

Abstract
Nipah virus (NiV) and Hendra virus (HeV) are highly pathogenic, bat-borne paramyxoviruses in the genus Henipavirus that cause severe and often fatal acute respiratory and/or neurologic diseases in humans and livestock. There are currently no approved antiviral therapeutics or vaccines for use in humans to treat or prevent NiV or HeV infection. To facilitate development of henipavirus antivirals, a high-throughput screening (HTS) platform was developed based on a well-characterized recombinant version of the nonpathogenic Henipavirus, Cedar virus (rCedV). Using reverse genetics, a rCedV encoding firefly luciferase (rCedV-Luc) was rescued and its utility evaluated for high-throughput antiviral compound screening. The luciferase reporter gene signal kinetics of rCedV-Luc in different human cell lines was characterized and validated as an authentic real-time measure of viral growth. The rCedV-Luc platform was optimized as an HTS assay that demonstrated high sensitivity with robust Z’ scores, excellent signal-to-background ratios and coefficients of variation. Eight candidate compounds that inhibited rCedV replication were identified for additional validation and demonstrated that 4 compounds inhibited authentic NiV-Bangladesh replication. Further evaluation of 2 of the 4 validated compounds in a 9-point dose response titration demonstrated potent antiviral activity against NiV-Bangladesh and HeV, with minimal cytotoxicity. This rCedV reporter can serve as a surrogate yet authentic BSL-2 henipavirus platform that will dramatically accelerate drug candidate identification in the development of anti-henipavirus therapies.
Keywords: High-throughput screening assay, antiviral, Cedar virus, Henipavirus, inhibitor, luciferase assay, reverse genetics
1. Introduction
Nipah virus (NiV) and Hendra virus (HeV), members of the genus Henipavirus, are highly pathogenic zoonotic paramyxoviruses capable of causing a severe and often fatal acute respiratory and/or neurological disease in a wide-range of naturally and experimentally infected mammalian hosts, including humans (reviewed in (Anonymous, 2018; Broder et al., 2016)). The natural host reservoirs for both NiV and HeV have been shown to be fruit bats in the genus Pteropus, and outbreaks of both NiV and HeV have coincided with the geographical distribution of these bats (Chua et al., 2002; Clayton et al., 2013; Halpin et al., 2011; Halpin et al., 2000).
HeV was discovered in 1994 as the cause of an outbreak of severe respiratory disease in horses and humans in the Brisbane suburb of Hendra, Australia (Murray et al., 1995; Selvey et al., 1995). In 1998 in Peninsular Malaysia, an outbreak of encephalitis among pig farmers, along with reports of respiratory and neurological illness in the farmed pigs, was later attributed to a novel virus genetically related to HeV, now known as NiV (Chua et al., 2000; Chua et al., 1999). Although the NiV strain, NiV-Malaysia (NiV-M), has not re-emerged in Malaysia, an outbreak of severe and fatal neurological disease attributed to NiV-M occurred in the Philippines in 2014 among people and horses with associated horse-to-human and human-to-human transmission (Ching et al., 2015). In 2001, a genetically similar but distinct strain of NiV, NiV-Bangladesh (NiV-B), was retrospectively identified as the causative agent of an outbreak of fatal encephalitis in people in Bangladesh and India (Harcourt et al., 2005; Harit et al., 2006; Hsu et al., 2004). Since then, there have been nearly annual occurrences of human NiV-B infections in this region (Nikolay et al., 2019), along with a significant outbreak in 2018 in Kerala, India having a case fatality rate of 91% (Arunkumar et al., 2019). Characterization of the human NiV isolate from Kerala revealed it to have 96.15% nucleotide identity to the NiV-B strain (Yadav et al., 2019). The most recent occurrences of HeV have been in 2019 (Anonymous, 2019) and again in June of 2020 (Anonymous, 2020a), while NiV appeared in early 2020 in Bangladesh (Anonymous, 2020b). The outbreaks of NiV in Bangladesh and India have been associated with high human case fatality rates and human-to-human transmission of infection, and the predominant source of infection in Bangladesh is the consumption of raw date palm sap (Homaira et al., 2010a; Homaira et al., 2010b; Luby et al., 2009; Nikolay et al., 2019; Rahman et al., 2012).
NiV and HeV are classified as biosafety level-4 (BSL-4) restricted pathogens and both remain transboundary threats to livestock and people throughout South Asia and Australia. NiV and henipaviral diseases have been categorized by the World Health Organization (WHO) as an epidemic threat needing urgent research and countermeasure development and are included in the WHO R&D Blueprint list of priority pathogens with epidemic potential (Sweileh, 2017). Presently, there are no prophylactic or therapeutic treatment options for henipavirus infections approved for use in humans. A subunit vaccine, Equivac® HeV, has been available for use in horses in Australia since 2012 (Middleton et al., 2014) for the prevention of HeV infection. This equine vaccine received full registration by the Australian Pesticides and Veterinary Medicines Authority in 2015, with all vaccinated horses receiving a microchip with a database being maintained and is the first commercially deployed vaccine against a BSL-4 agent (Amaya and Broder, 2020). Although a human monoclonal antibody (mAb) m102.4 has successfully completed a phase I safety trial in Australia (Playford et al., 2020), developing vaccines or therapeutics against henipaviruses for use in people remains a high priority.
A significant challenge in research and drug discovery for highly pathogenic viruses, such as NiV and HeV, is the requirement for BSL-4 containment suites for experimentation. The isolation of Cedar virus (CedV), a nonpathogenic phylogenetically related henipavirus from urine samples of P. alecto and P. poliocephalus in Australia (Marsh et al., 2012), provides an opportunity to utilize it as a surrogate to study henipavirus at BSL-2 facilities (Laing et al., 2018). Interestingly, unlike NiV and HeV that use ephrin-B2 and ephrin-B3 as entry receptors (Bishop et al., 2007; Bonaparte et al., 2005; Negrete et al., 2005; Negrete et al., 2006), CedV displays broader receptor usage by utilizing human ephrin-B2, ephrin-B1, ephrin-A2 and ephrin-A5 and murine-A1 receptors (Laing et al., 2019). CedV is nonpathogenic in experimental animal models of NiV and HeV infection and disease including the ferret (Marsh et al., 2012) and hamster (Schountz et al., 2019). CedV attenuation has been attributed to the lack of an RNA editing site in the P gene for the production of the interferon antagonist proteins, V and W (Ciancanelli et al., 2009; Kulkarni et al., 2009; Lieu et al., 2015; Marsh et al., 2012; Schountz et al., 2019; Shaw, 2009; Shaw et al., 2004; Uchida et al., 2018).
Here, a high-throughput screening (HTS) assay was developed based on recombinant infectious CedV (rCedV) to facilitate the identification of compounds capable of inhibiting the replication of henipaviruses. A reverse genetics approach was used to rescue a luciferase-expressing version of rCedV (rCedV-Luc) and demonstrated that luciferase expression is a faithful representation of virus growth. The luciferase reporter gene signal kinetics in different human cell lines were defined and adapted the HTS platform to 96- and 384-well formats with excellent signal-to-background (S/B) ratios and Z’ scores. The utility of the rCedV-Luc HTS platform was demonstrated by screening a large compound library resulting in the identification of several candidate compounds that inhibit the replication of both rCedV and infectious wild-type NiV-B. The low toxicities and high antiviral potencies of the identified anti-henipavirus compounds demonstrated that rCedV-Luc as a surrogate system can be used in BSL-2 laboratories to facilitate development of henipavirus antivirals.
2. Materials and methods
2.1. Cells and viruses
BSR-T7/5 (a BHK-derived cell line stably expressing T7 RNA polymerase (Buchholz et al., 1999), Vero E6 (ATCC CCL-81), Vero 76 (ATCC CRL-1587), HeLa (ATCC CCL-2) and HEK 293T (ATCC CRL-3216) cell lines were maintained in Dulbecco’s modified eagle media (DMEM) (Quality Biological; Gaithersburg, MD) supplemented with 10% cosmic calf serum (CCS) and 1% L-glutamine (Quality Biological) (cell culture medium). A549 cells (ATCC CCL-185) were maintained in Gibco F-12 Nutrient K (Kaighn’s Modification of Ham’s F-12) Medium (Thermo Fisher Scientific; Waltham, MA) supplemented with 10% CCS. All incubations were conducted at 37 °C, 5% CO2, unless otherwise stated. Infections with rCedV were conducted under BSL-2 biosafety conditions. Laboratory manipulation guidelines and standard operating procedures for rCedV under BSL-2 conditions were developed and this work was reviewed and approved by the Uniformed Services University (USU) Institutional Biosafety Committee in accordance with NIH guidelines. All studies with authentic infectious NiV-B and HeV were performed in the BSL-4 facility at the University of Texas Medical Branch at Galveston, TX. There were four mutations of sufficient frequency to note between the P2 stock of NiV-B and the reference sequence (GenBank Accession number AY988601.1). Of these, one was non-coding, and the other three led to single amino acid changes: one in the M protein and two in the F protein (Mire et al., 2016). HeV (GenBank Accession number NC_001906) was obtained from a patient from the 1994 outbreak in Australia and was provided by Dr. Thomas Ksiazek (Mire et al., 2019).
