in immature hippocampal neurons (Gu et al., 2016). Tonic inhibition in hippocampal neurons could also be enhanced during NMDAR-dependent inhibitory long-term potentiation (Wyroślak et al., 2021). Additionally, pathological activation of NMDARs during stroke decreases expression of extrasynaptic δ-GABAARs and reduces tonic currents in cortical neurons (Jaenisch et al., 2016). However, mechanistic understanding of the role of NMDAR-dependent regulation of tonic inhibition is lacking. Given that NMDAR subunit composition dictates its receptor properties, it is also unknown whether the subunit composition critically impacts tonic inhibition. Here, we employed genetic and pharmacological approaches to demonstrate the differential roles of GluN2A- and GluN2B-NMDARs in the regulation of extrasynaptic GABAAR trafficking and tonic inhibition in hippocampal neurons.
RESULTS
Overexpression of GluN2B inhibits tonic inhibition
To assess the role of GluN2 subunits in modulation of tonic inhibition, we first overexpressed GluN2A or GluN2B in cultured hippocampal neurons at days in vitro (DIV)11 and then measured miniature inhibitory postsynaptic currents (mIPSCs) and tonic inhibitory currents at DIV14 (Figure 1A). We observed that tonic currents were significantly reduced in neurons overexpressing GluN2B, whereas overexpression of GluN2A had no effect on tonic currents, compared to control neurons expressing GFP (Figure 1B). However, neither GluN2A nor GluN2B overexpression had a significant effect on mIPSCs (Figures S1A and S1B), indicating the existence of GluN2-dependent regulation of tonic, but not phasic, inhibition in neurons. Previous work has shown that extrasynaptic α5-GABAARs mediate a majority of tonic inhibition in hippocampal pyramidal neurons (Caraiscos et al., 2004; Glykys et al., 2008; Wu et al., 2021). To investigate whether overexpression of GluN2 subunits regulated α5-GABAAR surface abundance, we performed α5-GABAAR immunostaining assays in cultured neurons. Consistent with the observed effect on tonic currents (Figure 1B), overexpression of GluN2B, but not GluN2A or the control, GFP, reduced the surface expression of α5-GABAARs (Figure 1C).
Figure 1. Differential regulation of tonic inhibition by GluN2 subunits.
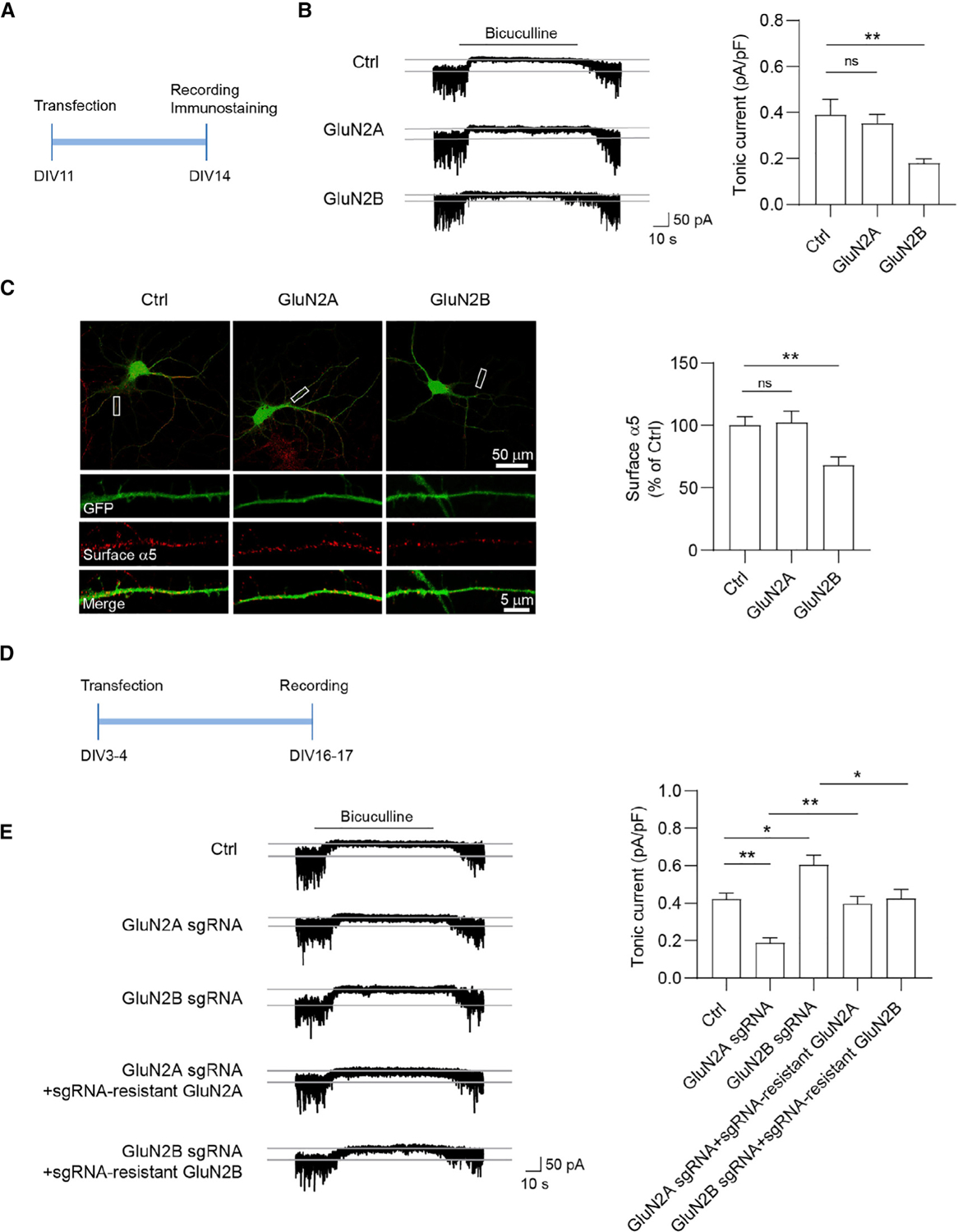
(A) Overexpression experiment design. Neurons were transfected at 11 days in vitro (DIV11) for 72 h and then recorded for tonic inhibitory currents at DIV14.
(B) GluN2B, but not GluN2A, overexpression decreased tonic currents in cultured neurons. n = 10–13 for each group, one-way ANOVA test, F(2,31) = 5.029, p = 0.0128 with Dunnett’s multiple comparisons test. Ctrl versus GluN2B: p = 0.0093.
(C) GluN2B, but not GluN2A, overexpression decreased surface α5 expression in cultured neurons. n = 28–31 for each group, one-way ANOVA test, F(2,87) = 6.369, p = 0.0026 with Dunnett’s multiple comparisons test, Ctrl versus GluN2B: p = 0.0071.
(D) Knockout (KO) experiment design. Neurons were transfected at DIV3–DIV4 and then recorded for tonic currents at DIV16–DIV17.
(E) GluN2A KO decreased tonic currents, whereas GluN2B KO increased tonic currents. The changes in tonic currents induced by either GluN2A or GluN2B KO were restored back to the control level by co-expression of corresponding sgRNA-resistant constructs, respectively. n = 8–10 for each group, one-way ANOVA, F(4,39) = 13.12, p < 0.0001 with Tukey’s multiple comparisons test, Ctrl versus GluN2A sgRNA: p = 0.0039; Ctrl versus GluN2B sgRNA: p = 0.0224: GluN2A sgRNA versus GluN2A sgRNA + sgRNA-resistant GluN2A: p = 0.0091; GluN2B sgRNA versus GluN2B sgRNA + sgRNA-resistant GluN2B: p = 0.0202.
*p < 0.05 and **p < 0.01. All data are presented as mean ± SEM.
See also Figure S1.
Knockout of GluN2A and GluN2B differentially regulates tonic inhibition
To complement overexpression experiments, we developed single-guide RNAs (sgRNAs) to perform single-cell genetic deletion of endogenous GluN2A or GluN2B in cultured hippocampal neurons and then measured tonic inhibitory currents (Figure 1D). The knockout (KO) efficacy was confirmed by western blot in co-transfected HEK293 cells (Figures S1C and S1D), as well as by NMDAR-mediated mEPSC and NMDA-evoked whole-cell current recordings in cultured neurons transfected with both sgRNAs (Figures S1E–S1G). Although KO of GluN2A significantly decreased tonic currents, we observed that GluN2B KO increased tonic currents, compared with control neurons expressing the empty vector (Figure 1E). Importantly, the changes in tonic currents induced by either GluN2A or GluN2B KO were restored by co-expression of the corresponding GluN2 sgRNA-resistant constructs (Figures 1E and S1D), suggesting that these effects were due to loss of endogenous GluN2A or GluN2B but not off-target effects.