2.2. Compound library
A library of 10,000 diverse small molecules was provided by the University of Illinois at Chicago Research Resources Center (UIC RRC). These compounds were carefully chosen from the ChemDiv (San Diego, CA) collection of screening compounds. The library contained compounds with drug-like properties and balance in terms of diversity of chemical scaffolds. The library included several representative analogs for each of the scaffolds and excluded pan-assay interference compounds (PAINS) (Baell and Holloway, 2010). Master compound stocks were prepared in dimethyl sulfoxide (DMSO).
2.3. Cloning and rescue of rCedV-Luc
A detailed account of the rCedV antigenome clone has been previously described (Laing et al., 2018). The pGL4–32-Luc2P plasmid was a kind gift from Dr. B. Schaefer, USU. To amplify the firefly (Photinus pyralis) luciferase (Luc) gene from pGL4–32-Luc2P, primers were designed to include CedV untranslated regions: transcriptional P stop, intergenic region, and transcriptional M start sequence (TTAGAAAAAACTTAGGATCCCAG). An additional start codon was added to the 5’ end of the luciferase open reading frame to maintain the “rule of six” (Pfaller et al., 2015). The Luc gene was inserted at the unique MluI restriction site between CedV P and M genes. The resultant antigenome was confirmed by sequencing. BSR-T7/5 cells were co-transfected with pCMV-CedV helper plasmids pCMV-CedV-N, pCMV-CedV -P and pCMV-CedV-L together with the pOLTV5-rCedV-Luc antigenome in accordance with established protocols (Laing et al., 2018). To propagate the rescued virus, supernatant was passaged onto Vero E6 cells. When maximal cytopathic effect (CPE) and/or syncytia were observed, viral supernatants were collected, clarified and concentrated by ultracentrifugation (131,101.2 × g, 27,000 rpm; 2 h) through a sucrose cushioned buffer. Stock rCedV-Luc was serially diluted and incubated with Vero E6 cells to determine infectious viral titer by CPE-based plaque assay (Laing et al., 2018; Weingartl et al., 2006). Briefly, 200 μl/well of virus inoculum was added to Vero E6 cells (5 × 105 cells/well) in 12-well cell culture plates and incubated for 1 h. Two ml of 2% carboxymethylcellulose sodium salt (medium viscosity) (Sigma-Aldrich; St. Louis, MO) + DMEM supplemented with 5% CCS and 1% L-glutamine (plaque assay medium) was then added to each well and incubated for 5 days. The plates were fixed with 4% formaldehyde and then stained with 0.5% crystal violet solution (80% methanol) in phosphate buffered saline (PBS). Plaques were counted to determine infectious viral titers as plaque forming units per ml (PFU/ml).
2.4. Replication kinetics of rCedV
Vero E6 cells (5 × 104 cells/well) in 96-well cell culture plates were infected at a multiplicity of infection (MOI) of 0.01 with either rCedV or rCedV-Luc. At 0, 24, 48 and 72 hours post infection (hpi) supernatants were collected for plaque assay and cells were lysed for intracellular luciferase activity. Plaque assays were performed as described in section 2.3. Intracellular luciferase activity was determined with the Steady-Glo® Luciferase Assay System (Promega; Madison, WI) in a 1:1 mixture with cell culture medium. After a 10 min incubation at room temperature, the homogenate was transferred to a white opaque 96-well cell culture plate, Nunc™ F96 MicroWell™ White Polystyrene Plate (ThermoFisher Scientific) and read using the GloMax® – Multi Detection System (Promega).
2.5. Optimization of the cell-based HTS assay in a 96-well format
HEK 293T, A549, HeLa and Vero E6 cells were seeded at a density of 2 × 104 cells/well in white opaque 96-well cell culture plates. The next day, cells were mock infected or infected with rCedV-Luc at varying MOIs (0.01, 0.1 or 1.0). At 0, 24, 48 and 72 hpi, cells were lysed and luciferase activity determined as described in section 2.4.
Murine monoclonal antibody (mAb) m14F3, is a CedV G specific virus neutralizing mAb of IgG2 subclass (Laing et al., 2018). Equal volumes of cell culture medium containing rCedV-Luc (MOI: 0.1) were incubated with or without m14F3 (20 μg/ml) for 1 h. The virus only or mAb-virus mixture was added to pre-seeded HEK 293T cells (2 × 104 cells/well) in white opaque 96-well cell culture plates and incubated for 1 h. Following removal of the overlay all cells were washed once with cell culture medium. Fresh cell culture medium with or without m14F3 (20 μg/ml) was added to the wells and incubated for an additional 48 h. Cells were then lysed and luciferase signal determined as described in section 2.4.
2.6. Automated high-throughput screening assay in a 384-well format, primary screen
Compounds and dilutions were prepared in cell culture medium with the high-throughput liquid handler in a tissue culture biosafety cabinet (PerkinElmer JANUS Automated Workstation). HEK 293T cells were seeded in white opaque 384-well culture plates (PerkinElmer; Waltham, MA) at 4 × 103 cells/well in 20 μl screen medium (DMEM supplemented with 10% fetal bovine serum (Gibco; Gaithersburg, MD), 100 μg/ml streptomycin, and 100 U penicillin (Invitrogen; Carlsbad, CA)). The next day, 10 μl of each compound from the 10,000 compound screening library (final concentration of 10 μM per well) followed by 10 μl of rCedV-Luc diluted in screen medium (MOI: 0.05), were added to the plates. In each plate, positive controls (rCedV-Luc with 20 μg/ml m14F3) and negative controls (rCedV-Luc) were placed in alternate wells in columns 1, 2, 23 and 24. Plates were further incubated for 48 h. Cells were lysed using the Neolite Reporter Gene Assay System (PerkinElmer) as per the manufacturer’s instructions. Luciferase activity was examined by reading the luminescent signal on an EnVision plate reader (PerkinElmer). An 80% inhibition of luciferase activity was used as the criterion for designating a compound as a “hit”. The data were normalized by the average signal of the negative control wells in each plate.
2.7. Confirmation screen of the automated high-throughput screening assay in a 384-well format
The hits (≥ 80% inhibition) identified in the primary screen were re-screened in a confirmation screen at a concentration of 10 μM. HEK 293T cells (4 × 103 cells/well) were seeded in 384-well plates in 20 μl screen medium. After an overnight incubation, 10 μl of each of the identified compounds was added to the cells, followed by 10 μl of the screen medium alone or in a mixture with rCedV-Luc (MOI: 0.05) and incubated for an additional 48 h. The Neolite substrate (20 μl) or CellTiter-Glo (20 μl) was added to the corresponding plates and rocked for 10 min at room temperature. Luminescent signal was measured as described in section 2.4.
2.8. Dose response titration, 50% inhibitory concentration (IC50) and 50% cytotoxic concentration (CC50) assays
Hit compounds were re-purchased from ChemDiv. For rCedV-Luc infection assays, HEK 293T cells (2 × 104 cells/well) or Vero E6 cells (1.5 × 104 cells/well) were seeded in 100 μl screen medium in 96-well plates. The next day, the screen medium was removed and 50 μl of the selected lead compound (3-fold serial dilution ranging from 15 nM to 100 μM) was added to the cells followed by 50 μl of either screen medium only or a mixture with rCedV-Luc (MOI: 0.05). After a 48 h incubation, the Neolite substrate (50 μl) or CellTiter-Glo (50 μl) was added to the plates and rocked for 10 min at room temperature. The CellTiter-Glo assay was used to determine viability of uninfected HEK 293T cells or Vero E6 cells exposed to 3-fold serial dilutions of the compounds for 48 h. Luminescent signal was measured as described in section 2.6. Sample signals were normalized by signals from the DMSO control wells. The 50% inhibitory concentration (IC50) was determined as the compound concentration at which there was a 50% reduction in luminescence of compound treated infected cells versus DMSO infected control cells. The 50% cytotoxic concentration (CC50) was determined as the compound concentration at which there was a 50% reduction in luminescence units of compound treated cells versus DMSO treated cells. The IC50 and CC50 values were calculated by dose-response curve fitting with GraphPad prism (GraphPad Software Inc.). For NiV-B and HeV infection assays, Vero 76 cells (1.5 × 104 cells/well) were treated with compounds C1 or F1 (15 nM to 100 μM) and then infected with NiV-B or HeV (MOI: 0.01). Control cultures were treated with DMSO. After an incubation period of 48 h, viral supernatants were collected and titrated by plaque assay to determine infectious viral titer (PFU/ml) as described in section 2.3. The percent reduction in progeny PFU/ml titers in the samples containing compounds was calculated by comparing titers obtained in parallel control cultures.