Pharmacological suppression of GluN2A- and GluN2B-NMDARs differentially modulates α5-GABAAR surface expression and tonic inhibition
To corroborate our genetic findings, we next employed a pharmacological approach to further determine the role of GluN2A and GluN2B in the regulation of tonic inhibition. Specifically, we treated cultured neurons at DIV14 with NVP-AAM077 (NVP, GluN2A-preferring antagonist; 100 nM), ifenprodil (Ifen, GluN2B-preferring antagonist; 5 μM) or APV (pan-NMDAR antagonist; 100 μM) for 24 h, and then measured tonic inhibitory currents (Figure 2A). We found that Ifen increased α5 surface expression and tonic inhibitory currents, whereas treatment with NVP induced opposite effects (Figures 2B and 2C). Interestingly, APV had little effect on α5 surface expression and tonic currents (Figures 2B and 2C), indicating that antagonism of both GluN2A- and GluN2B-NMDARs exerts a combinatory effect leading to little change in tonic inhibition. Taken together, these results suggest differential regulation of α5 surface expression and tonic inhibition by GluN2A- and GluN2B-NMDARs. Considering that δ-GABAARs also generate a tonic inhibitory conductance in the hippocampus (Glykys et al., 2008; Penna et al., 2014), we recorded THIP (3 μM, a GABAAR agonist with a preference for δ-GABAARs)-evoked currents in hippocampal neurons at DIV15 after 24-h treatment with NVP or Ifen. We found that neither NVP nor Ifen treatment affected THIP-evoked currents (Figure S2), suggesting that NMDARs do not regulate δ-GABAAR-mediated tonic inhibition in hippocampal neurons.
Figure 2. Pharmacological suppression of GluN2A- and GluN2B-NMDARs regulates tonic inhibition and α5-GABAAR internalization.
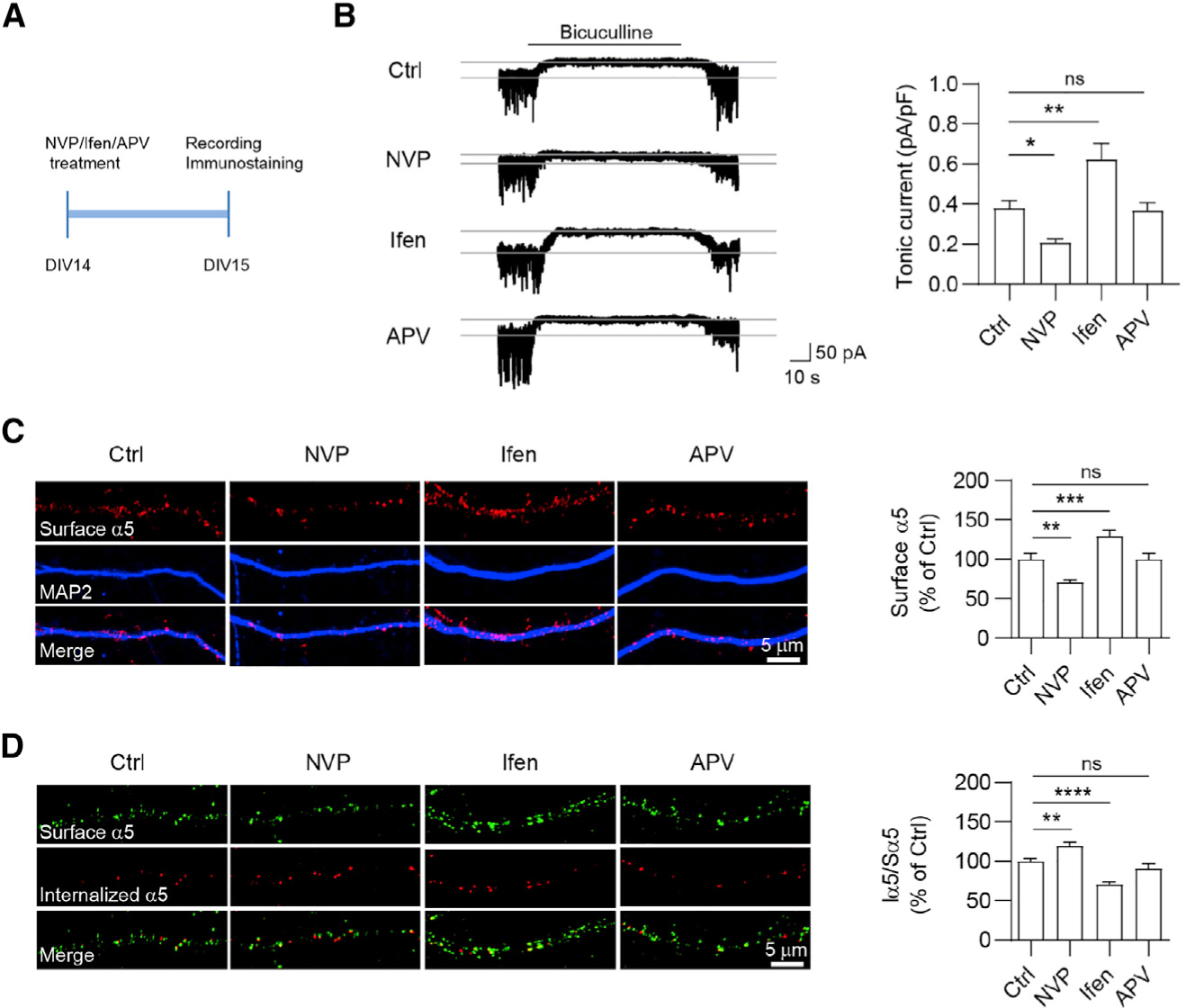
(A) Experimental design. Hippocampal neurons at DIV14 were treated with NVP (GluN2A-preferring antagonist NVP-AAM077, 100 nM), Ifen (GluN2B-preferring antagonist ifenprodil, 5 μM), or APV (broad-spectrum NMDAR antagonist, 100 μM) for 24 h and then recorded for tonic currents at DIV15.
(B) Ifen treatment increased tonic currents, whereas NVP treatment decreased tonic currents. n = 10–11 for each group, one-way ANOVA test, F(3,38) = 13.14, p < 0.0001 with Dunnett’s multiple comparisons test, Ctrl versus NVP, p = 0.031; Ctrl versus Ifen, p = 0.0023.
(C) Ifen treatment increased surface α5 expression, whereas NVP treatment decreased surface α5 expression. n = 33–43 for each group, Kruskal-Wallis test with Dunnett’s multiple comparisons test, Ctrl versus NVP, p = 0.0041; Ctrl versus Ifen, p = 0.0003.
(D) Endocytosis assay of α5-GABAARs in cultures. Surface α5-GABAARs (Sα5) were labeled in green, and internalized α5-GABAARs (Iα5) were in red. Bar graphs in the right showing that 24-h NVP treatment increased α5 internalization, whereas 24-h Ifen treatment decreased α5 internalization. n = 15–16 for each group, one-way ANOVA test, F(3,56) = 22.96, p < 0.0001 with Dunnett’s multiple comparisons test, Ctrl versus NVP: p = 0.0044; Ctrl versus Ifen: p < 0.0001.
*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. All data are presented as mean ± SEM.
See also Figures S2 and S3.
During development, NMDARs undergo a developmental switch from those incorporating the GluN2B subunit to the GluN2A subunit in neocortex and hippocampus (Dong et al., 2006; McKay et al., 2018; Sheng et al., 1994). Consistent with these previous studies, we confirmed this developmental switch from primarily GluN2B-NMDARs to primarily GluN2A-NMDARs in cultured hippocampal neurons (Figure S3A). In addition, both α5-GABAAR subunit expression and tonic currents increased during development within the first month in cultured neurons (Figures S3A and S3B), suggesting a possible link between developmental switch of NMDARs subunits and increase of α5-GABAAR expression within the first month in neuronal cultures. Thus, we tested the effects of GluN2A and GluN2B antagonists on tonic inhibition in immature and mature neurons. In immature neurons at DIV7–8, Ifen or APV increased α5 surface expression and tonic currents, whereas NVP had little effect (Figures S3C and S3D), consistent with higher expression of GluN2B in developing, immature neurons. By contrast, in mature neurons at DIV25–DIV-26, NVP and APV decreased α5 surface expression and tonic currents, whereas Ifen had little effect (Figures S3C and S3D). These results suggest that regulation of tonic inhibition by GluN2A- and GluN2B-NMDARs in neuronal culture is developmentally dependent.
Differential regulation of α5-GABAAR internalization by GluN2A- and GluN2B-NMDARs
Considering that opposing actions of GluN2A- and GluN2B-NMDAR antagonism on α5-GABAAR surface expression and tonic currents were observed at 2–3 weeks neurons, we then investigated the mechanism underlying the distinct effects at this developmental stage. The abundance of α5-GABAAR surface expression is dynamically controlled by the balance of receptor endocytosis and exocytosis. Because α5-GABAAR surface expression is differentially regulated by NMDAR GluN2 subunits, we hypothesized that α5-GABAAR endocytosis and/or exocytosis might be critically regulated by GluN2A- or GluN2B-NMDARs. To test this hypothesis, we first performed antibody-feeding experiments to label surface and internalized endogenous α5 in live hippocampal neurons at DIV15 and examined α5 endocytosis. We found that 24-h NVP treatment increased α5-GABAAR internalization, whereas 24-h Ifen treatment decreased α5-GABAAR internalization (Figure 2D). In addition, 24-h APV treatment did not alter α5-GABAAR internalization (Figure 2D). These results suggest that GluN2A and GluN2B have distinct roles in regulating α5-GABAAR internalization at this developmental stage. Next, we combined fluorescence recovery after photobleaching (FRAP) with fluorescence loss in photobleaching (FLIP) to investigate exocytosis of superecliptic pHluorin-tagged α5 (SEP-α5) as a measurement of the receptor exocytosis (Wu et al., 2021). In this experiment, repetitive photobleaching occurred at dendritic regions bilateral to the central FRAP area, thus excluding laterally diffusing SEP-α5 to the central area and allowing the measurement of newly exocytosed SEP-α5 (Figure S3E). We found that neurons treated with either selective NMDAR antagonists or pan-NMDAR antagonism exhibited similar fluorescence recovery after photobleaching (Figure S3F), indicating that neither GluN2A- nor GluN2B-NMDARs regulate α5-GABAAR exocytosis. The extrasynaptic abundance of α5-GABAAR is also affected by phosphorylation level of the actin-binding protein radixin, an ezrin/radixin/moesin (ERM)-family member (Hausrat et al., 2015). Specifically, phosphorylated radixin anchors α5-GABAARs at extrasynaptic sites (Hausrat et al., 2015). We found that antagonism of GluN2A- and GluN2B-NMDARs had little effect on ERM and phosphorylated ERM (p-ERM) levels (Figure S3G), suggesting that the regulation of α5-GABAARs by GluN2A- and GluN2B-NMDARs is not due to changes in the phosphorylation of radixin.
GluN2A-NMDARs are required for homeostatic potentiation of tonic inhibition
Chronic pharmacological manipulation of neuronal activity can induce homeostatic adaptions in both excitatory and inhibitory transmission, which are powerful mechanisms that control neuronal excitability and neural network function (Turrigiano, 2012). Our recent work demonstrated that tonic inhibition in hippocampal neurons exhibited homeostatic potentiation after prolonged elevation of neuronal activity (Wu et al., 2021). Consistent with this study, we found that surface α5 expression and tonic inhibition increased following 48-h bicuculline treatment (Figure 3). To determine the role of GluN2A- and GluN2B-NMDARs in homeostatic plasticity of tonic inhibition, we treated hippocampal neurons at DIV16 with bicuculline (40 μM) and 24 h later with NVP (100 nM), Ifen (5 μM), or APV (100 μM) (Figure 3A). 24 h later, we found that bicuculline-induced effects on tonic inhibitory currents (Figures 3B and 3C) and surface α5 expression (Figures 3D and 3E) were significantly diminished by treatment with NVP and APV, but not by Ifen. These observations support a GluN2A-, but not GluN2B-NMDAR-dependent mechanism in homeostatic potentiation of tonic inhibition.
Figure 3. GluN2A-NMDARs are required for homeostatic potentiation of tonic inhibition.
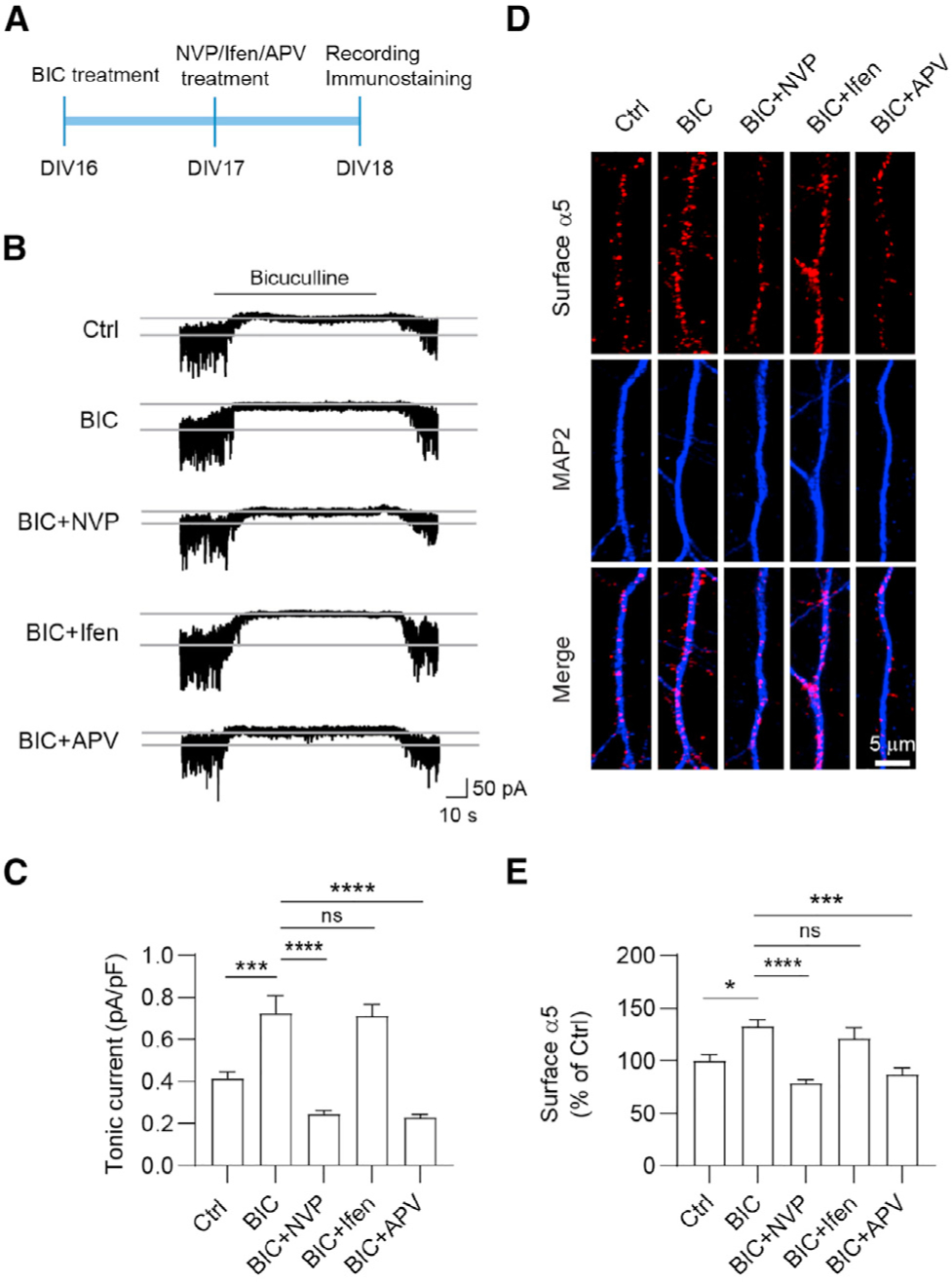
(A) Experimental design. Hippocampal neurons at DIV16 were treated with bicuculline (BIC, 40 μM), and at DIV17 they were treated with NVP (100 nM), Ifen (5 μM), or APV (100 μM) for 24 h before recording.
(B and C) NVP and APV treatment abolished BIC-induced potentiation of tonic currents. n = 10 for each group, one-way ANOVA test, F(4,45) = 25.32, p < 0.0001 with Tukey’s multiple comparison test, Ctrl versus BIC: p = 0.0004; BIC versus BIC+NVP: p < 0.0001; BIC versus BIC+APV: p < 0.0001.
(D and E) NVP and APV treatment abolished BIC-induced potentiation of surface α5 expression. n = 20–26 for each group, Kruskal-Wallis test with Dunn’s multiple comparisons test, Ctrl versus BIC: p = 0.0189; BIC versus BIC+NVP: p < 0.0001; BIC versus BIC+APV: p = 0.0002.
*p < 0.05, ***p < 0.001, and ****p < 0.0001. All data are presented as mean ± SEM.
GluN2A- and GluN2B-NMDARs regulate tonic inhibition in a KA-induced seizure model
Finally, we examined whether GluN2-dependent regulation of α5-GABAARs may occur during seizures based on evidence that deficits in tonic inhibition are potentially involved in epilepsy (Chen et al., 2007). To this end, we utilized an acute seizure model by systemically administering kainic acid (KA), an agonist for kainate- and AMPA-type ionotropic glutamate receptors. All mice injected with KA (20 mg/kg) showed epileptic behavior within 30 min and recovered 24 h after injection (Figure S4A). We then analyzed total and surface expression levels of GluN2A, GluN2B, and GABAARs in hippocampal slices prepared from mice at 1 h or 24 h after KA injection (Figure S4B). We found that KA administration had little effect on the total expression level of NMDAR and GABAAR subunits, comparing with saline-injected control (Figures 4A–4C). In contrast, 1-h post-KA injection significantly increased surface GluN2B, but not GluN2A expression (Figures 4A and 4B). Interestingly, 24 h after KA injection, surface GluN2A was increased, whereas surface GluN2B was unaffected (Figures 4A and 4B). As expected from the regulation of surface α5 expression by GluN2A- and GluN2B-NMDARs (Figure 2C), surface expression of α5-GABAAR was decreased at 1 h and then increased at 24 h after KA injection (Figure 4C). In addition, surface α1-GABAARs were increased at 24 h after KA injection (Figure S4C). These results suggest the expression of surface GluN2 subunits, synaptic and extrasynaptic GABAARs are dynamically regulated by KA-induced seizures in a time-dependent manner.
Figure 4. Pharmacological suppression of GluN2A- and GluN2B-NMDARs regulates tonic inhibition in the KA-induced seizure model.