2.9. Virus titer reduction assay
HEK 293T cells (2 × 104 cells/well) pre-seeded in a 96-well plate were incubated with the selected compounds (at the concentration 10 times exceeding the IC50 concentration) with or without rCedV (MOI: 0.05). For the NiV-B and HeV infection assays, Vero 76 cells (1.5 × 104 cells/well) were treated with 11.1 μM of the selected compounds and then infected with NiV-B (MOI: 0.01). Controls cells were treated with DMSO. After an incubation period of 48 h, viral supernatants were collected and titrated by plaque assay to determine infectious viral titer (PFU/ml) as described in section 2.3. Mock infected compound-containing cells were examined microscopically for CPE induced by compound treatment. The percent reduction in PFU/ml titers in the samples containing compounds was compared to that from parallel cultures with virus and DMSO vehicle only.
2.10. Fusion inhibition assay
CHO-K1 (ATCC CCL-61) cell lines were maintained in Dulbecco’s modified eagle media (DMEM) (Quality Biological) supplemented with 10% fetal bovine serum and 1% L-glutamine (Quality Biological). A CHO-K1 cell line stably expressing ephrin-B1 (CHO-B1) was generated by lentiviral transduction methods and selected for with 7.5 μg/ml Puromycin. All incubations were conducted at 37 °C, 5% CO2. The quantitative fusion inhibition assay was based on a dual-split reporter assay (Laing et al., 2019; Navaratnarajah et al., 2016). CHO-K1 cells seeded at a density of 7.5 × 105 cells/well in a 6-well plate were transfected with 750 ng each of CedV F and G and 500 ng of one half of the dual split reporter plasmid. CHO-B1 cells were seeded in a 96-well plate at a density of 1.5 × 104 cells/well and transfected with 120 ng of the other half of the DSP plasmid. Twenty-four h post transfection the CHO-K1 cells in the 6-well plate were re-suspended using Versene (Ethylenediaminetetraacetic acid or EDTA) and 5 × 104 cells and EnduRen (1:500 dilution) were overlaid on the 96-well plate in a total volume of 50 μl/well. Compounds were diluted in medium and added to a final concentration ranging from 5 μM to 20 μM. Controls cells were treated with DMSO.
2.11. Data normalization and analysis
Unless otherwise stated, graphs and images are the average of two independent experiments and are expressed as the arithmetic mean. Standard deviations were calculated and represented accordingly. All statistical analyses were performed with the unpaired, two-tailed Student t-test using GraphPad’s – QuickCalcs software (GraphPad Software Inc.). The quality of each screen was assessed by evaluating the signal-to-background (S/B) ratio, the coefficient of variation (CV), and the Z’ factor. For each plate, the parameters were calculated using the following equations: 1) ; 2) ; 3) . SD represents standard deviation. A robust high-throughput assay is defined as having a Z’ value 1 > Z’ ≥ 0.5 (Zhang et al., 1999) with minimal impact on assay sensitivity (Inglese et al., 2007). CV represents the signal deviation within an assay (Zhang et al., 1999) and should be ≤ 20% (Iversen et al., 2004). Percent inhibition of the compounds was calculated as follows: . Selectivity index was calculated as follows:
3. Results
3.1. Rescue and characterization of rCedV-Luc
A reverse genetics approach was used to generate rCedV expressing luciferase (rCedV-Luc). A firefly luciferase (Luc) gene was inserted between CedV P and M genes in the rCedV antigenome clone by standard restriction digest and ligation methods to yield the luciferase reporter rCedV full-length genome plasmid (Fig. 1A). Successful rescue of rCedV-Luc was demonstrated by observing multiple syncytia on Vero E6 cells inoculated with freeze-thawed cell culture supernatants from BSR-T7/5 cells transfected with rCedV-Luc antigenome and helper plasmids (yellow box in Fig. 1B). The growth kinetics of rCedV-Luc to rCedV was compared (Fig. 1C). Vero E6 cells were infected with either rCedV or rCedV-Luc at an MOI of 0.01. At 24 h intervals, supernatants from infected cells were collected and analyzed by plaque assay for infectious viral titers. At all-time points examined, no statistically significant differences were observed between rCedV and rCedV-Luc, indicating that the recombinant viruses have similar growth kinetics. In parallel, infected cells were lysed and analyzed for intracellular luciferase activity. As seen in Fig. 1C (blue axis), increasing luciferase activity was detected that reached saturation at ~1 × 108 relative luciferase units (RLU) at 72 hpi (blue dashed line). The increase in luciferase signal correlated with the increase in infectious viral titers. These results indicated that introduction of the luciferase reporter gene did not interfere with the growth of the recombinant virus and that luciferase activity is an indicator of viral genome expression.
Fig. 1.
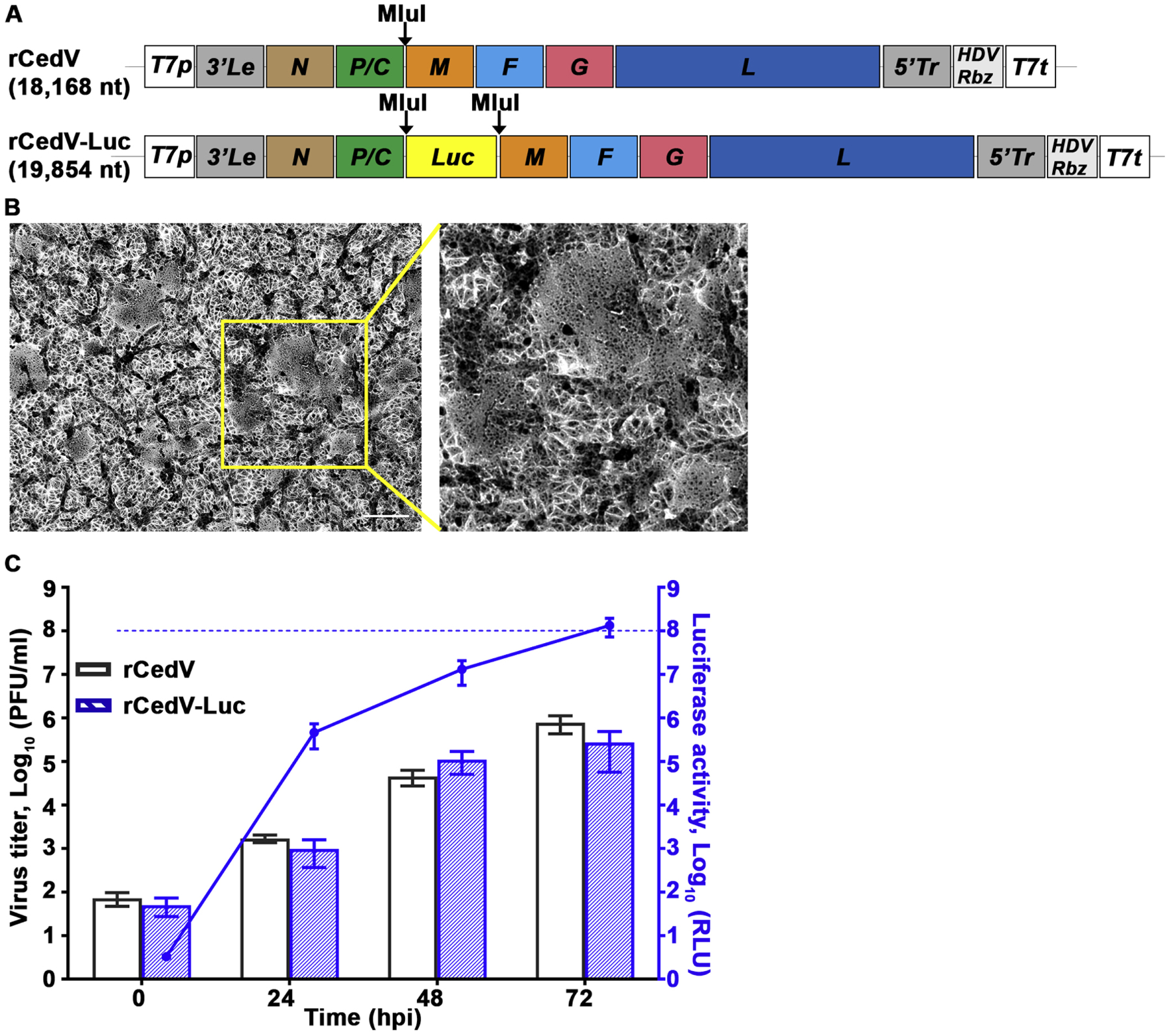
Characterization of recombinant CedV expressing luciferase. A) Schematic representation of rCedV antigenome in the pOLTV5 vector. The firefly luciferase (Luc) gene was inserted at the unique MluI restriction site between the P and M genes to produce rCedV-Luc. To preserve the “rule of six”, an additional start codon was added to the 5’ terminus of the Luc gene. B) At 48 hpi Vero E6 cells infected with rCedV-Luc were fixed with methanol and stained with 0.5% crystal violet solution (80% methanol). Images were captured with a Zeiss Axio Observer A1 inverted microscope using a 5X objective. Yellow box indicates giant cells, and scale bar represents 50 μm. Inset indicates zoomed-in region within the yellow box. C) Vero E6 cells in a 96-well plate were infected with either rCedV or rCedV-Luc at an MOI of 0.01. At 0, 24, 48 and 72 hpi supernatants were collected and viral titers determined by plaque assay and calculated as PFU/ml. Concurrently all infected cells were lysed and relative light units (RLU) were measured and calculated by subtracting the signal of rCedV infected cells from the signal of the rCedV-Luc infected cells and are represented on the right Y-axis in blue. The data represent mean ± standard error from two independent experiments. The blue dashed line represents the upper limit of detection of the luminometer. Viral titers and luciferase activity levels at 0 hpi indicate the lower limit of detection for the plaque assay and the luminometer, respectively.