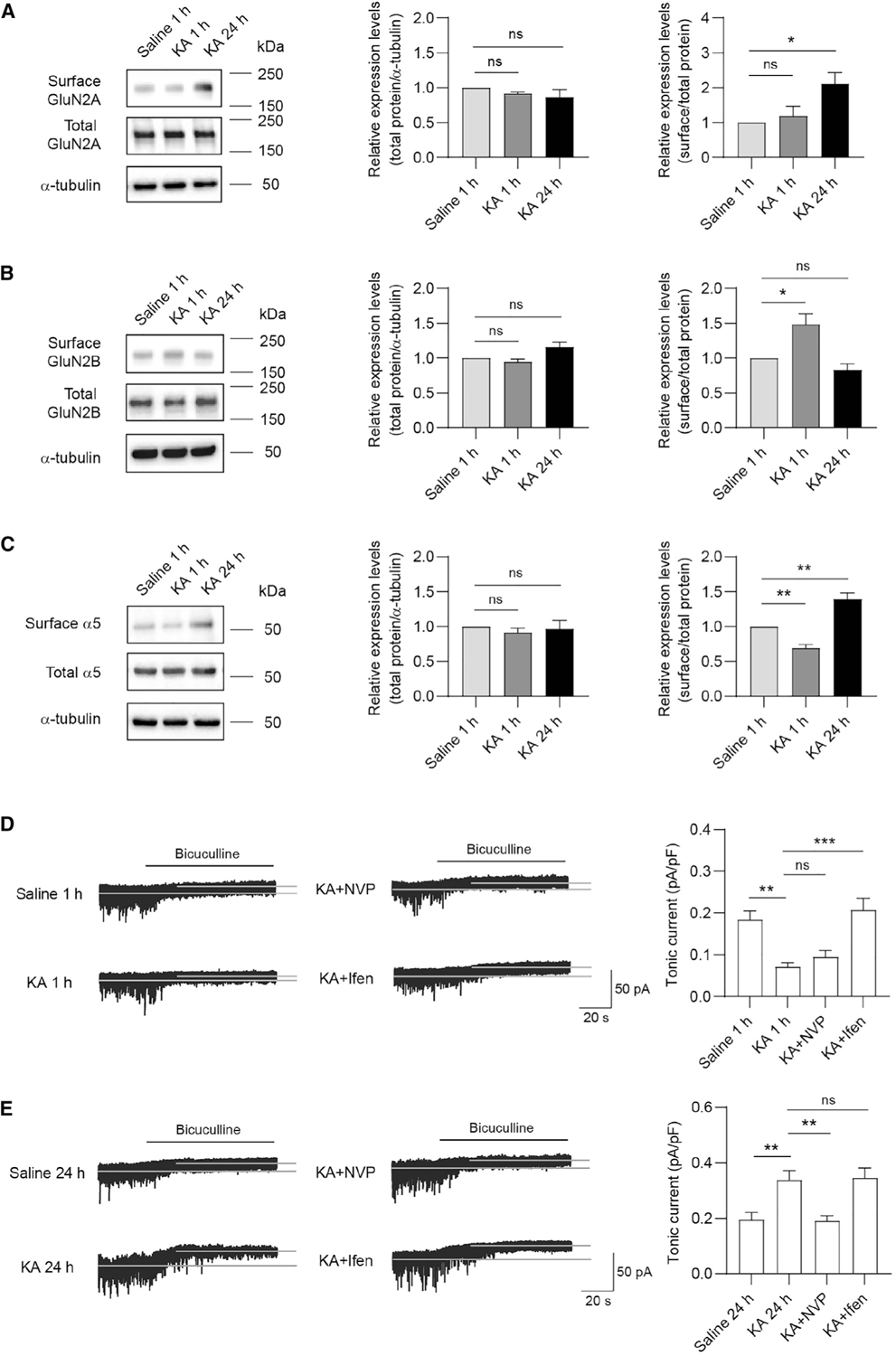
(A–C) Representative western blots and summary graphs from cell-surface biotinylation assays showing that surface and total GluN2A (A, n = 3 independent experiments, one-way ANOVA test, F(2,6) = 5.928, p = 0.0379 with Dunnett’s multiple comparisons test, p = 0.0321), GluN2B (B, n = 3 independent experiments, one-way ANOVA test, F(2,6) = 10.80, p = 0.0103 with Dunnett’s multiple comparisons test, p = 0.0283), and α5-GABAAR (C, n = 4 independent experiments, one-way ANOVA test, F(2,9) = 34.75, p < 0.0001 with Dunnett’s multiple comparisons test, saline 1 h versus KA 1 h: p = 0.0098; saline 1 h versus KA 24 h: p = 0.0022) expression in the KA-induced seizure model.
(D and E) Tonic currents in hippocampal CA3 neurons were decreased 1 h after KA injection, whereas increased 24 h after KA injection. Ifen or NVP treatment 1 h prior to KA injection, respectively, restored the decreased or increased tonic currents at corresponding time point after KA injection. (D) n = 10 for each group, one-way ANOVA test, F(3,36) = 11.42, p < 0.0001 with Dunnett’s multiple comparisons test, saline 1 h versus KA 1 h: p = 0.0014; KA 1 h versus KA + Ifen: p = 0.0001. (E) n = 10 for each group, one-way ANOVA test, F(3,36) = 8.925, p = 0.0001 with Dunnett’s multiple comparisons test, saline 24 h versus KA 24 h: p = 0.0065; KA 24 h versus KA+NVP: p = 0.0047.
*p < 0.05, **p < 0.01, and ***p < 0.001. All data are presented as mean ± SEM.
See also Figure S4.
It has been shown that the CA3 region of the hippocampus is substantially impacted in KA-induced seizures (Vincent and Mulle, 2009). We thus recorded tonic currents in CA3 pyramidal cells (Figure S4D). Consistent with KA-induced alterations in surface α5 expression, we found that tonic currents in hippocampal CA3 neurons at 1 h and 24 h post-injection in KA-administrated mice were decreased and increased, respectively, compared to saline controls (Figures 4D and 4E). Interestingly, Ifen (10 mg/kg) or NVP (10 mg/kg) treatment 1 h prior to KA injection respectively restored the decreased or increased tonic currents at corresponding time point after KA injection (Figures 4D and 4E). These results suggest that KA-induced changes in tonic inhibition are GluN2-dependent in a temporal-specific manner. In contrast, although the mIPSC amplitude was increased 24 h after KA treatment compared to control, antagonism of GluN2A- or GluN2B- NMDARs had little effect on this alteration (Figures S4E and S4F), indicating that KA-induced increases in phasic inhibition are independent of GluN2A- and GluN2B-NMDAR signaling.
DISCUSSION
In this study, we investigate the regulatory role of GluN2A- and GluN2B-NMDARs in tonic inhibition. Our genetic and pharmacological analyses indicate opposing actions of GluN2A- and GluN2B-NMDARs on α5-GABAAR internalization and tonic inhibition in hippocampal neurons. In addition, GluN2A-NMDARs, but not GluN2B-NMDARs, are required for homeostatic potentiation of tonic inhibition. Last, in a KA-induced acute seizure model, we observe a decrease in tonic inhibition during acute seizures, which is then increased 24 h later, and these alterations are dependent on the activity of distinct GluN2-NMDARs. Collectively, these data extend previous work showing the importance of NMDAR-dependent regulation of inhibitory synapse development and transmission (Chiu et al., 2018; Gu and Lu, 2018; Gu et al., 2016; Horn and Nicoll, 2018) and reveal important crosstalk between glutamatergic signaling and extrasynaptic GABAARs.
Differential regulation of tonic inhibition by NMDA receptor subtypes
It has been reported that extrasynaptic GABAAR-mediated tonic inhibition is regulated by various receptors such as GABAB (Connelly et al., 2013; Tao et al., 2013) and glycine receptors (Zou et al., 2019). In addition to these inhibitory receptors, tonic inhibition is modulated by glutamate receptors. For example, deletion of GluN1 augments tonic inhibition in immature hippocampal neurons in culture (Gu et al., 2016). In line with this study, here we found that pharmacological blockade of GluN2B-NMDARs, the predominant NMDAR subtype expressed in immature neuron or blockade of all NMDARs, enhanced tonic inhibition in immature neurons. Interestingly, we also found that the effect of blockade of GluN2B-NMDARs on tonic inhibition disappeared in mature neurons. The temporal-specific regulation of tonic inhibition is likely due to differential expression of NMDAR GluN2 subunits during development. Indeed, during the first 2 weeks after birth, NMDARs undergo a developmental switch from predominantly GluN2B- to GluN2A-NMDARs in the hippocampus (Dong et al., 2006). This may explain the lack of effects on tonic inhibition by antagonism of GluN2A-NMDARs in immature neurons. It is also important to mention that NMDAR subunits have been found to localize at GABAergic synapses in developing neurons (Cserép et al., 2012; Gundersen et al., 2004; Szabadits et al., 2011), providing anatomic proximity to regulate perisynaptic and extrasynaptic GABAARs. In contrast to a predominant effect of GluN2B-dependent regulation of tonic currents in immature neurons, we observed opposing actions of GluN2A- and GluN2B-NMDAR antagonism on tonic inhibition at 2–3 weeks neurons when both GluN2A and GluN2B are abundantly expressed. Interestingly, inhibition of all NMDARs by APV abolished the differential modulation of tonic inhibition by NMDAR subtypes, presumably due to normalizing the opposing effects of GluN2A- and GluN2B-NMDARs on α5-GABAARs. Of note, although both pharmacological inhibition and genetic deletion of GluN2A diminished tonic inhibition at 2–3 weeks neurons, overexpression of GluN2A had no significant effect on tonic inhibition, showing that further increase in GluN2A expression over endogenous levels at this developmental stage is not sufficient to alter tonic inhibitory currents.