3.2. Assay development and optimization for high-throughput-screening
The optimization of cell type, virus MOI, and incubation time was conducted to develop an assay suitable for high-throughput antiviral screening. Three common human cell lines (HEK 293T, A549 and HeLa cells) alongside Vero E6 cells were infected at various MOIs to determine which would yield the highest luciferase signal over the course of infection (Fig. 2). Non-infected cells were used as negative controls. An increase in luciferase signal from 0 hpi to 24 hpi for all cell lines, irrespective of MOI tested was recorded. HEK 293T cells infected at an MOI of 0.01 displayed the highest luciferase signal at all-time points examined, with peak luminescence of 4.7 × 107 RLU at 48 hpi followed by a slight decrease to 2.2 × 107 RLU by 72 hpi (Fig. 2A). Infected HeLa and A549 cells displayed maximum luciferase activity at 48 hpi as well, but at levels 1 – 2 logs lower when compared to the infected HEK 293T or Vero E6 cells. A similar trend was observed when cells were infected at an MOI of 0.1 (Fig. 2B). At the highest MOI examined, however, luciferase activity peaked at 24 hpi in all cell lines followed by a steady decrease by 72 hpi (Fig. 2C), suggesting rapid cell death due to CPE caused by the high concentration of virus used. Since the highest luciferase signal was observed in HEK 293T cells infected with the lowest MOI tested (0.01) for 48 h and because of the minimal difference in signal between 48 and 72 hpi, these parameters were chosen for subsequent assays.
Fig. 2.
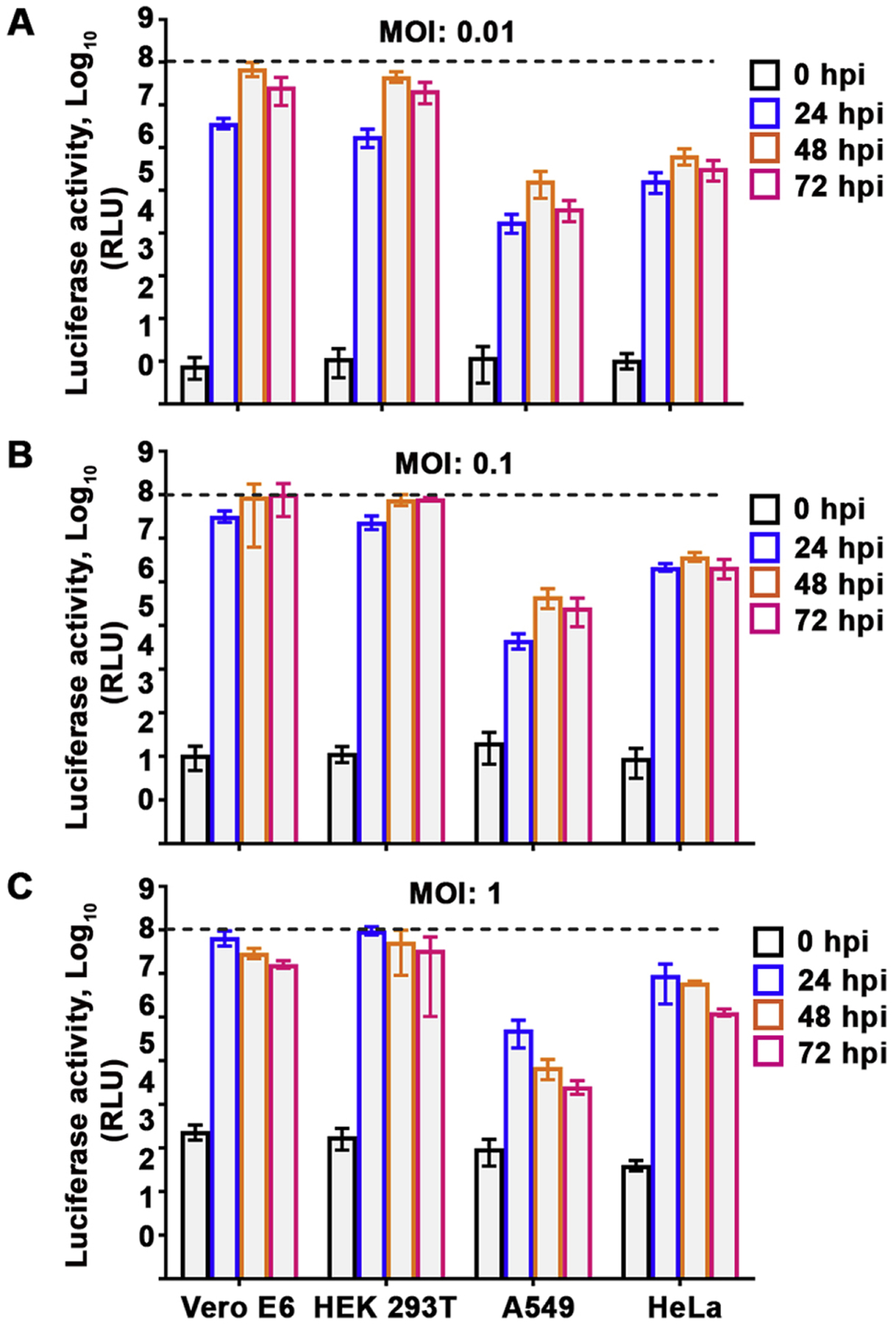
High-throughput screening parameters. Vero E6, HEK 293T, A549 and HeLa cells pre-seeded in a white opaque 96-well cell culture plate were either mock infected or infected with rCedV-Luc at MOI 0.01 (A), 0.1 (B) or 1 (C). At 0, 24, 48 and 72 hpi, cells were lysed and relative light units (RLU) in the rCedV-Luc infected cells were measured and normalized by subtracting the signal of mock infected cells from the signal of the rCedV-Luc infected cells. The data represent mean ± standard error from three independent experiments. Luciferase activity at 0 hpi indicate background levels of luminescence. The black dashed line represents the upper limit of detection of the luminometer.
We hypothesized that compounds capable of inhibiting rCedV-Luc can be identified by a reduction in luminescent signal compared to the DMSO control. It was previously shown that the CedV G attachment glycoprotein specific mouse mAb m14F3 inhibits rCedV entry into HeLa-USU cells (Laing et al., 2018), therefore mAb m14F3 was selected to serve as a suitable positive control for inhibition of rCedV luciferase activity. As shown in Fig. 3, incubation of rCedV-Luc with m14F3 (20 μg/ml) decreased luciferase activity by 4 logs, when compared to untreated infected cells. Thus, m14F3 can be used as a relevant control for virus entry inhibition.
Fig. 3.
Luciferase-expressing recombinant CedV as a tool for antiviral testing. rCedV-Luc (MOI: 0.01) was incubated with medium or an equal volume of m14F3 diluted in medium at a final concentration of 20 μg/ml for 1 h at 37 °C. The antibody-virus or virus only mixtures were then added to HEK 293T cells pre-seeded in a white opaque 96-well cell culture plate, such that 16 wells contained rCedV-Luc and 16 wells contained rCedV-Luc with m14F3. Mock infected cells were included as an additional control. The plate was incubated for 1 h at 37 °C, at which time the antibody-virus or virus only mixtures were removed, cells were washed once with medium, and fresh medium only or fresh medium containing 20 μg/ml m14F3 was added to the cells. After a 48 h incubation, all cells were lysed and relative light units (RLU) in the rCedV-Luc infected cells were measured. The data represent mean ± standard error from two independent experiments. * P<0.01.