Our data indicate that α5-GABAAR endocytosis, but not exocytosis, is differentially regulated by GluN2A- and GluN2B-NMDARs. Specifically, GluN2A-NMDARs inhibit, and GluN2B-NMDARs promote α5-GABAAR internalization. Currently, the molecular mechanisms underlying the distinct regulation of α5-GABAAR endocytosis by NMDARs remain unclear. One scenario is that GluN2A- and GluN2B-NMDARs are coupled to distinct downstream phosphatase and kinase pathways (Shipton and Paulsen, 2013; Sun et al., 2018; Wu and Tymianski, 2018), which in turn may differentially regulate α5-GABAAR trafficking and tonic inhibition. For instance, it has been reported that activation of GluN2A- and GluN2B-NMDARs differentially regulates ERK1/2 (Chen et al., 2007) and Akt signaling (Liu et al., 2007). However, it remains unknown whether these molecular pathways are involved in the regulation of α5-GABAAR trafficking and tonic inhibition. Interestingly, it has been shown that neuronal nitric oxide synthase (nNOS) is expressed at GABAergic synapses and can be activated by NMDAR activation (Szabadits et al., 2011). It is therefore plausible that NMDAR-dependent regulation of nNOS activity could modulate protein kinase activity, leading to the regulation of α5-GABAAR trafficking and tonic inhibition. Recently, a number of proteins such as gephyrin (Brady and Jacob, 2015), Shisa7 (Wu et al., 2021), and Clptm1 (Ge et al., 2018) have been identified to interact with the α5-GABAAR, which raise the possibility that GluN2A- and GluN2B-NMDARs might differentially regulate their interaction involved in α5-GABAAR trafficking. Radixin, an actin binding protein, also binds to α5-GABAARs, and phosphorylated radixin accumulates α5-GABAAR clustering at the extrasynaptic membrane (Hausrat et al., 2015; Loebrich et al., 2006). However, we found that antagonism of GluN2A- or GluN2B-NMDARs did not affect the phosphorylation of radixin, suggesting radixin is unlikely involved in NMDAR modulation of tonic inhibition. It is also worth noting that CaMKII activation can increase cell-surface α5β3-containing receptors and tonic currents (Saliba et al., 2012). Conversely, blockade of CaMKII activity reduces tonic currents in hippocampal neurons (Wu et al., 2021). Interestingly, GluN2A- and GluN2B-NMDARs can differentially regulate CaMKII activity (Barria and Malinow, 2005; Strack et al., 2000), which in turn might contribute to distinct modulation of α5-GABAAR trafficking and tonic inhibition. In the future, it will be important to determine how GluN2A- and GluN2B-NMDARs can distinctly modulate tonic inhibition involving further downstream signaling components.
Alterations of tonic inhibition in higher activity condition and epilepsy
It has been reported that GluN2B-NMDARs are involved in the pathophysiology of epilepsy (Morimoto et al., 2004; Waxman and Lynch, 2005). Indeed, selective GluN2B-NMDAR antagonists suppressed epileptogenesis in rodent seizure models (Gorlewicz et al., 2021; Mareš et al., 2021) and decreased neuronal excitability of neocortical pyramidal neurons from epileptic patients (Wang et al., 2017). Additionally, in the seizure model induced by pilocarpine, a cholinergic muscarinic agonist, surface accumulation of GluN2B-NMDARs is increased in the hippocampus (Müller et al., 2013; Naylor et al., 2013). Likewise, a reduction in α5-GABAARs in CA1 pyramidal cells of pilocarpine-treated rats has also been observed (Houser and Esclapez, 2003). In this study, we show that administration of KA in vivo induces upregulation of surface GluN2B and downregulation of surface α5-GABAARs 1 h after KA injection in the hippocampus. Together with the experiments demonstrating that GluN2B-NMDARs regulate α5-GABAAR internalization, our data indicate that an increase of GluN2B-NMDARs contributes to reduced α5-GABAAR surface expression by enhancing receptor endocytosis, resulting in downregulation of tonic inhibition during acute seizures. Given that the important role of tonic inhibition in regulating neural excitability (Belelli et al., 2009; Farrant and Nusser, 2005; Lee and Maguire, 2014), GluN2B-NMDAR-mediated reduction of tonic inhibition may contribute to KA-induced neuronal hyperexcitability at the early stage in this seizure model.
Our data also show that in the KA-induced acute seizure model, epileptic phenotypes disappear 24 h after KA induction with a corresponding increase in tonic inhibition in hippocampal CA3 neurons. Consistently, a recent study using a similar seizure model identified that neuronal hyperexcitability in cortical neurons returns to the basal level and tonic inhibition is increased 24 h after KA injection (Pan et al., 2018). Additionally, in the pilocarpine-induced seizure model, tonic current is enhanced in dentate gyrus granule cells 3 weeks after induction (Naylor et al., 2005). Considering the importance of tonic GABAergic signaling in regulating neural network excitability (Belelli et al., 2009; Farrant and Nusser, 2005; Lee and Maguire, 2014), enhanced tonic inhibition that occurs after acute seizures may act as a homeostatic adaptive response to KA-induced neuronal hyperexcitability and thus might help lessen the associated neurodegeneration. It is worth noting that the increase of tonic inhibition in hippocampal CA3 neurons 24 h after KA administration is abolished by blockade of GluN2A-NMDARs, showing an important role of GluN2A-NMDARs in control of tonic inhibition in the late stage of this seizure model. Interestingly, we found that homeostatic potentiation of tonic inhibition induced by prolonged elevation of neuronal activity is also dependent on the GluN2A subunit. Although the mechanisms underlying the regulation of tonic inhibition by GluN2A-NMDARs under these conditions remain unclear, it could be due to a variety of possibilities such as different electrophysiological properties or discrete intracellular signaling cascades that can be engaged (Paoletti et al., 2013). Additionally, in line with a previous report (Peng et al., 2010), we found that phasic inhibition could undergo homeostatic potentiation after KA administration. However, homeostatic potentiation of synaptic inhibition is independent of GluN2A, suggesting distinct mechanisms governing homeostatic plasticity of tonic and phasic inhibition induced by KA.
Taken together, we have uncovered a critical role for NMDAR-dependent regulation of α5-GABAAR trafficking and tonic inhibition during development, under higher activity, and in a KA seizure model. We have also revealed distinct roles for NMDAR subtypes in regulating tonic inhibition under a variety of conditions both in vitro and in vivo. As dysregulation of tonic inhibition has been shown to be a mechanism underlying a number of pathological brain states (Brickley and Mody, 2012; Hines et al., 2012), our findings also offer insight into crosstalk between glutamatergic and GABAergic systems, as well as how dysregulation of this crosstalk could be involved in epileptic conditions.
Limitations of study
Given the importance of assessing sex as a biological variable, one limitation of our study is that we only utilized male mice for these experiments. Because of the rising significance of neurosteroids, endogenous positive allosteric modulators of GABAARs (although with greater selectivity for δ-GABAARs), it has been demonstrated that differences in seizure susceptibility could be dependent on their production in different genders (Samba Reddy, 2017). Additionally, distributions of extrasynaptic GABAAR subtypes differ in a cell- and brain region-specific manner. Because our recordings were from hippocampal cultures and CA3 neurons in acute brain slices, it cannot be ruled out that our observed NMDAR-dependent mechanisms regulating tonic inhibition might not be the same in neurons in other brain regions. Last, although we demonstrate distinct roles for NMDAR subtypes, we did not address the downstream mechanisms and how they might differ between NMDAR subtypes to regulate α5-GABAAR surface expression and tonic inhibition.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Wei Lu (luw4@mail.nih.gov).
Materials availability
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animal handling was performed in accordance with animal protocols approved by the Institutional Animal Care and Use Committee (IACUC) at NIH/NINDS. All mice were housed and bred in a conventional vivarium with ad libitum access to food and water under a 12-h circadian cycle. Time-pregnant mice at E17.5–18.5 were used for dissociated hippocampal neuronal culture. 6–8 weeks old male mice were used for biochemical, electrophysiological, and behavioral experiments.
Cell Lines
HEK293T cells (ATCC, Cat# CRL-11268) were maintained with culture media containing 1% penicillin-streptomycin (GIBCO), 10% FBS (GIBCO) in Dulbecco’s Modified Eagle’s Medium (DMEM, GIBCO), in a humidified incubator at 37°C with 5% CO2.
Dissociated Hippocampal Neuronal Culture
Hippocampal neurons were prepared from E17.5–18.5 mice embryos of either sex as previously described (Wu et al., 2021). In brief, the hippocampi were dissected from embryonic brains and digested in the Hank’s Balanced Salt Solution (HBSS, GIBCO) containing 20 U/ml papain (Worthington) and 100 U/ml DNase I (Worthington) at 37°C for 45 min. After centrifugation for 5 min at 800 rpm, the pellet was resuspended in HBSS containing 100 U/ml DNase I, and was fully dissociated by pipetting up and down. Cells were then transferred into HBSS containing trypsin inhibitor (10 mg/ml, Sigma-Aldrich) and BSA (10 mg/ml, Sigma-Aldrich). After centrifugation for 10 min at 800 rpm, cells were resuspended in Neurobasal media (GIBCO) supplemented with 2% B27 (GIBCO) and 2 mM GlutaMAX (GIBCO) and were plated on poly-D-lysine (Sigma-Aldrich)-coated glass coverslips or 6-well plates. Cultures were maintained in Neurobasal media supplemented with 2% B27 and 2 mM GlutaMAX in a humidified incubator at 37°C with 5% CO2. Culture media were changed by half volume once a week.
METHOD DETAILS
Plasmids
pRK5-GFP-GluN2A and pRK5-GFP-GluN2B were gifts from Katherine Roche’s lab at NINDS, NIH. pSpCas9(BB)-2A-Puro (PX459) V2.0 and pSpCas9(BB)-2A-GFP (PX458) were purchased from Addgene. Custom oligonucleotides were generated (GluN2A forward, 5′ CACCGCGACGTGACAGAACGCGAAC 3′; and GluN2A reverse, 5′ AAACGTTCGCGTTCTGTCACGTCGC 3′; and GluN2B forward, 5′ CACCGTCTGACCGGAAGATCCAGG 3′; and GluN2B reverse, 5′ AAACCCTGGATCTTCCGGTCAGAC 3′; IDT), and cloned into pSpCas9-BB-2A-GFP (PX458) or pSpCas9(BB)-2A-Puro (PX459) V2.0 vector at the BbsI cutting site. The coding sequence of GluN2A and GluN2B point mutations for sgRNA resistant plasmid (GluN2A: AGCCACGACGTGACAGAACGCGAACTT to AGTCACGACGTGACTGAGAGAGAACTT; GluN2B: ATGTCTGACCGGAAGATCCAGGGG to ATGTCTGATCGTAAGATTCAAGGA) were generated by Q5 Site-Directed Mutagenesis Kit (NEB).
Cell Transfection
HEK293T cells were transfected with GluN2A or GluN2B, together with sgRNA using CalPhos Mammalian Transfection Kit (Takara). Western blot was performed 48 h after transfection. Hippocampal neurons at DIV3–4 were transfected with GluN2A sgRNA or GluN2B sgRNA using NeuroMag reagent (Oz Biosciences), and were recorded at DIV16–17. Hippocampal neurons at DIV11 were co-transfected with pCAG-IRES-GFP and GluN2A or GluN2B using NeuroMag reagent. Electrophysiological recordings or immunostaining were performed 72 h after transfection. All transfection kits were used according to the manufacturer’s instructions.
Electrophysiology
For recording in dissociated hippocampal cultures, neurons were continuously perfused with the extracellular solution containing (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 Glucose (pH 7.3; osmolality 300–310 mOsm). The internal solution contained (in mM): 70 CsMeSO4, 70 CsCl, 8 NaCl, 10 HEPES, 0.3 Na-GTP, 4 Mg-ATP and 0.3 EGTA (pH 7.3; osmolality 285–290 mOsm). Miniature inhibitory postsynaptic currents (mIPSCs) and tonic currents were recorded at −70 mV in the presence of 0.5 μM TTX (Alomone Labs) and 20 μM DNQX (Alomone labs) without exogenous GABA. To test the effects of GluN2A- or GluN2B-preferring antagonist on tonic currents, neurons were treated with 100 nM NVP (Ding et al., 2021) or 5 μM Ifen (Sibarov et al., 2016) for 24 h prior to electrophysiological recording. For recording THIP-evoked currents, THIP (3 μM, Santa Cruz) was added to the extracellular solution via a computer-controlled multi-barrel perfusion system (Automate Scientific). For recording NMDA mEPSCs at +40 mV, 0.5 μM TTX, 20 μM DNQX, and 50 μM picrotoxin (Sigma-Aldrich) were added into the extracellular solution. For recording NMDA-induced whole-cell currents, TTX (0.5 μM) were added into 0 Mg2+ extracellular solution. NMDA-induced current was recorded at −70 mV by rapid application/removal of NMDA (100 μM) using the perfusion system.
For recording in acute brain slices, transverse hippocampal slices (300 μm thickness) were prepared from 6–8 weeks old male mice in chilled high sucrose cutting solution, containing (in mM): 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 NaHCO3, 7 glucose, 210 sucrose and 1.3 ascorbic acid. The slices were recovered in artificial cerebrospinal fluid (ACSF) containing (in mM): 119 NaCl, 2.5 KCl, 26.2 NaHCO3, 1 NaH2PO4, 11 glucose, 2.5 CaCl2 and 1.3 MgSO4 (pH 7.3; osmolality 300–310 mOsm) at 32°C for 30 min and then were maintained at room temperature prior to recording. To record tonic currents, slices were transferred to a submersion chamber, continuously perfused with ACSF with 0.5 μM TTX, 20 μM DNQX and 5 μM GABA. The intracellular solution contained (in mM) 130 CsCl, 8.5 NaCl, 5 HEPES, 4 MgCl2, 4 Na-ATP, 0.3 Na-GTP and 1 QX-314 (pH 7.3; osmolality 285–290 mOsm).
To measure tonic inhibitory currents in neuronal cultures or in acute hippocampal slices, the GABAAR competitive antagonist bicuculline (20 μM, Abcam) was bath applied after obtaining a stable baseline recording at −70 mV. Igor Pro were used to determine the values of tonic currents. An all-points histogram was plotted for a 20 s period before and during bath-application of bicuculline, fitting the histogram with a Gaussian distribution gave the mean baseline holding currents, and the difference in baseline holding currents before and during bicuculline application was calculated to be the tonic currents. Tonic currents were normalized to membrane capacitance, to account for variability in cell size. Series resistance was monitored and not compensated, and cells in which series resistance was more than 25 MΩ or varied by 25% during a recording session were discarded. Whole-cell recordings were obtained from cells visualized with a fixed stage upright microscope (BX51WI, Olympus). Fluorescence-positive cells were identified by epifluorescence microscopy. Data were collected with a Multiclamp 700B amplifier (Axon Instruments), filtered at 2 kHz, and digitized at 10 kHz.
Immunostaining
For surface α5 receptor labeling, cultured hippocampal neurons at DIV15 on coverslips were incubated with anti-α5 antibody (1:500, Synaptic Systems) in culture medium for 15 min. Next, they were washed briefly with fresh culture medium and fixed with a solution containing 4% paraformaldehyde and 4% sucrose in PBS. Cultured neurons were subsequently incubated with Alexa 555-conjugated anti-rabbit secondary antibody (1:1000, Thermo Fisher Scientific) for the visualization of α5. For the endocytosis assay, cultured hippocampal neurons at DIV15 were incubated live with rabbit α5 antibody (1:500, Synaptic Systems) at 37°C for 10 min in conditioned culture medium. After incubation, the neurons were washed with PBS and then incubated in antibody-free medium to allow antibody-bound receptors to undergo internalization at 37°C for 30 min, followed by fixation with 4% paraformaldehyde and 4% sucrose in PBS. After fixation, neurons were washed and then blocked with 10% NGS for 1 h, exposed to Alexa 488-conjugated anti-rabbit secondary antibody (1:200, Jackson ImmunoResearch Labs) for 1 h under the nonpermeabilized condition, and then internalized α5 was labeled with Alexa 555-conjugated anti-rabbit secondary antibody (1:1000, Thermo Fisher Scientific) for 1h after permeabilization in PBS containing 0.25% Triton X-100 and blocking in 10% NGS. Coverslips were washed for three times with PBS and mounted with Fluoromount-G.
Fluorescence images were acquired on a Zeiss LSM 880 laser scanning confocal microscope with a 63 × 1.4 NA oil immersion objective. For quantification, sets of cells were prepared and stained simultaneously. Compared images were acquired at the same time using identical acquisition settings. The fluorescence intensity was analyzed using ImageJ. The results were based on the number of neurons from at least three independent experiments.
Surface cell biotinylation of hippocampal slices
Hippocampal slices were prepared from 6–8 weeks mice as described (Li et al., 2017). Surface expression of GluN2A, GluN2B, α1-GABAAR and α5-GABAAR was quantitated as described. Briefly, acute hippocampal slices were labeled for 30 min at 4°C with 1 mg/ml sulfo-NHS-SS biotin (ThermoFisher Scientific). Membranes were prepared and the biotinylated proteins were precipitated with streptavidin agarose resin (ThermoFisher Scientific) and detected by western blot.