Shown in Table 1 are the results of further optimization of the HTS assay and resulting parameters in both 96- and 384-well formats. Z’ scores ≥ 0.5, high S/B ratios and CV < 20% were recorded, indicating a robust sensitive high-throughput assay. These data demonstrated that the rCedV-Luc platform is suitable for HTS-based hit identification.
Table 1.
High throughput screening assay parameters
| Assay Parameters | 96-well format | 384-well format |
|---|---|---|
| Z’ | 0.6 ± 0.02 | 0.5 ± 0.2 |
| %CV | 13.3 ± 1.14 | 14 ± 0.05 |
| S/B | > 103 | > 40 |
3.3. High-throughput-screening for identifying rCedV-Luc inhibitors
A small molecule library of 10,000 compounds plated in a 384-well format was screened at 10 μM, as illustrated in Fig. 4. The data were normalized by luciferase signals from control wells (rCedV-Luc with DMSO vehicle). From the initial single-dose screen, using 80% inhibition as the cutoff, 151 hits (1.51% hit rate) were selected for further evaluation. To eliminate false positive hits due to cytotoxicity, a validation screen alongside a cellular toxicity assay (compound concentration of 33 μM) was carried out. The antiviral activities of 85 compounds were confirmed at 10 μM, and 47 hits showed only minimal cytotoxicity at 33 μM. Table 2 categorizes the resulting 47 nontoxic hits into 6 clusters (A-F), each of which includes at least 2 compounds with similar chemical structures, and group Z, which contains compounds with distinct scaffolds. Based on the compounds’ structures, antiviral potency and commercial availability, 8 compounds were prioritized for further evaluation. The chemical structures of the select compounds are shown in Fig. 5.
Fig. 4.
HTS workflow. Compounds diluted in medium were added to pre-seeded HEK 293T cells in a 384-well plate, followed by addition of rCedV-Luc (MOI: 0.05). Luminescence was measured 48 h later.
Table 2.
Top hit classification based on compound chemical structures
| Confirmed hits | For further evaluation | |
|---|---|---|
| Number of compounds | Number of compounds (ID) | |
| Hit cluster (A) | 6 | 3 (A1, A2, A3) |
| Hit cluster (B) | 6 | 2 (B1, B2) |
| Hit cluster (C) | 8 | 1 (C1) |
| Hit cluster (D) | 4 | 0 |
| Hit cluster (E) | 3 | 0 |
| Hit cluster (F) | 2 | 1 (F1) |
| Compounds with diverse scaffolds (Z) | 18 | 1 (Z) |
| Total | 47 | 8 |
Fig. 5.
Structures of the selected compounds used in the screening assays against rCedV-Luc infection in HEK 293T and Vero E6 cell lines. A1: 3-(1-(6-(cyclopropylamino)pyrimidin-4-yl)piperidin-4-yl)-N-(pyridin-3-ylmethyl)propenamide; A2: 3-(1-(6-(cyclopropylamino)pyrimidin-4-yl)piperidin-4-yl)-N-(2,4-difluorobenzyl)propenamide; A3: (1-(6-(cyclopropylamino)pyrimidin-4-yl)piperidin-4-yl)-N-(2-(thiophen-2-yl)ethyl)propenamide; B1: N-(3,4-dimethoxyphenyl)-3-(3-(4-(pyrrolidin-1-yl)phenyl)-1,2,4-oxadiazol-5-yl)propenamide; B2: N-(3-morpholinopropyl)-3-(3-(4-(pyrrolidin-1-yl)phenyl)-1,2,4-oxadiazol-5-yl)propenamide; C1: cyclobutyl(1-(4-ethoxyphenyl)-1,3,4,9-tetrahydro-2H-pyrido[3,4-b]indol-2-yl)methanone; F1: cyclobutyl(1-(4-ethoxyphenyl)-1,3,4,9-tetrahydro-2H-pyrido[3,4-b]indol-2-yl)methanone; Z: 3-(1-(4-bromo-2-fluorophenyl)-5-methyl-1H-1,2,3-triazol-4-yl)-N-(4-methoxyphenyl)-1,2,4-thiadiazol-5-amine.
The selected hits were assessed in 9-point dose-response titration assays at concentrations ranging from 15 nM to 100 μM in both HEK 293T cells and Vero E6 cells (Figs. 6 and S1). The IC50 and CC50 values of these compounds are summarized in Table 3. These inhibitors generally exhibited stronger antiviral activities against rCedV-Luc infection when tested in HEK 293T cells as compared with Vero E6 cells. Almost all compounds had no cytotoxicity in both cell lines, except F1, which displayed mild toxicity at 100 μM only in HEK 293T cells, with the estimated CC50 value of 85.8 μM. Remarkably, compounds with similar scaffolds shared similar antiviral profiles, as cluster A compounds (A1, A2, A3) had IC50 values of ~0.3 μM and cluster B compounds (B1, B2) had IC50 values of ~0.6 μM in HEK 293T cells. The most potent was compound Z with an IC50 value of 0.13 μM in HEK 293T cells and an IC50 value of 0.41 μM in Vero E6 cells. Overall, our lead compounds displayed IC50 values ≤ 12 μM with minimal cytotoxicity.
Fig. 6.
Anti-rCedV activities of lead compounds in HEK 293T and Vero E6 cells. Dose-response titrations were performed in HEK 293T (blue square) and Vero E6 (red diamond) cell lines against rCedV-Luc infection for lead compounds listed in Table 3. HEK 293T cells (2 × 104 cells/well) or Vero E6 cells (1.5 × 104 cells/well) were seeded in 96-well plates. The next day, the cells were infected with rCedV-Luc (MOI: 0.05) in the presence of increasing concentrations of compounds (15 nM to 100 μM; 1% DMSO). The virus infection was quantified after a 48 h incubation by measuring the luciferase activity with the Neolite reporter gene assay system. Sample signals were normalized by signals from the DMSO control wells. The data represent mean ± standard error from three independent experiments.
Table 3.
Antiviral profiles of 11 compounds against rCedV-Luc infection in HEK 293T and Vero E6 cell lines
| Comp. ID | IC50 (95% CI) (μM) | CC50 (95% CI) (μM) | Selectivity index (SI) | |||
|---|---|---|---|---|---|---|
| HEK 293T | Vero E6 | HEK 293T | Vero E6 | HEK 293T | Vero E6 | |
| A1 | 0.37 (0.33 to 0.41) | 2.37 (1.69 to 3.31) | >100 | >100 | >270 | >42.0 |
| A2 | 0.29 (0.25 to 0.32) | 0.73 (0.47 to 1.15) | >100 | >100 | >344 | >137 |
| A3 | 0.29 (0.26 to 0.31) | 1.29 (0.85 to 1.95) | >100 | >100 | >344 | >78.0 |
| B1 | 0.58 (0.40 to 0.85) | 0.35 (0.16 to 0.77) | >100 | >100 | >172 | >286 |
| B2 | 0.70 (0.58 to 0.86) | 1.58 (0.96 to 2.58) | >100 | >100 | >142 | >63.0 |
| C1 | 3.06 (2.72 to 3.45) | 7.82 (4.28 to 14.3) | >100 | >100 | >33.0 | >12.8 |
| F1 | 10.0 (9.29 to 10.7) | 11.8 (8.70 to 16.1) | 85.8 (59.1 to 124) | >100 | 8.58 | >8.50 |
| Z | 0.13 (0.07 to 0.22) | 0.41 (0.18 to 0.96) | >100 | >100 | >769 | >244 |
Note: All IC50 and CC50 values are estimated by nonlinear fit model from three independent experiments and shown as estimated values with 95% confidence interval (95% CI).
3.4. Inhibitory activity of hit compounds on henipavirus replication
The inhibitory activity of the select compounds on henipavirus replication was determined by viral titer reduction assay. HEK 293T cells treated with a concentration 10 times exceeding the IC50 concentration of each of the compounds were infected with rCedV, and collected supernatants were analyzed for infectious viral titers by plaque assay. A decrease, to varying degrees, in rCedV viral progeny production was recorded in all treated cells, in comparison to the DMSO control (Fig. 7). The 8 select compounds demonstrating inhibitory effects on rCedV infection and replication were then tested in viral titer reduction assays against infectious NiV-B. At a concentration of 11.1 μM, 4 out of 8 compounds reduced infectious NiV-B viral titers by 50% when compared to the DMSO control (Fig. 8A), indicating that inhibition of NiV-B viral titers paralleled inhibition of rCedV viral titers. Two of the validated compounds (C1 and F1) were then selected for further evaluation in a 9-point dose response titration with infectious NiV-B (Fig. 8B) and infectious HeV (Fig. 8C). Compounds C1 (cyclobutyl(1-(4-ethoxyphenyl)-1,3,4,9-tetrahydro-2H-pyrido[3,4-b]indol-2-yl)methanone) and F1 (cyclobutyl(1-(4-ethoxyphenyl)-1,3,4,9-tetrahydro-2H-pyrido[3,4-b]indol-2-yl)methanone) exhibited potent antiviral activity against NiV-B with IC50 values of 1.83 μM (SI >55) and 4.15 μM (SI >24), respectively (Table 4). Potent antiviral activity of compounds C1 (cyclobutyl(1-(4-ethoxyphenyl)-1,3,4,9-tetrahydro-2H-pyrido[3,4-b]indol-2-yl)methanone) and F1 (cyclobutyl(1-(4-ethoxyphenyl)-1,3,4,9-tetrahydro-2H-pyrido[3,4-b]indol-2-yl)methanone) was also observed against HeV with IC50 values of 9.87 μM (SI >10) and 19.6 μM (SI >5), respectively (Table 4), albeit with a ~5-fold lower potency in comparison to NiV-B. These data suggest that the tested compounds may be inhibiting henipavirus replication at various stages of the viral life cycle.
Fig. 7.
Lead compounds inhibit rCedV replication. HEK 293T cells seeded in a 96-well plate were treated for 1 h at the concentration 10 times exceeding the IC50 concentration of the selected compounds or DMSO. Medium only or medium containing rCedV (MOI: 0.05) was added to the cells. Viral supernatants were collected 48 hpi and titrated by plaque assay to determine infectious virus titer. The data represent mean ± standard error from two independent experiments indicating percent reduction in virus titer relative to the DMSO control (% Reduction in virus titer).
Fig. 8.
Inhibition of NiV-B and HeV replication by virus titer reduction assay. A) Compounds at 11.1 μM were first added to pre-seeded Vero 76 cells in a 96-well plate, followed by NiV-B infection at MOI of 0.01. Viral supernatants were collected 48 hpi and titrated by plaque assay to determine infectious virus titer. Values represent means and standard deviations indicating percent reduction in virus titer relative to the DMSO control (% Reduction in virus titer). The graph is representative of an independent experiment performed in duplicate (mean ± SD). The black dashed line represents 50% infection. Compounds C1 (blue square) and F1 (red diamond) were evaluated using NiV-B (B) or HeV (C) by virus titer reduction assay as in (A) at MOI of 0.01 by dose-response titration (15 nM to 100 μM; 1% DMSO). The data represent mean ± SD from a single experiment performed in duplicate.
Table 4.
Antiviral profiles of selected hits against NiV-B and HeV infection in Vero 76 cell line
| NiV-B | HeV | |||||
|---|---|---|---|---|---|---|
| Compound ID | IC50 (95% CI) (μM) | CC50 (μM) | Selectivity index (SI) | IC50 (95% CI) (μM) | CC50 (μM) | Selectivity index (SI) |
| C1 | 1.83 (0.96 to 3.50) | > 100 | > 55 | 9.87 (6.06 to 16.1) | > 100 | > 10 |
| F1 | 4.15 (1.89 to 9.07) | > 100 | > 24 | 19.6 (3.48 to 110) | > 100 | > 5 |
Note: All IC50 values are estimated by nonlinear fit model from one experiment with duplicates and shown as estimated values with 95% confidence interval (95% CI).
3.5. Select compounds inhibit a post-entry viral replication stage
A quantitative split-luciferase based reporter assay (Laing et al., 2019; Navaratnarajah et al., 2016) was used to determine if virus entry/fusion was specifically affected by the compounds. In this assay, content mixing between CHO-K1 effector cells expressing CedV recombinant F and G proteins (the rCedV fusion complex) and CHO-B1 target cells allows the two halves of Renilla luciferase to functionally interact. As shown in Fig. 9, selected compounds C1 and F1 did not inhibit cell-cell membrane fusion activity. These data suggest that these two compounds identified using the rCedV-Luc platform may in fact inhibit subsequent viral replication stages. Taken together, these data effectively demonstrate that rCedV-Luc is a suitable surrogate for antiviral discovery targeting NiV and further suggest a lower likelihood of detecting false positives in the context of HTS using the rCedV-Luc platform.
Fig. 9.
Select compounds do not inhibit fusion. CHO-B1 cells in a 96-well plate were transfected with a plasmid encoding one half of a split-luciferase reporter protein. Concurrently CHO-K1 cells in a 6-well were transfected with CedV F and G glycoproteins and with the other half of the split-luciferase. Twenty-four h post transfection the cells were re-suspended and overlaid on the receptor expressing CHO-K1 cells in the 96-well plate. Compounds C1 or F1 were added to a final concentration that ranged between 5 μM to 20 μM. The live-cell luciferase substrate EnduRen was used to monitor the level of cell-cell fusion at the indicated time points. The data represent mean ± standard error from three technical replicates.
4. Discussion
Here, an authentic replication-competent henipavirus assay system using rCedV was developed to safely and rapidly identify small molecule antivirals targeting henipavirus replication without the requirement for BSL-4 containment. The rCedV reporter-gene tool was adapted to a high-throughput screening assay that enabled the identification of compounds inhibiting henipavirus replication. Selected compounds that could inhibit rCedV also inhibited NiV-B replication. Using reverse genetics methods a rCedV encoding luciferase (rCedV-Luc) was rescued and characterized and the reporter signal kinetics determined in different human cell lines. The rCedV-Luc reporter-gene assay was optimized with Z’ ≥ 0.5, high S/B ratios and CV ≤ 20%, demonstrating a sensitive and robust assay suitable for HTS-based hit identification.
Using a cut-off of 80% decrease in luminescence, an initial HTS inhibitor hit rate of 1.51% was recorded and several compounds that inhibited rCedV replication and virus production were identified, and of these, 4 compounds were then shown to have antiviral activity against wild-type NiV-B. Compounds C1 and F1 were selected for further characterization and inhibited the replication of NiV-B by 69% and 56%, respectively in virus titer reduction assays, and were ~ 5-fold less effective against HeV. The compounds C1 and F1 demonstrated high antiviral potency against infectious NiV-B and HeV with low cytotoxicity values and inhibit a post-membrane fusion step of the viral replication cycle. Compounds C1 and F1 exhibited similar antiviral profiles between rCedV, NiV-B and HeV virus titer reduction assays suggesting that the inhibitory mechanism may be similar, such as targeting the viral polymerase complex.
A select few HTS platforms have been developed for henipavirus antiviral drug discovery suitable for use at BSL-2 (Porotto et al., 2009; Talekar et al., 2012). However, they only target specific stages of the replication cycle. A vesicular stomatitis virus (VSV) pseudotyped red fluorescent protein-expressing virus platform using the HeV or NiV G and F glycoproteins was used to identify inhibitors of the viral entry and/or cell-cell fusion stages (Porotto et al., 2009; Talekar et al., 2012). The Z’ values obtained in these studies (Z’ = 0.63 (Porotto et al., 2009) and Z’ = 0.72 (Talekar et al., 2012)) were comparable to the Z’ value reported here using the rCedV-Luc assay which can potentially identify inhibitors targeting any stage of henipavirus infection and replication. Also, a NiV mini-genome reporter gene assay that screened for inhibitors targeting transcription and replication by an enzymatic approach has been used (chloramphenicol acetyltransferase (CAT) enzyme-linked immunosorbent assay (ELISA)) (Freiberg et al., 2008). In contrast, the rCedV-based platform detailed here is as an authentic replication-competent henipavirus, recapitulating all stages of the henipavirus replication cycle. The rCedV assay system thus provides an opportunity to identify compounds that inhibit both virus entry and post-entry replication stages. In addition, the assay requires minimal manipulation and low sample volumes, and an automated endpoint detection is specific and selective, adaptable to a HTS approach with a fast turnaround time, and one that can be performed outside BSL-4 containment.
Taken together, the applicability of the rCedV-based platform as a robust henipavirus antiviral screening tool has been demonstrated. Although further target identification studies are warranted, the present data support the use of this assay as a suitable HTS platform for the identification of potential pan-henipavirus antiviral molecules. The compounds identified here will serve as suitable starting points for future viral specificity studies, mode of action evaluation and medicinal chemistry optimization, which together may lead to the development of effective therapeutics against NiV and HeV.
Supplementary Material
Highlights.
A high-throughput screening assay using the Henipavirus, Cedar virus, encoding luciferase (rCedV-Luc) was established.
A large diverse small molecule library was screened for compounds that inhibited rCedV-Luc infection and replication.
4 compounds that inhibited rCedV replication also inhibited authentic Nipah virus replication.