Kainic acid-induced seizure model
Young adult male mice (6–8 weeks old) were administered an intraperitoneal (i.p.) injection of kainic acid (KA, Abcam) dissolved in 0.9% saline solution at 20 mg/kg body weight. Seizure score was evaluated at 0 h, 0.5 h, 1 h and 24 h after KA injection according to the modified Racine scale (Racine, 1972): stage 0, normal behavior; stage 1, immobility and rigidity; stage 2, repetitive behaviors, head nodding or bobbing; stage 3, Forelimb clonus with partial or intermittent rearing; stage 4, continuous rearing and falling; stage 5, severe clonic-tonic seizures; stage 6, death. The expression levels of GluN2A, GluN2B and GABAARs in hippocampi were examined at 1 h or 24 h after KA injection. To examine the effects of GluN2A- and GluN2B-containing receptors on the tonic inhibition in KA-induced seizure model, mice were injected with 10 mg/kg NVP (Gordillo-Salas et al., 2018), 10 mg/kg Ifen (Raybuck et al., 2017) or saline 1 h prior to KA injection and were sacrificed for electrophysiological recordings at 1 h or 24 h after KA injection.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Statistical analysis was performed in GraphPad Prism 8.0 software. Normality distribution was tested by the Shapiro-Wilk test before carrying out a subsequent statistical test. Direct comparisons between two groups were made using two-tailed Student’s t test or Mann-Whitney U test. Multiple comparisons were performed using one-way ANOVA, Kruskal-Wallis test or two-way ANOVA with corrections for multiple comparisons test (see figure legends for specifics).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Chicken Polyclonal Anti-GFP | Abcam | Cat# ab13970, RRID: AB_300798 |
| Rabbit Polyclonal Anti-Glutamate Receptor NMDAR2A (GluN2A) | Sigma-Aldrich | Cat# M264, RRID: AB_260485 |
| Rabbit Polyclonal Anti-Glutamate Receptor NMDAR2B (GluN2B) | Sigma-Aldrich | Cat# M265, RRID: AB_260487 |
| Rabbit Polyclonal Anti-Phospho-Ezrin (Thr567)/Radixin(Thr564)/Moesin (Thr558) (p-ERM) | Cell Signaling Technology | Cat# 3141, RRID: AB_330232 |
| Rabbit Polyclonal Anti-Ezrin/Radixin/Moesin (ERM) | Cell Signaling Technology | Cat# 3142, RRID: AB_2100313 |
| Rabbit Polyclonal Anti-GABA(A) α5 Receptor | Synaptic Systems | Cat# 224503, RRID: AB_2619944 |
| Rabbit Polyclonal Anti-GABA(A) α1 Receptor | Millipore | Cat# 06–868, RRID: AB_310272 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| NeuroMag reagent | Oz Biosciences | Cat# NM51000 |
| CalPho Mammalian Transfection Kit | Takara | Cat# 631312 |
| Bicuculline | Abcam | Cat# ab120110 |
| D-APV | Abcam | Cat# ab120003 |
| DNQX | Alomone labs | Cat# D-131 |
| Tetrodotoxin (TTX) | Alomone Labs | Cat# T-550 |
| Picrotoxin | Sigma-Aldrich | Cat# P1675 |
| NVP-AAM077 Tetrasodium Hydrate | Sigma-Aldrich | Cat# 5.04528 |
| Ifenprodil (+)-tartrate salt | Sigma-Aldrich | Cat# I2892 |
| Kainic acid | Abcam | Cat# ab120100 |
| 4,5,6,7-tetrahydroisoxazolo(5,4-c) pyridin-3-ol (THIP) | Santa Cruz | Cat# SC204342 |
| Q5 Site-Directed Mutagenesis Kit | NEB | Cat# E0554S |
| EZ-Link Sulfo-NHS-SS-Biotin | Thermo Fisher Scientific | Cat# 21331 |
| Pierce Glutathione Agarose | Thermo Fisher Scientific | Cat# 16101 |
|
| ||
| Experimental models: cell lines | ||
|
| ||
| Primary cultures of hippocampal neurons | This paper | N/A |
| HEK293T | ATCC | Cat# CRL-1126 |
|
| ||
| Experimental models: organisms/strains | ||
|
| ||
| C57BL/6N mice | Charles River | Cat# 027 |
|
| ||
| Oligonucleotides | ||
|
| ||
| sgRNA targeting sequence: mouse GluN2A: CGACGTGACAGAACGCGAAC | This paper | N/A |
| sgRNA targeting sequence: mouse GluN2B: GTCTGACCGGAAGATCCAGG | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pRK5-GFP-GluN2A | Dr. Katherine Roche (NIH) | N/A |
| pRK5-GFP-GluN2B | Dr. Katherine Roche (NIH) | N/A |
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | Addgene | Cat# 62988 |
| pSpCas9(BB)-2A-GFP (PX458) | Addgene | Cat# 48138 |
| GluN2A sgRNA | This paper | N/A |
| GluN2B sgRNA | This paper | N/A |
| sgRNA-resistant GluN2A | This paper | N/A |
| sgRNA-resistant GluN2B | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism 8.0 | GraphPad | https://www.graphpad.com |
| Igor Pro | Wavemetrics | https://www.wavemetrics.com |
Highlights.
GluN2A- and GluN2B-NMDA receptors differentially regulate tonic inhibition
NMDA receptor subunit-specific modulation of α5-GABAAR trafficking
GluN2A-NMDARs are critical for homeostatic plasticity of tonic inhibition
Regulation of tonic inhibition by NMDA receptors in a kainate-induced seizure model
ACKNOWLEDGMENTS
We are grateful to all members from Lu laboratory for critical comments on the manuscript. We thank Dr. Ryan Shepard for editing the manuscript. This work was supported by the NIH/NINDS Intramural Research Program (to W.L.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109960.
REFERENCES
- Barria A, and Malinow R (2005). NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron 48, 289–301. [DOI] [PubMed] [Google Scholar]
- Belelli D, Harrison NL, Maguire J, Macdonald RL, Walker MC, and Cope DW (2009). Extrasynaptic GABAA receptors: form, pharmacology, and function. J. Neurosci. 29, 12757–12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady ML, and Jacob TC (2015). Synaptic localization of α5 GABA (A) receptors via gephyrin interaction regulates dendritic outgrowth and spine maturation. Dev. Neurobiol. 75, 1241–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley SG, and Mody I (2012). Extrasynaptic GABA(A) receptors: their function in the CNS and implications for disease. Neuron 73, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraiscos VB, Elliott EM, You-Ten KE, Cheng VY, Belelli D, Newell JG, Jackson MF, Lambert JJ, Rosahl TW, Wafford KA, et al. (2004). Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by alpha5 subunit-containing gamma-aminobutyric acid type A receptors. Proc. Natl. Acad. Sci. USA 101, 3662–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterton JE, Awobuluyi M, Premkumar LS, Takahashi H, Talantova M, Shin Y, Cui J, Tu S, Sevarino KA, Nakanishi N, et al. (2002). Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature 415, 793–798. [DOI] [PubMed] [Google Scholar]
- Chen Q, He S, Hu XL, Yu J, Zhou Y, Zheng J, Zhang S, Zhang C, Duan WH, and Xiong ZQ (2007). Differential roles of NR2A- and NR2B-containing NMDA receptors in activity-dependent brain-derived neurotrophic factor gene regulation and limbic epileptogenesis. J. Neurosci. 27, 542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Lu TJ, Chen XJ, Zhou Y, Chen Q, Feng XY, Xu L, Duan WH, and Xiong ZQ (2008). Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke 39, 3042–3048. [DOI] [PubMed] [Google Scholar]
- Chiu CQ, Martenson JS, Yamazaki M, Natsume R, Sakimura K, Tomita S, Tavalin SJ, and Higley MJ (2018). Input-Specific NMDAR-Dependent Potentiation of Dendritic GABAergic Inhibition. Neuron 97, 368–377.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly WM, Fyson SJ, Errington AC, McCafferty CP, Cope DW, Di Giovanni G, and Crunelli V (2013). GABAB Receptors Regulate Extrasynaptic GABAA Receptors. J. Neurosci. 33, 3780–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cserép C, Szabadits E, Szőnyi A, Watanabe M, Freund TF, and Nyiri G (2012). NMDA receptors in GABAergic synapses during postnatal development. PLoS ONE 7, e37753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F, Bai Y, Cheng Q, Yu S, Cheng M, Wu Y, Zhang X, Liang X, and Gu X (2021). Bidentatide, a Novel Plant Peptide Derived from Achyranthes bidentata Blume: Isolation, Characterization, and Neuroprotection through Inhibition of NR2B-Containing NMDA Receptors. Int. J. Mol. Sci. 22, 7977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong YN, Wu HY, Hsu FC, Coulter DA, and Lynch DR (2006). Developmental and cell-selective variations in N-methyl-D-aspartate receptor degradation by calpain. J. Neurochem. 99, 206–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, and Nusser Z (2005). Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat. Rev. Neurosci. 6, 215–229. [DOI] [PubMed] [Google Scholar]
- Gaïarsa JL (2004). Plasticity of GABAergic synapses in the neonatal rat hippocampus. J. Cell. Mol. Med. 8, 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambrill AC, and Barria A (2011). NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc. Natl. Acad. Sci. USA 108, 5855–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Kang Y, Cassidy RM, Moon KM, Lewis R, Wong ROL, Foster LJ, and Craig AM (2018). Clptm1 Limits Forward Trafficking of GABAA Receptors to Scale Inhibitory Synaptic Strength. Neuron 97, 596–610.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Mann EO, and Mody I (2008). Which GABA(A) receptor subunits are necessary for tonic inhibition in the hippocampus? J. Neurosci. 28, 1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonda S, Giesen J, Sieberath A, West F, Buchholz R, Klatt O, Ziebarth T, Räk A, Kleinhubbert S, Riedel C, et al. (2020). GluN2B but Not GluN2A for Basal Dendritic Growth of Cortical Pyramidal Neurons. Front. Neuroanat. 14, 571351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordillo-Salas M, Pilar-Cuéllar F, Auberson YP, and Adell A (2018). Signaling pathways responsible for the rapid antidepressant-like effects of a GluN2A-preferring NMDA receptor antagonist. Transl. Psychiatry 8, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlewicz A, Pijet B, Orlova K, Kaczmarek L, and Knapska E (2021). Epileptiform GluN2B–driven excitation in hippocampus as a therapeutic target against temporal lobe epilepsy. bioRxiv. 10.1101/2021.06.30.450508. [DOI] [PubMed] [Google Scholar]
- Gu X, and Lu W (2018). Genetic deletion of NMDA receptors suppresses GABAergic synaptic transmission in two distinct types of central neurons. Neurosci. Lett. 668, 147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Zhou L, and Lu W (2016). An NMDA Receptor-Dependent Mechanism Underlies Inhibitory Synapse Development. Cell Rep. 14, 471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen V, Holten AT, and Storm-Mathisen J (2004). GABAergic synapses in hippocampus exocytose aspartate on to NMDA receptors: quantitative immunogold evidence for co-transmission. Mol. Cell. Neurosci. 26, 156–165. [DOI] [PubMed] [Google Scholar]