2 compounds had IC50 concentrations of less than 10 μM for Nipah virus and less than 20 μM for Hendra virus.
rCedV-Luc is an authentic henipavirus platform that will accelerate the development of anti-henipavirus countermeasures.
Acknowledgments
The views expressed in this manuscript are solely those of the authors, and they do not represent official views or opinions of the Department of Defense or The Uniformed Services University of the Health Sciences. We thank Dr. B. Schaeffer for providing the pGL4-32-Luc2P plasmid and the UIC RRC for providing the screening library. We thank Glenn A. Marsh for kindly providing the pOLTV5 expression vector.
Funding
This work was partially supported by National Institutes of Health (NIH) grant AI137813 (CCB and LR) and AI157095 (LR and CCB). Operations support of the Galveston National Laboratory was funded by NIAID/NIH grant UC7AI094660.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interests
CCB is a US federal employee and co-inventor on US and foreign patents pertaining to Cedar Virus and Methods of Use, whose assignees are the US as represented by the Henry M Jackson Foundation for the Advancement of Military Medicine (Bethesda, MD). All other authors declare no competing interests.
References
- Amaya M, Broder CC, 2020. Vaccines to Emerging Viruses: Nipah and Hendra. Annu Rev Virol 7, 447–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anonymous, 2018. Henipavirus Gap Analysis Workshop Report. U.S. Department of Agriculture, Agricultural Research Service, Washington, DC. Available at http://go.usa.gov/xnHgR. [Google Scholar]
- Anonymous, 2019. Hendra virus - Australia: (New South Wales) horse, Pro-med. International Society for Infectious Diseases. June 19. archive no. 20190621.6531307. Available at www.promedmail.org.
- Anonymous, 2020a. Hendra virus - Australia: (New South Wales) Horse, Pro-Med. International Society for Infectious Diseases. June 2. archive no. 20200602.7420665. Available at www.promedmail.org.
- Anonymous, 2020b. Nipah virus - Bangladesh: (Khulna), Pro-Med. International Society for Infectious Diseases. January 19. archive no. 20200119.6897664. Available at www.promedmail.org.
- Arunkumar G, Chandni R, Mourya DT, Singh SK, Sadanandan R, Sudan P, Bhargava B, Nipah Investigators P, Health Study G, 2019. Outbreak Investigation of Nipah Virus Disease in Kerala, India, 2018. The Journal of Infectious Diseases 219, 1867–1878. [DOI] [PubMed] [Google Scholar]
- Baell JB, Holloway GA, 2010. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- Bishop KA, Stantchev TS, Hickey AC, Khetawat D, Bossart KN, Krasnoperov V, Gill P, Feng YR, Wang L, Eaton BT, Wang LF, Broder CC, 2007. Identification of hendra virus G glycoprotein residues that are critical for receptor binding. J Virol 81, 5893–5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaparte MI, Dimitrov AS, Bossart KN, Crameri G, Mungall BA, Bishop KA, Choudhry V, Dimitrov DS, Wang LF, Eaton BT, Broder CC, 2005. Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc Natl Acad Sci U S A 102, 10652–10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broder CC, Weir DL, Reid PA, 2016. Hendra virus and Nipah virus animal vaccines. Vaccine 34, 3525–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz UJ, Finke S, Conzelmann KK, 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J Virol 73, 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching PK, de Los Reyes VC, Sucaldito MN, Tayag E, Columna-Vingno AB, Malbas FF Jr., Bolo GC Jr., Sejvar JJ, Eagles D, Playford G, Dueger E, Kaku Y, Morikawa S, Kuroda M, Marsh GA, McCullough S, Foxwell AR, 2015. Outbreak of henipavirus infection, Philippines, 2014. Emerging Infectious Diseases 21, 328–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, Lam SK, Ksiazek TG, Rollin PE, Zaki SR, Shieh W, Goldsmith CS, Gubler DJ, Roehrig JT, Eaton B, Gould AR, Olson J, Field H, Daniels P, Ling AE, Peters CJ, Anderson LJ, Mahy BW, 2000. Nipah virus: a recently emergent deadly paramyxovirus. Science 288, 1432–1435. [DOI] [PubMed] [Google Scholar]
- Chua KB, Goh KJ, Wong KT, Kamarulzaman A, Tan PS, Ksiazek TG, Zaki SR, Paul G, Lam SK, Tan CT, 1999. Fatal encephalitis due to Nipah virus among pig-farmers in Malaysia. Lancet 354, 1257–1259. [DOI] [PubMed] [Google Scholar]
- Chua KB, Koh CL, Hooi PS, Wee KF, Khong JH, Chua BH, Chan YP, Lim ME, Lam SK, 2002. Isolation of Nipah virus from Malaysian Island flying-foxes. Microbes Infect 4, 145–151. [DOI] [PubMed] [Google Scholar]
- Ciancanelli MJ, Volchkova VA, Shaw ML, Volchkov VE, Basler CF, 2009. Nipah virus sequesters inactive STAT1 in the nucleus via a P gene-encoded mechanism. J Virol 83, 7828–7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton BA, Wang LF, Marsh GA, 2013. Henipaviruses: an updated review focusing on the pteropid reservoir and features of transmission. Zoonoses and Public Health 60, 69–83. [DOI] [PubMed] [Google Scholar]
- Freiberg A, Dolores LK, Enterlein S, Flick R, 2008. Establishment and characterization of plasmid-driven minigenome rescue systems for Nipah virus: RNA polymerase I- and T7-catalyzed generation of functional paramyxoviral RNA. Virology 370, 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin K, Hyatt AD, Fogarty R, Middleton D, Bingham J, Epstein JH, Rahman SA, Hughes T, Smith C, Field HE, Daszak P, The H, 2011. Pteropid Bats are Confirmed as the Reservoir Hosts of Henipaviruses: A Comprehensive Experimental Study of Virus Transmission. Am J Trop Med Hyg 85, 946–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin K, Young PL, Field HE, Mackenzie JS, 2000. Isolation of Hendra virus from pteropid bats: a natural reservoir of Hendra virus. J Gen Virol 81, 1927–1932. [DOI] [PubMed] [Google Scholar]
- Harcourt BH, Lowe L, Tamin A, Liu X, Bankamp B, Bowden N, Rollin PE, Comer JA, Ksiazek TG, Hossain MJ, Gurley ES, Breiman RF, Bellini WJ, Rota PA, 2005. Genetic characterization of Nipah virus, Bangladesh, 2004. Emerging Infectious Diseases 11, 1594–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harit AK, Ichhpujani RL, Gupta S, Gill KS, Lal S, Ganguly NK, Agarwal SP, 2006. Nipah/Hendra virus outbreak in Siliguri, West Bengal, India in 2001. Indian J Med Res 123, 553–560. [PubMed] [Google Scholar]
- Homaira N, Rahman M, Hossain MJ, Epstein JH, Sultana R, Khan MS, Podder G, Nahar K, Ahmed B, Gurley ES, Daszak P, Lipkin WI, Rollin PE, Comer JA, Ksiazek TG, Luby SP, 2010a. Nipah virus outbreak with person-to-person transmission in a district of Bangladesh, 2007. Epidemiol Infect 138, 1630–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homaira N, Rahman M, Hossain MJ, Nahar N, Khan R, Podder G, Nahar K, Khan D, Gurley ES, Rollin PE, Comer JA, Ksiazek TG, Luby SP, 2010b. Cluster of Nipah virus infection, Kushtia District, Bangladesh, 2007. PLoS One 5, e13570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu VP, Hossain MJ, Parashar UD, Ali MM, Ksiazek TG, Kuzmin I, Niezgoda M, Rupprecht C, Bresee J, Breiman RF, 2004. Nipah virus encephalitis reemergence, Bangladesh. Emerging Infectious Diseases 10, 2082–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglese J, Johnson RL, Simeonov A, Xia M, Zheng W, Austin CP, Auld DS, 2007. High-throughput screening assays for the identification of chemical probes. Nat Chem Biol 3, 466–479. [DOI] [PubMed] [Google Scholar]
- Iversen PW, Beck B, Chen YF, Dere W, Devanarayan V, Eastwood BJ, Farmen MW, Iturria SJ, Montrose C, Moore RA, Weidner JR, Sittampalam GS, 2004. HTS Assay Validation, in: Sittampalam GS, Grossman A, Brimacombe K, Arkin M, Auld D, Austin C, Baell J, Bejcek B, Caaveiro JMM, Chung TDY, Coussens NP, Dahlin JL, Devanaryan V, Foley TL, Glicksman M, Hall MD, Haas JV, Hoare SRJ, Inglese J, Iversen PW, Kahl SD, Kales SC, Kirshner S, Lal-Nag M, Li Z, McGee J, McManus O, Riss T, Trask OJ Jr., Weidner JR, Wildey MJ, Xia M, Xu X (Eds.), Assay Guidance Manual, Bethesda (MD). [Google Scholar]
- Kulkarni S, Volchkova V, Basler CF, Palese P, Volchkov VE, Shaw ML, 2009. Nipah virus edits its P gene at high frequency to express the V and W proteins. J Virol 83, 3982–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing ED, Amaya M, Navaratnarajah CK, Feng YR, Cattaneo R, Wang LF, Broder CC, 2018. Rescue and characterization of recombinant cedar virus, a non-pathogenic Henipavirus species. Virology Journal 15, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing ED, Navaratnarajah CK, Cheliout Da Silva S, Petzing SR, Xu Y, Sterling SL, Marsh GA, Wang LF, Amaya M, Nikolov DB, Cattaneo R, Broder CC, Xu K, 2019. Structural and functional analyses reveal promiscuous and species specific use of ephrin receptors by Cedar virus. Proc Natl Acad Sci U S A 116, 20707–20715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieu KG, Marsh GA, Wang LF, Netter HJ, 2015. The non-pathogenic Henipavirus Cedar paramyxovirus phosphoprotein has a compromised ability to target STAT1 and STAT2. Antiviral Research 124, 69–76. [DOI] [PubMed] [Google Scholar]
- Luby SP, Hossain MJ, Gurley ES, Ahmed BN, Banu S, Khan SU, Homaira N, Rota PA, Rollin PE, Comer JA, Kenah E, Ksiazek TG, Rahman M, 2009. Recurrent zoonotic transmission of Nipah virus into humans, Bangladesh, 2001–2007. Emerging Infectious Diseases 15, 1229–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh GA, de Jong C, Barr JA, Tachedjian M, Smith C, Middleton D, Yu M, Todd S, Foord AJ, Haring V, Payne J, Robinson R, Broz I, Crameri G, Field HE, Wang LF, 2012. Cedar virus: a novel Henipavirus isolated from Australian bats. PLoS Pathogens 8, e1002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton D, Pallister J, Klein R, Feng YR, Haining J, Arkinstall R, Frazer L, Huang JA, Edwards N, Wareing M, Elhay M, Hashmi Z, Bingham J, Yamada M, Johnson D, White J, Foord A, Heine HG, Marsh GA, Broder CC, Wang LF, 2014. Hendra virus vaccine, a one health approach to protecting horse, human, and environmental health. Emerging Infectious Diseases 20, 372–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mire CE, Chan YP, Borisevich V, Cross RW, Yan L, Agans KN, Dang HV, Veesler D, Fenton KA, Geisbert TW, Broder CC, 2020. A Cross-Reactive Humanized Monoclonal Antibody Targeting Fusion Glycoprotein Function Protects Ferrets Against Lethal Nipah Virus and Hendra Virus Infection. The Journal of Infectious Diseases 221, S471–S479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mire CE, Satterfield BA, Geisbert JB, Agans KN, Borisevich V, Yan L, Chan YP, Cross RW, Fenton KA, Broder CC, Geisbert TW, 2016. Pathogenic Differences between Nipah Virus Bangladesh and Malaysia Strains in Primates: Implications for Antibody Therapy. Scientific Reports 6, 30916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray K, Selleck P, Hooper P, Hyatt A, Gould A, Gleeson L, Westbury H, Hiley L, Selvey L, Rodwell B, et al. , 1995. A morbillivirus that caused fatal disease in horses and humans. Science 268, 94–97. [DOI] [PubMed] [Google Scholar]
- Navaratnarajah CK, Rosemarie Q, Cattaneo R, 2016. A Structurally Unresolved Head Segment of Defined Length Favors Proper Measles Virus Hemagglutinin Tetramerization and Efficient Membrane Fusion Triggering. J Virol 90, 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrete OA, Levroney EL, Aguilar HC, Bertolotti-Ciarlet A, Nazarian R, Tajyar S, Lee B, 2005. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature 436, 401–405. [DOI] [PubMed] [Google Scholar]
- Negrete OA, Wolf MC, Aguilar HC, Enterlein S, Wang W, Muhlberger E, Su SV, Bertolotti-Ciarlet A, Flick R, Lee B, 2006. Two key residues in ephrinB3 are critical for its use as an alternative receptor for Nipah virus. PLoS Pathogens 2, e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolay B, Salje H, Hossain MJ, Khan A, Sazzad HMS, Rahman M, Daszak P, Stroher U, Pulliam JRC, Kilpatrick AM, Nichol ST, Klena JD, Sultana S, Afroj S, Luby SP, Cauchemez S, Gurley ES, 2019. Transmission of Nipah Virus - 14 Years of Investigations in Bangladesh. N Engl J Med 380, 1804–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller CK, Mastorakos GM, Matchett WE, Ma X, Samuel CE, Cattaneo R, 2015. Measles Virus Defective Interfering RNAs Are Generated Frequently and Early in the Absence of C Protein and Can Be Destabilized by Adenosine Deaminase Acting on RNA-1-Like Hypermutations. J Virol 89, 7735–7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Playford EG, Munro T, Mahler SM, Elliott S, Gerometta M, Hoger KL, Jones ML, Griffin P, Lynch KD, Carroll H, El Saadi D, Gilmour ME, Hughes B, Hughes K, Huang E, de Bakker C, Klein R, Scher MG, Smith IL, Wang LF, Lambert SB, Dimitrov DS, Gray PP, Broder CC, 2020. Safety, tolerability, pharmacokinetics, and immunogenicity of a human monoclonal antibody targeting the G glycoprotein of henipaviruses in healthy adults: a first-in-human, randomised, controlled, phase 1 study. The Lancet. Infectious Diseases 20, 445–454. [DOI] [PubMed] [Google Scholar]
- Porotto M, Orefice G, Yokoyama CC, Mungall BA, Realubit R, Sganga ML, Aljofan M, Whitt M, Glickman F, Moscona A, 2009. Simulating henipavirus multicycle replication in a screening assay leads to identification of a promising candidate for therapy. J Virol 83, 5148–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MA, Hossain MJ, Sultana S, Homaira N, Khan SU, Rahman M, Gurley ES, Rollin PE, Lo MK, Comer JA, Lowe L, Rota PA, Ksiazek TG, Kenah E, Sharker Y, Luby SP, 2012. Date palm sap linked to Nipah virus outbreak in Bangladesh, 2008. Vector Borne and Zoonotic Diseases (Larchmont, N.Y.) 12, 65–72. [DOI] [PubMed] [Google Scholar]
- Schountz T, Campbell C, Wagner K, Rovnak J, Martellaro C, DeBuysscher BL, Feldmann H, Prescott J, 2019. Differential Innate Immune Responses Elicited by Nipah Virus and Cedar Virus Correlate with Disparate In Vivo Pathogenesis in Hamsters. Viruses.11(3):291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvey LA, Wells RM, McCormack JG, Ansford AJ, Murray K, Rogers RJ, Lavercombe PS, Selleck P, Sheridan JW, 1995. Infection of humans and horses by a newly described morbillivirus. Med J Aust 162, 642–645. [DOI] [PubMed] [Google Scholar]
- Shaw ML, 2009. Henipaviruses employ a multifaceted approach to evade the antiviral interferon response. Viruses 1, 1190–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw ML, Garcia-Sastre A, Palese P, Basler CF, 2004. Nipah virus V and W proteins have a common STAT1-binding domain yet inhibit STAT1 activation from the cytoplasmic and nuclear compartments, respectively. J Virol 78, 5633–5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweileh WM, 2017. Global research trends of World Health Organization’s top eight emerging pathogens. Global Health 13, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talekar A, Pessi A, Glickman F, Sengupta U, Briese T, Whitt MA, Mathieu C, Horvat B, Moscona A, Porotto M, 2012. Rapid screening for entry inhibitors of highly pathogenic viruses under low-level biocontainment. PLoS One 7, e30538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S, Horie R, Sato H, Kai C, Yoneda M, 2018. Possible role of the Nipah virus V protein in the regulation of the interferon beta induction by interacting with UBX domain-containing protein1. Scientific Reports 8, 7682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingartl HM, Berhane Y, Caswell JL, Loosmore S, Audonnet JC, Roth JA, Czub M, 2006. Recombinant nipah virus vaccines protect pigs against challenge. J Virol 80, 7929–7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav PD, Shete AM, Kumar GA, Sarkale P, Sahay RR, Radhakrishnan C, Lakra R, Pardeshi P, Gupta N, Gangakhedkar RR, Rajendran VR, Sadanandan R, Mourya DT, 2019. Nipah Virus Sequences from Humans and Bats during Nipah Outbreak, Kerala, India, 2018. Emerging Infectious Diseases 25, 1003–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR, 1999. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 4, 67–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.