- Han W, Shepard RD, and Lu W (2021). Regulation of GABAARs by Transmembrane Accessory Proteins. Trends Neurosci. 44, 152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausrat TJ, Muhia M, Gerrow K, Thomas P, Hirdes W, Tsukita S, Heisler FF, Herich L, Dubroqua S, Breiden P, et al. (2015). Radixin regulates synaptic GABAA receptor density and is essential for reversal learning and short-term memory. Nat. Commun. 6, 6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneberger C, Jüttner R, Schmidt SA, Walter J, Meier JC, Rothe T, and Grantyn R (2005). GluR- and TrkB-mediated maturation of GABA receptor function during the period of eye opening. Eur. J. Neurosci. 21, 431–440. [DOI] [PubMed] [Google Scholar]
- Hines RM, Davies PA, Moss SJ, and Maguire J (2012). Functional regulation of GABAA receptors in nervous system pathologies. Curr. Opin. Neurobiol. 22, 552–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holter NI, Zylla MM, Zuber N, Bruehl C, and Draguhn A (2010). Tonic GABAergic control of mouse dentate granule cells during postnatal development. Eur. J. Neurosci. 32, 1300–1309. [DOI] [PubMed] [Google Scholar]
- Horn ME, and Nicoll RA (2018). Somatostatin and parvalbumin inhibitory synapses onto hippocampal pyramidal neurons are regulated by distinct mechanisms. Proc. Natl. Acad. Sci. USA 115, 589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser CR, and Esclapez M (2003). Downregulation of the alpha5 subunit of the GABA(A) receptor in the pilocarpine model of temporal lobe epilepsy. Hippocampus 13, 633–645. [DOI] [PubMed] [Google Scholar]
- Jacob TC, Moss SJ, and Jurd R (2008). GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 9, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch N, Liebmann L, Guenther M, Hübner CA, Frahm C, and Witte OW (2016). Reduced tonic inhibition after stroke promotes motor performance and epileptic seizures. Sci. Rep. 6, 26173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee V, and Maguire J (2014). The impact of tonic GABAA receptor-mediated inhibition on neuronal excitability varies across brain region and cell type. Front. Neural Circuits 8, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Han W, Pelkey KA, Duan J, Mao X, Wang YX, Craig MT, Dong L, Petralia RS, McBain CJ, and Lu W (2017). Molecular Dissection of Neuroligin 2 and Slitrk3 Reveals an Essential Framework for GABAergic Synapse Development. Neuron 96, 808–826.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, and Wang YT (2007). NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci. 27, 2846–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loebrich S, Bähring R, Katsuno T, Tsukita S, and Kneussel M (2006). Activated radixin is essential for GABAA receptor alpha5 subunit anchoring at the actin cytoskeleton. EMBO J. 25, 987–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher B, Fuchs T, and Kilpatrick CL (2011). GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 70, 385–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareš P, Kozlová L, Mikulecká A, and Kubová H (2021). The GluN2B-Selective Antagonist Ro 25–6981 Is Effective against PTZ-Induced Seizures and Safe for Further Development in Infantile Rats. Pharmaceutics 13, 1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden KC, Beattie JB, Friedenthal J, and Carroll RC (2007). NMDA receptor activation potentiates inhibitory transmission through GABA receptor-associated protein-dependent exocytosis of GABA(A) receptors. J. Neurosci. 27, 14326–14337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBain CJ, and Mayer ML (1994). N-methyl-D-aspartic acid receptor structure and function. Physiol. Rev. 74, 723–760. [DOI] [PubMed] [Google Scholar]
- McKay S, Ryan TJ, McQueen J, Indersmitten T, Marwick KFM, Hasel P, Kopanitsa MV, Baxter PS, Martel MA, Kind PC, et al. (2018). The Developmental Shift of NMDA Receptor Composition Proceeds Independently of GluN2 Subunit-Specific GluN2 C-Terminal Sequences. Cell Rep. 25, 841–851.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, and Seeburg PH (1992). Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science 256, 1217–1221. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, and Seeburg PH (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540. [DOI] [PubMed] [Google Scholar]
- Morimoto K, Fahnestock M, and Racine RJ (2004). Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog. Neurobiol. 73, 1–60. [DOI] [PubMed] [Google Scholar]
- Müller L, Tokay T, Porath K, Köhling R, and Kirschstein T (2013). Enhanced NMDA receptor-dependent LTP in the epileptic CA1 area via upregulation of NR2B. Neurobiol. Dis. 54, 183–193. [DOI] [PubMed] [Google Scholar]
- Naylor DE, Liu H, and Wasterlain CG (2005). Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J. Neurosci. 25, 7724–7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor DE, Liu H, Niquet J, and Wasterlain CG (2013). Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol. Dis. 54, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan G, Chen Z, Zheng H, Zhang Y, Xu H, Bu G, Zheng H, and Li Y (2018). Compensatory Mechanisms Modulate the Neuronal Excitability in a Kainic Acid-Induced Epilepsy Mouse Model. Front. Neural Circuits 12, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, and Zhou Q (2013). NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400. [DOI] [PubMed] [Google Scholar]
- Peng YR, Zeng SY, Song HL, Li MY, Yamada MK, and Yu X (2010). Postsynaptic spiking homeostatically induces cell-autonomous regulation of inhibitory inputs via retrograde signaling. J. Neurosci. 30, 16220–16231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna A, Wang DS, Yu J, Lecker I, Brown PMGE, Bowie D, and Orser BA (2014). Hydrogen peroxide increases GABAA receptor-mediated tonic current in hippocampal neurons. J. Neurosci. 34, 10624–10634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrini EM, Ravasenga T, Hausrat TJ, Iurilli G, Olcese U, Racine V, Sibarita JB, Jacob TC, Moss SJ, Benfenati F, et al. (2014). Synaptic recruitment of gephyrin regulates surface GABAA receptor dynamics for the expression of inhibitory LTP. Nat. Commun. 5, 3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 32, 281–294. [DOI] [PubMed] [Google Scholar]
- Rajgor D, Purkey AM, Sanderson JL, Welle TM, Garcia JD, Dell’Acqua ML, and Smith KR (2020). Local miRNA-Dependent Translational Control of GABAAR Synthesis during Inhibitory Long-Term Potentiation. Cell Rep. 31, 107785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybuck JD, Hargus NJ, and Thayer SA (2017). A GluN2B-Selective NMDAR Antagonist Reverses Synapse Loss and Cognitive Impairment Produced by the HIV-1 Protein Tat. J. Neurosci. 37, 7837–7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba RS, Kretschmannova K, and Moss SJ (2012). Activity-dependent phosphorylation of GABAA receptors regulates receptor insertion and tonic current. EMBO J. 31, 2937–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samba Reddy D (2017). Sex differences in the anticonvulsant activity of neurosteroids. J. Neurosci. Res. 95, 661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda FJ, Bustos FJ, Inostroza E, Zúñiga FA, Neve RL, Montecino M, and van Zundert B (2010). Differential roles of NMDA Receptor Subtypes NR2A and NR2B in dendritic branch development and requirement of RasGRF1. J. Neurophysiol. 103, 1758–1770. [DOI] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, and Jan LY (1994). Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368, 144–147. [DOI] [PubMed] [Google Scholar]
- Shipton OA, and Paulsen O (2013). GluN2A and GluN2B subunit-containing NMDA receptors in hippocampal plasticity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibarov DA, Abushik PA, Giniatullin R, and Antonov SM (2016). GluN2A Subunit-Containing NMDA Receptors Are the Preferential Neuronal Targets of Homocysteine. Front. Cell. Neurosci. 10, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S, McNeill RB, and Colbran RJ (2000). Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem. 275, 23798–23806. [DOI] [PubMed] [Google Scholar]
- Sun Y, Xu Y, Cheng X, Chen X, Xie Y, Zhang L, Wang L, Hu J, and Gao Z (2018). The differences between GluN2A and GluN2B signaling in the brain. J. Neurosci. Res. 96, 1430–1443. [DOI] [PubMed] [Google Scholar]
- Szabadits E, Cserép C, Szonyi A, Fukazawa Y, Shigemoto R, Watanabe M, Itohara S, Freund TF, and Nyiri G (2011). NMDA receptors in hippocampal GABAergic synapses and their role in nitric oxide signaling. J. Neurosci. 31, 5893–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W, Higgs MH, Spain WJ, and Ransom CB (2013). Postsynaptic GABAB receptors enhance extrasynaptic GABAA receptor function in dentate gyrus granule cells. J. Neurosci. 33, 3738–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G (2012). Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol. 4, a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira M, Yong XLH, Roche KW, and Anggono V (2020). Regulation of NMDA glutamate receptor functions by the GluN2 subunits. J. Neurochem. 154, 121–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent P, and Mulle C (2009). Kainate receptors in epilepsy and excitotoxicity. Neuroscience 158, 309–323. [DOI] [PubMed] [Google Scholar]
- Vithlani M, Terunuma M, and Moss SJ (2011). The dynamic modulation of GABA(A) receptor trafficking and its role in regulating the plasticity of inhibitory synapses. Physiol. Rev. 91, 1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, He X, Li T, Shu Y, Qi S, and Luan G (2017). Anti-epileptic effect of ifenprodil on neocortical pyramidal neurons in patients with malformations of cortical development. Exp. Ther. Med. 14, 5757–5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, and Lynch DR (2005). N-methyl-D-aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist 11, 37–49. [DOI] [PubMed] [Google Scholar]
- Wu QJ, and Tymianski M (2018). Targeting NMDA receptors in stroke: new hope in neuroprotection. Mol. Brain 11, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K, Han W, Tian Q, Li Y, and Lu W (2021). Activity- and sleep-dependent regulation of tonic inhibition by Shisa7. Cell Rep. 34, 108899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyrošlak M, Lebida K, and Mozrzymas JW (2021). Induction of Inhibitory Synaptic Plasticity Enhances Tonic Current by Increasing the Content of α5-Subunit Containing GABAA Receptors in Hippocampal Pyramidal Neurons. Neuroscience 467, 39–46. [DOI] [PubMed] [Google Scholar]
- Zou G, Chen Q, Chen K, Zuo X, Ge Y, Hou Y, Pan T, Pan H, Liu D, Zhang L, and Xiong W (2019). Human Hyperekplexic Mutations in Glycine Receptors Disinhibit the Brainstem by Hijacking GABAA Receptors. iScience 19, 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.